

Abstract
This paper is based upon the “Charles Lieber Satellite Symposia” organized by Manuela G. Neuman at the Research Society on Alcoholism (RSA) Annual Meetings, 2013 and 2014. The present review includes pre-clinical, translational and clinical research that characterize alcoholic liver disease (ALD) and non-alcoholic steatohepatitis (NASH). In addition, a literature search in the discussed area was performed. Strong clinical and experimental evidence lead to recognition of the key toxic role of alcohol in the pathogenesis of ALD. The liver biopsy can confirm the etiology of NASH or alcoholic steatohepatitis (ASH) and assess structural alterations of cells, their organelles, as well as inflammatory activity. Three histological stages of ALD are simple steatosis, ASH, and chronic hepatitis with hepatic fibrosis or cirrhosis. These latter stages may also be associated with a number of cellular and histological changes, including the presence of Mallory's hyaline, megamitochondria, or perivenular and perisinusoidal fibrosis. Genetic polymorphisms of ethanol metabolizing enzymes such as cytochrome p450 (CYP) 2E1 activation may change the severity of ASH and NASH. Alcohol mediated hepatocarcinogenesis, immune response to alcohol in ASH, as well as the role of other risk factors such as its comorbidities with chronic viral hepatitis in the presence or absence of human deficiency virus are discussed. Dysregulation of hepatic methylation, as result of ethanol exposure, in hepatocytes transfected with hepatitis C virus (HCV), illustrates an impaired interferon signaling. The hepatotoxic effects of ethanol undermine the contribution of malnutrition to the liver injury. Dietary interventions such as micro and macronutrients, as well as changes to the microbiota are suggested. The clinical aspects of NASH, as part of metabolic syndrome in the aging population, are offered. The integrative symposia investigate different aspects of alcohol-induced liver damage and possible repair. We aim to (1) determine the immuno-pathology of alcohol-induced liver damage, (2) examine the role of genetics in the development of ASH, (3) propose diagnostic markers of ASH and NASH, (4) examine age differences, (5) develop common research tools to study alcohol-induced effects in clinical and pre-clinical studies, and (6) focus on factors that aggravate severity of organ-damage. The intention of these symposia is to advance the international profile of the biological research on alcoholism. We also wish to further our mission of leading the forum to progress the science and practice of translational research in alcoholism.
Keywords: Alcoholic hepatitis, Nonalcoholic steatohepatitis, Alcoholic liver disease, CYP2E1, Hangover, Hepatocarcinogenesis, Immunohistochemistry, Laboratory markers, Mallory–Denk bodies, Methylation, Mitochondrion, Micronutrients, Viral hepatitis, Human immunodeficiency virus
1. Lieber's and his colleagues' legacy
Manuela G. Neuman, Abraham Bautista.
Dr. Lieber broke new ground of biological research on alcohol by discovering the cytochrome p450 (CYP) 2E1-dependent microsomal ethanol oxidizing system (MEOS) (Lieber and DeCarli, 1968, 1970). The clarification of MEOS has contributed to the recognition of mechanisms accountable for alcohol–drug interactions (Lieber, 1988a), alcohol–methylation pathways (Lieber et al., 1994), alcohol-mediated fibrinogenesis (Lieber et al., 2006), molecular basis of alcohol-induced injury to liver and other tissues (Lieber, 1988b), alcohol-induced fatty liver, hepatitis and cirrhosis in sub-human primates (Lieber et al., 1975; Rubin and Lieber, 1974) and alcohol-related hepatotoxicity (Lieber, 1978, 1997; Lieber and DeCarli, 1991).
Alcohol-induced hepatic injury has been called as steatonecrosis (Harinasuta et al., 1967) and steatohepatitis (Ludwig et al., 1980). The injury has characteristic histologic, clinical, and biochemical features (Christoffersen and Nielsen, 1972; French, 1981; French et al., 1993; Ishak et al., 1991; Maher and Friedman, 1995; Mendenhall, 1981; Rojkind, 1985).
The classical clinical syndrome of alcoholic steatohepatitis (ASH) consists of jaundice, varying degrees of hepatic failure, abdominal distress, fever, and leukocytosis, although patients with the histological features of the entity may be asymptomatic and anicteric (Birschbach et al., 1974; Green, 1965). The syndrome appears in patients who have been consuming excess amounts of alcohol for periods of 1–5 years or more (Boitnott and Maddrey, 1981; Edmonson et al., 1963; Gregory and Levi, 1972; Helman et al., 1971; Schaffner and Popper, 1970).
Factors that affect the development of liver injury include the dose, duration, and type of alcohol consumption, drinking patterns, sex, age, ethnicity, as well as associated risk factors such as obesity, iron overload, concomitant infections, and genetics (Bautista, 2001; Martini and Bode, 1970; Zimmerman, 1999). Recognition of hepatic injury due to alcohol in the presence of antimicrobial agents may result from the hepatic involvement of bacterial, viral, or parasitic infections (Benhamou, 1986).
The assumption that the malnutrition so commonly manifested in alcoholics was responsible for the cirrhosis was supported by the therapeutic benefits of dietary treatment (Schenker and Halff, 1993). Moreover, the protein malnutrition plays an important role in determining the outcome of patients with alcoholic liver disease (ALD) (Zimmerman, 1955). Mortality increases in proportion to malnutrition, achieving 80% in patients with severe malnutrition (Mendenhall et al., 1995). Also the role of micronutrients and unsaturated or saturated dietary fat influences the severity of the ALD (Leevy and Moroianu, 2005; Lieber, 1988a; Mezey, 1998). Cirrhosis, including evidence of hepatic failure, as well as portal hypertension, should be treated with awareness to other organ damage associated with alcohol (Zimmerman, 1999). ALD may coexist with other organ damage related to alcohol misuse, including cardio-myopathy (Klatsky et al., 2005; Lazarević et al., 2000), muscle wasting (Preedy et al., 2001), and alcoholic neurotoxicity (Estruch et al., 1993). All of these concomitant organ dysfunctions should be considered in alcohol-induced injury (Anderson et al., 1993; Zimmerman, 1986).
Multiple laboratory markers establish alcohol as the etiology of liver disease. Furthermore, alcohol may be one of a number of factors causing liver injury, and the specific contributory role of alcohol alone may be difficult to assess in a patient with multiple causes of liver disease (O'Shea et al., 2010; Zimmerman, 1999).
Many researchers continue to work in this fascinating area of investigation. The purpose of the modern research is to use new technologies, innovative ideas, and integrative teams of basic and clinical researchers to elucidate different aspects of alcohol-induced liver damage. Since 2009, we meet each year before the Research of Alcoholism annual meeting to celebrate new achievements in understanding alcohol and non-alcohol liver damage.
We reviewed the “Alcohol and liver damage” symposia 2009–2012 in two publications (Neuman et al., 2013, 2014). The present review summarizes multi-factorial aspects of the pathological consequences of acute and chronic alcohol abuse exposed at the 2013 and 2014 symposia. The symposia addressed clinical issues such as pathology, diagnosis and treatments for ASH and NASH as well as nutritional and life style interventions. Investigating the common and different mechanisms in ASH and NASH, the symposia also proposed in vivo and in vitro models for ASH, NASH, hepatocellular carcinoma (HCC) and co-morbidities. We also intended to elucidate polymorphisms of genes involved in ALD and the role of microbiota in regulating endotoxin-mediated release of cytokines that have been associated with ALD. In addition to the original data, we searched the literature (2013–2014) for the latest publications on the described subjects. In order to obtain the updated data we used the usual engines (PubMed and Google Scholar).
2. The hallmarks of alcoholic hepatitis compared to non-alcoholic steatohepatitis
Samuel W. French, Barbara A. French, Hong Shen, Brittany Tillman, Jun Li, Hui Liu.
Alcoholic steatosis can occur after large alcohol consumption. It represents a direct effect of alcohol per se and can occur despite an adequate nutritional state. Early changes seen under the electron microscope include accumulation of membrane-bound fat droplets, proliferation of smooth endoplasmic reticulum, and gradual distortion of mitochondria. Lipid accumulation in ALD is macrovesicular and composed of neutral triglycerides. However, small droplets of such triglycerides may appear like microvesicular droplets. A liver biopsy is an important component of the diagnostic evaluation in patients with suspected ALD, because it can grade the severity and exclude other diagnoses. In addition, some histologic findings, such as perivenular fibrosis and the presence Mallory–Denk bodies (MDB) may be associated with an unfavorable prognosis in patients who have steatosis but have not yet developed cirrhosis.
Many years ago, Hans Popper speculated that, since ASH and NASH had a lot in common, their comparison would help us understand both disease processes. The differences and similarities are numerous. So how do we distinguish one from the other? We define them by their hallmarks, many of which we discuss here.
2.1. Clinical aspects
A constellation of abnormalities forms the background for NASH development including obesity, type 2 diabetes, hypertension, polycystic ovarian syndrome and acanthosis nigricans. ASH however develops as the consequence of alcohol abuse. Symptoms of NASH are absent or include upper quadrant abdominal pain in one-third to one half of patients. ASH may include fatigue and anorexia. Signs in NASH include hepatomegaly often missed because of obesity. ASH may include fever and jaundice. If cirrhosis has developed, muscle wasting, spider angiomata and encephalopathy occur.
A number of laboratory abnormalities, including elevated serum aminotransferases, have been reported in patients with alcoholic liver injury, and used to diagnose ALD. Serum aspartate aminotransferase (AST) is less than 500 IU/L in alcoholic hepatitis. Laboratory findings in NASH include alanine aminotransferase (ALT) normal to 300 U/L, ALT > AST (1.5:1 ratio) (Sorbi et al., 1999), and AST > ALT if there is cirrhotic. Alkaline phosphatase (ALP) is usually normal. Total bilirubin is normal, prothrombin time is normal, white blood count (WBC) is normal, and mean corpuscular volume (MCV) of erythrocytes is normal. Ferritin may be elevated (Kowdley et al., 2012). Laboratory findings in ASH differ from NASH as follows: AST > ALT often by more than a 2:1 ratio, ALP may be elevated, total bilirubin is elevated, prothrombin time may be elevated, WBC is elevated up to 50,000/mm2, and the MCV may be elevated.
Carbohydrate-deficient transferrin (CDT) levels remain elevated for 1 to 2 weeks after alcohol abstinence in ASH. Therefore it is a powerful measurement that documents the exclusion of ASH from NASH (Delanghe et al., 2007; Ohtsuka et al., 2005). When CDT levels are combined with gamma glutamate transferase (γGT) levels (level of cholestasis) the ability to differentiate ASH from NASH is improved (Chen et al., 2003). Serum levels of cytokeratin (CK) 19 and CK18 are elevated in both ASH and NASH when MDBs are formed (Gonzalez-Quintela et al., 2006; Tsutsui et al., 2010). The sensitivity and specificity of the biomarkers AST, ALT, MCV, CDT, or the combination of CDT with γGT or CDT + γGT + MCV to distinguish between harmful or heavy alcohol consumption showed that CDT + γGT gave the best results i.e., sensitivity of 83–90% and specificity of 95–98%.
2.2. Adduct formation
4-Hydroxynonenal (4-HNE) adduct aggresomes form in hepatocytes and macrophages independent of MDBs in the livers of both ASH and NASH just as M30 [fragments of cytokeratin (CK18)] are found in MDBs (Amidi et al., 2007). M30 were found in 100% of MBDs in both NASH and ASH. MDBs were found in 45% of ASH and 40% of NASH. 4-HNE aggresomes were found in 55% of ASH and 73% of NASH patients.
Liver complement factors increase.C1Q, C3 and C4 accumulate in the NASH livers (Rensen et al., 2009). We have found that C1Q, C3 and C5 accumulate in the livers of ASH (Fig. 1). We measured the increase by measuring the fluorescent antibody intensity morphometrically (Fig. 2A and B). C1Q, C3 and C5 were significantly increased in ASH compared to normal controls.
Fig. 1.
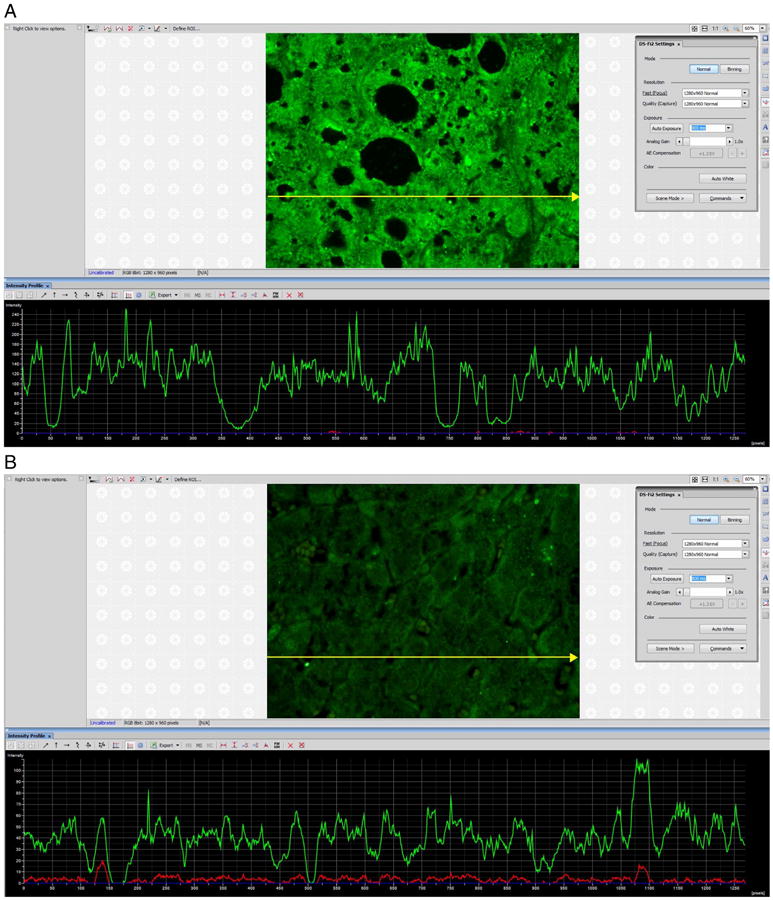
A) SNIP of morphometric quantitative immunofluorescence of FITC labeled antibody to C1Q in a liver of a patient with alcoholic hepatitis. The scale on the left is in arbitrary units of intensity tracing (197 compared with the control (B) of 78 p < 0.01) along the yellow line/arrow x918. B) SNIP of morphometric semi quantitative immunofluorescence of FITC labeled antibody to C1Q in the liver of a control patient. The fluorescent intensity scale on the left is lower than that shown in panel A. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.

A) Flow chart for TLR4/TLR3 in liver biopsies from patients with alcoholic hepatitis (ASH). B) Flow chart for TLR4/TLR3 in liver biopsies from patients with non-alcoholic hepatitis (NASH).
2.3. Histopathology of NASH and ASH compared
Comparison of the different histologic changes seen in the livers of NASH and ASH have been published (Tannafel et al., 2011) as follows: ASH steatosis+, NASH++; ballooning ASH++, NASH++; lobular inflammation ASH++, NASH−/+; MDBs ASH+++, NASH+; satellitosis ASH+++, NASH−; acute cholestasis ASH+, NASH−; perisinusoidal fibrosis ASH+, NASH+; sclerosing hyaline necrosis ASH++, NASH−, venoocclusive disease ASH++, NASH−. Balloon degeneration should be required to make the diagnosis in both NASH and ASH (French et al., 1989, 1993).
2.4. Toll like receptor (TLR)3/4 signaling pathways in NASH compared with ASH
Liver biopsies from NASH and ASH patients subjected to PCR analysis of the expression of toll like receptors (TLRs) showed that TLR3 and 4 were up regulated in ASH, TLR4 more than TLR3. In contrast TLR3 and 4 were up regulated equally in NASH. MyD88 was greatly up regulated in NASH whereas MyD88 was up regulated less in ASH. IRF3 and 7 were up regulated in NASH (Liu et al., 2014a). The chemokine expression end point of the TLR3/4 pathways also differed. CXCR4 was up regulated in NASH and CXCR7 was up regulated in NASH. NFκB was only up regulated in NASH. Thus the ASH TLR3/4 pathway differed from NASH. In ASH the TLR4-MyD88 dependent pathway and the TLR3 independent pathway were up regulated (Fig. 2A) where as in NASH the TLR3, IRF3 and 7 pathways were also increased (Fig. 2B). FAT10 was up regulated in ASH but not in NASH (Fig. 2A & B).
2.5. The role of FATylation and ubiquitylation pathways in protein quality control in ASH and NASH
Based on the down regulation of FATylation and ubiquitylation in the protein quality control in the DDC mouse model of MDB aggresome formation (Liu et al., 2014b), we assayed the expression of this system in the livers of patients who had ASH and NASH. UBLs and ligases 1 and 2 in the ubiquitylation and FATylation pathways of protein quality control were measured using quantitated real time PCR on formalin fixed and paraffin embedded liver biopsies. Ufc1, Ufm1, Uba5, Uba6 and FAT10 were measured. Biopsies from NASH, cirrhosis, ASH and normal controls were studied. Ufc1, Ufm1, Uba5 and 6 were down regulated. FAT10 was markedly up regulated in ASH and cirrhosis. Uba6 and Uba5 were down regulated in NASH. Uba6 is an Ubl protein essential for FATylation and Ub5 is involved in Ufmylation. Using morphometric quantitation of protein levels in the liver cell cytoplasm of liver biopsies from ASH, showed a selective loss of Uba6 in balloon degenerated MDB forming hepatocytes. From these changes it was concluded that the loss of normal protein quality control due to deficiency of function of the FATylation and ubiquitylation pathways accounted for the accumulation of misfolded proteins and MDB formation in the cytoplasm of balloon cells in NASH and ASH (Fig. 3).
Fig. 3.

Flow chart for FATylation, ubiquitylation and ufmylation in liver biopsies from patients with alcoholic hepatitis (AH) and non alcoholic hepatitis (NASH).
3. Induction of cytochrome P4502E1 in alcoholic liver disease and the role of iron overload
Helmut K. Seitz and Sebastian Mueller.
Centre of Alcohol Research, University of Heidelberg, Heidelberg, Germany.
The increase in CYP2E1 has been reported not only in liver, but also in extrahepatic tissues such as the mucosal cells of the gastrointestinal tract (Seitz et al., 1978, 1982). In addition age plays a significant role in modeling the liver damage (Seitz and Stickel, 2007a). Expression of CYP2E1 is increased by chronic ethanol consumption accounting for increased hepatic ethanol oxidation observed in this setting. In addition, a peroxisomal catalase also involved in ethanol oxidation may further contribute to liver injury. Non-oxidative ethanol metabolism involves formation of fatty acid ethyl esters from free fatty acids and ethanol. Fatty acid ethyl esters can also contribute to the cell injury.
The induction of CYP2E1 by chronic alcohol consumption results in a variety of complex cellular effects with enormous clinical significance such as an increase of ethanol metabolism and increased production of reactive oxygen species (ROS) with increased cellular toxicity resulting in ALD and stimulating carcinogenesis. The effect of ROS on lipid peroxidation is leading to lipid peroxidation products such as 4-HNE and malondialdehyde. 4-HNE by itself may bind to nucleotide bases such as cytosine and adenosine resulting in the generation of exocyclic etheno DNA adducts which are highly mutagenic and carcinogenic (Wang et al., 2009).
