

Abstract
Objectives
Recently, recurrent mutations in regulatory DNA regions, such as promoter mutations in the TERT gene were identified in melanoma. Subsequently, Weinhold et al. reported SDHD promoter mutations occurring in 10% of melanomas and being associated with a lower overall survival rate. Our study analyzes the mutation rate and clinico-pathologic associations of SDHD promoter mutations in a large cohort of different melanoma subtypes.
Methods
451 melanoma samples (incl. 223 non-acral cutaneous, 38 acral, 33 mucosal, 43 occult, 43 conjunctival and 51 uveal melanoma) were analyzed for the presence of SDHD promoter mutations by Sanger-sequencing. Statistical analysis was performed to screen for potential correlations of SDHD promoter mutation status with various clinico-pathologic criteria.
Results
The SDHD promoter was successfully sequenced in 451 tumor samples. ETS binding site changing SDHD promoter mutations were identified in 16 (4%) samples, of which 5 mutations had not been described previously. Additionally, 5 point mutations not located in ETS binding elements were identified. Mutations in UV-exposed tumors were frequently C>T. One germline C>A SDHD promoter mutation was identified. No statistically significant associations between SDHD promoter mutation status and various clinico-pathologic variables or overall patient survival were observed.
Conclusions
Melanomas harbor recurrent SDHD promoter mutations, which occur primarily as C>T alterations in UV-exposed melanomas. In contrast to the initial report and promoter mutations in the TERT gene, our analysis suggests that SDHD promoter mutations are a relatively rare event in melanoma (4% of tumors) of unclear clinical and prognostic relevance.
Keywords: melanoma, SDHD, promoter mutations
INTRODUCTION
Melanoma continues to be a major health burden worldwide [1, 2]. Effective removal of the tumor at an early stage remains the only reliable curative treatment. Although an impressive number of new systemic therapeutic approaches have become available in recent years [3–9], the long-term outlook for patients with metastatic disease remains poor.
A number of landmark genetic studies, primarily focusing on analyzing protein coding genes, have identified a large number of recurrently mutated genes in melanoma [10, 11]. In contrast to previously recognized mutations such as BRAF and NRAS, most of these genes are mutated less frequently (i.e. NF1, RAC1, ARID1A, etc.); their function and the clinical implications of these mutations are still poorly understood.
Recent efforts have moved beyond focusing primarily on protein coding genes and have identified mutations in non-protein coding areas. Potentially the most relevant such mutation identified to date was the finding of TERT promoter mutations in a large proportion of melanoma samples (30–70%) [12, 13]. These mutations were found to generate novel transcription factor binding sites increasing telomerase expression and have been associated with poor prognosis [14]. In search of novel recurrent mutations that alter transcription factor binding sites, Weinhold et al. recently reported recurrent mutations of the succinate dehydrogenase complex subunit D (SDHD) promoter in melanoma [15].
SDHD is one of two mitochondrial transmembrane subunits of the four-subunit succinate dehydrogenase (SDH) protein. SDH is an enzyme with two important functions. First, it acts as part of the citric acid cycle converting succinate to fumarate. Succinate functions as an oxygen sensor in the cell and stimulates cell growth in a hypoxic environment, in particular by stabilizing hypoxia-inducible factor (HIF), which controls several genes involved in cell division and the formation of new blood vessels [16–18]. Loss of SDH enzyme activity can lead to abnormal hypoxia signaling, leading to proliferation and tumor formation. The second known function of SDH is oxidative phosphorylation, an important process for the cell's energy budget.
Mutations in SDHD have been described in gastrointestinal stromal tumor (GIST) [19], paraganglioma [20, 21] and pheochromocytoma [22, 23]. Promoter mutations of SDHD in 13 of 128 (10%) melanomas were recently described by Weinhold et al. in a genome-wide analysis screening for mutations in noncoding regulatory regions of the DNA [15]. All three described recurrent hotspot mutations in the SDHD promoter region substitute a cytosine for a thymine nucleotide (C>T). The mutations are located at chr.11:111,957,523 (TTCC>TTTC), chr.11:111,957,541 (TTCC>TTTC) and chr.11:111,957,544 (CTTCC>TTTCC). The TTCC response element is highly conserved for E26 transformation-specific (ETS) transcription factors. The mutations alter existing ETS binding motifs predicted to lead to a reduced expression of the SDHD gene.
The aim of our study was to further evaluate the incidence of SDHD promoter mutations in a large cohort of melanoma samples of various subtypes and to investigate associations of SDHD mutation status with clinico-pathologic variables and other common oncogenic mutations in melanoma, such as BRAF, NRAS, KIT and TERT promoter mutations.
RESULTS
Tumors and patients
The SDHD promoter was successfully sequenced in 451 melanoma samples available for analysis, including 223 non-acral cutaneous, 38 acral, 33 mucosal, 43 occult, 43 conjunctival and 51 uveal melanoma samples. Based on lack of detected SDHD promoter mutations (addressed below), the 51 primary uveal melanoma samples were excluded from further statistical analyses. The 400 non-uveal melanoma samples included 167 primary tumors, 158 metastases, 5 recurrences, 43 occult and 27 not-classified tumor samples. The samples originated from 230 male and 170 female patients with a median age of 60 years (range 12–90 years) and a median follow-up time of 30 months (range 0.3–375 months) The clinico-pathologic information is summarized in Table 1.
