

Background: Thyroid hormone (TH) and FOXO1 share similar transcriptional networks. However, TH regulation of FOXO1 activity is not well understood.
Results: TH decreased RICTOR acetylation and MTORC2/AKT activity by SIRT1 activation and reduced FOXO1 phosphorylation.
Conclusion: TH co-regulated transcription of FOXO1 target genes via RICTOR deacetylation.
Significance: Downstream metabolic effects by TH can post-translationally activate other transcription factors.
Keywords: carbohydrate metabolism, gene regulation, gluconeogenesis, thyroid hormone, transcription coregulator, AKT, MTORC2, RICTOR, insulin signaling
Abstract
MTORC2-AKT is a key regulator of carbohydrate metabolism and insulin signaling due to its effects on FOXO1 phosphorylation. Interestingly, both FOXO1 and thyroid hormone (TH) have similar effects on carbohydrate and energy metabolism as well as overlapping transcriptional regulation of many target genes. Currently, little is known about the regulation of MTORC2-AKT or FOXO1 by TH. Accordingly, we performed hepatic transcriptome profiling in mice after FOXO1 knockdown in the absence or presence of TH, and we compared these results with hepatic FOXO1 and THRB1 (TRβ1) ChIP-Seq data. We identified a subset of TH-stimulated FOXO1 target genes that required co-regulation by FOXO1 and TH. TH activation of FOXO1 was directly linked to an increase in SIRT1-MTORC2 interaction and RICTOR deacetylation. This, in turn, led to decreased AKT and FOXO1 phosphorylation. Moreover, TH increased FOXO1 nuclear localization, DNA binding, and target gene transcription by reducing AKT-dependent FOXO1 phosphorylation in a THRB1-dependent manner. These events were associated with TH-mediated oxidative phosphorylation and NAD+ production and suggested that downstream metabolic effects by TH can post-translationally activate other transcription factors. Our results showed that RICTOR/MTORC2-AKT can integrate convergent hormonal and metabolic signals to provide coordinated and sensitive regulation of hepatic FOXO1-target gene expression.
Introduction
FOXO1 has been identified to be an important downstream target of the insulin/IGF-1/PI3K signaling pathway that modulates various metabolic pathways, including carbohydrate and insulin signaling (1–5). FOXO transcription factors have wide, but variable, expression in tissues throughout the body. In particular, FOXO1 is highly expressed in insulin-responsive tissues such as adipose tissue, muscle, and liver (5, 6). Furthermore, FOXO1 is localized in different subcellular compartments and is sequestered in the cytoplasm in its inactive state. Its nuclear localization and transcriptional activity are regulated by a complex set of post-translational modifications involving dephosphorylation and deacetylation that are controlled by AKT/PKB and SIRT1 activities, respectively (5, 7), whereas mechanistic target of rapamycin complex 2 (MTORC2) regulates the action of insulin on AKT through RICTOR (8). FOXOs also can associate with a variety of unrelated transcription factors/co-regulators (such as HNF4A, CEBP, ESR1, and ESRRA, etc.) to regulate transcription of its target genes (9). Thus, the particular complement of transcription factors expressed in a particular cell type may potentially influence FOXO1 activity. Moreover, FOXO1-mediated transcription can be regulated by other signaling pathways and/or transcription factors. Although the regulation of FOXO1 phosphorylation by insulin during normal carbohydrate metabolism and diabetes is well characterized (1, 5); currently, little is known about FOXO1 regulation by nuclear hormones.
Thyroid hormones (THs,2 active form-T3, and prohormone-T4) are master regulators of energy metabolism in vertebrates (10–12). TH controls carbohydrate metabolism, energy expenditure, and body lipid content (13, 14). Excess TH leads to a hypermetabolic state that is characterized by increased resting energy expenditure, weight loss, lipolysis, gluconeogenesis, decreased cholesterol, and hepatic insulin resistance. Additionally, thyroid dysfunction results in altered intrahepatic as well as systemic lipid and carbohydrate metabolism of nutrients (11). The actions of TH are mainly mediated through TH receptors (THRs) that belong to the nuclear receptor superfamily (15). THRs are intracellular DNA-binding proteins that function as hormone-responsive transcription factors by binding to thyroid hormone-response elements (TREs) located in the promoter regions of target genes (15). Hormone-bound THRs then activate transcription through recruitment of co-activators and RNA polymerase II. There are two major isoforms of THR, THRA1 (TRα1) and THRB1, that are differentially expressed. Although THRA1 and THRB1 are ubiquitously expressed, THRA1 mRNA is predominantly found in the cardiac and skeletal muscles, whereas THRB1 mRNA is more highly expressed in brain, kidney, and liver (16, 17). TH directly, and via other co-regulators such as PPARGC1A and FOXO1, increases the transcription of genes involved in various metabolic pathways such as gluconeogenesis, β-oxidation, and fatty acid synthesis (11).
Although the physiological effects of TH on metabolism have been known for decades, mechanistic studies on the molecular mechanisms of TH-mediated metabolism have been limited (11, 12, 18). In this connection, it is interesting to note that FOXO1 and TH both regulate several of the same key genes involved in gluconeogenesis (19, 20). Currently, little is known about how TH interacts with other cell signaling pathways to activate transcription factors such as FOXO1. This issue is particularly germane because TH regulates the transcription of a large number of genes; however, only a subset of these genes have bona fide TREs suggesting that these genes may be co-regulated by other transcription factors that are modulated by TH (21–24). Therefore, we hypothesized that TH and FOXO1 worked in concert or TH activated FOXO1 to direct its action on metabolic target genes.
Here, we show that TH regulates transcription of a subset of hepatic FOXO1 target genes by SIRT1-MTORC2-AKT signaling to activate the FOXO1 transcription factor. Our findings demonstrate a novel transcriptional pathway mediated by TH to integrate cell metabolic status with the regulation of FOXO1 transcription factor activity.
Experimental Procedures
Reagents
Triiodothyronine (T3), LY-294002 hydrochloride, Ex527 (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide), insulin, fetal bovine serum, and DAPI were purchased from Sigma. Phenol red-free cell culture media were purchased from Invitrogen. FLAG® immunoprecipitation kit (FLAGIPT1) was from Sigma. For Western blotting, antibodies recognizing p-AKT (4060), AKT (4691), p-FOXO1 (9261), FOXO1 (2880), RICTOR (2114), MTOR (2983), and GAPDH (5174) were purchased from Cell Signaling Technologies, and antibodies recognizing THRB1 (sc-737x and 738x), histone H3 (sc-8654), β-actin (sc-8432), as well as HRP-conjugated secondary antibodies recognizing mouse (sc-2954) and rabbit (sc-2955) IgG were from Santa Cruz Biotechnology. Antibody recognizing SIRT1 (6137-100) was from BioVision. For immunofluorescence and immunoprecipitation, antibodies recognizing FOXO1 (sc-11350 X) were from Santa Cruz Biotechnology, and for immunoprecipitation, IgG (I5006), and acetyl-lysine (ab21623) were from Abcam.
Cell Culture and Maintenance
In this study we used HepG2 cells, a well characterized cell line derived from a human hepatocellular carcinoma that retains many liver- specific functions. HepG2 cells display a detectable response to thyroid hormone confirming that the necessary auxiliary machinery exists in these cells (21). We used isogenic HepG2 cell lines that ectopically express THRB1 (THRB1-HepG2 cells) or empty expression plasmid that contains no receptor (NR-HepG2 cells) (a kind gift from Prof. Martin L. Privalsky, University of California at Davis). These HepG2 cells were previously characterized and used to study TH action on its target genes (19, 21, 25). Stable ectopic expression of THRB1 in HepG2 cells resulted in enhanced T3 response on gene activation when compared with empty (no receptor; NR) expression plasmids (21). All HepG2 cell lines were cultured and maintained as described earlier (19). For T3 treatments, all cells were grown for at least 3 days in DMEM containing 10% Dowex-stripped FBS and 1× penicillin/streptomycin (normal T3-depleted DMEM) before adding T3 (100 nm; unless mentioned otherwise) with or without other compounds (LY292004, 5 μm) at indicated durations in the respective figures. Primary mouse hepatocytes were isolated from male C57BL/6 mice (8–10 weeks old) using standard two-step collagenase perfusion method, as mentioned previously (26), and cultured in normal T3-depleted DMEM as mentioned above.
Animals
Male C57BL/6 mice (6–8 weeks old) were purchased and housed in hanging polycarbonate cages under a 12-h light/12-h dark cycle at 23 °C with food and water available ad libitum. All cages contained shelters and nesting material. Hyperthyroidism was induced and confirmed as described earlier (19). During the course of treatment, animals were monitored daily for their general health and weight. The THRB null (Thrb−/−) mice, which lack both THRB1 and THRB2 isoforms, were in a C57BL/6:129sv mixed background (27). Thrb−/− mice and wild type (Thrb+/+) mice of the same strain were treated with TH (a mix of T3 = 6.5 μg per mouse and thyroxine = 7.8 μg per mouse) for 5 days (28). Liver-specific Thrb−/− mouse liver samples were generated as described elsewhere (29, 30) and used in co-immunoprecipitation experiments.
All mice were maintained according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication 1.0.0; revised 2011), and experiments were approved by the IACUCs at the University of Pennsylvania, the NCI (National Institutes of Health), and the Duke-NUS Graduate Medical School.
Immunofluorescence Staining and Confocal Imaging
Prior to treatment, cells were seeded on chambered slides. Following treatment, cells were washed in PBS, fixed for 15 min in 4% formaldehyde, washed, and blocked in PBST containing 1% normal goat serum for 1 h. Cells were then incubated with FOXO1 antibody overnight at 4 °C, washed three times with PBS, and then incubated for 2 h at room temperature with Alexa Fluor® 594 secondary antibody (Molecular Probes, Invitrogen). Cells were washed once and then treated with DAPI at 1:3000 dilution in PBS for 15 min. Coverslips were mounted using Vectashield mounting media (Invitrogen) and visualized and captured using an LSM710 Carl Zeiss confocal microscope. After acquisition, pseudo colors were given to the pictures to get better overlay visualization in ZEN 2012 SP1 (black edition) (Carl Zeiss) software.