We performed liver biopsies in a group of 97 (31 women and 66 men) non-cirrhotic ALD patients that are drinking >60 g ethanol/day. Their age varied between 24 and 72 years. AST/ALT ratio was >1.5. γGT was 35–5760 U/L. Their histology showed steatohepatitis and fibrosis with various degree of severity (Kleiner et al., 2005). Immunohistochemistry staining identified CYP2E1 and exocyclic etheno-DNA adducts (Frank et al., 2004). The biopsies showed an increased induction of CYP2E1 associated with a similar increase not only in 4-HNE but also in the adducts such as N6-etheno-deoxyadenosine and N2,3-etheno-deoxycytidine. We also found a significant correlation between etheno DNA adducts and CYP2E1. Moreover, a significant positive correlation exists between CYP2E1 and fibrosis. We therefore conclude that CYP2E1 activity plays an important role in the progression of ALD.
The use of non-invasive tests to determine the severity of the liver injury in ALD is in continuous search for innovative methods. Non-invasive tests for liver fibrosis are based on imaging techniques such as ultrasound, computed tomography and magnetic resonance imaging. For example, transient elastography uses ultrasound techniques with wave vibrations in order to measure liver stiffness, which reflects the extent of fibrosis (Foucher et al., 2006; Ziol et al., 2005). These techniques are currently performed predominantly when a diagnosis of cirrhosis is suspected, as they can detect cirrhosis, but are unable to distinguish accurately between other stages (Seitz and Stickel, 2007a, 2007b).
Another factor contributing to the severity of ALD is iron overload. Alcohol drinking increases the body iron stores. Even moderate consumption leads to the elevation of the iron concentration, ferritin and transferrin saturation in serum and the hepatic iron content. Both iron and alcohol cause the oxidative stress and lipid peroxidation leading to the liver injury. The excessive iron accumulation can be one of the reasons involved in ALD. Body iron stores affect the indicators of liver function. In addition, there are two conditions that may accompany ALD: hemochromatosis and porphyria cutanea tarda (Zimmerman, 1999). There is an association between alcoholism and the two iron overload diseases. Twenty-five to 50% of patients with hemochromatosis take alcohol in excess (Zimmerman and Ishak, 1996), and ahigh proportion of patients with porphyria cutanea tarda also are heavy drinkers (Hift and Kirsch, 1995; Lefkowitch and Grossman, 1983).
The degree of CYP2E1 induction can be correlated with generation of ROS. Moreover, altered cell-mediated immunity increased postoperative infection rate in alcoholic patients (Sander et al., 2002; Spies et al., 2004). Seitz and Suter (2001) showed also the role of nutrition in alcohol-induced HCC. In addition to viral infections, there are cofactors by which alcohol may enhance the development of hepatocellular malignant transformation. These include environmental carcinogens, activation of pro-carcinogens via induction of CYP2E1, and iron-overload. It has become evident that excess body iron may complicate ALD and lead to HCC. Alcohol may contribute to inappropriately low secretion of hepcidin. Cirrhosis is almost always present when HCC is diagnosed. Iron loading of the liver results and may be complicated by malignant transformation of the liver.
In summary, a major impact of alcohol on CYP2E1 and formation of exocyclic etheno-DNA adducts associated with a increase in 4-HNE is undisputed. Dietary and environmental factors favor tumor development and expansion probably by a mechanism in which alcohol compromises antitumor immune surveillance.
4. Non-alcoholic steatohepatitis in octogenariens in Israel
Steve Malnick.
Non-alcoholic fatty liver disease (NAFLD) is one of the major causes of hepatic disease worldwide (Musso, 2012). It is a major cause of cirrhosis and its complications including HCC (Ascha et al., 2010). NAFLD is considered to be the hepatic manifestation of the metabolic syndrome (Chalasani et al., 2012). The metabolic syndrome is defined by the National Cholesterol Education Program (NCEP) criteria although other criteria are accepted for non-Caucasian populations (Eckel et al., 2005).
There have been three large population-based studies to determine the prevalence of NAFLD (Bellentani et al., 2010; Lazo et al., 2013). The prevalence of NAFLD is approximately 21% and the more severe and clinically important subtype of steatohepatitis is approximately 10% (Chalasani et al., 2012). The components of the metabolic syndrome have increased concomitant with the increase in obesity in developed countries (Younossi et al., 2011).
NAFLD has a much higher prevalence in the aged population. Our group has reported a prevalence rate of 46% detected by ultrasound, in a cohort of healthy octogenerians admitted to the rehabilitation wards of a geriatric hospital (Kagansky et al., 2004). This rate was significantly higher than that found in a younger population. There was, however, no correlation between NAFLD and the metabolic syndrome, cardiovascular disease and cirrhosis. A study from Rotterdam of 2811 participants, with an average age of 76 years, found a prevalence of 35.1% based on ultrasound (Koehler et al., 2012). There were 265 patients over the age of 85 years. The prevalence of NAFLD decreased with advancing age. The reason for this is unclear but may be due to selective mortality. Furthermore, the association between the number of components of the metabolic syndrome and fatty liver decreased with advancing age.
A study from Shanghai of 2201 participants aged 50–83 with a diagnosis of fatty liver based on ultrasound revealed a prevalence of 22.8% in females and 16.03% in males (Yang et al., 2011). The prevalence of fatty liver was significantly higher in the group aged greater than 70 years of age (20.22%) compared to the group aged 50–59 years of age (17%). In addition, serum adiponectin levels were significantly higher in the group aged more than 70 years of age compared to the group aged between 50–59 years. Adiponectin levels are linked to insulin sensitivity and hypoadiponectinemia is linked to insulin resistance. Aging is also a risk factor for NAFLD in premenopausal women. Thus there are differences between NAFLD in younger and aged populations.
The components of the metabolic syndrome also change with age (Hildrum et al., 2007). According to the International Diabetes Federation definition of obesity, the prevalence of the metabolic syndrome increased from 9.2% in women and 11% in men in those aged 20 to 29 years of age, to 64.4% in women and 47.2% in men in those aged 80–89 years of age.
NAFLD encompasses a spectrum of histology ranging from steatosis through steatohepatitis to fibrosis. Liver-related morbidity and mortality are linked to the severity of the liver damage. NAFLD has a relatively benign prognosis but NAFLD-related cirrhosis may be complicated by portal hypertension, HCC and death from liver disease. Three easily assessed clinical parameters help us to identify individuals with cryptogenic hepatitis who are likely to have advanced fibrosis on liver biopsy. These are age older than 45–50 years, overweight or obese body mass index and type 2 diabetes (Angulo et al., 1999). The probability of having bridging fibrosis or cirrhosis on liver biopsy is approximately 66% in patients with cryptogenic cirrhosis who are older, overweight/obese and diabetic. This may reflect referral bias to hepatologists in tertiary referral centers.
Other groups have looked at obese and diabetic individuals who were not selected on the basis of elevated liver enzymes. Gastric bypass surgery is usually not offered to patients with significant liver disease. However, intra-operative liver biopsies demonstrate advanced bridging fibrosis (F3) or cirrhosis (F4) in 12% of these morbidly obese patients (Beymer et al., 2003). Similarly, when liver biopsy was offered to a group of middle-aged or older diabetics who were noted incidentally to have fatty liver on ultrasound, the prevalence of F3-4 fibrosis was about 20% (Gupte et al., 2004).
The connection between NAFLD and mortality is unclear. Earlier studies have found that liver disease was the third leading cause of death (Adams et al., 2005; Söderberg et al., 2010). However, a recent re-assessment of the NHANES archived videotapes of US performed on the gallbladders of 11,371 adults from 1988–1994 showed no increased risk of death from all causes, cardiovascular or liver disease, or cancer (Lazo et al., 2011).
Once again these differences may be related to referral bias between population-based studies and liver clinics in tertiary referral centers. Thus, together with the risk factors for the metabolic syndrome, the prevalence of NAFLD increases with age. Despite the comorbidity associated with these risk factors, there is no change in the mortality related to NAFLD. This is similar to the obesity paradox in heart failure (Chase et al., 2014).
The possible explanation for this is unclear. It is possible that changes in the fecal microbiome play a role. The intestinal microbiota play an important role in the maintenance of host health by providing energy, nutrients and immunological protection (Blaut et al., 2002; Nicholson et al., 2012). The fecal microbiota composition changes from childhood to old age (Mariat et al., 2009). There are also differences across age and geography (Yatsunenko et al., 2012). There is an influence of the intestinal microbiome on liver disease including NAFLD. This has recently been reviewed (Schnabl and Brenner, 2014). Furthermore, the effect of metformin on insulin resistance may also be related to the effect on the fecal microbiome (Burcelin, 2014). The cohort of aged people were born and grew up either before or at the beginning of the antibiotic era and had a very different exposure to antibiotics than younger people.
In summary, although NAFLD is a common cause of cirrhosis and end-stage liver disease, it is more common in the population of aged people than in younger groups. The challenge is to understand this paradox and the natural history of patients with NAFLD.
5. Alcoholic hepatitis
Ramon Bataller.
Acute alcoholic hepatitis (AH) is a clinical syndrome characterized by new onset jaundice and/or ascites in the setting of ongoing alcohol abuse and underlying ALD (Lucey et al., 2009). Population based studies estimate approximately 4.5 hospitalizations for AH per 100,000 persons each year, with a slight male predominance (Sandahl et al., 2011). Patients typically present with rapidly progressive jaundice, which can be accompanied by fever, abdominal pain, anorexia, and weight loss. In severe cases, portal hypertension is severe, and the patient presents with ascites, encephalopathy, or variceal bleeding. There are no reliable non-invasive markers that estimate the presence of AH in patients with decompensated ALD. A cardinal featureof AH is anelevated ratio of AST to ALT, typically greater than 2:1, and both are usually <300 IU/L. The diagnosis of AH is made on clinical grounds, based on a history of excessive alcohol use with the typical physical exam and laboratory findings. Liver biopsy may be helpful in establishing the presence of ASH and has been endorsed in recent clinical practice guidelines (Mathurin et al., 2012; Murray and Carithers, 2005).
Several models have been developed to help predict outcomes of patients with AH and to guide therapy. The most widely used is Maddrey's discriminant function (DF). Additional predictive models include the Model for End-Stage Liver Disease (MELD), the Glasgow AH score, the ABIC score, and the Lille model (Altamirano and Bataller, 2011; Dominguez et al., 2008). Clinical practice guidelines therefore recommend stopping corticosteroids after one week in those with an unfavorable Lille score, as the risks of continued therapy likely outweigh the benefits (Murray and Carithers, 2005; Mathurin et al., 2012). Recently, we performed a large multicentric study to develop a histological scoring system capable of predicting short-term survival in patients with AH. The resulting Alcoholic Hepatitis Histological Score (AHHS) comprises 4 parameters that are independently associated with patients' survival: fibrosis stage, polymorphonuclear infiltration, type of bilirubinostasis and presence of megamitochondria. By combining these parameters in a semiquantitative manner, we were able to stratify patients into low, intermediate, or high risk for death within 90 days (Altamirano et al., 2014).
The management of patients with AH has not evolved much in the last decades. Besides general measures, patients with a high risk of death (DF ≥ 32, or poor prognosis based on other scoring systems) should be treated with specific therapy. Prednisolone and pentoxifylline are considered the two first-line therapies for severe AH. In most centers, pentoxifylline is typically reserved as a second-line agent for patients with contraindications to corticosteroid therapy (1). Moreover, promising results were recently obtained with N-acetylcysteine, a powerful antioxidant substance that potentiates the beneficial effects of prednisolone. However, many patients do not respond to medical therapy and novel targeted therapies are urgently needed. Recently, salvage liver transplantation in highly selected patients has been shown to improve survival significantly, but is not available in the vast majority of transplant centers (Mathurin et al., 2011).
In the last years, translational studies have identified several potential cellular and molecular targets to treat patients with advanced ALD including AH (Gao and Bataller, 2011). An incomplete maturation of hepatic progenitor cells is associated with bad prognosis, suggesting a role for maneuvers aimed at promoting the differentiation of these cells (Sancho-Bru et al., 2012). Molecular targets include the CXC chemokine family, tumor necrosis factor receptor superfamily member 12A (Fn14), osteopontin (OPN), CCl20, members of the inflammasome, interleukin-22, the Hedgehog signaling pathway and macrophage migration inhibitory factor (MIF) (Altamirano and Bataller, 2011). The development of animal models of true AH is urgently needed to translate these recent studies into the clinical practice.
6. Dysregulation of hepatic methylation reaction impact on alcohol and HCV
Natalia A. Osna, Paul G. Thomas, Murali Ganesan, Kusum K. Kharbanda.
Findings in our laboratory have demonstrated that ethanol feeding lowers the hepatocellular S-adenosylmethionine to S-adenosylhomocysteine ratio. This decrease causes serious functional consequences including reductions in essential methylation reactions and impaired interferon signaling in the context of hepatitis C virus (HCV) infection. Further, increasing methylation demand by an exogenously supplied methyl-group consuming compound in combination with ethanol generates more pronounced liver injury than either agent alone. Thus, chronic alcohol consuming patients should be advised against increased dietary intake of methyl-consuming compounds even for a short period of time.
ALD is caused by excessive intake of alcohol (Gao and Bataller, 2011; Orman et al., 2013). A majority of alcoholics (>90%) develop hepatic steatosis, an early stage of ALD, but progression to steatohepatitis occurs only in 20% of the patients. Further, only a fraction of patients (20 to 50%) with ASH develop cirrhosis. These observations and subsequent animal studies have led to the conclusions that excessive intake of alcohol is the first hit to induce hepatic steatosis, and the second hit is required for the development of steatohepatitis and of cirrhosis. Second hits have been categorically divided into nutritional, agonistic/ xenobiotic/pharmacologic, hemodynamic, HCV and genetic groups (Tsukamoto et al., 2009).
In our ongoing investigation, we have reported that chronic ethanol intake causes defects in methionine metabolism that ultimately lower the hepatocellular S-adenosylmethionine (SAM) to S-adenosylhomocysteine (SAH) ratio. The reduced ratio decreases the cellular methylation potential and significantly impairs many liver methylation reactions catalyzed by specific SAM-dependent methyltransferases (Kharbanda, 2009, 2013; Kharbanda et al., 2007). One such methyltransferase, guanidinoacetate methyltransferase (GAMT) that catalyzes the last step in creatine synthesis is considerably impaired after chronic alcohol consumption (Kharbanda et al., 2014a). GAMT is a major consumer of methyl groups and utilizes as much as 40% of the SAM-derived groups to convert guanidinoacetate (GAA) to creatine (Mudd et al., 2007).
Over the past decade, exposure to methyl-group consuming compounds has substantially increased, introducing additional stresses on the cellular methylation potential (Zhou et al., 2011). Thus, we sought to investigate whether increased ingestion of a methyl-group consumer, GAA, could be a second hit that could exacerbate the progression of alcoholic liver injury. Adult male Wistar rats were pair-fed the Lieber DeCarli control or ethanol diet in the presence or absence of GAA for 2 weeks. At the end of the feeding regimen, biochemical and histological analyses were conducted. We observed that 2 weeks of GAA – or ethanol – alone treatment increases hepatic triglyceride accumulation by 4.5 and 7-fold, respectively as compared with the pair-fed controls. However, supplementing GAA in the ethanol diet produced panlobular macro- and micro-vesicular steatosis, a marked decrease in the methylation potential and a 28-fold increased triglyceride accumulation. These GAA-supplemented ethanol diet-fed rats displayed inflammatory changes and significantly increased liver toxicity compared to the other groups (Kharbanda et al., 2014b). In conclusion, increased methylation demand superimposed on chronic ethanol consumption causes more pronounced liver injury. Thus, alcoholic patients should be cautioned for increased dietary intake of methyl-group consuming compounds even for a short period of time.
Another “second hit” that drives ALD progression is the persistence of HCV infection that occurs due to compromised IFNα signaling (Gale and Foy, 2005). Interferon signaling is crucial for induction of antiviral activity through the activation of interferon-sensitive genes (ISGs). One of the requirements for successful transduction of IFN signal through the Jak–STAT1 pathway is STAT1 methylation (Duong et al., 2004, 2006). Previously, we have shown that IFN signaling in liver cells is altered by ethanol metabolism (Osna et al., 2005, 2007). We hypothesize that ethanol-induced impairments of IFN signaling in HCV-infected cells are due to dysregulation of methylation reactions. To test the hypothesis, we treated HCV (JFH1)-infected and non-infected Huh 7.5 cells with acetaldehyde-generating system (AGS), using ethanol, yeast alcohol dehydrogenase (ADH) and NAD, which produces ∼200 μM acetaldehyde (Ach). These levels are comparable to those generated by alcohol dehydrogenase-transfected hepatoma cells exposed to ethanol (Donohue et al., 2006). We observed that exposure to AGS lowered the hepatocellular SAM:SAH ratio, reduced the cellular methylation potential as well as suppressed STAT1 phosphorylation on both tyrosine and serine residues. In addition, Ach decreased accumulation of pSTAT1 in the nuclear fraction. Furthermore, as a result of Ach-induced hypomethylation, STAT1 formed complexes with protein inhibitor of activated STAT1 (PIAS1), thereby preventing the attachment of STAT1 to DNA and decreasing ISG activation. All these Ach-induced defects were more pronounced in HCV-infected cells compared with non-infected cells. The effects of Ach on STAT-1 phosphorylation, PIASI-STAT-1 complex formation, and attachment of STAT1 to DNA were all reversed by betaine treatment indicating that these events are methylation-dependent. Further evidence linking Ach-induced hypomethylation and impaired IFN signaling was provided by treatment with a pan-methylation inhibitor, tubercidin and the specific inhibitors of arginine- and lysine methylation. These methylation inhibitors mimicked all of the above-mentioned effects of Ach treatment on IFN signaling and lowered the expression of 2′,5′-oligoadenylate synthetase-like (OASL), an anti-viral protein that is encoded by ISGs. In conclusion, Ach, via impairing methylation reactions, suppresses IFN signaling at multiple levels in both non-infected and HCV-infected cells. However, the suppression is more pronounced in HCV-infected cells. Betaine treatment prevents all investigated Ach-induced changes in the IFN-induced Jak-STAT1 pathway, demonstrating the importance of normal methylation status in the promotion of anti-viral response.
7. Laboratory diagnosis of alcohol drinking
Radu M. Nanau and Manuela G. Neuman.
In order to monitor the patient's alcohol-related damages, numerous biochemical and psychological tests and devices have been developed and suggested to uncover alcohol consumption. Biological tests can provide direct measurements to clinicians and toxicologists.
The quantitative, measurable detection of drinking is important for the successful treatment of alcohol misuse. Also detection is imperative in liver transplantation of patients with alcohol disorders, people living with human deficiency virus that need to adhere to medication, as well as, special occupational hazard offenders, many of whom continually deny drinking (Allen et al., 2013; Maenhout et al., 2013). The accurate identification of alcohol consumption via biochemical tests contributes significantly to the monitoring of drinking behavior.
