

Abstract
Introduction
The endocannabinoid (eCB) system plays an important role in the control of mood, and its dysregulation has been implicated in several psychiatric disorders. Targeting the eCB system appears to represent an attractive and novel approach to the treatment of depression and other mood disorders. However, several failed clinical trials have diminished enthusiasm for the continued development of eCB-targeted therapeutics for psychiatric disorders, despite of the encouraging preclinical data and promising preliminary results obtained with the synthetic cannabinoid nabilone for treating post-traumatic stress disorder (PTSD).
Areas covered
This review describes the eCB system’s role in modulating cell signaling within the brain. There is a specific focus on eCB’s regulation of monoamine neurotransmission and the stress axis, as well as how dysfunction of this interaction can contribute to the development of psychiatric disorders. Additionally, the review provides discussion on compounds and drugs that target this system and might prove to be successful for the treatment of mood-related psychiatric disorders.
Expert opinion
The discovery of increasingly selective modulators of CB receptors should enable the identification of optimal therapeutic strategies. It should also maximize the likelihood of developing safe and effective treatments for debilitating psychiatric disorders.
1. Introduction
Cannabis sativa, or marijuana, is one of the most widely used illicit drugs, known to promote relaxation, euphoria, and a feeling of contentment [1–3]. Chronic use, however, can be accompanied by dysphoria, depressive mood, and increased anxiety [4] with the severity of symptoms being exacerbated by exposure to greater concentrations and increased frequency of usage [5]. Cessation of the clinical trial of the cannabinoid receptor antagonist, Rimonabant, due to serious side effects of depression and suicidal ideation brought increased attention to the role of the endocannabinoid (eCB) system on the regulation of mood [6]. CB receptors are widely distributed in the CNS and the eCB system modulates several key physiological systems involved in the control of mood. Recent studies have indicated that cannabis impacts stress-integrative neural circuits resulting in adaptive or maladaptive responses that may partially explain its biphasic effect on mood and emotional state. Targeting specific components of the eCB system may represent a novel and attractive approach to the treatment of multiple psychiatric disorders. The purpose of the present review is to (1) summarize current knowledge regarding cell signaling of the eCB system in brain, (2) present evidence for eCB regulation of monoamine transmitters and its implications for the regulation of mood, (3) discuss data showing the eCB system’s role in the regulation of the stress response and how its dysfunction could lead to psychiatric disorders, and (4) highlight compounds, in various stages of development, that target this system and may provide therapeutic benefit for the treatment of mood-related psychiatric disorders.
2. Principles of Endocannabinoid Signaling
The first pharmacologically active compound identified in Cannabis sativa, Δ-9-tetrahydrocannabinol (THC), was characterized in 1964 and has since been established as its primary psychoactive component [7, 8]. Since its discovery, over 100 additional active components of Cannabis sativa have been identified [8]. The first cannabinoid receptor, CB1r, was identified in 1988 and a second receptor, CB2r, was characterized in 1993 [3, 9]. Both are Gi/o protein-coupled receptors with distinct distributions in the body [10]. CB1 receptors are one of the most abundant G protein coupled receptors (GPCR) in the brain and their activation most commonly results in the inhibition of neurotransmitter release [10, 11]. CB2 receptors are most prevalent in the immune system [10, 12]; however, recent studies suggest a presence in the central nervous system as well, showing CB2 receptor localization in the hippocampus, substantia nigra, periaqueductal gray matter, and parvocellular reticular nucleus [13, 14]. Following the identification of the CB receptors, endogenous cannabinoid ligands, or endocannabinoids (eCBs), were discovered. The first was N-arachidonylethanolamide (AEA), which was named “anandamide” after the Sanskrit word meaning “bliss” [3, 15]. Another well characterized eCB is 2-arachidonoylglycerol (2-AG), first isolated from canine intestines by Mechoulam et al. in 1995 [16]. It is now generally accepted that 2-AG is a full CB1r and CB2r agonist, whereas AEA, which is less potent, is a partial agonist [17]. Initially, the mechanism proposed for eCB release involved a depolarization-induced event followed by retrograde signaling and binding of the endogenous ligand to presynaptically distributed receptors [10, 18]. New evidence suggests that eCB can regulate synaptic transmission via non-retrograde and autocrine mechanisms, with CB1 receptors having been discovered postsynaptically [19]. Furthermore, eCBs can bind and activate transient receptor potential vanilloid receptor type 1 (TRPV1) receptors [10].
The rate of eCB synthesis and degradation determines their signaling profiles. Two primary mechanisms are known to be responsible for 2-AG synthesis: increases in intracellular Ca2+ via postsynaptic depolarization and activation of phospholipase C β (PLCβ) via stimulation of group I metabotropic glutamate receptors. PLCβ forms diacylglycerol from the hydrolysis of phosphatidylinositol, which diacylglycerol lipase α (DGLα) then converts to 2-AG [10]. Monoacylglycerol lipase (MGL) is the main enzyme responsible for breaking down 2-AG, rendering it inactive and thus controlling the strength and duration of its modulatory activity [10, 20]. The synthesis and degradation of AEA is more complex. Though it is known that increases in intracellular Ca2+ and postsynaptic depolarization stimulate AEA formation, the mechanism underlying this process has yet to be elucidated [21]. N-acyl-phosphatidylethanolamine-hydrolyzing phospholipase-D (NAPE-PLC) has been identified as a contributor to AEA synthesis, but other synthetic pathways have also been reported [22]. AEA is primarily degraded by fatty acid amide hydrolase (FAAH) and, similar to MGL for 2-AG, FAAH controls the spatiotemporal profile for AEA signaling [10]. In contrast to the activity-dependent classical eCB signaling, tonic eCB release has been observed in several brain regions [10].
3. Targeting the Endocannabinoid System
There are a wide variety of ways in which the eCB system can be targeted and modulated (Table 1). The most direct method is by utilizing CB1r or CB2r agonists and antagonists to increase or decrease eCB signaling, respectively. Numerous selective and nonselective agonists and antagonists have been synthesized and characterized (Table 1) and have been useful tools in elucidating the role of the eCB system. Several of these compounds have advanced to clinical trials over the past decade or two, though predominantly they have been tested for the treatment of pain, inflammation, neurodegenerative disorders, nausea, obesity, and nicotine and alcohol dependence (reviewed in [23]).
Table 1.
Summary of drugs and compounds that are known to target the eCB system. Each class of drug is described with respect to its putative target and effect on eCB signaling.
 |
 |
eCB signaling can also be modified by targeting the catabolic and metabolic enzymes of AEA and 2-AG (Table 1). By inhibiting FAAH and MGL, eCB levels can be increased, allowing for greater signaling to occur. Conversely, by inhibiting eCB-synthesis, eCB levels are decreased resulting in less signaling. Finally, the eCB degradative enzymes are located intracellularly, so by blocking eCB uptake into the pre- or post-synaptic cell, eCB levels will remain high and signaling will be increased [3]. These methods allow more fine-tuning of the eCB system as opposed to the CB1r agonists and antagonists, and many of these approaches have also been utilized to test the effects of altered eCB signaling in preclinical models of psychiatric disorders. More recently, eCB-based drugs have begun clinical testing for the treatment of various psychiatric disorders, including schizophrenia, posttraumatic stress disorder (PTSD), and depression (Table 2).
Table 2.
Summary of the clinical progression of eCB-based drugs tested for the treatment of psychiatric disorders.
| TARGET | DRUG | FOR THE TREATMENT OF | CLINICAL PROGRESSION |
|---|---|---|---|
|
CB1r/CB2r AGONISTS |
Dronabinol | Schizophrenia [83] | Phase IIa |
| Fear extinction [156] | Phase II (completed) | ||
| THC | PTSD [157] | Phase IV | |
| Nabilone | PTSD (nightmares) [101] | Phase II | |
| Sativex (THC & CBD) |
Cannabis dependence w/ anxiety [158] | Phase I | |
| Multiple Sclerosis (cognitive function & mood) [159] |
Phase IV | ||
|
CB1r/CB2r ANTAGONISTS |
Cannabidiol (CBD) |
Schizophrenia (psychotic symptoms) [160] | Phase II (completed) |
| GWP42003 (CBD) | Schizophrenia [84] | Phase II | |
|
CB1r ANTAGONIST |
AVE-1625 | Schizophrenia (cotreatment w/ anti- psychotics) [161] |
Phase II (terminated) |
|
FAAH INHIBITORS |
URB597 | Schizophrenia [162] | Phase I |
| SSR411298 | Major Depressive Disorder [153] | Phase II |
4. The Endocannabinoid System and Mood Regulation
Recognition of the involvement of the eCB system in the regulation of mood and specifically its role in depression and anxiety arose, in part, from observations obtained from symptomatic individuals [24, 25]. A significant increase in CB1r density and efficacy was reported in the dorsolateral prefrontal cortex (PFC) of depressed suicide victims, suggesting that altered functioning of the eCB system in the PFC could contribute to depression [24, 26]. Several other studies examined dysregulation of the eCB system in individuals suffering from post-traumatic stress disorder (PTSD) and discovered that PTSD patients had greater CB1r availability throughout the brain as well as a significant decrease in AEA plasma concentrations [25, 27].
Genetic manipulations of the eCB system in animal models, particularly CB1r knockout (KO) mice, have provided insight into how eCB signaling affects behavior. CB1 KO mice exhibit an increase in passive behaviors compared to wild type (WT) mice in the forced swim test (FST), which is typically interpreted as a depressive phenotype [28]. They also show an increase in immobility time compared to WT mice in another animal model of depression, the tail suspension test (TST) [29]. Additionally, when exposed to chronic mild stress, KO mice develop anhedonia at a faster rate than WT mice, suggesting an increase in vulnerability to chronic stress [30]. In behavioral paradigms measuring anxiety-like behaviors, such as the elevated plus-maze, open-field test and light-dark box, CB1r KO mice exhibited increased anxiety-like behaviors [24]. CB1r KO mice display hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis and higher levels of circulating corticosterone following stressor exposure, a response that is also commonly observed in depressed patients [31].
Pharmacological approaches also support a role for the eCB system in mediating depression and anxiety. Acute administration of CB1r agonists decreases the amount of behavioral despair observed in the FST, and similar anti-depressant-like effects are observed in the FST and TST following chronic administration [32, 33]. In support of this, injection of CB1r agonists directly into brain regions that are known to be involved with emotion reduce the depressive phenotype [24]. Consistent with these preclinical findings, Rimonabant, a CB1r antagonist originally intended as an anti-obesity drug was withdrawn from clinical trials due to significant undesirable side effects including depression and suicidal ideation [6]. Concerns about mood-altering side effects resulted in the withdrawal of several other CBr antagonists from clinical trials, including Taranabant and Otenabant from phase III trials and Ibipinabant and Surinabant from phase II trials [34]. These results provide evidence for a potential protective role of the eCB system in the development and treatment of depression and anxiety.
