

Summary
Nucleotide pool imbalance has been proposed to drive genetic instability in cancer. Here, we show that slowing replication forks by depleting nucleotide pools with hydroxyurea (HU) can also give rise to both transient and permanent epigenetic instability of a reporter locus, BU-1, in DT40 cells. HU induces stochastic formation of Bu-1low variants in dividing cells, which have lost the H3K4me3 present in untreated cells. This instability is potentiated by an intragenic G quadruplex, which also promotes local H2Ax phosphorylation and transient heterochromatinization. Genome-wide, gene expression changes induced by HU significantly overlap with those resulting from loss of the G4-helicases FANCJ, WRN, and BLM. Thus, the effects of global replication stress induced by nucleotide pool depletion can be focused by local replication impediments caused by G quadruplex formation to induce epigenetic instability and changes in gene expression, a mechanism that may contribute to selectable transcriptional changes in cancer.
Keywords: G quadruplexes, replication stress, epigenetic instability, transcriptional memory
Graphical Abstract
Highlights
-
•
Hydroxyurea (HU) stochastically perturbs gene expression
-
•
G quadruplex (G4) formation potentiates HU-induced epigenetic changes
-
•
HU induces G4-dependent DNA damage and heterochromatin formation
-
•
HU and G4 helicase mutations cause similar changes in expression
Slowing replication forks by depleting nucleotide pools enhances the ability of G quadruplexes to stochastically perturb gene expression during replication. Papadopoulou et al. find that a common global replication stressor interacts with local DNA secondary structures to cause epigenetic instability.
Introduction
The term replication stress describes the slowing or stalling of replication forks by endogenously or exogenously derived impediments to DNA polymerization (Zeman and Cimprich, 2014). Replication stressors can be local factors, such as DNA damage or secondary structures that affect forks randomly as they are encountered, or global ones, such as nucleotide pool depletion or imbalance that simultaneously slows all forks (Poli et al., 2012, Anglana et al., 2003). It is now recognized that replication stress induced by nucleotide pool imbalance is an important consequence of the activation of some oncogenes, which drive cells into S phase without upregulation of nucleotide supply (Bester et al., 2011). The resulting loss of polymerase processivity is thought to lead to localized uncoupling of the replicative helicase and polymerase and formation of tracts of single-stranded DNA (Byun et al., 2005, Pacek and Walter, 2004). While this normally induces checkpoint activation and senescence (Bartkova et al., 2006, Di Micco et al., 2006), in cells that can bypass the checkpoint, such replication stress provides a fertile source of genetic instability, particularly in the vicinity of fragile sites and sites capable of forming secondary structures (De and Michor, 2011, Tsantoulis et al., 2008).
In addition to the extensive genetic changes that have been well documented in many types of cancer, there are also extensive local and global alterations in histone and DNA modifications. The consequent changes in chromatin structure are accompanied by significant dysregulation of gene expression (Timp and Feinberg, 2013, Berdasco and Esteller, 2010), which, since it is not accompanied by changes in the DNA sequence, may be considered epigenetic (Berger et al., 2009). These epigenetic changes could act alongside genetic instability to produce clonal variation within a tumor, upon which selective pressure can act, and so may contribute to tumor evolution. Mutations in histone and DNA-modifying enzymes, and even histone proteins themselves, have been found in several cancers and are likely to explain at least some of the observed epigenetic instability (Timp and Feinberg, 2013). However, it is not clear that mutations in histone-modifying enzymes account for all the alterations observed in different cancer types.
We recently provided evidence that deficiencies in enzymes responsible for replicating G quadruplex (G4) structures, such as the specialized DNA polymerase REV1 and helicases FANCJ, WRN, and BLM, can lead to localized changes in histone modifications and gene expression (Sarkies et al., 2010, Sarkies et al., 2012, Schiavone et al., 2014). G4s can form within motifs comprising four short runs of dG bases, separated by linker sequences. The dG bases in the motif form planar Hoogsteen-bonded quartet structures that can stack on top of each other, resulting in an often highly thermodynamically stable secondary structure, the G4 (reviewed in Maizels and Gray, 2013). We proposed that persistent replication fork stalling at G4s in mutants such as rev1 or fancj leads to pathologically long daughter strand gap formation, resulting in local uncoupling of DNA synthesis from parental histone recycling. This, in turn, leads to loss of the histone modifications present on the parental chromatin, which, if in the vicinity of a gene promoter, results in changes in transcription (Sarkies et al., 2010, Sarkies et al., 2012, Schiavone et al., 2014). A prediction of this model is that global replication stressors that lead to loss of processive DNA polymerization with uncoupling of the replicative helicase and polymerase also should promote epigenetic instability by dissociating DNA synthesis from histone recycling.
Here we test this hypothesis by examining the effect of hydroxyurea (HU)-induced nucleotide pool depletion on the epigenetic stability of a sensitive reporter locus, BU-1, in chicken DT40 cells (Sarkies et al., 2012, Schiavone et al., 2014). We show that chronic treatment with low-dose HU induces stochastic instability of BU-1 expression, characterized by loss of the chromatin marks H3K4me3 and H3K9/14ac seen in the normally active locus. This instability depends significantly on the presence of a G4 motif 3′ of the promoter, oriented to stall the leading strand of a fork heading toward the transcription start site (TSS). The presence of this G4 motif not only increases the rate at which BU-1 expression is lost, but is additionally associated with phosphorylation of H2Ax and appearance of the heterochromatic mark H3K9me3. This is consistent with the G4 acting to focus DNA damage induced by the global replication stress imposed by HU, with the damage leading to repression of the locus. Further, we show that, across the genome, chronic exposure to HU results in an altered pattern of gene expression similar to that seen in cells lacking the G4-unwinding helicases FANCJ, WRN, and BLM, and that affected genes are enriched in G4 motifs. Together, these observations indicate that nucleotide depletion can combine with naturally occurring DNA secondary structures to promote epigenetic instability.
Results
Induction of Chronic Replicative Stress in DT40 Cells with HU
We first sought conditions in which we could culture DT40 cells in low-dose HU such that replication is slowed but completed (Alvino et al., 2007). We therefore exposed wild-type DT40 cells to a range of HU concentrations and monitored their doubling time. The cells were able to proliferate for over a week in up to 150 μM HU (Figure 1A). At this dose, their doubling time increased from 12.3 to 32.7 hr, recovering when the HU was washed out (Figure 1A). To determine the effect of low-dose HU on replication dynamics, we performed DNA molecular combing after pulse labeling the cells with halogenated nucleotides (Figure S1A) 3 days after initiating culture in HU. Average fork velocity decreased from 1.26 to 0.71 kb/min (Figure 1B), with a compensatory decrease in average interorigin distance from 72 to 40 kb (Figure 1B). Consistent with these perturbed replication dynamics, cell-cycle profiles revealed a significant accumulation of cells in S phase while in HU (Figure S1B).
