

Abstract
The BCL-2 protein family plays a critical role in regulating cellular commitment to mitochondrial apoptosis. Pro-apoptotic Bcl-2-associated X protein (BAX) is an executioner protein of the BCL-2 family that represents the gateway to mitochondrial apoptosis. Following cellular stresses that induce apoptosis, cytosolic BAX is activated and translocates to the mitochondria, where it inserts into the mitochondrial outer membrane to form a toxic pore. How the BAX activation pathway proceeds and how this may be inhibited is not yet completely understood. Here we describe synthetic antibody fragments (Fabs) as structural and biochemical probes to investigate the potential mechanisms of BAX regulation. These synthetic Fabs bind with high affinity to BAX and inhibit its activation by the BH3-only protein tBID (truncated Bcl2 interacting protein) in assays using liposomal membranes. Inhibition of BAX by a representative Fab, 3G11, prevented mitochondrial translocation of BAX and BAX-mediated cytochrome c release. Using NMR and hydrogen-deuterium exchange mass spectrometry, we showed that 3G11 forms a stoichiometric and stable complex without inducing a significant conformational change on monomeric and inactive BAX. We identified that the Fab-binding site on BAX involves residues of helices α1/α6 and the α1-α2 loop. Therefore, the inhibitory binding surface of 3G11 overlaps with the N-terminal activation site of BAX, suggesting a novel mechanism of BAX inhibition through direct binding to the BAX N-terminal activation site. The synthetic Fabs reported here reveal, as probes, novel mechanistic insights into BAX inhibition and provide a blueprint for developing inhibitors of BAX activation.
Keywords: antibody engineering, B cell lymphoma 2 (Bcl-2) family, BAX, mitochondrial apoptosis, protein structure, structure-function, protein targeting, synthetic antibody, phage display
Introduction
Apoptosis plays a critical role in maintaining normal tissue homoeostasis in multicellular organisms, and its deregulation results in an imbalance of homeostasis contributing to several diseases (1, 2) The BCL-2 protein family plays a central role in regulating the mitochondrial pathway of apoptosis (3, 4). Mitochondrial outer membrane permeabilization (MOMP)5 is considered the key event that is regulated by the complex network of protein-protein interactions between pro-apoptotic and anti-apoptotic members of the BCL-2 family. Activation of the pro-apoptotic members Bcl-2-associated X protein (BAX) and/or Bcl2-antagonist/killer 1 (BAK) is required for induction of MOMP, whereas the anti-apoptotic or survival proteins, such as BCL-2, BCL-XL, and MCL-1, inhibit the pro-apoptotic proteins and prevent MOMP. Activation of BAX and BAK or inhibition of anti-apoptotic BCL-2 proteins is regulated by direct interaction with the BH3-only proteins.
The activation pathway of BAX represents the gateway to apoptosis, and understanding the function of BAX and its regulation mechanisms is an area of intensive investigation. BAX is found predominantly in the cytosolic compartment in an inactive conformation (5, 6). Upon cellular stress, BAX is triggered and undergoes a series of conformational changes that enable its translocation to the mitochondrial membrane and oligomerization, leading to MOMP induction (7, 8). The structure of the BAX monomer in the inactive conformation has been determined previously by NMR spectroscopy (9). The inactive BAX structure adopts a typical BCL-2-fold, consisting of nine α helices linked with variable loops. In contrast to BAK and anti-apoptotic BCL-2 proteins that reside at the mitochondrial outer membrane, the structure of BAX was determined with its hydrophobic C-terminal helix α9 bound to the canonical hydrophobic groove. When the C-terminal α9 helix dissociates from the canonical hydrophobic groove, it binds to the mitochondrial outer membrane, facilitating the mitochondrial translocation of BAX (9). Structural analysis of a hydrocarbon-stapled BH3 helix from Bcl-2 interacting mediator of cell death (BIM) bound to monomeric BAX uncovered an activation site at the N-terminal surface of BAX (10). This activation site regulates the trigger mechanism for conformational activation of cytosolic BAX, leading to the release of the hydrophobic α9 helix and exposure of the hydrophobic α2 helix (BH3 domain) (10–12). Mitochondrial translocated BAX undergoes further conformational changes on the membrane that induce BAX oligomerization and MOMP (13–16) or is inhibited by anti-apoptotic Bcl-2 proteins (17–20). Despite these insights, we still lack a complete understanding of the possible BAX conformations in the cytosol or the mitochondrial membrane and the interactions involved in the protein-protein complexes of BAX with the BCL-2 proteins. This information is critical for understanding the mechanisms that regulate BAX-mediated apoptosis and how it can be modulated with drugs for therapeutic purposes (7, 21).
To probe novel conformations and functional regions on the surface of BAX, we used synthetic antibody technology, applied here for the first time to a BCL-2 family protein. Synthetic antibody discovery utilizes diversity targeted to the antibody complementarity-determining regions (CDRs) that is encoded by designed synthetic oligonucleotides and screened by phage or yeast display (22, 23). Because library construction and selection are performed entirely in vitro, the state of the target can be controlled through the antibody discovery process. Therefore, the specificity of the output antibodies can be tuned to user-specified stringency (24, 25). Using a phage display screen, we aimed to identify synthetic antibodies that bind with high affinity and specificity to BAX. Although BAX can be expressed and purified readily, and studied by NMR and other biophysical techniques, it is prone to aggregation under some conditions (such as detergents). This aggregation phenomenon is presumably linked to its function of forming oligomeric pores in the mitochondrial membrane but presents a formidable challenge in raising conformation-specific antibodies using techniques requiring immunization. Therefore, there was a strong rationale for utilizing in vitro selection.
Here we identified antibodies that can be used as structural and biochemical probes to dissect key regulatory mechanisms and conformations of BAX (26, 27). We present the discovery of 14 novel synthetic antibody fragments (Fabs) that specifically target BAX. These synthetic Fabs have no significant homology in the CDR sequences, suggesting a diversity of molecular interactions with BAX. They bind to BAX with nanomolar affinities, and six of the Fabs tested occupy overlapping binding sites on BAX. Additionally, the synthetic Fabs inhibit BAX in assays using liposomal membranes. Further analysis with a representative Fab, 3G11, using isolated mitochondria suggests that the Fabs bind to cytosolic BAX and inhibit its ability to translocate and insert onto the mitochondrial outer membrane. Structural studies using NMR and hydrogen-deuterium exchange mass spectrometry showed that 3G11 forms a stoichiometric and stable complex with monomeric and inactive BAX, with a binding site that involves residues of helices α1/α6 and of the α1-α2 loop. Therefore, binding of 3G11 overlaps with the N-terminal activation site of BAX, suggesting a novel mechanism of BAX inhibition through direct binding to the BAX N-terminal activation site. These Fabs provide new tools for probing BAX activity in an unparalleled manner and provide a strategy for therapeutic inhibition of BAX in disease.