It has been postulated that cognitive impairment is associated with various psychiatric disorders [35]. Work by Robbe and Buzsaki has shown that cannabinoid administration causes memory impairment by altering the synchrony and temporal coordination in both the hippocampus and the thalamocortical region, and this impairment represents another potential mechanism for cannabinoid involvement in psychiatric disorders [36, 37]. Recent studies have also shown that the eCB system, particularly via CB2r actions on glial cells and microglia, interacts with the immune system whereby inflammation produces an increase in 2-AG production [38]. Additionally, various pro-inflammatory cytokines that are known to be upregulated in depression are reduced following cannabinoid treatment as well as antidepressants [38]. While this link between CB2r and the inflammatory response in depression and other psychiatric disorders should not be overlooked, for the scope of this review, the remainder of the paper will focus on how the eCB system modulates monoamine transmission and the stress response.
Putative mechanisms underlying a neuroprotective role of the eCB system arise from studies showing that increases in eCB signaling can stimulate neurogenesis [24, 39, 40] by increasing brain-derived neurotrophic factor (BDNF) levels [41]. As decreased expression of hippocampal BDNF has been linked to a depression, agents that increase eCB signaling may stimulate hippocampal neurogenesis thereby engaging neural circuitry mediating the regulation of mood [24, 39, 40]. Interestingly, THC administration has also been shown to increase hippocampal BDNF levels [41]. Compounds that increase eCB neurotransmission have also been shown to attenuate the neuroendocrine response to stress and increase adaptive coping behaviors [42]. This effect is mediated, in part, by eCB modulation of noradrenergic circuitry [24]. Moreover, the ability of the eCB system to more broadly affect monoaminergic neurotransmission may underlie, in part, cannabinoids effects on mood. For example, FAAH inhibitors and CB1r antagonists enhance serotonergic neurotransmission [32, 33], CB1r activation can stimulate the release of norepinephrine (NE) and cannabinoid receptor agonists stimulate DA efflux in the cortex [33]. These findings indicate that eCBs may be neuroprotectective, not only with respect to neurodegeneration but also against the development of psychiatric disorders. The next section of the review will focus on evidence supporting a role for the eCB system in the modulation of monoaminergic systems and the HPA axis.
5. The Endocannabinoid System and Monoamines
5.1 eCB and 5-HT
Serotonin (5-HT) is one of the major monoamines implicated in depressive disorders and selective-serotonin reuptake inhibitors (SSRIs) are a commonly prescribed antidepressant [8]. Recent research shows that the eCB system is positioned to modulate serotonergic transmission. Dorsal Raphe (DR)-5-HT neurons contain CB1r and the synthetic and catabolic eCB enzymes, suggesting that the eCB system is positioned to modulate DR-5-HT signaling [43]. Electrophysiological studies have shown that eCBs are synthesized and released in an activity-dependent fashion within the DR [44, 45]. In vivo extracellular recordings indicate that administration of the CB1r agonist, WIN 55,212-2, and FAAH inhibitors result in increased DR-5-HT firing [32, 33]. Studies have also shown that eCB-induced increases in serotonergic signaling involve CB1r activation in the PFC, as lesions of the PFC abrogate eCB-induced excitatory responses in DR-5HT neurons [32]. In vitro studies provide a more direct role for eCB regulation of the DR-5-HT system. Several studies have shown that CB1r stimulation and administration of WIN 55,212-2 decrease glutamatergic signaling in DR-5-HT neurons [44, 45]; however, other findings indicate that eCB tone regulates DR-5-HT signaling by modulating GABAergic inputs to serotonergic neurons [46]. Neuroanatomical studies examining the synaptic relationship of CB1r with GABAergic and glutamatergic neurons are needed to better define sites of action of eCBs in the modulation of DR-5-HT signaling [46].
The 5-HT system plays a role in the neuroendocrine response to stress and eCB modulation of 5-HT circuits impact the regulation of mood [43]. 5-HT1A receptor activation decreases stress-induced ACTH and corticosterone production, and pharmacological antagonism of these receptors enhances the stress response and contributes to an anxiety phenotype [47, 48]. For example, stress-induced increases in anxiety-like behaviors are attenuated by cannabidiol (CBD) administration and this involves 5-HT neurotransmission as WAY100635, a 5-HT1A receptor antagonist, prevents the anxiolytic effect [49]. Additionally, in an adolescent mouse model of social withdrawal, hippocampal CB1 and 5-HT1A receptor densities were increased and HPA stress responses were exaggerated, further illustrating how the eCB and 5-HT systems are both involved in anxiety-like behaviors [50]. Further, one study using micro-positron emission tomography observed that both non-stressed CB1 KO mice and chronically stressed wild type mice exhibited similar decreases in 5-HT transporter levels, showing an interplay between the stress, 5-HT, and eCB systems in the development of mood disorders [51]. In contrast, chronic exposure to cannabinoids may elevate stress responses via an increase in 5-HT2A receptor expression in the paraventricular nuclei (PVN) resulting in increased production of prolactin and corticosterone [52]. Taken together, these data illustrate that the eCB system modulates serotonergic neurotransmission to impact behavior but the extent to which the exposure produces an anxiogenic or depressive response depends on duration of exposure and the involvement of stress-integrative circuitry.
5.2 eCB and NE
High levels of NE have been correlated with an increased duration of remission in previously depressed patients, implicating a potentially protective role of NE [53, 54]. It is well accepted that NE signaling is important in the pathophysiology of depression [54] and compounds that increase NE levels, such as serotonin-norepinephrine reuptake inhibitors (SNRIs), tricyclic antidepressants and MAO inhibitors are effective antidepressants. The locus coeruleus (LC) provides NE to the PFC, and high resolution neuroanatomical studies have demonstrated co-existence of CB1r with noradrenergic axon terminals in this brain region [55]. CB1 receptors are localized both pre- and post-synaptically in the LC [56]. Most of the pre-synaptic CB1r were localized to symmetric synapses, indicating that they are most likely regulating GABAergic transmission [54]. The presence of CB1r in noradrenergic neurons [56] further suggests that the eCB system may modulate noradrenergic activity directly without pre-synaptic modulation of amino acid signaling [54], potentially acting as a subsequent brake on LC activation. The opposing effects of CB1 receptors on noradrenergic terminals decreasing NE release versus CB1 receptors on GABAergic and serotonergic terminals increasing NE release also demonstrates the importance of local eCB levels in alteration of monoamine neurotransmission [57].
Additional studies support eCB regulation of NE signaling. CB receptor agonists CP55940 and WIN 55,212-2 increase spontaneous firing and stimulate immediate early gene c-Fos expression in LC-NE neurons [58, 59]. Additionally, an increase in Fos expression was observed in dopaminergic neurons following treatment with CBr agonists; however, this increase was blocked by co-treatment with either an α1-AR antagonist or an α2-AR agonist, indicating that the CB agonist-induced increase in dopaminergic activation is likely due to LC-NE activation [54, 59]. The dose-dependent increase in LC-NE firing observed after both systemic and central CB agonist administration is blocked by co-administration with SR141716A, a CB1r antagonist [58, 60]. Interestingly, SR141716A administration by itself results in a decrease in LC activity, suggesting that tonic eCB production controls the LC under basal conditions [54]. Also, administration of a FAAH inhibitor increases the spontaneous firing rate of LC-NE neurons, supporting the notion of tonic eCB regulation of LC neurons [33]. Finally, CBs also alter NE neurotransmission in other noradrenergic circuits, such as the nucleus of the solitary tract (NTS), that influence limbic circuitry including the amygdala and the HPA axis via the hypothalamus [61]. As the NTS is involved in interoceptive responses to stress, a better understanding of how the eCB system is positioned to influence neuronal activity in this region would help elucidate how potential cannabinoid-based therapeutics might alter stress responses in humans.
Following CB exposure, increases in NE levels have been observed and may involve mechanisms other than disinhibition of LC noradrenergic neurons [62]. For example, in vitro studies have shown that CBs can inhibit monoamine oxidase (MAO) [63]. MAO metabolizes monoamine neurotransmitters, so inhibition would produce increased NE levels. CB-induced decreases in α2-AR expression in the LC have been observed which would result in an increase in NE release at postsynaptic targets [54]. Increases in NE efflux in the PFC have been observed following acute and chronic CB administration and pretreatment with SR141716A blocks CB-induced increases in NE levels [64, 65]. Taken together, these data illustrate alterations in NE signaling following CB administration in regions where dysregulation is associated with stress and depressive- and anxiety-like effects.
5.3 eCB and DA
There is evidence that the eCB system is also positioned to modulate and alter dopamine (DA) neurotransmission. Several eCBs, including 2-AG and AEA have been found in dopaminergic brain regions and CB1r expression has been observed in the substantia nigra (SN) and ventral tegmental area (VTA), as well as the dorsal and ventral striatum, and various other targets of dopamine innervation [3, 66–68]. Unlike serotonergic neurons in the DR and noradrenergic neurons in the LC, CB1r expression is not very prominent in tyrosine hydroxylase positive, dopamine-producing neurons [69]. eCB modulation of dopaminergic signaling most likely occurs indirectly via eCB modulation of glutamatergic or GABAergic neurons [69]. Exogenous cannabinoids also appear to affect dopaminergic transmission differently than eCBs. It has shown that cannabinoid agonists increase the spontaneous firing of dopaminergic cells within the VTA through CB1r signaling [69, 70]. In olfactory-bulbectomized rats, which is an animal model of depression, self-administration of WIN 55,212-2 was increased, suggesting that the increase in the CB1r agonist administration could be an attempt to compensate for the decreased DA neurotransmission and rewarding effects observed in some animal models of depression [71]. Additionally, several other studies have similarly reported that cannabinoid agonists cause an increase in DA efflux within the cortex [72, 73]. As mentioned previously, increases in monoamine levels in the PFC represent one putative mechanism underlying the effectiveness of antidepressant therapeutics [74].
