

Abstract
Actinobacillus pleuropneumoniae is a Gram-negative bacterium belonging to the Pasteurellaceae family and the causative agent of porcine pleuropneumonia, a highly contagious lung disease causing important economic losses. Surface polysaccharides, including lipopolysaccharides (LPS) and capsular polysaccharides (CPS), are implicated in the adhesion and virulence of A. pleuropneumoniae, but their role in biofilm formation is still unclear. In this study, we investigated the requirement for these surface polysaccharides in biofilm formation by A. pleuropneumoniae serotype 1. Well-characterized mutants were used: an O-antigen LPS mutant, a truncated core LPS mutant with an intact O antigen, a capsule mutant, and a poly-N-acetylglucosamine (PGA) mutant. We compared the amount of biofilm produced by the parental strain and the isogenic mutants using static and dynamic systems. Compared to the findings for the biofilm of the parental or other strains, the biofilm of the O antigen and the PGA mutants was dramatically reduced, and it had less cell-associated PGA. Real-time PCR analyses revealed a significant reduction in the level of pgaA, cpxR, and cpxA mRNA in the biofilm cells of the O-antigen mutant compared to that in the biofilm cells of the parental strain. Specific binding between PGA and LPS was consistently detected by surface plasmon resonance, but the lack of O antigen did not abolish these interactions. In conclusion, the absence of the O antigen reduces the ability of A. pleuropneumoniae to form a biofilm, and this is associated with the reduced expression and production of PGA.
INTRODUCTION
Actinobacillus pleuropneumoniae is a Gram-negative bacterium belonging to the Pasteurellaceae family and is the causative agent of porcine pleuropneumonia, a disease causing important economic losses to the swine industry worldwide (1). Several virulence factors of A. pleuropneumoniae have been identified. These factors include the Apx toxins, iron uptake systems, and surface polysaccharides. These polysaccharides are divided into three categories: the lipopolysaccharides (LPS), the capsular polysaccharides (CPS), and the poly-N-acetyl-d-glucosamine polymer (PGA) present in the biofilm matrix (2–4).
LPS are large biomolecules composed of three well-defined regions: (i) lipid A, anchored in the outer membrane; (ii) the core oligosaccharide; and (iii) the O antigen, a polysaccharide consisting of repeating units. Altman and coworkers described the structure of the A. pleuropneumoniae serotype 1 O antigen to be branched tetrasaccharide repeating units composed of two l-rhamnopyranosyl residues, one d-glycopyranosyl residue, and one 2-acetamido-2-deoxy-d-glucose residue (5). The LPS inner core oligosaccharide is relatively conserved among A. pleuropneumoniae serotype 1, 2, 5a, and 5b strains and is composed of 2-keto-3-deoxyoctulosonic acid and heptose, whereas the outer core oligosaccharide is variable among serotypes and is generally made of a variable number of branching hexoses (6). The A. pleuropneumoniae LPS has been identified to be an adhesin, and the polysaccharide part of the LPS, but not the lipid A, is implicated in the adhesion to porcine respiratory tract cells and mucus (7, 8). Furthermore, an intact LPS core is essential for adhesion and virulence (9, 10).
Another major surface component of A. pleuropneumoniae serotype 1 is the capsular polysaccharide, composed of a repeating disaccharide unit of 2-acetamido-2-deoxy-d-glucose and d-galactose (11). CPS is encoded by the cpsABCD and cpxDCBA genes, which also encode an ABC transport system that is responsible for the export of the CPS (12) and is considered a crucial virulence factor of A. pleuropneumoniae (13, 14). A capsule mutant had enhanced adhesion to porcine epithelial cells (13, 15). Thus, it is thought that the CPS is not involved in adherence but, rather, appears to mask the adhesins required for attachment (13, 16).
Recent literature indicates the presence of a third category of surface polysaccharides in A. pleuropneumoniae, an exopolysaccharide (EPS) that covers and holds bacteria together and that is composed of PGA (17, 18). In A. pleuropneumoniae, this polymeric matrix has been shown to be involved in the formation of a robust biofilm in a very short period of time (4). Biofilms are sessile communities of microorganisms attached to a biotic or abiotic surface and enclosed in a self-produced extracellular polymer matrix (19, 20). These are more frequently associated with chronic diseases due particularly to an increased resistance to both conventional biocides and the host immune system (20–22). In this context, we have shown that A. pleuropneumoniae biofilm cells are 100 to 30,000 times more tolerant to antimicrobials than their planktonic counterparts (23).
The position of surface polysaccharides at the bacterium/surface interface is crucial for adhesion (24) and, consequently, likely plays a key role in biofilm formation by facilitating attachment, microcolony formation, and/or subsequent maturation of the biofilm. Indeed, several genetic determinants of A. pleuropneumoniae have also been associated with biofilm formation in microtiter plates or drip-flow reactors, and genes associated with LPS, CPS, and PGA biosynthesis have been identified in transposon mutants and by transcriptomic analysis (25–27). In this study, we investigated the role of surface polysaccharides in the biofilm formation of A. pleuropneumoniae. We specifically used well-characterized LPS, CPS, and PGA isogenic mutants of A. pleuropneumoniae serotype 1 strain 4074 (9, 13, 15, 25) and investigated the influence of these structural mutations on functional biofilm formation under static conditions or low-shear-force conditions.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
A. pleuropneumoniae strains were grown on brain heart infusion (BHI) agar or broth (Oxoid Ltd., Basingstoke, Hampshire, United Kingdom) supplemented with 15 μg/ml β-NAD (BHI-NAD) at 37°C with 5% CO2. All bacterial strains used in the present study are listed in Table 1.
TABLE 1.
Actinobacillus pleuropneumoniae strains used in the present study
| Strain | Relevant traits | Affected gene | Putative gene function | Reference |
|---|---|---|---|---|
| 4074Nalr | Serotype 1 (Nalra), parent strain | 15 | ||
| 44.1b | LPS O-antigen mutant | rfbN | Rhamnosyltransferase | 15 |
| CG3b | LPS core oligosaccharide mutant still expressing a complete O antigen | lbgB | d-Glycero-d-manno-heptosyl transferase | 9 |
| 33.2b | Acapsular mutant | cpxC | Capsule polysaccharide export inner membrane protein | 13 |
| ΔpgaCc | Exopolysaccharide PGA mutant | pgaC | Integral membrane glycoside transferase enzyme | 25 |
Nalr, nalidixic acid resistant.
Derived from A. pleuropneumoniae serotype 1 4074Nalr.
Derived from A. pleuropneumoniae serotype 1 4074.
Microtiter plate biofilm assay.
We used a microtiter plate biofilm assay as described previously (4). Briefly, the wells of a 96-well microtiter plate (Costar 3599; Corning, Corning, NY, USA) were filled in triplicate with a dilution (1/100) of an overnight bacterial culture. Following an incubation of 4 h at 37°C with 5% CO2, the culture medium was removed by aspiration and the plate was washed once by immersion in water. The wells were then filled with 100 μl of crystal violet (0.1%, wt/vol), and the plate was incubated for 2 min at room temperature. After removal of the crystal violet solution, the plate was washed and dried in a 37°C incubator for 30 min and 100 μl of ethanol (70% vol/vol) was added to the wells. The absorbance at 590 nm was measured using a spectrophotometer (PowerWave; BioTek Instruments, Winooski, VT, USA). The surface attachment of bacteria to polystyrene after a short incubation time of 2 h was measured as described before (26). Images of the adherent bacteria were captured with a microscope (DMI 4000 B; Leica Microsystems Inc., Richmond Hill, ON, Canada) using a DFC490 digital camera (Leica) and documented using the associated software (version 2.4.0). These images were analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA); the average number of particles (bacterial cells) from three fields of view across two independent biological replicates is reported.
Drip-flow biofilm reactor.
A drip-flow biofilm reactor (model DFR 110-4; BioSurface Technologies Corporation, Bozeman, MT, USA) was used to cultivate biofilms under a continuous low shear force on a glass slide as described before (27) with some modification; the flow rate was set to 50 ml/h per channel. After 24 h, the attached cells were scraped off the glass slide and resuspended in 1 ml of phosphate-buffered saline (PBS). After centrifugation, the supernatant was discarded and pellets were dried using a DNA 120 Speed Vac apparatus (Thermo Scientific, Ottawa, ON, Canada). When possible, the dry weight of the biofilms was measured and the dried biofilms were kept for further analyses.