Table 1. Characteristics of all tumor samples in regard to SDHD promoter status (n = 400).
| All samples | ||||||||
|---|---|---|---|---|---|---|---|---|
| Total (All samples) | SDHD WT | SDHD mut | ||||||
| N | % | N | % | N | % | P | ||
| Sex | Female | 170 | 42.5 | 163 | 41 | 7 | 2.3 | 0.98 |
| Male | 230 | 57.5 | 221 | 55 | 9 | 1.7 | ||
| Age at Diagnosis | Median | 60 | ||||||
| Range | 12–90 | |||||||
| <=60 years | 191 | 47.5 | 174 | 44 | 6 | 1.5 | 0.45 | |
| >60 years | 172 | 42.8 | 174 | 44 | 7 | 1.8 | ||
| Missing data | 39 | 9.7 | 36 | 9 | 3 | 0.8 | ||
| Mutant oncogene** | WT | 150 | 40 | 145 | 39 | 5 | 1.3 | 0.89 |
| BRAF* | 142 | 38 | n = 134 | 35 | 8 | 2 | ||
| NRAS* | 82 | 22 | 79 | 21 | 3 | 0.8 | ||
| KIT | 4 | 1 | 4 | 1 | 0 | 0 | ||
| TERT prom. WT (underscribt) | 115 | 55 | 112 | 53 | 3 | 1.4 | 0.38 | |
| TERT prom. mut | 95 | 45 | 89 | 42 | 6 | 2.9 | ||
| BRAF and NRAS | Either mutant* | 222 | 59 | n = 211 | 56 | 11 | 2.9 | 0.42 |
| Both WT | 154 | 41 | 149 | 40 | 5 | 1.3 | ||
| Stage at diagnosis# | I | 46 | 12 | 44 | 11 | 2 | 0.5 | 0.88 |
| II | 122 | 31 | 118 | 30 | 4 | 1 | ||
| III | 122 | 31 | 118 | 30 | 4 | 1 | ||
| IV | 29 | 7 | 27 | 7 | 2 | 0.5 | ||
| Missing data | 81 | 20 | 77 | 19 | 4 | 1 | ||
| Anatomic distribution of primary | Non-acral skin | 223 | 56 | 214 | 54 | 9 | 2.3 | 0.9 |
| Acral | 38 | 10 | 38 | 9 | 0 | 0 | ||
| Mucosal | 33 | 8 | 32 | 8 | 1 | 0.3 | ||
| Occult | 43 | 11 | 40 | 10 | 3 | 0.8 | ||
| Eye (conj.) | 43 | 11 | 42 | 11 | 1 | 0.3 | ||
| Missing data | 20 | 5 | 18 | 5 | 2 | 0.5 | ||
| Anatomic site of skin and acral tumors | Head & neck | n = 40 | 10% | n = 39 | 9.8% | 1 | 0.3 | 0.5 |
| Upper limbs | 23 | 6 | 22 | 6 | 1 | 0.3 | ||
| Trunk | n = 94 | 24 | n = 88 | 22% | n = 6 | 1.5% | ||
| Lower limbs | 64 | 16 | 63 | 16 | 1 | 0.3 | ||
| Acral | 38 | 10 | 38 | 10 | 0 | 0 | ||
| Occult | 43 | 11 | n = 40 | 9 | 3 | 0.8 | ||
| Missing data | n = 22 | 6 | n = 20 | 5% | 2 | 0.5 | ||
| Histologic type | ALM | 30 | 8 | 29 | 7 | 1 | 0.3 | 0.9 |
| LMM | 4 | 1 | 4 | 1 | 0 | 0 | ||
| NM | 77 | 23 | 75 | 19 | 2 | 0.5 | ||
| SSM | 46 | 12 | 43 | 11 | 3 | 0.8 | ||
| Unclassified | 243 | 61 | 233 | 58 | 10 | 2.1 | ||
| Breslow thickness | Median | 3 | ||||||
| Range | 0.1–55.0 | |||||||
| 0.01–1.00mm | 36 | 9 | 34 | 8,5 | 2 | 0.5 | 0.58 | |
| 1.01–2.00mm | 42 | 11 | 39 | 10 | 3 | 0.8 | ||
| 2.01–4.00mm | 82 | 21 | 80 | 20 | 2 | 0.5 | ||
| >4.00mm | 93 | 24 | 91 | 23 | 2 | 0.5 | ||
| Missing data | 147 | 37 | 140 | 35 | 7 | 1.8 | ||
| Clark level (skin tumors only) | I | 0 | 0 | 0 | 0 | 0 | 0 | 0.89 |
| II | 5 | 2 | 5 | 2 | 0 | 0 | ||
| III | 37 | 17 | 34 | 15 | 3 | 1.3 | ||
| IV | 74 | 33 | 72 | 32 | 2 | 0.9 | ||
| V | 21 | 9 | 20 | 9 | 1 | 0.5 | ||
| Unknown | 86 | 22% | n = 83 | 21% | 3 | 1.3 | ||
| Sample type sequenced | Primary | 167 | 42 | 163 | 41 | 4 | 1 | 0.34 |
| Metastasis | 158 | 40 | 149 | 37 | 9 | 2.3 | ||
| Recurrence | 5 | 1 | 5 | 1 | 0 | 0 | ||
| Occult | 43 | 11 | 40 | 10 | 3 | 0.8 | ||
| Missing data | 27 | 7 | 27 | 7 | 0 | 0 | ||
| Ulceration | Absent | 62 | 16 | 59 | 15 | 3 | 0.8 | 0.48 |
| Present | 88 | 22 | 87 | 22 | 1 | 0.3 | ||
| Unknown | 250 | 63 | 238 | 60 | 12 | 3.1 | ||
| SLN | Negative | 78 | 20 | 75 | 19 | 3 | 0.8 | 0.97 |
| Positive | 84 | 21 | 81 | 20 | 3 | 0.8 | ||
| Not done | 238 | 60 | 228 | 57 | 10 | 2.5 | ||
WT = wild-type; mut = mutant; ALM = acral lentiginous melanoma; NM = nodular melanoma; SSM = superficial spreading melanoma; LMM = lentigo maligna melanoma; SLN = sentinel lymph node; conj. = conjunctival; prom. = promoter
p-values are derived from chi-squared or Fisher exact tests, as appropriate
Staging according to the American Joint Committee on Cancer (AJCC) Melanoma Staging System 2009[37]
2 cases harbored a BRAF and a NRAS mutation.
