

Abstract
Purpose
To identify specific mutations causing North Carolina Macular Dystrophy (NCMD).
Study Design
Whole genome sequencing coupled with RT-PCR analysis of gene expression in human retinal cells.
Subjects
141 members of 12 families with NCMD and 261 unrelated control individuals.
Methods
Genome sequencing was performed on eight affected individuals from three families affected with chromosome-6-linked NCMD (MCDR1) and two individuals affected with chromosome-5-linked NCMD (MCDR3). Variants observed in the MCDR1 locus with frequencies of less than 1% in published databases were confirmed using Sanger sequencing. Confirmed variants absent from all published databases were sought in affected individuals from 8 additional MCDR1 families and the 261 controls. RT-PCR analysis of selected genes was performed in stem-cell-derived human retinal cells.
Main Outcome Measure
Cosegregation of rare genetic variants with disease phenotype.
Results
Five sequenced individuals with MCDR1-linked NCMD shared a haplotype of 14 rare variants that spanned one megabase of the disease-causing allele. One of these variants (V1) was absent from all published databases and all 261 controls, but was found in five additional NCMD kindreds. This variant lies in a DNase 1 hypersensitivity site (DHS) upstream of both the PRDM13 and CCNC genes. Sanger sequencing of 1000 base pairs centered on V1 was performed in the remaining four NCMD probands and two additional novel single nucleotide variants (V2 in three families and V3 in a single family) were identified in the DHS within 134 base pairs of the location of V1. A complete duplication of the PRDM13 gene was also discovered in a single family (V4). RT-PCR analysis of PRDM13 expression in developing retinal cells revealed marked developmental regulation. Next generation sequencing of two individuals affected with chromosome-5-linked NCMD revealed a 900kb duplication that included the entire IRX1 gene (V5). The five mutations V1–V5 segregated perfectly in the 102 affected and 39 unaffected members of the 12 NCMD families.
Conclusion
We have identified five rare mutations that are each capable of arresting the development of the human macula. Four of these strongly implicate the involvement of the gene PRDM13 in macular development, while the pathophysiologic mechanism of the fifth remains unknown but may involve the developmental dysregulation of IRX1.
Introduction
Few tissues in the human body are as important to the well-being of a person as the central three millimeters of the human retina. The ability to drive a car, recognize friends in public and see words on a computer, cell phone or printed page are just a few of the many activities of daily living that depend heavily upon the normal function of the macula.
For all but a few people, the macula functions very well for the first six or seven decades of life; but in older individuals, the macula is quite prone to a genetically and mechanistically diverse group of disorders that are known collectively as age-related macular degeneration (AMD). For many years, the neovascular complications of AMD were the most common cause of irreversible blindness in developed countries1–4. However, the recent advent of anti-VEGF drugs5–8 has dramatically reduced the vision loss from neovascularization, thereby increasing the fraction of blindness caused by geographic atrophy of the macula.
There are at least two approaches that one could envision for reducing the burden of blindness caused by geographic atrophy of the macula. The first would be to understand the pathophysiologic mechanisms of AMD in sufficient detail that one could detect the disease at a very early stage, perhaps even as an asymptomatic genetic predisposition, and deliver a safe and effective preventive therapy to those at risk, much as statins are now used to reduce the risk of heart disease. Another strategy would be to rebuild an injured macula with new stem-cell-derived retinal cells9,10. Molecular genetics will play an important role in both of these approaches.
In the 1990s, scientists sought the genetic causes of several Mendelian forms of human macular disease for at least two reasons. First, it was possible that mild mutations in the genes responsible for these early onset conditions might prove to be responsible for a significant subset of the age-related forms of the disease. Second, it was thought that by discovering how relatively minor alterations of individual genes could cause clinical findings similar to AMD, one would gain valuable insight into the normal function of the macula. Twenty years later, it is clear that none of the genes that cause the classic Mendelian macular dystrophies cause a significant fraction of the late onset disease; and, none of the genes that have been shown to predispose people to typical AMD cause any meaningful fraction of early onset Mendelian macular disease.
The first of the classic macular dystrophies to have its gene mapped to a chromosome11, North Carolina Macular Dystrophy (NCMD) is the last to have its specific disease-causing mutations identified. The reason for this delay – the unusual developmental mechanism of this disease – may ultimately make NCMD the most relevant of the Mendelian macular dystrophies to the treatment of AMD. NCMD was first described in a large kindred from North Carolina by Lefler, Wadsworth and Sidbury12 and later described in more detail by Frank et al.13. The cross-sectional nature of these studies led the investigators to believe that the disease was slowly progressive. However, Small and co-workers reexamined the original Lefler kindred almost 20 years later and realized that NCMD is in fact a non-progressive developmental disorder with widely variable expressivity14.
In the decades since the MCDR1 locus was mapped, many additional families with NCMD have been described15–20 including two families that link to a separate locus on chromosome 5 (MCDR3)21,22. The critical region on chromosome 6 has been considerably narrowed23,24, and all of the coding regions of genes within this interval have been exhaustively studied by us and others25. The failure of these experiments to identify plausible disease-causing mutations in any of these kindreds suggested that the mutations were likely to exist in non-exomic DNA and to affect the expression of a nearby gene or genes rather than the structure of its gene product. The purpose of this study was to take advantage of recent advances in whole genome sequencing to comprehensively screen the non-exomic sequences within the MCDR1 and MCDR3 loci to identify disease-causing mutations in families affected with these diseases.
Methods
Human Subjects
All subjects provided written informed consent for this research study, which was approved by the Institutional Review Board of the University of Iowa and adhered to the tenets set forth in the Declaration of Helsinki. Blood samples were obtained from all subjects and DNA was extracted using a nonorganic protocol as previously described26.