CLSM.
Biofilms were grown in the microtiter plate biofilm assay as described above and stained with wheat germ agglutinin (WGA)-Oregon Green 488 (Invitrogen, Eugene, OR, USA), BOBO-3 (Invitrogen), FilmTracer SYPRO Ruby biofilm matrix stain (Invitrogen), the stain from a FilmTracer LIVE/DEAD biofilm viability kit (Invitrogen), or FilmTracer FM1-43 stain (Invitrogen) as recommended by the manufacturer. On the basis of the information provided by the manufacturer, WGA binds to N-acetyl-d-glucosamine and N-acetylneuraminic acid residues, BOBO-3 is a cell-impermeant extracellular DNA (eDNA) stain, and SYPRO Ruby stain labels most classes of proteins. FilmTracer FM1-43 is a green cell dye that stains bacterial membranes. The stained biofilms were visualized by confocal laser scanning microscopy (CLSM; FV1000 IX81; Olympus, Markham, ON, Canada), and images were acquired using FluoView software (Olympus). Biomass analysis of the biofilms was carried using the z-stack images obtained from areas selected randomly. The biomass and average thickness of biofilms were determined using Image-Pro software (Media Cybernetics, MD, USA) as described before (27).
Enzymatic treatments.
A biofilm dispersion assay was performed as described previously (28). Biofilms were grown in microtiter plates, and after the 4 h of incubation, 50 μl of DNase I (500 μg/ml in 150 mM NaCl, 1 mM CaCl2), 50 μl of dispersin B (100 μg/ml in PBS; Kane Biotech Inc., Winnipeg, MB, Canada), or 50 μl of proteinase K (500 μg/ml in 50 mM Tris-HCl, pH 7.5, 1 mM CaCl2) was added to the wells. Control wells were treated with 50 μl of the buffer without the enzyme. Wells treated with dispersin B were incubated for 5 min at 37°C, and those treated with proteinase K or DNase I were incubated for 1 h at 37°C. After the treatments, the biofilms were stained with crystal violet as described above.
Semiquantitative detection of PGA.
A. pleuropneumoniae strains were grown either in 6-well plates (Costar 3516) or in the drip-flow reactor. Culture supernatants were collected, centrifuged to remove the bacteria, and used for immunodetection of PGA. Biofilms were scraped off, resuspended in PBS, centrifuged, and dried as described above in “Drip-flow biofilm reactor.” The dried biofilms were resuspended in water to a concentration of 10 mg/ml and were then treated with proteinase K (100 μg/ml) for 1 h at 37°C. The treated biofilm suspensions were serially diluted, and these 2-fold dilutions were transferred to a nitrocellulose membrane (Millipore, Billerica, MA, USA) using a 96-well dot blot vacuum manifold system (Bio-Rad, Hercules, CA, USA). The membrane was air dried and then blocked with 5% (wt/vol) skim milk in Tris-buffered saline with Tween 20 (TTBS; 145 mM NaCl, 100 mM Tris-HCl, pH 7.4, supplemented with 0.15% [vol/vol] Tween 20) for 1 h. The blocked membrane was incubated with a goat anti-PGA antibody (1:2,500; kindly supplied by G. Pier, Harvard Medical School, Boston, MA, USA) in TTBS containing 5% (wt/vol) skim milk for 1 h, followed by three washes with TTBS. The membrane was then incubated with a rabbit anti-goat horseradish peroxidase-conjugated antibody (1:10,000; Jackson ImmunoResearch Laboratories West Grove, PA, USA) in TTBS containing 5% (wt/vol) skim milk for 30 min and then washed twice with TTBS. The membrane was developed with the substrate tetramethylbenzidine (Sigma-Aldrich). Water was added to wells as a negative control. The experiments were repeated three times.
Extraction and analysis of eDNA in biofilm matrix.
A 1/100 dilution of an overnight culture of A. pleuropneumoniae strains was incubated in 6-well plates (Costar 3516) for 4 h at 37°C in 5% CO2. After this time, the medium was removed and the biofilms were washed, gently scraped from the surface, and resuspended in 1.5 ml of 0.9% NaCl. For each strain, the biofilms from five wells were pooled to represent one sample. The biofilms were resuspended and treated with 20 μg/ml of dispersin B to promote the release of eDNA. Cells were then separated from the supernatant containing the eDNA fraction by centrifugation at 14,000 × g for 2 min. The eDNA and intracellular DNA (iDNA) from the supernatant and cell pellet, respectively, were extracted using the phenol-chloroform method as described before (29, 30). The DNA concentration and purity were determined with a NanoDrop spectrophotometer, and the eDNA/iDNA ratio was calculated. eDNA and iDNA were also visualized on agarose (0.8%) gels.
qRT-PCR analysis.
Parental and surface polysaccharide mutant strains were grown in 6-well plates (Costar 3516) for 4 h. Planktonic and biofilm cells were collected separately using ice-cold methanol to prevent changes in transcript levels. RNA extraction and a two-step quantitative real-time reverse transcriptase (RT) PCR (qRT-PCR) were performed as described elsewhere (27). The primers used to amplify the pgaA, cpxR, cpxA, dspB, and 16S rRNA genes are listed in Table 2. A Maxima SYBR green kit (Thermo Scientific Fermentas, Glen Burnie, MD, USA) was used for SYBR green labeling analysis (16-place Cepheid smart cycler system). Triplicates of qRT-PCR mixtures (25 μl) containing 0.75 ng of cDNA were prepared for each experiment. Relative expression was normalized using the 16S rRNA gene as the endogenous control, and the results were analyzed by a threshold cycle (ΔΔCT) method to calculate the fold change in gene expression (31). The expression in each sample was measured in duplicate, and the experiment was repeated in its entirety three times.
TABLE 2.
Primers used for quantitative real-time RT-PCR
| Primer | Sequence | Fragment size (bp) |
|---|---|---|
| 16SrRNA_Fw | GGAATAACTGGGCGTAAAGG | 200 |
| 16SrRNA_Dw | GCTCAGTACATTCCCAAGG | |
| pgaA_Fw | GATAAAGCAAGCCAGTTCTTAGGT | 215 |
| pgaA_Dw | GCTGTTTGATGAGAAATACCGA | |
| cpxR_Fw | TTGATGTTAAGCGCCAGAGA | 185 |
| cpxR_Dw | CCGGAGACTGGTTGGAATAA | |
| cpxA_Fw | CGGATATTTCGCATGAACTG | 187 |
| cpxA_Dw | GCGGGTCAAATGCTGATTG | |
| dspB_Fw | CATTCAAGCCGGCATTTC | 216 |
| dspB_Dw | TTGAGCGTCACCGTCGTA |
LPS extraction.
LPS of parental strain 4074Nalr and O-antigen mutant 44.1 were isolated from dried cell masses by the Darveau-Hancock method (32). Briefly, disrupted cells were treated with DNase, RNase, pronase, and sodium dodecyl sulfate (SDS). Treated cells were subjected to MgCl2 precipitation and high-speed centrifugation. All samples were lyophilized, and the LPS purity was evaluated on SDS-polyacrylamide gels stained with silver (9).
Surface plasmon resonance (SPR).