BRAF, NRAS, KIT were screened in n = 376; the TERT promoter in n = 210 cases
Oncogene mutations
BRAF mutations occurred in 38% (142/376) of the tumor samples, including 128 (90%) V600E and 8 (6%) V600K. Additionally, individual (1%) V600G, V600D, K601N, K601E, G469A and D594N mutations were identified. NRAS mutations were found in 82 (22%) tumor samples, including 37 (45%) Q61R, 25 (25%) Q61K, 14 (17%) Q61L, 4 (5%) Q61H and 2 (1%) G12D mutations. KIT mutations were found in 4 (1%) cases. TERT promoter mutations occurred in 95 of 210 analyzed cases (45%), including 52 (55%) chr.5:1,295,250C>T, 31 (33%) chr.5:1,295,228C>T, 11 (12%) chr.5:1,295,242_1,295,243CC>TT and 1 (1%) chr.5:1,295,228_1,295,229CC>TT mutations (Table 1).
SDHD promoter mutation analysis
ETS binding site affecting SDHD promoter mutations were identified in 16 of 400 tumors (4%), obtained from 7 female and 9 male patients. Eleven mutations were identified at the previously described locations [15]: chr.11:111,957,523 (TTCC>TTTC), chr.11:111,957,541 (TTCC>TTTC) and chr.11:111,957, 544 (CTTCC>TTTCC). Furthermore, we found five mutations occurring at two hotspots (Figure 1) located at chr.11:111,957,542 (TTCC>TTCA) and chr.11:111,957, 547 (CTTCC>CTTTC or CTTCC>CCTAC), which were not yet described, but also result in sequence alterations of the ETS-binding element (Figure 1). In the remaining manuscript, only the last three digits of the chromosome location nomenclature will be used for annotating the mutations location (i.e. 523C>T). The SDHD promoter mutations identified included 5 (1.3%) 523C>T cases, 3 (0.8%) 541C>T, 3 (0.8%) 542C>A, 3 (0.8%) 544C>T cases, 1 (0.3%) 547C>T and 1 (0.3%) 547C>A case (Table 2).
Figure 1. Recurrent SDHD promoter mutations altering ETS binding elements.
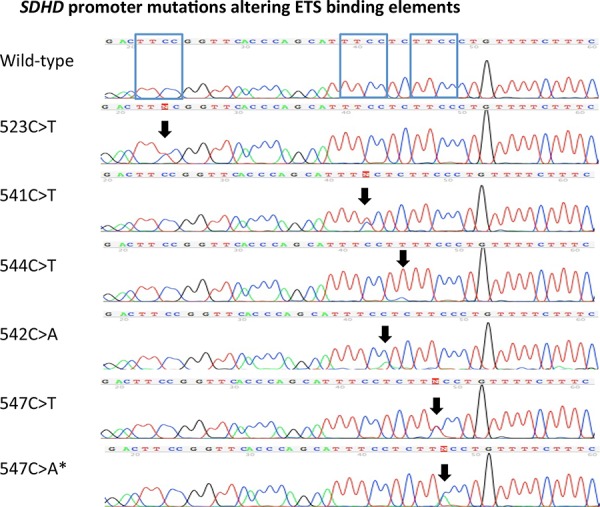
Sanger sequencing chromatograms of the identified recurrent SDHD promoter mutations located at chr.11:111,957,523C>T(TTCC>TTTC), chr.11:111,957,541C > T (TTCC>TTTC), chr.11:111,957,544C>T (CTTCC>TTTCC), chr.11:111,957,542C>A (TTCC>TTCA) and chr.11:111,957,547 (CTTCC>CTTTC and CTTCC>CTTAC) (according to human genome assembly [hg19]). The mutations locations are highlighted with black arrows. A wild-type promoter sequence for comparison is shown on the top. The blue boxes in the wild-type SDHD promoter sequence show the three different ETS binding elements. * signifies the mutation also identified in a germ-line sample.
Table 2. SDHD promoter mutations in association to clinico-pathologic variables.