Genetic Knockdown in Vivo and in Vitro Using siRNA
SiStablein vivo siRNA for mouse FOXO1 (GAGCGUGCCCCUACUUCAAGUU) or siGenome non-targeting siRNA (Dharmacon; Thermo Scientific) was used to knock down FOXO1 preferentially in the liver as described earlier (11). Stealth siRNA duplex oligoribonucleotides (a set of three siRNAs) targeting FOXO1, SIRT1, and RICTOR were procured from Invitrogen (Life Technologies, Inc.) and used per the manufacturer's instructions. Transfections were carried out in HepG2 cells in a 6-well plate using 10 nm of the above indicated siRNAs and negative control siRNA with Lipofectamine RNAiMAX (Invitrogen, Life Technologies, Inc.) following the reverse transfection protocol as provided by the manufacturer. For immunoprecipitation analysis in siRNA knockdown conditions, HepG2 cells expressing THRB1 were transfected in 100-mm culture dishes, using Lipofectamine RNAiMAX with the above-mentioned siRNAs according to the manufacturer's protocol. After 48 h of transfection, cells were subjected to T3 (100 nm) treatment in normal T3-depleted medium. After 24 h of treatment, total RNA or protein was isolated for further analysis.
Gene Overexpression Studies in Vitro
To overexpress FOXO1, SIRT1, RICTOR, and myrAKT in HepG2 cells expressing THRB1, we used FLAG-FKHR, FLAG-FKHR AAA (gift from Kunliang Guan, Addgene plasmids 13507 and 13508), FLAG-SIRT1 (gift from Michael Greenberg; Addgene plasmid 1791), myc-RICTOR corrected (gift from David Sabatini; Addgene plasmid 11367), and 1036 pcDNA3 Myr HA Akt1 (gift from William Sellers; Addgene plasmid 9008), respectively. All overexpression constructs were described previously (31–34) and purchased from Addgene (non-profit repository). Transfections were carried out in HepG2 cells in a 12-well plate using 1 μg of the above indicated plasmids or empty vector along with Lipofectamine 3000 (Invitrogen; Life Technologies, Inc.) following the reverse transfection protocol as provided by the manufacturer. After 48 h of transfection, cells were subjected to T3 (100 nm) treatment in normal T3-depleted medium as described above. After 24 h of treatment, total RNA or protein was isolated for further analysis. For co-immunoprecipitation of FLAG-SIRT1, transfection was done in 100-mm culture dishes using the above-mentioned protocol.
Pharmacological Inhibition of SIRT1 and AKT
Ex527 (intraperitoneal 0.8 mg/day/100 g body weight, for 3 days) was used to inhibit SIRT1 activity in vivo (35). After the first injection of Ex527, mice were injected subcutaneously with T3 (10 μg/kg body weight/day for 2 days) along with Ex527. After 3 days, animals were euthanized, and the liver tissues were subjected to Western blot analysis. LY-294,002 hydrochloride was used (5 mm) for 24 h to inhibit AKT in THRB1-HepG2 cells with or without T3 (100 nm).
Western Blotting
Cultured cells or 50 mg of liver tissues were lysed using CelLyticTM mammalian cell lysis/extraction reagent (C2978; Sigma). An aliquot was removed, and protein concentrations were measured using the BCA kit (Bio-Rad). Western blotting was performed using standard protocol. Laemmli sample buffer (1×; Bio-Rad) added to the protein samples was then heated to 100 °C for 5 min. These denatured samples were separated by SDS-PAGE and transferred immediately onto polyvinylidene difluoride membranes (Bio-Rad) using 1× Towbin buffer (25 mmol/liter Tris, pH 8.8, 192 mmol/liter glycine, 15% v/v methanol) using Trans-Blot® TurboTM transfer system (Bio-Rad). Membranes were blocked in 5% milk and subsequently were incubated in 1% w/v bovine serum albumin in TBST (1× TBS with 0.1% Tween 20) with specific antibodies overnight at 4 °C. After overnight incubation in primary antibodies, membranes were washed three times in TBST and subsequently incubated with species-appropriate, peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 h. Blots were washed three times with TBST and once with TBS without Tween and developed using an enhanced chemiluminescence system (GE Healthcare). Images were captured on the Gel-Doc system (Bio-Rad), and densitometry analysis was performed using ImageJ software (National Institutes of Health). Integrated density of phospho-modification of target proteins was normalized with its total protein levels, and the target protein was normalized with the indicated internal controls (β-actin or GAPDH); the mean was plotted in graphs.
Co-immunoprecipitation (Co-IP) Analysis
Immunoprecipitation was performed using the immunoprecipitation starter pack (GE Healthcare) as per the manufacturer's protocol. 100 nm trichostatin A was added in non-denaturing lysis buffer to inhibit histone deacetylase activity other than the SIRTs in lysate. Antibodies against IgG, and acetyl-lysine were used for pulldown assay. For co-immunoprecipitation, FLAG-SIRT1 was overexpressed in HepG2 cells expressing THRB1, and Co-IP was performed using FLAG® immunoprecipitation kit (FLAGIPT1, Sigma) as per the manufacturer's protocol. Western blot analysis was performed as described above. In acetyl-lysine pulldown assays, Western blot for IgG only was used for normalization.
RNA Isolation and RT-qPCR Analysis for Transcript Expression
Total RNA isolation and RT-qPCR were performed as described previously (19). ACTB expression was taken for normalization, and fold change was calculated using 2−ΔΔCt. SYBR Green-optimized primers from Sigma (KiCqStartTM SYBR® Green primer, KSPQ12012) were used for the target gene analysis unless mentioned otherwise.
Transcriptome Analysis, FOXO1-ChIP-Seq, and THRB1-ChIP-Seq Data Generation
Gene expression microarray profiling was performed using MouseWG-6 version 2.0 Expression BeadChip kit (Illumina) by hybridizing RNA from liver tissues of wild type (euthyroid), wild type + T3 (hyperthyroid), and FOXO1 knockdown + T3 (Foxo1 KD hyperthyroid) mouse (n = 3). cRNA generation, labeling, and hybridization were performed at Duke-NUS Genome Biology Facility, Duke-NUS Graduate Medical School, Singapore. Gene expression signals were quantile normalized, and differentially expressed genes were identified via analysis of variance, using treatment-specific contrasts (Partek Genomics Suite software, version 6.6). Statistical significance of differentially expressed genes was ascertained in terms of the false discovery rate (36). Complete dataset was submitted to the GEO repository (GSE68803). Principal components analysis based on gene expression demonstrated a clear separation between the three experimental groups and no outliers. Microarray-derived transcript expression was validated by RT-qPCR of top hit genes. A list of putative FOXO1 target genes was extracted from the FOXO1-ChIP-Seq data reported by Shin et al. (37) and compared with gene expression data from euthyroid, hyperthyroid, and FOXO1 knockdown + hyperthyroid groups of mice. Pathway enrichment analysis was conducted via the Gene Set Enrichment Analysis tool (38) using a list of KEGG pathways extracted from the Molecular Signatures Database (39) and a custom pathway constructed from the list of FOXO1 target genes (37). Pathways with greater than 260 genes or less than 10 genes were excluded from the analysis. All analyses were conducted using mouse gene symbols. Significance of pathway enrichment was ascertained by permutation testing of gene sets and calculation of the false discovery rate (FDR q value <0.050). THRB1-ChIP-Seq data set was generated and used as described previously (37, 40).
Analysis of NAD+/NADH Ratio
NAD+/NADH cell-based assay kit (Cayman chemicals) was used to measure the NAD+/NADH ratio in cultured cells in a 96-well plate format according to the manufacturer's protocol.
Oxygen Consumption Rate (OCR) Measurement
To determine cellular oxygen consumption in HepG2 cells, the Seahorse Extracellular Flux Analyzer XF24 (Seahorse Bioscience, Billerica, MA), which measures the OCR, was used. To analyze mitochondrial ATP turnover, XF Cell Mito Stress test kit was used as per the manufacturer's protocol. In brief, prior to assay, T3-depleted medium was removed and replaced by 500 μl of assay medium at 37 °C in air with 5% CO2. Oligomycin (1 μm), which inhibits the F0 proton channel of the F0F1-ATP synthase, was employed to determine the oligomycin-independent leak of the OCR. The mitochondrial uncoupler carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP; 1 μm) was added to determine the total respiratory capacity of the mitochondrial electron transport chain. Rotenone (1 μm) and antimycin A (1 μm) was added to block complex I and complex III of electron transport chain. The following mitochondrial functional parameters were calculated as follows: (i) basal O2 consumption = baseline oxygen consumption reading per well, before compounds are injected; (ii) ATP turnover = baseline oxygen consumption reading per well (before compounds are injected) subtracted from oxygen consumption reading per well after oligomycin injection; (iii) maximum respiratory capacity = oxygen consumption reading per well (after FCCP injection) subtracted from oxygen consumption reading per well after oligomycin injection. Computed data were plotted as bar graphs.
Statistical Analysis
Individual culture experiments were performed in duplicate or triplicate and repeated at least three times independently using matched controls; the data were pooled. Results were expressed as mean ± S.D. for all in vitro experiments and expressed as mean ± S.E. for all in vivo experiments. The statistical significance of differences (p < 0.05) was assessed by two-tailed Student's t test or analysis of variance.
Results
Thyroid Hormone (T3)-regulated Expression of a Subset of FOXO1 Target Genes
TH and FOXO1 regulate overlapping genes, particularly those involved in glucose metabolism. To better understand the extent of this overlapping transcriptional regulation, and their potential cross-talk at the genomic level, we performed gene expression microarrays in livers from euthyroid, hyperthyroid, and Foxo1 siRNA knockdown hyperthyroid mice (Fig. 1A) to observe global changes in gene expression regulated by both T3 and FOXO1. Additionally, we determined the identities and number of FOXO1 target genes containing FOXO1 binding in their promoters that also were regulated by T3. Accordingly, we compared the microarray data set from Foxo1 knockdown mice (±T3) with a Foxo1-ChIP-Seq genomic data set from hepatic chromatin as reported previously by Shin et al. (37). Based on their results, we created a group of 222 genes containing Foxo1 binding as a “custom Foxo1 ChIP-Seq gene pathway” and subjected it to gene-set enrichment analysis together with the standard set of KEGG pathways downloaded from MSigDB (Fig. 1B). The custom pathway was the third most enriched pathway based upon the normalized pathway enrichment scores (false discovery rate; FDR q value = 0.000) when hepatic gene expression from hyperthyroid mice was compared with hyperthyroid Foxo1 knockdown mice. These results show that Foxo1 knockdown affected target gene expression in hyperthyroid mice. This custom Foxo1 ChIP-Seq gene pathway also was significantly enriched (FDR q value = 0.045) when hepatic gene expressions from hyperthyroid mice were compared with euthyroid mice, suggesting that T3 regulated the expression of genes containing Foxo1-binding sites.
FIGURE 1.