Figure 1.

Chronic Replication Stress Induces Epigenetic Instability of BU-1
(A) Doubling time of wild-type DT40 cells in different concentrations of HU. Recovery indicates the growth rate of cells that had been cultured for 1 week in 150 μM HU and then transferred to drug-free medium. Error bars indicate SD of three experiments.
(B) Replication dynamics of DT40 cells after 3 days of growth in 150 μM HU determined by DNA molecular combing. See also Figure S1A. For fork speed, the bin size is 0.2 kb/min with untreated wild-type cells in blue overlaid in red with results from HU-treated cells. For the inter-origin distance, the bin size is 15 kb. The median fork rate and interorigin distance are given ± SEM. The probability that the HU-treated distribution is different from untreated was calculated with the Mann-Whitney test.
(C) Schematic of the model for G4-induced, replication-dependent epigenetic instability of the Bu-1 locus (adapted from Schiavone et al., 2014). The BU-1 locus is bidirectionally replicated (Schiavone et al., 2014).
(D) Bu-1alow cells appear stochastically as a function of time as cells divide in HU. Flow cytometry for Bu-1a was performed daily on a population of wild-type DT40 cells growing in 150 μM HU.
(E) Fluctuation analysis for Bu-1a loss in multiple parallel cultures grown in 150 μM HU. Each symbol represents the percentage of Bu-1alow cells in an individual culture after 7-day growth with or without HU (150 μM).
(F) Fluctuation analysis for Bu-1a loss following 7-day growth in low-dose (150 μM) aphidicolin.
HU and Aphidicolin Induce Instability of BU-1 Expression
We have reported previously that replication-dependent transcriptional instability associated with G4 motifs can be monitored by following expression of a surface marker, Bu-1a, in DT40 cells (Sarkies et al., 2012, Schiavone et al., 2014). The BU-1 locus contains prominent G4 motifs 3.5 kb downstream of the TSS and 3 kb upstream. Both are orientated to be G-rich on the feature strand with respect to the BU-1 transcript (Figure 1C). Epigenetic instability of BU-1 in rev1 cells is entirely dependent on the +3.5 G4 motif, and it requires the motif to be orientated such that its G-rich strand forms on the leading strand of a replication fork entering the locus from the 3′ end (Figure 1C; Schiavone et al., 2014). We have reported previously that the BU-1 locus is bidirectionally replicated, meaning that during any given S phase there is a 50% probability of the +3.5 G4 being replicated on the leading strand template (Schiavone et al., 2014).
Growth of wild-type DT40 cells in 150 μM HU resulted in the appearance of a Bu-1alow population as cells divided over the course of 7 days (Figure 1D). Surface expression of Bu-1a correlates closely with transcript abundance (Sarkies et al., 2012), and this held true for Bu-1alow clones recovered after HU treatment (Figure S1C). To estimate the rate at which Bu-1alow variants are formed in HU, we performed a fluctuation analysis by expanding multiple parallel populations of 104 Bu-1ahigh cells in HU for 7 days, after which we monitored the appearance of Bu-1alow variants. This revealed a striking degree of expression instability despite the small number of cell cycles through which the cells had passed (Figure 1E). Using our previously described Monte Carlo simulation of Bu-1a loss as a replication-dependent phenomenon (Schiavone et al., 2014), we estimated a per-division probability of generating of a Bu-1alow state during culture in HU of c. 0.15. To obtain additional evidence that this induced transcriptional instability of BU-1 reflected decreased DNA polymerase processivity, we asked whether Bu-1low variants could be induced by aphidicolin. Aphidicolin slows replication by directly inhibiting DNA polymerases, particularly Polα (Oguro et al., 1979), and a low dose induces replication stress (Pacek et al., 2006). DT40 cells were able to proliferate in up to 150 μM aphidicolin for 10 days and, as with low-dose HU, this resulted in substantial instability of Bu-1a expression (Figure 1F).
Replication Stress-Induced Instability of Bu-1 Expression Is Potentiated by the +3.5 G4 Motif
We have shown previously that removal of the +3.5 G4 motif from both alleles of BU-1 in REV1-deficient cells results in complete stabilization of expression of the locus (Schiavone et al., 2014). We therefore examined the extent to which this motif also accounted for the observed HU-induced instability of Bu-1a expression in wild-type DT40. We grew wild-type cells lacking the +3.5 G4 on both alleles, BU-1ΔG4 (Schiavone et al., 2014), in HU and assessed the frequency of Bu-1alow variants after 7 days by fluctuation analysis (see Figure E3 in Schiavone et al., 2014). This revealed that removal of the +3.5 G4 motif resulted in a significant reduction in the rate at which Bu-1alow variants were generated. However, it did not result in complete stabilization of the locus (Figures 2A and 2B, i and ii). We considered the possibility that the residual instability could be due to the −3.0 G4 upstream of the TSS. However, deleting this motif (Figure S2) had no impact on HU-induced instability (Figure 2B, iii). To confirm the contribution of the +3.5 G4 motif, we re-introduced it to its native position. This resulted in the return of the high-level HU-induced instability of BU-1 expression observed in wild-type cells (Figure 2C, i). However, this was not seen if the motif was mutated to render it incapable of forming an intramolecular G4 (Figure 2C, ii) or when it was inverted so that the G4 structure would form on the lagging strand template (Figure 2C, iii). Thus, HU treatment alone can induce instability of Bu-1a expression, but its effect is significantly potentiated by the presence of a G4-forming sequence orientated to stall the leading strand replication of a fork heading toward the TSS.
Figure 2.

Chronic Replication Stress Induces Stochastic Instability of Bu-1a Expression that Is Largely Dependent on the +3.5 G4 Motif
(A) Example flow cytometry plots for Bu-1a expression in wild-type and BU-1ΔG4 cells, before and after 7 days culture in 150 μM HU, are shown.
(B) Potentiation of HU-induced instability of Bu-1a expression by the +3.5 G4, but not the −3.0 G4. Wild-type cells (i), BU-1Δ+3.5G4 cells (ii), and BU-1Δ-3.0G4 cells (iii) are shown.