Dysregulation of dopaminergic neurotransmission has been implicated in major psychiatric disorders such as schizophrenia. Low levels of dopamine in the mesocortical pathway and increased levels of dopamine in the mesolimbic pathway are thought to underlie the pathophysiology of schizophrenia [75, 76]. Based on evidence showing that cannabinoids can increase dopaminergic signaling in the mesolimbic pathway, contributing to the positive symptoms of schizophrenia, a cannabinoid hypothesis of schizophrenia has been proposed [77]. This theory suggests that overeactivity of the eCB system could play a role in the development of schizophrenia [77]. In support of this, several studies have shown that paranoid schizophrenic patients, without a drug history, have heightened AEA levels in their cerebrospinal fluid (CSF) [78–80]. Also, postmortem examination of schizophrenic patients revealed that an increase in CB1r binding in several brain regions, including the dorsolateral PFC and cingulate cortex [77]. Additionally, many studies have indicated that adolescent cannabis use is correlated with an earlier onset of schizophrenic symptoms and more severe psychosis [81]. More recently, however, there has been an abundance of compelling data gathered that disproves this hypothesis. First, CB1r antagonism using Rimonabant proved unsuccessful in clinical trials at alleviating psychotic symptoms [77]. Second, AEA levels in the CRF of schizophrenic patients have been negatively correlated with psychosis [79, 80]. Electrophysiological and behavioral studies have shown that increasing AEA levels further increases the search for reward in mice, counteracting the effects of dopaminergic dysfunction [68]. Therefore, this increase in AEA levels might indicate a compensatory mechanism in schizophrenic patients, since it is thought that alterations in reward-related processes could contribute to the emotional disturbances seen in schizophrenic individuals [68]. Finally, direct measurement of CB1r expression in the dorsolateral PFC showed a decrease in receptor density [82], suggesting that the eCB system could be exploited to protect against schizophrenic symptoms. Current CBr agonists are being tested in clinical trials and could hopefully become efficacious drugs for alleviating the psychosis associated with schizophrenia [83, 84].
5.4 eCB and MAO Inhibition
Monoamine oxidase (MAO) is the main enzyme responsible for metabolizing catecholamines and other neurotransmitters, resulting in their inactivation and termination of their signaling [74]. It exists as two different isoforms. MAO-A is responsible for metabolizing NE and 5-HT while MAO-B is responsible for breaking down benzylamine [74]. Both MAO-A and –B metabolize DA and tyramine; however, since MAO-B is the predominant isoform in the basal ganglia, it plays a much greater role in the metabolism of DA compared to MAO-A [74, 85]. In the 1950s and 60s, it was discovered serendipitously that treatment of tuberculosis with isoniazid resulted in patients experiencing euphoria and antidepressant phenotypes [74]. Subsequent studies then showed that isoniazid also functions to inhibit MAO, which lead to the development of the first class of antidepressant medications and the monoamine hypothesis of depression, suggesting that an increase in monoamine levels can alleviate symptoms [74]. While these drugs are very successful at treating depression, they also can result in extreme side effects, such as liver toxicity and hypertension due to dietary tyramine intake (the cheese reaction) [86]. Reversible MAO inhibitors have since been developed, which help alleviate the drug-food interactions by allowing tyramine to compete for the binding site [74].
In 2010, Fisar reported that several cannabinoids alter MAO functioning in vitro. By performing radiochemical assays on the mitochondria from pig brains, the effects of THC, AEA and WIN 55,212-2 on MAO-A and –B activities were determined. All three were nonselective and weak MAO inhibitors, with THC having the greatest potency for both enzymes. AEA has the lowest potency of the tested cannabinoids, and competitively binds to MAO-A while noncompetitively binding to MAO-B. Finally, WIN 55,212-2 had equal potency to THC for MAO-A; however, it is uncompetitive at MAO-B, not fully inhibiting its functioning, even when at high concentrations [63]. It is worth noting that the IC50 values of the cannabinoids were much larger than that of iproniazid, indicating that only at very high concentrations, typically larger than physiologically active levels, do these compounds function to inhibit MAO. Since cannabinoids are very lipophilic and MAO is localized to the outer membrane of the mitochondria, it is possible that they can affect MAO activity by accumulating in the mitochondrial membrane following chronic administration [63, 74]. Considering that strong MAO inhibition results in many negative side effects, potentially weaker inhibition by physiological eCB levels could result in a slight increase in monoamine levels without causing toxicity.
6. Effects of Cannabinoids on the Stress Response
HPA hyperactivity is very common in individuals suffering from depression and anxiety [31, 42]. While glucocorticoid release initially is beneficial, priming the body physiologically and metabolically to deal with threats, long-term secretion can result in maladaptive cardiovascular, metabolic, and even neurological conditions [87]. There are negative feedback mechanisms in place, allowing glucocorticoids to attenuate HPA axis activity, and studies have shown the PFC to be the critical site for this termination [88]. Immunohistochemical data and electron microscopy provide evidence that CB1r in layer V of the prelimbic PFC region are found on GABAergic terminals [88]. Genetic deletion of CB1r and injection of CB1r antagonists directly into the PFC produce an increase in the stress response and corticosterone levels, further implicating the importance of the eCB system in the negative feedback mechanisms on the HPA axis [88]. Activation of the GABAergic circuits located in either the prelimbic PFC or the bed nucleus of the stria terminalis (BNST) result in a decrease in corticotropin-releasing factor (CRF) release from the paraventricular nucleus (PVN) [89].
In vivo rat studies have demonstrated that peripheral injection of corticosterone causes a swift escalation of eCB levels in the PVN, indicating that stress upregulates hypothalamic eCB levels via a glucocorticoid-mediated mechanism [90]. This has led Hill et al. to propose a model for the influence of the eCB system on the temporal phases observed in glucocorticoid feedback. It is known that stress causes the production and release of glucocorticoids into the circulation. According to the model, rapid attenuation of the HPA axis occurs via an increase in eCB synthesis and release in the PVN, resulting in the suppression of glutamatergic signaling on CRF-releasing neurons [88]. A longer, time-delayed feedback loop centers on eCB production in the mPFC. Circulating glucocorticoids stimulate 2-AG synthesis and release in the prelimbic mPFC, which then binds to CB1r on GABAergic neurons. This results in the disinhibition of projection neurons that synapse with GABAergic neurons in the BNST, ultimately causing a decrease in signals projecting to the PVN and a subsequent decrease in CRF release [88]. While it appears that eCBs are produced on demand in the above pathways, it has been proposed that tonic AEA signaling in the basolateral amygdala (BLA) occurs [91]. This is the basis for a gatekeeper function, in which tonic eCB tone results in basal inhibition of the HPA axis [90]. During acute stress, there is an increase in FAAH activity, resulting in a decrease in AEA levels [91]. This causes a disinhibition of the principal neurons located in the BLA, subsequently leading to an increased amygdalar output to various regions including the PVN, stimulating the HPA axis [91].
The amygdala is a key structure involved in the regulation of fear and emotional memory, and the eCB system plays a role in regulating the amygdala’s response to stress. As previously mentioned, stress causes a rapid decrease in AEA levels within the amygdala however, when stress is absent and corticosterone is administered, an increase in AEA levels within the amygdala is observed [90, 91]. While this might seem contradictory, such a mechanism may be adaptive. Activation of the HPA axis is important for escaping and managing an acute stress; however, problems arise from over-activation of the HPA axis. Therefore, though stress initially causes a decrease in amygdalar AEA levels via a glucocorticoid-independent pathway, the subsequent glucocorticoid release caused by HPA axis stimulation feeds back to increase amygdalar AEA levels, attenuating the HPA axis activity [90]. While acute stressors only affect the production of AEA in the amygdala, repeated chronic stressors can increase amygdalar 2-AG signaling temporarily, with levels beginning to return to normal one hour after the stressful event [92]. Therefore, elevated levels of 2-AG following repeated stressors represents another mechanism by which eCBs protect from HPA axis overactivation. Consistent with this hypothesis, injection of a CB1r antagonist locally into the BLA reverses this stress habituation [93].
7. Effects of Cannabinoids on Fear and PTSD
A direct role for eCB modulation of the emotional components of amygdalar function has been observed in animal studies. Classical associative fear conditioning and extinction behavioral models show that eCB levels are increased in the amygdala during the extinction session; however, these increases were not observed during the initial fear condition, and fear behaviors persisted longer in the extinction sessions in CB1r KO mice compared to WT mice [94]. Rats exposed to a footshock followed by situational reminders, which is a potential model for PTSD, exhibit impaired extinction of the traumatic memory and increased CB1r levels in the hippocampus (CA1) and PFC, and these alterations were prevented by WIN 55,212-2 administration following exposure to the traumatic event [95]. Combined with results from other studies, it has been concluded that amygdalar eCB signaling is critical for both within- and between-session habituation and adaption of fear-related behaviors [94, 96]. These data suggest that the eCB system is essential for regulating amygdalar function and that the amygdala is a nucleus where eCB signaling can affect both neuroendocrine and stress adaptation behaviors [90].
In addition to affecting fear related behaviors, the eCB system is also involved in the consolidation, retrieval, and extinction of emotionally charged and distressing memories [97]. As previously mentioned, glucocorticoids that are released following a stressor can stimulate eCB synthesis, which in turn inhibits GABAergic neurotransmission. This disinhibition of GABAergic projections from the BLA to the LC results in increased NE release and its subsequent binding to β-ARs, causing the consolidation of stressful and potentially traumatic memories [91]. Administration of WIN 55,212 or glucocorticoid receptor antagonist RU-486 into the BLA before exposure to a stressful stimuli prevented the enhancement in memory consolidation that is normally observed [98]. Since stress also leads to rapid increases in FAAH levels, FAAH inhibitors prevent the degradation of AEA, which in turn promotes long-term fear extinction in animal models via CB1r binding in the BLA and provides protection against stress-induced alterations to eCB signaling [99]. It has been suggested that since eCBs are released within the BLA during fear extinction, the resulting eCB-dependent negative feedback on the HPA axis is critical for the extinction of traumatic memories [94, 100]. Therefore, compounds that enhance the eCB system could serve as therapeutics for PTSD.
8. Therapeutic Potential for the Treatment of Psychiatric Disorders
The ability of the eCB system to modulate monoaminergic neurotransmission and the HPA axis make it a potentially attractive therapeutic target for the treatment of numerous psychiatric disorders, which involve abnormal function of these systems. As described earlier in this review, there is an extensive body of preclinical data demonstrating that various compounds and manipulations that increase CB signaling produce effects in behavioral assays that are predictive of therapeutic efficacy. Although several CB compounds have been evaluated in clinical trials for non-psychiatric disorders such as obesity and pain, it is only recently that some of these compounds have begun to be tested for psychiatric disorders including schizophrenia, PTSD and depression (Table 2) [101, 102]. While only a limited number of studies have released information on the results of their trials, some of them seem particulalry promising. For example, cannabidiol resulted in relief from psychotic symptoms in acute schizophrenic patients that was comparable to a potent antipsychotic while resulting in fewer side effects. In a study of PTSD, Nabilone, a synthetic cannabinoid, greatly improved the quality of sleep and decreased the number of daytime flashbacks in treatment resistant patients [101]. Nabilone also significantly improved PTSD-associated insomnia, chronic pain, and nightmares in a retrospective study of 104 mentally ill men [103]. A second study found that THC treatment, twice a day over the course of three weeks, decreased the number of nightmares and increased sleep quality in ten patients suffering from chronic PTSD [104]. PTSD patients are plagued with debilitating flashbacks of a horrific event, potentially due to dysfunctional retrieval and extinction of emotional memories [105, 106]. These results are consistent with clinical studies showing that many individuals afflicted with PTSD self-medicate with cannabis to help alleviate their symptoms [107]. Cannabis use is correlated with both the onset and severity of PTSD symptoms [108, 109]. Since it is known that the eCB system is involved in these processes and that people suffering from PTSD often self-medicate with cannabis, other compounds that increase eCB signaling could prove to be therapeutic as well.