Experimental Procedures
Production of Recombinant BAX
Human full-length (1–192) wild-type BAX, truncated α9 ΒΑΧ (ΒΑΧ ΔC26), and BAX mutants were cloned in the pTYB1 vector (New England Biolabs) and fused at the N terminus of chitin protein using the restriction sites NdeI and SapI. Fresh transformants in Escherichia coli were expressed in BL21 CodonPlus (DE3)-RIPL cells and grown in Luria broth medium at an optical density of 0.8–1.0 following induction of expression at 30 °C with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 4 h. Cells were harvested by centrifugation at 5000 rpm for 25 min at 4 °C and then resuspended in cold lysis buffer containing 20 mm Tris-HCl (pH 7.6), 500 mm NaCl, 1 mm EDTA, and Roche protease inhibitor mixture. Cells were disrupted using a microfluidizer, and then the supernatant was separated by ultracentrifugation at 45,000 rpm for 1 h at 4 °C and loaded onto a disposable gravity column (Bio-Rad) containing chitin beads (New England Biolabs) pre-equilibrated in lysis buffer. The beads were washed with lysis buffer and then with lysis buffer containing 50 mm DTT. Chitin beads were left overnight at 4 °C for cleavage of the chitin fusion protein. BAX was eluted from the column with at least 10 bed volumes of lysis buffer. The protein was concentrated with a Centricon spin concentrator (Millipore) and then loaded onto a gel filtration column (Superdex 75, 10/300 GL, GE Healthcare Life Sciences) that was pre-equilibrated with gel filtration buffer (20 mm HEPES and 150 mm KCl (pH 7.2)) at 4 °C. Fractions containing BAX monomer were eluted at ∼12 ml buffer volume, pooled, and then concentrated using a 10-kDa cutoff Centricon spin concentrator (Millipore) for prompt use in biochemical and structural studies. To ensure that BAX was in monomeric conformation in our assays, we purified BAX by gel filtration and used the single homogenous monomeric fraction within hours (0–48 h) of purification.
Phage Display Selections
Biopanning, direct phage ELISAs, and competitive phage ELISAs were performed as described previously (25). Briefly, in sorting for BAX specific Fabs, phage pools representing a phage-displayed synthetic antibody library (Library F) were cycled through three rounds of binding selection with BAX immobilized on 96-well enzyme immunoassay/radio immunoassay plate (Corning Incorporated, Corning, NY) as the capture target. BAX protein was immobilized either through direct binding to the plate or through biotinylation and subsequent binding to streptavidin-coated plate. BAX biotinylation was carried out using the EZ-Link® Maleimide-PEG2-Biotin Kit (Thermo Scientific, Pierce Biotechnology, Rockford, IL) following product instructions. 1 μg BAX per well (in 100 μl PBS, pH 7.0) was used for the selection process. After selection, 48 clones from round 2 selection output of using biotinylated BAX and another 48 clones from round 3 selection output of using non-biotinylated BAX were grown 20–24 h in 96-well deep well plates with 2YT broth supplemented with carbenicillin and M13K07 helper phage. The culture supernatants were used directly in phage ELISAs to identify high-affinity clones specifically targeting BAX. Clones exhibiting phage ELISA signals at least tenfold higher than those of the control (3% BSA) were subjected to DNA sequence analysis.
Fab Protein Expression
The phage display vectors for the 14 identified clones were converted to Fab protein expression vectors by insertion of a stop codon and a hexahistidine tag upstream of the P3 gene fusion. Fab proteins were expressed periplasmically in E. coli BL21(DE3) (New England Biolabs) by growth in low-phosphate media at 30 °C for 18–22 h. The cells were harvested by centrifugation and lysed by using BugBuster lysis reagent according to the instructions of the manufacturer (Novagen, Madison, WI). The lysate was subjected to centrifugation, and the supernatant was applied to a nickel column (Ni-NTA resin, Qiagen, Valencia, CA) pre-equilibrated with TBS (pH 7). The Fab-bound Ni-NTA column was then washed with 20 column volumes of TBS buffer with 20 mm imidazole. Next the protein was eluted with TBS buffer with 250 mm imidazole. The eluent was dialyzed into PBS at pH 8.0 and applied to a protein A affinity column (the beads were from Pierce Thermo Scientific, Rockford, IL) for further purification. The Fab-bound beads were washed with PBS (pH 8.0, 15 column volumes), and the Fab proteins were eluted with 100 mm glycine (pH 2.0). The eluted Fab proteins were immediately neutralized to ∼pH 7 using 1 m Tris buffer (pH 8.0). Fractions containing the Fab proteins were buffer-exchanged into PBS (pH 7.0) and used directly in the following binding and activity assays or flash-frozen and stored at 80 °C for future use. The Fab protein concentrations were determined by measuring the absorbance at 280 nm. The extinction coefficients at 280 nm were calculated from the Fab protein sequences using ExPASy.
Direct Binding ELISAs
BAX (1 μg/well) was first immobilized on a 96-well enzyme immunoassay/radio immunoassay plate (Corning Inc., Corning, NY) at room temperature for 1 h or at 4 °C overnight. PBS containing 3% BSA was used to block the wells after BAX immobilization (incubation for 1.5 h at room temperature). The Fabs were diluted into PBS buffer (pH 7.0), applied to the wells, and incubated for 1 h at room temperature. The plates were then washed with PBS and incubated for 1 h with horseradish peroxidase/anti-FLAG M2 antibody conjugate (binding to the FLAG tag at the C terminus of the Fab light chain). The wells were washed with PBS, developed with 3,3′,5,5′-tetramethylbenzidine substrate, and quenched with 0.5 m H2SO4. The absorbance at 450 nm was determined. The data were fit to a standard four-parameter logistic equation by using GraphPad Prism (GraphPad Software, La Jolla, CA). The EC50 values were obtained from the inflection point of the curve.