In 8708 adult participants in the third U.S. National Health and Nutrition Examination Survey Liangpunsakul et al., (2010) analyzed the relationship between the amount of alcohol drinking and γGT, AST:ALT ratio, MCV of erythrocytes, and apolipoprotein A1 and B. When tested alone or in combination, and adjusted for potential liver injury risk factors, the sensitivity and positive predictive values for these blood tests were too low to be clinically useful in identifying the subjects in the heavier drinking category (Liangpunsakul et al., 2010).
Alcohol ingestion may directly be measure using blood alcohol concentrations and a breath test (Roiu et al., 2013). Because alcohol is rapidly eliminated from the circulation, the time for detection by this analysis is in the range of hours. Alcohol consumption can alternatively be detected by direct measurement of ethanol concentration in blood or urine. A ratio of 2100:1 between the blood alcohol concentration and the breath alcohol concentration, which is the standard ratio used in law enforcement, was achieved after 30 min in a sample of healthy volunteers, and this remained relatively stable through almost 3 h post-ingestion (Grubb et al., 2012). Several markers have been proposed to extend the detection interval and sensitivities, including ethyl glucuronide and ethyl sulfate in urine, phosphatidyl-ethanol in blood, and ethyl glucuronide and fatty acid ethyl esters in hair, among others (Lahmek et al., 2012). Moreover, CDT, which reflects longer lasting consumption of higher amounts of alcohol, should be better standardized (Anton et al., 2002; Daves et al., 2011; Jeppsson et al., 2007). There is a need to correlate CDT, with serum γGT, another long term indirect bio-markers that is routinely used in monitoring alcohol-induced liver damage and is standardized in laboratory medicine.
This work describes the laboratory methods that are used to determine alcohol levels in different matrices (breath, blood, urine, hair, saliva). The laboratory standardization is needed to ensure the large use of different methods. Moreover, since some tests are influenced by ethnicity, gender or the presence of other co-morbidities, the laboratory medicine specialist and the clinician should communicate in order to ensure that the diagnosis is correct.
Single serum biomarkers that are involved in fibrosis physiopathology of the ECM can be used as indicators of fibrosis (Castera et al., 2000; Neuman et al., 2012a). However, most of these markers are not liver-specific, and can be affected by other clinical conditions, such as inflammation. They are often used in combination in order to generate a score according to an algorithm, which is then used to give a fibrosis prediction.
Most indirect markers used in the algorithms of scores are simple parameters, readily available in the current clinical chemistry such as AST, ALT, cholesterol, γGT, bilirubin, gamma globulin and platelet counts. Other tests may use α2-macroglobulin, haptoglobin and apolipoprotein A1. Platelet counts decrease as liver fibrosis extends because of the low level of thrombopoietin produced by the liver. Neopterin and interleukin 8 are predictors for alcohol induced cirrhosis (Homann et al., 2000). α2-Macroglobulin is an inhibitor of endoproteases synthesized in the liver. Its serum levels increase with the degree of liver fibrosis (Rossi et al., 2007). Haptoglobin is an acute phase protein whose concentration increases in inflammatory conditions. Its level decreases with increasing stages of fibrosis (Lee et al., 2010). Apolipoprotein A1 is the major protein component of high-density lipoproteins. Its levels decrease as the fibrosis progresses.
Direct biochemical markers include cytokines involved in the fibrogenetic process such as TGF-β1, components of the ECM such as collagen, glycoproteins, proteoglycans and glycosaminoglycans, and molecules involved in the wound-healing process of the liver such as matrix metalloproteins (MMP) and tissue inhibitors of metalloproteinase (TIMP). Collagen markers include pro-collagen peptides, as well as type I, III and IV collagen, and laminin. The most extensively studied collagen marker is the N-terminal peptide of procollagen type III (PIIINP), cleaved from procollagen III during its secretion from fibroblasts (Hayasaka et al., 1990; Neuman et al., 2012a; Nøjgaard et al., 2003; Schaefer et al., 2003). Hyaluronic acid is a structural glycosaminoglycan present in the ECM. It has been used on its own as a single fibrosis marker or, more recently, in combination with other markers. Liver fibrosis causes elevations in hyaluronic acid levels in serum. The specific importance of hyaluronic acid measurement was in correlation with other investigators' opinion. MMPs and TIMPs are proteins involved in the regulation of fibrogenesis and fibrolysis. The excess collagen deposition in the hepatic tissue, a characteristic of fibrosis, results from increased collagen synthesis and decreased collagen degradation, mediated by increased TIMPs. MMPs and TIMPs are not currently assessed in routine clinical laboratories (Neuman et al., 2012a).
8. Alcohol misuse and therapeutic-alcohol interactions
Manuela G. Neuman, Lawrence E. Cohen.
A variety of morphologic and functional changes reflect organ injury produced by alcohol. The present review describes several laboratory approaches in alcohol misuse, with a particular emphasis on the mechanisms by which alcohol interacts with therapeutics and leads to unwanted adverse effects. The main pathologies associated with alcohol-induced organ damage are severe steatosis, cirrhosis, chronic pancreatitis, cardiomyopathy and cerebellar atrophy (Neuman et al., 2012a, 2012b). The high rate of potentially lethal adverse drug reactions associated with alcohol misuse highlights the need of laboratory monitoring. Understanding the molecular mechanisms by which alcohol leads to adverse drug reactions can help tailor therapeutic interventions that will prevent patient mortality.
Possible factors that affect the development of injury include the dose, duration and type of alcohol consumption, drinking patterns, sex, and ethnicity (Wechsler and Austin, 1998). In addition, there are associated risk factors such as obesity, concomitant infections and the interaction between therapeutics with alcohol, and genetic factors (Neuman et al., 2012c).
The diagnosis of ALD may appear straightforward in the patient with a documented history of alcohol abuse and compatible clinical features. However, patients misusing alcohol can have concurrent forms of liver disease, or have liver disease unrelated to alcohol. A high prevalence (25 to 65%) of HCV infection has been recognized in individuals that abuse alcohol. Accelerated fibrosis in ALD patients with In Wilson's disease, copper overload, is rarely seen. There are several characteristic laboratory abnormalities in patients with ALD but none is diagnostic. The most common pattern of biochemical abnormalities is a disproportionate elevation of serum AST compared to ALT. This ratio is usually greater than 2.0, a value that is rarely seen in other forms of liver disease. For example, the average AST/ALT ratio was 2.85 in patients with ALD versus 0.9 in those with NASH and a ratio below 1.0 in patients with chronic viral hepatitis (Cohen and Kaplan, 1979; Sorbi et al., 1999; Williams and Hoofnagle, 1988).
Using World Health Organization-International Federation of Clinical Chemistry and Laboratory Medicine (WHO-IFCC) recommendation and methods our laboratory defines normal values in adults as follows: ALT 5–40 U/L for men and 5–32 U/L for women; AST 5–38 U/L for men and 5–32 U/L for women; γGT 2–50 U/L for men and 2–35 U/L for women. Since γGT shows the function of cholangiocyte epithelia, if the value is higher the differential diagnosis of cholestasis (primary biliary cirrhosis, primary sclerosing cholangitis) should be ruled out. Higher concentrations of serum AST and ALT than 500 U/L should raise the suspicion of concurrent liver injury due to viral or ischemic hepatitis, hypersensitivity syndrome to therapeutics taken concomitantly, even at therapeutic doses. In addition to the laboratory features common to all forms of ALD, alcoholic hepatitis is typically associated with elevations in serum ALP and γGT concentrations and with hyperbilirubinemia. These changes may persist for weeks after the aminotransferase concentrations have returned to normal. Alcoholic foamy degeneration, which is characterized by jaundice and hyperlipidemia, can elevate the aminotransferase with the AST > ALT (Uchida et al., 1983). Higher values of AST (>5000) can be observe in the combination of acetaminophen (APAP) and alcohol. Relevant to the hepatotoxic potential of APAP is its metabolic disposition of APAP. The hepatic injury is produced by accumulation of its toxic metabolite. Normally, a small fraction of APAP is converted to an active metabolite which binds with glutathione (GSH) and is then excreted as mercapturic acid. Large doses of APAP lead to increased formation of the active metabolite (Neuman et al., 1994, 1998). Metabolite, in excess of the available GSH, binds covalently to cytoplasmic proteins and, as a consequence, leads to necrosis. Only when the amount of toxic metabolite exceeds the available GSH, toxicity occurs (Katz et al., 2001). Since alcohol competes with the same GST detoxification pathway and is metabolized by the same CYP isoenzyme, accordingly, susceptibility to the hepatotoxic effects of a dose of APAP depends on dose, rate of biotrans-formation of APAP and hepatic content of GSH. Toxicity is enhanced by agents that enhance activity of the CYP system or deplete stores of GSH. Alcohol and APAP synergistically induce CYP2E1, increasing the rate of metabolism of the drug and compete for GSH. Administration of acetylcysteine, which increase the GSH deposits, during the first 16 h after a toxic dose can prevent hepatic injury.
Seeff et al. (1986) described 25 ALD patients who developed mild to moderate jaundice, mild to severe coagulopathy, and highly abnormal aminotransferase levels after moderate doses of acetaminophen. Zimmerman and Maddrey (1995) described 67 heavy drinkers (alcohol 60–80 g/day) who developed liver damage after ingesting therapeutic doses of acetaminophen (4–6 g/day). AST levels ranged from 3000–48,000 IU/L in more than 90% of cases, and almost 20% of the patients died (Zimmerman and Maddrey, 1995).
The alcohol-associated APAP syndrome in the patients with alcohol misuse has appeared after repetitive tolerable doses in a short period for headache or hangover. In APAP-alcohol interaction, the AST levels can be extremely high (40 to 1000 times normal). Levels of ALT are lower. The syndrome is frequently accompanied by metabolic acidosis (Zimmerman and Maddrey, 1995).
The interaction between alcohol and the anti-tuberculosis drug isoniazid also presents clinical importance, since the metabolism of this drug involves acetylation. Since acyl transferase, the enzyme that catalyzes this reaction, is polymorphic, individuals who possess an acyl transferase with low activity may accumulate an intermediate, which is then activated by CYP2E1 to hepatotoxins. Thus, only individuals with low acyltransferase and high CYP2E1 activity develop liver injury (Huang et al., 2003). Values for AST and ALT have been very high. ALP levels have been less than threefold elevation in 80% of recorded cases.
In addition to signs of hepatic injury, concurrent hematologic abnormalities are common in moderate to severe alcoholic hepatitis. Macrocytosis indicates a longstanding disease and may reflect poor nutritional status, cobalamin or folate deficiency, toxicity of alcohol, and/or MCV.
Alcohol consumption interacts with medication used in cardiology. For example reserpine, a beta adrenergic inhibitor is intensified by alcohol, while clonidine hypotensive effect is reversed by alcohol ingestion. Moreover, alcohol reduces the metabolism of warfarin and increases its blood levels and therefore its anticoagulant effect predisposing the patient to hemorrhage (Zimmerman, 1999). As a result, the prothrombin time of the patients who consume alcohol should be monitored. Nitroglycerine, a drug used to treat heart disease, could lead to elevations of aminotransferases and ALP levels up to three times in the presence of alcohol. Also, the vasodilator effect of nitroglycerin is enhanced by alcohol, such that it leads to a drop of blood pressure cardiovascular medication patients using alcohol. Amiodarone, an anti-arrhythmic medication per se, is known to induce hepatotoxicity.
In the presence of alcohol consumption, the medication is more hepatotoxic. Adderall (amphetamine, dextroamphetamine mixed salts) is a prescribed stimulant for the treatment of attention-deficit/ hyperactivity disorder in children and adolescents. Nonmedical use of Adderall is prevalent in high school and college students without intention to overdose. However, 3 cases have been reported in the pediatric literature of acute myocardial infarction in adolescents without cardiovascular risk factors, who took the total prescribed daily dose of Adderall one time while consuming alcohol (Cousins et al., 2014).
Tricyclic antidepressants should be administrated cautiously in alcohol withdraw since they can produce synergistic effects with alcohol leading to hypotension. The combination of morphine, codeine or heroin with alcohol dramatically increased the sedation profile of these agents (Zimmerman, 1999).
9. Hangover adventure and misadventure
Manuela G. Neuman, Mihai Voiculescu, Mihai Opris.
An additional aspect which can lead to acute hepatitis and needs specific laboratory tools is the “hangover” phenomenon that represents the post drinking effects of alcohol. Alcohol hangover develops when the blood alcohol concentration falls considerably and peaks when it returns to almost zero. It may last up to 36 h. Several symptoms characterize the hangover including nausea, cognitive impairment and mood changes. Some of the symptoms of the hangover are similar to those of alcohol withdrawal syndrome, but the term “hangover” is usually reserved for the after-effects of a single drinking episode and does not necessarily imply any other alcohol use disorder (Allen and Litten, 2001).
The processes by which alcohol is metabolized in the cells generate a variety of molecules that can be toxic to the brain and liver. For example, alcohol breakdown leads to the formation of toxic acetaldehyde and ROS. In addition, alcohol might add to dysregulation of gut microbiota, leading to increased gut permeability as well as increased endotoxin levels in the blood. Endotoxin produces nausea and detrimental cardiovascular outcomes. In addition, heavy drinking occasions lead to increase in traumatic injury. Drinking to intoxication is a form of acute heavy drinking. The impossibility to detoxify the breakdown products of alcohol results in “hangover” (Neuman, 2001).
The economic consequence of the hangover encompasses loss of work time and poor job performance. Wiese et al. (2000) reviewing the literature between 1966–1999 concluded that “in the United States, related absenteeism cost $148 billion annually (average annual cost per working adult, $2000)”. Individuals with a hangover may pose substantial risk to themselves and others despite having normal blood alcohol levels (Bennett et al., 2004). The knowledge of different aspects of this scenario could enable us to generate greater awareness around this issue for the general public, thus reducing this phenomenon all the while improving livelihood. This project might in turn create incentives for medicine to promote analysis of circulatory alcohol metabolites in irresponsible individuals that consider working during the hangover. Moreover, prevention and education should be designed to improve work climate and alcohol outcomes.
The link between alcohol and infectious diseases, such as tuberculosis and human deficiency virus was reviewed recently by Neuman et al. (2012d). Emerging evidence of links between harmful alcohol consumption levels and some infectious diseases is noted in the WHO Global Status Report on Alcohol (Rehm et al., 2004). Neuman et al. (2006) show a link between alcohol misuse and other drugs use for HIV. Moreover, there is an increase mortality due to drinking problems in specific regions (Leon et al., 2009; Neuman et al., 2012d; Nicholson et al., 2005).
Changes in cognition are not prominent in the early phases of alcoholic intoxication. However, these changes become important during the hangover. Clinical-pathological data suggest at least three subtypes of intoxication: disinhibited, apathetic, and stereotypic. The disinhibited subtype is characterized by purposeless over-activity, unconcern, profound social breakdown, and a behavioral disorder that is more prominent than the cognitive problems (Bobak et al., 2004). The apathetic subtype is characterized by unconcern (Goodglass and Kaplan, 1983). The stereotypic subtype is characterized by pronounced compulsive traits. However, there is substantial overlap in symptomatology among subtypes, especially with progression of the hangover. Marked changes in personality are prominent in individuals during hangovers and include altered mood, decreased concern for human life, exuberance, euphoria, lack of judgment, and poor judgment (Bobak et al., 2004).
The hangover is a post-intoxication state comprising the immediate after-effects of drinking alcoholic beverages in excess (Neuman, 2001). Non-ethanol components of alcoholic beverages may be involved in the etiology. The additional substances such as amines, amides, polyphenols, histamines, esters, furfural, and tannins can be hepatotoxic (Neuman, 2001). Such congeners may worsen alcohol hangover severity. Also, there are differences between several types of alcoholic beverages. Vodka and whisky are both spirits but differ in their composition and in their effect on the drinkers. Moreover, diverse types of whisky (brandy, Scotch, American or Canadian) vary significantly in composition. Bourbon whisky has more than 3 times as much congeners as Canadian whisky. Therefore, differences in types of spirits may have different effects on the way in which they contribute to hangover severity. In addition, preparation techniques for distilled spirits and the manner of their preservation in wood, glass or copper may introduce new toxic elements into the equation (Neuman, 2001). Becker et al. (2002) found lower risk for alcohol-induced cirrhosis in wine drinkers, therefore showing that an alcohol from grapes might not present the same toxicity as the distilled alcohol. In addition dietary iron overload has been described in rural Black Africans in at least 15 sub-Saharan African countries. The condition may affect as many as 15% of the adult male population. It results from the consumption, over time, of large volumes of homemade beer, which has a high iron content as a consequence of it being homebrewed in iron devices (Kew and Asare, 2007).
Clinical features of hangover may include headache, vertigo, gastric disorder, insomnia, tremors of the hands and raised or lowered blood pressure. Psychological symptoms include acute anxiety, depression, or irritability. The amount of alcohol needed to produce a hangover varies with the mental and physical conditions of the individual, though generally the higher the blood alcohol level during the period of intoxication, the more intense the subsequent symptoms will be. The symptoms vary also with social attitude versus the women (Morojele et al., 2006; Seedat et al., 2009). An additional factor is genetic background; Caucasians are less affected by hangovers when compared with Asians (Neuman et al., 2014).
9.1. Hangover and work-related impediment
Frone (2006) found that 9.23% of U.S. workers reported to work with a hangover in the past year, making it the most common form of alcohol-related workplace impairment in the survey. Additionally, there is a significant relationship between alcohol consumption and next-day workplace absenteeism. A survey among 280 employees revealed a two-fold increased likelihood of absenteeism the day after alcohol consumption (McFarlin and Fals-Stewart, 2002). An alcohol screening program used to detect chronic alcohol misuse should employ a combination of biomarkers including CDT, MCV, γGT and AST/ALT).
9.1.1. Professional drivers
Verster et al. (2014a) showed that more than half of the professional drivers who consume alcohol, and who reported occasionally having a hangover (56.4%), acknowledge that they have driven while in a hangover state during the past year: 26.5% only when driving privately, 2.6% only when driving professionally, and 27.4% both privately and professionally. During an alcohol hangover, professional drivers rated their driving style as significantly less relaxed, less safe, less responsible and less responsive. Driving with a hangover is a common phenomenon, and professional drivers acknowledge that their driving is impaired. The purpose of another study by Verster et al. (2014b) was to examine the effects of alcohol hangover on driving performance of forty-two social drinkers. The participants were tested on simulated highway driving in the morning, following an evening of exaggerated consumption of alcohol (on average 10.2 alcoholic drinks – SD = 4.2). This situation represented an alcohol hangover. Participants were also tested on a control day, when no alcohol was previously consumed. Subjects performed a standardized 100-km highway driving test in the STISIM driving simulator. Self-reported driving quality and driving style were scored, as well as mental effort to perform the test, sleepiness before and after driving, and hangover severity. The study shows that driving performance was impaired during alcohol hangover. Participants reported their driving quality to be poorer and less safe. They noted being more tense while driving and extra effort was needed to perform the driving test. Additionally, there was an important interaction with total sleep time and hangover effects on sleep disorders.