(C) Determinants of BU-1 instability induced by the +3.5 G4 motif. (i) The +3.5 G4 motif was knocked back into BU-1ΔG4 cells. (ii) Knockin of the +3.5 G4 motif mutated to prevent intramolecular G4 formation (Schiavone et al., 2014). (iii) Knockin of the +3.5 G4 motif inverted so that the G4 forms on the lagging-strand template.
G4-Dependent and -Independent Changes in Histone Modification at the BU-1 Promoter Induced by HU
We next investigated the basis for the HU-induced generation of Bu-1alow variants. To test whether the Bu-1low state is permanent, we isolated five Bu-1low clones at the end of 1-week growth in HU and cultured them for a further 3 weeks in HU-free medium. The clones remained stably Bu-1low with no evidence of reversion to Bu-1high, suggesting that this was a permanent change. We considered the possibility that Bu-1low cells resulted from genetic changes in the locus, although the observed rate of mutation would be extraordinarily high for this to be the case. We therefore sequenced the region around the +3.5G4 to look for mutation of the motif and used PCR with restriction digestion to detect larger deletions (Figure S3). Neither assay revealed any evidence of genetic instability consistent with the formation of Bu-1low variants being an epigenetic event.
We therefore examined the pattern of histone modification at the BU-1 promoter by chromatin immunoprecipitation (ChIP) from bulk populations of cells exposed to HU. BU-1 is a transcriptionally active locus characterized by high levels of H3K4me3 around its TSS. After 48-hr treatment with HU, we observed a small but not significant loss of H3K4me3 at the BU-1 promoter (Figure 3A), consistent with the size of the population of Bu-1alow cells generated by this time point (Figure 1D). However, after 7-day treatment, we observed a more significant loss of H3K4me3 correlating with the much larger population of Bu-1alow cells at this time point (Figures 1D and 2A). The loss of H3K4me3 was accompanied by a reduction in H3K9/14 acetylation (Figure S4). Seven days of HU treatment also induced a marked increase in H3K9me3 at the BU-1 promoter (Figure 3A). We considered two potential explanations for this observation. It has been proposed previously that HU-induced displacement of parental H3/4 and its buffering by the histone chaperone Asf1 may lead to unscheduled heterochromatinization by ectopic deposition of pre-marked histones upon their release from Asf1 (Jasencakova et al., 2010, Schwab et al., 2013). Alternatively, the appearance of H3K9me3 may result from DNA damage-induced heterochromatinization, which has been observed following double-strand breaks (Ayrapetov et al., 2014, Shanbhag et al., 2010). Breaks can arise from fork collapse in HU (Petermann et al., 2010), and this may be exacerbated by the +3.5 G4 motif. Thus, if unscheduled incorporation of H3 with K9 methylation was responsible, then an increase in H3K9me3 would be observed irrespective of whether the +3.5 G4 motif was present. However, if localized G4-induced DNA damage was responsible, then the appearance of H3K9me3 would be dependent on the +3.5 G4 motif. We therefore examined H3K4me3 and H3K9me3 at the promoter of BU-1 in cells lacking the +3.5 G4 motif. After 7 days in HU, H3K4me3 was reduced (Figure 3B), but to a lesser extent than in wild-type cells (Figure 3A), consistent with the reduced rate at which Bu-1alow variants are generated in cells lacking the +3.5 G4 motif (Figure 2B). However, we observed no associated increase in H3K9me3 (Figure 3B). To monitor the extent to which HU induced DNA damage in the two situations, we performed ChIP for phosphorylated H2Ax (γH2Ax) (Rogakou et al., 1998) at the BU-1 promoter. γH2Ax was enriched 2.5-fold at the BU-1 promoter in wild-type cells after 7 days in HU, but not enriched in cells lacking the +3.5 G4 motif grown under the same conditions (Figure 3C). This favors a model in which heterochromatinization of BU-1 in HU is promoted by DNA damage, likely from fork collapse associated with the +3.5 G4 motif.
Figure 3.

Epigenetic Changes in the BU-1 Promoter Are Dependent on HU and the +3.5 G4 Motif
(A) H3K4me3 and H3K9me3 in untreated wild-type cells and cells treated with 150 μM for 48 hr and for 7 days. The enrichment for each mark is normalized to total H3 and then to untreated. The enrichment with non-specific IgG is shown as a control. Positive and negative controls for each mark are shown with ChIP at a constitutively active locus, GAS41, and a heterochromatinized locus, LYSC, respectively. Error bars show SD for three independent ChIPs.
(B) H3K4me3 and H3K9me3 in BU1ΔG4 cells, either untreated or treated with 150 μM for 7 days.
(C) Enrichment of γH2Ax in wild-type and BU1ΔG4 after treatment with HU for 7 days. γH2Ax signal is normalized to H3 and then to the level in untreated cells of each condition. Error bars show SD for four independent ChIPs for wild-type and five for BU1ΔG4 cells.
(D) H3K4me3 in Bu-1alow clones isolated after 7 days in 150 μM HU. Each point represents the results of a single ChIP from a single clone of either untreated (solid circles) or treated (open circles) cells. The central bar shows the mean and whiskers the SD for H3K4me3 in the five samples normalized to H3 and then to the mean of the untreated wild-type.
(E) H3K9me3 in the same clones as shown in (D). Normalization and error bars are as in (D).
We next examined the extent to which HU-induced changes in histone modifications were permanent by performing ChIP at the BU-1 promoter in the five stable Bu-1alow clones discussed above. This revealed that promoter H3K4me3 remained low, showing that loss of this mark was permanent (Figure 3D). However, the enrichment of H3K9me3 was not preserved (Figure 3E). Thus, while H3K9me3 is induced by HU in cells containing the +3.5 G4 motif, this mark is not essential to maintain the Bu-1alow state. This may be because it is installed only transiently during repair of HU-induced DNA damage in the locus, or because cells in which H3K9me3 persists are growth disadvantaged and are lost from the population.