9. Novel Modulators of Cannabinoid Receptors
Recently, allosteric CB1r agonists have been identified, which has important implications for drug discovery, as allosteric compounds allow for the modulation of signaling without completely inducing or blocking receptor responses as traditional agonists and antagonists would do [110]. Prince et al identified the first allosteric CB1r modulators in 2005. They discovered three Organon compounds that all enhanced agonist affinity for the CB1r; however, they function as insurmountable antagonists, decreasing the Emax value for CB1r agonists and increasing the length of time it takes for the agonists to dissociate from the receptor [111]. Subsequent research has shown that Org-27569 might in fact function as a biased ligand, decreasing coupling to cAMP and β-arrestin signaling while increasing Gα-independent ERK phosphorylation and stimulating receptor internalization [112]. This compound was tested in several in vivo rodent studies but failed to alter CB1-mediated effects of AEA, CP 55,940 (a CB1r agonist), and THC in anti-nociception, catalepsy, and hypothermia [113]; however, other eCBs were not tested in conjunction with Org-27569, nor were psychiatric effects evaluated. PSNCBAM-1 appears to have a similar profile to Org-27596, functioning as a negative allosteric CB1r modulator. In vivo studies, though, have shown it to be effective in an acute food-intake model [114]. Additionally, several positive allosteric modulators of CB1r activity have been identified and in particular, carbozamides have been found to selectively enhance CB1r activity [115]. Lipoxin A4 enhances AEA induced nociception and catalepsy in various mouse models [116, 117]. Finally, RTI-371, a selective DAT inhibitor, has also been found to increase CP 55940 signaling in vitro in a concentration-dependent fashion [118].
10. Conclusion
There is a growing body of evidence that the eCB system plays an important role in the control of mood. Anatomical, electrophysiological and in vivo behavioral studies have revealed that the eCB system can modulate both the monoamine systems and the HPA axis. Since current antidepressant therapy is designed to modulate monoamine transmission and dysfunction of the HPA axis is a hallmark of depression, targeting the eCB system appears to represent an attractive and novel approach to the treatment of depression and other mood disorders. In support of this notion, studies in animal models of depression and anxiety predict that activation of CB receptors is likely to be efficacious in the treatment of these disorders. However, there are reports that chronic use of cannabis can actually result in dysphoria and panic. This suggests that over-activation of the eCB system may have undesirable consequences and it will be critical to take this into consideration when targeting the eCB system. One approach with the potential to produce therapeutic levels of CB activation without risking the undesirable side effects associated with over-activation is inhibition of the degradation of the endogenous eCBs, AEA and 2-AG. In theory, this should result in indirect, activity-dependent increases in CB receptor stimulation due to elevation of endogenous eCB levels while minimizing the likelihood of over-activation. More specifically, inhibition of FAAH, the enzyme that degrades AEA, would appear to be particularly promising, because AEA is a partial agonist that would be even less likely to produce over-activation of CB receptors. It is worth noting that virtually all current antidepressants rely on activity-dependent mechanisms to increase monoamine transmission. Interestingly, cannabidiol, which is an inhibitor of AEA degradation, has been reported to have therapeutic effects in a clinical trial for schizophrenia. One possible concern with this strategy is that AEA and 2-AG are not completely selective for CB receptors and have been shown to bind to and activate TRPV1 receptors. The recent development of allosteric modulators of CBr1 may provide an attractive alternative to modulation of eCB levels. Both positive and negative allosteric modulators of CB1r have been identified and compounds that appear to be biased agonists of CB1r have also been reported. These classes of compounds have the potential to specifically activate CB1r in an activity-dependent manner and minimize the risk of over-activation or undesirable side effects.
11. Expert Opinion
The eCB and noradrenergic systems are significantly and dynamically impacted by stress [119–123] and noradrenergic transmission is responsible for cannabinoid-induced activation of the HPA axis [124]. Under conditions of acute stress, NE is increased centrally and peripherally [120, 125–130] while the eCB system tonically constrains activation of neural circuits, including the hypothalamic-pituitary-adrenal (HPA) axis [123, 131]. However, disrupted noradrenergic and eCB signaling is associated with an inability to adapt to chronic stress [121, 123, 127, 132–134]. Our work indicates a different consequence to the regulation of NE by cannabinoids under stress conditions. Specifically, stress-induced increases in cortical NE levels are significantly attenuated by prior treatment with a cannabinoid receptor agonist suggesting complex actions of cannabinoids on noradrenergic circuitry that vary under basal vs stress conditions. The working model posits that, under basal conditions, decreased signaling of pre-synaptically distributed CB1r localized to noradrenergic afferents contribute to local increases in cortical NE and adrenergic receptor (AR) desensitization. Under conditions of stress where NE levels are elevated, increased release of eCB from cortical neurons attenuates pre-synaptic release of NE potentially leading to AR sensitization.
There is significant potential for establishing cannabinoid-adrenergic interactions as a novel target in the development of improved treatment strategies for stress-induced anxiety. The pathophysiology underlying anxiety disorders, and specifically PTSD, may be related to an inability to extinguish aversive memories [135]. Increased salience of aversive memories due to activation of limbic circuits and poor cognitive inhibition/flexibility due to decreased cortical activity may contribute to the behavioral expression of anxiety. Understanding the cellular mechanism responsible for extinction of fear memories may provide the basis for more effective forms of clinical treatment of anxiety. Patients with PTSD suffer from recurrent retrieval of traumatic memories in the form of context-induced flashbacks and repeated nightmares. Repeated re-consolidation of fear memories in limbic circuits and inability to extinguish fear memories [136] are thought to underlie the pathophysiology of PTSD. Consolidation of emotionally arousing memories involves, in part, noradrenergic circuits targeting the amygdala [126, 137], while extinction of memory is dependent on the mPFC [138, 139]. Pharmacological manipulation of AR systems has provided symptomatic relief in PTSD patients [140, 141] suggesting that therapeutic improvement may result, in part, from attenuation of signaling of sensitized ARs. Moreover, the cannabinoid receptor agonist, nabilone, has recently been reported to be effective in management of symptoms of PTSD [101]. Taken with recent evidence that the EC and noradrenergic systems interact in stress-related memory consolidation [122, 142], targeting interactions between these two systems may represent a novel approach for the treatment of stress-induced anxiety disorders. Elucidating reciprocal interactions between the cannabinoid-adrenergic systems in stress-integrative circuits is vital for demonstrating that interaction of the two is important in modulating stress-induced anxiety and extinction of conditioned fear. Given that the PFC represents a critical region in mediating the extinction of traumatic/aversive memories, treatments that target this region may help alleviate symptoms of anxiety disorders by increasing extinction of such memories. Achieving the proper balance in frontal cortical activity by targeting cannabinoid-adrenergic interactions may result in enhancing extinction of aversive memories and diminish anxiety-like behaviors that are precipitated by stress.
The increasing availability of different classes of compounds that target discrete aspects of the eCB system provide a unique opportunity to more thoroughly evaluate the importance of cannabinoid-adrenergic interactions on anxious behaviors in both preclinical and clinical studies. As mentioned earlier, the preliminary results obtained with the synthetic cannabinoid Nabilone as well as the natural cannabinoid THC and cannabis itself for the treatment of PTSD have been very promising. Additional insight provided by detailed preclinical studies and the discovery of increasingly selective modulators of CB receptors should enable the identification of optimal therapeutic strategies and maximize the likelihood of developing safe and effective treatments for debilitating disorders such as PTSD.
Figure 1.
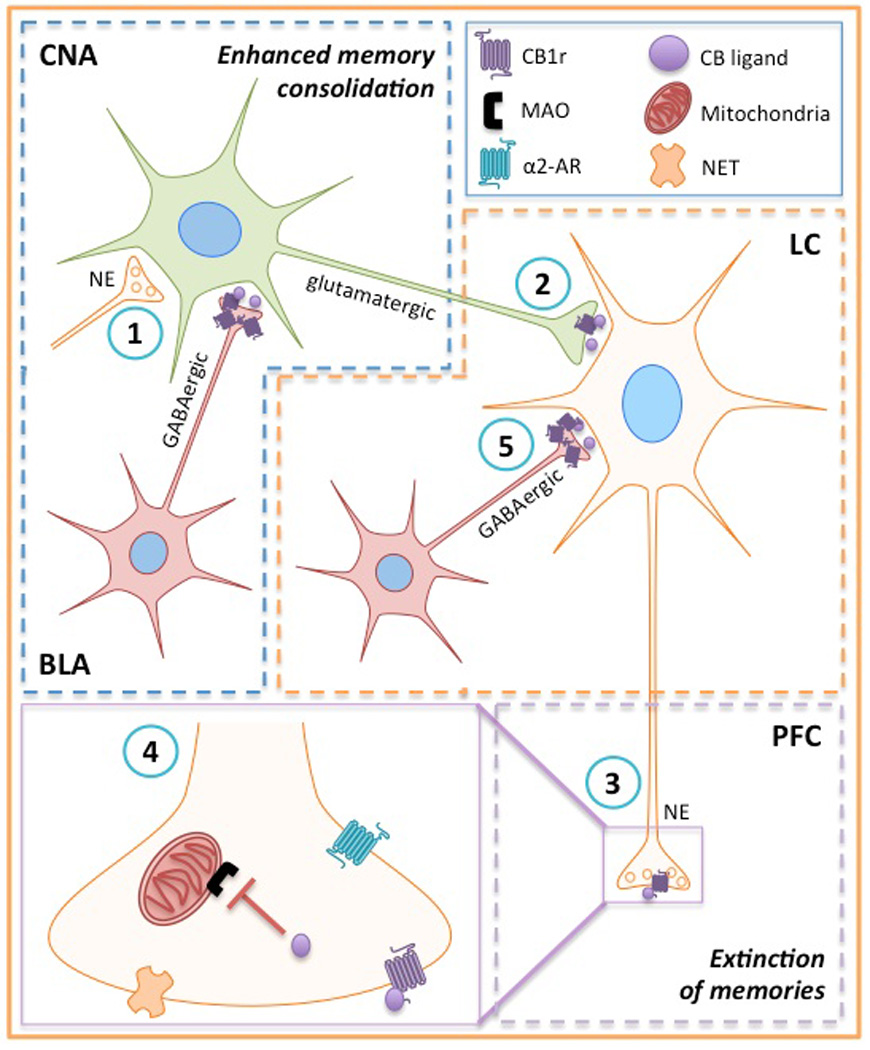
Schematic diagram depicting cannabinoid-adrenergic interactions in stress-integrative circuitry. The basolateral complex of the amygdala (BLA) has been implicated in the consolidation of emotionally arousing experiences and involves glucocorticoid-mediated increases in eCB release and interactions with norepinephrine [146]. (1) eCBs are posited to increase BLA activity by decreasing GABAergic neurotransmission [147]. (2) Disinhibition of GABAergic interneurons results in an increase of glutamatergic signaling in the central nucleus of the amygdala (CNA), a source of excitatory afferents to the LC [148]. (3) Activation of the LC causes an increase in noradrenergic signaling and norepinephrine (NE) release in postsynaptic targets, such as the prefrontal cortex (PFC). Given that the PFC represents a critical region in mediating the extinction of traumatic/aversive memories, treatments involving the eCB system that target this region may help alleviate symptoms of anxiety disorders by increasing extinction of such memories. For example, (4) CBs have been shown to inhibit monoamine oxidase (MAO), representing another mechanism in which CB signaling can regulate NE levels. (5) Targeting GABAergic projections to the LC with CB ligands can potentially modulate LC afferent activity to the PFC. Achieving the proper balance in frontal cortical activity by targeting cannabinoid-adrenergic interactions may result in enhancing extinction of aversive memories and diminish anxiety-like behaviors that are precipitated by stress.