| Affecting ETS domains | Not affecting ETS domains | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SDHD promoter mutation | 523C>T | 541C>T | 542C>A | 544C>T | 547C>T | 547C>A | Total | 529C>T | 538C>A | 550T>A | 556C>T | Total | |
| N | N | N | N | N | N | N | N | N | N | N | N | ||
| Sex | Female | 1 | 3 | 2 | 0 | 1 | 0 | 7 | 0 | 1 | 1 | 1 | 3 |
| Male | 4 | 0 | 1 | 3 | 0 | 1 | 9 | 1 | 1 | 0 | 0 | 2 | |
| DNA type | Primary | 3 | 1 | 0 | 0 | 0 | 0 | 4 | 0 | 0 | 0 | 0 | 0 |
| Metastasis | 2 | 2 | 1 | 3 | 1 | 0 | 9 | 1 | 1 | 1 | 1 | 4 | |
| Occult | 0 | 0 | 2 | 0 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | |
| Missing data | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | |
| Mutant oncogene | BRAF | 2 | 1 | 2 | 2 | 1 | 0 | 8 | 0 | 0 | 1 | 0 | 1 |
| NRAS | 1 | 0 | 0 | 1 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | |
| KIT | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| TERT prom. mut | 0 | 2 | 2 | 2 | 0 | 0 | 6 | 1 | 1 | 1 | 0 | 3 | |
| BRAF and NRAS | Either mutant | 3 | 1 | 2 | 2 | 1 | 0 | 9 | 0 | 0 | 1 | 0 | 1 |
| Both WT | 2 | 2 | 1 | 1 | 0 | 1 | 7 | 1 | 2 | 0 | 1 | 4 | |
| Clinical stage at diagnose# | I | 1 | 0 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| II | 0 | 3 | 0 | 1 | 0 | 0 | 4 | 1 | 1 | 1 | 0 | 3 | |
| III | 2 | 0 | 1 | 1 | 0 | 0 | 4 | 0 | 0 | 0 | 1 | 1 | |
| IV | 0 | 0 | 1 | 0 | 0 | 1 | 2 | 0 | 0 | 0 | 0 | 0 | |
| Missing data | 2 | 0 | 1 | 0 | 1 | 0 | 4 | 0 | 1 | 0 | 0 | 1 | |
| Anatomic distribution of primary | Non-acral | 3 | 3 | 1 | 2 | 0 | 0 | 9 | 1 | 1 | 1 | 1 | 4 |
| Acral | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| Mucosal | 0 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Occult | 0 | 0 | 1 | 1 | 0 | 1 | 3 | 0 | 0 | 0 | 0 | 0 | |
| Eye (conj.) | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| Missing data | 1 | 0 | 0 | 0 | 1 | 0 | 2 | 0 | 1 | 0 | 0 | 1 | |
| Germline | Analyzed samples | 4 | 1 | 3 | 2 | 1 | 1 | 12/16 | 0 | 2 | 1 | 1 | 4/5 |
| Mutant (+/−) | − | − | − | − | − | + | − | − | − | − | |||
WT = wild-type; mut = mutant; prom. = promoter; conj. = conjunctival
Staging according to the American Joint Committee on Cancer (AJCC) Melanoma Staging System 2009[37]
In addition to the ETS binding site affecting mutations, SDHD promoter mutations not affecting the ETS binding elements were identified in 5 tumor samples. These mutations included individual chr.11:111,957,529 (TTCA>TTTA), chr.11:111,957,550 (CCCT>CCCA) and chr.11:111,957,556 (TTCT>TTTT) mutations, as well as two chr.11:111,957,538 (TTTCC>ATTCC) mutations (Figure 2).
Figure 2. SDHD promoter mutations outside of ETS binding elements.
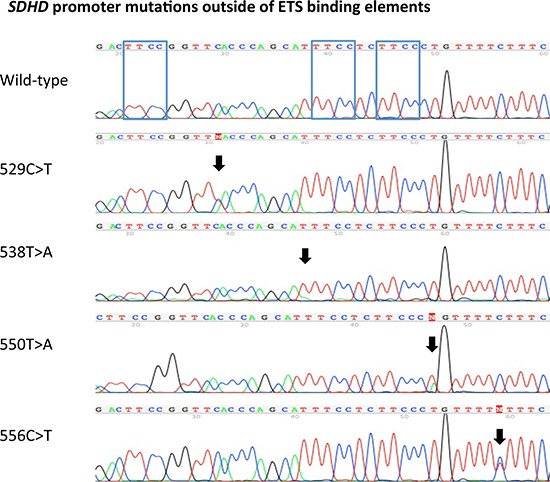
Sanger sequencing chromatograms of the identified SDHD promoter mutations located at chr.11:111,957,529 (TTCA>TTTA), chr.11:111,957,538 (TTTCC>ATTCC) chr.11:111,957,550 (CCCT>CCCA), and chr.11:111,957,556 (TTCT>TTTT) (according to human genome assembly [hg19]). The location of the mutations is highlighted with black arrows. The top chromatogram demonstrates a wild-type promoter sequence for comparison. The blue boxes in the wild-type SDHD promoter sequence show the three different ETS binding elements.
None of the 51 uveal tumor samples analyzed harbored a SDHD promoter mutation.
Germline SDHD promoter analysis
For tumors in which SDHD promoter mutations were identified, matching constitutional DNA from peripheral blood mononuclear cells was analyzed if available. This was the case in 12 of the 16 tumors with SDHD promoter mutations affecting ETS binding sites and 4 of 5 tumor samples with SDHD promoter mutations not affecting ETS binding sites. One of the ETS binding site affecting mutations (547C>A) was found to be present in the germline (Figure 3). In all other cases, SDHD promoter mutations detected in the tumor were not present in the constitutional DNA, confirming they were acquired somatically (Table 2).
Figure 3. Germ-line SDHD promoter mutation.

Sanger sequence chromatograms show a chr.11:111,957,547C > A SDHD promoter mutation present both in the patient's melanoma sample as well as the germ-line. PBMC = peripheral blood mononuclear cells.
Clinico-pathologic correlations of SDHD promoter mutant tumors
For the analyses presented in the paper, only those SDHD promoter mutations affecting the ETS domains were deemed to be relevant (n = 16). However, no significant differences were observed when the samples harboring SDHD promoter mutations not affecting ETS binding domains were also included in the analysis (data not shown).
ETS binding site affecting SDHD promoter mutations were detected in 9 metastatic and 4 primary tumor samples (Table 1). Non-acral cutaneous melanomas harbored 9 of the 16 mutations (56%). One mutation each was found in a conjunctival and mucosal melanoma tumor sample. Three mutations occurred in occult tumor samples and in two cases anatomic site information was not available (Table 2). Of the 5 SDHD promoter mutations not affecting the ETS binding domains, 4 were in metastatic samples of non-acral cutaneous melanoma. No anatomic site information was available for the remaining case.