G4 Stabilization Potentiates the Effect of HU on Instability of BU-1
The data thus far were consistent with a working hypothesis that reduced polymerase processivity increases the probability of G4 formation at the +3.5 G4 motif through exposure of more single-stranded DNA within the replisome, which, in turn, focuses replication stalling at this site. Implicit in this model is the idea that the +3.5 G4 can form during replication but that it is usually rapidly resolved to maintain fork progression. We therefore reasoned that trapping the G4 structure using a G4-binding ligand also might induce instability of Bu-1a expression in otherwise wild-type cells. Further, we predicted that G4 ligands and HU would act synergistically to destabilize expression of the locus. To test these ideas, we treated cells with the G4 ligand N-methyl mesoporphyrin IX (NMM) (Nicoludis et al., 2012). We first identified the maximum dose at which the cells retained normal viability and global replication dynamics. At 2 μM NMM, the fork rate, as assessed by molecular combing, was 1.13 kb/min compared with 1.26 kb/min in wild-type cells, with no significant change in the inter-origin distance (Figure S5). Nonetheless, fluctuation analysis for Bu-1a loss in wild-type and BU-1ΔG4 cells cultured for 7 days in 2 μM NMM revealed instability in Bu-1a expression in wild-type cells, but not cells lacking the +3.5 G4 motif (Figure 4A). Since 2 μM NMM does not in itself significantly reduce global fork rates and the agent will only interact with the formed G4 structure, not with just the linear DNA sequence (Ren and Chaires, 1999), this observation is consistent with transient formation of G4s during normal replication.
Figure 4.

A G4-Binding Ligand and Replication Stress Synergistically Destabilize Bu-1a Expression
(A) NMM induces instability of Bu-1a expression that is dependent on the +3.5 G4 motif.
(B) Fluctuation analysis of wild-type and BU1ΔG4 cells grown with or without 2 μM NMM and HU act synergistically to destabilize Bu-1a expression. Fluctuation analysis of parallel cultures of ten cells grown in 75 μM HU, 1 μM NMM, or both drugs for 7 days.
We next asked whether combining NMM-induced G4 stabilization with HU-induced reduction in polymerase processivity led to a further destabilization of BU-1 expression. Interestingly, use of both drugs together resulted in significant toxicity, meaning that we had to reduce the dose of each drug by 50% in order to carry out the fluctuation analysis. As expected, growth of cells in HU at 75 μM or NMM at 1 μM individually had little effect on stability of Bu-1a expression (Figure 4B). However, the combination of NMM and HU at these doses resulted in a significant increase in Bu-1a instability, revealing a marked synergy between HU-induced replication stress and G4 stabilization.
Replication Stress-Induced Epigenetic Instability Is Potentiated by a Wide Range of G4 Motifs
We next asked whether other G4 motifs could potentiate HU-induced epigenetic instability. We replaced the natural +3.5 G4 motif with a series of four G4 motifs of varying in vitro thermal stabilities (Schiavone et al., 2014). All four motifs (G4 1–4) potentiated the formation of Bu-1alow variants upon treatment with HU (Figure 5A). Interestingly, we observed no correlation between the degree of potentiation by the motifs and the in vitro melting temperature of the equivalent oligonucleotides (Figure 5B). However, there was a significant trend toward greater potentiation of Bu-1a loss being associated with longer non-G loops in the range of 1 to 9 bp (Figure 5C, solid line). To explore this further, we also tested a single repeat of the G4 motif containing human CEB1 mini-satellite (Piazza et al., 2012), which has 18 bp between its first three and last run of dGs. This G4 motif, but not a mutated form that is incapable of forming a G4 structure in vitro (Piazza et al., 2012), also potentiated Bu-1a instability after treatment with HU. However, this was not to a greater extent than the natural +3.5 G4 DNA with its central 9-bp loop, suggesting that there may be a limit after which lengthening the loop has no further effect.
Figure 5.

HU-Induced Instability Can Be Potentiated by a Wide Range of G4 Motifs
(A) Fluctuation analyses for Bu-1a loss in lines carrying G4 motifs of differing in vitro thermal stabilities and potential loop lengths in place of the +3.5 G4 motif on BU1A. The BU1B +3.5 G4 motif is deleted. The knocked-in sequence is shown along with the measured in vitro thermal stability and maximum non-G loop length (Ribeyre et al., 2009, Schiavone et al., 2014).
(B) Correlation of in vitro G4 melting temperature with potentiation of Bu-1a expression instability. The y axis shows the additional mean Bu-1a loss over that seen in the BU1ΔG4 cells.
(C) Correlation of maximum non-G loop length with potentiation of Bu-1a expression instability. The linear regression line fits the points from G4 1–4 and the +3.5 G4 motif.
HU Treatment and Loss of the G4 Processing Helicase FANCJ Results in Very Similar Patterns of Transcriptional Dysregulation
Finally, we asked whether we could detect genome-wide evidence of an interaction between HU and G4s. We therefore performed Affymetrix expression microarray analysis on cells before and after culture in 150 μM HU. Three parallel cultures of DT40 were treated with 150 μM HU for 7 days, or mock treated, and then recovered into normal medium for 7 days, after which RNA was prepared for array hybridization. Despite this relatively short treatment, a total of 2,937 of 12,920 unique genes exhibited a change in expression of >0.25 log2 units with p < 0.05, with an approximately equal number of genes being upregulated and downregulated (Figure 6A).
Figure 6.

Similarity in the Pattern of Genome-wide Changes Induced by HU Treatment and Disruption of fancj or wrn/blm
(A) Up- and downregulated genes in HU-treated cells are shown.
(B) Overlap in genes dysregulated in HU-treated cells, fancj cells, and wrn/blm cells. (Left) Venn diagram shows the number of genes in each set. (Right) Venn diagram shows the percentage of genes in each overlap whose expression changes in the same direction. The p value was calculated with Fisher’s hypergeometric distribution.
(C) Correlation of magnitude and direction of change in expression between fancj, wrn/blm mutants and HU-treated wild-type cells.
We previously have observed a similarly large number of dysregulated genes in cells deficient in the 5′-3′ G4-unwinding helicase FANCJ and in double mutants for the 3′-5′ helicases WRN and BLM (Sarkies et al., 2012). Further, we found a highly significant overlap in the identity of dysregulated genes in the two sets, the direction in which their expression changed, and the association of the dysregulated genes with G4 motifs (Sarkies et al., 2012). We anticipated that if transcriptional dysregulation by HU was linked with G4s that there might be a significant similarity in the gene set altered by HU and the sets altered by loss of FANCJ and WRN/BLM. Indeed, the overlap in the identities of the genes dysregulated in all three conditions was highly significant (Figure 6B), as were the pairwise correlations in the direction of the change in expression (Figure 6C). Nearly 68% of the 6,061 genes within the Venn diagram in Figure 6B have a G4 motif within 1 kb upstream of the TSS and the end of the body of the gene in comparison with 59% of the 6,859 genes in the remainder of the array (p < 1 × 10−13) (Table S1).