Competitive Binding ELISAs
Six Fab clones (3H1, 3G11, 3E8, 2A5, 2D9, and 2B1) were chosen for the competition ELISAs. Two different approaches were used. In the first approach, non-biotinylated Fabs were used to compete with the binding of biotinylated 3G11 (b3G11). To immobilized BAX on the 96-well EIA/RIA plate (Corning Inc.), a mixture of 50 nm b3G11 and increasing amounts of each of the six selected Fabs were added. After washes, only the binding of b3G11 was monitored using horseradish peroxidase/streptavidin conjugate and 3,3′,5,5′-tetramethylbenzidine substrate. In the second approach, purified Fab proteins were used to compete with Fabs displayed at the phage surface. Similarly, to immobilized BAX on the 96-well EIA/RIA plate (Corning Inc.), a mixture of Fab-displayed phage (with a titer of ∼1012 pfu/ml) and 100 nm of each of the six selected Fab proteins was added. Only Fab-phage complex binding was monitored using horseradish peroxidase/anti-M13 antibody conjugate and 3,3′,5,5′-tetramethylbenzidine substrate.
Biolayer Interferometry
The forteBio BLItz system (Pall Corp., Menlo Park, CA) was used to determine the binding kinetics and affinity between BAX and Fab proteins. Ni-NTA biosensors (Pall Corp.) were used for initial Fab protein immobilization, which was followed by BAX association and dissociation interaction analysis. 10 μg/ml Fab was used for immobilization at pH 7. For each Fab protein, at least three different BAX concentrations, varying from 30–200 nm, were used, and, subsequently, global fitting was used to generate the ka (association rate constant), kd (dissociation rate constant) and KD (equilibrium dissociation constant) values.
Size Exclusion Chromatography
Superdex 75 10/300 GL and 200 10/300 GL (GE Healthcare Life Sciences) columns were used for size exclusion chromatography of recombinant BAX and the Fab-BAX complex. Proteins were injected in columns equilibrated with a buffer containing 20 mm HEPES (pH 7.2) and 150 mm KCl.
Liposome Permeabilization Assay
The following lipids, at the indicated ratios, were mixed in a total of 4 mg, dried and resuspended in 0.2 mm EDTA, 10 mm HEPES (pH 7), 200 mm KCl, and 5 mm MgCl2 buffer with 12.5 mm 8-aminonaphthalene-1,3,6-trisulfonic acid (ANTS) dye and 45 mm p-xylene-bis-pyridinium bromide (DPX) quencher (Molecular Probes) using a water bath sonicator: phosphatidylcholine, 48%; phosphatidylinositol, 10%; dioleoyl phosphatidylserine, 10%; phosphatidylethanolamine, 28%; and tetraoleoyl cardiolipin, 4% (Avanti Polar Lipids). Liposomes were formed by extrusion of the suspension using polycarbonate membranes of 0.2-μm pore size (Avanti Polar Lipids). ANTS/DPX-encapsulated liposomes were purified from non-encapsulated ANTS/DPX by gel filtration over a 10-ml CL2B-Sepharose (GE Healthcare Life Sciences) gravity flow column. In 96-well format (Corning), Fabs were added at the indicated concentrations with and without BAX (400 nm) or BAX (400 nm) and tBID (30 nm), and then liposomes were added (10 μl from 1 ml of lipid stock) in assay buffer (10 mm HEPES (pH 7), 200 mm KCl, and 1 mm MgCl2) to a final volume of 100 μl. ANTS/DPX release was quantified on the basis of the increase in fluorescence intensity that occurs when the ANTS fluorophore is separated from the DPX quencher upon release from the liposomes into solution. Fluorescence (λex = 355 nm and λem = 520 nm) was measured over time at 32 °C using a Tecan Infinite M1000 plate reader. The percentage release of ANTS/DPX at 90 min was calculated as percentage release = ((F − F0) / (F100 − F0)) × 100, where F0 and F100 are the baseline and maximal fluorescence, respectively. 1% Triton treatment was used to determine the maximum amount of liposomal release per assay, and this value set the 100% value for the kinetic curve figures, whereas the calculated percent release value for the bar graphs was calculated by using the percent release value of tBid-induced, BAX-mediated ANTS/DPX release as 100%.
BAX Translocation Assay
Mitochondria from the livers of Bak−/− mice (0.75 mg/ml) were resuspended in experimental buffer (125 mm KCl, 10 mm Tris-MOPS (pH 7.4), 5 mm glutamate, 2.5 mm malate, 1 mm K3PO4, and 0.1 mm EGTA-Tris (pH 7.4)), treated with the indicated concentrations of 3G11 recombinant BAX (200 nm) and tBid (100 nm), singly and in combination, and incubated at room temperature for 20 min. The supernatant fractions were isolated by centrifugation at 5500 × g for 10 min, and the mitochondrial pellets were resuspended and washed with 0.1 m sodium carbonate (pH 11.5) for 20 min, centrifuged at 13,000 × g for 10 min at 4 °C, and then solubilized in 1% Triton X-100/PBS for 1 h at 4 °C. The mitochondrial supernatant and pellet fractions were separated by 4–12% NuPage (Invitrogen) gels and analyzed by immunoblotting with anti-BAX antibody (Cell Signaling Technology, catalog no. 2772).
Mitochondrial Release Assay
Mitochondria from the livers of Bak−/− mice (1.5 mg/ml) were resuspended in experimental buffer (125 mm KCl, 10 mm Tris-MOPS (pH 7.4), 5 mm glutamate, 2.5 mm malate, 1 mm K3PO4, 0.1 mm and EGTA-Tris (pH 7.4)), treated with the indicated concentrations of 3G11 recombinant BAX (400 nm) and tBid (30 nm), singly and in combination, and incubated at room temperature for 60 min. The supernatants were isolated by centrifugation at 5500 × g for 10 min, and the mitochondrial pellets were solubilized in 1% Triton X-100/PBS. The mitochondrial supernatant and pellet fractions were separated by 4–12% NuPage (Invitrogen) gels and analyzed by immunoblotting with anti-cytochrome c antibody (BD Biosciences, catalog no. 556433).