No statistically significant associations of SDHD promoter mutation status with available clinical parameters including stage at diagnosis, Breslow thickness, Clark level, presence of ulceration, histologic type and mutation status (BRAF, NRAS, KIT, TERT promoter) were identified (Table 1, Supplemental Table 1 + 2). This was the case regardless of whether selectively ETS binding site mutations (Table 1) or all identified mutations were considered (data not shown).
In our cohort, 123 patients died, on average 52 months (range 2–375) after diagnosis. Six patients with SDHD promoter mutation died during the follow-up period. A statistically significant association between SDHD promoter mutation and overall survival was not found (Figure 4).
Figure 4. Survival based on stage at diagnosis and SDHD promoter mutation status.
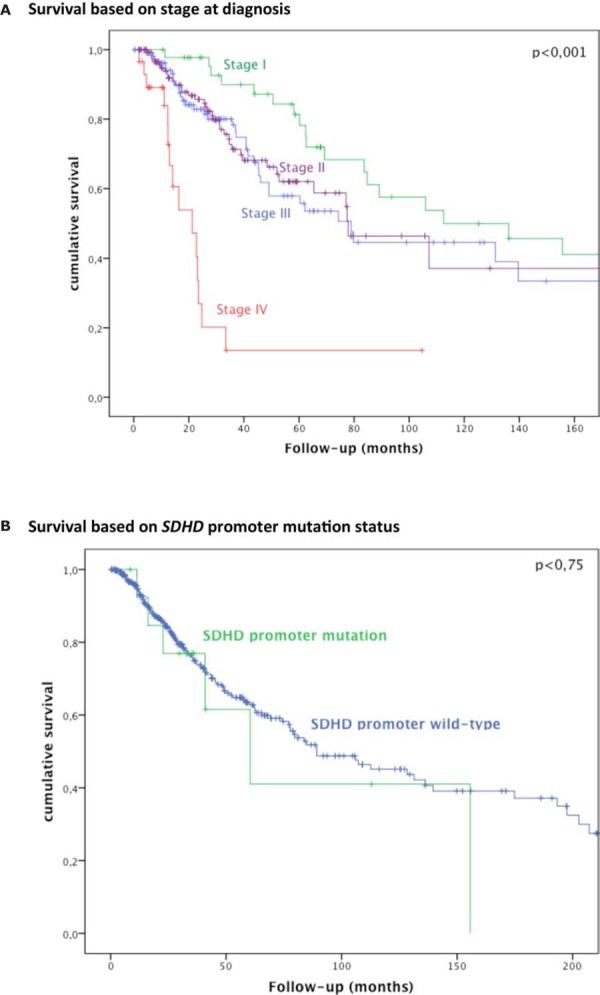
Kaplan-Meier curves of overall survival in 400 patients with melanoma according to A. stage at diagnosis; B. SDHD promoter status (mutant vs wild-type).
SDHD promoter mutations in existing exome data
To explore the general frequency of SDHD promoter mutations and potential relevance for therapy resistance in melanoma, we re-analyzed exomes sequenced from 69 melanoma patients under MAPKi therapy [24]. DNA outside the targeted coding regions can be assessed if pulled down along with the targets during the capture process. Of 183 available exomes, sufficient coverage in the SDHD promoter region was obtained in 126 (69%) cases (min. 10x, max. 72.8x, average 22.7x). In the 92 tumor exomes with sufficient coverage we detected 4 SDHD promoter variants (4.3%, Supplemental Table 3). No mutations were detected in germline samples.
DISCUSSION
In our study of a large cohort of ocular, cutaneous, mucosal and occult melanomas, SDHD promoter mutations affecting recurrent ETS binding elements were identified in only 4% (16/400) of the samples. Most mutations detected were at previously reported hotspots (n = 11) [15], with a C>T UV-signature [25, 26]. Additionally, 10 other mutations were identified, five of which altered the sequence of ETS transcription factor binding elements. Our study validates the finding of recurrent mutations in the SDHD promoter; however these mutations were considerably rarer than previously reported and showed no association with prognosis or the clinico-pathologic variables that were analyzed.
Most of the identified SDHD promoter mutations altering the TTCC element of the ETS transcription binding sites were found in one of the three previously described mutation hotspots located at chr.11:111,957,523, chr.11:111,957,541 and chr.11:111,957,544 (Figure 1). However, 5 additional mutations were identified at two previously undescribed hotspots, three mutations at Chr.11:111,957,542 (TTCC>TTCA) and two mutations at Chr.11:111,957,547 (CTTCC>CTTTC, CTTCC>CTTAC) (Figure 1). All of these newly identified mutations alter the ETS binding site core element (TTCC). In contrast, the previously reported recurrent 544C>T mutation is located just outside the core element (CTTCC>TTTCC, Figure 1). The altered nucleotide in this setting is conserved in a number of ETS transcription factors including ELF1 [15]. It would appear logical that the various mutations exert differing effects on SDHD gene transcription and protein translation. Detailed functional studies will be required to elucidate the extent to which these mutations differ in terms of their effect on transcriptional regulation of SDHD.