Next-generation sequencing of MCDR1 patients
A targeted genome capture of the linked region was performed on three members of Family A (two affected and one unaffected), two members of Family K, and one member of Family B. Libraries prepared from these captures were sequenced on an Illumina HiSeq. Additionally, 30x whole genomes were obtained from five affected individuals: two from Family A, one from Family K, and two from Family L. These libraries were sequenced on an Illumina HiseqX. All of these individuals are noted in blue in Supplemental Figure 1 (available at http://aaojournal.org).
Bioinformatic analysis of next-generation sequencing data
Sequences were analyzed as described previously27. Briefly, sequences were aligned to the reference genome using BWA-mem, and single nucleotide variants and small indels were identified using a GATK-based pipeline28,29. Variants mapping outside the MCDR1-linked region and those found at a frequency of 1% or greater in public databases30–32 were removed. Variants were then filtered, requiring that all affected individuals with a given haplotype shared the heterozygous variant, and all other individuals did not share the variant. Copy number variants (CNV) were investigated using Pindel and manual inspection of the aligned sequence data using the Integrative Genome Viewer (IGV)33,34. As a control, the identified genes were screened for CNVs using Conifer35 in an internal database of 953 whole exomes of eye disease patients.
Confirmation of Whole Genome Sequencing
Variants identified by whole genome sequencing were confirmed using automated bidirectional DNA sequencing with dye termination chemistry on an ABI3730 sequencer.
Screening of Control Subjects
Two hundred and sixty-one normal control subjects were screened for the presence of V1–V3 (Table 1) using unidirectional automated DNA sequencing. To evaluate these controls for the presence of V4 and V5 (Table 1), oligonucleotide primers were designed to amplify across the novel junctions created by these tandem duplications (Supplemental Table 1, available at http://aaojournal.org) and the products of these amplifications were evaluated by electrophoresis on 6% non-denaturing polyacrylamide gels followed by silver staining as previously described36.
Table 1.
| Family | Previously Published Family ID (reference) |
Haplotype | Variant # |
HG19 Chromosomal Location |
Nucleotide Change |
# Affected |
# Unaffected |
Total Samples |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| A |
765 (11–14,24,40–45) | North Carolina | V1 | Chr6_100040906 | G>T | 51 | 22 | 73 |
| B |
702 (45) | North Carolina | V1 | Chr6_100040906 | G>T | 4 | 5 | 9 |
C
|
768 (17,24,45) | North Carolina | V1 | Chr6_100040906 | G>T | 1 | 1 | 2 |
D
|
772 (24,45) | North Carolina | V1 | Chr6_100040906 | G>T | 4 | 0 | 4 |
E
|
1193 (24,45) | North Carolina | V1 | Chr6_100040906 | G>T | 3 | 0 | 3 |
F
|
1292 (24,45) | N/A | V1 | Chr6_100040906 | G>T | 2 | 1 | 3 |
| G |
769 (16,24,45) | French | V2 | Chr6_100040987 | G>C | 1 | 0 | 1 |
H
|
718 (45) | N/A | V2 | Chr6_100040987 | G>C | 1 | 0 | 1 |
I
|
1574 | N/A | V2 | Chr6_100040987 | G>C | 11 | 5 | 16 |
J
|
709 (45) | N/A | V3 | Chr6_100041040 | C>T | 2 | 0 | 2 |
K
|
1463 (19,45) | Belize | V4 | Chr6_100020205-100143306 | 123101bp Tandem Duplication | 11 | 4 | 15 |
L
|
MCDR3 (22) | Danish | V5 | Chr5_3587901-4486027 | 898126bp Tandem Duplication | 11 | 1 | 12 |
|
| ||||||||
| 102 | 39 | 141 | ||||||
Mutations in the promoter of the PRDM13 gene cause North Carolina Macular Dystrophy and suggest that this retinal transcription factor is an important regulator of human macular development.
iPSC Generation and 3D differentiation
Human dermal fibroblasts were isolated from skin biopsies obtained from normal individuals following informed consent. Cultured fibroblasts were reprogrammed via viral transduction of the transcription factors OCT4, SOX2, KLF4, and c-MYC as previously described27,37,38. Human induced pluripotent stem cells (iPSCs) were maintained in Essential 8 media (Life Technologies, Carlsbad, CA) on Laminin 521 coated plates (Corning Life Sciences, Tewksbury, MA). To initiate differentiation, iPSCs were removed from the culture substrate via incubation with TrypLE Express Enzyme (Life Technologies) dissociated into a single cell suspension and subsequently differentiated via the 3D differentiation protocol previously published by Eiraku et al.39.
RNA isolation and RT-PCR
Total RNA was extracted from normal human iPSCs isolated at 0, 30, 60 and 100 days post-differentiation using the RNeasy Mini-kit (Qiagen, Germantown, MD) per the manufacturers instructions. 100ng of RNA was amplified via SuperScript III One-Step RT-PCR System with Platinum Taq DNA Polymerase (Life Technologies, Carlsbad, CA) using the gene-specific primers described in Supplemental Table 1 (available at http://aaojournal.org).