Western Blotting
Samples from the mitochondria and translocation assay were separated electrophoretically on 4–12% NuPage (Invitrogen) gels, transferred to Imobilon-FL PVDF membranes (Millipore), and subjected to immunoblotting. For visualization of proteins with the Odyssey infrared imaging system (LI-COR Biosciences), membranes were blocked in PBS containing 3% milk powder. Primary BAX antibody (Cell Signaling Technology, catalog no. 2772S) was incubated overnight at 4 °C in a 1:1000 dilution. After washing, membranes were incubated with an IRdye800-conjugated goat anti-rabbit IgG secondary antibody (LI-COR Biosciences) in a 1:10,000 dilution. Proteins were detected with the Odyssey infrared imaging system.
NMR Samples and Spectroscopy
Uniformly 15N-labeled full-length human BAX was generated as described previously (10). Protein samples of BAX and BAX-3G11 mixtures at the indicated concentrations were prepared in 25 mm sodium phosphate, 50 mm NaCl solution (pH 6.0) in 5% D2O. Correlation 1H,15N Heteronuclear Single Quantum Coherence (HSQC) and 1H,15N Transverse Relaxation Optimized Spectroscopy (TROSY) spectra were acquired at 25 °C on a Bruker 600 MHz NMR spectrometer equipped with a cryogenic probe, processed using Topsin, and analyzed with CCPNMR. BAX wild-type cross-peak assignments were applied as reported previously (9).
Hydrogen-Deuterium Exchange Mass Spectrometry
Prior to hydrogen-deuterium exchange experiments, the quench condition for the best sequence coverage of BAX was optimized as described previously (28). Briefly, 3 μl of stock solution of BAX at 1.0 mg/ml was mixed with 9 μl of H2O buffer (8.3 mm Tris and 150 mm NaCl in H2O (pH7.2)) at 0 °C and then quenched with 18 μl of ice-cold quench solutions of 0.8% formic acid, 16% glycerol, and guanidine hydrochloride at final concentrations of 0.05, 0.5, 1.0, and 2.0 m. The quenched samples were frozen on dry ice and then subjected to an immobilized pepsin column (1 × 20 mm, 30 mg/ml porcine pepsin (Sigma)) for online digestion for 40 s. The resulting peptides were collected on a C18 trap (Michrom MAGIC C18AQ, 0.2 × 2) and separated using a reverse-phase C18 column (Michrom MAGIC C18AQ, 0.2 × 50, 3 μm, 200 Å) with a 30-min linear gradient of 0.046% (v/v) trifluoroacetic acid, 6.4% (v/v) acetonitrile to 0.03% (v/v) trifluoroacetic acid, 38.4% (v/v) acetonitrile. The effluent was directed into an OrbiTtrap Elite mass spectrometer (Thermo-Fisher Scientific) for MS analysis. The instrument was operated in positive ESI mode, and the resolution of the instrument was set at 60,000. Proteome Discoverer software (Thermo Fisher Scientific) was used to identify the sequence of the resulting peptides. The optimal quench condition with the best coverage map of BAX (0.08 m guanidinium hydrochloride in 0.8% formic acid) was used for subsequent functionally deuterated studies. Hydrogen-deuterium exchange reactions were initiated by diluting 3 μl of prechilled protein stock solution (free BAX, 1 mg/ml, or antibody-bound BAX, 2 mg/ml) into 9 μl of D2O buffer (8.3 mm Tris and 150 mm NaCl in D2O, pD 7.2 (is the pH in D2O). The samples were incubated at 0 °C for 10, 100, and 1000 s. The exchange reaction was terminated by the addition of 18 μl of optimized quench solution at 0 °C, and samples were frozen immediately on dry ice and stored at −80 °C. In addition, undeuterated samples and equilibrium-deuterated control samples were also prepared as described previously (29). The deuterated samples were then loaded onto the abovementioned instrument for HXMS analysis. The centroids of the isotopic envelopes of undeuterated, functionally deuterated, and equilibrium-deuterated peptides were measured using HDExaminer and then converted to the corresponding deuteration levels with corrections for back-exchange (30).
Structure Calculations
Structure calculations were performed with Crystallography and NMR System Solve (CNS) within the High Ambiguity Driven Biomolecular Docking (HADDOCK) web server using the Guru interface (31). HADDOCK calculations generated models of the complex that are in agreement with experimental distance restraints and have optimal electrostatic and van der Waals interactions on the basis of a combination of molecular dynamics and energy minimization. HADDOCK docking was performed using the lowest energy structure from the BAX NMR structural ensemble from the PDB (PDB code 1F16). The 3G11 structural model was generated using prediction of immunoglobulin structure software on the basis of structural homology modeling of known Fab structures of the same family that have different amino acid compositions of CDR regions (32). Calculations were performed with ambiguous interaction restraints derived from the hydrogen-deuterium exchange mass spectrometry data and residues of the CDR regions of 3G11. For BAX ambiguous interaction restraint calculations, only residues that exhibited significant protection of hydrogen-deuterium exchange upon titration and a solvent accessibility over 50%, as determined by the program NACCESS, were defined as active residues. Passive residues were automatically assigned by the HADDOCK web interface as residues surrounding the active residues. For 3G11, ambiguous interaction restraints were assigned on the basis of residues involved in CDRH1, CDRH2, CDRH3, and CDRL3. Specifically, active residues for BAX included solvent-accessible residues from 19–45 and 123–142. Active residues for 3G11 were defined as heavy chain (26–32 (loop 1), 52–54 (loop 2), and 96–101 (loop 3)) and light chain (141–148, 165–168, and 206–211). In each HADDOCK structure calculation, 1000 orientations/structure of the complex were generated by rigid body docking energy minimization of the individual structures. The 200 lowest energy structures were semiflexibly refined in torsion angle space and then refined in explicit solvent. 177 structures formed the most populous cluster, 1, with root-mean-square deviation from the overall lowest energy structure (0.7 ± 0.4 Å) calculated on the backbone (CA, C, N, O) atoms. Cluster 2 contained the remaining 23 structures and had a significantly lower HADDOCK score, higher Z score, and higher restraint violation energy. Importantly, structures in both clusters showed interaction of 3G11 with helices 1 and 6 of BAX and part of the loop between helices 1 and 2, the N-terminal activation site, which is consistent with HXMS and mutagenesis data. A folder with the calculations, including protocols, PDB structures, and structural analysis from the HADDOCK calculations is provided in the supplemental data. Ribbon diagrams and molecular models were depicted using PyMOL (Schroedinger, LLC). Software was available through the SBGrid collaborative network (33).