Binding between purified PGA (generously provided by Irina Sadovskaya, Université du Littoral-Côte d'Opale, Boulogne-sur-Mer, France) and LPS (both parental and O-antigen mutant 44.1 LPS) was examined using a Biacore 3000 system (GE Healthcare Bio-Sciences AB, Uppsala, Sweden). All lyophilized stocks were resuspended to 1 mg/ml in PBS. Experiments were performed on HPA sensor chips (Biacore) at 25°C using filtered (pore size, 0.2 μm) and degassed running buffers: HBS-N (10 mM HEPES, pH 7.4, 150 mM NaCl) and/or PBS-E (1 mM EDTA in 10 mM sodium phosphate dibasic, 1.8 mM potassium phosphate monobasic, 137 mM NaCl, 2.7 mM potassium chloride; EDTA is believed to favor the smallest 1,000-kDa form of LPS, as noted in Sigma [St. Louis, MO, USA] product data sheet L-2630). All chemicals were reagent-grade quality, including protein-grade n-octyl-β-d-glucopyranoside (nOG; 10% [vol/vol] nOG stock; catalog number 0311; Affymetrix-Anatrace, IL, USA), fatty acid-free bovine serum albumin (BSA; a 10-mg/ml BSA stock prepared in water; catalog number A-8806; Sigma, St. Louis, MO, USA), and glucocerebrosides (a 2-mg/ml glucocerebroside stock prepared in chloroform-methanol [2:1] solution; catalog number 1322; Matreya Pleasant Gap, PA, USA). Immobilized surfaces were prepared via hydrophobic adsorption to HPA sensors as previously described (33). Briefly, the flow cells were prewashed (5 μl/min, 25 μl, 2 times) with nOG (10% stock, 1/9 dilution in water = ∼40 mM). Active biomolecules (e.g., 1 mg/ml PGA in PBS) and reference biomolecules (e.g., 0.7 mg/ml glucocerebroside in 70% ethanol, 1 mg/ml BSA in buffer) were injected at 5 μl/min until surface densities of ∼1,000 relative units (RU) were obtained. The surfaces were washed with NaOH (5 mM in buffer; 50 μl/min, 25 μl) before blocking with BSA (0.5 mg/ml in buffer; 50 μl/min, 250 μl). The surfaces were finally stabilized with NaCl (1 M in buffer; 50 μl/min, 25 μl, 3 times) before use. For the reversed analyte-ligand orientation, active surfaces (1 mg/ml parental LPS or O-antigen mutant LPS in PBS) and reference surfaces (e.g., 1 mg/ml PGA in PBS) were prepared in a fashion similar to that described above.
To screen for binding specificity, fixed concentrations of LPS (parental or O-antigen mutant), PGA, and/or BSA (negative control) were injected over the reference and active surfaces in tandem at 10 to 25 μl/min (e.g., 1 min association, 1 min dissociation; multicycle mode using HBS-N or PBS-E running buffer). Between sample injections, the surfaces were regenerated at 50 μl/min using two 30-s pulses of solution I (1 M imidazole, pH 9.5) and solution II (1 M guanidine, pH 2.0), followed by routine washing procedures (i.e., extraclean and wash options). To further examine the binding kinetics, parental/mutant LPS and PGA were titrated over the sensor surfaces at 10 to 25 μl/min (e.g., 1 min association, 1 to 5 min dissociation; single-cycle mode using HBS-N or PBS-E running buffer); between titrations, the surfaces were regenerated as noted above. The double-referenced data (34) presented are representative of those from duplicate injections acquired from three independent trials. Due to the inherent sample heterogeneity with both PGA and LPS (i.e., each is a mixture of species with highly variable molecular masses), the binding kinetics could not be fit to a 1:1 titration model in the BIAevaluation software.
Statistical analysis.
Data were analyzed with GraphPad Prism (version 5.0) software (GraphPad Inc., San Diego, CA) using one-way analysis of variance (ANOVA) followed by Dunnett's multiple-comparison test. A P value of 0.05 or less was considered statistically significant.
RESULTS
The absence of O antigen and PGA reduces biofilm formation.
Biofilm formation by the parental strain and surface polysaccharide mutants was assessed using static and low-shear-force models. In a static 96-well microtiter plate assay, the biofilm formed by the pgaC mutant was, as expected, barely detected when the amount of biofilm formed was compared to that formed by the parental strain (Fig. 1A). The amount of biofilm formed by O-antigen mutant 44.1 was significantly reduced (P < 0.05) compared to that formed by parental strain 4074Nalr, LPS core mutant CG3, and capsule mutant 33.2 (Fig. 1A). Our results indicate that the absence of either PGA or O antigen negatively impacts biofilm formation by A. pleuropneumoniae.
FIG 1.

(A) Optical density of biofilm formation in 96-well microtiter plates by A. pleuropneumoniae parental strain 4074Nalr, O-antigen mutant 44.1, LPS core mutant CG3, capsule mutant 33.2, and the pgaC mutant. (B) Dry weight of 24-h-old biofilms (average weight [in milligrams] ± SD from 3 independent experiments) formed on a glass slide in the drip-flow reactor system by A. pleuropneumoniae parental strain 4074Nalr (11.6 ± 2.5 mg), O-antigen mutant 44.1 (for which biofilm formation was barely detectable), LPS core mutant CG3 (12.2 ± 4.6 mg), capsule mutant 33.2 (14.9 ± 4.5 mg), and the pgaC mutant (for which biofilm formation was not detected). Statistical analysis was performed using ANOVA. *, P < 0.05 compared to parental strain 4074Nalr.
To further analyze the LPS, CPS, and PGA mutants, we examined biofilm formation in the drip-flow reactor, a continuous-flow model with an air-liquid interface and low shear force considered to be more representative of the lung environment (35). While the O-antigen mutant barely formed detectable thin biofilms, parental strain 4074Nalr, LPS core mutant CG3, and capsule mutant 33.2 formed similar amounts of thick, rough biofilms (Fig. 1B). The pgaC mutant was unable to form a biofilm in this system. These results cross-validate the finding that the absence of the O antigen or PGA reduces the ability of A. pleuropneumoniae serotype 1 to form a mature biofilm, whereas truncation of the core oligosaccharide with an intact O antigen or the absence of the capsule polysaccharide does not affect biofilm formation under static (i.e., 96-well microtiter plate assay) and low-shear-force (i.e., drip-flow reactor) conditions.
The absence of O antigen and PGA does not affect the initial attachment of A. pleuropneumoniae to an abiotic surface.
Given that the initial attachment of bacteria to a surface is a critical step for biofilm formation, we examined the attachment of A. pleuropneumoniae to the plastic surface of 96-well plates (see Fig. S1 in the supplemental material). Image analysis showed that the overall number of attached cells for both O-antigen mutant 44.1 (see Fig. S1B in the supplemental material) and LPS core mutant CG3 (see Fig. S1C in the supplemental material) appeared to be similar to that for the parental strain (see Fig. S1A in the supplemental material). The pgaC mutant (see Fig. S1E in the supplemental material) had fewer cells; however, this difference was not significant (P > 0.05). In contrast, more attached cells were detected for capsule mutant 33.2 (see Fig. S1D in the supplemental material). It is unlikely that the initial rate of A. pleuropneumoniae attachment accounts for the inability of both the O-antigen and PGA mutants to form a biofilm.
Biofilm morphology and composition.
The biofilm architecture was visualized by CLSM using a WGA fluorescent probe specifically targeted to PGA, the major component of the biofilm matrix of A. pleuropneumoniae. While O-antigen mutant 44.1 formed only small, patchy microcolonies after 4 h of incubation in microtiter plates (Fig. 2B), LPS core mutant CG3 (Fig. 2C) and capsule mutant 33.2 (Fig. 2D) formed biofilms similar to those formed by the parental strain (Fig. 2A). As quantified from the confocal images, the biofilm biomass of O-antigen mutant 44.1 was significantly reduced (i.e., under 0.1 μm3/μm2) compared to the biofilm biomasses of the parental strain, LPS core mutant CG3, and capsule mutant 33.2 (i.e., 4.50 ± 0.84, 5.26 ± 1.1, and 4.44 ± 0.89 μm3/μm2, respectively). Since the pgaC mutant is deficient in PGA production, the staining with WGA, which recognizes N-acetyl-d-glucosamine and N-acetylneuraminic acid residues, was negative (data not shown). Therefore, we used FilmTracer FM1-43, a fluorescent probe that targets bacterial membranes, to visualize the biofilm of the pgaC mutant. As observed by CLSM, the biofilm of the pgaC mutant was characterized by isolated clusters (Fig. 3). Similarly to the O-antigen mutant, the pgaC mutant was deficient in biofilm formation. Therefore, the CLSM data were consistent with the biofilm results obtained from the microtiter plate (crystal violet staining) and drip-flow (dry weight measurement) assays. In addition to PGA, the biofilm of the A. pleuropneumoniae parental strain was also stained with cell-impermeant dyes to determine the overall matrix composition. Significant amounts of eDNA (stained with BOBO-3) and proteins (stained with SYPRO Ruby) were detected within the parental biofilm (data not shown).
FIG 2.

CLSM images of biofilms formed in 96-well microtiter plates by A. pleuropneumoniae parental strain 4074Nalr (A), O-antigen mutant 44.1 (B), LPS core mutant CG3 (C), and capsule mutant 33.2 (D) stained with WGA-Oregon Green 488. (Top) x-y planes of the biofilms; (bottom) three-dimensional (3D) images. The numerical values represent the biomass (in cubic micrometers per square micrometer) of each biofilm.
FIG 3.