Immunocytochemistry of 3D iPSC-derived eyecups
3D iPSC-derived eyecups were embedded in 4% agarose, sectioned at a thickness of 100μm using a Leica VT1000 S vibratome (Leica Microsystems, Wetzlar, Germany) and labeled with primary antibodies targeted against: mouse anti-SOX2 (#MAB2018; 1:1000; R&D Systems, Minneapolis, MN), rabbit anti-PAX6 (#901301; 1:1000; BioLegend, San Diego, CA), goat anti-biotinylated-OTX2 (#BAF1979; 1:500; R&D Systems, Minneapolis, MN), rabbit anti-Ki67 (#ab15580; 1:500; Abcam, Cambridge, MA), rabbit anti-TUJ1 (neuron-specific class III beta-tubulin; #T2200; 1:500; Sigma-Aldrich; 1:500), goat anti-biotinylated-NRL (#BAF2945; 1:500; R&D Systems), mouse anti-HuC/D (#A-21271; 1:500; Thermo Fisher Scientific, Waltham, MA) and rabbit anti-recoverin (#AB5585; 1:2000; EMD Millipore, Billerica, MA). To detect F-actin, eyecups were stained with Alexa Fluor® 488 Phalloidin (Life Technologies, Madison, WI; #A12379; 1:500). Primary antibodies were detected using fluorescently-conjugated Alexa Fluor® secondary antibodies (Life Technologies). Cell nuclei were counterstained using DAPI. Sectioned eyecups were imaged using a Leica DM 2500 SPE confocal microscope (Leica Microsystems).
Results
Twelve families manifesting the clinical features of NCMD were studied, all but one of which have been previously published11–14,16,17,19,24,40–44. Six of these families share a haplotype of short tandem repeat polymorphisms in the MCDR1 locus on chromosome 6 suggestive of a common founder45, while five others have been linked to MCDR1 but exhibit a different marker haplotype. The remaining family has been previously linked to the MCDR3 locus on chromosome 522. DNA samples from 102 affected and 39 unaffected members of these families were available for this study. The family structures and specific individuals included in this study are shown in Supplemental Figure 1 (available at http://aaojournal.org).
Subject 7043 in Family A has been followed by the authors for more than 30 years43 and is an excellent example of the cardinal clinical features of NCMD. She was first seen at two years and nine months of age and displayed a visual acuity of 20/40 OD and 20/60 OS with line pictures. Fundus exam revealed small areas of atrophy surrounded by drusen-like deposits in both eyes. A prism cover test revealed unmaintained fixation OS and a trial of part time occlusion was begun. Two months later, her vision had improved to 20/40 OU and patching was discontinued. At age 6, her acuity had fallen slightly to 20/50 OU. Two small red dots suggestive of hemorrhage were observed on the nasal edge of the atrophy in the OS (Figure 1A and B), but fluorescein angiography revealed no evidence of active neovascularization on that visit (Figure 1C and D, Supplemental Figure 2, available at http://aaojournal.org). At age 8, her acuity remained 20/50 OU and a new subretinal fibrotic scar was noted in the OS extending from 1 o’clock to 7 o’clock around the central patch of atrophy (Figure 1E). Two years later the acuity and fundus appearance were unchanged (Figure 1F), but the following year, at age 11, the scar in the OS had extended another three clock hours (Figure 1G) with little change in acuity (20/50-2). When last seen at age 33, her visual acuity was 20/60-1 OD and 20/70-1 OS. The fundus appearance (Figure 1H) was very similar to her visit 22 years earlier. Optical coherence tomography of the OS revealed an abrupt termination of the photoreceptors, RPE and choroid at the one edge of the atrophic lesion that was not distorted by the fibrotic scar (Figure 1I and J).
Figure 1.

Retinal images spanning 30 years from the left eye of an affected member of Family A: color fundus photograph (A), red free fundus photograph (B), early phase fluorescein angiogram (C) and late phase fluorescein angiogram (D) at age 6; color fundus photographs at ages 8 (E), 10 (F), 11 (G) and 33 years (H); optical coherence tomogram at age 33 years (I, J). This patient has been previously reported (Table 1). Stereo images of panels B–D are provided in Supplemental Figure 2 (available at http://aaojournal.org).
The original linkage of the NCMD phenotype to chromosome 6p11 and the subsequent narrowing of the MCDR1 interval24 depended heavily on Families A and J. Detailed genotyping of additional members of these families revealed an unaffected recombinant individual (Supplemental Figure 1K, available at http://aaojournal.org) that narrowed the centromeric end of the interval to the genetic marker D6S1717 (Figure 2). A genomic fragment capture of the narrowed disease interval and next generation sequencing were then performed in one unaffected and two affected members of Family A. However, only 85% of the nucleotides in the disease interval were successfully sequenced in this experiment and therefore two additional affected members of the same family were subjected to whole genome sequencing. Analysis of the sequence data from these four affected individuals (noted in blue, Supplemental Figure 1A, available at http://aaojournal.org) revealed a haplotype of 14 rare variants that spanned one megabase of the disease-causing allele (Figure 2). One of these variants (V1 – Table 1) was absent from all published databases and 261 normal controls, but was found in all affected members of five of ten additional NCMD kindreds (Families B-F, Supplemental Figure 1, available at http://aaojournal.org) that were known or suspected to map to MCDR1. This variant lies in a DNase 1 hypersensitivity site (DHS) upstream of both the PRDM13 and CCNC genes (Figure 2). Sanger sequencing of 1000 base pairs centered on V1 was performed in the probands of the remaining five NCMD families and two additional novel single nucleotide variants (V2 in Families G-I and V3 in Family J, Table 1, Supplemental Figure 1, available at http://aaojournal.org) were identified within 134 base pairs of the location of V1 (Figure 2). Whole genome sequencing of an affected individual from the remaining MCDR1 family (Family K, Supplemental Figure 1, available at http://aaojournal.org) was performed and a 123 kb tandem duplication (V4 – Table 1) containing the entire coding sequence of PRDM13 was identified (Figure 2; Supplemental Figure 3A, available at http://aaojournal.org). Collectively, V1–V4 were present in 91 of 91 affected members of these eleven families, absent from 38 of 38 unaffected members and also absent from 261 unrelated control individuals (522 chromosomes). In addition, a review of the Databases of Genome Variants46 revealed no instances of duplication of the entire PRDM13 coding sequence in normal individuals.