Mutation frequencies among melanoma subtypes did not vary greatly: non-acral cutaneous 4% (9 of 223), mucosal 3% (1 of 33), acral 0% (0 of 38), conjunctival 2% (1 of 43), occult 7% (3 of 43). The majority of ETS binding site altering SDHD promoter mutations (9 of 16 = 56%) were identified in the non-acral cutaneous melanoma. C>T mutations, which are a marker of UV-exposure, were observed only in these tumors. A single mucosal melanoma (1 of 33, 3%) harbored a 542C>A alteration; no mutations in acral (n = 38) melanomas were identified (Table 2). In contrast to C>T mutations, C>A alterations are not typical for UV induction. Additionally, no mutations were identified in uveal melanoma samples (n = 51), a tumor also lacking association with UV exposure [27]. The type of mutations identified does support SDHD promoter mutations in UV-exposed tumors (i.e. non-acral cutaneous) being primarily UV-induced, whereas the 542C>A mutation occurring in a mucosal, non UV-exposed melanoma probably developed in a UV-independent fashion. The overall lack of mutations in uveal melanoma further supports their being genetically distinct from cutaneous, mucosal and conjunctival melanoma [28–30].
The significance of the 5 SDHD promoter mutations identified outside of the three existing ETS binding elements (Figure 2) is unclear. Given that melanoma has a particularly high frequency of mutations [31], many of unclear functional relevance, it is possible that the identified alterations are simply passenger mutations. The only recurrent mutation, chr.11:111,957,538 (TTTCC>ATTCC), identified in two tumors, is located just outside of the ETS core element, similar to the previously described 544C>T mutation, however resulting in a different sequence (ATTCC versus TTTCC, respectively). Detailed functional studies will be required to determine the significance of these alterations. To exclude the possibility that these mutations might have (as yet unknown) functional importance, all statistical analyses were also performed including all 21 samples with SDHD promoter mutations (independent of their known effects on ETS binding sites). The results were similar in that no significant associations with various clinico-pathologic parameters including survival were observed.
The identification of a germline SDHD promoter mutation altering the ETS binding site is intriguing. The patient with the 547C>A germline mutation was a 79-old male presenting with stage IV disease upon initial diagnosis of an occult Melanom, which harbored an NRAS Q61K mutation (in addition to the 547C>A SDHD promoter mutation). The patient died of melanoma 4.5 months after diagnosis. Unfortunately, no detailed information on the patient and his family were available. Although the age of the affected patient argues against the role of this mutation in increasing the risk of melanoma, larger numbers of patients will be needed to adequately address this question.
There is a difference in mutation frequency observed between our study (∼4%) and the previous one by Weinhold et al. (10%). This could simply be due to differences in cohort characteristics and sample sizes (Weinhold et al. analyzed 128 samples). Another relevant factor may be the difference in experimental approaches applied. Weinhold et al. analyzed existing next generation sequencing data, searching for recurrent transcription factor mutations in the promoter region of genes, without sequencing validation. Our approach relied on targeted Sanger-sequencing. Both approaches have advantages and disadvantages. Sanger-sequencing does have a detection limit of ∼20%, and the potential to miss low-level mutations. On the other hand, next-generation sequencing approaches are not error-free, and can report incorrect results based on sequencing errors or various bioinformatic analytic hurdles.
To compare our Sanger sequencing results to NGS data, we explored an existing exome dataset detecting SDHD promoter variants in ∼4% of tumors. These variants were equally distributed in pre-treatment and recurrent tumors, showing no obvious association of SDHD variants with therapy resistance. Filter criteria for NGS analyses probably influenced the higher frequency of SDHD promoter variants in Weinhold's study. Their Bayesian methods identified the SDHD promoter as regionally recurrent in 5′UTRs (5 of 128 samples, with a FDR of 0.0016). However, the unique coverage at mutated positions and mutational frequency in the sequencing reads was not listed. Repeating our analysis without requiring a minimum of 2 unique reads to support a SDHD variant, we detected 11 variants in 92 tumors (12%). Strikingly, the additional variants were represented by a single sequencing read each and could likely represent false positives due to sequencing error. Even if one assumes some of the mutation calls could be correct, the question arises what biologic relevance such low frequency mutations may have. We believe that Sanger-sequencing remains a particularly robust form of sequence analysis and should be used whenever possible to confirm the presence of sequence variations before reporting newly-identified mutations.
No association of SDHD promoter mutation status with overall survival was seen (Figure 4). Admittedly, the number of mutated samples in our cohort is low (n = 16, or n = 21 if including SDHD promoter mutations not affecting ETS binding sites), meaning larger studies will be required to convincingly assess survival. However, Weinhold et al. reported a statistically significant (p = 0.005) survival difference with poorer prognosis for SDHD promoter mutations analyzing less mutant cases (n = 12). Given the discrepancy in our findings and taking into account Weinhold et al.′s relatively small sample size, we believe that the prognostic association of SDHD promoter mutations is yet to be unequivocally established and should be explored further in future studies.
It would be interesting to determine to which extent expression levels of SDHD protein are actually affected by SDHD promoter mutations. Considering the mutations are assumed to disrupt promoter binding sites, tumors with promoter mutations would be expected to show lower SDHD protein expression [15]. As the number of SDHD promoter mutated samples we found is very low, one can expect future studies will be required to screen large cohorts of tumors to allow a convincing statistical analysis of promoter mutation status and protein expression to be performed.
Overall, our study validates the finding of recurrent mutations in the SDHD promoter, which are enriched for mutations inactivating ETS transcription binding sites. However, in contrast to the initial report, the overall frequency of SDHD promoter mutation we identified is low (∼4%) and showed no association with poorer survival. Should these findings be validated in additional cohorts, they argue that compared to the much more frequent and prognostically relevant TERT promoter mutations, SDHD promoter mutations play a relatively minor role in melanoma.
MATERIALS AND METHODS
Sample selection
451 melanoma samples were obtained from patients treated in the Department of Dermatology or Ophthalmology of the University Hospital Essen, Germany. The samples included 223 non-acral cutaneous, 38 acral, 33 mucosal, 43 occult, 43 conjunctival and 51 uveal melanomas (Table 1). The study was performed in accordance with the guidelines put forth by the ethics committee of the University of Duisburg-Essen.