CLSM images of biofilms formed in 96-well microtiter plates by A. pleuropneumoniae parental wild-type (WT) strain 4074 and the pgaC mutant stained with FilmTracer FM1-43, a fluorescent probe that stains bacterial membranes. The three-dimensional images were obtained by using FluoView software.
To further examine the contribution of PGA, proteins, and eDNA to the matrix architecture, preformed biofilms were treated with dispersin B, proteinase K, and DNase I. Dispersin B, a biofilm-releasing glycoside hydrolase that degrades PGA, was able to significantly disperse the preformed biofilms (P < 0.05) of the parental strain, the LPS core mutant, and the capsule mutant (Fig. 4). Given that the O-antigen and pgaC mutants produced biofilms in amounts barely above the limit of detection of the assay, dispersal of their biofilms by enzymatic digestion was difficult to detect and, thus, not observable (data not shown). Indicative of the fact that eDNA and proteins do not play a major role in the architecture of static A. pleuropneumoniae biofilms, digestions with proteinase K, a broad-spectrum serine protease, and DNase I, a site-specific nuclease, were not able to disperse the mature biofilms. The small amount of eDNA in the biofilm matrices (range, 26 to 45 ng/μl; approximately 1/10 of the intracellular DNA content, based upon our equalized parental, CG3, and 33.2 strain biofilm extractions) suggests that eDNA does not contribute to the architecture of A. pleuropneumoniae biofilms.
FIG 4.

Dispersion of biofilms formed in 96-well microtiter plates by A. pleuropneumoniae parental strain 4074Nalr, LPS core mutant CG3, and capsule mutant 33.2 by DNase I, proteinase K, and dispersin B (DspB). The means ± SDs from 3 independent experiments are shown. Statistical analysis was performed using ANOVA. *, P < 0.05 compared to the control (Ctr).
Specific binding between PGA and LPS.
Label-free, real-time SPR was utilized to test if PGA specifically interacts with LPS. With PGA (net positive charge at pH 7) immobilized to HPA sensor surfaces (Fig. 5A), fixed concentrations of LPS from the parental strain and O-antigen mutant 44.1 (both with a net negative charge in HBS-N running buffer) generated significant binding responses. In contrast, there was little or no signal change when BSA, a negatively charged protein at pH 7 (36), was injected as a nonbinding control. Moreover, glucocerebrosides (net neutral charge) were chosen as the corresponding reference surfaces in the assays whose results are shown in Fig. 5A to ensure that there was minimal nonspecific binding of parental/mutant LPS to the sensors in the absence of PGA. Due to the inherent heterogeneity of the parental/mutant LPS and PGA samples tested (i.e., they form aggregates of various sizes), the observed binding profiles deviated from simple 1:1 kinetics, as anticipated (e.g., see the heterogeneous O-antigen binding profile in Fig. 5A). Regardless, fixed concentrations of PGA (in PBS-E running buffer) once again generated significant binding responses against LPS from both the parental strain and O-antigen mutant 44.1 when the analyte-ligand orientation was reversed (Fig. 5B). Using the optimized assay orientation (i.e., for the ease of regeneration of sensor surfaces when LPS was injected over immobilized PGA), additional kinetic titrations (Fig. 5C) demonstrated that the binding of both the parental strain and O-antigen mutant LPS to PGA was dose dependent. Although equimolar concentrations of parental and mutant 44.1 LPS were anticipated to yield similar SPR signal changes (i.e., similar molecular masses) when interacting with PGA, differences in their intrinsic properties (e.g., exposed binding interfaces) likely account for fluctuations in the absolute binding responses (e.g., see Fig. 5A and C, which show a larger signal shift with mutant 44.1 LPS, and Fig. 5B, which shows a larger signal shift with the parental strain LPS). While the inherent heterogeneity of both PGA and LPS (i.e., each is a mixture of species with highly variable molecular masses) did not allow us to assess overall binding affinities, specific dose-dependent binding between PGA and LPS was consistently detected using multiple assay orientations, running buffers, and reference surfaces. A lack of interaction between LPS and PGA does not account for the inability of O-antigen mutant 44.1 to form mature biofilms.
FIG 5.
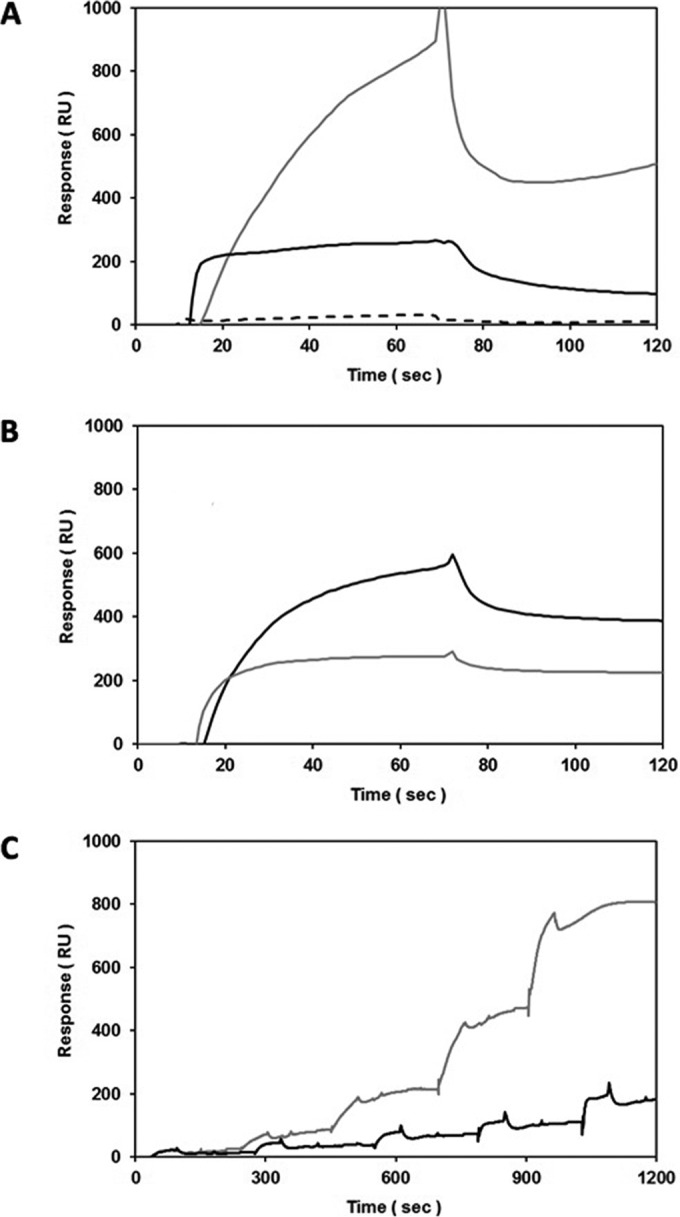
Specific, dose-dependent binding between PGA and the parental strain LPS or LPS from O-antigen mutant 44.1, as assessed by SPR using HPA sensors. (A) Screening of BSA (dashed line; 0.1 mg/ml = 1 μM; 66 kDa), parental strain LPS (black line; 1 mg/ml; 1 μM if the molecular mass is 1,000 kDa), and the LPS of mutant 44.1 (gray line; 1 mg/ml; 1 μM if the molecular mass is 1,000 kDa) binding to immobilized PGA (1,400 RU; in-line reference subtraction = 1,400 RU glucocerebrosides) at 25 μl/min in HBS-N running buffer; (B) screening of solution-phase PGA (1 mg/ml) binding to immobilized parental strain LPS and LPS of mutant 44.1 (1,000 RU each; black and gray lines, respectively; in-line reference subtraction = 1,000 RU immobilized PGA) at 25 μl/min in PBS-E running buffer; (C) single-cycle titrations of parental strain LPS and mutant 44.1 LPS (0.0313, 0.0625, 0.125, 0.25, and 0.5 mg/ml; black and gray lines, respectively) binding to immobilized PGA (1,000 RU; in-line reference subtraction = 1,000 RU BSA) at 25 μl/min in PBS-E running buffer.
The O-antigen mutant produces less PGA.