Highlights Box.
Dysregulation of the eCB system has been implicated in several psychiatric disorders.
Much evidence has been gathered in support of eCB regulation of monoamine neurotransmission and its ability to alter mood.
Stress response is regulated by the eCB system, and its dysfunction can lead to HPA axis dysregulation and the development of psychiatric disorders.
Many compounds have been developed that target the eCB system and could prove to be successful therapeutics for psychiatric disorders, especially the more selective modulators of CB receptors.
The cannabinoid-adrenergic interaction represents a promising novel target for the symptomatic treatment of stress-induced anxiety and PTSD, since these systems are heavily involved in memory consolidation and extinction.
Acknowledgments
The authors are supported by the National Institute of Drug Abuse, grant number DA020129.
Footnotes
Financial and Competing Interests Disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Green B, Kavanagh D, Young R. Being stoned: a review of self-reported cannabis effects. Drug and Alcohol Review. 2003;22:453–460. doi: 10.1080/09595230310001613976. [DOI] [PubMed] [Google Scholar]
- 2.Velez CN, Johnson J, Cohen PA. A longitudinal analysis of selected risk factors for childhood psychopathology. Journal of the American Academy of Child and Adolescent Psychiatry. 1989;28:861–864. doi: 10.1097/00004583-198911000-00009. [DOI] [PubMed] [Google Scholar]
- 3.Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nature Reviews Drug Discovery. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- 4.Reilly D, Didcott P, Swift W, et al. Long-term cannabis use: characteristics of users in an Australian rural area. Addiction. 1998;93:837–846. doi: 10.1046/j.1360-0443.1998.9368375.x. [DOI] [PubMed] [Google Scholar]
- 5.Lee KS, Conigrave KM, Patton GC, et al. Cannabis use in remote Indigenous communities in Australia: endemic yet neglected. Medical Journal of Australia. 2009;190:228–229. doi: 10.5694/j.1326-5377.2009.tb02379.x. [DOI] [PubMed] [Google Scholar]
- 6.Nissen SE, Nicholls SJ, Wolski K, et al. Effect of rimonabant on progression of atherosclerosis in patients with abdominal obesity and coronary artery disease: the STRADIVARIUS randomized controlled trial. JAMA. 2008;299:1547–1560. doi: 10.1001/jama.299.13.1547. [DOI] [PubMed] [Google Scholar]
- 7.Gaoni Y, Mechoulam R. Isolation, structure and partial synthesis of an active constituent of hashish. Journal of the American Chemical Society. 1964;86:1646–1647. [Google Scholar]
- 8.Micale V, Di Marzo V, Sulcova A, et al. Endocannabinoid system and mood disorders: Priming a target for new therapies. Pharmacology & Therapeutics. 2013;138(1):18–37. doi: 10.1016/j.pharmthera.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Devane WA, Dysarz FA, III, Johnson MR, et al. Determination and characterization of a cannabinoid receptor in rat brain. Molecular Pharmacology. 1988;34:605–613. [PubMed] [Google Scholar]
- 10.Castillo PE, Younts TJ, Chavez AE, et al. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herkenham M, Lynn AB, Little MD, et al. Cannabinoid receptor localization in brain. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:1932–1936. doi: 10.1073/pnas.87.5.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Sickle MD, Duncan M, Kingsley PJ, et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005;310:329–332. doi: 10.1126/science.1115740. [DOI] [PubMed] [Google Scholar]
- 13.Brusco A, Tagliaferro PA, Saez T, et al. Ultrastructural localization of neuronal brain CB2 cannabinoid receptors. Annals of the New York Academy of Sciences. 2008;1139:450–457. doi: 10.1196/annals.1432.037. [DOI] [PubMed] [Google Scholar]
- 14.Onaivi ES, Ishiguro H, Gu S, et al. CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. Journal of Psychopharmacology. 2012;26(1):92–103. doi: 10.1177/0269881111400652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 16.Mechoulam R, Ben-Shabat S, Hanus L, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochemical Pharmacology. 1995;50(1):83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 17.Sugiura T. Physiological roles of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Biofactors. 2009;35:88–97. doi: 10.1002/biof.18. [DOI] [PubMed] [Google Scholar]
- 18.Wang H, Lupica CR. Release of endogenous cannabinoids from ventral tegmental area dopamine neurons and the modulation of synaptic processes. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014;52:24–27. doi: 10.1016/j.pnpbp.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bacci A, Huguenard JR, Prince DA. Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature. 2004;431:312–316. doi: 10.1038/nature02913. [DOI] [PubMed] [Google Scholar]
- 20.Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chemical Biology. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Di Marzo V. Endocannabinoid signaling in the brain: biosynthetic mechanisms in the limelight. Nature Neuroscience. 2011;14:9–15. doi: 10.1038/nn.2720. [DOI] [PubMed] [Google Scholar]
- 22.Okamoto Y, Wang J, Morishita J, et al. Biosynthetic pathways of the endocannabinoid anandamide. Chemistry & Biodiversity. 2007;4:1842–1857. doi: 10.1002/cbdv.200790155. [DOI] [PubMed] [Google Scholar]
- 23.Ligresti A, Petrosino S, Di Marzo V. From endocannabinoid profiling to ‘endocannabinoid therapeutics’. Current Opinion in Chemical Biology. 2009;13(3):321–331. doi: 10.1016/j.cbpa.2009.04.615. [DOI] [PubMed] [Google Scholar]
- 24.Parolaro D, Realini N, Vigano D, et al. The endocannabinoid system and psychiatric disorders. Experimental Neurology. 2010;224:3–14. doi: 10.1016/j.expneurol.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 25.Hauer D, Kaufmann I, Strewe C, et al. The role of glucocorticoids, catecholamines and endocannabinoids in the development of traumatic memories and posttraumatic stress symptoms in survivors of critical illness. Neurobiology of Learning and Memory. 2013:1–7. doi: 10.1016/j.nlm.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 26.Hungund BL, Vinod KY, Kassir SA, et al. Upregulation of CB1 receptors and antagonist-stimulated [35S]GTPgammaS binding in the prefrontal cortex of depressed suicide victims. Molecular Psychiatry. 2004;9:184–190. doi: 10.1038/sj.mp.4001376. [DOI] [PubMed] [Google Scholar]
- 27.Neumeister A, Normandin MD, Pietrzak RH, et al. Elevated brain cannabinoid CB receptor availability in post-traumatic stress disorder: a positron emission tomography study. Molecular Psychiatry. 2013;18:1034–1040. doi: 10.1038/mp.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steiner MA, Wanisch K, Monory K, et al. Impaired cannabinoid receptor type 1 signaling interferes with stress-coping behavior in mice. Pharmacogenomics Journal. 2008;8:196–208. doi: 10.1038/sj.tpj.6500466. [DOI] [PubMed] [Google Scholar]
- 29.Aso E, Ozaita A, Valdizan EM, et al. BDNF impairment in the hippocampus is related to enhanced despair behavior in CB1 knockout mice. Journal of Neurochemistry. 2008;105:565–572. doi: 10.1111/j.1471-4159.2007.05149.x. [DOI] [PubMed] [Google Scholar]
- 30.Martin M, Ledent C, Parmentier M, et al. Involvement of CB1 cannabinoid receptors in emotional behaviour. Psychopharmacology (Berlin) 2002;159:379–387. doi: 10.1007/s00213-001-0946-5. [DOI] [PubMed] [Google Scholar]
- 31.Uriguen L, Perez-Rial S, Ledent C, et al. Impaired action of anxiolytic drugs in mice deficient in cannabinoid CB1 receptors. Neuropharmacology. 2004;46:966–973. doi: 10.1016/j.neuropharm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 32.Bambico FR, Katz N, Debonnel G, et al. Cannabinoids elicit antidepressant-like behavior and activate serotoninergic neurons through the medial prefrontal cortex. The Journal of Neuroscience. 2007;27:11700–11711. doi: 10.1523/JNEUROSCI.1636-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gobbi G, Bambico FR, Mangieri R, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Foll B, Gorelick DA, Goldberg SR. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology. 2009;205:171–174. doi: 10.1007/s00213-009-1506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Millan MJ, Agid Y, Brune M, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nature Reviews Drug Discovery. 2012;11:141–168. doi: 10.1038/nrd3628. [DOI] [PubMed] [Google Scholar]
- 36.Robbe D, Montgomery SM, Thome A, et al. Cannbinoids reveal importance of spike timing coordination in hippocampal function. Nature Neuroscience. 2006;9(12):1526–1533. doi: 10.1038/nn1801. [DOI] [PubMed] [Google Scholar]
- 37.Robbe D, Buzsaki G. Alternation of theta timescale dynamics of hippocampal place cells by a cannabinoid is associated with memory impairment. The Journal of Neuroscience. 2009;29(40):12597–12605. doi: 10.1523/JNEUROSCI.2407-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zajkowska ZE, Englund A, Zunszain PA. Towards a personalized treatment in depression: endocannabinoids, inflammation and stress response. Pharmacogenomics. 2014;15(5):687–698. doi: 10.2217/pgs.14.40. [DOI] [PubMed] [Google Scholar]
- 39.Jiang W, Zhang Y, Xiao L, et al. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. Journal of Clinical Investigation. 2005;115:3104–3116. doi: 10.1172/JCI25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biological Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Rubino T, Vigano D, Premoli F, et al. Changes in the expression of G protein-coupled receptor kinases and beta-arrestins in mouse brain during cannabinoid tolerance: a role for RAS-ERK cascade. Molecular Neurobiology. 2006;33:199–213. doi: 10.1385/MN:33:3:199. [DOI] [PubMed] [Google Scholar]
- 42.Patel S, Roelke CT, Rademacher DJ, et al. Endocannabinoid signaling negatively modulates stress-induced activation of the hypothalamic-pituitary-adrenal axis. Endocrinology. 2004;145:5431–5438. doi: 10.1210/en.2004-0638. [DOI] [PubMed] [Google Scholar]
- 43. Haj-Dahmane S, Shen R-Y. Modulation of the serotonin system by endocannabinoid signaling. Neuropharmacology. 2011;61:414–420. doi: 10.1016/j.neuropharm.2011.02.016. (This review provides further information on eCB modulation of 5-HT signaling.)