Thyroid hormone (T3)-regulated gene expression of a subset of Foxo1 target genes. A, Western blot analysis to confirm Foxo1 knockdown in mouse liver tissue. Lane I, control siRNA injected euthyroid (EuTH) mice; lane II, control siRNA injected hyperthyroid (HyperTH) mice; lane III, Foxo1 siRNA injected HyperTH mice. B, gene ontology-KEGG Pathway analysis of the genes obtained by custom Foxo1-ChIP-Seq genes (as described under “Experimental Procedures”) indicating nominal significance and false discovery rate. C, Venn diagram for overlap analysis of core-enriched significantly differentially expressed genes under custom Foxo1-ChIP-Seq gene pathway data set in HyperTH versus EuTH (TH); HyperTH versus Foxo1 knockdown + HyperTH (Foxo1). Venn diagram was formed as described under “Experimental Procedures” by integrating the data from core-enriched genes from gene expression microarray data. Green circle shows Foxo1 up-regulated genes (75 genes). Red circle shows TH up-regulated genes (37). Brown circle shows TH-Foxo1 co-regulated genes (27). D, overlap analysis of TH-only regulated (10) and TH-Foxo1 co-regulated genes (27) in the liver tissues from HyperTH versus EuTH (TH) and HyperTH versus Foxo1 knockdown + HyperTH mice (Foxo1); and the genes have THRB1 binding to their locus (Thrb1, black circle). The data were obtained by comparisons from transcriptome analysis, Foxo1-ChiP-Seq, and THRB1-Chip-seq analysis. Venn diagram was formed as described under “Experimental Procedures” by integrating the data from core-enriched genes from gene expression microarray data. Brown circle shows TH-Foxo1 co-regulated genes (16 + 11 = 27). Red circle shows TH regulated genes (8 + 2 = 10). Black circle shows THRB1 binding in the genes (2 + 16 = 18). E, transcript expression validation by RT-qPCR for T3 as well as Foxo1 target genes in the liver tissues of mice as described in A. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.E. F and G, transcript expression validation by RT-qPCR for T3 alone (G) or Foxo1 alone (H) regulated genes in the liver tissues of mice as described in A. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.E. H, transcript expression by RT-qPCR for T3 and Foxo1 target genes in the primary mouse hepatocytes and human hepatic cell line HepG2 expressing THRB1. Statistical significance was calculated as *, p < 0.05, and error bars represent ±S.D. NS, not significant.
Upon inspection, we found that a total of 75 genes within the custom Foxo1 ChIP-seq gene pathway were core-enriched (i.e. up-regulated) in hyperthyroid mice versus hyperthyroid Foxo1 knockdown mice (green circle, Fig. 1C), whereas 37 genes were core-enriched in hyperthyroid versus euthyroid mice (red circle). These latter genes were either directly or indirectly activated by T3, and they contained one or more Foxo1-binding sites in their promoter region. When we compared the two data sets of 75 and 37 genes, we found 27 overlapping genes that were co-regulated by both Foxo1 and T3 (based on their common occurrence in both data sets) (Fig. 1C). Upon examining the non-overlapping genes, 48 out of 75 genes (64.0%) in the Foxo1 knockdown hyperthyroid mice were regulated primarily by Foxo1 in the presence of T3, and 10 out of 37 genes in the T3-dependent hyperthyroid group (27.0%) were regulated by T3 alone despite having Foxo1-binding sites.
We further examined the 37 genes that either were regulated by T3 alone (subset of 10 genes) or co-regulated by both T3 as well as Foxo1 (subset of 27 genes) by analyzing a database containing hepatic Thrb1-binding sites generated by our previous ChIP-seq study (Fig. 1D; Table 1) (40). Among the 10 genes regulated by T3 alone, only two genes had Thrb1 binding within the proximal promoter or in the gene body (500 bp upstream of the transcription start site to the transcription end site) (Table 2) suggesting that other T3-regulated transcription factors/co-regulators may indirectly activate transcription of those genes. Of note, our previous study showed that almost all functional binding sites of T3-regulated target genes occurred in this proximal region of the promoter (40). Interestingly, among the 27 genes that were co-regulated by both T3 and Foxo1, we observed that 16 (60%) of these co-regulated genes had Thrb1-binding sites, and the remaining 11 (40%) genes appeared to be independent of Thrb1 binding in their proximal promoters and gene bodies. These target genes could be regulated by T3 effects on Foxo1 without Thrb1 binding or by T3-dependent activation of other co-regulators or transcription factors that act in concert with Foxo1 to regulate target gene transcription.
TABLE 1.
Thrb1-ChIP-Seq analysis in FOXO1-TH up-regulated genes (27 co-regulated genes)
Thrb1-ChIP-Seq analysis in previously known Foxo1 target genes (37) and co-regulated by TH showed two distinct groups of genes. One has Thrb1 binding in the proximal promoter region, and the other does not, yet both gene groups are co-regulated by TH and Foxo1. NA means not applicable.
| Serial no. | Gene | Thrb1 binding | No. of peaks |
|---|---|---|---|
| 1 | Cyp17a1 | + | 2 |
| 2 | Tsku | + | 16 |
| 3 | Igfbp1 | − | NA |
| 4 | Ela2a | − | NA |
| 5 | Pck1 | + | 2 |
| 6 | Eif4ebp3 | + | 2 |
| 7 | Dhcr7 | + | 1 |
| 8 | Pim3 | − | NA |
| 9 | Rab43 | + | 8 |
| 10 | Sc5d | − | NA |
| 11 | Gys2 | + | 1 |
| 12 | Mgll | + | 91 |
| 13 | Pdk4 | + | 2 |
| 14 | Tomm40l | − | NA |
| 15 | Acacb | + | 4 |
| 16 | Gdf9 | − | NA |
| 17 | Ctcfl | − | NA |
| 18 | Elmod3 | + | 4 |
| 19 | Slc25a33 | − | NA |
| 20 | Abcg5 | + | 2 |
| 21 | Lgals6 | − | NA |
| 22 | Slc24a6 | + | 111 |
| 23 | Dusp28 | − | NA |
| 24 | Sult1a1 | + | 2 |
| 25 | Klf15 | + | 1 |
| 26 | Als2cl | + | 78 |
| 27 | Map1lc3b | − | NA |
TABLE 2.
Thrb1-ChIP-Seq analysis in Foxo1-TH up-regulated genes that were regulated by TH but did not require Foxo1 binding
Thrb1-ChIP-Seq analysis in previously known Foxo1 target genes (37) that were only regulated by TH showed two distinct groups of genes. One has Thrb1 binding in the proximal promoter region, and the other does not, and both gene groups are regulated by TH. NA means not applicable.
| Serial no. | Gene | Thrb1 binding | No. of peaks |
|---|---|---|---|
| 1 | Cnih | − | NA |
| 2 | Itpa | − | NA |
| 3 | Ankrd27 | + | 2 |
| 4 | Tdg | − | NA |
| 5 | Osgin1 | + | 4 |
| 6 | Apoa4 | − | NA |
| 7 | Sqstm1 | − | NA |
| 8 | Rnf215 | − | NA |
| 9 | Cnpy3 | − | NA |
| 10 | Nodal | − | NA |
We next employed RT-qPCR analysis to validate the gene expression of several genes that were regulated by both T3 and Foxo1 and were top hits in the common data set. They included Cyp17a1, Tsku, Igfbp1, Pck1, Pim3, and Pdk4 (Fig. 1E). T3 significantly induced the mRNA expression of these genes when compared with euthyroid mice. Foxo1 knockdown in mouse liver completely inhibited T3-induced mRNA expression of these genes. Furthermore, we also analyzed top hit genes from the T3-only regulated set (i.e. Cnih, Osgin1, and Apoa4) as well as Foxo1-only regulated set (i.e. Abcg8, Fbxo31, and Retsat) (Fig. 1, F and G). Moreover, these Foxo1 target genes also were validated in primary mouse hepatocytes as well as in a HepG2 cell line that overexpressed THRB1, which retained many liver-specific metabolic functions (41) and also has been shown to have enhanced T3 response in vitro (Fig. 1H) (25).
We then examined the gene expression of PCK1 and IGFBP1, two of the 27 genes co-regulated by FOXO1 and T3. PCK1 had both THRB1- and FOXO1-binding sites in the proximal promoter and gene body, whereas IGFBP1-only had FOXO1 binding (Table 1) (37). These two genes thus are representative of the two classes of target genes that are co-regulated by both FOXO1 and T3. We then knocked down FOXO1 in HepG2 cells (either expressing no receptor control vector, NR-HepG2; or expressing THRB1, THRB1-HepG2) using siRNA as well as overexpressed wild type FOXO1 and phospho-mutant FOXO1 (AAA) (T24A/S256A/S319A) (Fig. 2, A and B). Knockdown of FOXO1 decreased induction of PCK1 and IGFBP1 gene expression by T3 (Fig. 2, C and D), whereas FOXO1 overexpression further increased T3 induction of PCK1 and IGFBP1 gene expression (Fig. 1, E and F). Overexpression of mutant FOXO1 (AAA) produced a large induction of transcription that was only slightly enhanced by T3 treatment. These data showed that T3 regulated FOXO1-mediated transcription of these two genes and further confirmed the proposed co-regulation of the overlapping group of 27 genes by T3 and FOXO1.
FIGURE 2.

Thyroid hormone (T3)-regulated gene expression of PCK1 and IGFBP1 in a FOXO1-dependent manner. A and B, FOXO1 knockdown and overexpression in HepG2 cells expressing control vector containing no receptor (NR-HepG2) or THRB1 (THRB1-HepG2) confirmed by Western blotting. ACTB, β-actin. Overexpression of wild type FOXO1 (FLAG-FOXO1) as well as phosphorylation mutant of FOXO1 (FLAG-FOXO1AAA) (T24A/S256A/S319A) in these HepG2 cells was performed. C and E show results for NR-HepG2 cells, ands D and F show results for THRB1-HepG2 cells. C and D show PCK1 and IGFBP1 transcripts expression by RT-qPCR in NR-HepG2 and THRB1-HepG2, respectively, under FOXO1 knocked down conditions. Data were calculated by comparing with siRNA control in panels C and D. T3 was used at 100 nm concentration. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. E and F, FOXO1 target genes PCK1 and IGFBP1 transcripts expression by RT-qPCR during FOXO1 overexpression in NR-HepG2 and THRB1-HepG2, respectively. Data were calculated by comparing with empty vector in panels E and F. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. NS, not significant.