Biofilms grown under both static and drip-flow conditions were collected, their amounts were normalized according to dry weight, and the relative amounts of associated PGA were semiquantified by immunoblotting. In both cases, biofilms of O-antigen mutant 44.1 had less PGA associated with the biofilm cells, whereas similar amounts of PGA were found to be associated with the biofilm cells for parental strain 4074Nalr, LPS core mutant CG3, and capsule mutant 33.2 (Fig. 6). Under static conditions, the amount of PGA in the biofilm supernatant was also measured to verify that the PGA produced was not released into the growth medium. As with the biofilm/cell-associated PGA results, less PGA was also detected in the O-antigen mutant supernatant (see Fig. S2 in the supplemental material). Taken together, our data indicate that O-antigen mutant 44.1 produced less PGA than the parental strain. It should be noted that the barely detectable biofilm formed by the pgaC mutant did not allow us to perform immunoblotting with this mutant.
FIG 6.

Detection of PGA in the biofilm matrix isolated from parental strain 4074Nalr and mutant strains cultured in a drip-flow reactor for 24 h. Starting wells (1/1) contained 75 μg, and then the contents were serially diluted (1/2 to 1/512).
Analysis of pgaA, cpxR, cpxA, and dspB expression by real-time RT-PCR.
To verify the effect of the LPS and CPS biosynthetic gene mutations on PGA production, the expression of pgaA (one of the genes in the pgaABCD operon encoding PGA production) was measured in mature biofilms. The expression of pgaA was downregulated (P < 0.05) in O-antigen mutant 44.1 (Fig. 7A), whereas expression in core oligosaccharide mutant CG3 and acapsular mutant 33.2 was not significantly different from that in parental strain 4074Nalr. Given that cpxR and cpxA (37–39) are differentially regulated in A. pleuropneumoniae biofilms (27), their expression was also measured. Both genes were downregulated (P < 0.05) in O-antigen mutant 44.1, whereas no significant differences in expression were observed for LPS core oligosaccharide mutant CG3 and acapsular mutant 33.2 (Fig. 7B). The expression of dspB, coding for the dispersin B enzyme, was also measured. No difference in expression of the dspB gene was detected between O-antigen mutant 44.1 and the parental strain (data not shown). Once again, it should be noted that the barely detectable biofilm formed by the pgaC mutant did not allow us to perform qRT-PCR with this mutant.
FIG 7.
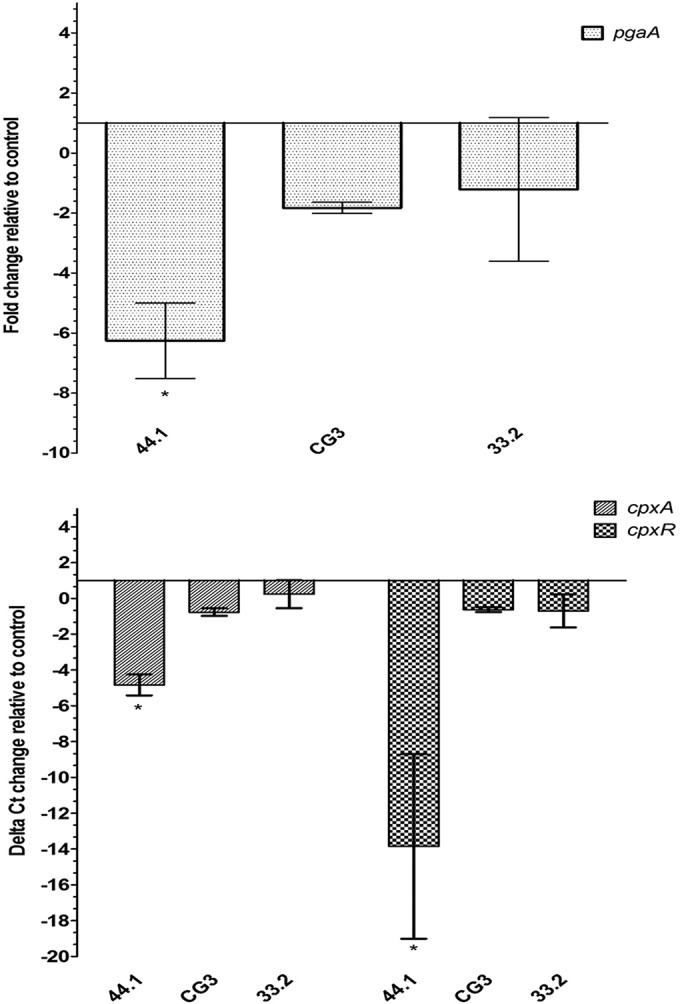
Expression of pgaA (top) and cpxA and cpxR (bottom) of O-antigen mutant 44.1, LPS core mutant CG3, and capsule mutant 33.2 relative to that of parental strain 4074Nalr. The means ± SDs from 3 independent experiments are shown. Statistical analyses were performed using 2−ΔΔCT values, and all results with an asterisk were statistically significant (P < 0.05).
DISCUSSION
Surface polysaccharides, including LPS, CPS, and PGA, have been linked to biofilm formation in several bacterial species (17, 25, 40–51); thus, investigation of their unassessed role in the biofilm formation of A. pleuropneumoniae serotype 1 was the focus of our present study. LPS and CPS have been implicated in both the adhesion and virulence of A. pleuropneumoniae, but their role in biofilm formation is still unclear. For example, LPS and CPS can affect the adhesion of A. pleuropneumoniae to epithelial cells and mucus (9, 13, 15). Furthermore, as previously demonstrated by our laboratory, the core oligosaccharide of LPS is required for its adherence to porcine respiratory tract cells (9). To our knowledge, this is the first report that an O-antigen mutant of A. pleuropneumoniae serotype 1 is severely defective in biofilm formation in both static and low-shear-force models (Fig. 1). Confocal microscopy with PGA-specific staining confirmed that the O-antigen mutant biofilms were patchy, small microcolonies, whereas parental strain biofilms were dense and thick (Fig. 2). These findings indicate that a lack of the O antigen significantly impairs the biofilm formation of A. pleuropneumoniae serotype 1. At the same time, the adherence of O-antigen mutant 44.1 to an abiotic surface was not affected (see Fig. S1 in the supplemental material), and on the basis of our previous work, the absence of the O chains does not affect the overall surface hydrophobicity or charge of O-antigen mutant 44.1 (15). Overall, the defective biofilm formation that we observed with the O-antigen mutant of A. pleuropneumoniae serotype 1 is likely due to its inability to progress from microcolonies into a mature biofilm. Our findings are consistent with those for other bacterial species where a loss of O antigen has profound effects. For example, the biofilm formation and EPS accumulation of Xanthomonas oryzae pv. Oryzicola were affected by the loss of O antigen (40). Li and Wang observed that mutations in the wxacO and rfbC genes (involved in the biosynthesis of Xanthomonas citri subsp. citri O-antigen LPS) impaired biofilm formation on plastic surfaces and host plant leaves (41). The formation of Stenotrophomonas maltophilia biofilms requires an intact LPS, and a mutation in O-antigen biosynthesis affects EPS production and biofilm formation (42). Clifford and coworkers showed that the O antigen of Xylella fastidiosa is needed for biofilm maturation and surface attachment (50). It was recently demonstrated that mutations in the waaWVL operon, encoding LPS biosynthesis genes, lead to a shorter LPS species and a decreased ability of adherent-invasive Escherichia coli (AIEC) strain LF82 to form biofilms (51). On the other hand, Ciornei and coworkers have demonstrated that the ability of Pseudomonas aeruginosa to form a mature biofilm is dependent on a shorter form of LPS or a complete loss of O antigen (52). Additionally, Coulon and coworkers found that the extracellular matrix of P. aeruginosa biofilms contains significant amounts of O-antigen polysaccharides (53).
The pgaC mutant, which lacks an integral membrane glycoside transferase enzyme (PgaC) catalyzing the polymerization of PGA from UDP-GlcNAc monomers, was also deficient in biofilm formation, like the O-antigen mutant. The biofilms produced by the pgaC and O-antigen mutants were barely detectable in vitro and were characterized by few dispatched colonies attached to the surface; however, adherence to an abiotic surface was not affected for either mutant. Izano et al. previously showed that an A. pleuropneumoniae serotype 5 pgaC mutant was deficient in EPS, and this mutant failed to form a biofilm in vitro (54). Our results indicate that both PGA and O antigen are important for biofilm production by A. pleuropneumoniae serotype 1.