Figure 2.

Discovery of NCMD-causing variants in MCDR1. The critical region of MCDR1 was narrowed to 883kb by a single unaffected recombinant individual (Supplemental Figure 1J, asterisk, available at http://aaojournal.org). Genome sequencing revealed 14 rare variants (violet vertical bars) across this region, one of which has never been observed in normal individuals (V1). This novel variant falls within a DNAse hypersensitivity site (pink) upstream of the PRDM13 gene (green) that was later found to include other rare variants in NCMD families (V2 and V3). Additionally, a 123kb tandem duplication containing the PRDM13 gene (yellow – V4) was discovered in one NCMD family.
To determine if PRDM13 and CCNC are expressed during retinal development, iPSCs were used to generate retinal tissue via 3D differentiation. After 30 days of differentiation (D30), 3D iPSC-derived eyecup-like structures are polarized with highly organized filamentous actin (F-actin) networks comprised of actively proliferating Ki67-positive cells (Figure 3A). At this stage of development, 3D eyecups predominantly contain cells that express the early retinal-specific markers, SOX2, PAX6 and OTX2 (Figure 3B). PAX6, a master regulator of retinal development, is expressed throughout the eyecup and helps to drive the expression of the photoreceptor precursor cell-specific transcription factor, OTX2. PAX6 and OTX2 are co-expressed in most cells at this stage of development (Figure 3B). After 60 days of differentiation, PAX6 expression becomes restricted to presumptive RPE cells and pockets of presumptive photoreceptor cells that express OTX2 independently of PAX6 arise (Figure 3C). After 100 days of differentiation, 3D eyecups are laminated with an inner layer containing retinal neurons that express the ganglion cell-specific marker, HuC/D and an outer layer containing photoreceptor cells that robustly express the phototransduction protein, recoverin (Figure 3D). Analysis of RNA isolated from iPSCs at 0, 30, 60 and 100 days post-differentiation revealed that expression of PRDM13 is negatively correlated with retinal development (Figure 4). Specifically, as cells progress from a pluripotent stem cell state to mature retinal neurons, PRDM13 transcript is down regulated. Interestingly, CCNC is consistently expressed across all developmental time points (Figure 4).
Figure 3.

Using normal human iPSCs to model retinal development. A–D: Immunocytochemical analysis of iPSC derived eyecup-like structures targeted against F-actin (Phalloidin), SOX2, PAX6, OTX2, HuC/D and recoverin (RCVRN). After 30 days of differentiation (D30) polarized neural epithelia (A, F-Actin - green) comprised of proliferating cells (A, Ki67 - red) positive for the early retinal progenitor cell markers SOX2 (B, green), PAX6 (B, red) and OTX2 (B, white) are present. After 60 days of differentiation, PAX6 (C, red) expression is restricted to OTX2 negative presumptive RPE while OTX2 (C, white) is restricted to PAX6 negative photoreceptor precursor cells. After 100 days of differentiation, eyecups are laminated with HuC/D-positive (D, green) ganglion cell like neurons in the inner layer and recoverin-positive (D, Red) photoreceptor precursor cells in the outer layer. Insets depict individual fluorescent channels. A & D: 40X magnification. B & C: 20X magnification.
Figure 4.
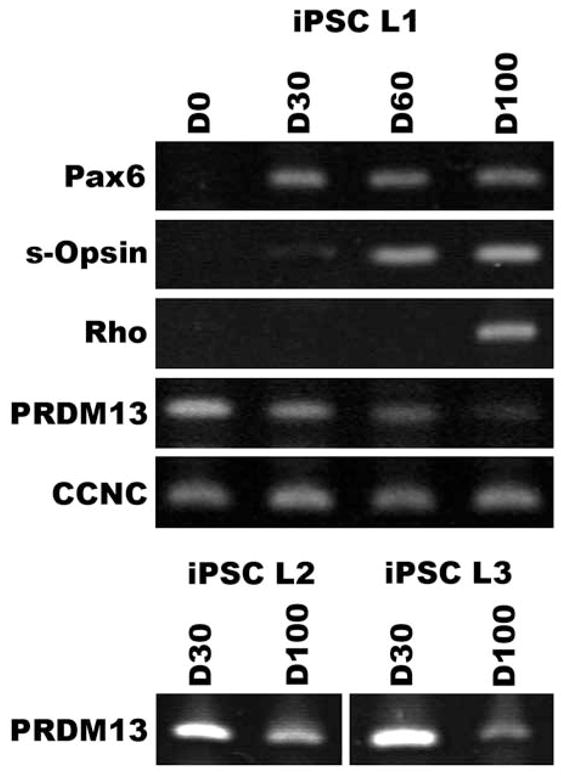
Retinal expression of PRDM13 is developmentally regulated. RT-PCR analysis of iPSCs after 0, 30, 60 and 100 days of retinal differentiation using primers targeted against the retinal lineage markers PAX6, s-Opsin, and Rhodopsin, and genes within the MCDR1 locus, PRDM13 and CCNC. As iPSCs progress from a pluripotent state to immature PAX6-expressing retinal progenitor cells to mature s-Opsin-expressing cone and rhodopsin-expressing rod photoreceptor cells, PRDM13 expression decreases (PRDM13 iPSC-L1, iPSC-L2 and iPSC-L3). iPSC-L1 – Control iPSC line 1. iPSC-L2 - Control iPSC line 2. iPSC-L3 - Control iPSC line 3.