From the laboratory point of view, the evidence that the alcohol metabolites contribute actively to hangover is shown by Høiseth et al. (in press). The group investigated the prevalence and concentrations of the two ethanol metabolites, namely ethyl glucuronide (EtG) and ethyl sulfate (EtS), in blood during hangovers. The study was performed among 146 apprehended drivers, in which no psychoactive substances, including alcohol, were detected. Among the “impaired drivers,” EtG and EtS were detected in 18%, while among “not impaired drivers;” they were detected in 5% of the cases. Also the concentrations of both EtG and EtS were significantly higher in the group of impaired drivers compared with the not-impaired drivers. There was a statistically significant positive correlation between the concentrations of EtG and EtS and the degree of impairment. The results indicate that hangover symptoms are relevant for traffic safety.
9.1.2. Health professionals
Edvardsen et al. (2014) analyzed the prevalence of alcohol among a sample of 916 (81.1% women) health professionals and pharmacists in Norway. In addition to a self-reported absence from or impairment at work due to alcohol and/or drug use, this study analyzed samples of oral fluid. Alcohol was not detected in any of the samples. EtG was found in 0.3% of the collected samples. Illicit drugs and medicinal drugs were identified in 0.6% and 7.3% of the samples, respectively. Both analytical results and self-reported use of alcohol were analyzed for 12 months. Reduced efficiency at work due to alcohol use during the 12 month period was reported by 12.2% of the participants. Hangover related to the use of alcohol appeared to be an important issue in this population.
9.1.3. Does a hangover episode make a difference in overall drinking behavior?
The term “delayed alcohol-induced headache” is often used synonymously with alcohol hangover as a cluster. The objective of Zlotnik et al. (2014) was to compare alcohol hangover symptoms in students suffering from migraines as a hangover symptom and the ones who do not usually have migraines during hangover. In this cross-sectional study, university students were asked to fill structured questionnaires assessing headache history, alcoholic consumption, and hangover symptoms using the Hangover Symptom Scale. Subjects were classified as suffering from migraine with or without aura and non-sufferers from headache according to the International Classification of Headache Disorders. The students vulnerable to migraine-like hangover symptoms consume less alcohol, especially beer and liquors than the students that are not. The findings demonstrate that the tendency to develop migraine attacks affects post hangover behavior.
In another study, Epler et al. (2014) recruited 385 frequent drinkers. Each individual carried electronic diaries for 21 days and reported on their drinking behaviors. Analysis from 2276 drinking episodes was performed. There were 463 episodes of self-reported hangovers in the morning diary entries. Apart from stress after the drinking episode, the hangover was the only predictor of typical drinking frequency. The findings suggest that the hangover has a modest but inconsistent influence on the timing of subsequent alcohol use among frequent drinkers.
9.1.4. Adolescents
The impact of an alcohol hangover on daily activities can be profound. A survey among Dutch university students showed that more than half of them reported being unable to study when experiencing an alcohol hangover often or always. Noteworthy is that the average hangover frequency in this case is 2.7 days/month. Several experimental studies confirmed that memory functioning is impaired during an alcohol hangover. Hernandez et al. (2014) examined the psychometric properties of the original version of the Drinking Index in a sample of 740 adolescents (mean age = 15.26; 58.5% males) during an emergency department visit. Results demonstrated that there was a correlation between four-factor interpersonal, social, psychological, and physical indicators. This was also associated with outcomes such as hangover, alcohol withdrawal, and substance use.
9.2. Mechanism After
The hangover mechanism remains to be identified. The role of acetaldehyde, formic acid, alcohol dehydrogenase and acetaldehyde dehydrogenase polymorphism and intoxication is described. The deficiency of the active classes of these isoenzymes, frequent among Orientals, accounts for increased concentration of acetaldehyde and accounts for the decreased tolerance for alcohol (Neuman et al., 2014).
Additionally, mitochondrial dysfunction is a recognized cause of alcohol-induced damage. Mitochondrial malfunctioning may depend on the direct toxicity of the reactive metabolite itself or by generating a bio-activation process which inhibits mitochondrial beta oxidation and depletes cellular GSH. Peripheral blood monocytes of patients with alcohol-induced liver damage has been reported to generate cytokines such as TNF-α, IL-1β and IL-6 at rates that are three to six times those of monocytes from controls (Neuman et al., 2012a). Moreover, studies in several laboratories have focused on the identification, involvement and mode of action of TNF-α in regulating hepatic acute phase reaction similar with the acute episodes of intoxication from binge drinking.
9.3. Further Research
Serum concentrations of cytokines correlate with prognostic factors in AH and with increases in C-reactive protein levels (Neuman et al., 2012a).
Researchers should aim to (1) further determine the clinical pathology of the alcohol hangover, (2) examine the role of immunogenetics in susceptible individuals, (3) monitor metabolic components of alcohol in blood that characterize the alcohol hangover, (4) examine sex and age differences on molecular and clinical pathology aspects of the phenomenon, (5) develop common laboratory measurements, methodologies and cut-off levels, (6) focus on toxic substances that aggravate hangover severity (e.g. congeners, concomitant use of drugs), and (7) show the possible toxic effects of false hangover remedies that may lead to additional clinical toxicity.
The need for continual education and awareness cannot be understated. This phenomenon should be recognized not only by laboratory medicine but also by clinicians, as well as the legal system. Public transportation legislators should make mandatory the analysis of alcohol metabolites in the blood of professional drivers, pilots, flight attendants, as well as health professionals.
10. MicroRNA profiling to identify promoters of HCC progression in mice
Kyle J. Thompson, Ph.D. & Iain H. McKillop, Ph.D.
Department of Surgery, Carolinas Medical Center, Charlotte, NC 28203, USA.
HCC comprises approximately 80% of all primary tumors of the liver and represents the fifth most commonly diagnosed cancer in the world (McKillop and Schrum, 2009; Siegel et al., 2013). HCC is (primarily) attributed to exposure to known risk factors of which, hepatitis B and C infection, aflatoxin exposure, chronic high ethanol consumption, and obesity, are the most common (Mittal and El-Serag, 2013), and these risk factors act synergistically. Of increasing concern is the obesity epidemic in developed nations, where 30–40% of the population are obese and a strong link to risk for HCC development has been established, particularly in men (Calle et al., 2003). Recent epidemiological findings confirm these links; a 7.19 relative risk in HCC development for ethanol use and obesity (BMI > 30) having been reported (Loomba et al., 2013). Given the prevalence of obesity in developed countries, concomitant with higher relative ethanol consumption in these countries, we may be on the verge of an HCC epidemic. Given the long latency periods for HCC to manifest it is highly desirable to identify both the obese patients most at risk for developing HCC, as well as accurate disease staging in this patient population.
MicroRNAs (miRNAs) are small (18–25 nucleotide), non-coding RNAs that are principally involved in gene expression, and can become dysregulated in a variety of pathological states, including liver disease and cancer (Bala et al., 2009). Additionally, miRNAs have the potential to be minimally invasive biomarkers, often expressed inversely with miRNA target tissue levels (Lakner et al., 2012). We sought to test the relationship between obesity and ethanol consumption in a mouse model of HCC development, and to identify miRNAs as potential bio-markers for HCC in the setting of obesity and obesity with ethanol. To achieve this, we utilized male C57BL/6 mice, a strain noted for their relative susceptibility to diet-induced obesity (DIO) and voluntary ethanol consumption, yet relatively resistant to HCC when compared with other mouse strains (Brandon-Warner et al., 2012a). Mice were injected at 21–24 days old with vehicle or 5 mg/kg DEN and placed with adlibitum access on control diet (CD; 10% kcal%/fat) or high fat diet (HFD; 60% kcal%/fat) at 5-weeks of age and maintained until 35-weeks old. At this point mice were placed on in voluntary ethanol in drinking water (EtOH-DW; 10%/20% alternating days) or drinking water alone (DW) for an additional 6-weeks (Thompson et al., 2013).
Mice on HFD alone had spontaneous tumor formation (30%), an effect exacerbated by DEN administration (89%). However, mice on CD with DEN formed tumors at a lesser rate (60%) and these tumors were smaller than HFD counterparts (9). Introduction and maintenance on EtOH-DW with the CD regimen resulted in reduced tumor formation (44%) and no significant difference for those on a HFD. This is in contrast to previous results from our group utilizing the same EtOH-DW regimen in B6C3F1 mice, supporting inter-strain differences in susceptibility to hepatic tumor formation and progression, despite similar blood alcohol content levels (12–15 mM/L) in the two strains (Brandon-Warneret al., 2012b; Thompson et al., 2013).
To identify potential biomarkers for HCC, a GeneChip miRNA 2.0 Array was performed on liver tissue excised following sacrifice. For mice on HFD–DEN (DW and EtOH-DW), the large tumor burden permitted analysis of miRs from paired tumor and non-tumor liver (NTL) tissue. Screening revealed numerous dysregulated miRs in HFD compared to CD alone; however, noteworthy were seven miRs consistently dysregulated in HFD–DEN and HFD–EtOH-DW–DEN tumors compared to non-tumor tissue (Table 1). These findings were confirmed by qRT-PCR using Exiqon miRCURY LNA assays.
Table 1.
MicroRNAs dysregulated between non-tumor and tumor liver in a model of obesity-associated HCC.
| miRNA | Dysregulation vs. non-tumor liver | Function(s) |
|---|---|---|
| miR-182 | Down | FOX proteins (Segura et al., 2009), metastasis (Hirata et al., 2013) |
| miR-27a | Up | Lipid metabolism (Fernández-Hernando et al., 2011), Fox proteins (Guttilla and White, 2009) |
| miR-125a-5p | Up | Tumor suppressor (Kim et al., 2013), insulin resistance (Herrera et al., 2009) |
| miR-139-5p | Down | Tumor suppressor (Gu et al., 2014), c-Fos regulation (Fan et al., 2013) |
| miR-455 | Down | Poorly characterized |
| miR-378 | Down | Lipogenesis (Fernández-Hernando et al., 2011), CYP2E1 (Mohri et al., 2010), Liver regeneration (Song et al., 2010) |
| miR-193a | Down | Poorly characterized |
Individual miR analysis confirmed array results for 6/7 miRs (miR-139-5p was down regulated in contrast to array data). MicroRNAs 182 (down regulated versus NTL), 27a (up regulated vs NTL) and 125a (up regulated vs NTL) had the most significant changes detected and may represent the best candidates for biomarker status in human patients. Further studies in patients comparing tumor, NTL and sera expression of miR-182, -27a and -125a will be conducted in healthy, obese, normal BMI with HCC and obese with HCC.
11. Macronutrients, micronutrients, and probiotics in ALD development and treatment
Irina A. Kirpich and Craig McClain.
Clinically important ALD develops only in a subset of people who drink heavily. Dietary factors likely play important roles in both ALD pathogenesis and treatment. This short review summarizes the effects of macronutrients (dietary fat), micronutrients (Zn), and probiotics (specifically Lactobacillus rhamnosus GG (LGG)) in ALD development and treatment.
Dietary fat and alcohol both play important roles in the pathogenesis of ALD. Several studies have shown that dietary saturated fat protects against alcohol-induced liver disease in rodents, whereas dietary unsaturated fat, enriched in linoleic acid (LA) in particular, promotes alcohol-induced liver damage (Nanji and French, 1989; Nanji et al., 1989, 1995b, 2001). The deleterious effects of dietary unsaturated fatty acids are thought to be mediated through induction of oxidative stress, potentiated by inducing CYP2E1 (Nanji et al., 1994b, 1994c, 1995a), elevated endotoxin levels, and increased production of proinflammatory cytokines (Nanji, 2004). However, the exact mechanisms by which the combination of LA and alcohol promotes liver injury are not fully understood. This is particularly relevant because LA is a major unsaturated fatty acid in the Western diet (2005), and LA consumption has dramatically increased during the 20th century (Blasbalg et al., 2011). Our group has recently demonstrated that 8 weeks of EtOH feeding significantly increase liver injury, steatosis and inflammation in mice fed unsaturated fat (USF + EtOH, corn oil/LA enriched) compared to pair-fed mice (Kirpich et al., 2012). These effects of EtOH were blunted by a saturated fat (SF) diet containing medium chain triglycerides. Hepatic TLR (TLR 1, 2, 3, 4, 7, 8, 9) mRNA expression was significantly increased compared to control in the livers of USF + EtOH fed animals, but not in the livers of the SF + EtOH group. In parallel with liver injury, increased gut permeability and elevated endotoxemia were observed in response to USF + EtOH but not SF + EtOH. Intestinal inflammation was positively correlated with the EtOH + USF triggered disruption of the intestinal tight junctions (TJ). Importantly, USF diet alone resulted in down-regulation of intestinal TJ protein mRNA expression compared to SF. Alcohol further suppressed TJ proteins in USF + EtOH, but did not affect intestinal TJ in SF + EtOH group. Additionally, USF + EtOH, but not SF + EtOH, resulted in alterations of the intestinal mucus layer and intestinal antimicrobial defense (Kirpich et al., 2013). Therefore, unsaturated, but not saturated, fat was a significant contributing factor to EtOH-mediated intestinal pro-inflammatory response, dysregulated intestinal tight junctions, endotoxemia and consequent liver injury. The oxidized metabolites of LA may be possible metabolites underlying pathogenic effects of EtOH and unsaturated dietary fat on intestinal and liver injury. This hypothesis needs to be further investigated.
Zinc is an essential trace element required for a broad range of biological activities including the function of hundreds of zinc finger proteins. In the United States, the Recommended Dietary Allowance (RDA) is 8 mg/d for women and 11 mg/d for men. Zinc and dietary protein intake directly correlate with each other. Alcoholics, and especially those with ALD, have poor diets that are low in protein and low in zinc. Absorption of zinc is concentration dependent and occurs throughout the small intestine (mainly the jejunum). Absorption may be impaired in cirrhosis, and typically there is increased urinary excretion of zinc in cirrhosis (reviewed by Mohammad et al. (2012)). Zinc status and the serum zinc levels drop with low dietary zinc intake. There normally are multiple mechanisms in place to protect against zinc deficiency, including increased absorption and decreased excretion via modification of zinc transporters (Mohammad et al., 2012). Zinc status is typically assessed by plasma/serum zinc concentration. However, inflammation/ stress hormones (e.g. LPS, TNF) may cause a decrease in serum zinc level, with an internal redistribution of the zinc and a potential loss of zinc from critical zinc finger proteins.
Recent animal and in vitro studies provide major new insights into the molecular mechanisms of altered zinc metabolism in the development and progression of experimental ALD. In both acute and chronic alcohol-induced hepatotoxicities, alcohol intake and oxidative stress disrupt tight junctions in the intestine, which leads to translocation of bacterial products, such as endotoxin (reviewed by Mohammad et al. (2012)). Endotoxin activates TLR-4 with subsequent TNF production, oxidative stress and liver injury. Endotoxin and TNF also play critical roles in liver fibrosis. Disruption of tight-junction proteins occurs not only in the intestine, but also in the lung and likely at the blood–brain barrier, thus potentially predisposing to lung injury and hepatic encephalopathy. Zinc treatment in experimental animals with ALD attenuated the increased gut permeability, endotoxemia, TNF production, oxidative stress, and liver injury, while improving activity of key zinc transcription factors (reviewed by Mohammad et al. (2012)). Thus, zinc supplementation targets most postulated mechanisms for the development of ALD (Fig. 4).
Fig. 4.
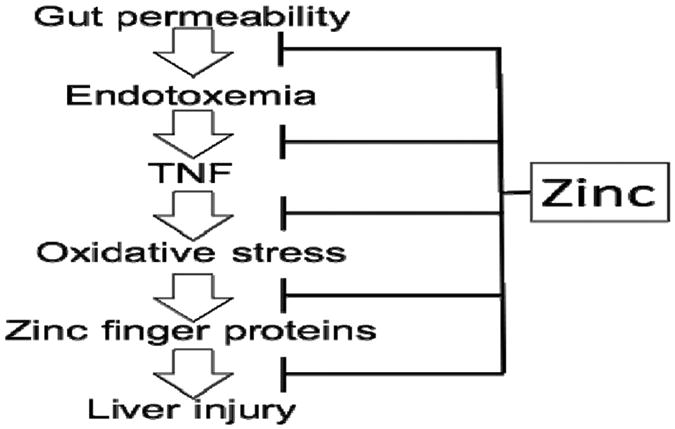
Zinc therapy positively impacts multiple mechanisms of ALD.
As highlighted above, zinc deficiency has been implicated in the development of ALD, and zinc mobilization and loss from zinc proteins due to oxidative stress are important putative etiologic factors in the development/progression of ALD. A mouse model of ALD showed a decrease in the hepatic zinc level even though a zinc-adequate diet was fed (Kang et al., 2009; Zhou et al., 2005). Zinc participates in cell functions mainly through binding to thousands of zinc proteins including metalloenzymes and transcription factors (Maret, 2005). Removal of zinc from zinc metalloenzymes has been shown to either inactivate (e.g. alcohol dehydrogenase) or activate (e.g. caspase-3) enzyme activities. It is well known that zinc is the key element in zinc finger structure that is required for DNA binding activity of zinc finger transcription factors. Oxidative stress can release zinc from zinc finger transcription factors, leading to loss of DNA binding activity. We found that chronic alcohol exposure inhibits hepatocyte nuclear factor-4α(HNF-4α) function without affecting its protein level, suggesting a zinc release from the protein which was then further validated by our group (Kang et al., 2008, 2009). Zinc deprivation also inactivated other zinc proteins, such as insulin-like growth factor-I (IGF-I) and IGF binding protein-1 (IGFBP-1) (Kang et al., 2008). These results support the concept that oxidative stress with inactivation of zinc finger proteins is an important molecular mechanism in the development of ALD, and yet another reason to provide zinc supplementation in ALD (recommended dose 220 mg zinc sulfate = 50 mg elemental zinc/day).
The importance of the gut:liver axis in alcohol-mediated multi-organ pathology is well recognized (Keshavarzian et al., 2009; Parlesak et al., 2000). It is generally accepted that enteric dysbiosis plays an important role in the pathogenesis of ALD. Recent studies have demonstrated favorable effects of modulating gut microbiota by probiotics in clinical (Kirpich et al., 2008) and experimental ALD (Forsyth et al., 2009; Nanji et al., 1994a). Results from our recent study have demonstrated that in a mouse model of ALD, chronic alcohol feeding leads to a significant change in the intestinal bacterial community structure over time (Bull-Otterson et al., 2013). After 8 weeks of EtOH feeding, dietary USF and EtOH caused a decline in the abundance of both Bacteriodetes and Firmicutes phyla, with a proportional increase in the gram negative Proteobacteria and gram positive Actinobacteria phyla; the bacterial genera that showed the biggest expansion were the gram negative alkaline tolerant Alcaligenes and gram positive Corynebacterium. In parallel with these changes in microbiome, EtOH caused an increase in plasma endotoxin, fecal pH, and hepatic inflammation and injury. The ethanol-induced pathogenic changes in the microbiome and the liver were prevented by 2 weeks of LGG treatment (Bull-Otterson et al., 2013). Our recent mechanistic study has showed that LGG supplementation markedly decreased lived injury by significant reduction in TNF-α expression (Wang et al., 2013). We also have demonstrated that LGG supplementation prevents alcohol-induced decrease in the expression of intestinal trefoil factor (ITF) and its transcriptional regulator hypoxia-inducible factor-2α (HIF-2α) attenuating intestinal TJ alterations and barrier dysfunction (Wang et al., 2011). Importantly, not only LGG live bacteria, but also LGG supernatant has demonstrated protective effects against alcohol-mediated intestinal and liver injury in an acute alcohol animal model (Huang et al., 2012).