While other groups have shown that the LPS core oligosaccharide is essential for biofilm formation by E. coli, Klebsiella pneumoniae, and S. maltophilia (42–44), our LPS core mutant, strain CG3, was still able to produce biofilms similarly to the parental A. pleuropneumoniae strain (Fig. 1). Other groups have also shown that capsule expression can affect the biofilm formation of pathogens such as Vibrio vulnificus (45) and Porphyromonas gingivalis (46). For example, capsule synthesis genes (wza and wzc for K. pneumoniae and kpsM for E. coli) were recently shown to negatively affect biofilm formation (47, 48). In contrast, our acapsular mutant, strain 33.2, produced a biofilm similar to that formed by the parental A. pleuropneumoniae strain, even though the former exhibited enhanced adherence. These results are consistent with those of a previous study demonstrating that acapsular mutant 33.2 adheres significantly more to piglet tracheal frozen sections than the parental strain (13) and also with those of a study by Karwacki and coworkers, who showed that biofilms of an acapsular A. pleuropneumoniae serotype 5 mutant were not affected on 96-well polystyrene microtiter plates compared to the wild-type strain (49).
As the importance of surface polysaccharide in biofilm formation continues to be recognized in the literature, our data and the studies noted above reinforce the potential role of O antigen and PGA, but not the core oligosaccharide and the capsule, in the biofilm formation and maturation of A. pleuropneumoniae serotype 1 strains. Treatment with dispersin B completely dispersed preformed biofilms, thereby confirming that PGA is a key scaffolding component of the matrix (4, 26). Within the biofilms of P. aeruginosa and Staphylococcus aureus, eDNA is required for the attachment, aggregation, and stabilization of microcolonies, and DNase treatment removes the biofilms (55, 56). While we detected eDNA within the biofilm matrix of our parental and mutant A. pleuropneumoniae strains, DNase I treatment did not disperse the preformed biofilms. Since proteinase K treatment also did not lead to a dispersion of the biofilms, our data indicate that PGA, eDNA, and proteins are all part of the matrix, but only PGA is critical for biofilm formation by A. pleuropneumoniae serotype 1.
It has been shown that LPS mediates interactions with EPS. For example, binding has been observed between LPS and PGA from E. coli (43) and between LPS and cellulose, an EPS in the biofilm of Pseudomonas fluorescens (57). Amini and coworkers showed that modified E. coli LPS (which has less negatively charged phosphate groups) decreases its interaction with the positively charged exopolysaccharide PGA (43). The findings by Amini et al. highlight the importance of electrostatic interactions between LPS and EPS for biofilm development, strength, and integrity (43).
While significantly less is known about interactions between surface polysaccharides in A. pleuropneumoniae, the findings of the studies with P. fluorescens and E. coli prompted us to hypothesize that PGA may interact directly with LPS in A. pleuropneumoniae. Using SPR, multiple assay designs (i.e., assays with different coupling orientations, running buffers, and reference surfaces) consistently detected specific binding between the two surface polysaccharides (Fig. 5). In comparison to other LPS studies in the SPR literature, our assays were performed using a stringent flow rate (i.e., 25 μl/min, like that used by Bahl et al. [58]) and avoided the use of complicating carrier proteins, such as BSA, in the running buffer (59). Like the high-affinity interaction reported by Bahl et al. (58) that mapped specific LPS-binding hot spots on hemoglobin, the slow dissociation kinetics that we observed suggest that the PGA-LPS interaction is not weak (e.g., there was no immediate return of the SPR signal to the baseline at 70 to 120 s in Fig. 5A and B). These results indicate that the SPR binding detected between LPS and PGA is specific (i.e., it does not result from random electrostatic or hydrophobic interactions or the use of a low, nonstringent flow rate of 5 μl/min). Therefore, our current study provides the first direct evidence of a specific binding between PGA and LPS in A. pleuropneumoniae serotype 1.
Our subsequent evaluation of PGA production by immunoblotting demonstrated that O-antigen mutant 44.1 had less cell-associated PGA than the parental strain, core mutant CG3, or capsule mutant 33.2 (Fig. 6). Complementary qRT-PCR analyses also revealed a downregulation of the pgaA gene in O-antigen mutant 44.1 (Fig. 7), thereby corroborating the defect in PGA production observed by immunoblotting. This defect likely accounts for the inability of the O-antigen mutant to form robust biofilms. It is important to note that the O-antigen mutant that we used in this study, mutant 44.1, has a transposon insertion in the rfbN gene which encodes a rhamnosyl transferase involved in the assembly of the O chain (15). Recently, a mutant with a rhamnosyl transferase (wxocB) mutation, which resulted in a complete loss of the O chain in X. oryzae pv. Oryzicola, produced significantly less EPS (40).
Considering the critical position of LPS at cell-cell and cell-surface interfaces, we investigated the putative effect of the O-antigen truncation on the two-component regulatory system CpxRA. It is well established that CpxRA senses envelope stress, such as overproduction or misfolding of membrane molecules (37), and the Cpx system controls genes involved in biofilm formation by E. coli strains (38, 39). In several Gram-negative bacteria, the Cpx envelope stress response is activated by pH, osmolarity, alterations in inner membrane composition, and surface adhesion (37). Indeed, Otto and Silhavy showed that the E. coli Cpx system is highly induced in mature biofilms (60). Using a cpxR mutant, Beloin and coworkers demonstrated that E. coli K-12 was unable to produce mature biofilms, with only dispatched microcolonies being observed by confocal microscopy (39). In addition, previous transcriptomic analyses from our group have demonstrated the differential regulation of cpxR or cpxA when A. pleuropneumoniae grew as biofilms (27). In the present study, we now show that cpxA and cpxR expression in O-antigen mutant 44.1 is significantly downregulated compared to their levels of expression in LPS core mutant CG3, capsule mutant 33.2, or the parental strain (Fig. 7). Therefore, it is conceivable that the absence of O antigen results in the downregulation of cpxRA. With the knowledge that both the regulators σE (a positive regulator) and H-NS (a negative regulator) independently regulate the expression of the pga operon (25), further investigations will be required to determine how the Cpx system in A. pleuropneumoniae directly or indirectly affects the pgaABCD operon.
In conclusion, our current study provides important new insights about surface polysaccharides in A. pleuropneumoniae. While truncation of the LPS core oligosaccharide or the absence of CPS did not have any effect, the absence of PGA or O antigen markedly reduced the ability of A. pleuropneumoniae serotype 1 strains to form a mature biofilm. This finding was linked for the O-antigen mutant to a reduced level of pgaA expression and, consequently, a reduced level of PGA production. Moreover, we have successfully demonstrated for the first time a direct binding interaction between LPS and PGA in A. pleuropneumoniae serotype 1, and this may represent a new strategy to prevent biofilm formation. However, our study demonstrated that the absence of the LPS O antigen did not abolish this binding interaction and the defect in A. pleuropneumoniae biofilm formation was associated with the reduced expression and production of PGA. Interestingly, the O-antigen mutant also exhibited reduced cpxRA expression; the link between the CpxRA system and biofilm formation in A. pleuropneumoniae warrants further investigation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Irina Sadovskaya and G. Pier for kindly providing PGA and anti-PGA antibodies, respectively.
We declare that we have no conflicts of interest with the contents of this article.
S.H., Y.D.N.T., and M.J. designed the study. S.H., Y.D.N.T., M.A.H., and M.J. wrote the manuscript. S.H. performed the biofilm assays, CLSM analysis, qRT-PCR, and immunodetection of PGA. Y.D.N.T. optimized the PGA immunodetection and early adhesion assays. J.L. helped with the biofilm and the adhesion assays. M.A.H. carried out the SPR experiments. P.R.L. and J.T.B. designed and created the pgaC mutant. All authors analyzed the results and approved the final version of the manuscript.
Funding Statement
The Canada Foundation for Innovation provided funding to the McGill Surface Plasmon Resonance-Mass Spectrometry Facility. The Swine and Poultry Infectious Diseases Research Center (CRIPA) provided a scholarship to S.H.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00912-15.