T3 Increased Nuclear Localization and Inhibited AKT-dependent Phosphorylation of FOXO1
We next investigated the mechanism for T3 activation of transcription of this group of genes that were co-regulated by T3 and FOXO1. We considered whether FOXO1 nucleo-cytoplasmic localization might be regulated by T3 treatment because phosphorylation of FOXO1 by insulin signaling retains it in the cytoplasm. Interestingly, we observed that T3 markedly increased FOXO1 nuclear localization in HepG2 cells expressing THRB1 when compared with untreated controls using double label immunocytochemistry (Fig. 3, A and B). We further confirmed these results by performing nuclear fractionation, and we found that FOXO1 was significantly enriched in the nuclear fraction of T3-treated HepG2 cells in a time-dependent manner (Fig. 3C).
FIGURE 3.

Thyroid hormone (T3) increased FOXO1 nuclear localization and inhibited AKT phosphorylation. A, immunofluorescence confocal microscopy analysis of FOXO1 nuclear localization. FOXO1 was stained with Alexa Fluor® 594 secondary antibody (Molecular Probes, Invitrogen) and nucleus with DAPI and then given pseudo colors (red for nucleus and green for FOXO1) for better overlay visualization in ZEN 2012 SP1 (black edition; Carl Zeiss) software. Images were captured at ×40 magnification. Image shown in inset is enlarged view of indicated parent image. B, quantification of confocal microscopy data for the ratio of nuclear FOXO1 to total FOXO1 in HepG2 cells by computing percent co-localization, with or without T3 (100 nm) treatment. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. C, Western blot analysis of cytoplasmic and nuclear fractions to estimate FOXO1 nuclear localization and AKT/FOXO1 phosphorylation after T3 treatment in THRB1-HepG2 cells at given time points. GAPDH was used as loading control for cytosolic fraction and histone H3 for nuclear fraction. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D.
We next analyzed AKT phosphorylation status because AKT-dependent phosphorylation of FOXO1 is necessary for its cytoplasmic localization (3), and T3 significantly reduced both AKT and FOXO1 phosphorylation, which happened exclusively in the cytoplasm as we did not detect any phosphorylated AKT or FOXO1 in the nuclear fraction (Fig. 3C). We further confirmed that T3 significantly reduced AKT and FOXO1 phosphorylation in mouse liver tissues from mice treated with or without T3, primary mouse hepatocytes and HepG2 cells (Fig. 4, A–C). Furthermore, T3 decreased phosphorylation of AKT and FOXO1 in a dose- and time-dependent manner that corroborates very well with the increase in PCK1 and IGFBP1 transcription by T3 (Fig. 4, D–G).
FIGURE 4.

Thyroid hormone (T3) inhibited AKT-dependent phosphorylation and increased FOXO1 target gene expression. A–C, Western blot analysis to analyze phosphorylated Akt and Foxo1 in liver tissues (A) from euthyroid (EuTH) and hyperthyroid (HyperTH) mice, primary mouse hepatocytes (B) and HepG2 cells expressing THRB1 (C). Bar graphs below each Western blot data set represent relative densitometric measurements. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. D and E, Western blot analysis showing dose response (D) and time course (E) for T3 treatment on phosphorylated AKT and FOXO1 in THRB1-HepG2 cells. Line graphs below each Western blot data set represent relative densitometric measurements. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. ACTB, β-actin. F and G, graph showing dose response (F) and time course (G) of FOXO1 target genes PCK1 and IGFBP1 transcript expression by RT-qPCR in THRB1-HepG2, respectively. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D.
THRB1 Is Required for T3-mediated Decrease in AKT Phosphorylation and Induction of FOXO1 Target Gene Expression
We then determined whether the decreased AKT phosphorylation by T3 required THRB1 by examining phosphorylation of Akt and Foxo1 in livers from WT (Thrb+/+) and Thrb null mice (Thrb−/−) that were treated with T3 or vehicle. We observed that phosphorylation of Akt as well as Foxo1 was decreased in hyperthyroid Thrb+/+ mouse liver when compared with hypothyroid Thrb+/+ mouse liver tissues (Fig. 5, A and B). However, there was no significant difference in Akt or Foxo1 phosphorylation in the livers from hypo- and hyperthyroid Thrb−/− mice. This effect was further confirmed in control HepG2 cells that were stably transfected with a control vector that did not express receptor (NR-HepG2) versus stably transfected HepG2 cells expressing human THRB1 (THRB1-HepG2) (Fig. 5, C and D). AKT and FOXO1 phosphorylation was significantly reduced in THRB1-expressing HepG2 cells treated with T3 in NR-HepG2 cells but had little decrease after T3 treatment. Furthermore, similarly we found that T3 had a mild but significant increase in mRNA expression of FOXO1 target genes (PCK1 and IGFBP1) in NR-HepG2 cells and that was much higher in THRB1-expressing HepG2 cells (Fig. 5E). Taken together, these findings strongly suggest that THRB1 was required for reduction of AKT/FOXO1 phosphorylation and induction of FOXO1 target genes by T3.
FIGURE 5.

Thyroid hormone (T3) decreased AKT/FOXO1 phosphorylation and increased FOXO1 target gene expression in THRB1-dependent manner. A and B, Western blot analysis to confirm Thrb1 knock-out (A) in mouse liver tissues and phosphorylated Akt/Foxo1 (B) in liver tissues from hypothyroid (HypoTH) and hyperthyroid (HyperTH) of Thrb+/+ and Thrb−/− mice, respectively. Bar graphs below each Western blot data set represent relative densitometric measurements. Statistical significance was calculated as *, p < 0.05, and error bars represent ± S.E. C and D, Western blot analysis to confirm stable overexpression of THRB1 in HepG2 cells (C) and analysis of phosphorylated AKT.FOXO1 in NR-HepG2 and THRB1-HepG2 cells (D). Bar graph represents relative densitometric measurements of phosphorylated AKT and FOXO1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. E, transcript expression of FOXO1 target genes (PCK1 and IGFBP1) by RT-qPCR analysis in NR-HepG2 Versus HepG2 cells expressing THRB1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D.
T3 Increased FOXO1 Target Gene Expression in an AKT-dependent Manner
We next examined whether T3-mediated induction of FOXO1 target genes was dependent upon AKT by inhibiting AKT activity using LY292004. As seen earlier, T3 decreased AKT and FOXO1 phosphorylation. AKT inhibition by LY292004 further decreased AKT as well as FOXO1 phosphorylation by T3 (Fig. 6A). These effects correlated well with the induction of endogenous FOXO1 target gene expression. LY294002 significantly increased PCK1 and IGFBP1 mRNA levels by 2.51- and 2.18-fold, respectively (Fig. 6B). In combination with T3, LY294002 further increased their gene expression by 19.43- and 21.69-fold, respectively. We next confirmed the critical role of AKT in T3-induced activation of FOXO1-mediated transcription in cells overexpressing constitutively active AKT, i.e. myristoylated AKT (myrAKT). myrAKT overexpression significantly inhibited basal as well as T3-induced PCK1 and IGFBP1 mRNA expression (Fig. 6, C and D).
FIGURE 6.
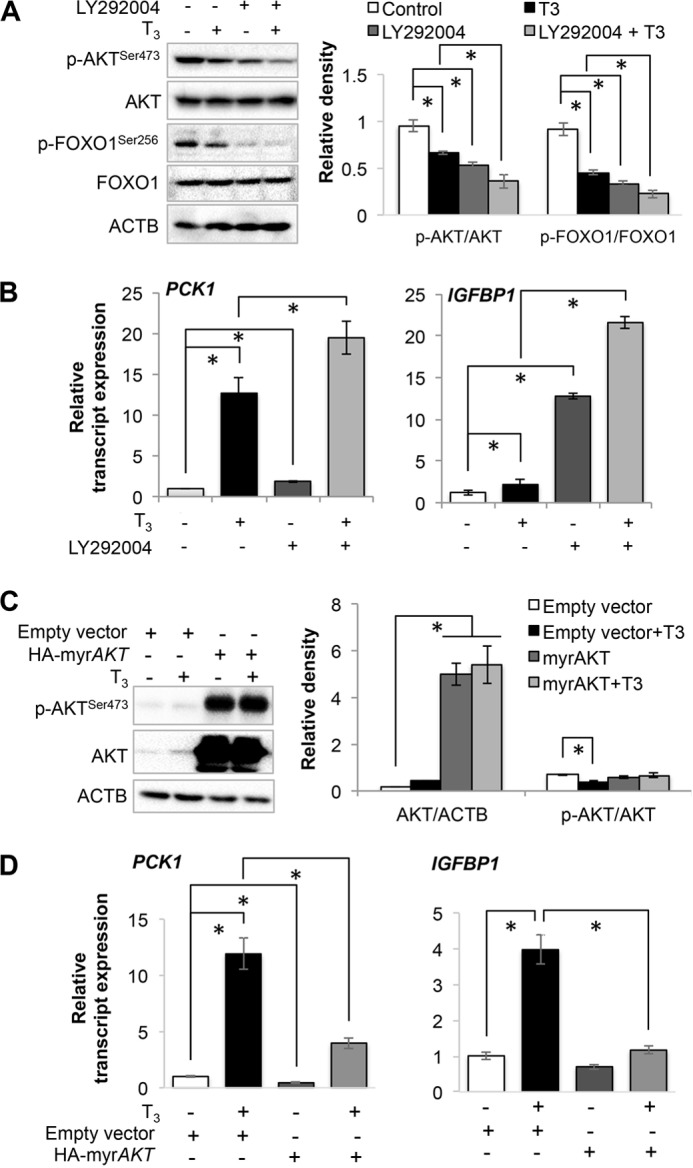
Thyroid hormone (T3) induced FOXO1 target genes in an AKT-dependent manner. A, Western blot analysis for phosphorylated AKT and FOXO1 in HepG2 cells. LY292004 compound (5 μm) was used to inhibit AKT in HepG2 cells along with or without T3 (100 nm). Bar graph represents relative densitometric measurements of phosphorylated AKT and FOXO1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. B, transcript expression of PCK1 and IGFBP1 in HepG2 cells treated with LY292004 compound and/or T3. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. C, Western blot analysis of myristoylated AKT (myrAKT), a constitutively active form of AKT, overexpression in THRB1-HepG2 cells. Bar graph represents relative densitometric measurements of total as well as phosphorylated AKT. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. D, transcript expression of PCK1 and IGFBP1 in HepG2 cells overexpressing myrAKT and treated with or without T3. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. ACTB, β-actin.