12. Genetics of alcoholic liver disease
Devanshi Seth.
Evidence for a genetic component associated with the risk of ALD comes from earlier twin studies showing that the concordance rate for alcoholic cirrhosis was three times higher in monozygotic than in dizygotic twin pairs (Hrubec and Omenn, 1981). A subsequent study showed susceptibility genes for alcoholic cirrhosis independently affected their prevalence and provided evidence for a genetic predisposition to organ-specific complications of alcoholism (Reed et al., 1996). This study also showed that approximately 50% of phenotypic variation in ALD in heavy drinkers was due to genetic modifiers. Further indirect evidence comes from the wide inter-ethnic variation in ALD-related mortality that is not entirely explained by variations in the prevalence of alcohol abuse (Stinson et al., 2001). For example, in the USA, death rate from ALD was highest in Hispanic whites followed by African Americans, non-Hispanic whites and Hispanic African Americans (Said et al., 2004). Mortality rates in Black and Asian men is 3.8 times greater compared to White British men (Fisher et al., 2002); and 8-fold higher in indigenous population compared to non-indigenous Australians (Liang et al., 2010).
A battery of genes known to impact pathogenic processes during ALD progression such as oxidative stress, inflammation, impairment of hepatic regeneration and fibrosis, has been tested for associations with ALD. Genetic studies investigating mutations in genes related to alcohol metabolism (ADH, ALDH, CYP2E1), oxidative stress (GST, superoxide dismutase [SOD]), endotoxin (TNF-a, CD14, TLR4), inflammatory cytokines/chemokines (IL-10, MCP-1), immune (cytotoxic T-lymphocyte antigen-4) and fibrosis (collagen, MMPs, osteopontin, TGF-β) have had limited success in providing evidence for association with ALD. Studies found allelic variability in alcohol and acetaldehyde-metabolizing genes ALDH2*2, ALDH2*1/1, ADH1B*2, ADH1B*1, ADH1C*2, ADH1C*2, and CYP2E1. The enzyme activity induces susceptibility of the host to liver damage (Zakhari and Li, 2007). Individuals with more active ADH1B*2 and ADH1C*1 alleles were at increased risk of developing alcoholic liver injury due to a higher acetaldehyde exposure, but other studies were inconsistent due to ADH1B*2 being a rare allele in Caucasians, leading to ethnic variability in the populations studied.
There are several polymorphic loci within the human CYP2E1 gene with two mutations in linkage disequilibrium giving rise to c1 and c2 alleles. The CYP2E1*5 (c2) allele is associated with ∼10-fold higher mRNA, protein and enzyme activity than c1 allele potentially causing a higher exposure of the liver to acetaldehyde and ROS (Watanabe et al., 1994). Both ADH and CYP2E1 are minor risk factors for ALD progression but there are reports of their significant contribution in individuals homozygous for the alleles CYP2E1 c1 and ADH1C*1/ADH1C*1 to develop hepatoma (Homann et al., 2006). Manganese-dependent SOD2 is a key player in ALD pathogenesis since it protects mitochondria from peroxidative damage. A single nucleotide polymorphism at position —9 in the precursor protein to alanine (Ala) or valine (Val) results in an enhanced translocation into mitochondria and higher concentration of active MnSOD for Ala genotype. In ALD patients stratified for the degree of liver damage, Ala/Ala was found to be associated with severe ALD with odds ratio of 9.6 although not confirmed in a larger study (Stewart et al., 2002). Partial deletions in the GSTT1 and GSTM1 result in the absence of enzyme activity and increase levels of toxic intermediates. Significant increase in the frequency of GSTM1 ‘null allele’ was observed in patients with advanced ALD and an increased risk for ALD in individuals with combined carriage of GSTM1 and GSTT1 ‘null’ genotype (Ladero et al., 2005). CD14, a lipopolysaccharide (LPS) receptor on monocytes, macrophages and neutrophils enhances signaling through TLR4, another LPS receptor. A C/T polymorphism at position —159 in the CD14 promoter region, is associated with increased levels of soluble and membrane CD14 and the TT genotype is associated with advanced ALD (Järveläinen et al., 2001). However, this has not been confirmed in other studies. Polymorphism in IL-10 promoter-region at position —627 from C → A has association with ALD (Grove et al., 2000). An excess of the rare TNFα-A allele at position —238 was found in patients with ALD. The OR for this variant versus non-diseased individuals was 3.5 for cirrhosis and 4 for ASH (Grove et al., 1997). A few studies have found associations between the development of alcoholic cirrhosis and certain genotypes with poylmorphisms of IL-1β, T-lymphocyte antigen-4 gene (CTLA-4) associated with the titer of anti-CYP2E1 antibodies and genes involved in fibrogenesis (collagen α1, α2 chains) (Weiner et al., 1988). There are a number of obvious candidates such as, those encoding collagen I, matrix metalloprotein (MMP)-3, osteopontin and transforming growth factor (TGF)-β1, but so far remain unconfirmed (Stickel and Hampe, 2012; Stickel and Osterreicher, 2006).
To date, no significant associations with ALD for the polymorphisms chosen have been reported. A search for single nucleotide polymorphismsin a hypothesis-driven candidate gene approach has been largely disappointing in identifying risk factors for ALD due to inter-ethnic variability, small sample size and inappropriate study design, introducing type I and II errors. GWA in NAFLD identified locus rs738409 in PNPLA3 to be associated with hepatic fat, inflammation and ALT levels. The mutation in rs738409 from C to G changing a highly conserved isoleucine to methionine also shows a clear association with alcoholic steatosis, increased ALT and severity of cirrhosis (Childs Pugh) increasing the relative risk by 1.79 per G allele. Subsequent studies by several independent groups have shown association of rs738409 variant in PNPLA3 with ALD (Seth et al., 2010; Stickel et al., 2011; Tian et al., 2010), and are testimony to a genetic basis for this disease. PNPLA3, also called adiponutrin, is a phospholipase suspected to have lipogenic transacetylase activity with involvement in energy metabolism and lipid storage. The mutation from C to G at residue 148 in rs738409, changes a highly conserved isoleucine to methionine. This change from isoleucine to methionine is thought to be a gain of function enhancing lipid accumulation leading to hepatocyte injury and inflammation. A clear association with higher fat content and increased ALT. in homozygous carriers of G allele was observed in these studies. Recent functional data show PNPLA3 hydrolyses the triglicerides. The presence of homozygous GG reduces triglyceride hydrolysis in vitro thus increasing the fat content (Lake et al., 2005). In transgenic mice overexpressing GG allele there is higher hepatic lipid accumulation compared to wild type controls (He et al., 2010).
Most recently, polymorphism in the neurocan (NCAN) has been associated with NAFLD, and is a risk factor in ALD, related HCC. Association of genes in ALD and NAFLD presents increasing evidence of parallel mechanisms operating in alcoholic and non-ALD.
13. Concluding remarks
The symposia presented epidemiologic, experimental, and clinical evidence that clearly demonstrated once more that alcohol is toxic to the liver. The manner in which alcohol induces liver injury has become increasingly clear. The toxic role for ethanol in the pathogenesis of liver disease is evident. However, there is a contributory role for malnutrition. The possible interplay of these factors with environmental agents in producing the liver disease of alcoholism is demonstrated by clinical and experimental data. Chronic alcohol abuse remains an important risk factor for malignant transformation of hepatocytes, frequently in association with alcohol-induced cirrhosis.
The effect of alcohol on the adverse effects of therapeutics, secondary to ethanol induction of CYP2E1 is an important additional factor of alcohol induced-liver injury, and suggests an important additional pathway for alcohol-associated hepatic disease. Also relevant is the bioactivation of other toxic agents whose metabolism is enhanced by induction of CYP2E1 by ethanol. An associated change is the reduction of hepatic GSH content, in part because of acetaldehyde binding to the molecule and inpart because alcohol intake interferes with GSH synthesis. Methylation in response to therapy in alcohol and viral infections plays a crucial role. Genetic factors in susceptibility to ALD and NASH are an increasingly studied subject. The interplay of these factors may explain the differential susceptibility to the development of ALD. In addition, it is essential to use adequate laboratory tools.
Acknowledgments
S. French and his team thank Adriana Flores for typing the manuscript and thank NIH for the grant support (AAUOI-021848-02 and P50-11999 Morphology Core).
Drs. Osna and Kharbanda acknowledge that their research reported here was supported by Merit Review grants BX001673 (NAO) and BX001155 (KKK) from the Department of Veterans Affairs, Office of Research and Development (Biomedical Laboratory and Development).
Drs. Seitz and Muller acknowledge the contribution of Dietmar Hopp Foundation, Germany.
Drs. Neuman and Cohen thank In Vitro Drug Safety and Biotechnology and Muhaffy Grant, Sunnybrook HSC for the funding.
Dr. Neuman is grateful to Debra Sharp, Director RSA, for the continuous support provided for all Charles Lieber's symposia.
Abbreviations
- ADH
alcohol dehydrogenase
- AH
acute alcoholic hepatitis
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- ASH
alcoholic steato-hepatitis
- AST
aspartate aminotransferase
- CDT
carbohydrate-deficient transferrin
- CYP
cytochrome p450
- GAA
guanidinoacetate
- γGT
gamma glutamyl transpeptidase
- HCC
hepatocellular carcinoma
- HCV
hepatitis C virus
- HFD
high fat diet
- 4-HNE
4-hydroxynonenal
- IGF
insulin-like growth factor
- IL
interleukin
- LA
linoleic acid
- LGG
Lactobacillus rhamnosus GG
- LPS
lipopolysaccharide
- MCV
mean corpuscular volume of erythrocytes
- MDB
Mallory–Denk body
- MDF
Maddrey discriminant function
- MELD
Model for End-Stage Liver Disease
- MEOS
microsomal ethanol oxidizing system
- MMP
matrix metalloprotein
- MRI
magnetic resonance imaging
- NAFLD
non-alcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PTU
propylthiouracil
- ROS
reactive oxygen species
- SAH
S-adenosylhomocysteine
- SAM
S-adenoslmethionine
- SF
saturated fat
- TIMP
tissue inhibitor of metalloproteinase
- TLR
toll like receptor
- TNF
tumor necrosis factor
- USF
unsaturated fat
Footnotes
Conflict of interest statement: All the authors contributed actively to the review. All authors disclose that they have no actual or potential conflict of interest including any financial, personal or other relationships with other people or organizations that could inappropriately influence, or be perceived to influence their work.
References
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Allen JP, Litten RZ. The role of laboratory testing in alcoholism treatment. J Subst Abus Treat. 2001;20:81–85. doi: 10.1016/s0740-5472(00)00144-6. [DOI] [PubMed] [Google Scholar]
- Allen JP, Wurst FM, Thon N, Litten RZ. Assessing the drinking status of liver transplant patients with alcoholic liver disease. Liver Transpl. 2013;19:369–376. doi: 10.1002/lt.23596. [DOI] [PubMed] [Google Scholar]
- Altamirano J, Bataller R. Alcoholic liver disease: pathogenesis and new targets for therapy. Nat Rev Gastroenterol Hepatol. 2011;8:491–501. doi: 10.1038/nrgastro.2011.134. [DOI] [PubMed] [Google Scholar]
- Altamirano J, Miquel R, Katoonizadeh A, Abraldes JG, Duarte-Rojo A, Louvet A, Augustin S, Mookerjee RP, Michelena J, Smyrk TC, Buob D, Leteurtre E, Rincón D, Ruiz P, García-Pagán JC, Guerrero-Marquez C, Jones PD, Barritt AS, IV, Arroyo V, Bruguera M, Bañares R, Ginès P, Caballería J, Roskams T, Nevens F, Jalan R, Mathurin P, Shah VH, Bataller R. A histologic scoring system for prognosis of patients with alcoholic hepatitis. Gastroenterology. 2014;146:1231–1239. doi: 10.1053/j.gastro.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amidi F, French BA, Chung D, Halsted CH, Medici V, French SU. M30 and 4HNE are sequestered in different aggresomes in the same hepatocytes. Exp Mol Pathol. 2007;83:296–300. doi: 10.1016/j.yexmp.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Cremona A, Paton A, Turner C, Wallace P. The risk of alcohol. Addiction. 1993;88:1493–1508. doi: 10.1111/j.1360-0443.1993.tb03135.x. [DOI] [PubMed] [Google Scholar]
- Angulo P, Keach JC, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology (Baltim Md) 1999;30:1356–1362. doi: 10.1002/hep.510300604. [DOI] [PubMed] [Google Scholar]
- Anton RF, Lieber C, Tabakoff B. Carbohydrate-deficient transferrin and gamma-glutamyltransferase for the detection and monitoring of alcohol use: results from a multisite study. Alcohol Clin Exp Res. 2002;26:1215–1222. doi: 10.1097/01.ALC.0000023986.42254.F5. [DOI] [PubMed] [Google Scholar]
- Ascha MS, Hanouneh IA, Lopez R, Tamimi TAR, Feldstein AF, Zein NN. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology. 2010;51:1972–1978. doi: 10.1002/hep.23527. [DOI] [PubMed] [Google Scholar]
- Bala S, Marcos M, Szabo G. Emerging role of microRNAs in liver diseases. World J Gastroenterol. 2009;155:5633–5640. doi: 10.3748/wjg.15.5633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista AP. Free radicals, chemokines, and cell injury in HIV-1 and SIV infections and alcoholic hepatitis. Free Radic Biol Med. 2001;31:1527–1532. doi: 10.1016/s0891-5849(01)00745-6. [DOI] [PubMed] [Google Scholar]
- Becker U, Grønbæk M, Johansen D, Sørensen TIA. Lower risk for alcohol-induced cirrhosis in wine drinkers. Hepatology. 2002;35:868–875. doi: 10.1053/jhep.2002.32101. http://dx.doi.org/10.1053/jhep.2002.32101. [DOI] [PubMed] [Google Scholar]
- Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis (Basel Switz) 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- Benhamou JP. Drug-induced hepatitis: clinical aspects. In: Fillastre JP, editor. Hepatotoxicity of Drugs. University de Rouen; Rouen: 1986. pp. 23–30. [Google Scholar]
- Bennett JB, Patterson CR, Reynolds GS, Wiitala WL, Lehman WE. Team awareness, problem drinking, and drinking climate: workplace social health promotion in a policy context. Am J Health Promot. 2004;19:103–113. doi: 10.4278/0890-1171-19.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beymer C, Kowdley KV, Larson A, Edmonson P, Dellinger EP, Flum DR. Prevalence and predictors of asymptomatic liver disease in patients undergoing gastric bypass surgery. Arch Surg. 2003;138:1240–1244. doi: 10.1001/archsurg.138.11.1240. [DOI] [PubMed] [Google Scholar]
- Birschbach HR, Harinasuta U, Zimmerman HJ. Alcoholic steatonecrosis. II. Prospective study of prevalence of Mallory bodies in biopsy specimens and comparison of severity of hepatic disease in patients with and without this histologic feature. Gastroenterology. 1974;66:1195–1202. [PubMed] [Google Scholar]
- Blasbalg TL, Hibbeln JR, Ramsden CE, Majchrzak SF, Rawlings RR. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am J Clin Nutr. 2011;93:950–962. doi: 10.3945/ajcn.110.006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaut M, Collins MD, Welling GW, Doré J, van Loo J, de Vos W. Molecular biological methods for studying the gut microbiota: the EU human gut flora project. Br J Nutr. 2002;87:S203–S211. doi: 10.1079/BJNBJN/2002539. [DOI] [PubMed] [Google Scholar]
- Bobak M, Room R, Pikhart H, Kubinova R, Malyutina S, Pajak A, Kurilovitch S, Topor R, Nikitin Y, Marmot M. Contribution of drinking patterns to differences in rates of alcohol related problems between three urban populations. J Epidemiol Community Health. 2004;58:238–242. doi: 10.1136/jech.2003.011825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitnott JK, Maddrey WC. Alcoholic liver disease. I. Interrelationships among histologic features and the histologic effects of prednisolone therapy. Hepatology. 1981;1:599–610. doi: 10.1002/hep.1840010607. [DOI] [PubMed] [Google Scholar]
- Brandon-Warner E, Schrum LW, Schmidt CM, McKillop IH. Rodent models of alcoholic liver disease: of mice and men. Alcohol. 2012a;46:715–725. doi: 10.1016/j.alcohol.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon-Warner E, Walling TL, Schrum LW, McKillop IH. Chronic ethanol feeding accelerates hepatocellular carcinoma progression in a sex-dependent manner in a mouse model of hepatocarcinogenesis. Alcohol Clin Exp Res. 2012b;36:641–653. doi: 10.1111/j.1530-0277.2011.01660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull-Otterson L, Feng W, Kirpich I, Wang Y, Qin X, Liu Y, Gobejishvili L, Joshi-Barve S, Ayvaz T, Petrosino J, Kong M, Barker D, McClain C, Barve S. Metagenomic analyses of alcohol induced pathogenic alterations in the intestinal microbiome and the effect of Lactobacillus rhamnosus GG treatment. PLoS One. 2013;8:e53028. doi: 10.1371/journal.pone.0053028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burcelin R. The antidiabetic gutsy role of metformin uncovered? Gut. 2014;63:706–707. doi: 10.1136/gutjnl-2013-305370. [DOI] [PubMed] [Google Scholar]