In 2010, Rosenberg and co-workers mapped the disease-causing mutation of a Danish kindred (Family L, Supplemental Figure 1, available at http://aaojournal.org) with an NCMD phenotype to an 8 cM interval on chromosome 5 (MCDR3; Figure 5A)22 that had been previously identified by Michaelides, et al.21. We performed whole genome sequencing of two affected individuals in this family and identified a 900 kb tandem duplication (V5 – Table 1) that included the entire coding sequence of IRX1 (Figure 5A; Supplemental Figure 3B, available at http://aaojournal.org). This duplication was present in all eleven affected members of the family, absent from one unaffected member, absent from the Database of Genome Variants46 and also absent from 261 unrelated controls. However, some much smaller duplications that include in some cases the entire coding sequence of IRX1 have been observed in normal individuals46 suggesting that the disease-causing element in this large duplication is not the IRX1 coding sequence itself. Also, unlike PRDM13 (Figure 3D), RT-PCR analysis of IRX1 in normal iPSC-derived human retinal cells revealed no variation in expression in the first 100 days of development (Figure 5B).
Figure 5.

Discovery of the NCMD-causing variant in MCDR3. A: Using whole genome sequencing, a 900kb duplication (yellow – V5) containing the gene IRX1 (green) was found in a family mapped to MCDR3. B: RT-PCR of developing iPSC-derived photoreceptor precursor cells revealed that unlike PRDM13, IRX1 expression is consistent across all developmental time points tested.
Discussion
The technological advancements that have occurred in the field of human genomics since the North Carolina Macular Dystrophy locus on chromosome 6 was first identified11 have been breathtaking. Few investigators who studied inherited eye diseases in the 1990s would have imagined that in less than 25 years, whole genome sequencing of individual patients would be so commonplace that the sequence of thousands of unrelated individuals would be freely available on public databases30–32 and that the President of the United States would launch a precision medicine initiative based upon these new molecular capabilities and data47. However, the most valuable data in both the original linkage study and the present study were not molecular; the most valuable data were the detailed clinical observations that allowed several families with a very rare and unusual phenotype to be correctly separated from thousands of other members of hundreds of other families with similar diseases caused by genes at other loci.
Although counterintuitive to many people, it is a fact that as genomic tools become more powerful and less expensive, very accurate and detailed clinical information become more necessary for the correct interpretation of the resulting molecular data. There are both quantitative and qualitative reasons for this. Now that tens of thousands of genes can be assessed in a single patient, there are tens of thousands of additional opportunities to observe a plausible disease-causing variant by chance than if one investigated only a single gene. By using clinical data to focus the hypothesis to just a few genes, one can overcome the large multiple measurements problem inherent in whole genome data.
The qualitative reason that molecular data have become more difficult to interpret as they have become easier and less expensive to acquire is embodied in the difference between the coding and noncoding portions of genes. Coding sequences exist in groups of three nucleotides known as codons that each specify a single amino acid in the resulting proteins. The universality of the genetic code allows one to predict the structural effect of a given coding sequence mutation on the resulting protein with much greater accuracy than one could if the same mutation occurred in the noncoding portion of a gene where its effect would be tempered by the actions of DNA binding proteins, DNA methylation, noncoding RNA molecules, the proximity to coding sequences and other factors that are incompletely understood at the present time.
There are ten genes in the MCDR1 locus and individuals from multiple unrelated kindreds affected with MCDR1-linked NCMD have been extensively screened for mutations in the coding sequences of these genes with no plausible disease-causing variants identified. We therefore expected that NCMD-causing mutations would eventually be found in the noncoding portions of the MCDR1 locus and we took advantage of two valuable resources and one genetic fact to detect these mutations among the many functionally neutral polymorphisms that exist in the noncoding sequences of all individuals: 1) multiple unrelated families exhibiting a classic NCMD phenotype, 2) public genome databases with sequences of thousands of individuals30–32 and 3) the fact that mutations that cause high-penetrance autosomal dominant diseases should be no more common in the general population than the disease itself.
The data supporting the pathogenicity of V1–V4 are compelling. In Family A, the original NCMD family and the largest one ascertained to date, V1 is the only nucleotide in the 883 kb MCDR1 locus that is absent from all public databases and therefore of similar population frequency to NCMD itself. This variant lies in a 255 bp region of DNase I hypersensitivity that is upstream of a gene encoding a retinal transcription factor, PRDM13. It is noteworthy that PRDM13 is the only gene in the MCDR1 critical region that is solely expressed in the neural retina48,49. DNAse I hypersensitivity is an indicator of chromatin accessibility that is often associated with transcription factor binding sites50. V1 was later found in five independently ascertained NCMD kindreds, shown to segregate perfectly among 65 affected and 29 unaffected members of these six families, and shown to be absent from 261 unrelated individuals ascertained in Iowa. The latter individuals were sequenced just to make sure that there was not an artifactual gap in the public genome data. Conventional sequencing of this DNase I hypersensitivity site (DHS) in five V1-negative NCMD families revealed four to harbor point mutations (V2 and V3) within 134 base pairs of V1. Whole genome sequencing of the fifth V1-negative family revealed a tandem duplication containing the DHS and the entire coding sequence of PRDM13 (V4). V2–V4 were found to segregate perfectly among the 26 affected and 9 unaffected members of these five families and were absent from all public databases and the 261 control individuals from Iowa.