Results
Discovery of 14 Novel Synthetic Antibodies Targeting Monomeric BAX
We used a restricted diversity phage antigen-binding fragment (Fab) library (“Library F”) to select a panel of Fabs that bind to monomeric BAX (9). Library F was designed to contain mostly binomial Tyr/Ser diversity in CDR-H1 and H2 and expanded diversity encoding the nine amino acids Tyr/Ser/Gly/Ala/Phe/Trp/His/Pro/Val in a 5/4/4/2/1/1/1/1/1 ratio, at CDR-L3 and H3 (34). In addition, the CDR-L3 and H3 loops lengths vary in size. The expanded diversity in CDR-L3 and H3 in Library F was designed to mimic the distribution of amino acids in functional CDR-H3 segments of natural antibodies. We screened Library F against freshly purified monomeric BAX (10). After two to three rounds of selection, a panel of 14 BAX-specific Fabs with diverse CDR sequences was obtained (Fig. 1A). These clones were isolated from two distinct panning regimes, one in which biotinylated BAX was immobilized onto streptavidin-coated wells and another in which BAX was coated directly on the wells. The properties of the selected clones were similar from either screening regime and, therefore, analyzed further as a single conglomerated panel. Sequence comparison between the 14 Fabs revealed a high diversity of residues and loop sizes with no apparent sequence homology in CDR segments from any two clones, demonstrating the diverse modes of interaction with BAX.
FIGURE 1.

Discovery of synthetic antibody fragments that bind with high affinity to BAX at overlapping binding sites. A, synthetic Fabs that bind to BAX with the corresponding variable sequences of CDRs and EC50 values as determined by ELISA. B, top panel, KD, ka, and kd values for select synthetic Fabs that bind to BAX and BAX ΔC26, as determined by biolayer interferometry. Bottom panel, a representative biolayer interferometry experiment using immobilized 3G11 and three different BAX concentrations. C, synthetic Fabs, except 2A5, compete with b3G11 for binding to BAX using a competitive ELISA. D, binding of selected phage-bound expressed Fabs (Φ) to BAX as competitively inhibited by the corresponding free synthetic Fab proteins (100 nm). All synthetic Fabs, except 2A5, show at least 40% competition efficacy to different phage-bound expressed Fabs. The data shown in B–D represent the mean ± S.D. from at least three independent experiments.
Fabs were expressed and purified, and the EC50 titers for BAX were determined using ELISA. The EC50 values ranged from 2.3 nm (2B1) to 250 nm (2A5), with most Fabs having a single- or double-digit nanomolar EC50 value (Fig. 1A). The selected Fabs 3G11, 3E8, 2A5, and 2B1 were evaluated for binding kinetics to monomeric BAX using biolayer interferometry (Fig. 1B). The binding affinities and ranking of the Fab proteins on the basis of binding correlate with the EC50 values from the ELISA. In both assays, Fabs 3G11, 3E8, and 2B1 were the highest-affinity binders to BAX, whereas Fab 2A5 was the lowest-affinity binder (Fig. 1, A and B). Therefore, an array of 14 high-affinity antibodies against monomeric BAX was discovered, which will significantly expand the probes for monitoring BAX activity.
To explore whether the Fabs engage overlapping or distinct epitopes on BAX, we performed a competitive ELISA. We chose six of the clones (3H1, 3G11, 3E8, 2A5, 2D9, and 2B1) whose EC50 values cover the entire affinity range. A competitive ELISA was performed in which Fab 3G11 was first biotinylated (b3G11), and then binding of b3G11 to BAX in the presence of other non-biotinylated Fab proteins was analyzed. All Fabs except Fab 2A5 demonstrated competitive inhibition of b3G11 binding, with IC50 values in nanomolar range (Fig. 1C). Fab 2A5 does not compete with b3G11 binding, likely because it is a much weaker binder than 3G11. Furthermore, we also studied how binding of phage-expressed Fabs (Φ) can be inhibited competitively by free Fab proteins. The data showed that phage-expressed Fab binding was inhibited to varying degrees in the presence of 100 nm Fab protein for many of the clones and that, in each case, the phage-expressed Fab for a particular clone could be inhibited by its own Fab protein, with the exception of 3H1 (Fig. 1D). Again, Fab 2A5, the lowest-affinity clone (EC50, 250 nm) had little effect on Fab phage binding for all other clones, but all other Fab proteins could significantly inhibit Fab 2A5 phage binding. On the contrary, Fabs 3E8 and 2B1, the highest-affinity clones (EC50, 2.6 and 2.3 nm, respectively) showed the strongest inhibition of all Fab phage binding. Therefore, the competitive ELISA data match predictions on the basis of ELISA half-maximal binding titers and KD values. These results indicate that, to a large extent, Fabs have overlapping epitopes on the BAX surface with a range of affinities, but many are in the low nanomolar range.
Synthetic Antibodies Inhibit BH3-triggered BAX Activation and MOMP
To examine how binding to BAX by Fabs modulates BAX function, we performed liposomal assays that explicitly evaluated how BH3-triggered BAX forms a pore in a membrane environment that mimics the mitochondrial membrane without the contribution of other mitochondrial factors (35). Fabs have the potential to activate BAX by engaging one of the activation sites of BAX either at its N-terminal or C-terminal surface (10, 15, 36, 37) or to inhibit BAX activation by inhibiting the BAX binding surface of the activator protein tBID or conformational changes associated with BAX activation (12, 15, 16, 19, 20). Therefore, we examined the capacity of the Fabs to either activate BAX or inhibit BAX activation induced by tBID. None of the 14 Fabs had an effect on liposomal integrity alone (data not shown) and neither does an unrelated VEGF-specific Fab (YADS1, negative control). Furthermore, none of the Fab proteins activated BAX and induced liposomal release (Fig. 2A). However, when tBID and BAX were combined with liposomes, liposomal release was robust, as expected (Fig. 2, A and B). In contrast, high-affinity Fabs, when combined with tBID and BAX, inhibited tBID-triggered BAX activation significantly or completely at 2 μm in all cases, with the exception of the lower-affinity clone Fab 2A5 and the negative control YADS1 (Fig. 2B). Three of the highest-binding Fabs, 3G11, 3E8, and 2B1, yielded dose-responsive and time-dependent inhibition of liposomal release, whereas the weaker binder, 2A5, had no effect, even at 2μΜ (Fig. 2, C–F).