To ascertain whether the overlaps in the identity of dysregulated genes reflected the perturbation of common pathways in the three datasets, we analyzed the functional annotation terms associated with the genes in each set and in the overlap sets using DAVID (https://david.ncifcrf.gov; Huang et al., 2009a, Huang et al., 2009b). While treatment with HU resulted in dysregulation of genes with gene ontology (GO) terms associated with cellular stress and nucleotide metabolism, a large number of miscellaneous GO terms also were enriched to a similar degree (Table S2). Significantly, despite the large number of genes overlapping in the three datasets, there was no evidence of their being members of common pathways (Figure S6). This is consistent with much of the dysregulation of expression resulting from processes that are not related to a coordinated physiological response either to treatment with HU or ablation of FANCJ or WRN and BLM helicases. Nonetheless, the degree of overlap in the dysregulated transcriptomes in these three conditions suggest that cells treated with HU and cells lacking FANCJ and WRN/BLM face similar challenges. However, the enrichment of G4s in affected genes, while statistically significant, is still relatively modest, suggesting that other factors, such as secondary effects or other DNA secondary structures, may be contributing as well.
Discussion
Numerous lines of evidence have linked replication stress with genetic instability (Halazonetis et al., 2008, Zeman and Cimprich, 2014). Imbalanced or depleted nucleotide pools during replication are an important cause of such stress and can arise from the expression of oncogenes uncoupling entry into S phase from upregulation of nucleotide supply (Bester et al., 2011). Importantly, the DNA damage resulting from replication stressors like HU or aphidicolin that act directly on the replicative DNA polymerases is not randomly distributed across the genome, but is instead focused on sites that often have features that make them potentially problematic to replicate even under ideal conditions (Tsantoulis et al., 2008). Many of these hotspots also correspond to classical fragile sites, in which chromosome breaks are observed after replication stress. Such sites have been linked to regions depleted in replication origins, meaning that single forks have to traverse long distances (Letessier et al., 2011). Thus, a combination of regions of low fork density and problematic structures may focus sites of fork collapse under conditions of global replication stress (Wickramasinghe et al., 2015).
Mechanisms of Replication Stress-Induced Epigenetic Instability
The mechanisms by which replication stress leads to epigenetic changes are less well explored. Alterations in chromatin composition and structure are common features of cancer cells (Berdasco and Esteller, 2010, Hansen et al., 2011, Timp and Feinberg, 2013) and are particularly associated with G4-dense breakpoint hotspots (De and Michor, 2011). Although the cell line we used in this study, DT40, is itself transformed, we found no evidence of significant stress to the DNA replication program under normal growth conditions. However, growth of the cells in low-dose HU recapitulated the key features of the acutely stressed replication observed in oncogene-expressing primary cells (Bester et al., 2011, Neelsen et al., 2013). Through genetic manipulation of a reporter locus, we have been able to explore directly the interaction between global replication stress induced by nucleotide depletion and a DNA secondary structure to demonstrate how they conspire to exacerbate replication-dependent epigenetic instability. We have provided evidence that two parallel epigenetic perturbations contribute to permanent and transient epigenetic changes following an episode of replication stress.
The first mechanism relates to the uncoupling of the activity of replicative helicase and polymerase (Byun et al., 2005, Pacek and Walter, 2004) during HU treatment. This has been shown to lead to interruption of the normal flow of histones from the parental to the nascent daughter strands, with the histone chaperone Asf1 buffering the displaced H3/H4 (Jasencakova et al., 2010). A key question is what then happens to these displaced histones. Groth and colleagues suggested that their release from Asf1 might lead to local alterations in epigenetic state of chromatin due to unscheduled incorporation of inappropriately marked histones (Jasencakova and Groth, 2010, Jasencakova et al., 2010). Schwab et al. (2013) invoked this model to explain an increase in heterochromatin formation in cells deficient in FANCJ, suggesting that failure to unwind lagging-strand template G4 structures in FANCJ-deficient cells led to unscheduled deposition of histones bearing marks that would lead to H3K9me3 and heterochromatin formation. However, this model does not adequately explain the bidirectional changes in gene expression changes seen either in fancj cells or in wild-type cells treated with HU (Sarkies et al., 2012; Figure 6). In contrast, the model we have developed previously, in which loss of processive replication at G4s leads to localized loss of parental histone mark recycling, could explain both derepression of loci, such as ρ-globin (Sarkies et al., 2010), and loss of activation, as can be observed in the BU-1 locus (Sarkies et al., 2012, Schiavone et al., 2014). However, since the mechanism by which H3K4me3 is maintained during replication is poorly understood, the precise mechanisms by which replication impediments disturb the maintenance of this mark remain to be fully elucidated.
The second mechanism relates to the induction of H3K9me3. This mark is induced alongside the loss of H3K4me3 when the +3.5 G4 motif is present and cells are exposed to HU. Importantly, the appearance of this mark of heterochromatin is accompanied by H2Ax phosphorylation, a marker of DNA damage (Rogakou et al., 1998). G4 motifs have been linked to hotspots of genetic instability and translocation (De and Michor, 2011). Further, the G4 ligand pyridostatin, which acts similarly to NMM, leads to localized γH2Ax accumulation at G4 motifs across the genome, suggesting the formation of DNA breaks (Rodriguez et al., 2012). DNA breaks have been shown to induce transcriptional repression (Shanbhag et al., 2010) and to induce H3K9me3 even in a normally euchromatic locus (Ayrapetov et al., 2014). Thus, the appearance of γH2Ax and H3K9me3 in the BU-1 locus only in HU-treated cells containing the +3.5 G4 is consistent with collapse or incision of replication forks, already stressed by nucleotide depletion, that have stalled at the G4. We therefore propose that nucleotide depletion can give rise to loss of parental H3K4me3 and the appearance of H3K9me3 by distinct mechanisms. H3K4me3 is lost stochastically as a result of interruption of parental histone recycling, a mechanism that is locally exacerbated by the presence of a G4 motif. In contrast, we propose that H3K9me3 may reflect protective transient heterochromatinization of the locus during repair of breaks resulting from HU-induced fork collapse at G4 structures (Figure 7).