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- Castera L, Hartmann DJ, Chapel F, Guettier C, Mall F, Lons T, Richardet JP, Grimbert S, Morassi O, Beaugrand M, Trinchet JC. Serum laminin and type IV collagen are accurate markers of histologically severe alcoholic hepatitis in patients with cirrhosis. J Hepatol. 2000;32:412–418. doi: 10.1016/s0168-8278(00)80391-8. [DOI] [PubMed] [Google Scholar]
- Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55:2005–2023. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- Chase PJ, Davis PG, Bensimhon DR. The obesity paradox in chronic heart failure: what does it mean? Curr Heart Fail Rep. 2014;11:111–117. doi: 10.1007/s11897-013-0184-2. [DOI] [PubMed] [Google Scholar]
- Chen J, Conigrave KM, Macaskill P, Whilfield P, Irwig L. Combining carbohydrate-deficient transferrin and gamma-glutamyltransferase to increase diagnostic accuracy for problem drinking alcohol. Alcoholism. 2003;38:574–582. doi: 10.1093/alcalc/agg113. [DOI] [PubMed] [Google Scholar]
- Christoffersen P, Nielsen K. Histological changes in human liver biopsies from chronic alcoholics. Acta Pathol Microbiol Scand A. 1972;80:557–565. doi: 10.1111/j.1699-0463.1972.tb00316.x. [DOI] [PubMed] [Google Scholar]
- Cohen JA, Kaplan MA. The SGOT/SGPT ratio — an indicator of alcoholic liver disease. Dig Dis Sci. 1979;24:835–838. doi: 10.1007/BF01324898. [DOI] [PubMed] [Google Scholar]
- Cousins G, Galvin R, Flood M, Kennedy MC, Motterlini N, Henman MC, Kenny RA, Fahey T. Potential for alcohol and drug interactions in older adults: evidence from the Irish longitudinal study on ageing. BMC Geriatr. 2014;14:57. doi: 10.1186/1471-2318-14-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daves M, Cemin R, Floreani M, Pusceddu I, Cosio G, Lippi G. Comparative evaluation of capillary zone electrophoresis and HPLC in the determination of carbohydrate-deficient transferrin. Clin Chem Lab Med. 2011;49:1677–1680. doi: 10.1515/CCLM.2011.630. [DOI] [PubMed] [Google Scholar]
- Delanghe JR, Helander A, Wielders JPM, Pekelharing JM, Roth HJ, Schellenberg F, Born C, Yagmur E, Gentzer W, Althaus H. Development and multicenter evaluation of the N latex CDT direct immunonephelometric assay for serum carbohydrate-deficient transferrin. Clin Chem. 2007;53:1116–1121. doi: 10.1373/clinchem.2006.084459. [DOI] [PubMed] [Google Scholar]
- Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids. Institute of Medicine of the National Academy of Sciences. Standing Committee on the Scientific Evaluation of Dietary Reference Intakes, National Academy Press.; Washington, DC: 2005. [Google Scholar]
- Dominguez M, Rincón D, Abraldes JG, Miquel R, Colmenero J, Bellot P, García-Pagán JC, Fernández R, Moreno M, Bañares R, Arroyo V, Caballería J, Ginès P, Bataller R. A new scoring system for prognostic stratification of patients with alcoholic hepatitis. Am J Gastroenterol. 2008;103:2747–2756. doi: 10.1111/j.1572-0241.2008.02104.x. [DOI] [PubMed] [Google Scholar]
- Donohue TM, Osna NA, Clemens DL. Recombinant Hep G2 cells that express alcohol dehydrogenase and cytochrome P450 2E1 as a model of ethanol-elicited cytotoxicity. Int J Biochem Cell Biol. 2006;38:92–101. doi: 10.1016/j.biocel.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126:263–277. doi: 10.1053/j.gastro.2003.10.076. [DOI] [PubMed] [Google Scholar]
- Duong FH, Christen V, Filipowicz M, Heim MH. S-adenosylmethionine and betaine correct hepatitis C virus induced inhibition of interferon signaling in vitro. Hepatology. 2006;43:796–806. doi: 10.1002/hep.21116. [DOI] [PubMed] [Google Scholar]
- Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. doi: 10.1016/S0140-6736(05)66378-7. [DOI] [PubMed] [Google Scholar]
- Edmonson HA, Peters RL, Reynolds TB, Kyzma OT. Sclerosing hyaline necrosis of the liver in the chronic alcoholic. A recognizable clinical syndrome. Ann Intern Med. 1963;59:646–649. doi: 10.7326/0003-4819-59-5-646. [DOI] [PubMed] [Google Scholar]
- Edvardsen HM, Karinen R, Moan IS, Oiestad EL, Christophersen AS, Gjerde H. Use of alcohol and drugs among health professionals in Norway: a study using data from questionnaires and samples of oral fluid. J Occup Med Toxicol. 2014;9:8. doi: 10.1186/1745-6673-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epler AJ, Tomko RL, Piasecki TM, Wood PK, Sher KJ, Shiffman S, Heath AC. Does hangover influence the time to next drink? An investigation using ecological momentary assessment. Alcohol Clin Exp Res. 2014;38:1461–1469. doi: 10.1111/acer.12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch R, Nicolas JM, Villegas E, Junque A, Urbano-Marquez A. Relationship between ethanol-related diseases and nutritional status in chronically alcoholic men. Alcohol Alcohol. 1993;28:543–550. [PubMed] [Google Scholar]
- Fan Q, He M, Deng X, Wu WK, Zhao L, Tang J, Wen G, Sun X, Liu Y. Derepression of c-Fos caused by microRNA-139 down-regulation contributes to the metastasis of human hepatocellular carcinoma. Cell Biochem Funct. 2013;31:319–324. doi: 10.1002/cbf.2902. [DOI] [PubMed] [Google Scholar]
- Fernández-Hernando C, Suárez Y, Rayner KJ, Moore KJ. MicroRNAs in lipid metabolism. Curr Opin Lipidol. 2011;22:86–92. doi: 10.1097/MOL.0b013e3283428d9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher NC, Hanson J, Phillips A, Rao JN, Swarbrick ET. Mortality from liver disease in the West Midlands, 1993–2000: observational study. BMJ. 2002;325:312–313. doi: 10.1136/bmj.325.7359.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsyth CB, Farhadi A, Jakate SM, Tang Y, Shaikh M, Keshavarzian A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol. 2009;43:163–172. doi: 10.1016/j.alcohol.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foucher J, Chanteloup E, Vergniol J, Castéra L, Le Bail B, Adhoute X, Bertet J, Couzigou P, de Lédinghen V. Diagnosis of cirrhosis by transient elastography (FibroScan): a prospective study. Gut. 2006;55:403–408. doi: 10.1136/gut.2005.069153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank A, Seitz HK, Bartsch H, Frank N, Nair J. Immunohistochemical detection of 1, N6-ethenodeoxyadenosine in nuclei of human liver affected by diseases predisposing to hepato-carcinogenesis. Carcinogenesis. 2004;25:1027–1031. doi: 10.1093/carcin/bgh089. [DOI] [PubMed] [Google Scholar]
- French SW. The Mallory body: structure, composition and pathogenesis. Hepatology. 1981;1:76–81. doi: 10.1002/hep.1840010113. [DOI] [PubMed] [Google Scholar]
- French SW, Eidus LB, Freeman J. Nonalcoholic fatty hepatitis. An important clinical condition. Can J Gastroenterol. 1989;3:189–197. [Google Scholar]
- French SW, Nash J, Shitabata P, Kachi K, Hara C, Chedid A, Mendenhall CL. Pathology of alcoholic liver disease. VA Cooperative Study Group 119. Semin Liver Dis. 1993;13:154–169. doi: 10.1055/s-2007-1007346. [DOI] [PubMed] [Google Scholar]
- Frone MR. Prevalence and distribution of alcohol use and impairment in the workplace: a U.S. national survey. J Stud Alcohol. 2006;67:147–156. doi: 10.15288/jsa.2006.67.147. [DOI] [PubMed] [Google Scholar]
- Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141:1572–1585. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Quintela A, Garcia J, Campos J, Perez LE, Alende MR, Otero E, Abdulkader I. Serum cytokeratins in alcoholic liver disease: contrasting levels of cytokeratin-18 and cytokeratin-19. Alcohol. 2006;38:45–49. doi: 10.1016/j.alcohol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Goodglass H, Kaplan E. The Assessment of Aphasia and Related Disorders. Lea & Febiger; Philadelphia: 1983. [Google Scholar]
- Green JA. Subclinical acute liver disease of the alcoholic. Australas Ann Med. 1965;14:111–124. doi: 10.1111/imj.1965.14.2.111. [DOI] [PubMed] [Google Scholar]
- Gregory DH, Levi DF. The clinical-pathologic spectrum of alcoholic hepatitis. Am J Dig Dis. 1972;17:479–488. doi: 10.1007/BF02231202. [DOI] [PubMed] [Google Scholar]
- Grove J, Daly AK, Bassendine MF. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic hepatitis. Hepatology. 1997;26:143–146. doi: 10.1002/hep.510260119. [DOI] [PubMed] [Google Scholar]
- Grove J, Daly AK, Bassendine MF. Interleukin 10 promoter region polymorphism and susceptibility to advanced alcoholic liver disease. Gut. 2000;46:540–545. doi: 10.1136/gut.46.4.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb D, Rasmussen B, Linnet K, Olsson SG, Lindberg L. Breath alcohol analysis incorporating standardization to water vapour is as precise as blood alcohol analysis. Forensic Sci Int. 2012;216:88–91. doi: 10.1016/j.forsciint.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Gu W, Li X, Wang J. miR-139 regulates the proliferation and invasion of hepatocellular carcinoma through the WNT/TCF-4 pathway. Oncol Rep. 2014;31:397–404. doi: 10.3892/or.2013.2831. [DOI] [PubMed] [Google Scholar]
- Gupte P, Amarapurkar D, Agal S, Baijal R, Kulshrestha P, Pramanik S, Patel N, Madan A, Amarapurkar A, Hafeezunnisa Non-alcoholic steatohepatitis in type 2 diabetes mellitus. J Gastroenterol Hepatol. 2004;19:854–858. doi: 10.1111/j.1440-1746.2004.03312.x. [DOI] [PubMed] [Google Scholar]
- Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284:23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harinasuta U, Chomet B, Ishak KG, Zimmerman HJ. Steatonecrosis—Mallory body type. Medicine. 1967;46:141–162. doi: 10.1097/00005792-196703000-00008. [DOI] [PubMed] [Google Scholar]
- Hayasaka A, Schuppan D, Ohnishi K, Okuda K, Hahn EG. Serum concentrations of the carboxyterminal cross-linking domain of procollagen type IV (NC1) and the aminoterminal propeptide of procollagen type III (PIIIP) in chronic liver disease. J Hepatol. 1990;10:17–22. doi: 10.1016/0168-8278(90)90067-2. [DOI] [PubMed] [Google Scholar]
- He S, McPhaul C, Li JZ, Garuti R, Kinch L, Grishin NV, Cohen JC, Hobbs HH. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J Biol Chem. 2010;285:6706–6715. doi: 10.1074/jbc.M109.064501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helman RA, Temko MH, Nye FW, Fallon HJ. Alcoholic hepatitis. Natural history and evaluation of prednisolone therapy. Ann Intern Med. 1971;74:311–321. doi: 10.7326/0003-4819-74-3-311. [DOI] [PubMed] [Google Scholar]
- Hernandez L, Salas-Wright CP, Graves H, Cancilliere MK, Spirito A. A confirmatory approach to understanding the four-factor structure of the Adolescent Drinking Index: evidence for a brief version. J Subst Abus Treat. 2014;47:239–244. doi: 10.1016/j.jsat.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera BM, Lockstone HE, Taylor JM, Wills QF, Kaisaki PJ, Barrett A, Camps C, Fernandez C, Ragoussis J, Gauguier D, McCarthy MI, Lindgren CM. MicroRNA-125a is over-expressed in insulin target tissues in a spontaneous rat model of type 2 diabetes. BMC Med Genom. 2009;2:54. doi: 10.1186/1755-8794-2-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hift R, Kirsch R. Porphyria cutanea tarda. In: Hall P, editor. Alcoholic Liver Disease, Second ed Pathology and Pathogenesis. Edward Arnold; London: 1995. pp. 219–231. [Google Scholar]
- Hildrum B, Mykletun A, Hole T, Midthjell K, Dahl AA. Age-specific prevalence of the metabolic syndrome defined by the International Diabetes Federation and the National Cholesterol Education Program: the Norwegian HUNT 2 study. BMC Public Health. 2007;7:220. doi: 10.1186/1471-2458-7-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, Ueno K, Shahryari V, Deng G, Tanaka Y, Tabatabai ZL, Hinoda Y, Dahiya R. MicroRNA-182-5p promotes cell invasion and proliferation by down regulating FOXF2, RECK and MTSS1 genes in human prostate cancer. PLoS One. 2013;8:e55502. doi: 10.1371/journal.pone.0055502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Høiseth G, Fosen JT, Liane V, Bogstrand ST, Mørland J. Alcohol hangover as a cause of impairment in apprehended drivers. Traffic Inj Prev. 2014 doi: 10.1080/15389588.2014.938324. http://dx.doi.org/10.1080/15389588.2014.938324 (in press) [DOI] [PubMed]
- Homann C, Benfield TL, Graudal NA, Garred P. Neopterin and interleukin-8—prognosis in alcohol-induced cirrhosis. Liver. 2000;20:442–449. doi: 10.1034/j.1600-0676.2000.020006442.x. [DOI] [PubMed] [Google Scholar]
- Homann N, Stickel F, Konig IR, Jacobs A, Junghanns K, Benesova M, Schuppan D, Himsel S, Zuber-Jerger I, Hellerbrand C, Ludwig D, Caselmann WH, Seitz HK. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int J Cancer. 2006;118:1998–2002. doi: 10.1002/ijc.21583. [DOI] [PubMed] [Google Scholar]
- Hrubec Z, Omenn GS. Evidence for genetic predisposition to alcoholic cirrhosis and psychosis: twin concordances for alcoholism and its biological end points by zygosity among male veterans. Alcohol Clin Exp Res. 1981;5:207–215. doi: 10.1111/j.1530-0277.1981.tb04890.x. [DOI] [PubMed] [Google Scholar]
- Huang YS, Chern HD, Su WJ, Wu JC, Chang SC, Chiang CH, Chang FY, Lee SD. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology. 2003;37:924–930. doi: 10.1053/jhep.2003.50144. [DOI] [PubMed] [Google Scholar]
- Huang HL, Wang YJ, Zhang QY, Liu B, Wang FY, Li JJ, Zhu RZ. Hepatoprotective effects of baicalein against CCl(4)-induced acute liver injury in mice. World J Gastroenterol. 2012;18:6605–6613. doi: 10.3748/wjg.v18.i45.6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishak KG, Zimmerman HJ, Ray MB. Alcoholic liver disease: pathologic, pathogenetic and clinical aspects. Alcohol Clin Exp Res. 1991;15:45–66. doi: 10.1111/j.1530-0277.1991.tb00518.x. [DOI] [PubMed] [Google Scholar]
- Järveläinen HA, Orpana A, Perola M, Savolainen VT, Karhunen PJ, Lindros KO. Promoter polymorphism of the CD14 endotoxin receptor gene as a risk factor for alcoholic liver disease. Hepatology. 2001;33:1148–1153. doi: 10.1053/jhep.2001.24236. [DOI] [PubMed] [Google Scholar]
- Jeppsson JO, Arndt T, Schellenberg F, Wielders JP, Anton RF, Whitfield JB, Helander A. Toward standardization of carbohydrate-deficient transferrin (CDT) measurements: I. Analyte definition and proposal of a candidate reference method. Clin Chem Lab Med. 2007;45:558–562. doi: 10.1515/CCLM.2007.107. [DOI] [PubMed] [Google Scholar]
- Kagansky N, Levy S, Keter D, Rimon E, Taiba Z, Fridman Z, Berger D, Knobler H, Malnick S. Non-alcoholic fatty liver disease—a common and benign finding in octogenarian patients. Liver Int. 2004;24:588–594. doi: 10.1111/j.1478-3231.2004.0969.x. [DOI] [PubMed] [Google Scholar]
- Kang X, Song Z, McClain CJ, Kang YJ, Zhou Z. Zinc supplementation enhances hepatic regeneration by preserving hepatocyte nuclear factor-4alpha in mice subjected to long-term ethanol administration. Am J Pathol. 2008;172:916–925. doi: 10.2353/ajpath.2008.070631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang X, Zhong W, Liu J, Song Z, McClain CJ, Kang YJ, Zhou Z. Zinc supplementation reverses alcohol-induced steatosis in mice through reactivating hepatocyte nuclear factor-4alpha and peroxisome proliferator-activated receptor-alpha. Hepatology. 2009;50:1241–1250. doi: 10.1002/hep.23090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz GG, Shear NH, Malkiewicz IM, Valentino K, Neuman MG. Signaling for ethanol-induced apoptosis and repair in vitro. Clin Biochem. 2001;34:218–235. doi: 10.1016/s0009-9120(01)00218-1. [DOI] [PubMed] [Google Scholar]
- Keshavarzian A, Farhadi A, Forsyth CB, Rangan J, Jakate S, Shaikh M, Banan A, Fields JZ. Evidence that chronic alcohol exposure promotes intestinal oxidative stress, intestinal hyperpermeability and endotoxemia prior to development of alcoholic steatohepatitis in rats. J Hepatol. 2009;50:538–547. doi: 10.1016/j.jhep.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew MC, Asare GA. Dietary iron overload in the African and hepatocellular carcinoma. Liver Int. 2007;27:735–741. doi: 10.1111/j.1478-3231.2007.01515.x. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK. Alcoholic liver disease and methionine metabolism. Sem Liver Dis. 2009;29:155–165. doi: 10.1055/s-0029-1214371. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK. Methionine metabolic pathway in alcoholic liver injury. Curr Opin Clin Nutr Metab Care. 2013;16:89–95. doi: 10.1097/MCO.0b013e32835a892a. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Mailliard ME, Baldwin CR, Beckenhauer HC, Sorrell MF, Tuma DJ. Betaine attenuates alcoholic steatosis by restoring phosphatidylcholine generation via the phosphatidylethanolamine methyltransferase pathway. J Hepatol. 2007;46:314–321. doi: 10.1016/j.jhep.2006.08.024. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Todero SL, Moats JC, Harris RM, Osna NA, Thomes PG, Tuma DJ. Alcohol consumption decreases rat hepatic creatine biosynthesis via altered guanidinoacetate methyltransferase activity. Alcohol Clin Exp Res. 2014a;38:641–648. doi: 10.1111/acer.12306. [DOI] [PubMed] [Google Scholar]
- Kharbanda KK, Todero SL, Thomes PG, Orlicky DJ, Osna NA, French SW, Tuma DJ. Increased methylation demand exacerbates ethanol-induced liver injury. Exp Mol Pathol. 2014b;97:49–56. doi: 10.1016/j.yexmp.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Kim JK, Noh JH, Jung KH, Eun JW, Bae HJ, Kim MG, Chang YG, Shen Q, Park WS, Lee JY, Borlak J, Nam SW. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology. 2013;57:1055–1067. doi: 10.1002/hep.26101. [DOI] [PubMed] [Google Scholar]