T3-induced FOXO1 Activation Required RICTOR (Regulatory Component of MTORC2) and Its Deacetylation in a SIRT1-dependent Manner
RICTOR is a key component of MTOR complex-2 (MTORC2) that phosphorylates AKT at Ser-473 (42). Accordingly, we examined whether RICTOR was required for reduction of AKT phosphorylation and FOXO1 activation by T3, and we performed RICTOR siRNA knockdown in THRB1-HepG2 cells. We observed that RICTOR knockdown significantly decreased basal AKT and FOXO1 phosphorylation (Fig. 7A). However, RICTOR knockdown did not further augment T3-mediated reduction in AKT or FOXO1 phosphorylation. Similar observations also were found in the mRNA expression of FOXO1 target genes. RICTOR knockdown increased mRNA expression of PCK1 and IGFBP1 by 14.77- and 3.35-fold, respectively (Fig. 7B), whereas T3 was unable to stimulate the gene expression of these FOXO1 target genes in the absence of RICTOR. To better understand the role of RICTOR in T3-induced deactivation of AKT, we overexpressed RICTOR in these HepG2 cells. RICTOR overexpression significantly increased AKT phosphorylation and partially blocked T3-induced reduction in AKT phosphorylation (Fig. 7C). We also confirmed that RICTOR overexpression inhibited basal (0.26- and 0.51-fold respectively) as well as T3-induced mRNA expression of PCK1 and IGFBP1 (Fig. 7D). These results demonstrated that RICTOR was involved in T3-mediated induction of these target genes.
FIGURE 7.

RICTOR was required for thyroid hormone (T3)-induced FOXO1 target genes. A, Western blot analysis for RICTOR knockdown in THRB1-HepG2 cells and phosphorylation of AKT as well as FOXO1. Bar graph represents relative densitometric measurements of phosphorylated AKT and FOXO1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. B, RT-qPCR analysis was performed to measure PCK1 and IGFBP1 transcript expression in RICTOR knockdown THRB1-HepG2 cells with/without T3 (100 nm). Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. C, Western blot analysis for RICTOR overexpression in HepG2 cells and AKT phosphorylation. Bar graph represents relative densitometric measurements of phosphorylated AKT. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. D, PCK1 and IGFBP1 transcripts expression in THRB1-HepG2 cells overexpressing RICTOR and treated with or without T3. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. E–G, immunoprecipitation-Western blot (IP-WB) analysis to observe RICTOR acetylation in NR-HepG2 versus THRB1-HepG2 cells (E), liver tissues (F) from euthyroid (EuTH) and hyperthyroid (HyperTH) mice and from Thrb+/+ and Thrb−/− mice (G), respectively. IgG-heavy chain Western blot was used to normalize acetylated lysine pulldown in E–G. Bar graph represents relative densitometric measurements for acetylated RICTOR. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. (E) or mean ± S.E. (F and G). ACTB, β-actin; NS, not significant.
Recently, RICTOR acetylation was shown to be necessary for MTORC2 activation and AKT phosphorylation (43); thus, we examined RICTOR acetylation after T3 treatment in vitro and in vivo. Interestingly, T3 decreased RICTOR acetylation in HepG2 cells as well as in mouse liver in a THRB1-dependent manner, (Fig. 7, E–G). These results suggest that T3-induced RICTOR deacetylation likely decreased MTORC2 activity and AKT phosphorylation.
We recently showed that T3 stimulated SIRT1-dependent FOXO1 deacetylation (19); thus, we considered whether SIRT1 was playing a role in T3-dependent RICTOR deacetylation, so we used two-step IP-WB and found that T3 decreased RICTOR acetylation in a SIRT1-dependent manner in THRB1-HepG2 cells (Fig. 8A). Furthermore, this effect was confirmed in vivo because the Sirt1 inhibitor, Ex527, blocked the T3-mediated decrease in Rictor acetylation (Fig. 8B). Furthermore, to observe whether SIRT1 interacted with MTORC2 complex, we thus performed co-immunoprecipitation of FLAG-SIRT1 with the MTORC2 components, MTOR and RICTOR (Fig. 8C). We observed that T3 increased the physical interaction of MTOR and RICTOR with SIRT1, thereby raising the possibility that the latter might deacetylate RICTOR. When taken together with the previous data, these data strongly support the notion that TH stimulates FOXO1 target gene transcription by activating FOXO1 through decreased phosphorylation by AKT that is mediated by SIRT1-RICTOR- (MTORC2)-AKT signaling.
FIGURE 8.

Thyroid hormone (T3)-induced RICTOR deacetylation was SIRT1-dependent. A, IP-WB analysis to observe RICTOR acetylation in SIRT1 knockdown NR-HepG2 versus THRB1-HepG2 cells treated with or without T3. B, IP-WB analysis to observe Rictor acetylation in liver tissues from euthyroid (EuTH), hyperthyroid (HyperTH), and Ex527-treated hyper-TH mice. IgG-heavy chain Western blot was used to normalize acetylated lysine pulldown in A and B. Bar graph represents relative densitometric measurements for acetylated RICTOR. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. (A) or mean ± S.E. (B). C, co-IP-WB of FLAG-tagged SIRT1 to observe SIRT1 interaction with MTORC2 components (MTOR and RICTOR) in THRB1-HepG2 cells treated with or without T3. Bar graph represents relative densitometric measurements for MTOR and RICTOR normalized to total SIRT1 pulled down. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. D, NAD+/NADH measurement in THRB1-HepG2 cells treated with or without T3. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. E, measurement of OCR in THRB1-HepG2 cells treated with or without T3 for mentioned time points using Seahorse extracellular flux analyzer. Oligomycin (Oligo.; 1 μm; F0F1-ATP synthase inhibitor), FCCP (1 μm; mitochondrial uncoupler) and rotenone (R; 1 μm) + antimycin A (A; 1 μm) (complex I and complex III inhibitors in electron transport chain, respectively) was used to analyze various parameters of mitochondrial OCR. F, computational analysis of functional parameters of mitochondrial OCR as under “Experimental Procedures,” and the data were plotted as a bar graph. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.E.
NAD+/NADH, the Key Determinant of SIRT1 Activity, Was Increased After T3 Treatment
SIRT1 is activated by NAD+ and inhibited by NADH, and the ratio of their concentration is a critical determinant of SIRT1 activity. Therefore, we next analyzed whether T3 induced intracellular changes in the ratio of NAD+/NADH concentrations because SIRT1 is a NAD+/NADH-sensitive deacetylase (44, 45). Interestingly, T3 significantly increased NAD+/NADH by 1.5-fold (Fig. 8D). Because ATP synthesis during mitochondrial oxidative phosphorylation is known to increase NAD+/NADH (46), we measured OCR by Seahorse Extracellular Flux Analyzer in THRB1-HepG2 cells (Fig. 8, E and F). T3 significantly increased basal oxygen consumption, mitochondrial ATP turnover, as well as maximum respiratory capacity in a time-dependent manner. Collectively, our data strongly suggest that increased mitochondrial ATP turnover and increased NAD+/NADH ratio by T3 led to SIRT1 activation.
T3-mediated Decrease in AKT Phosphorylation was SIRT1-mediated
Given the effects of T3 on the stimulation of SIRT1 activity and its deacetylation of RICTOR, we examined the effect of T3 and SIRT1 on MTORC2-dependent AKT and FOXO1 phosphorylation. Accordingly, we treated mice with Ex527, a pharmacological compound used to specifically inhibit Sirt1 or knocked down SIRT1 by siRNA in HepG2 cells in combination with T3 treatment. As shown in Fig. 9A, phosphorylation of Akt and Foxo1 was significantly decreased in livers from hyperthyroid mice when compared with those from euthyroid mice. Interestingly, Ex527 treatment blocked this reduction in Akt and Foxo1 phosphorylation by T3. Similar effects also were observed in THRB1-HepG2 cells as SIRT1 knockdown blocked the T3-mediated decrease in AKT and FOXO1 phosphorylation (Fig. 9B). The mRNA expression of FOXO1 target genes PCK1 and IGFBP1 also was not significantly induced by T3 under these conditions (Fig. 9C), suggesting that SIRT1 is involved in both T3-mediated induction of target gene expression as well as reduction of AKT and FOXO1 phosphorylation.
FIGURE 9.
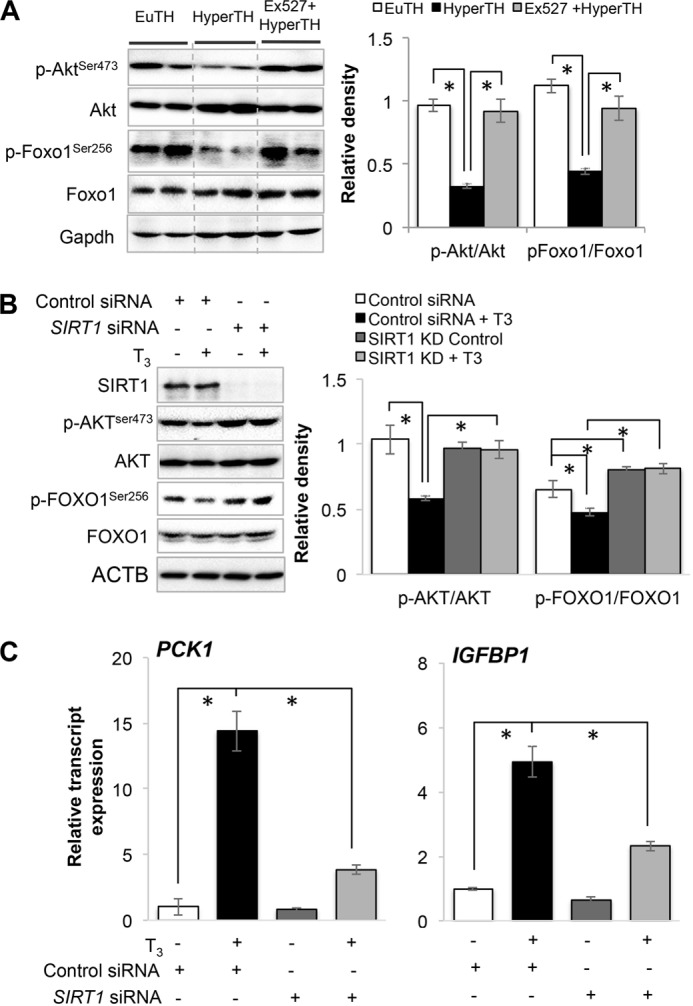
SIRT1 inhibition inversely affects thyroid hormone (T3)-dependent decrease in AKT/FOXO1 phosphorylation and FOXO1 activation. A, Western blot analysis of Akt as well as Foxo1 phosphorylation in liver tissues from euthyroid (EuTH), hyperthyroid (HyperTH), and Ex527-treated HyperTH mouse. Ex527 compound was used to inhibit Sirt1 activity in vivo. Bar graph represents relative densitometric measurements of phosphorylated Akt and Foxo1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.E. B, Western blot analysis of SIRT1 knockdown in THRB1-HepG2 cells and phosphorylation and AKT and FOXO1 in SIRT1 knockdown cells treated with or without T3 (100 nm). Bar graph represents relative densitometric measurements of phosphorylated AKT and FOXO1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. C, transcripts expression of FOXO1 target genes PCK1 and IGFBP1 using RT-qPCR in SIRT1 knockdown cells treated with or without T3. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D.