- 44.Haj-Dahmane S, Shen R-Y. The wake-promoting peptide orexin-B inhibits glutamatergic transmission to dorsal raphe nucleus serotonin neurons through retrograde endocannabinoid signaling. The Journal of Neuroscience. 2005;25:896–905. doi: 10.1523/JNEUROSCI.3258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haj-Dahmane S, Shen R-Y. Endocannabinoids suppress excitatory synaptic transmission to dorsal raphe serotonin neurons through the activation of presynaptic CB1 receptors. The Journal of Pharmacology and Experimental Therapeutics. 2009;331:186–196. doi: 10.1124/jpet.109.153858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mendiguren A, Pineda J. Effect of the CB1 receptor antagonists rimonabant and AM251 on the firing rate of dorsal raphe nucleus neurons in rat brain slices. British Journal of Pharmacology. 2009;158:1579–1587. doi: 10.1111/j.1476-5381.2009.00434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carrasco GA, Van de Kar LD. Neuroendocrine pharmacology of stress. European Journal of Pharmacology. 2003;463:235–272. doi: 10.1016/s0014-2999(03)01285-8. [DOI] [PubMed] [Google Scholar]
- 48.Gingrich JA, Hen R. Dissecting the role of the serotonin system in neuropsychiatric disorders using knockout mice. Psychopharmacology. 2001;155:1–10. doi: 10.1007/s002130000573. [DOI] [PubMed] [Google Scholar]
- 49.Resstel LBM, Tavares RF, Lisboa SFS, et al. 5-HT1A receptors are involved in the cannabidiol-induced attenuation of behavioral and cardiovascular responses to acute restraint stress in rats. British Journal of Pharmacology. 2009;156:181–188. doi: 10.1111/j.1476-5381.2008.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gould GG, Burke TF, Osorio MD, et al. Enhanced novelty-induced corticosterone spike and upregulated serotonin 5-HT1A and cannabinoid CB1 receptors in adolescent BTBR mice. Psychoneuroendocrinology. 2014;39:158–169. doi: 10.1016/j.psyneuen.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Burokas A, Martin-Garcia E, Gutierrez-Cuesta J, et al. Relationships between serotonergic and cannabinoid system in depressive-like behavior: a PET study with [11C]-DASB. Journal of Neurochemistry. 2014;130:126–135. doi: 10.1111/jnc.12716. [DOI] [PubMed] [Google Scholar]
- 52.Franklin JM, Mathew M, Carrasco GA. Cannabinoid-induced upregulation of serotonin 2A receptors in the hypothalamic paraventricular nucleus and anxiety-like behaviors in rats. Neuroscience Letters. 2013;548:165–169. doi: 10.1016/j.neulet.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Johnston TG, Kelly CB, Stevenson MR, et al. Plasma norepinephrine and prediciton of outcome in major depressive disorder. Biological Psychiatry. 1999;46:1253–1258. doi: 10.1016/s0006-3223(99)00134-1. [DOI] [PubMed] [Google Scholar]
- 54.Carvalho AF, Van Bockstaele EJ. Cannabinoid modulation of noradrenergic circuits: Implications for psychiatric disorders. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2012;38(1):59–67. doi: 10.1016/j.pnpbp.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sara SJ. The locus coeruleus and noradrenergic modulation of cognition. Nature Reviews Neuroscience. 2009;10:211–223. doi: 10.1038/nrn2573. [DOI] [PubMed] [Google Scholar]
- 56.Scavone JL, Mackie K, Van Bockstaele EJ. Characterization of cannabinoid-1 receptors in the locus coeruleus: relationship with mu-opioid receptors. Brain Research. 2010;1312:18–31. doi: 10.1016/j.brainres.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirilly E, Hunyady L, Bagdy G. Opposing local effects of endocannbinoids on the activity of noradrenergic neurons and release of noradrenaline: relevance for their role in depression and in the actions of CB1 receptor antagonists. Biological Psychiatry. 2013;120:177–186. doi: 10.1007/s00702-012-0900-1. [DOI] [PubMed] [Google Scholar]
- 58.Muntoni AL, Pillolla G, Melis M, et al. Cannabinoids modulate spontaneous neuronal activity and evoked inhibition of locus coeruleus noradrenergic neurons. European Journal of Pharmacology. 2006;23:2385–2394. doi: 10.1111/j.1460-9568.2006.04759.x. [DOI] [PubMed] [Google Scholar]
- 59.Patel KD, Hillard CJ. Cannabinoid-induced fos expression within A10 dopaminergic neurons. Brain Research. 2003;963:15–25. doi: 10.1016/s0006-8993(02)03797-6. [DOI] [PubMed] [Google Scholar]
- 60.Mendiguren A, Pineda J. Systemic effect of cannabinoids on the spontaneous firing rate of locus coeruleus neurons in rats. European Journal of Pharmacology. 2006;534:83–88. doi: 10.1016/j.ejphar.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 61.Carvalho AF, Mackie K, Van Bockstaele EJ. Cannabinoid modulation of limbic forebrain noradrenergic circuitry. European Journal of Pharmacology. 2010;31:286–301. doi: 10.1111/j.1460-9568.2009.07054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jentsch JD, Andrusiak E, Tranm A, et al. Delta-9-tetrahydrocannabinol increases prefrontal cortical catecholaminergic utilization and impairs spatial working memory in the rat: blockade of dopaminergic effects with HA966. Neuropsychopharmacology. 1997;16:426–432. doi: 10.1016/S0893-133X(97)00018-3. [DOI] [PubMed] [Google Scholar]
- 63.Fisar Z. Inhibition of monoamine oxidase activity by cannabinoids. Naunyn-Schmiedeberg Archives of Pharmacology. 2010;381:563–572. doi: 10.1007/s00210-010-0517-6. [DOI] [PubMed] [Google Scholar]
- 64.Oropeza VC, Page ME, Van Bockstaele EJ. Systemic administration of WIN 55,212-2 increases norepinephrine release in the rat frontal cortex. Brain Research. 2005;1046:45–54. doi: 10.1016/j.brainres.2005.03.036. [DOI] [PubMed] [Google Scholar]
- 65.Page ME, Oropeza VC, Sparks SE, et al. Repeated cannabinoid administration increases indices of noradrenergic activity in rats. Pharmacology, Biochemistry, and Behavior. 2007;86:162–168. doi: 10.1016/j.pbb.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marsicano G, Lutz B. Expression of the cannabinoid receptor CB1 in distinct neuronal subpopulations in the adult mouse forebrain. European Journal of Neuroscience. 1999;11:4213–4224. doi: 10.1046/j.1460-9568.1999.00847.x. [DOI] [PubMed] [Google Scholar]
- 67.Tsou K, Brown S, Sanudo-Pena MC, et al. Immunohistochemical distrobution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience. 1998;83:393–411. doi: 10.1016/s0306-4522(97)00436-3. [DOI] [PubMed] [Google Scholar]
- 68.Laricchiuta D, Musella A, Rossi S, et al. Behavioral and electrophysiological effects of endocannabinoid and dopaminergic systems on salient stimuli. Frontiers in Behavioral Neuroscience. 2014;8:1–11. doi: 10.3389/fnbeh.2014.00183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.El Khoury MA, Gorgievski V, Moutsimilli L, et al. Interactions between the endocannabinoid and dopaminergic systems: evidence from animal studies. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2012;38(1):36–50. doi: 10.1016/j.pnpbp.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Matricon J, Giuffrida A. Cannabinoid modulation of dopaminergic circuits in neurodegenerative and neuropsychiatric disorders. In: Van Bockstaele EJ, editor. Endocannabinoid Regulation of Monoamines in Psyciatric and Neurological Disorders. New York, NY: Springer; 2013. pp. 73–101. [Google Scholar]
- 71.Amchova P, Kucerova J, Giugliano V, et al. Enhanced self-administration of the CB1 receptor agonist WIN55,212-2 in olfactory bulbectomized rats: evaluation of possible serotonergic and dopaminergic underlying mechanisms. Frontiers in Pharmacology. 2014;5:1–15. doi: 10.3389/fphar.2014.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen J, Paredes W, Lowinson JH, et al. Delta 9-tetrahydrocannabinol enhances presynaptic dopamine efflux in medial prefrontal cortex. European Journal of Pharmacology. 1990;190:259–262. doi: 10.1016/0014-2999(90)94136-l. [DOI] [PubMed] [Google Scholar]
- 73.Pistis M, Ferraro L, Pira L, et al. Delta(9)-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Research. 2002;948:155–158. doi: 10.1016/s0006-8993(02)03055-x. [DOI] [PubMed] [Google Scholar]
- 74.Youdim MBH, Bakhle YS. Monoamine oxidase: isoforms and inhibitors in Parkinson’s disease and depressive illness. British Journal of Pharmacology. 2006;147:S287–S296. doi: 10.1038/sj.bjp.0706464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Laruelle M, Kegeles LS, Abi-Dargham A. Glutamate, dopamine, and schizophrenia: from pathophysiology to treatment. Annals of the New York Academy of Sciences. 2003;1003:138–158. doi: 10.1196/annals.1300.063. [DOI] [PubMed] [Google Scholar]
- 76.Toda M, Abi-Dargham A. Dopamine hypothesis of schizophrenia: making sense of it all. Current Psychiatry Reports. 2007;9:329–336. doi: 10.1007/s11920-007-0041-7. [DOI] [PubMed] [Google Scholar]
- 77.Muller-Vahl KR, Emrich HM. Cannabis and schizophrenia: towards a cannabinoid hypothesis of schizophrenia. Expert Review of Neurotherapeutics. 2008;8:1037–1048. doi: 10.1586/14737175.8.7.1037. [DOI] [PubMed] [Google Scholar]
- 78.Leweke FM, Giuffrida A, Wurster U, et al. Elevated endogenous cannabinoids in schizophrenia. Neuroreport. 1999;10:1665–1669. doi: 10.1097/00001756-199906030-00008. [DOI] [PubMed] [Google Scholar]
- 79.Giuffrida A, Leweke FM, Gerth CW, et al. Cerebrospinal anandamide levels are elevated in acute schizophrenia and are inversely correlated with psychotic symptoms. Neuropsychopharmacology. 2004;29:2108–2114. doi: 10.1038/sj.npp.1300558. [DOI] [PubMed] [Google Scholar]
- 80.Koethe D, Giuffrida A, Schreiber D, et al. Anandamide elevation in cerebrospinal fluid in initial prodromal states of psychosis. British Journal of Psychiatry. 2009;194:371–372. doi: 10.1192/bjp.bp.108.053843. [DOI] [PubMed] [Google Scholar]
- 81.Malone DT, Hill MN, Rubino T. Adolescent cannbis use and psychosis: epidemiology and neurodevelopmental models. British Journal of Pharmacology. 2010;160:511–522. doi: 10.1111/j.1476-5381.2010.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Eggan SM, Hashimoto T, Lewis DA. Reduced cortical cannabinoid 1 receptor messenger RNA and protein expression in schizophrenia. Archives of General Psychiatry. 2008;65:772–784. doi: 10.1001/archpsyc.65.7.772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schwarcz G, Karajgi B, McCarthy R. Synthetic d-9-tetrahydrocannabinol (Dronabinol) can improve the symptoms of schizophrenia. Journal of Clinical Psychopharmacology. 2009;29(3):255–258. doi: 10.1097/JCP.0b013e3181a6bc3b. [DOI] [PubMed] [Google Scholar]
- 84.A study of GWP42003 as an adjunctive therapy in the first line treatment of schizophrenia or related psychotic disorder. U.S. National Institutes of Health; GW Pharmaceuticals Ltd. Bethesda, MD: National Library of Medicine (US); 2014. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/show/NCT02006628. NLM Identifier: NCT02006628. [Google Scholar]
- 85.Collins GGS, Sandler M, Williams ED, et al. Multiple forms of human brain monoamine oxidase. Nature. 1970;225:817–820. doi: 10.1038/225817a0. [DOI] [PubMed] [Google Scholar]
- 86.Fisar Z. Cannabinoids and monoamine neurotransmission with focus on monoamine oxidase. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2012;38:68–77. doi: 10.1016/j.pnpbp.2011.12.010. [DOI] [PubMed] [Google Scholar]
- 87.McEwen BS. Central effects of stress hormones in health and disease: understanding the protective and damaging effects of stress and stress mediators. European Journal of Pharmacology. 2008;583:174–185. doi: 10.1016/j.ejphar.2007.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hill MN, McLaughlin RJ, Pan B, et al. Recruitment of prefrontal cortical endocannabinoid signaling by glucocorticoids contributes to termination of the stress response. The Journal of Neuroscience. 2011;31(29):10506–10515. doi: 10.1523/JNEUROSCI.0496-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Herman JP, Ostrander MM, Mueller NK, et al. Limbic system mechanisms of stress regulation: hypothalamo-pituitary-adrenocortical axis. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2005;29:1201–1213. doi: 10.1016/j.pnpbp.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 90. Hill MN, Patel S, Campolongo P, et al. Functional interactions between stress and the endocannabinoid system: from synaptic signaling to behavioral output. The Journal of Neuroscience. 2010;30(45):14980–14986. doi: 10.1523/JNEUROSCI.4283-10.2010. (This review gives additional information on the interactions between the eCB and stress systems.)