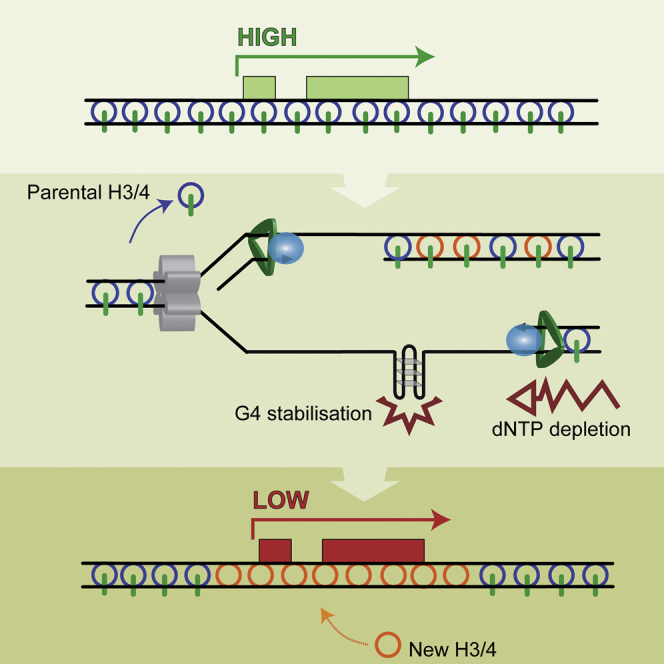
Figure 7.

A Model for the Mechanism of the Collaborative Effect of Nucleotide Depletion and G4 Formation in Promoting Epigenetic Instability
(A) During normal replication H3/4 tetramers are recycled, with their marks, from the parental to nascent daughter strands. This process is closely coupled to the advancing fork and, thus, the registration between the marks and the underlying DNA sequence in maintained. Newly synthesized H3/4 tetramers also are incorporated into the nascent strands where they receive copies of the parental marks, a process dependent on the availability of nearby parental tetramers.
(B) In HU the replicative helicase and polymerase become uncoupled, leading to formation of regions of single-stranded DNA. The displaced tetramers that cannot be incorporated into these regions are buffered by Asf1.
(C) The single-stranded DNA is filled in late, DNA synthesis being uncoupled from histone recycling.
(D) There is thus biased incorporation of newly synthesized H3/4 and the potential for loss of parental histone modifications and epigenetic information.
(E and F) Formation of G4 structures is promoted by the single-stranded DNA formed during HU treatment, and the struggling polymerases stall preferentially at these sites (E). This can lead to localized loss of epigenetic information (C) or to fork collapse (F). In the latter case, we propose that there is transient formation of heterochromatin while the collapsed fork is repaired. In the longer term, this leaves loss of the parental marks, or cells with persistent breaks and/or extensive ectopic heterochromatin die.
In the case of loci like BU-1, in which HU-induced epigenetic instability is linked to G4 formation, an interesting question is whether HU results in a greater opportunity for G4 formation during replication or diminished G4 resolution. It is not currently possible to formally distinguish these possibilities and indeed it is likely that elements of both are true. Notably, the observation that the G4-binding ligand NMM can induce Bu-1a expression instability at a dose that does not significantly impact on global replication dynamics provides strong evidence that G4 structures form readily during normal replication and that they are usually promptly resolved.
The Potential Clinical Importance of Dysregulated Gene Expression Caused by Replication Stress and Structured DNA
The synergy between replication stress caused by nucleotide depletion and structured DNA is of considerable potential importance to understanding the development of cancer. Epigenetic changes are prevalent in many cancer types, although their origin is unclear, and likely complex (Berdasco and Esteller, 2010, Timp and Feinberg, 2013). Recently, several instances of epigenetic instability in cancer have been linked to mutations in histone or DNA-modifying enzymes. However, the widespread and often apparently random nature of epigenetic changes in tumors suggests that other processes also may be at work. We have suggested previously that delayed replication of G quadruplex structures could contribute to the epigenetic diversity of cancer (Sarkies and Sale, 2012). However, mutations in enzymes that may cause this form of epigenetic instability, for example REV1, FANCJ, WRN, and BLM (Sarkies et al., 2010, Sarkies et al., 2012), are rarely observed in sporadic cancers. Replication stress, on the other hand, is emerging as an important feature of cancer cells, particularly in the early stages of their evolution (Bester et al., 2011, Di Micco et al., 2006, Halazonetis et al., 2008). Thus, we suggest that some of the epigenetic changes seen in tumors may be explained by problems managing replication blocks. Consistent with this idea, both copy number variations and changes in DNA methylation patterns in cancer have been linked to G4 motifs (De and Michor, 2011).
Finally, it is worth noting that HU is used extensively in treatment of hemoglobinopathies, such as sickle cell disease and thalassaemia, as it can re-induce expression of the fetal γ-globin gene, ameliorating the effects of the defective adult globins found in these disorders (Platt et al., 1984). However, the mechanism by which HU does this is unclear. Importantly, the effect of HU on γ-globin expression is unlikely to be specific, since chronic exposure to the drug leads to quite widespread changes in erythroid gene expression (Flanagan et al., 2012). Although the γ-globin locus in humans has no G4 motifs in the immediate vicinity of its promoter, its key transcriptional regulator, BCL11a (Bauer et al., 2013), has a high density of G4 motifs on both sides of its TSS. It will, therefore, be interesting to explore whether the mechanisms we propose here could help explain the action of HU on fetal globin expression.
Experimental Procedures
DT40 Cell Culture, Constructs, and Gene Targeting
DT40 cells culture and the strategy for removing and replacing the +3.5 G4 motif in the BU-1 locus have been described previously (Schiavone et al., 2014). Genetic manipulation of the +3.5 G4 motif was performed in the BU-1A allele of cells in which the motif had been removed from the BU-1B allele to avoid the transvection-like effect between the alleles (Schiavone et al., 2014). Oligonucleotides are listed in Table S3.
Drug Treatments and Fluctuation Analysis for Generation of Bu-1alow Variants
For fluctuation analysis, 150 μM HU (Sigma-Aldrich, H8627) was added to 1 × 104 cells in a 24-well plate. After 7 days, cells at a concentration between 0.2 and 1 × 106 were stained for 20 min at 37°C with anti-Bu-1a-phycoerythrin (1:100, Santa Cruz Biotechnology clone 5K98, 70447). Bu-1a expression was assessed by flow cytometry using an LSRII cytometer (Becton Dickinson). Experiments with aphidicolin (Sigma-Aldrich, A0781) and NMM (Frontier Scientific, NMM580) were conducted in 96-well plates starting with ten cells expanded for 10 days. Bu-1alow cells were isolated after HU treatment using a MoFlo sorting cytometer (Dako-Cytomation).