Clinical and pathologic parameters
All clinical and pathologic parameters were obtained from patient records. The following parameters were assessed: sex, age, anatomic location of the tumor, pathologic stage, histologic subtype, Breslow thickness, Clark level, sentinel lymph node status, overall survival, and correlation with other gene mutations (incl. BRAF, NRAS, KIT, TERT promoter).
DNA isolation and direct (Sanger) sequencing
Five ten-micrometer-thick sections were cut from paraffin-embedded tumor tissues and were deparaffinized. Genomic DNA was isolated using the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Polymerase chain reaction (PCR) was preformed to amplify the SDHD promoter region with the following primers: SDHD-F: ACC TTC CGA CAG CTG TGT TT, and SDHD-R: CTC AAG GTC ATC CAC CAA CC amplifying a 151-bp fragment. The PCR products were Sanger sequenced, as previously described [32]. For sequence-data analysis Chromas software was applied (version 2.01, University of Sussex, Brighton, UK). BRAF exon 15, NRAS exon 1 and 2, KIT exon 9, 11, 13, 17, 18 and the TERT promoter were PCR amplified and sequenced as previously described [32, 33]. Sequencing for BRAF, NRAS and KIT was generally performed sequentially; NRAS sequenced in BRAF wild-type samples, KIT in BRAF and NRAS wild-type samples. SDHD PCR and Sanger sequencing of peripheral blood mononuclear cells (PBMC) derived constitutional DNA was performed in 15 patients with SDHD promoter mutated tumor samples. DNA was isolated from PBMC, as described previously [34].
Statistical analyses
Associations of SDHD promoter mutations and clinico-pathologic variables, such as age, sex, primary tumor location, TNM status, histologic type, mutation status (for BRAF, NRAS, KIT and TERT promoter mutations), Breslow thickness, Clark level, ulceration and sentinel lymph node status were explored using chi-square or Fisher exact tests as appropriate. A p value < 0.05 was considered statistically significant. For all statistical analysis, SPSS Statistics software (version 22.0; SPSS Chicago, IL) was applied.
SDHD promoter analysis in existing exome data
We re-analyzed a dataset of 116 tumor and 67 germline exomes from 69 patients under MAPK inhibition (MAPKi) therapy [24] with respect to the SDHD promoter region. Bam files were indexed using samtools [35] and the hg19 human genome reference. Coverage was obtained using samtools-1.0 mpileup and the unix tools awk [36] and sed (http://www.gnu.org/software/sed/). Variant calls were performed using samtools-1.0 mpileup and bcftools query at positions 111,957,519 to 111,957,551 of chromosome 11 (32bp). A minimum average coverage of 10 unique reads across the 32bp region of the SDHD promoter was required and variants called when supported by at least 2 unique sequence reads.
SUPPLEMENTARY TABLES
Acknowledgments
We would like to thank Nadine Stadler and Nicola Bielefeld for their excellent technical support. We thank Elizer Van Allen for providing access to exome data.
Abbreviations
- NGS
next generation sequencing
- ETS
E26 transformation-specific
- SDHD
Succinate dehydrogenase complex subunit D
Footnotes
FUNDING
The research was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) Grant Nr. GR 3671/3–1. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions
Literature search: S.L.S., K.G.G., H.W., R.M., S.H. M.S., D.S. Study design: S.L.S., K.G.G., S.H., M.S., H.W., D.S. Data collection: K.G.G., H.W., S.H., M.S., A.S., I.M., C.M. Data analysis: S.l.S., S.H., M.S., K.G.G., I.M., A.S., E.L., W.S., H.W., D.S. Data interpretation: S.L.S., K.G.G., M.S., S.H., H.R., E.L., B.S., L.Z., R.M., B.S., E.L., M.Z., C.M., K.P.S., H.W., A.P. Manuscript writing: all authors.
CONFLICTS OF INTEREST
Elisabeth Livingstone has received honoraria from Roche, Bristol-Myers Squibb, Amgen, Boehringer-Ingelheim, Merck Sharp & Dohme and Merck, and travel support from Bristol-Myers Squibb. Lisa Zimmer has honoraria from Roche, Bristol-Meyers Squibb, and Amgen, and travel support from Merck Sharp & Dohme and Bristol-Meyers Squibb. Bastian Schilling has received honoraria from Roche and travel support as well research funding from Bristol-Myers Squibb. Dirk Schadendorf is on the advisory board or has received honoraria from Roche, Genentech, Novartis, Amgen, GlaxoSmithKline, Bristol-Myers Squibb, Boehringer Ingelheim, and Merck Sharp & Dohme. All other authors have nothing to declare.