- 91.Hill MN, McEwen BS. Endocannabinoids: the silent partner of glucocorticoids in the synapse. Proceedings of the National Academy of Sciences. 2009;106:4579–4580. doi: 10.1073/pnas.0901519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Patel S, Kingsley PJ, Mackie K, et al. Repeated homotypic stress elevates 2-arachidonoylglycerol levels and enhanves short-term endocannabinoid signalling at inhibitory synapses in basolateral amygdala. Neuropsychopharmacology. 2009;34:2699–2709. doi: 10.1038/npp.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hill MN, McLaughlin RJ, Bingham B, et al. Endogenous cannabinoid signaling is essential for stress adaptation. Proceedings of the National Academy of Sciences. 2010;107:9406–9411. doi: 10.1073/pnas.0914661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Marsicano G, Wotjak CT, Azad SC, et al. The endogenous cannabinoid system controls extinction of aversive memories. Nature. 2002;481:530–534. doi: 10.1038/nature00839. [DOI] [PubMed] [Google Scholar]
- 95.Korem N, Akirav I. Cannabinoids prevent the effects of a footshock followed by situational reminders on emotional processing. Neuropsychopharmacology. 2014 doi: 10.1038/npp.2014.132. published online 2 July 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kamprath K, Marsicano G, Tang J, et al. Cannabinoid CB1 receptor mediates fear extinction via habituation-like processes. Journal of Neuroscience. 2006;26:6677–6686. doi: 10.1523/JNEUROSCI.0153-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Atsak P, Hauer D, Campolongo P, et al. Glucocorticoids interact with the hippocampal endocannabinoid system in impairing retrieval of contextual fear memory. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(9):3504–3509. doi: 10.1073/pnas.1200742109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ramot A, Akirav I. Cannabinoid receptors activation and glucocorticoid receptors deactivation in the amygdala prevent the stress-induced enhancement of a negative learning experience. Neurobiology of Learning and Memory. 2012;97:393–401. doi: 10.1016/j.nlm.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 99.Gunduz-Cinar O, Hill MN, McEwen BS, et al. Amygdala FAAH and anandamide: mediating protection and recovery from stress. Trends in Pharmacological Science. 2013;34(11):637–644. doi: 10.1016/j.tips.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.de Bitencourt R, Pamplona FA, Takahashi RN. A current overview of cannabinoids and glucocorticoids in facilitating extinction of aversive memories: potential extinction enhancers. Neuropharmacology. 2013;64:389–395. doi: 10.1016/j.neuropharm.2012.05.039. [DOI] [PubMed] [Google Scholar]
- 101.Fraser GA. The use of a synthetic cannabinoid in the management of treatment-resistant nightmares in posttraumatic stress disorder (PTSD) CNS Neuroscience & Therapeutics. 2009;15(1):84–88. doi: 10.1111/j.1755-5949.2008.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Leweke FM, Piomelli D, Muhl D, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Translational Psychiatry. 2012;2(e94):1–7. doi: 10.1038/tp.2012.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cameron C, Watson D, Robinson J. Use of a synthetic cannbinoid in a correctional population for posttraumatic stress disorder-related insomnia and nightmares, chronic pain, harm reduction, and other indications: a retrospective evaluation. Journal of Clinical Psychopharmacology. 2014;5:559–564. doi: 10.1097/JCP.0000000000000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roitman P, Mechoulam R, Shalev A. Israel Society for Biological Psychiatry abstract book. 2013 [Google Scholar]
- 105.Nemeroff B, Bremner JD, Foa EB, et al. Posttraumatic stress disorder: a state-of-the-science review. Journal of Psychiatric Research. 2006;40:1–21. doi: 10.1016/j.jpsychires.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 106. Akirav I. Cannabinoids and glucocorticoids modulate emotional memory after stress. Neuroscience and Biobehavioral Reviews. 2013;37(10):2554–2563. doi: 10.1016/j.neubiorev.2013.08.002. (This review elaborates on the interaction between the eCB and stress systems, and how they modulate emotional memory and learning.)
- 107.Passie T, Emrich HM, Karst M, et al. Mitigation of post-traumatic stress symptoms by Cannabis resin: a review of the clinical and neurobiological evidence. Drug Testing and Analysis. 2012;4:649–659. doi: 10.1002/dta.1377. [DOI] [PubMed] [Google Scholar]
- 108.Cougle JR, Bonn-Miller MO, Vujanovic AA, et al. Posttraumatic stress disorder and cannabins use in a nationally representative sample. Psychology of Addictive Behaviors. 2011;25:554–558. doi: 10.1037/a0023076. [DOI] [PubMed] [Google Scholar]
- 109.Potter CM, Vujanovic AA, Marshall-Berenz EC, et al. Posttraumatic stress and marijuana use coping motives: the mediating role of distress tolerance. Journal of Anxiety Disorders. 2011;25:437–443. doi: 10.1016/j.janxdis.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.May LT, Leach K, Sexton PM, et al. Allosteric modulation of G protein-coupled receptors. Annual Review of Pharmacology and Toxicology. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 111.Price MR, Baillie GL, Thomas A, et al. Allosteric modulation of the cannabinoid CB1 receptor. Molecular Pharmacology. 2005;68(5):1484–1495. doi: 10.1124/mol.105.016162. [DOI] [PubMed] [Google Scholar]
- 112.Ahn K, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces a CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization and Gi-independent ERK1/2 activation. Journal of Biological Chemistry. 2012;287:12070–12082. doi: 10.1074/jbc.M111.316463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gamage TF, Ignatowska-Jankowska BM, Wiley JL, et al. In-vivo pharmacological evaluation of the CB1-receptor allosteric modulator Org-27569. Behavioral Pharmacology. 2014;25(2):182–185. doi: 10.1097/FBP.0000000000000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Horswill JG, Bali U, Shaaban S, et al. PSNCBAM-1, a novel allosteric antagonist at cannbinoid CB1 receptors with hypophagic effects in rats. British Journal of Pharmacology. 2007;152:805–814. doi: 10.1038/sj.bjp.0707347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Piscitelli F, Ligresti A, La Regina G, et al. Indole-2-carboxamides as allosteric modulators of the cannabinoid CB1 receptor. Journal of Medicinal Chemistry. 2012;55:5627–5631. doi: 10.1021/jm201485c. [DOI] [PubMed] [Google Scholar]
- 116.Pamplona FA, Ferreira J, Menezes de Lima J, Octavio, et al. Anti-inflammatory lipoxin A4 is an endogenous allosteric enhancer of CB1 cannabinoid receptor. Proceedings of the National Academy of Sciences. 2012;109(51):21134–21139. doi: 10.1073/pnas.1202906109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pertwee RG. Lipoxin A4 is an allosteric endocannabinoid that strengthens anandamide-induced CB1 receptor activation. Proceedings of the National Academy of Sciences. 2012;109(51):20781–20782. doi: 10.1073/pnas.1218529110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Navarro HA, Howard JL, Pollard GT, et al. Positive allosteric modulation of the human cannabinoid (CB1) receptor by RTI-371, a selective inhibitor of the dopamine transporter. British Journal of Pharmacology. 2009;156:1178–1184. doi: 10.1111/j.1476-5381.2009.00124.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shinba T, Ozawa N, Yoshii M, et al. Delayed increase of brain noradrenaline after acute footshock stress in rats. Neurochem Res. 2010;35(3):412–417. doi: 10.1007/s11064-009-0070-1. [DOI] [PubMed] [Google Scholar]
- 120.Cassens G, Roffman M, Kuruc A, et al. Alterations in brain norepinephrine metabolism induced by environmental stimuli previously paired with inescapable shock. Science. 1980;209(4461):1138–1140. doi: 10.1126/science.7403874. [DOI] [PubMed] [Google Scholar]
- 121.Flugge G, Van Kampen M, Mijnster MJ. Perturbations in brain monoamine systems during stress. Cell Tissue Res. 2004;315(1):1–14. doi: 10.1007/s00441-003-0807-0. [DOI] [PubMed] [Google Scholar]
- 122.Hill MN, McEwen BS. Involvement of the endocannabinoid system in the neurobehavioural effects of stress and glucocorticoids. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34(5):791–797. doi: 10.1016/j.pnpbp.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gorzalka BB, Hill MN, Hillard CJ. Regulation of endocannabinoid signaling by stress: implications for stress-related affective disorders. Neurosci Biobehav Rev. 2008;32(6):1152–1160. doi: 10.1016/j.neubiorev.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 124.McLaughlin RJ, Hill MN, Gorzalka BB. Monoaminergic neurotransmission contributes to cannabinoid-induced activation of the hypothalamic-pituitary-adrenal axis. European journal of pharmacology. 2009;624(1–3):71–76. doi: 10.1016/j.ejphar.2009.09.055. [DOI] [PubMed] [Google Scholar]
- 125.Abercrombie ED, Jacobs BL. Single-unit response of noradrenergic neurons in the locus coeruleus of freely moving cats. II. Adaptation to chronically presented stressful stimuli. J Neurosci. 1987;7(9):2844–2848. doi: 10.1523/JNEUROSCI.07-09-02844.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ferry B, Roozendaal B, McGaugh JL. Role of norepinephrine in mediating stress hormone regulation of long-term memory storage: a critical involvement of the amygdala. Biol Psychiatry. 1999;46(9):1140–1152. doi: 10.1016/s0006-3223(99)00157-2. [DOI] [PubMed] [Google Scholar]