ChIP and Antibodies
ChIP was performed as described previously (Nelson et al., 2006) with modifications. Following a 10-min incubation at room temperature with 1% (v/v) formaldehyde, glycine was added to 0.2 M for 5 min. The extracted nuclei were sonicated at 4°C using a Bioruptor water bath sonicator (Diagenode) with 30 cycles of 30 s separated by 30-s intervals. Sheared chromatin samples were resuspended in dilution buffer (1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris [pH 8.0], 167 mM NaCl supplemented with PMSF, and a protease inhibitor cocktail). For immunoprecipitation, lysates were incubated overnight with the following antibodies at 4°C: histone H3 (1:100, Cell Signaling Technology, 2650), H3K4me3 (1:100, Cell Signaling Technology, 9727), H3K9/14ac (1:200, Millipore, 17-615), H3K9me3 (1:200, Abcam, ab8898), γH2AX (1:50, Abcam, ab2893), and the negative control normal rabbit IgG (Millipore). Following overnight incubation at 4°C with tumbling and four washing steps, the DNA was purified.
The qPCR was performed with Power SYBR Green Master Mix (Applied Biosystems, 4367659) on an ABI Prism real-time cycler with the following cycle times: 50°C for 2 min, 90°C for 10 min, 45 cycles of 90°C for 15 s plus 60°C for 1 min. The qPCR reactions were performed in triplicate. Primers used are listed in Table S4.
qRT-PCR
The cDNA was made from 5 mg mRNA with Super RT (HT Biotechnology) and oligodT primer in a final volume of 40 μl. β-actin was used as a control and primers are listed in Table S4.
DNA Combing
DNA molecular combing was conducted 3 days into culture with 150 μM HU. It was performed and analyzed as previously described (Guilbaud et al., 2011).
Microarray Analysis
RNA was extracted from three independent wild-type cell populations treated for 7 days with HU and allowed to recover for another 7 days, as well as from three untreated parallel controls. Extraction was performed using Trizol as previously described. Microarray hybridizations were performed using total RNA and the Affymetrix Chicken Genome array. Microarray analysis was performed using R (http://www.R-project.org/) and its Bioconductor packages (Gentleman et al., 2004). Raw CEL files were processed using the robust multichip average (RMA) algorithm available in the affy package (Gautier et al., 2004). A total of 12,920 unique genes from the galgal4 genome build were analyzed. Genes that showed a change of >0.25 log2 units relative to the mean wild-type intensity, with a p value of < 0.05 (t test), were identified as exhibiting statistically significant transcriptional dysregulation. Custom written R scripts were used to identify and plot genes co-dysregulated between different mutants. Venn diagrams were generated with the limma package (Smyth, 2004), and significance for the overlaps was calculated using Fisher’s hypergeometric distribution.
Acknowledgments
The authors thank Maria Daly and Fan Zhang in the LMB Flow Cytometry Facility for cell sorting and members of the J.E.S. lab for discussions. Work in the lab is supported by a central grant to the LMB from the Medical Research Council (U105178808). C.P. was supported by a Wellcome Trust TMAT studentship, D.S. was funded by The Association for International Cancer Research (now Worldwide Cancer Research, 11-0514) and an EMBO Long Term Fellowship, and G.G. was supported by The Fanconi Anemia Research Fund and the Medical Research Council.
Published: December 10, 2015
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, six figures, and four tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2015.11.039.
Contributor Information
Guillaume Guilbaud, Email: guilbaud@mrc-lmb.cam.ac.uk.
Julian E. Sale, Email: jes@mrc-lmb.cam.ac.uk.
Accession Numbers
The accession number for the array experiments reported in this paper is ArrayExpress: E-MTAB-3930.
Supplemental Information
References
- Alvino G.M., Collingwood D., Murphy J.M., Delrow J., Brewer B.J., Raghuraman M.K. Replication in hydroxyurea: it’s a matter of time. Mol. Cell. Biol. 2007;27:6396–6406. doi: 10.1128/MCB.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglana M., Apiou F., Bensimon A., Debatisse M. Dynamics of DNA replication in mammalian somatic cells: nucleotide pool modulates origin choice and interorigin spacing. Cell. 2003;114:385–394. doi: 10.1016/s0092-8674(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Ayrapetov M.K., Gursoy-Yuzugullu O., Xu C., Xu Y., Price B.D. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc. Natl. Acad. Sci. USA. 2014;111:9169–9174. doi: 10.1073/pnas.1403565111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L.-V.F., Kolettas E., Niforou K., Zoumpourlis V.C. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- Bauer D.E., Kamran S.C., Lessard S., Xu J., Fujiwara Y., Lin C., Shao Z., Canver M.C., Smith E.C., Pinello L. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342:253–257. doi: 10.1126/science.1242088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M., Esteller M. Aberrant epigenetic landscape in cancer: how cellular identity goes awry. Dev. Cell. 2010;19:698–711. doi: 10.1016/j.devcel.2010.10.005. [DOI] [PubMed] [Google Scholar]
- Berger S.L., Kouzarides T., Shiekhattar R., Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bester A.C., Roniger M., Oren Y.S., Im M.M., Sarni D., Chaoat M., Bensimon A., Zamir G., Shewach D.S., Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun T.S., Pacek M., Yee M.-C., Walter J.C., Cimprich K.A. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De S., Michor F. DNA secondary structures and epigenetic determinants of cancer genome evolution. Nat. Struct. Mol. Biol. 2011;18:950–955. doi: 10.1038/nsmb.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre’ M., Nuciforo P.G., Bensimon A. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- Flanagan J.M., Steward S., Howard T.A., Mortier N.A., Kimble A.C., Aygun B., Hankins J.S., Neale G.A., Ware R.E. Hydroxycarbamide alters erythroid gene expression in children with sickle cell anaemia. Br. J. Haematol. 2012;157:240–248. doi: 10.1111/j.1365-2141.2012.09061.x. [DOI] [PubMed] [Google Scholar]
- Gautier L., Cope L., Bolstad B.M., Irizarry R.A. affy--analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20:307–315. doi: 10.1093/bioinformatics/btg405. [DOI] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud G., Rappailles A., Baker A., Chen C.-L., Arneodo A., Goldar A., d’Aubenton-Carafa Y., Thermes C., Audit B., Hyrien O. Evidence for sequential and increasing activation of replication origins along replication timing gradients in the human genome. PLoS Comput. Biol. 2011;7:e1002322. doi: 10.1371/journal.pcbi.1002322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis T.D., Gorgoulis V.G., Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008;319:1352–1355. doi: 10.1126/science.1140735. [DOI] [PubMed] [Google Scholar]