REFERENCES
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA: a cancer journal for clinicians. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2013;380:2095–2128. doi: 10.1016/S0140-6736(12)61728-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS, Dronca R, Gangadhar TC, Patnaik A, Zarour H, Joshua AM, Gergich K, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. The New England journal of medicine. 2013;369:134–144. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, et al. Nivolumab plus ipilimumab in advanced melanoma. The New England journal of medicine. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lipson EJ, Sharfman WH, Drake CG, Wollner I, Taube JM, Anders RA, Xu H, Yao S, Pons A, Chen L, Pardoll DM, Brahmer JR, Topalian SL. Durable cancer regression off-treatment and effective reinduction therapy with an anti-PD-1 antibody. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:462–468. doi: 10.1158/1078-0432.CCR-12-2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32:1020–1030. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ascierto PA, Schadendorf D, Berking C, Agarwala SS, van Herpen CM, Queirolo P, Blank CU, Hauschild A, Beck JT, St-Pierre A, Niazi F, Wandel S, Peters M, Zubel A, Dummer R. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: a non-randomised, open-label phase 2 study. The lancet oncology. 2013;14:249–256. doi: 10.1016/S1470-2045(13)70024-X. [DOI] [PubMed] [Google Scholar]
- 8.Long GV, Stroyakovskiy D, Gogas H, Levchenko E, de Braud F, Larkin J, Garbe C, Jouary T, Hauschild A, Grob JJ, Chiarion Sileni V, Lebbe C, Mandala M, Millward M, Arance A, Bondarenko I, et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. The New England journal of medicine. 2014;371:1877–1888. doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 9.Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, Hauschild A, Lorigan P, Wolter P, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. The New England journal of medicine. 2015;372:30–39. doi: 10.1056/NEJMoa1412690. [DOI] [PubMed] [Google Scholar]
- 10.Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, Ariyan S, Narayan D, Dutton-Regester K, Capatana A, Holman EC, Bosenberg M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nature genetics. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, Schadendorf D, Kumar R. TERT promoter mutations in familial and sporadic melanoma. Science. 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- 13.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science. 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, Carrera C, Schimming T, Moller I, Schwamborn M, Sucker A, Hillen U, Badenas C, Malvehy J, Zimmer L, Scherag A, et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. Journal of the National Cancer Institute. 2014;106 doi: 10.1093/jnci/dju246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nature genetics. 2014;46:1160–1165. doi: 10.1038/ng.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 17.Gimenez-Roqueplo AP, Favier J, Rustin P, Mourad JJ, Plouin PF, Corvol P, Rotig A, Jeunemaitre X. The R22X mutation of the SDHD gene in hereditary paraganglioma abolishes the enzymatic activity of complex II in the mitochondrial respiratory chain and activates the hypoxia pathway. American journal of human genetics. 2001;69:1186–1197. doi: 10.1086/324413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, et al. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Human molecular genetics. 2005;14:2231–2239. doi: 10.1093/hmg/ddi227. [DOI] [PubMed] [Google Scholar]
- 19.Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, Boikos SA, Ferrando B, Pacak K, Assie G, Baudin E, Chompret A, Ellison JW, Briere JJ, Rustin P, Gimenez-Roqueplo AP, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. European journal of human genetics : EJHG. 2008;16:79–88. doi: 10.1038/sj.ejhg.5201904. [DOI] [PubMed] [Google Scholar]
- 20.Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH, 3rd, Myers EN, Ferrell RE, Rubinstein WS. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. Journal of medical genetics. 2002;39:178–183. doi: 10.1136/jmg.39.3.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 22.Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. American journal of human genetics. 2001;69:49–54. doi: 10.1086/321282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Astuti D, Douglas F, Lennard TW, Aligianis IA, Woodward ER, Evans DG, Eng C, Latif F, Maher ER. Germline SDHD mutation in familial phaeochromocytoma. Lancet. 2001;357:1181–1182. doi: 10.1016/S0140-6736(00)04378-6. [DOI] [PubMed] [Google Scholar]
- 24.Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, Hodis E, Rosenberg M, McKenna A, Cibulskis K, Farlow D, Zimmer L, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer discovery. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brash DE, Rudolph JA, Simon JA, Lin A, McKenna GJ, Baden HP, Halperin AJ, Ponten J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, Stern MH, Marais R. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer discovery. 2013;3:1122–1129. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Griewank K, Westekemper H, Murali R, Mach M, Schilling B, Wiesner T, Schimming T, Livingstone E, Sucker A, Grabellus F, Metz C, Susskind D, Hillen U, Speicher MR, Woodman SE, Steuhl KP, et al. Conjunctival Melanomas harbor BRAF and NRAS Mutations and Copy Number Changes Similar to Cutaneous and Mucosal Melanomas. Clinical cancer research: an official journal of the American Association for Cancer Research. 2013 doi: 10.1158/1078-0432.CCR-13-0163. [DOI] [PubMed] [Google Scholar]
- 29.Griewank KG, Murali R, Schilling B, Scholz S, Sucker A, Song M, Susskind D, Grabellus F, Zimmer L, Hillen U, Steuhl KP, Schadendorf D, Westekemper H, Zeschnigk M. TERT promoter mutations in ocular melanoma distinguish between conjunctival and uveal tumours. British journal of cancer. 2013;109:497–501. doi: 10.1038/bjc.2013.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spendlove HE, Damato BE, Humphreys J, Barker KT, Hiscott PS, Houlston RS. BRAF mutations are detectable in conjunctival but not uveal melanomas. Melanoma research. 2004;14:449–452. doi: 10.1097/00008390-200412000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houben R, Becker JC, Kappel A, Terheyden P, Brocker EB, Goetz R, Rapp UR. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. Journal of carcinogenesis. 2004;3:6. doi: 10.1186/1477-3163-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Griewank KG, Westekemper H, Murali R, Mach M, Schilling B, Wiesner T, Schimming T, Livingstone E, Sucker A, Grabellus F, Metz C, Susskind D, Hillen U, Speicher MR, Woodman SE, Steuhl KP, et al. Conjunctival melanomas harbor BRAF and NRAS mutations and copy number changes similar to cutaneous and mucosal melanomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19:3143–3152. doi: 10.1158/1078-0432.CCR-13-0163. [DOI] [PubMed] [Google Scholar]
- 34.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic acids research. 1988;16:1215. doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Genome Project Data Processing S The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aho AV, Kernighan BW, Weinberger PJ. Awk — a pattern scanning and processing language. Software: Practice and Experience. 2006;9:267–279. [Google Scholar]
- 37.Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC, Jr, et al. Final version of 2009 AJCC melanoma staging and classification. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:6199–6206. doi: 10.1200/JCO.2009.23.4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.