FIGURE 2.

Synthetic Fabs inhibit BAX activation induced by pro-apoptotic tBID in liposomal assays. A, synthetic BAX-binding Fabs and a VEGF-specific Fab (YADS1) at 2 μm have no capacity to induce BAX-mediated liposomal ANT/DPX release, whereas tBID induced potent BAX-mediated liposomal release. B, BAX-binding Fabs at 2 μm, except 2A5 and YADS1, inhibit tBID-induced BAX-mediated liposomal ANTS/DPX release. Error bars represent mean ± S.D. at 90 min normalized to tBID-induced maximum ANTS/DPX release at 90 min. C–F, representative liposomal ANTS/DPX release experiments in kinetic representation showing the inhibitory activity of Fabs 3G11, 2B1, and 3E8 at 0.5, 1, and 2 μm Fab. 2A5 had no inhibitory effect. A–F, the experiments were performed with 400 nm BAX, 30 nm tBID, and up to 2 μm Fabs. Data represent mean ± S.D. from triplicates normalized using either tBid-induced BAX mediated ANTS/DPX release as 100% (A and B) or 1% Triton release as 100% (C–F). Data shown are representative of at least three independent experiments.
To examine whether Fabs can inhibit BAX activation in the presence of mitochondrial membranes loaded with anti-apoptotic BCl-2 or other mitochondrial proteins, we isolated mouse liver mitochondria from Bak−/− mice to perform a mitochondrial release assay. A combination of tBID and BAX was exposed to several doses of Fab 3G11, which demonstrated a dose-responsive inhibition of mitochondrial cytochrome c release induced by activated BAX, as assessed by separation of the supernatant and mitochondrial fractions and Western blot analysis (Fig. 3A). Moreover, using the mitochondrial release assay, BAX localization was determined to examine whether inhibition by Fab 3G11 was due to prevention of BAX translocation to the membrane or blocking of the membrane integration and oligomerization on the mitochondrial outer membrane. Fab 3G11 dose-responsively inhibited the capacity of BAX for mitochondrial translocation, suggesting that the high-affinity binding of 3G11 to the monomeric BAX either competes with binding of tBID or prevents the conformational changes of BAX required for its mitochondrial translocation (Fig. 3B). Taken together, these data highlight the fact that we discovered Fab proteins that inhibit BH3-triggered BAX activation and MOMP by restraining the mitochondrial membrane translocation of BAX.
FIGURE 3.
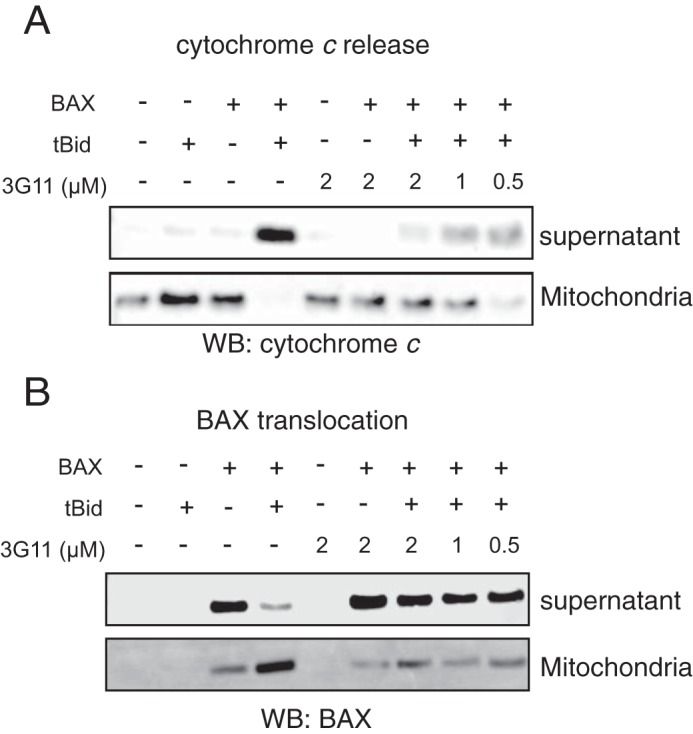
Synthetic Fab 3G11 inhibits BAX-mediated cytochrome c release and BAX mitochondrial translocation induced by pro-apoptotic tBID in isolated mitochondria. A, Fab 3G11 inhibits, dose-responsively, tBID-induced BAX-mediated cytochrome c release from isolated BAK−/− mitochondria. WB, Western blot. B, 3G11 Fab inhibits, dose-responsively, tBID-induced BAX mitochondrial translocation in isolated BAK−/− mitochondria. The data shown are representative of at least three independent experiments.
Fab 3G11 Forms a Stable and Stoichiometric Complex with BAX
To investigate the effect of synthetic Fabs on the structure of BAX, we first studied the stability and stoichiometry of several Fab-BAX complexes upon mixing the proteins at various concentrations and analyzing them by size exclusion chromatography. On the basis of size exclusion chromatography and SDS-PAGE analysis, binding of Fab 3G11 to BAX resulted in a stable 1:1 stoichiometric complex (data not shown). We focused on 3G11 for further structural analysis because of its high affinity and low dissociation constant (Fig. 1B). Next, the structural effects of 3G11 were analyzed on the basis of 1H,15N HSQC spectra of the full-length BAX monomer at several doses of 3G11 (Fig. 4, A–D). The HSQC spectra are in agreement with 3G11 and BAX forming a stable complex with a 1:1 stoichiometry and in slow exchange on the NMR timescale, as evidenced by the dose-dependent loss of intensity of monomeric BAX cross-peaks and lack of new chemical shifts upon 3G11 titration. Despite the 3G11-BAX complex formation evidenced by the NMR titration, it is not possible to observe the 1H,15N cross-peaks of the complex in the 1H,15N HSQC spectra because of the size of the complex (70 kDa).
FIGURE 4.

NMR analysis of the 15N-labeled BAX monomer upon 3G11 titration. A–D, NMR HSQC analysis of 15N-labeled BAX (100 μm) upon titration of Fab 3G11 up to a ratio of 1:1.5 BAX:3G11, as indicated for each overlaid spectrum of unbound BAX and 3G11-bound BAX. Unbound BAX HSQC spectra are shown with blue cross-peaks, and 3G11-bound BAX HSQC spectra are shown with red cross-peaks. Dose-dependent loss of the intensity of HSQC cross-peaks of 15N-labeled BAX spectra was observed upon 3G11 binding to BAX. At a 1:1 ratio of BAX:3G11, the vast majority of 15N-labeled BAX cross-peaks disappear because of the formation of a stoichiometric 3G11-BAX complex. No further changes in the 15N-labeled BAX spectra are observed at 1:1.5 ratio of BAX:3G11.