SIRT1 deacetylates FOXO1 to increase its DNA binding affinity, and AKT phosphorylates FOXO1 to retain FOXO1 in the cytoplasm (3). We thus considered whether T3 stimulation of SIRT1 effects on FOXO1 transcription was upstream or downstream of AKT phosphorylation. Accordingly, we overexpressed SIRT1 in THRB1-HepG2 cells and found that SIRT1 significantly decreased both AKT and FOXO1 phosphorylation (Fig. 10A). T3 treatment further enhanced this effect. We then co-transfected constitutively active AKT (myrAKT) and SIRT1 and measured the mRNA expression of FOXO1 target genes, PCK1 and IGFBP1. SIRT1 overexpression significantly increased basal as well as T3-induced mRNA expression of these genes (Fig. 10B). As shown previously, overexpression of myrAKT significantly inhibited basal as well as T3-induced mRNA expression of PCK1 and IGFBP1. Interestingly, co-overexpression of both myrAKT and SIRT1 inhibited the effect of SIRT1 overexpression alone. T3 induced PCK1 and IGFBP1 mRNA expression by 48.95- and 6.3-fold, whereas during co-expression of myrAKT and SIRT1, T3 only induced their expression by 15.64- and 2.15-fold. This indicates that AKT most likely functions downstream of SIRT1, although SIRT1-dependent FOXO1 deacetylation is also critical for TH-induced FOXO1 activation (19).
FIGURE 10.
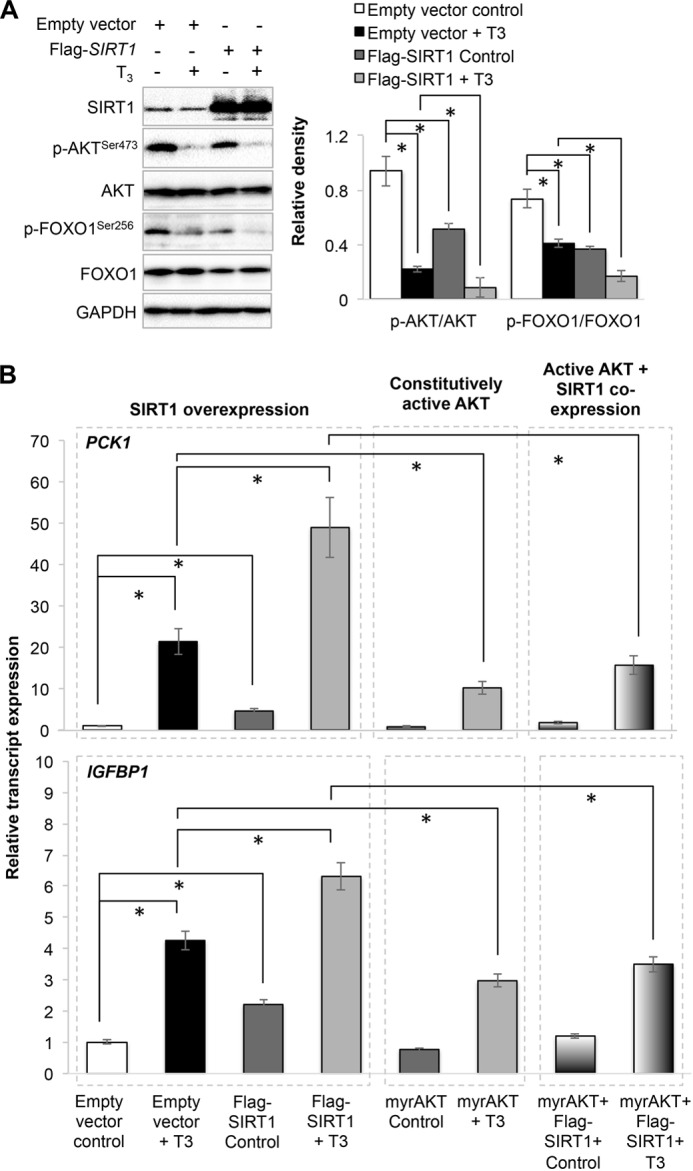
AKT activation inhibited SIRT1-mediated thyroid hormone (T3)-induced dephosphorylation of AKT/FOXO1 and FOXO1 activation. A, Western blot analysis of FLAG-tagged SIRT1 overexpression and AKT/FOXO1 phosphorylation in THRB1-HepG2 cells treated with or without T3 (100 nm). Bar graph represents relative densitometric measurements of phosphorylated AKT and FOXO1. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D. B, PCK1 and IGFBP1 transcripts expression after co-overexpression of FLAG-SIRT1 and myrAKT in these HepG2 cells with or without T3. Statistical significance was calculated as *, p < 0.05, and error bars represent mean ± S.D.
Discussion
FOXO1 is a metabolically regulated transcription factor that belongs to the mammalian forkhead box O (FOXO) family that is conserved throughout evolution. FOXO proteins regulate the expression of target genes involved in a variety of biological processes, including cell cycle arrest, apoptosis, energy metabolism, and longevity (1, 37). Studies using genetically modified mouse models have documented the physiological role of FOXO1 in insulin-mediated suppression of hepatic glucose production (7, 37). FOXO1 is primarily regulated by phosphorylation of multiple residues by AKT (7, 34); however, no role for TH on AKT and FOXO1 phosphorylation has been described thus far. Additionally, although FOXO1 and TH may regulate a significant number of common target genes (3–5, 9, 11, 13), it is not known whether their transcriptional regulation occurs in parallel or by transcriptional co-regulation by TH and FOXO1.
To investigate potential TH co-regulation of FOXO1 target genes, we performed hepatic transcriptome profiling of livers from euthyroid, hyperthyroid, and Foxo1 siRNA knockdown hyperthyroid mice, and we compared the results with a list of genes identified previously as having functional Foxo1-binding sites based on Foxo1-ChIP-Seq and transcriptome analysis (37). We observed that 7% (15 out of 213 genes) of the total genes were regulated by TH and FOXO1 together.3 Interestingly, among the genes previously identified as having functional FOXO1-binding sites, we identified a subset of target genes that were co-regulated by both TH and FOXO1 (31%; 27 out of 85 genes) (Fig. 1). Furthermore, comparison of these genes with Thrb1-ChIP-Seq data showed that 60% of the TH-FOXO1-co-regulated genes had THRB1 binding at thyroid hormone-response elements (TREs) within or in the proximal regions of their genomes in addition to FOXO1 binding. Surprisingly, however, ∼40% of TH-FOXO1 co-regulated target genes did not have THRB1-binding sites in the proximal promoter regions. Although the possibility of long range interactions between FOXO1 and THRs binding to distal sites cannot be eliminated, it is possible that FOXOs also can regulate target genes by interacting with other specific co-regulators and transcription factors (including nuclear hormone receptors) that are regulated by TH (9, 47). Moreover, enrichment analysis of co-regulatory protein-binding sites near the Foxo1-binding sites that were identified by Foxo1-ChIP-Seq (37) showed that Hnf4A- and Cebpb-binding motifs mapped near the Foxo1-binding sites in 35% of target genes, whereas Esrra- and Nr3c1-binding motifs were enriched near the other Foxo1 target genes (65%). Of note, our TH-Foxo1 microarray also showed significant increases in Hnf4a and Cebpb mRNA expression by T3, whereas there was no significant change in Esrra or Nr3c1 (Table 3). Additionally, it is possible that both FOXO1 and/or TH may activate signaling pathways that, in turn, regulate the expression of some of these genes. Identifying these pathways would further enhance our understanding of both FOXO1 and TH action. Interestingly, we also identified a subset of FOXO1 target genes that were regulated by T3 and did not depend upon FOXO1 for T3-mediated transcription in our microarray analysis, strongly suggesting that TH can specifically regulate a subset of FOXO1 target genes in a manner that is independent of FOXO1 (Tables 1 and 2). Interestingly, the majority of these genes did not bind THRB1 to TREs in the promoter region and likely are regulated indirectly by transcription factors that are stimulated by TH (Table 2).
TABLE 3.
Change in gene expression of Foxo1 co-regulatory proteins in liver by TH
Microarray analysis of euthyroid (EuTH) and hyperthyroid (HyperTH) mouse liver tissues showed changes in gene expression of previously reported (37) Foxo1 co-regulators by TH.
| Gene expression | Gene | Co-regulatory protein name | Log ratio (hyper-TH vs. EuTH) | Fold change (hyper-TH vs EuTH) | p value |
|---|---|---|---|---|---|
| ↑ | Hnf4a | Hnf4α | 0.5068667 | 1.41 | 0.0018231 |
| ↑ | Cebpb | Cebpβ | 0.8745 | 1.74 | 0.0000948 |
| ˜ | Esrra | Errα | −0.30049 | 1.23 | 0.0629851 |
| ˜ | Nr3c1 | Gr | 0.01199 | 1.00 | 0.887947 |
FOXO1 is phosphorylated by AKT/PKB (3) and is retained in the cytoplasm and is transcriptionally inactive when it is in the phosphorylated state. We found that TH significantly decreased FOXO1 phosphorylation by AKT which, in turn, increased its nuclear localization. Previously Zhu et al. (48) showed that basal Akt phosphorylation was increased in Thra1−/−Thrb1−/− mice compared with Thra1+/+Thrb1+/+ mice, and they showed that THRs can act as tumor suppressors in a mouse model of metastatic follicular thyroid carcinoma. Similarly, Thra1−/−Thrb1−/− deletion in murine epidermis showed increased ERK and Akt activity and increased expression of downstream targets, such as AP-1 components (49). Introduction of Thra0/0 deletion in the ApoE−/− background in mice accelerated the appearance of plaques in aorta macrophages and correlated with an activation of the Akt/nuclear factor κB pathway in these cells (29). However, non-genomic effects of TH also have been reported in skin fibroblasts and endothelial cells in which T3 rapidly increased AKT phosphorylation (10–30 min)3 (24, 50). We also observed similar acute results in HepG2 cells as T3 acutely increased AKT phosphorylation within 30 min. However, AKT phosphorylation was reduced after 60 min of T3 treatment and continued to decrease in a time-dependent manner (8–24 h) (Fig. 2H). Recently, Kalyanaraman et al. (51) have also shown similar results in primary osteoblasts in which they showed that T3 rapidly stimulated the phosphorylation of Src, ERK1/2, and Akt within 2–30 min and then returned to its basal amounts after 30 min. The reduction of AKT phosphorylation by TH led to decreased FOXO1 phosphorylation and increased nuclear localization in HepG2 cells (Figs. 3 and 4). Moreover, we observed that TH reduction of AKT phosphorylation was THRB1-dependent (Fig. 5).