REFERENCES
- 1.Gottschalk M. 2012. Actinobacillosis, p 653–669. In Karriker L, Ramirez A, Schwartz K, Stevenson G, Zimmerman J (ed), Diseases of swine, 10th ed John Wiley & Sons, Inc, Hoboken, NJ. [Google Scholar]
- 2.Jacques M. 2004. Surface polysaccharides and iron-uptake systems of Actinobacillus pleuropneumoniae. Can J Vet Res 68:81–85. [PMC free article] [PubMed] [Google Scholar]
- 3.Bossé JT, Janson H, Sheehan BJ, Beddek AJ, Rycroft AN, Kroll JS, Langford PR. 2002. Actinobacillus pleuropneumoniae: pathobiology and pathogenesis of infection. Microb Infect 4:225–235. doi: 10.1016/S1286-4579(01)01534-9. [DOI] [PubMed] [Google Scholar]
- 4.Labrie J, Pelletier-Jacques G, Deslandes V, Ramjeet M, Auger E, Nash JH, Jacques M. 2010. Effects of growth conditions on biofilm formation by Actinobacillus pleuropneumoniae. Vet Res 41:3. doi: 10.1051/vetres/2009051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Altman E, Brisson JR, Perry MB. 1986. Structure of the O-chain of the lipopolysaccharide of Haemophilus pleuropneumoniae serotype 1. Biochem Cell Biol 64:1317–1325. doi: 10.1139/o86-173. [DOI] [PubMed] [Google Scholar]
- 6.Michael FS, Brisson JR, Larocque S, Monteiro M, Li J, Jacques M, Perry MB, Cox AD. 2004. Structural analysis of the lipopolysaccharide derived core oligosaccharides of Actinobacillus pleuropneumoniae serotypes 1, 2, 5a and the genome strain 5b. Carbohydr Res 339:1973–1984. doi: 10.1016/j.carres.2004.04.019. [DOI] [PubMed] [Google Scholar]
- 7.Paradis SE, Dubreuil D, Rioux S, Gottschalk M, Jacques M. 1994. High-molecular-mass lipopolysaccharides are involved in Actinobacillus pleuropneumoniae adherence to porcine respiratory tract cells. Infect Immun 62:3311–3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Belanger M, Rioux S, Foiry B, Jacques M. 1992. Affinity for porcine respiratory tract mucus is found in some isolates of Actinobacillus pleuropneumoniae. FEMS Microbiol Lett 76:119–125. [DOI] [PubMed] [Google Scholar]
- 9.Rioux S, Galarneau C, Harel J, Frey J, Nicolet J, Kobisch M, Dubreuil JD, Jacques M. 1999. Isolation and characterization of mini-Tn10 lipopolysaccharide mutants of Actinobacillus pleuropneumoniae serotype 1. Can J Microbiol 45:1017–1026. doi: 10.1139/w99-107. [DOI] [PubMed] [Google Scholar]
- 10.Ramjeet M, Deslandes V, St Michael F, Cox AD, Kobisch M, Gottschalk M, Jacques M. 2005. Truncation of the lipopolysaccharide outer core affects susceptibility to antimicrobial peptides and virulence of Actinobacillus pleuropneumoniae serotype 1. J Biol Chem 280:39104–39114. doi: 10.1074/jbc.M502852200. [DOI] [PubMed] [Google Scholar]
- 11.Altman E, Brisson JR, Perry MB. 1986. Structural studies of the capsular polysaccharide from Haemophilus pleuropneumoniae serotype 1. Biochem Cell Biol 64:707–716. doi: 10.1139/o86-097. [DOI] [PubMed] [Google Scholar]
- 12.Ward CK, Inzana TJ. 1997. Identification and characterization of a DNA region involved in the export of capsular polysaccharide by Actinobacillus pleuropneumoniae serotype 5a. Infect Immun 65:2491–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rioux S, Galarneau C, Harel J, Kobisch M, Frey J, Gottschalk M, Jacques M. 2000. Isolation and characterization of a capsule-deficient mutant of Actinobacillus pleuropneumoniae serotype 1. Microb Pathog 28:279–289. doi: 10.1006/mpat.1999.0347. [DOI] [PubMed] [Google Scholar]
- 14.Bandara AB, Lawrence ML, Veit HP, Inzana TJ. 2003. Association of Actinobacillus pleuropneumoniae capsular polysaccharide with virulence in pigs. Infect Immun 71:3320–3328. doi: 10.1128/IAI.71.6.3320-3328.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Labrie J, Rioux S, Wade MM, Champlin FR, Holman SC, Wilson WW, Savoye C, Kobisch M, Sirois M, Galarneau C, Jacques M. 2002. Identification of genes involved in biosynthesis of Actinobacillus pleuropneumoniae serotype 1 O-antigen and biological properties of rough mutants. J Endotoxin Res 8:27–38. doi: 10.1179/096805102125000065. [DOI] [PubMed] [Google Scholar]
- 16.Van Overbeke I, Chiers K, Charlier G, Vandenberghe I, Van Beeumen J, Ducatelle R, Haesebrouck F. 2002. Characterization of the in vitro adhesion of Actinobacillus pleuropneumoniae to swine alveolar epithelial cells. Vet Microbiol 88:59–74. doi: 10.1016/S0378-1135(02)00080-9. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan JB, Velliyagounder K, Ragunath C, Rohde H, Mack D, Knobloch JK, Ramasubbu N. 2004. Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J Bacteriol 186:8213–8220. doi: 10.1128/JB.186.24.8213-8220.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, Preston JF III, Romeo T. 2004. The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol 186:2724–2734. doi: 10.1128/JB.186.9.2724-2734.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Costerton JW. 1999. Introduction to biofilm. Int J Antimicrob Agents 11:217–221. doi: 10.1016/S0924-8579(99)00018-7. [DOI] [PubMed] [Google Scholar]
- 20.Jacques M, Aragon V, Tremblay YD. 2010. Biofilm formation in bacterial pathogens of veterinary importance. Anim Health Res Rev 11:97–121. doi: 10.1017/S1466252310000149. [DOI] [PubMed] [Google Scholar]
- 21.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 22.Gardner A, Percival L, Cochrane C. 2011. Biofilms and role to infection and disease in veterinary medicine, p 111–128. In Percival L, Knottenbelt D, Cochrane C (ed). Biofilms and veterinary medicine. Springer, Berlin, Germany. [Google Scholar]
- 23.Archambault M, Harel J, Goure J, Tremblay YD, Jacques M. 2012. Antimicrobial susceptibilities and resistance genes of Canadian isolates of Actinobacillus pleuropneumoniae. Microb Drug Resist 18:198–206. doi: 10.1089/mdr.2011.0150. [DOI] [PubMed] [Google Scholar]
- 24.Jacques M. 1996. Role of lipo-oligosaccharides and lipopolysaccharides in bacterial adherence. Trends Microbiol 4:408–409. doi: 10.1016/0966-842X(96)10054-8. [DOI] [PubMed] [Google Scholar]
- 25.Bossé JT, Sinha S, Li MS, O'Dwyer CA, Nash JH, Rycroft AN, Kroll JS, Langford PR. 2010. Regulation of pga operon expression and biofilm formation in Actinobacillus pleuropneumoniae by σE and H-NS. J Bacteriol 192:2414–2423. doi: 10.1128/JB.01513-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grasteau A, Tremblay YD, Labrie J, Jacques M. 2011. Novel genes associated with biofilm formation of Actinobacillus pleuropneumoniae. Vet Microbiol 153:134–143. doi: 10.1016/j.vetmic.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 27.Tremblay YD, Deslandes V, Jacques M. 2013. Actinobacillus pleuropneumoniae genes expression in biofilms cultured under static conditions and in a drip-flow apparatus. BMC Genomics 14:364. doi: 10.1186/1471-2164-14-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tremblay YD, Lamarche D, Chever P, Haine D, Messier S, Jacques M. 2013. Characterization of the ability of coagulase-negative staphylococci isolated from the milk of Canadian farms to form biofilms. J Dairy Sci 96:234–246. doi: 10.3168/jds.2012-5795. [DOI] [PubMed] [Google Scholar]
- 29.Shields RC, Mokhtar N, Ford M, Hall MJ, Burgess JG, Elbadawey MR, Jakubovics NS. 2013. Efficacy of a marine bacterial nuclease against biofilm forming microorganisms isolated from chronic rhinosinusitis. PLoS One 8:e55339. doi: 10.1371/journal.pone.0055339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakao R, Ramstedt M, Wai SN, Uhlin BE. 2012. Enhanced biofilm formation by Escherichia coli LPS mutants defective in Hep biosynthesis. PLoS One 7:e51241. doi: 10.1371/journal.pone.0051241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Darveau RP, Hancock RE. 1983. Procedure for isolation of bacterial lipopolysaccharides from both smooth and rough Pseudomonas aeruginosa and Salmonella typhimurium strains. J Bacteriol 155:831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taillon C, Hancock MA, Mourez M, Dubreuil JD. 2012. Biochemical and biological characterization of Escherichia coli STb His12 to Asn variant. Toxicon 59:300–305. doi: 10.1016/j.toxicon.2011.11.015. [DOI] [PubMed] [Google Scholar]