Fab 3G11 Binds the N-terminal Surface of BAX to Prevent BAX Activation
To further analyze the Fab-BAX interaction, we analyzed the changes on the BAX structure by measuring the solvent accessibility and hydrogen-deuterium exchange of the backbone amide hydrogens using hydrogen-deuterium exchange mass spectrometry (HXMS). First, we analyzed the deuterium exchange of unbound BAX in solution, which underlined the different deuterium exchange rates for exposed or unfolded regions (N terminus, α1-α2 loop) and more solvent-protected or structured regions, including α2, α3-α4, α5, and α6-α8 (Fig. 5A). Upon 3G11-BAX complex formation at stoichiometric levels, deuterium exchange, sample digestion, preparation, and analysis were performed under the same conditions as with free BAX (Fig. 5B). Interestingly, HXMS analysis of the Fab-bound BAX in solution highlighted significant solvent protection in helix α1 and α6 and the α1-α2 loop, whereas other regions of the BAX structure had little to no change in deuterium incorporation upon interaction with 3G11 (Fig. 5C). Furthermore, HXMS showed a modest increase in solvent accessibility for residues in helices α7, α8, and, partially, α9 and α2, which are found at the C-terminal surface of BAX. Moreover, binding of the 3G11 Fab to full-length BAX (BAX WT) and to the C-terminal helix α9-truncated BAX (BAX ΔC26) was determined with a similar KD (Fig. 1B), suggesting that major contacts of 3G11 occur elsewhere from the α9 or the canonical hydrophobic groove of BAX.
FIGURE 5.

3G11 binds to the N-terminal surface of BAX. A, percent deuterium incorporation of unbound BAX conformation in solution over 10, 100, and 1000 s. HXMS analysis suggests increased rates of deuterium exchange in the N-terminal region of BAX, the α1-α2 loop, helix α2, and part of helices α5, α8, and α9. B, percent deuterium incorporation of 3G11-bound BAX in solution over 10, 100, and 1000 s. C, the relative difference of percent deuterium incorporation of BAX conformation bound to 3G11 minus the percent deuterium incorporation of BAX conformation alone over 10, 100, and 1000 s. HXMS analysis suggests increased protection from deuterium incorporation in residues of helices α1 and α6, part of helix α5, and the α1-α2 loop compared with the unbound BAX. C, the regions of significant protection from deuterium incorporation (>−20%) are highlighted in blue on the ribbon representation of the full-length BAX structure (PDB code 16F6) and on the amino acid sequence of BAX. The ribbon representation of BAX also highlights regions with moderate protection (−20–0%) in light blue, regions with no change in deuterium incorporation in gray, and regions with moderately increased deuterium incorporation (0–20%) in light red. The positions of the Lys-21 residue in helix 1 and the Arg-134 residue in helix 6 are shown with yellow stars on the ribbon structure and the amino acid sequence of BAX.
The HXMS analysis suggests that the binding region of 3G11 is localized on the N-terminal surface of the BAX structure and overlaps with the N-terminal activation site of BAX (α1/α6) that controls a series of conformational changes upon BH3 domain activation (11–12) and with the binding epitope (residues 12–24) that is recognized by the 6A7 antibody only on the conformationally active BAX (Fig. 5C). Interestingly, the α1-α2 loop, whose displacement was determined to be essential for the initiation of conformational changes upon BH3-triggered BAX activation (12), is protected by the Fab binding interaction. We found that the binding interaction of 3G11 to BAX is localized to the N-terminal surface of BAX, overlaps with the N-terminal activation site, and is consistent with preventing conformational changes that lead to activation of monomeric BAX.
Although recent structural studies on the basis of peptides from the cytomegalovirus protein vMIA (39) and BCL-2 (20) have suggested potential sites that intervene with BAX activation, interestingly, none of these peptides directly block the N-terminal activation site of the soluble BAX or the C-terminal activation site of the mitochondrion-associated BAX. To further confirm this novel interaction of 3G11 bound to the N-terminal surface of BAX, we used a protein-protein structure calculation approach (40). HADDOCK structure calculations were performed using ambiguous interaction restraints between BAX residues determined by HXMS to be most protected from solvent upon 3G11 binding (residues of α1 and α6) and residues of the CDRs present in the 3G11 protein sequence (see details under “Experimental Procedures”). HADDOCK structure calculations generated structures that fit into two clusters. An ensemble of 10 structures overlaid on the structure of BAX and the lowest-energy structure of each cluster are shown in Fig. 6, A–D. Cluster 1 has a more favorable Haddock score and Z score, a more populous cluster with 177 structures of 200, and structures with lower-energy terms (Fig. 6E). Structures in both clusters are consistent with 3G11 binding to BAX helices α1 and α6 and the closed conformation of the loop between helices α1-α2 (10, 12) (Fig. 6, C and D). However, 3G11 in cluster 2 has its orientation rotated ∼180° around the z axis compared with cluster 1. In all structures, 3G11 makes interactions of a hydrophobic and hydrophilic nature with the solvent-exposed hydrophobic residues and polar/charged residues of helices α1 and α6.
FIGURE 6.

3G11 binding to BAX blocks the N-terminal activation site. A and B, structural ensemble of the 3G11-BAX complex, as calculated with HADDOCK, showing the 10 lowest-energy structures of cluster 1 and cluster 2. Structures from both clusters are centered on the interface of 3G11 (cyan) and BAX (light gray) and overlaid using the BAX structure from the lowest-energy complex structure. The N-terminal interaction surface of BAX with α1 and α6 is shown in blue. C and D, ribbon representations of the 3G11-BAX lowest-energy structures for each cluster highlighting the binding of 3G11 (cyan) using residues in the CDRs (red) to the N-terminal interaction surface (blue) of BAX (light gray). The Lys-21 and Arg-134 residues of BAX are shown as blue sticks and predicted to interact with 3G11 CDR residues. The N-terminal and C-terminal residues of 3G11 are shown by N and C, respectively. E, statistics of HADDOCK calculations for clusters 1 and 2.