To further understand TH regulation of AKT-dependent FOXO1 activation, we analyzed the upstream regulator of AKT, MTORC2 (42). MTORC2 is known to be a master regulator of AKT by virtue of its phosphorylation of the AKT activation site at Ser-473. Because MTORC2 is a multiprotein complex, we decided to analyze RICTOR, a regulatory protein of MTORC2 that activates AKT. RICTOR is expressed in a wide variety of tissues and has been implicated in growth and metabolism (52, 53). Conditional deletion of Rictor in skeletal myocytes, hepatocytes, pancreatic β cells, and adipocytes impaired insulin signaling and led to systemic metabolic defects in glucose and lipid homeostasis (8, 52). RICTOR overexpression increased AKT phosphorylation and reduced TH stimulation of FOXO1 transcriptional activity. This demonstration that RICTOR expression levels altered TH regulation of FOXO1 transcriptional activity confirmed the critical role of RICTOR in mediating the TH effect on AKT phosphorylation (Fig. 7).
Protein modifications such as phosphorylation and acetylation of RICTOR have been associated with MTORC2 activity. Although the role of RICTOR phosphorylation in the regulation of MTORC2 has been known previously (33, 54), only recently has the effect of RICTOR acetylation on MTORC2 activity been appreciated (43). Because we previously reported that TH induced FOXO1 deacetylation by SIRT1 (19), we examined whether TH similarly could stimulate RICTOR deacetylation by SIRT1. Indeed, we found that TH decreased RICTOR acetylation in cultured hepatic cells and in mouse livers (Fig. 7, E–G) via activation of SIRT1 (Fig. 8, A–C). Furthermore, we demonstrated the critical role for SIRT1 itself in TH-mediated activation of FOXO1 target genes in hepatic cells, primary hepatocytes, and in vivo by pharmacologic blockade and siRNA knockdown. Additionally, we found that SIRT1/MTORC2 may form a complex because they could be co-immunoprecipitated from hepatic cell lysate.
SIRT1 is an NAD+/NADH-sensitive deacetylase that regulates acetylation of histones as well as cellular metabolic co-activators and transcription factors such as PPARGC1A and FOXO proteins (44, 45). During oxidative phosphorylation, mitochondrial ATP turnover is associated with an increase in NAD+/NADH that stimulates SIRT1 deacetylase activity (55, 56). We observed that TH increased both mitochondrial ATP turnover and NAD+/NADH (Fig. 8, D–F) which, in turn, most likely stimulated SIRT1 activity. Thus, TH-mediated changes in the energy and redox status of the cell may play a critical role in regulating the SIRT1/MTORC2/AKT pathway's effects on FOXO1 target gene transcription.
Because TH decreases FOXO1 phosphorylation and acetylation (57), it would be expected that all FOXO1 target genes should be activated; however, it is surprising that only a subset of genes that contained functional FOXO1 binding were activated by T3 (Fig. 1). Currently, the reason(s) for this limited co-regulation of FOXO1 target genes by TH is not known. However, the presence of other tissue-specific or metabolically regulated repressors may limit the expression of FOXO1 target genes in the liver. Furthermore, TH has been reported to activate PPARGC1A-dependent gene transcription (20); thus, raising the possibility that TH co-regulates FOXO1 target genes through its activation or repression of other transcriptional co-regulators. These issues notwithstanding, our results clearly show that metabolic effects mediated by a nuclear hormone receptor such as THR (e.g. oxidative phosphorylation and increased NAD+/NADH concentration) can activate intracellular signaling pathways that affect activities of other metabolic enzymes (e.g. SIRT1, AKT, and RICTOR) as well as other transcription factors such as FOXO1 to regulate FOXO1 target genes.
Our findings show that TH co-regulated FOXO1 target genes directly via THRB1 binding to TREs located in their promoter regions. TH also co-regulated some FOXO1 target genes indirectly without THRB1 binding because some target genes, such as IGFBP1, do not appear to have bona fide TREs. This latter group includes genes that require binding of other co-regulators/transcription factors to their promoters to stimulate their transcription. These co-regulators (e.g. HNF4A and CEBPB) may or may not be regulated by TH. For other target genes, FOXO1 binding alone was sufficient for TH-mediated transcription. In such cases, it is possible that TH may play a permissive role in the transcription of these target genes.
In summary, we showed that FOXO1 activity is regulated by TH-dependent activation of a SIRT1-RICTOR (MTORC2)-AKT signaling pathway that leads to decreased FOXO1 phosphorylation. This pathway can stimulate the expression of gluconeogenic genes such as PCK1 and IGFBP1. Additionally, this regulation of FOXO1 phosphorylation occurs in parallel with TH stimulation of FOXO1 deacetylation by SIRT1 (19). Such a multistep regulation of FOXO1 transcriptional activity enables more complex, yet finer, control of glucose homeostasis within the cell. Finally, the convergence of TH and insulin signaling on AKT phosphorylation enables dual hormonal regulation of FOXO1 activity. In the future, we anticipate the discovery of similar types of cell signaling pathways that are metabolically regulated by nuclear hormone receptors, in addition to those that they regulate directly at the transcriptional level.
Author Contributions
B. K. S., R. A. S., J. Z., K. O., and M. W. performed the experiments. B. K. S., R. A. S., and P. M. Y. designed the study and wrote manuscript. M. T. and S. G. performed and analyzed the gene expression microarray data. I. A. and A. N. H. performed and analyzed the Thrb1-ChIP-seq data. K. G. shared Thrb−/− mice and provided constructive suggestions on the manuscript.
Acknowledgments
We thank Dr. Parmeen Akhtar, Benjamin Livingston Farah (Cardiovascular and Metabolic Disorders Program, Duke-NUS Graduate Medical School), Dr. Ronny Lesmana (Universitas Padjadjaran, Indonesia and Cardiovascular and Metabolic Disorders Program, Duke-NUS Graduate Medical School), and Cyrielle Billon (Institut de Génomique Fonctionnelle de Lyon, Université de Lyon, Lyon, France) for their helpful advice and constructive comments. We also acknowledge Prof. Martin L. Privalsky for the kind gift of HepG2 cells ectopically expressing THRB1 or no receptor vector as used in this study.
This work was supported by Duke-NUS Graduate Medical School Faculty Funds (to P. M. Y.), National Medical Research Council/Early Development Grant 1044/2011, National Medical Research Council/Clinical Investigator Research Grant 1340/2012, and National Medical Research Council/Clinician Scientist Award Grant MH95:03/1-8, Ministry of Health, Ministry of Education, and Ministry of Trade, Singapore, and A*StaR. The authors declare that they have no conflicts of interest with the contents of this article.
B. K. Singh and P. M. Yen, unpublished results.
- TH
- thyroid hormone
- T3
- triiodothyronine
- EuTH
- euthyroid
- hyper-TH
- hyperthyroid
- THR
- thyroid hormone receptor
- TRE
- thyroid hormone response element
- qPCR
- quantitative PCR
- IP
- immunoprecipitation
- WB
- Western blot
- OCR
- oxygen consumption rate
- NR
- no receptor
- FDR
- false discovery rate
- FCCP
- carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone.