- Kirpich IA, Solovieva NV, Leikhter SN, Shidakova NA, Lebedeva OV, Sidorov PI, Bazhukova TA, Soloviev AG, Barve SS, McClain CJ, Cave M. Probiotics restore bowel flora and improve liver enzymes in human alcohol-induced liver injury: a pilot study. Alcohol. 2008;42:675–678. doi: 10.1016/j.alcohol.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirpich IA, Feng W, Wang Y, Liu Y, Barker DF, Barve SS, McClain CJ. The type of dietary fat modulates intestinal tight junction integrity, gut permeability, and hepatic toll-like receptor expression in a mouse model of alcoholic liver disease. Alcohol Clin Exp Res. 2012;36:835–846. doi: 10.1111/j.1530-0277.2011.01673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirpich IA, Feng W, Wang Y, Liu Y, Beier JI, Arteel GE, Falkner KC, Barve SS, McClain CJ. Ethanol and dietary unsaturated fat (corn oil/linoleic acid enriched) cause intestinal inflammation and impaired intestinal barrier defense in mice chronically fed alcohol. Alcohol. 2013;47:257–264. doi: 10.1016/j.alcohol.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatsky AL, Chartier D, Udaltsova N, Gronningen S, Brar S, Friedman GD, Lundstrom RJ. Alcohol drinking and risk of hospitalization for heart failure with and without associated coronary artery disease. Am J Cardiol. 2005;96:346–351. doi: 10.1016/j.amjcard.2005.03.073. [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Koehler EM, Schouten JN, Hansen BE, van Rooij FJ, Hofman A, Stricker BH, Janssen HL. Prevalence and risk factors of non-alcoholic fatty liver disease in the elderly: results from the Rotterdam study. J Hepatol. 2012;57:1305–1311. doi: 10.1016/j.jhep.2012.07.028. [DOI] [PubMed] [Google Scholar]
- Kowdley KV, Belt P, Wilson LA, Yeh MV, Nenschwander-Tetri BA, Chalassani N, Sanyal AJ, Nelson JE. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology. 2012;55:77–85. doi: 10.1002/hep.24706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladero JM, Martinez C, Garcia-Martin E. Polymorphisms of the glutathione S-transferases mu-1 (GSTM1) and theta-1 (GSTT1) and the risk of advanced alcoholic liver disease. Scand J Gastroenterol. 2005;40:348–353. doi: 10.1080/00365520510012109. [DOI] [PubMed] [Google Scholar]
- Lahmek P, Michel L, Diviné C, Meunier N, Pham B, Cassereau C, Aussel C, Aubin HJ. Ethyl glucuronide for detecting alcohol lapses in patients with an alcohol use disorder. J Addict Med. 2012;6:35–38. doi: 10.1097/ADM.0b013e3182249b93. [DOI] [PubMed] [Google Scholar]
- Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, Gimeno RE. Expression, regulation, and triglyceride hydrolase activity of Adiponutrin family members. J Lipid Res. 2005;46:2477–2487. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- Lakner AM, Steuerwald NM, Walling TL, Ghosh S, Li T, McKillop IH, Russo MW, Bonkovsky HL, Schrum LW. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology. 2012;56:300–310. doi: 10.1002/hep.25613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarević AM, Nakatani S, Nesković AN, Marinković J, Yasumura Y, Stojicić D, Miyatake K, Bojić M, Popović AD. Early changes in left ventricular function in chronic asymptomatic alcoholics: relation to the duration of heavy drinking. J Am Coll Cardiol. 2000;35:1599–1606. doi: 10.1016/s0735-1097(00)00565-9. [DOI] [PubMed] [Google Scholar]
- Lazo M, Hernaez R, Bonekamp S, Kamel IR, Brancati FL, Guallar E, Clark JM. Non-alcoholic fatty liver disease and mortality among US adults: prospective cohort study. BMJ. 2011;343:d6891. doi: 10.1136/bmj.d6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, Koteish A, Brancati FL, Clark JM. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol. 2013;178:38–45. doi: 10.1093/aje/kws448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, Seo YS, Um SH, Won NH, Yoo H, Jung ES, Kwon YD, Park S, Keum B, Kim YS, Yim HJ, Jeen YT, Chun HJ, Kim CD, Ryu HS. Usefulness of non-invasive markers for predicting significant fibrosis in patients with chronic liver disease. J Korean Med Sci. 2010;25:67–74. doi: 10.3346/jkms.2010.25.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevy CM, Moroianu SA. Nutritional aspects of alcoholic liver disease. Clin Liver Dis. 2005;9:67–81. doi: 10.1016/j.cld.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Lefkowitch JH, Grossman ME. Hepatic pathology in porphyria cutanea tarda. Liver. 1983;3:19–29. doi: 10.1111/j.1600-0676.1983.tb00846.x. [DOI] [PubMed] [Google Scholar]
- Leon DA, Shkolnikov VM, McKee M. Alcohol and Russian mortality: a continuing crisis. Addiction. 2009;104:1630–1636. doi: 10.1111/j.1360-0443.2009.02655.x. [DOI] [PubMed] [Google Scholar]
- Liang W, Chikritzhs TN, Pascal R, Binns CW. Mortality rate of alcoholic liver disease and risk of hospitalisation for alcoholic liver cirrhosis, alcoholic hepatitis, and alcoholic liver failure in Australia between 1993 and 2005. Int Med J. 2010;41:34–41. doi: 10.1111/j.1445-5994.2010.02279.x. [DOI] [PubMed] [Google Scholar]
- Liangpunsakul S, Qi R, Crabb DW, Witzmann F. Relationship between alcohol drinking and aspartate aminotransferase:alanine aminotransferase (AST:ALT) ratio, mean corpuscular volume (MCV), gamma-glutamyl transpeptidase (GGT), and apolipoprotein A1 and B in the U.S. population. J Stud Alcohol Drugs. 2010;71:249–252. doi: 10.15288/jsad.2010.71.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS. Pathogenesis and early diagnosis of alcoholic liver injury. N Engl J Med. 1978;298:888–892. doi: 10.1056/NEJM197804202981606. [DOI] [PubMed] [Google Scholar]
- Lieber CS. Metabolic effects of ethanol and its interaction with other drugs, hepatoxic agents, vitamins and carcinogens: a 1988 update. Semin Liver Dis. 1988a;8:47–68. doi: 10.1055/s-2008-1040528. [DOI] [PubMed] [Google Scholar]
- Lieber CS. Biochemical and molecular basis of alcohol-induced injury to liver and other tissues. N Engl J Med. 1988b;319:1639–1644. doi: 10.1056/NEJM198812223192505. [DOI] [PubMed] [Google Scholar]
- Lieber CS. Ethanol metabolism, cirrhosis and alcoholism. Clin Chim Acta. 1997;257:59–84. doi: 10.1016/s0009-8981(96)06434-0. [DOI] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM. Ethanol oxidation by hepatic microsomes: adaptive increase after ethanol feeding. Science. 1968;162:917–918. doi: 10.1126/science.162.3856.917. [DOI] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM. Hepatic microsomal ethanol oxidizing system. In vitro characteristics and adaptive properties in vivo. J Biol Chem. 1970;245:2505–2512. [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM. Hepatotoxicity of ethanol. J Hepatol. 1991;12:394–398. doi: 10.1016/0168-8278(91)90846-4. [DOI] [PubMed] [Google Scholar]
- Lieber CS, DeCarli LM, Rubin E. Sequential production of fatty liver, hepatitis and cirrhosis in sub-human primates fed ethanol with adequate diets. Proc Natl Acad Sci U S A. 1975;72:437–441. doi: 10.1073/pnas.72.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber CS, Robins SJ, Leo MA. Hepatic phosphatidylethanolamine methyltransferase activity is decreased by ethanol and increased by phosphatidylcholine. Alcohol Clin Exp Res. 1994;18:592–595. doi: 10.1111/j.1530-0277.1994.tb00915.x. [DOI] [PubMed] [Google Scholar]
- Lieber CS, Weiss D, Morgan T, Paronetto F. Aspartate aminotransferase (AST) to platelet ratio index (APRI) in patients with alcoholic liver fibrosis. Am J Gastroenterol. 2006;101:1500–1508. doi: 10.1111/j.1572-0241.2006.00610.x. [DOI] [PubMed] [Google Scholar]
- Liu H, Li J, French BA, Morgan T, Tillman B, French SW. TLR3/4 signaling is mediated via the NFκB-CXCR4/7 pathway in human alcoholic hepatitis and non-alcoholic hepatitis which formed Mallory–Denk bodies. Exp Mol Pathol. 2014a;97:234–240. doi: 10.1016/j.yexmp.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li J, Tillman B, French BA, French SW. Ufmylation and FATylation pathways are down regulated in human alcoholic and nonalcoholic steatohepatitis and mice fed DDC where Mallory–Denk bodies (MDBs) form. Exp Mol Pathol. 2014b;97:81–88. doi: 10.1016/j.yexmp.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Yang HI, Su J, Brenner D, Barrett-Connor E, Iloeje U, Chen CJ. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: a prospective cohort study. Am J Epidemiol. 2013;177:333–342. doi: 10.1093/aje/kws252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey MR, Mathurin P, Morgan TR. Alcoholic hepatitis. N Engl J Med. 2009;360:2758–2769. doi: 10.1056/NEJMra0805786. [DOI] [PubMed] [Google Scholar]
- Ludwig J, Viggiano TR, McGill DB, Oh BJ. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc. 1980;55:434–438. [PubMed] [Google Scholar]
- Maenhout TM, Poll A, Wuyts B, Lecocq E, Van Vlierberghe H, De Buyzere ML, Delanghe JR. Microheterogeneity of serum β-hexosaminidase in chronic alcohol abusers in a driver's license regranting program. Alcohol Clin Exp Res. 2013;37:1264–1270. doi: 10.1111/acer.12112. [DOI] [PubMed] [Google Scholar]
- Maher JJ, Friedman SL. Pathogenesis of hepatic fibrosis. In: Hall P, editor. Alcoholic Liver Disease Pathobiology, Epidemiology and Clinical Aspects. Edward Arnold; London: 1995. pp. 71–78. [Google Scholar]
- Maret W. Zinc coordination environments in proteins determine zinc functions. J Trace Elem Med Biol. 2005;19:7–12. doi: 10.1016/j.jtemb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Mariat D, Firmesse O, Levenez F, Guimarăes V, Sokol H, Doré J, Corthier G, Furet JP. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123. doi: 10.1186/1471-2180-9-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini GA, Bode CH. The epidemiology of cirrhosis of the liver. In: Engel A, Larsson T, editors. Alcoholic Cirrhosis and Other Toxic Hepatopathies. Nordiska Bokhandelns Forlag; Stockholm: 1970. pp. 315–335. [Google Scholar]
- Mathurin P, Moreno C, Samuel D, Dumortier J, Salleron J, Durand F, Castel H, Duhamel A, Pageaux GP, Leroy V, Dharancy S, Louvet A, Boleslawski E, Lucidi V, Gustot T, Francoz C, Letoublon C, Castaing D, Belghiti J, Donckier V, Pruvot FR, Duclos-Vallée JC. Early liver transplantation for severe alcoholic hepatitis. N Engl J Med. 2011;365:1790–1800. doi: 10.1056/NEJMoa1105703. [DOI] [PubMed] [Google Scholar]
- Mathurin P, Hadengue A, Bataller R, Addolorato G, Burra P, Burt A, Caballeria J, Cortez-Pinto H, Day CP, Forrest EH, Gual A, Leon DA, Lligoña A, Jepsen P, Mueller S, Pageaux GP, Roskams T, Seitz HK, Stickel F, Thursz M, Naveau S, Morgan T, Nevens F. European association for the study of liver clinical practical guidelines: management of alcoholic liver disease. J Hepatol. 2012;57(2):399–420. doi: 10.1016/j.jhep.2012.04.004. [DOI] [PubMed] [Google Scholar]
- McFarlin SK, Fals-Stewart W. Workplace absenteeism and alcohol use: a sequential analysis. Psychol Addict Behav. 2002;16:17–21. doi: 10.1037//0893-164x.16.1.17. [DOI] [PubMed] [Google Scholar]
- McKillop IH, Schrum LW. Role of alcohol in liver carcinogenesis. Semin Liver Dis. 2009;29:222–232. doi: 10.1055/s-0029-1214377. [DOI] [PubMed] [Google Scholar]
- Mendenhall CL. Alcoholic hepatitis. Clin Gastroenterol. 1981;10:417–444. [PubMed] [Google Scholar]
- Mendenhall C, Roselle GA, Gartside P, Moritz T. Relationship of protein calorie malnutrition to alcoholic liver disease: a reexamination of data from two Veterans Administration Cooperative Studies. Alcohol Clin Exp Res. 1995;19:635–641. doi: 10.1111/j.1530-0277.1995.tb01560.x. [DOI] [PubMed] [Google Scholar]
- Mezey E. Dietary fat and alcoholic liver disease. Hepatology. 1998;28:901–905. doi: 10.1002/hep.510280401. [DOI] [PubMed] [Google Scholar]
- Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47:S2–S6. doi: 10.1097/MCG.0b013e3182872f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohammad MK, Zhou Z, Cave M, Barve A, McClain CJ. Zinc and liver disease. Nutr Clin Pract. 2012;27:8–20. doi: 10.1177/0884533611433534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohri T, Nakajima M, Fukami T, Takamiya M, Aoki Y, Yokoi T. Human CYP2E1 is regulated by miR-378. Biochem Pharmacol. 2010;79:1045–1052. doi: 10.1016/j.bcp.2009.11.015. [DOI] [PubMed] [Google Scholar]
- Morojele NK, Kachieng'a MA, Mokoko E, Nkoko MA, Parry CD, Nkowane AM, Moshia KM, Saxena S. Alcohol use and sexual behaviour among risky drinkers and bar and she-been patrons in Gauteng province, South Africa. Soc Sci Med. 2006;62:217–227. doi: 10.1016/j.socscimed.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Mudd SH, Brosnan JT, Brosnan ME, Jacobs RL, Stabler SP, Allen RH, Vance DE, Wagner C. Methyl balance and transmethylation fluxes in humans. Am J Clin Nutr. 2007;85:19–25. doi: 10.1093/ajcn/85.1.19. [DOI] [PubMed] [Google Scholar]
- Murray KF, Carithers RL., Jr AASLD practice guidelines: evaluation of the patient for liver transplantation. Hepatology. 2005;41:1407–1432. doi: 10.1002/hep.20704. [DOI] [PubMed] [Google Scholar]
- Musso G. NAFLD: old issues and emerging concepts. Semin Liver Dis. 2012;32:1–2. doi: 10.1055/s-0032-1306426. [DOI] [PubMed] [Google Scholar]
- Nanji AA. Role of different dietary fatty acids in the pathogenesis of experimental alcoholic liver disease. Alcohol. 2004;34:21–25. doi: 10.1016/j.alcohol.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Nanji AA, French SW. Dietary linoleic acid is required for development of experimentally induced alcoholic liver injury. Life Sci. 1989;44:223–227. doi: 10.1016/0024-3205(89)90599-7. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Mendenhall CL, French SW. Beef fat prevents alcoholic liver disease in the rat. Alcohol Clin Exp Res. 1989;13:15–19. doi: 10.1111/j.1530-0277.1989.tb00276.x. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Khettry U, Sadrzadeh SM. Lactobacillus feeding reduces endotoxemia and severity of experimental alcoholic liver (disease) Proc Soc Exp Biol Med. 1994a;205:243–247. doi: 10.3181/00379727-205-43703. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Zhao S, Lamb RG, Dannenberg AJ, Sadrzadeh SM, Waxman DJ. Changes in cytochromes P-450, 2E1, 2B1, and 4A, and phospholipases A and C in the intragastric feeding rat model for alcoholic liver disease: relationship to dietary fats and pathologic liver injury. Alcohol Clin Exp Res. 1994b;18:902–908. doi: 10.1111/j.1530-0277.1994.tb00058.x. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Zhao S, Sadrzadeh SM, Dannenberg AJ, Tahan SR, Waxman DJ. Markedly enhanced cytochrome P450 2E1 induction and lipid peroxidation is associated with severe liver injury in fish oil-ethanol-fed rats. Alcohol Clin Exp Res. 1994c;18:1280–1285. doi: 10.1111/j.1530-0277.1994.tb00119.x. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Griniuviene B, Sadrzadeh SM, Levitsky S, McCully JD. Effect of type of dietary fat and ethanol on antioxidant enzyme mRNA induction in rat liver. J Lipid Res. 1995a;36:736–744. [PubMed] [Google Scholar]
- Nanji AA, Sadrzadeh SM, Yang EK, Fogt F, Meydani M, Dannenberg AJ. Dietary saturated fatty acids: a novel treatment for alcoholic liver disease. Gastroenterology. 1995b;109:547–554. doi: 10.1016/0016-5085(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Nanji AA, Jokelainen K, Tipoe GL, Rahemtulla A, Dannenberg AJ. Dietary saturated fatty acids reverse inflammatory and fibrotic changes in rat liver despite continued ethanol administration. J Pharmacol Exp Ther. 2001;299:638–644. [PubMed] [Google Scholar]
- Neuman MG. Mechanism of alcoholic liver disease. Clin Biochem. 2001;34:163–167. [Google Scholar]
- Neuman MG, Shear NH, Oh P, Zimmerman HJ. Increased hepatotoxicity from acetaminophen in human hepatocytes pretreated with ethanol in cell culture. Clin Invest Med. 1994;17(Suppl. B14) Abstract 85. [Google Scholar]
- Neuman MG, Shear NH, Bellentani S, Tiribelli C. Role of cytokines in ethanol-induced hepatocytotoxicity in HepG2 cells. Gastroenterology. 1998;114:157–169. doi: 10.1016/s0016-5085(98)70377-4. [DOI] [PubMed] [Google Scholar]
- Neuman MG, Monteiro M, Rehm Drug interactions between psychoactive substances and antiretroviral therapy in individuals infected with human immunodeficiency and hepatitis viruses. Drugs Use Misuse. 2006;41(10–12):1395–1463. doi: 10.1080/10826080600846235. http://dx.doi.org/10.1080/10826080600846235. [DOI] [PubMed] [Google Scholar]
- Neuman MG, Schmilowitz-Weiss H, Hilzenrat N, Bourliere M, Marcelin P, Trepo C, Mazulli T, Moussa G, Patel A, Baig AA, Cohen L. Markers of inflammation and fibrosis in alcoholic hepatitis and viral hepatitis C. Int J Hepatol. 2012a;2012:231210. doi: 10.1155/2012/231210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman MG, Schneider M, Nanau RM, Parry C. HIV-antiretroviral therapy induced liver, gastrointestinal, and pancreatic injury. Int J Hepatol. 2012b;2012:760706. doi: 10.1155/2012/760706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman MG, Schneider M, Nanau RM, Parry C. Alcohol consumption, progression of disease and other comorbidities, and responses to antiretroviral medication in people living with HIV. AIDS Res Treat. 2012c;2012:751827. doi: 10.1155/2012/751827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman MG, Schneider M, Nanau RM, Parry C, Chersich M. Public Health — Social and Behavioral Health. InTech Open Access Publisher; Rijeka: 2012d. The relationship between alcohol consumption and human immunodeficiency virus infection and risk behaviour: a systematic literature review of high-risk groups, with a focus on South Africa; pp. 243–292. [Google Scholar]
- Neuman MG, French SW, Casey CA, Kharbanda KK, Nanau RM, Rasineni K, McVicker BL, Kong V, Donohue TM., Jr Changes in the pathogenesis of alcohol-induced liver disease — preclinical studies. Exp Mol Pathol. 2013;95:376–384. doi: 10.1016/j.yexmp.2013.10.006. [DOI] [PubMed] [Google Scholar]