While the association between these variants and the disease phenotype is extraordinarily strong (p < 10−29 by Fisher’s exact test), the mechanism by which they cause disease is far from established. For example, the gene CCNC, which encodes a ubiquitous cell cycle controller, lies in the opposite orientation of PRDM13 on the opposite side of the DHS and thus could in principle also be affected by these mutations and therefore involved in the pathogenesis of NCMD. One argument against CNCC as an NCMD gene, in addition to its ubiquitous expression, is the configuration of the DHSs in the tandem duplication of Family K. The entire coding region of PRDM13 is duplicated in this mutation and both DHSs are immediately adjacent to a PRDM13 gene. In contrast, only one of the DHSs is adjacent to the unduplicated CCNC gene (Supplemental Figure 4, available at http://aaojournal.org).
The observation that NCMD is a developmental abnormality is also consistent with PRDM13 being the responsible gene. PRDM13 is a member of a large family of “helix-loop-helix” DNA-binding proteins that play key roles in controlling gene expression during development51. Since the formation of the macula is accompanied by differential expression of an array of genes involved in axon guidance and inhibition of angiogenesis52, this process likely relies on a precise interaction between transcription factors (like PRDM13) and their target genes. Thus, a change in the abundance of a transcription factor due to mutations in its own regulatory regions could plausibly lead to impaired cell fate specifications in the developing macula. It is therefore notable that both PRDM13 and IRX1 are proteins with important roles in regulating gene expression.
One of the great advantages of induced pluripotent stem cells is their ability to differentiate ex vivo into any cell type of the three embryonic germ layers. For many organ systems, iPSC differentiation faithfully recapitulates the various cell fate decisions made during embryonic development39,53–56. Being able to obtain embryonic tissue from adult somatic cells affords researchers with the ability to determine if and when in cellular development specific genes are expressed. In this study, human iPSC-derived retinal tissue was used to demonstrate that PRDM13 is developmentally regulated while other genes in the MCDR1 locus, i.e. CCNC, are not. To demonstrate this finding in the absence of the pluripotent stem cell technology one would have to obtain retinal tissue from human fetuses at different points in development, an approach that would be logistically difficult and raise serious ethical concerns. The capability of iPSCs to generate otherwise inaccessible tissues such as the retina also gives researchers the ability to investigate the pathophysiologic effect of newly identified gene defects on cell health and function. This will be especially useful in the modern gene-sequencing era when trying to determine the mechanistic effects of non-coding genetic variants such as those identified in this study. In future studies, it will be interesting to generate retinal tissue from patients with each of the mutations described in this study and to determine their effect on gene expression, as well as cellular differentiation, maturation, health and function.
The tandem duplication in the MCDR3 locus is very likely to be the disease-causing mutation in Family L simply because it is very unlikely that the largest duplication involving IRX1 currently known to exist among the thousands of currently available human genome sequences46 would occur by chance in the very small portion of the genome that has been previously implicated in the disease21,22. However, unlike MCDR1, no additional mutations have yet been identified in different MCDR3 families to corroborate this finding and to narrow the mechanistic possibilities. Also unlike MCDR1, where PRDM13 exhibits dramatic expression differences in the first 100 days of retinal development, IRX1 is constitutively expressed in normal individuals. Perhaps the large duplication alters the evolutionarily conserved chromosome conformation of the IRXA gene cluster57. Future experiments with retinal cells generated from NCMD patients themselves will likely clarify the mechanism of both MCDR loci significantly.
A very practical outcome of this work is that one can detect every mutation reported in this paper using only three PCR-based sequencing reactions (Supplemental Table 1, available at http://aaojournal.org). The availability of a simple genetic test for this disease will likely result in the diagnosis of many additional individuals, which will not only allow physicians to provide much more accurate genetic and prognostic information than was possible before, but it will likely accelerate the discovery of additional disease-causing variants as well as additional clinical manifestations of the known mutations. Both of these will help unravel the precise mechanisms through which these loci contribute to the formation of the normal macula.
In conclusion, we have identified five rare mutations that are each capable of arresting the development of the human macula. Four of these strongly implicate the involvement of the gene PRDM13 in macular development, while the pathophysiologic mechanism of the fifth remains unknown but may involve the developmental dysregulation of IRX1.
Supplementary Material
Pedigrees of families included in this study. Individuals from whom DNA was available in sufficient quantity and quality are indicated in green, individuals who were studied using next-generation sequencing are indicated in blue. The individual marked with the asterisk in Family K harbored the interval defining recombination.
Stereo images of panels B–D from Figure 1.
Schematic illustration and chromatograms illustrating the breakpoints and regions of duplication in MCDR1 Family K (A) and MCDR3 Family L (B).
Schematic illustration of the relative positions of the DNase hypersensitivity sites and the PRDM13 and CCNC genes in Family K.
Supplemental Table 1. Oligonucleotide Primers
Mutations in the promoter of the PRDM13 gene cause North Carolina Macular Dystrophy and suggest that this retinal transcription factor is an important regulator of human macular development.
Acknowledgments
The authors thank the members of the families reported here for their valuable participation in this study and Jeaneen Andorf for her excellent technical assistance and proofreading of the manuscript. This work is dedicated to the memory of our colleague Maurice Rabb, M.D.