References
- 1.Barthel A., Schmoll D., and Unterman T. G. (2005) FoxO proteins in insulin action and metabolism. Trends Endocrinol. Metab. 16, 183–189 [DOI] [PubMed] [Google Scholar]
- 2.Calnan D. R., and Brunet A. (2008) The FoxO code. Oncogene 27, 2276–2288 [DOI] [PubMed] [Google Scholar]
- 3.Huang H., and Tindall D. J. (2007) Dynamic FoxO transcription factors. J. Cell Sci. 120, 2479–2487 [DOI] [PubMed] [Google Scholar]
- 4.Webb A. E., and Brunet A. (2014) FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci. 39, 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gross D. N., Wan M., and Birnbaum M. J. (2009) The role of FOXO in the regulation of metabolism. Curr. Diab. Rep. 9, 208–214 [DOI] [PubMed] [Google Scholar]
- 6.Gross D. N., van den Heuvel A. P., and Birnbaum M. J. (2008) The role of FoxO in the regulation of metabolism. Oncogene 27, 2320–2336 [DOI] [PubMed] [Google Scholar]
- 7.Eijkelenboom A., and Burgering B. M. (2013) FOXOs: signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 14, 83–97 [DOI] [PubMed] [Google Scholar]
- 8.Hagiwara A., Cornu M., Cybulski N., Polak P., Betz C., Trapani F., Terracciano L., Heim M. H., Rüegg M. A., and Hall M. N. (2012) Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 15, 725–738 [DOI] [PubMed] [Google Scholar]
- 9.van der Vos K. E., and Coffer P. J. (2008) FOXO-binding partners: it takes two to tango. Oncogene 27, 2289–2299 [DOI] [PubMed] [Google Scholar]
- 10.Oppenheimer J. H. (1985) Thyroid hormone action at the nuclear level. Ann. Intern. Med. 102, 374–384 [DOI] [PubMed] [Google Scholar]
- 11.Sinha R. A., Singh B. K., and Yen P. M. (2014) Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol. Metab. 25, 538–545 [DOI] [PubMed] [Google Scholar]
- 12.McAninch E. A., and Bianco A. C. (2014) Thyroid hormone signaling in energy homeostasis and energy metabolism. Ann. N.Y. Acad. Sci. 1311, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y. Y., and Brent G. A. (2010) Thyroid hormone crosstalk with nuclear receptor signaling in metabolic regulation. Trends Endocrinol. Metab. 21, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brent G. A. (2012) Mechanisms of thyroid hormone action. J. Clin. Invest. 122, 3035–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yen P. M. (2001) Physiological and molecular basis of thyroid hormone action. Physiol. Rev. 81, 1097–1142 [DOI] [PubMed] [Google Scholar]
- 16.Ortiga-Carvalho T. M., Sidhaye A. R., and Wondisford F. E. (2014) Thyroid hormone receptors and resistance to thyroid hormone disorders. Nat. Rev. Endocrinol. 10, 582–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yen P. M. (2003) Molecular basis of resistance to thyroid hormone. Trends Endocrinol. Metab. 14, 327–333 [DOI] [PubMed] [Google Scholar]
- 18.Mullur R., Liu Y. Y., and Brent G. A. (2014) Thyroid hormone regulation of metabolism. Physiol. Rev. 94, 355–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh B. K., Sinha R. A., Zhou J., Xie S. Y., You S. H., Gauthier K., and Yen P. M. (2013) FoxO1 deacetylation regulates thyroid hormone-induced transcription of key hepatic gluconeogenic genes. J. Biol. Chem. 288, 30365–30372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thakran S., Sharma P., Attia R. R., Hori R. T., Deng X., Elam M. B., and Park E. A. (2013) Role of sirtuin 1 in the regulation of hepatic gene expression by thyroid hormone. J. Biol. Chem. 288, 807–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chan I. H., and Privalsky M. L. (2009) Isoform-specific transcriptional activity of overlapping target genes that respond to thyroid hormone receptors α1 and β1. Mol. Endocrinol. 23, 1758–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yen P. M., Feng X., Flamant F., Chen Y., Walker R. L., Weiss R. E., Chassande O., Samarut J., Refetoff S., and Meltzer P. S. (2003) Effects of ligand and thyroid hormone receptor isoforms on hepatic gene expression profiles of thyroid hormone receptor knockout mice. EMBO Rep. 4, 581–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suh J. H., Sieglaff D. H., Zhang A., Xia X., Cvoro A., Winnier G. E., and Webb P. (2013) SIRT1 is a direct coactivator of thyroid hormone receptor β1 with gene-specific actions. PLoS One 8, e70097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hiroi Y., Kim H. H., Ying H., Furuya F., Huang Z., Simoncini T., Noma K., Ueki K., Nguyen N. H., Scanlan T. S., Moskowitz M. A., Cheng S. Y., and Liao J. K. (2006) Rapid nongenomic actions of thyroid hormone. Proc. Natl. Acad. Sci. U.S.A. 103, 14104–14109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sinha R. A., You S. H., Zhou J., Siddique M. M., Bay B. H., Zhu X., Privalsky M. L., Cheng S. Y., Stevens R. D., Summers S. A., Newgard C. B., Lazar M. A., and Yen P. M. (2012) Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J. Clin. Invest. 122, 2428–2438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farah B. L., Sinha R. A., Wu Y., Singh B. K., Zhou J., Bay B. H., and Yen P. M. (2014) β-Adrenergic agonist and antagonist regulation of autophagy in HepG2 cells, primary mouse hepatocytes, and mouse liver. PLoS One 9, e98155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gauthier K., Chassande O., Plateroti M., Roux J. P., Legrand C., Pain B., Rousset B., Weiss R., Trouillas J., and Samarut J. (1999) Different functions for the thyroid hormone receptors TRα and TRβ in the control of thyroid hormone production and post-natal development. EMBO J. 18, 623–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gauthier K., Billon C., Bissler M., Beylot M., Lobaccaro J. M., Vanacker J. M., and Samarut J. (2010) Thyroid hormone receptor β (TRβ) and liver X receptor (LXR) regulate carbohydrate-response element-binding protein (ChREBP) expression in a tissue-selective manner. J. Biol. Chem. 285, 28156–28163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Billon C., Canaple L., Fleury S., Deloire A., Beylot M., Dombrowicz D., Del Carmine P., Samarut J., and Gauthier K. (2014) TRα protects against atherosclerosis in male mice: identification of a novel anti-inflammatory property for TRα in mice. Endocrinology 155, 2735–2745 [DOI] [PubMed] [Google Scholar]
- 30.Selmi-Ruby S., Bouazza L., Obregon M. J., Conscience A., Flamant F., Samarut J., Borson-Chazot F., and Rousset B. (2014) The targeted inactivation of TRβ gene in thyroid follicular cells suggests a new mechanism of regulation of thyroid hormone production. Endocrinology 155, 635–646 [DOI] [PubMed] [Google Scholar]
- 31.Brunet A., Sweeney L. B., Sturgill J. F., Chua K. F., Greer P. L., Lin Y., Tran H., Ross S. E., Mostoslavsky R., Cohen H. Y., Hu L. S., Cheng H. L., Jedrychowski M. P., Gygi S. P., Sinclair D. A., et al. (2004) Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 32.Ramaswamy S., Nakamura N., Vazquez F., Batt D. B., Perera S., Roberts T. M., and Sellers W. R. (1999) Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc. Natl. Acad. Sci. U.S.A. 96, 2110–2115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sarbassov D. D., Ali S. M., Kim D. H., Guertin D. A., Latek R. R., Erdjument-Bromage H., Tempst P., and Sabatini D. M. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 14, 1296–1302 [DOI] [PubMed] [Google Scholar]
- 34.Tang E. D., Nuñez G., Barr F. G., and Guan K. L. (1999) Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 274, 16741–16746 [DOI] [PubMed] [Google Scholar]
- 35.Nie Y., Erion D. M., Yuan Z., Dietrich M., Shulman G. I., Horvath T. L., and Gao Q. (2009) STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat. Cell Biol. 11, 492–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Storey J. D., and Tibshirani R. (2003) Statistical significance for genome-wide studies. Proc. Natl. Acad. Sci. U.S.A. 100, 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shin D. J., Joshi P., Hong S. H., Mosure K., Shin D. G., and Osborne T. F. (2012) Genome-wide analysis of FoxO1 binding in hepatic chromatin: potential involvement of FoxO1 in linking retinoid signaling to hepatic gluconeogenesis. Nucleic Acids Res. 40, 11499–11509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., and Mesirov J. P. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liberzon A. (2014) A description of the Molecular Signatures Database (MSigDB) Web site. Methods Mol. Biol. 1150, 153–160 [DOI] [PubMed] [Google Scholar]
- 40.Ramadoss P., Abraham B. J., Tsai L., Zhou Y., Costa-e-Sousa R. H., Ye F., Bilban M., Zhao K., and Hollenberg A. N. (2014) Novel mechanism of positive versus negative regulation by thyroid hormone receptor β1 (TRβ1) identified by genome-wide profiling of binding sites in mouse liver. J. Biol. Chem. 289, 1313–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Javitt N. B. (1990) Hep G2 cells as a resource for metabolic studies: lipoprotein, cholesterol, and bile acids. FASEB J. 4, 161–168 [DOI] [PubMed] [Google Scholar]
- 42.Sarbassov D. D., Guertin D. A., Ali S. M., and Sabatini D. M. (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 [DOI] [PubMed] [Google Scholar]
- 43.Glidden E. J., Gray L. G., Vemuru S., Li D., Harris T. E., and Mayo M. W. (2012) Multiple site acetylation of Rictor stimulates mammalian target of rapamycin complex 2 (mTORC2)-dependent phosphorylation of Akt protein. J. Biol. Chem. 287, 581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang H. C., and Guarente L. (2014) SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 25, 138–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rehan L., Laszki-Szcza̦chor K., Sobieszczańska M., and Polak-Jonkisz D. (2014) SIRT1 and NAD as regulators of ageing. Life Sci. 105, 1–6 [DOI] [PubMed] [Google Scholar]
- 46.Kakkar P., and Singh B. K. (2007) Mitochondria: a hub of redox activities and cellular distress control. Mol. Cell. Biochem. 305, 235–253 [DOI] [PubMed] [Google Scholar]
- 47.Zhao H. H., Herrera R. E., Coronado-Heinsohn E., Yang M. C., Ludes-Meyers J. H., Seybold-Tilson K. J., Nawaz Z., Yee D., Barr F. G., Diab S. G., Brown P. H., Fuqua S. A., and Osborne C. K. (2001) Forkhead homologue in rhabdomyosarcoma functions as a bifunctional nuclear receptor-interacting protein with both coactivator and corepressor functions. J. Biol. Chem. 276, 27907–27912 [DOI] [PubMed] [Google Scholar]
- 48.Zhu X. G., Zhao L., Willingham M. C., and Cheng S. Y. (2010) Thyroid hormone receptors are tumor suppressors in a mouse model of metastatic follicular thyroid carcinoma. Oncogene 29, 1909–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Contreras-Jurado C., García-Serrano L., Gómez-Ferrería M., Costa C., Paramio J. M., and Aranda A. (2011) The thyroid hormone receptors as modulators of skin proliferation and inflammation. J. Biol. Chem. 286, 24079–24088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao X., Kambe F., Moeller L. C., Refetoff S., and Seo H. (2005) Thyroid hormone induces rapid activation of Akt/protein kinase B-mammalian target of rapamycin-p70S6K cascade through phosphatidylinositol 3-kinase in human fibroblasts. Mol. Endocrinol. 19, 102–112 [DOI] [PubMed] [Google Scholar]
- 51.Kalyanaraman H., Schwappacher R., Joshua J., Zhuang S., Scott B. T., Klos M., Casteel D. E., Frangos J. A., Dillmann W., Boss G. R., and Pilz R. B. (2014) Nongenomic thyroid hormone signaling occurs through a plasma membrane-localized receptor. Sci. Signal. 7, ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kocalis H. E., Hagan S. L., George L., Turney M. K., Siuta M. A., Laryea G. N., Morris L. C., Muglia L. J., Printz R. L., Stanwood G. D., and Niswender K. D. (2014) Rictor/mTORC2 facilitates central regulation of energy and glucose homeostasis. Mol. Metab. 3, 394–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Masui K., Cavenee W. K., and Mischel P. S. (2014) mTORC2 in the center of cancer metabolic reprogramming. Trends Endocrinol. Metab. 25, 364–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Boulbes D., Chen C. H., Shaikenov T., Agarwal N. K., Peterson T. R., Addona T. A., Keshishian H., Carr S. A., Magnuson M. A., Sabatini D. M., and Sarbassov dos D. (2010) Rictor phosphorylation on the Thr-1135 site does not require mammalian target of rapamycin complex 2. Mol. Cancer Res. 8, 896–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Santidrian A. F., Matsuno-Yagi A., Ritland M., Seo B. B., LeBoeuf S. E., Gay L. J., Yagi T., and Felding-Habermann B. (2013) Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J. Clin. Invest. 123, 1068–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cantó C., and Auwerx J. (2012) Targeting sirtuin 1 to improve metabolism: all you need is NAD(+)? Pharmacol. Rev. 64, 166–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mouradian M., and Abourizk N. (1983) Diabetes mellitus and thyroid disease. Diabetes Care 6, 512–520 [DOI] [PubMed] [Google Scholar]