- 34.Myszka DG. 1999. Improving biosensor analysis. J Mol Recognit 12:279–284. doi:. [DOI] [PubMed] [Google Scholar]
- 35.Goeres DM, Hamilton MA, Beck NA, Buckingham-Meyer K, Hilyard JD, Loetterle LR, Lorenz LA, Walker DK, Stewart PS. 2009. A method for growing a biofilm under low shear at the air-liquid interface using the drip flow biofilm reactor. Nat Protoc 4:783–788. doi: 10.1038/nprot.2009.59. [DOI] [PubMed] [Google Scholar]
- 36.Barbosa LR, Ortore MG, Spinozzi F, Mariani P, Bernstorff S, Itri R. 2010. The importance of protein-protein interactions on the pH-induced conformational changes of bovine serum albumin: a small-angle X-ray scattering study. Biophys J 98:147–157. doi: 10.1016/j.bpj.2009.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vogt S, Raivio T. 2012. Just scratching the surface: an expanding view of the Cpx envelope stress response. FEMS Microbiol Lett 326:2–11. doi: 10.1111/j.1574-6968.2011.02406.x. [DOI] [PubMed] [Google Scholar]
- 38.Price NL, Raivio TL. 2009. Characterization of the Cpx regulon in Escherichia coli strain MC4100. J Bacteriol 191:1798–1815. doi: 10.1128/JB.00798-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beloin C, Valle J, Latour-Lambert P, Faure P, Kzreminski M, Balestrino D, Haagensen JA, Molin S, Prensier G, Arbeille B, Ghigo JM. 2004. Global impact of mature biofilm lifestyle on Escherichia coli K-12 gene expression. Mol Microbiol 51:659–674. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Vinogradov EV, Bogdanove AJ. 2013. Requirement of the lipopolysaccharide O-chain biosynthesis gene wxocB for type III secretion and virulence of Xanthomonas oryzae pv. Oryzicola. J Bacteriol 195:1959–1969. doi: 10.1128/JB.02299-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Wang N. 2011. The wxacO gene of Xanthomonas citri ssp. citri encodes a protein with a role in lipopolysaccharide biosynthesis, biofilm formation, stress tolerance and virulence. Mol Plant Pathol 12:381–396. doi: 10.1111/j.1364-3703.2010.00681.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang TP, Somers EB, Wong AC. 2006. Differential biofilm formation and motility associated with lipopolysaccharide/exopolysaccharide-coupled biosynthetic genes in Stenotrophomonas maltophilia. J Bacteriol 188:3116–3120. doi: 10.1128/JB.188.8.3116-3120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amini S, Goodarzi H, Tavazoie S. 2009. Genetic dissection of an exogenously induced biofilm in laboratory and clinical isolates of E. coli. PLoS Pathog 5:e1000432. doi: 10.1371/journal.ppat.1000432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balestrino D, Ghigo JM, Charbonnel N, Haagensen JA, Forestier C. 2008. The characterization of functions involved in the establishment and maturation of Klebsiella pneumoniae in vitro biofilm reveals dual roles for surface exopolysaccharides. Environ Microbiol 10:685–701. doi: 10.1111/j.1462-2920.2007.01491.x. [DOI] [PubMed] [Google Scholar]
- 45.Joseph LA, Wright AC. 2004. Expression of Vibrio vulnificus capsular polysaccharide inhibits biofilm formation. J Bacteriol 186:889–893. doi: 10.1128/JB.186.3.889-893.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davey ME, Duncan MJ. 2006. Enhanced biofilm formation and loss of capsule synthesis: deletion of a putative glycosyltransferase in Porphyromonas gingivalis. J Bacteriol 188:5510–5523. doi: 10.1128/JB.01685-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ong CL, Ulett GC, Mabbett AN, Beatson SA, Webb RI, Monaghan W, Nimmo GR, Looke DF, McEwan AG, Schembri MA. 2008. Identification of type 3 fimbriae in uropathogenic Escherichia coli reveals a role in biofilm formation. J Bacteriol 190:1054–1063. doi: 10.1128/JB.01523-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu MC, Lin TL, Hsieh PF, Yang HC, Wang JT. 2011. Isolation of genes involved in biofilm formation of a Klebsiella pneumoniae strain causing pyogenic liver abscess. PLoS One 6:e23500. doi: 10.1371/journal.pone.0023500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Karwacki MT, Kadouri DE, Bendaoud M, Izano EA, Sampathkumar V, Inzana TJ, Kaplan JB. 2013. Antibiofilm activity of Actinobacillus pleuropneumoniae serotype 5 capsular polysaccharide. PLoS One 8:e63844. doi: 10.1371/journal.pone.0063844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clifford JC, Rapicavoli JN, Roper MC. 2013. A rhamnose-rich O-antigen mediates adhesion, virulence and host colonization for the xylem-limited phytopathogen, Xylella fastidiosa. Mol Plant Microbe Interact 26:676–685. doi: 10.1094/MPMI-12-12-0283-R. [DOI] [PubMed] [Google Scholar]
- 51.Chassaing B, Garénaux E, Carriere J, Rolhion N, Guérardel Y, Barnich N, Bonnet R, Darfeuille-Michaud A. 2015. Analysis of the σE regulon in Crohn's disease-associated Escherichia coli revealed involvement of the waaWVL operon in biofilm formation. J Bacteriol 197:1451–1465. doi: 10.1128/JB.02499-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ciornei CD, Novikov A, Beloin C, Fitting C, Caroff M, Ghigo JM, Cavaillon JM, Adib-Conquy M. 2010. Biofilm-forming Pseudomonas aeruginosa bacteria undergo lipopolysaccharide structural modifications and induce enhanced inflammatory cytokine response in human monocytes. Innate Immun 16:288–301. doi: 10.1177/1753425909341807. [DOI] [PubMed] [Google Scholar]
- 53.Coulon C, Vinogradov E, Filloux A, Sadovskaya I. 2010. Chemical analysis of cellular and extracellular carbohydrates of a biofilm-forming strain Pseudomonas aeruginosa PA14. PLoS One 5:e14220. doi: 10.1371/journal.pone.0014220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izano EA, Sadovskaya I, Vinogradov E, Mulks MH, Velliyagounder K, Ragunath C, Kher WB, Ramasubbu N, Jabbouri S, Perry MB, Kaplan JB. 2007. Poly-N-acetylglucosamine mediates biofilm formation and antibiotic resistance in Actinobacillus pleuropneumoniae. Microb Pathog 43:1–9. doi: 10.1016/j.micpath.2007.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewenza S. 2013. Extracellular DNA-induced antimicrobial peptide resistance mechanisms in Pseudomonas aeruginosa. Front Microbiol 4:21. doi: 10.3389/fmicb.2013.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Izano EA, Amarante MA, Kher WB, Kaplan JB. 2008. Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl Environ Microbiol 74:470–476. doi: 10.1128/AEM.02073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spiers AJ, Rainey PB. 2005. The Pseudomonas fluorescens SBW25 wrinkly spreader biofilm requires attachment factor, cellulose fibre and LPS interactions to maintain strength and integrity. Microbiology 151:2829–2839. doi: 10.1099/mic.0.27984-0. [DOI] [PubMed] [Google Scholar]
- 58.Bahl N, Du R, Winarsih I, Ho B, Tucker-Kellogg L, Tidor B, Ding JL. 2011. Delineation of lipopolysaccharide (LPS)-binding sites on hemoglobin: from in silico predictions to biophysical characterization. J Biol Chem 286:37793–37803. doi: 10.1074/jbc.M111.245472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clark RW, Cunningham D, Cong Y, Subashi TA, Tkalcevic GT, Lloyd DB, Boyd JG, Chrunyk BA, Karam GA, Qiu X, Wang IK, Francone OL. 2010. Assessment of cholesteryl ester transfer protein inhibitors for interaction with proteins involved in the immune response to infection. J Lipid Res 51:967–974. doi: 10.1194/jlr.M002295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Otto K, Silhavy TJ. 2002. Surface sensing and adhesion of Escherichia coli controlled by the Cpx-signaling pathway. Proc Natl Acad Sci U S A 99:2287–2292. doi: 10.1073/pnas.042521699. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.