Footnotes
The authors have no proprietary/financial interests to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Friedman DS, O’Colmain BJ, Muñoz B, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004;122:564–572. doi: 10.1001/archopht.122.4.564. [DOI] [PubMed] [Google Scholar]
- 2.Klein R, Klein BE, Cruickshanks KJ. The prevalence of age-related maculopathy by geographic region and ethnicity. Progress in Retinal and Eye Research. 1999;18:371–389. doi: 10.1016/s1350-9462(98)00025-1. [DOI] [PubMed] [Google Scholar]
- 3.Wong TY, Wong T, Chakravarthy U, et al. The natural history and prognosis of neovascular age-related macular degeneration: a systematic review of the literature and meta-analysis. Ophthalmology. 2008;115:116–126. doi: 10.1016/j.ophtha.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 4.WLW, XS, XL, et al. ArticlesGlobal prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. The Lancet Global Health. 2014;2:e106–e116. doi: 10.1016/S2214-109X(13)70145-1. [DOI] [PubMed] [Google Scholar]
- 5.Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355:1419–1431. doi: 10.1056/NEJMoa054481. [DOI] [PubMed] [Google Scholar]
- 6.Rosenfeld PJ, Moshfeghi AA, Puliafito CA. Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for neovascular age-related macular degeneration. Ophthalmic Surg Lasers Imaging. 2005;36:331–335. [PubMed] [Google Scholar]
- 7.CATT Research Group. Martin DF, Maguire MG, et al. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364:1897–1908. doi: 10.1056/NEJMoa1102673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bressler NM, Doan QV, Varma R, et al. Estimated cases of legal blindness and visual impairment avoided using ranibizumab for choroidal neovascularization: non-Hispanic white population in the United States with age-related macular degeneration. Arch Ophthalmol. 2011;129:709–717. doi: 10.1001/archophthalmol.2011.140. [DOI] [PubMed] [Google Scholar]
- 9.Patient-specific iPSC-derived photoreceptor precursor cells as a means to investigate retinitis pigmentosa. 2013;2:e00824. doi: 10.7554/eLife.00824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Transplantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in degenerative mice. 2011;6:e18992. doi: 10.1371/journal.pone.0018992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Small KW, Weber JL, Roses A, et al. North Carolina macular dystrophy is assigned to chromosome 6. Genomics. 1992;13:681–685. doi: 10.1016/0888-7543(92)90141-e. [DOI] [PubMed] [Google Scholar]
- 12.Lefler WH, Wadsworth JA, Sidbury JB. Hereditary macular degeneration and amino-aciduria. Am J Ophthalmol. 1971;71:224–230. doi: 10.1016/0002-9394(71)90394-1. [DOI] [PubMed] [Google Scholar]
- 13.Frank HR, Landers MB, Williams RJ, Sidbury JB. A new dominant progressive foveal dystrophy. Am J Ophthalmol. 1974;78:903–916. doi: 10.1016/0002-9394(74)90800-9. [DOI] [PubMed] [Google Scholar]
- 14.Small KW. North Carolina macular dystrophy, revisited. Ophthalmology. 1989;96:1747–1754. doi: 10.1016/s0161-6420(89)32655-8. [DOI] [PubMed] [Google Scholar]
- 15.Pauleikhoff D, Sauer CG, Müller CR, et al. Clinical and genetic evidence for autosomal dominant North Carolina macular dystrophy in a German family. Am J Ophthalmol. 1997;124(3):412–415. doi: 10.1016/s0002-9394(14)70842-6. [DOI] [PubMed] [Google Scholar]
- 16.Small KW, Puech B, Mullen L, Yelchits S. North Carolina macular dystrophy phenotype in France maps to the MCDR1 locus. Mol Vis. 1997;3:1. [PubMed] [Google Scholar]
- 17.Small KW, Garcia CA, Gallardo G, et al. North Carolina macular dystrophy (MCDR1) in Texas. Retina (Philadelphia, Pa) 1998;18:448–452. [PubMed] [Google Scholar]
- 18.Rohrschneider K, Blankenagel A, Kruse FE, et al. Macular function testing in a German pedigree with North Carolina macular dystrophy. Retina (Philadelphia, Pa) 1998;18:453–459. doi: 10.1097/00006982-199805000-00013. [DOI] [PubMed] [Google Scholar]
- 19.Rabb MF, Mullen L, Yelchits S, et al. A North Carolina macular dystrophy phenotype in a Belizean family maps to the MCDR1 locus. Am J Ophthalmol. 1998;125:502–508. doi: 10.1016/s0002-9394(99)80191-3. [DOI] [PubMed] [Google Scholar]
- 20.Reichel MB, Kelsell RE, Fan J, et al. Phenotype of a British North Carolina macular dystrophy family linked to chromosome 6q. Br J Ophthalmol. 1998;82:1162–1168. doi: 10.1136/bjo.82.10.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Michaelides M, Johnson S, Tekriwal AK, et al. An early-onset autosomal dominant macular dystrophy (MCDR3) resembling North Carolina macular dystrophy maps to chromosome 5. Investigative Ophthalmology & Visual Science. 2003;44:2178–2183. doi: 10.1167/iovs.02-1094. [DOI] [PubMed] [Google Scholar]
- 22.Rosenberg T, Roos B, Johnsen T, et al. Clinical and genetic characterization of a Danish family with North Carolina macular dystrophy. Mol Vis. 2010;16:2659–2668. [PMC free article] [PubMed] [Google Scholar]
- 23.Sauer CG, Schworm HD, Ulbig M, et al. An ancestral core haplotype defines the critical region harbouring the North Carolina macular dystrophy gene (MCDR1) Journal of medical genetics. 1997;34:961–966. doi: 10.1136/jmg.34.12.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Small KW, Udar N, Yelchits S, et al. North Carolina macular dystrophy (MCDR1) locus: a fine resolution genetic map and haplotype analysis. Mol Vis. 1999 [PubMed] [Google Scholar]
- 25.Yang Z, Tong Z, Chorich LJ, et al. Clinical characterization and genetic mapping of North Carolina macular dystrophy. Vision Research. 2008;48:470–477. doi: 10.1016/j.visres.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braun TA, Mullins RF, Wagner AH, et al. Non-exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Hum Mol Genet. 2013;22:5136–5145. doi: 10.1093/hmg/ddt367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tucker BA, Scheetz TE, Mullins RF, et al. Exome sequencing and analysis of induced pluripotent stem cells identify the cilia-related gene male germ cell-associated kinase (MAK) as a cause of retinitis pigmentosa. 2011;108:E569–E576. doi: 10.1073/pnas.1108918108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Exome Aggregation Consortium (ExAC) Available at: http://exac.broadinstitute.org.