- Neuman MG, Cohen L, Zakhari S, Nanau RM, Mueller S, Schneider M, Parry C, Isip R, Seitz HK. Alcoholic liver disease: a synopsis of the Charles Lieber's Memorial Symposia 2009–2012. Alcohol Alcohol. 2014;49:373–380. doi: 10.1093/alcalc/agu021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson A, Bobak M, Murphy M, Rose R, Marmot M. Alcohol consumption and increased mortality in Russian men and women: a cohort study based on the mortality of relatives. Bull World Health Organ. 2005;83:812–819. [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science. 2012;336:1262–1267. doi: 10.1126/science.1223813. [DOI] [PubMed] [Google Scholar]
- Nøjgaard C, Johansen JS, Christensen E, Skovgaard LT, Price PA, Becker U. Serum levels of YKL-40 and PIIINP as prognostic markers in patients with alcoholic liver disease. J Hepatol. 2003;39:179–186. doi: 10.1016/s0168-8278(03)00184-3. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Tsutsumi M, Fukumara M, Tsuchischima M, Takase S. Use of serum carbohydrate-deficient transferrin values to exclude alcoholic hepatitis from nonalcoholic steatohepatitis: a pilot study. Alcohol Clin Exp Res. 2005;29:236S–239S. doi: 10.1097/01.alc.0000190659.85025.b3. [DOI] [PubMed] [Google Scholar]
- Orman ES, Odena G, Bataller R. Alcoholic liver disease: pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28(Suppl. 1):77–84. doi: 10.1111/jgh.12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Shea RS, Dasarathy S, McCullough AJ. Alcoholic liver disease. Hepatology. 2010;51:307–328. doi: 10.1002/hep.23258. [DOI] [PubMed] [Google Scholar]
- Osna NA, Clemens DL, Donohue TM., Jr Ethanol metabolism alters interferon gamma signaling in recombinant HepG2 cells. Hepatology. 2005;42:1109–1117. doi: 10.1002/hep.20909. [DOI] [PubMed] [Google Scholar]
- Osna NA, White RL, Todero S, McVicker BL, Thiele GM, Clemens DL, Tuma DJ, Donohue TM., Jr Ethanol-induced oxidative stress suppresses generation of peptides for antigen presentation by hepatoma cells. Hepatology. 2007;45:53–61. doi: 10.1002/hep.21442. [DOI] [PubMed] [Google Scholar]
- Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32:742–747. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- Preedy VR, Adachi J, Ueno Y, Ahmed S, Mantle D, Mullatti N, Rajendram R, Peters TJ. Alcoholic skeletal muscle myopathy: definitions, features, contribution of neuropathy, impact and diagnosis. Eur J Neurol. 2001;8:677–687. doi: 10.1046/j.1468-1331.2001.00303.x. [DOI] [PubMed] [Google Scholar]
- Reed T, Page WF, Viken RJ, Christian JC. Genetic predisposition to organ-specific endpoints of alcoholism. Alcohol Clin Exp Res. 1996;20:1528–1533. doi: 10.1111/j.1530-0277.1996.tb01695.x. [DOI] [PubMed] [Google Scholar]
- Rehm J, Room R, Monteiro M, Gmel G, Graham K, Rehn N, Sempos CT, Frick U, Jernigan D. Alcohol use. In: Ezzati M, Lopez AD, Rodgers A, Murray CJL, editors. Comparative Quantification of Health Risks Global and Regional Burden of Disease Attributable to Selected Major Risk Factors. Vol. 1. World Health Organization; Geneva: 2004. pp. 959–1108. [Google Scholar]
- Rensen SS, Slaats Y, Driessen A, Peutz-Kootstra CJ, Nijhuis J, Steffensen R, Greve JW, Buurman WA. Activation of the complement system in human non-alcoholic fatty liver disease. Hepatology. 2009;50:1809–1817. doi: 10.1002/hep.23228. [DOI] [PubMed] [Google Scholar]
- Roiu I, Birngruber CG, Spencer VC, Wollersen H, Dettmeyer R, Verhoff MA. A comparison of breath- and blood-alcohol test results from real-life policing situations: a one-year study of data from the Central Hessian Police District in Germany. Forensic Sci Int. 2013;232:125–130. doi: 10.1016/j.forsciint.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Rojkind M. Collagen metabolism in the liver. In: Hall P, editor. Alcoholic Liver Disease Pathobiology, Epidemiology and Clinical Aspects. Edward Arnold; London: 1985. pp. 90–112. [Google Scholar]
- Rossi E, Adams LA, Bulsara M, Jeffrey GP. Assessing liver fibrosis with serum marker models. Clin Biochem Rev. 2007;28:3–10. [PMC free article] [PubMed] [Google Scholar]
- Rubin E, Lieber CS. Fatty liver, alcoholic hepatitis and cirrhosis produced by alcohol in primates. N Engl J Med. 1974;290:128–135. doi: 10.1056/NEJM197401172900303. [DOI] [PubMed] [Google Scholar]
- Said A, Williams J, Holden J, Remington P, Musat A, Lucey MR. The prevalence of alcohol-induced liver disease and hepatitis C and their interaction in a tertiary care setting. Clin Gastroenterol Hepatol. 2004;2:928–934. doi: 10.1016/s1542-3565(04)00393-3. [DOI] [PubMed] [Google Scholar]
- Sancho-Bru P, Altamirano J, Rodrigo-Torres D, Coll M, Millán C, José Lozano J, Miquel R, Arroyo V, Caballería J, Ginès P, Bataller R. Liver progenitor cell markers correlate with liver damage and predict short-term mortality in patients with alcoholic hepatitis. Hepatology. 2012;55:1931–1941. doi: 10.1002/hep.25614. [DOI] [PubMed] [Google Scholar]
- Sandahl TD, Jepsen P, Thomsen KL, Vilstrup H. Incidence and mortality of alcoholic hepatitis in Denmark 1999–2008: a nationwide population based cohort study. J Hepatol. 2011;54:760–764. doi: 10.1016/j.jhep.2010.07.016. [DOI] [PubMed] [Google Scholar]
- Sander M, Irwin M, Sinha P, Naumann E, Kox WJ, Spies CD. Suppression of interleukin-6 to interleukin-10 ratio in chronic alcoholics: association with postoperative infections. Intensive Care Med. 2002;28:285–292. doi: 10.1007/s00134-001-1199-9. [DOI] [PubMed] [Google Scholar]
- Schaefer B, Rivas-Estilla AM, Meraz-Cruz N, Reyes-Romero MA, Hernandez-Nazara ZH, Dominguez-Rosales JA, Schuppan D, Greenwel P, Rojkind M. Reciprocal modulation of matrix metalloproteinase-13 and type I collagen genes in rat hepatic stellate cells. Am J Pathol. 2003;162:1771–1780. doi: 10.1016/S0002-9440(10)64312-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner F, Popper H. Alcoholic hepatitis in the spectrum of ethanol-induced liver injury. Scand J Gastroenterol Suppl. 1970;7:69–78. [PubMed] [Google Scholar]
- Schenker S, Halff GA. Nutritional therapy in alcoholic liver disease. Semin Liver Dis. 1993;13:196–199. doi: 10.1055/s-2007-1007349. [DOI] [PubMed] [Google Scholar]
- Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146:1513–1524. doi: 10.1053/j.gastro.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedat M, Van Niekerk A, Jewkes R, Suffla S, Ratele K. Violence and injuries in South Africa: prioritising an agenda for prevention. Lancet. 2009;374:1011–1022. doi: 10.1016/S0140-6736(09)60948-X. [DOI] [PubMed] [Google Scholar]
- Seeff LB, Cuccherini BA, Zimmerman HJ, Adler E, Benjamin SB. Acetaminophen hepatotoxicity in alcoholics. A therapeutic misadventure. Ann Intern Med. 1986;104:399–404. doi: 10.7326/0003-4819-104-3-399. [DOI] [PubMed] [Google Scholar]
- Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S, Zakrzewski J, Blochin E, Rose A, Bogunovic D, Polsky D, Wei J, Lee P, Belitskaya-Levy I, Bhardwaj N, Osman I, Hernando E. Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci U S A. 2009;106:1814–1819. doi: 10.1073/pnas.0808263106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seitz HK, Stickel F. Alcoholic liver disease in the elderly. Clin Geriatr Med. 2007a;23:905–921. doi: 10.1016/j.cger.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Stickel F. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer. 2007b;7:599–612. doi: 10.1038/nrc2191. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Suter P. Ethanol toxicity and nutritional status. In: Kotsonis FM, McKey M, editors. Nutritional Toxicology. Taylor and Francis; London: 2001. pp. 122–154. [Google Scholar]
- Seitz HK, Garro AJ, Lieber CS. Effect of chronic ethanol ingestion on intestinal metabolism and mutagenicity of benzo(α)pyrene. Biochem Biophys Res Commun. 1978;85:1061–1066. doi: 10.1016/0006-291x(78)90650-2. [DOI] [PubMed] [Google Scholar]
- Seitz HK, Boesche J, Czygan P, Veith S, Kommerell B. Microsomal ethanol oxidation in the colonic mucosa of the rat. Effect of chronic ethanol ingestion. Naunyn Schmiedeberg's Arch Pharmacol. 1982;320:81–84. doi: 10.1007/BF00499078. [DOI] [PubMed] [Google Scholar]
- Seth D, Daly AK, Haber PS, Day CP. PNPLA3 — a case in point linking genetic susceptibility for alcoholic and non-alcoholic liver disease. Hepatology. 2010;51:1463–1465. doi: 10.1002/hep.23606. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Söderberg C, Stål P, Askling J, Glaumann H, Lindberg G, Marmur J, Hultcrantz R. Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology. 2010;51:595–602. doi: 10.1002/hep.23314. [DOI] [PubMed] [Google Scholar]
- Song G, Sharma AD, Roll GR, Ng R, Lee AY, Blelloch RH, Frandsen NM, Willenbring H. MicroRNAs control hepatocyte proliferation during liver regeneration. Hepatology. 2010;51:1735–1743. doi: 10.1002/hep.23547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorbi D, Boyriton J, Lindro KD. The ratio of aspartate aminotransferase to alanine aminotransferase: potential value in differentiating nonalcoholic steatohepatitis from alcoholic liver disease. Am J Gastroenterol. 1999;94:1018–1022. doi: 10.1111/j.1572-0241.1999.01006.x. [DOI] [PubMed] [Google Scholar]
- Spies CD, von Dossow V, Eggers V, Jetschmann G, El-Hilali R, Egert J, Fischer M, Schroder T, Hoflich C, Sinha P, Paschen C, Mirsalim P, Brunsch R, Hopf J, Marks C, Wernecke KD, Pragst F, Ehrenreich H, Muller C, Tonnesen H, Oelkers W, Rohde W, Stein C, Kox WJ. Altered cell-mediated immunity and increased postoperative infection rate in long-term alcoholic patients. Anesthesiology. 2004;100:1088–1100. doi: 10.1097/00000542-200405000-00010. [DOI] [PubMed] [Google Scholar]
- Stewart SF, Leathart JB, Chen Y, Daly AK, Rolla R, Vay D, Mottaran E, Vidali M, Albano E, Day CP. The valine–alanine manganese superoxide dismutase polymorphism is not associated with increased oxidative stress or susceptibility to advanced alcoholic liver disease. Hepatology. 2002;36:1355–1360. doi: 10.1053/jhep.2002.36940. [DOI] [PubMed] [Google Scholar]
- Stickel F, Hampe J. Genetic determinants of alcoholic liver disease. Gut. 2012;61:150–159. doi: 10.1136/gutjnl-2011-301239. [DOI] [PubMed] [Google Scholar]
- Stickel F, Osterreicher CH. The role of genetic polymorphisms in alcoholic liver disease. Alcohol Alcohol. 2006;41:209–224. doi: 10.1093/alcalc/agl011. [DOI] [PubMed] [Google Scholar]
- Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, Rietschel M, Schafmayer C, Braun F, Hinrichsen H, Günther R, Arlt A, Seeger M, Müller S, Seitz HK, Soyka M, Lerch M, Lammert F, Sarrazin C, Kubitz R, Häussinger D, Hellerbrand C, Bröring D, Schreiber S, Kiefer F, Spanagel R, Mann K, Datz C, Krawczak M, Wodarz N, Völzke H, Hampe J. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in Caucasians. Hepatology. 2011;53:86–95. doi: 10.1002/hep.24017. [DOI] [PubMed] [Google Scholar]
- Stinson FS, Grant BF, Dufour MC. The critical dimension of ethnicity in liver cirrhosis mortality statistics. Alcohol Clin Exp Res. 2001;25:1181–1187. [PubMed] [Google Scholar]
- Tannafel A, Denk H, Dienes HP, Langner C, Schirmacher P, Trauner M, Flott-Rahmel B. Histopathological diagnosis of non-alcoholic and alcoholic fatty liver disease. Vichow Arch. 2011;458:511–523. doi: 10.1007/s00428-011-1066-1. [DOI] [PubMed] [Google Scholar]
- Thompson KJ, Swan RZ, Walling TL, Iannitti DA, McKillop IH, Sindram D. Obesity, but not ethanol, promotes tumor incidence and progression in a mouse model of hepatocellular carcinoma in vivo. Surg Endosc. 2013;27:2782–2791. doi: 10.1007/s00464-013-2808-8. [DOI] [PubMed] [Google Scholar]
- Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet. 2010;42:21–23. doi: 10.1038/ng.488. [DOI] [PubMed] [Google Scholar]
- Tsukamoto H, Machida K, Dynnyk A, Mkrtchyan H. “Second hit” models of alcoholic liver disease. Semin Liver Dis. 2009;29:178–187. doi: 10.1055/s-0029-1214373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsui M, Tanaka N, Kawakubo M, Sheena DE, Horiuchi A, Kamatsu M, Nagaya T, Joshita S, Imemura T, Ichijo T, Matsumoto A, Yoshizawa K, Aoyama T, Tanaka E, Sno K. Serum fragmented cytokeratin 18 levels reflect the histologic activity score of nonalcoholic fatty liver disease more accurately than serum alanine aminotransferase levels. J Clin Gastroenterol. 2010;44:440–447. doi: 10.1097/MCG.0b013e3181bdefe2. [DOI] [PubMed] [Google Scholar]
- Uchida T, Kao H, Quispe-Sjogren M, Peters RL. Alcoholic foamy degeneration—a pattern of acute alcoholic injury of the liver. Gastroenterology. 1983;84:683–692. [PubMed] [Google Scholar]
- Verster JC, Van Der Maarel MA, McKinney A, Olivier B, De Haan L. Driving during alcohol hangover among Dutch professional truck drivers. Traffic Inj Prev. 2014a;15:434–438. doi: 10.1080/15389588.2013.833329. [DOI] [PubMed] [Google Scholar]
- Verster JC, Bervoets AC, de Klerk S, Vreman RA, Olivier B, Roth T, Brookhuis KA. Effects of alcohol hangover on simulated highway driving performance. Psychopharmacology (Berl) 2014b;231:2999–3008. doi: 10.1007/s00213-014-3474-9. [DOI] [PubMed] [Google Scholar]
- Wang Y, Millonig G, Nair J, Patsenker E, Stickel F, Mueller S, Bartsch H, Seitz HK. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology. 2009;50:453–461. doi: 10.1002/hep.22978. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kirpich I, Liu Y, Ma Z, Barve S, McClain CJ, Feng W. Lactobacillus rhamnosus GG treatment potentiates intestinal hypoxia-inducible factor, promotes intestinal integrity and ameliorates alcohol-induced liver injury. Am J Pathol. 2011;179:2866–2875. doi: 10.1016/j.ajpath.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Liu Y, Kirpich I, Ma Z, Wang C, Zhang M, Suttles J, McClain C, Feng W. Lactobacillus rhamnosus GG reduces hepatic TNFalpha production and inflammation in chronic alcohol-induced liver injury. J Nutr Biochem. 2013;24:1609–1615. doi: 10.1016/j.jnutbio.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe J, Hayashi S, Kawajiri K. Different regulation and expression of the human CYP2E1 gene due to the RsaI polymorphism in the 50-flanking region. J Biochem. 1994;116:321–326. doi: 10.1093/oxfordjournals.jbchem.a124526. [DOI] [PubMed] [Google Scholar]
- Wechsler H, Austin SB. Binge drinking: the five/four measure. J Stud Alcohol. 1998;59:122–124. doi: 10.15288/jsa.1998.59.122. [DOI] [PubMed] [Google Scholar]
- Weiner FR, Eskries DS, Compton KV, Orrego H, Zern M. Haplotype analysis of a type 1 collagen gene and its association with alcoholic cirrhosis in man. Mol Aspects Med. 1988;10:159–168. doi: 10.1016/0098-2997(88)90020-9. [DOI] [PubMed] [Google Scholar]
- Wiese JG, Shlipak MG, Browner WS. The alcohol hangover. Ann Intern Med. 2000;132:897–902. doi: 10.7326/0003-4819-132-11-200006060-00008. [DOI] [PubMed] [Google Scholar]
- Williams AL, Hoofnagle JH. Ratio of serum aspartate aminotranferase in chronic hepatitis. Relationship to cirrhosis. Gastroenterology. 1988;95:734–739. doi: 10.1016/s0016-5085(88)80022-2. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wang X, Wen J, Ye Z, Li Q, He M, Lu B, Ling C, Wu S, Hu R. Prevalence of non-alcoholic fatty liver disease and its relation to hypoadiponectinaemia in the middle-aged and elderly Chinese population. Arch Med Sci. 2011;4:665–672. doi: 10.5114/aoms.2011.24137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi ZM, Stepanova M, Afendy M, Fang Y, Younossi Y, Mir H, Srishord M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9:524–530. doi: 10.1016/j.cgh.2011.03.020. [DOI] [PubMed] [Google Scholar]
- Zakhari S, Li TK. Determinants of alcohol use and abuse: impact of quantity and frequency patterns on liver disease. Hepatology. 2007;46:2032–2039. doi: 10.1002/hep.22010. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Wang L, Song Z, Saari JT, McClain CJ, Kang YJ. Zinc supplementation prevents alcoholic liver injury in mice through attenuation of oxidative stress. Am J Pathol. 2005;166:1681–1690. doi: 10.1016/S0002-9440(10)62478-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou SS, Zhou YM, Li D, Lun YZ. Dietary methyl-consuming compounds and metabolic syndrome. Hypertens Res. 2011;34:1239–1245. doi: 10.1038/hr.2011.133. [DOI] [PubMed] [Google Scholar]
- Zimmerman HJ. The evolution of alcoholic cirrhosis. Med Clin N Am. 1955;39:249–251. doi: 10.1016/s0025-7125(16)34773-3. [DOI] [PubMed] [Google Scholar]
- Zimmerman HJ. Effects of alcohol on other hepatotoxins. Alcohol Clin Exp Res. 1986;10:3–15. doi: 10.1111/j.1530-0277.1986.tb05605.x. [DOI] [PubMed] [Google Scholar]
- Zimmerman HJ. Hepatotoxicity Adverse Effects of Drugs and Other Chemicals on the Liver. Second. Lippincott Williams & Wilkins; Philadelphia: 1999. [Google Scholar]
- Zimmerman HJ, Ishak KG. Nonalcoholic steatohepatitis and other forms of pseudoalcoholic liver disease. In: Hall P, editor. Alcoholic Liver Disease, Second ed Pathology and Pathogenesis. Edward Arnold; London: 1996. pp. 175–196. [Google Scholar]
- Zimmerman HJ, Maddrey WC. Acetaminophen (paracetamol) hepatotoxicity with regular intake of alcohol: analysis of instances of therapeutic misadventure. Hepatology. 1995;22:767–773. [PubMed] [Google Scholar]
- Ziol M, Handra-Luca A, Kettaneh A, Christidis C, Mal F, Kazemi F, de Lédinghen V, Marcellin P, Dhumeaux D, Trinchet JC, Beaugrand M. Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology. 2005;41:48–54. doi: 10.1002/hep.20506. [DOI] [PubMed] [Google Scholar]
- Zlotnik Y, Plakht Y, Aven A, Engel Y, Am NB, Ifergane G. Alcohol consumption and hangover patterns among migraine sufferers. J Neurosci Rural Pract. 2014;5:128–134. doi: 10.4103/0976-3147.131652. [DOI] [PMC free article] [PubMed] [Google Scholar]