- Hansen K.D., Timp W., Bravo H.C., Sabunciyan S., Langmead B., McDonald O.G., Wen B., Wu H., Liu Y., Diep D. Increased methylation variation in epigenetic domains across cancer types. Nat. Genet. 2011;43:768–775. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Jasencakova Z., Groth A. Replication stress, a source of epigenetic aberrations in cancer? BioEssays. 2010;32:847–855. doi: 10.1002/bies.201000055. [DOI] [PubMed] [Google Scholar]
- Jasencakova Z., Scharf A.N.D., Ask K., Corpet A., Imhof A., Almouzni G., Groth A. Replication stress interferes with histone recycling and predeposition marking of new histones. Mol. Cell. 2010;37:736–743. doi: 10.1016/j.molcel.2010.01.033. [DOI] [PubMed] [Google Scholar]
- Letessier A., Millot G.A., Koundrioukoff S., Lachagès A.-M., Vogt N., Hansen R.S., Malfoy B., Brison O., Debatisse M. Cell-type-specific replication initiation programs set fragility of the FRA3B fragile site. Nature. 2011;470:120–123. doi: 10.1038/nature09745. [DOI] [PubMed] [Google Scholar]
- Maizels N., Gray L.T. The G4 genome. PLoS Genet. 2013;9:e1003468. doi: 10.1371/journal.pgen.1003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen K.J., Zanini I.M.Y., Herrador R., Lopes M. Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J. Cell Biol. 2013;200:699–708. doi: 10.1083/jcb.201212058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson J.D., Denisenko O., Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 2006;1:179–185. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- Nicoludis J.M., Barrett S.P., Mergny J.-L., Yatsunyk L.A. Interaction of human telomeric DNA with N-methyl mesoporphyrin IX. Nucleic Acids Res. 2012;40:5432–5447. doi: 10.1093/nar/gks152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro M., Suzuki-Hori C., Nagano H., Mano Y., Ikegami S. The mode of inhibitory action by aphidicolin on eukaryotic DNA polymerase alpha. Eur. J. Biochem. 1979;97:603–607. doi: 10.1111/j.1432-1033.1979.tb13149.x. [DOI] [PubMed] [Google Scholar]
- Pacek M., Walter J.C. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004;23:3667–3676. doi: 10.1038/sj.emboj.7600369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek M., Tutter A.V., Kubota Y., Takisawa H., Walter J.C. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol. Cell. 2006;21:581–587. doi: 10.1016/j.molcel.2006.01.030. [DOI] [PubMed] [Google Scholar]
- Petermann E., Orta M.L., Issaeva N., Schultz N., Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol. Cell. 2010;37:492–502. doi: 10.1016/j.molcel.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza A., Serero A., Boulé J.-B., Legoix-Né P., Lopes J., Nicolas A. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012;8:e1003033. doi: 10.1371/journal.pgen.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt O.S., Orkin S.H., Dover G., Beardsley G.P., Miller B., Nathan D.G. Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia. J. Clin. Invest. 1984;74:652–656. doi: 10.1172/JCI111464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli J., Tsaponina O., Crabbé L., Keszthelyi A., Pantesco V., Chabes A., Lengronne A., Pasero P. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 2012;31:883–894. doi: 10.1038/emboj.2011.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J., Chaires J.B. Sequence and structural selectivity of nucleic acid binding ligands. Biochemistry. 1999;38:16067–16075. doi: 10.1021/bi992070s. [DOI] [PubMed] [Google Scholar]
- Ribeyre C., Lopes J., Boulé J.-B., Piazza A., Guédin A., Zakian V.A., Mergny J.-L., Nicolas A. The yeast Pif1 helicase prevents genomic instability caused by G-quadruplex-forming CEB1 sequences in vivo. PLoS Genet. 2009;5:e1000475. doi: 10.1371/journal.pgen.1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez R., Miller K.M., Forment J.V., Bradshaw C.R., Nikan M., Britton S., Oelschlaegel T., Xhemalce B., Balasubramanian S., Jackson S.P. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012;8:301–310. doi: 10.1038/nchembio.780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- Sarkies P., Sale J.E. Cellular epigenetic stability and cancer. Trends Genet. 2012;28:118–127. doi: 10.1016/j.tig.2011.11.005. [DOI] [PubMed] [Google Scholar]
- Sarkies P., Reams C., Simpson L.J., Sale J.E. Epigenetic instability due to defective replication of structured DNA. Mol. Cell. 2010;40:703–713. doi: 10.1016/j.molcel.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkies P., Murat P., Phillips L.G., Patel K.J., Balasubramanian S., Sale J.E. FANCJ coordinates two pathways that maintain epigenetic stability at G-quadruplex DNA. Nucleic Acids Res. 2012;40:1485–1498. doi: 10.1093/nar/gkr868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavone D., Guilbaud G., Murat P., Papadopoulou C., Sarkies P., Prioleau M.-N., Balasubramanian S., Sale J.E. Determinants of G quadruplex-induced epigenetic instability in REV1-deficient cells. EMBO J. 2014;33:2507–2520. doi: 10.15252/embj.201488398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab R.A., Nieminuszczy J., Shin-ya K., Niedzwiedz W. FANCJ couples replication past natural fork barriers with maintenance of chromatin structure. J. Cell Biol. 2013;201:33–48. doi: 10.1083/jcb.201208009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shanbhag N.M., Rafalska-Metcalf I.U., Balane-Bolivar C., Janicki S.M., Greenberg R.A. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–981. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article 3. [DOI] [PubMed] [Google Scholar]
- Timp W., Feinberg A.P. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat. Rev. Cancer. 2013;13:497–510. doi: 10.1038/nrc3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsantoulis P.K., Kotsinas A., Sfikakis P.P., Evangelou K., Sideridou M., Levy B., Mo L., Kittas C., Wu X.-R., Papavassiliou A.G., Gorgoulis V.G. Oncogene-induced replication stress preferentially targets common fragile sites in preneoplastic lesions. A genome-wide study. Oncogene. 2008;27:3256–3264. doi: 10.1038/sj.onc.1210989. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe C.M., Arzouk H., Frey A., Maiter A., Sale J.E. Contributions of the specialised DNA polymerases to replication of structured DNA. DNA Repair (Amst.) 2015;29:83–90. doi: 10.1016/j.dnarep.2015.01.004. [DOI] [PubMed] [Google Scholar]
- Zeman M.K., Cimprich K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.