- 127.Nestler EJ, Alreja M, Aghajanian GK. Molecular control of locus coeruleus neurotransmission. Biol Psychiatry. 1999;46(9):1131–1139. doi: 10.1016/s0006-3223(99)00158-4. [DOI] [PubMed] [Google Scholar]
- 128.Page ME, Valentino RJ. Locus coeruleus activation by physiological challenges. Brain Res Bull. 1994;35(5–6):557–560. doi: 10.1016/0361-9230(94)90169-4. [DOI] [PubMed] [Google Scholar]
- 129.Sands SA, Strong R, Corbitt J, et al. Effects of acute restraint stress on tyrosine hydroxylase mRNA expression in locus coeruleus of Wistar and Wistar-Kyoto rats. Brain Res Mol Brain Res. 2000;75(1):1–7. doi: 10.1016/s0169-328x(99)00255-7. [DOI] [PubMed] [Google Scholar]
- 130.Valentino RJ, Curtis AL, Page ME, et al. Activation of the locus ceruleus brain noradrenergic system during stress: circuitry, consequences, and regulation. Adv Pharmacol. 1998;42:781–784. doi: 10.1016/s1054-3589(08)60863-7. [DOI] [PubMed] [Google Scholar]
- 131.Steiner MA, Wotjak CT. Role of the endocannabinoid system in regulation of the hypothalamic-pituitary-adrenocortical axis. Prog Brain Res. 2008;170:397–432. doi: 10.1016/S0079-6123(08)00433-0. [DOI] [PubMed] [Google Scholar]
- 132.Hill MN, Carrier EJ, Ho WS, et al. Prolonged glucocorticoid treatment decreases cannabinoid CB1 receptor density in the hippocampus. Hippocampus. 2008;18(2):221–226. doi: 10.1002/hipo.20386. [DOI] [PubMed] [Google Scholar]
- 133.Hill MN, Gorzalka BB. Enhancement of anxiety-like responsiveness to the cannabinoid CB(1) receptor agonist HU-210 following chronic stress. Eur J Pharmacol. 2004;499(3):291–295. doi: 10.1016/j.ejphar.2004.06.069. [DOI] [PubMed] [Google Scholar]
- 134.Wong ML, Kling MA, Munson PJ, et al. Pronounced and sustained central hypernoradrenergic function in major depression with melancholic features: relation to hypercortisolism and corticotropin-releasing hormone. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(1):325–330. doi: 10.1073/pnas.97.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lehner M, Wisłowska-Stanek A, Płaznik A. [Extinction of emotional response as a novel approach of pharmacotherapy of anxiety disorders] Psychiatr Pol. 2009;43(6):639–653. [PubMed] [Google Scholar]
- 136.Jovanovic T, Ressler KJ. How the neurocircuitry and genetics of fear inhibition may inform our understanding of PTSD. Am J Psychiatry. 2010;167(6):648–662. doi: 10.1176/appi.ajp.2009.09071074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.McGaugh JL, Cahill L, Roozendaal B. Involvement of the amygdala in memory storage: interaction with other brain systems. Proc Natl Acad Sci U S A. 1996;93(24):13508–13514. doi: 10.1073/pnas.93.24.13508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mueller D, Cahill SP. Noradrenergic modulation of extinction learning and exposure therapy. Behav Brain Res. 2010;208(1):1–11. doi: 10.1016/j.bbr.2009.11.025. [DOI] [PubMed] [Google Scholar]
- 139.Mueller D, Porter JT, Quirk GJ. Noradrenergic signaling in infralimbic cortex increases cell excitability and strengthens memory for fear extinction. J Neurosci. 2008;28(2):369–375. doi: 10.1523/JNEUROSCI.3248-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Taylor HR, Freeman MK, Cates ME. Prazosin for treatment of nightmares related to posttraumatic stress disorder. Am J Health Syst Pharm. 2008;65(8):716–722. doi: 10.2146/ajhp070124. [DOI] [PubMed] [Google Scholar]
- 141.Byers MG, Allison KM, Wendel CS, et al. Prazosin versus quetiapine for nighttime posttraumatic stress disorder symptoms in veterans: an assessment of long-term comparative effectiveness and safety. J Clin Psychopharmacol. 2010;30(3):225–229. doi: 10.1097/JCP.0b013e3181dac52f. [DOI] [PubMed] [Google Scholar]
- 142.Campolongo P, Roozendaal B, Trezza V, et al. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc Natl Acad Sci USA. 2009;106(12):4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Thomas A, Baillie GL, Phillips AM, et al. Cannabidiol displays unexpectidly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro . British Journal of Pharmacology. 2007;150:613–623. doi: 10.1038/sj.bjp.0707133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hayakawa K, Mishima K, Hazekawa M, et al. Cannabidiol potentiates the pharmacological effects of Delta(9)-tetrahydrocannabinol via CB(1) receptor-dependent mechanism. Brain Research. 2008;1188:157–164. doi: 10.1016/j.brainres.2007.09.090. [DOI] [PubMed] [Google Scholar]
- 145.Bisogno T, Hanus L, De Petrocellis L, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysi of anandamide. British Journal of Pharmacology. 2001;134:845–852. doi: 10.1038/sj.bjp.0704327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Campolongo P, Roozendaal B, Trezza V, et al. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(12):4888–4893. doi: 10.1073/pnas.0900835106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Duvarci S, Pare D. Glucocorticoids enhance the excitability of principal basolateral amygdala neurons. Journal of Neuroscience. 2007;27:4482–4491. doi: 10.1523/JNEUROSCI.0680-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Valentino RJ, Van Bockstaele EJ. Convergent regulation of locus coeruleus activity as an adaptive response to stress. European Journal of Pharmacology. 2008;583(2–3):194–203. doi: 10.1016/j.ejphar.2007.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Di Marzo V. Targeting the endocannabinoid system: to enahnce or reduce? Nature Reviews Drug Discovery. 2008;7:438–455. doi: 10.1038/nrd2553. [DOI] [PubMed] [Google Scholar]
- 150.Saito VM, Wotjak CT, Moreira FA. Pharmacological exploitation of the endocannabinoid system: new perspectives for the treatment of depression and anxiety disorders? Revista Brasileira de Psiquiatria. 2010;32:S7–S14. [PubMed] [Google Scholar]
- 151.Murataeva N, Mackie K, Straiker A. The CB2-preferring agonist JWH015 also potently and efficaciously activates CB1 in autaptic hippocampal neurons. Pharmacological Research. 2012;66(5):437–442. doi: 10.1016/j.phrs.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ross RA, Brockie HC, Stevenson LA, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. British Journal of Pharmacology. 1999;126(3):665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.An Eight-week Study of SSR411298 as Treatment for Major Depressive Disorder in Elderly Patients (FIDELIO) U.S. National Institutes of Health; Sanofi. Bethesda, MD: National Library of Medicine (US); 2013. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/ct2/show/NCT00822744. NLM Identifier: NCT00822744. [Google Scholar]
- 154.Long JZ, Li W, Booker L, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nature Chemical Biology. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bisogno T, Cascio MG, Saha B, et al. Development of the first potent and specific inhibitors of endocannabinoid biosynthesis. Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 2006;1761(2):205–212. doi: 10.1016/j.bbalip.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 156.Rabinak CA, Angstadt M, Sripada CS, et al. Cannabinoid facilitation of fear extinction memory recall in humans. Neuropharmacology. 2013;64:396–402. doi: 10.1016/j.neuropharm.2012.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Add on Study on d9-THC Treatment for Posttraumatic Stress Disorders (PTSD) (THC_PTSD) U.S. National Institutes of Health; Hadassah Medical Organization. Bethesda, MD: National Library of Medicine (US); 2012. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/show/NCT00965809. NLM Identifier: NCT00965809. [Google Scholar]
- 158.Integrated CBT for Cannabis Dependence With Co-occurring Anxiety Disorders. U.S. National Institutes of Health; Louisiana State University and A&M College. Bethesda, MD: National Library of Medicine (US); 2013. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/ct2/show/NCT01875796. NLM Identifier: NCT01875796. [Google Scholar]
- 159.A Randomized Study of Sativex on Cognitive Function and Mood: Multiple Sclerosis Patients. U.S. National Institutes of Health; GW Pharmaceuticals Ltd. Bethesda, MD: National Library of Medicine (US); 2014. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/ct2/show/NCT01964547. NLM Identifier: NCT01964547. [Google Scholar]
- 160.Evaluation of the antipsychotic efficacy of cannabidiol in acute schizophrenic psychosis (CBD-CT1) U.S. National Institutes of Health; University of Cologne. Bethesda, MD: National Library of Medicine (US); 2008. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/show/NCT00628290. NLM Identifier: NCT00628290. Bethesda, MD: National Library of Medicine (US) [Google Scholar]
- 161.Efficacy and Safety of AVE1625 as a Co-treatment With Antipsychotic Therapy in Schizophrenia (CONNECT) U.S. National Institutes of Health; Sanofi. Bethesda, MD: National Library of Medicine (US); 2010. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/ct2/show/NCT00439634. NLM Identifier: NCT00439634. [Google Scholar]
- 162.Evaluation Study of New Compounds With Potential Use in Schizophrenia (EICAS) U.S. National Institutes of Health; Central Institute of Mental Health, Mannheim. Bethesda, MD: National Library of Medicine (US); 2013. [[Last accessed 7 July 2014]]. Available at: http://clinicaltrials.gov/ct2/show/NCT00916201. NLM Identifier: NCT00916201. [Google Scholar]