- 31.Exome Variant Server. [Accessed June 27, 2012];NHLBI Exome Sequencing Project (ESP) Available at: http://evs.gs.washington.edu/EVS/
- 32.1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye K, Schulz MH, Long Q, et al. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Briefings in bioinformatics. 2012 doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012 doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mykytyn K, Nishimura DY, Searby CC, et al. Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human obesity syndrome. Nat Genet. 2002;31:435–438. doi: 10.1038/ng935. [DOI] [PubMed] [Google Scholar]
- 37.Tucker BA, Anfinson KR, Mullins RF, et al. Use of a Synthetic Xeno-Free Culture Substrate for Induced Pluripotent Stem Cell Induction and Retinal Differentiation. Stem Cells Translational Medicine. 2013;2:16–24. doi: 10.5966/sctm.2012-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burnight ER, Wiley LA, Drack AV, et al. CEP290 gene transfer rescues Leber congenital amaurosis cellular phenotype. 2014:1–11. doi: 10.1038/gt.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eiraku M, Takata N, Ishibashi H, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 40.Fetkenhour CL, Gurney N, Dobbie JG, Choromokos E. Central areolar pigment epithelial dystrophy. Am J Ophthalmol. 1976;81:745–753. doi: 10.1016/0002-9394(76)90357-3. [DOI] [PubMed] [Google Scholar]
- 41.Hermsen VM, Judisch GF. Central areolar pigment epithelial dystrophy. Ophthalmologica. 1984;189:69–72. doi: 10.1159/000309388. [DOI] [PubMed] [Google Scholar]
- 42.Small KW, Killian J, McLean WC. North Carolina’s dominant progressive foveal dystrophy: how progressive is it? Br J Ophthalmol. 1991;75:401–406. doi: 10.1136/bjo.75.7.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Small KW, Hermsen V, Gurney N, et al. North Carolina macular dystrophy and central areolar pigment epithelial dystrophy. One family, one disease. Arch Ophthalmol. 1992;110:515–518. doi: 10.1001/archopht.1992.01080160093040. [DOI] [PubMed] [Google Scholar]
- 44.Keithahn MA, Huang M, Keltner JL, et al. The variable expressivity of a family with central areolar pigment epithelial dystrophy. Ophthalmology. 1996;103:406–415. doi: 10.1016/s0161-6420(96)30678-7. [DOI] [PubMed] [Google Scholar]
- 45.Small KW. North Carolina macular dystrophy: clinical features, genealogy, and genetic linkage analysis. Transactions of the American Ophthalmological Society. 1998;96:925–961. [PMC free article] [PubMed] [Google Scholar]
- 46.MacDonald JR, Ziman R, Yuen RKC, et al. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–92. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Collins FS, Varmus H. A new initiative on precision medicine. N Engl J Med. 2015;372:793–795. doi: 10.1056/NEJMp1500523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whitmore SS, Wagner AH, DeLuca AP, et al. Transcriptomic analysis across nasal, temporal, and macular regions of human neural retina and RPE/choroid by RNA-Seq. Experimental Eye Research. 2014:1–14. doi: 10.1016/j.exer.2014.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melé M, Ferreira PG, Reverter F, et al. Human genomics. The human transcriptome across tissues and individuals. Science. 2015;348:660–665. doi: 10.1126/science.aaa0355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thurman RE, Rynes E, Humbert R, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fog CK, Galli GG, Lund AH. PRDM proteins: important players in differentiation and disease. BioEssays. 2012;34:50–60. doi: 10.1002/bies.201100107. [DOI] [PubMed] [Google Scholar]
- 52.Kozulin P, Natoli R, O’Brien KMB, et al. Differential expression of anti-angiogenic factors and guidance genes in the developing macula. Mol Vis. 2009;15:45–59. [PMC free article] [PubMed] [Google Scholar]
- 53.Lancaster MA, Renner M, Martin C-A, et al. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Spence JR, Mayhew CN, Rankin SA, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470:105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia Y, Sancho-Martinez I, Nivet E, et al. The generation of kidney organoids by differentiation of human pluripotent cells to ureteric bud progenitor-like cells. Nat Protoc. 2014;9:2693–2704. doi: 10.1038/nprot.2014.182. [DOI] [PubMed] [Google Scholar]
- 56.Zhong X, Gutierrez C, Xue T, et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nature Communications. 2014;5:4047. doi: 10.1038/ncomms5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tena JJ, Alonso ME, la Calle-Mustienes de E, et al. An evolutionarily conserved three-dimensional structure in the vertebrate Irx clusters facilitates enhancer sharing and coregulation. Nature Communications. 2011;2:310. doi: 10.1038/ncomms1301. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pedigrees of families included in this study. Individuals from whom DNA was available in sufficient quantity and quality are indicated in green, individuals who were studied using next-generation sequencing are indicated in blue. The individual marked with the asterisk in Family K harbored the interval defining recombination.
Stereo images of panels B–D from Figure 1.
Schematic illustration and chromatograms illustrating the breakpoints and regions of duplication in MCDR1 Family K (A) and MCDR3 Family L (B).
Schematic illustration of the relative positions of the DNase hypersensitivity sites and the PRDM13 and CCNC genes in Family K.
Supplemental Table 1. Oligonucleotide Primers