To validate the direct interaction of 3G11 with the N-terminal activation site, we tested whether mutations on BAX can disrupt binding to 3G11 and affect its inhibitory activity of 3G11 in liposomal ANTS/DPX release assays. We used the 3G11-BAX structural ensemble and selected residues on BAX that are predicted to form contacts with residues of 3G11 using a 3-Å cutoff. Residues Lys-21 in helix α1 and Arg-134 in helix α6 have been shown previously to interact with the stapled BIM BH3 peptide that activates BAX through the N-terminal activation site (10). Further analysis of all structural models showed that the Arg-134 residue may form hydrogen bonds with up to four residues of CDR loops, whereas the Lys-21 residue is predicted to form hydrogen bonds with one or two residues in cluster 1 structures (Fig. 6, C and D). Consistent with the predicted contributions of each residue to the interaction with the 3G11 Fab, liposomal ANTS/DPX release experiments showed that the inhibition effect of 3G11 on BAX WT activation by tBID is weakened in the presence of the K21E mutation but that it was abolished completely with the R134E mutation (Fig. 7, A–E). Likewise, the double mutation R134E/K21E also abolished the capacity of 3G11 to inhibit BAX activation (Fig. 7, D and E). The inability of 3G11 to inhibit activation of these BAX mutants is consistent with the decreased affinity of 3G11 to these BAX mutants, as determined by ELISA binding experiments (Fig. 7F). Therefore, our data suggest that 3G11 binds and blocks the N-terminal activation site of BAX and are consistent with inhibition of BAX activation and its associated conformational changes. In accordance, mapping the predicted BAX-interacting residues on the basis of the 3G11-BAX structural model and residues of the trigger site that interact with the stapled BIM BH3 peptide, as determined by NMR and biochemical studies, demonstrates an extensive overlap (Fig. 8, A and B) (10). Taken together, these results suggest a novel mechanism of BAX inhibition and demonstrate the capacity of the reported Fabs as structural and functional probes of BAX.
FIGURE 7.

Select BAX mutants inhibit functional inhibition and binding to BAX by 3G11. A–D, representative liposomal ANTS/DPX release experiments in kinetic representation showing the inhibitory activity of 3G11 (2 μm) with BAX WT, which is weakened by the K21E mutation and completely abolished by the R134E mutation or the double mutation R134E/K21E. The experiments were performed with 400 nm BAX WT or BAX mutants, 30 nm tBID, and 2 μm 3G11. E, percent inhibition on the basis of the maximum tBID-induced BAX activation for the BAX WT and mutants at 90 min in the presence or absence of 3G11 Fab. F, ELISA binding profiles for 3G11 binding to BAX WT and mutants. EC50 values were determined as follows: 8 ± 2 nm for BAX WT, 29 ± 4 for K21E, 89 ± 2 nm for the R134E mutant, and 91 ± 2 nm for the R134E/K21E double mutant. OD, optical density. The data shown in A–F, represent the mean ± S.D. from triplicates and two independent experiments.
FIGURE 8.
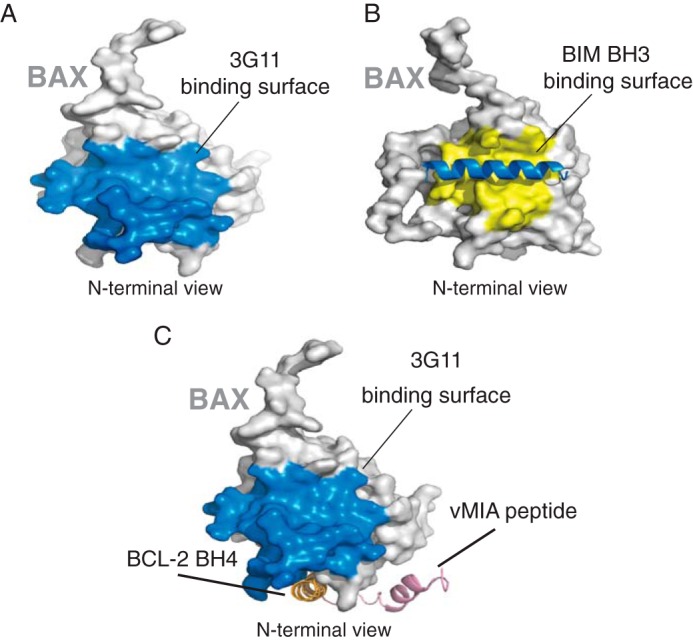
Overlap of the 3G11-binding surface and the BIM BH3-binding surface with the N-terminal trigger site of BAX. A, surface representation of BAX (light gray) highlighting the 3G11-binding surface to BAX (blue) as determined in this study. B, surface representation of BAX (light gray) highlighting the binding surface (yellow) of BIM BH3 (blue helix) in contact with BAX, as determined previously (PDB code 2K7W). C, surface representation of BAX (light gray) highlighting the 3G11-binding surface to BAX (blue) compared with the binding sites of peptides from cytomegalovirus protein vMIA (pink) and BCL-2 BH4 domain (orange).
Discussion
Decision of cellular life or death through the mitochondrial apoptotic pathway, under physiological or disease conditions, is mainly controlled by the interactions among the BCL-2 family proteins. Activation of pro-apoptotic BAX is essential for apoptosis to proceed through mitochondrial dysfunction and caspase activation. Probing and understanding the different BAX conformations during the BAX activation pathway, from the inactive cytosolic monomer to the mitochondrial membrane oligomer, is a critical area of investigation. Our understanding of the different conformations and binding surfaces using structural and biochemical insights is prerequisite for developing effective pharmacological approaches to modulate cell death in diseases with either excess of cell survival, as in cancer, or loss of vital cells, as in myocardial infarction and stroke. Here we harnessed synthetic antibody technology to generate high-affinity BAX binding Fabs to use as structural and biochemical probes. Our screen identified 14 different Fabs with sequence diversity in CDRs that bind with nanomolar affinity to BAX. Interestingly, competitive ELISAs confirmed that six representative Fabs of the 14 Fab proteins bind to overlapping regions on the BAΧ surface. These Fabs bind full-length BAX, which represents cytosolic BAX (9). We also showed that 3G11 binds α9-truncated BAX, which mimics some function of the mitochondrially associated BAX (15). Mitochondrially inserted BAX oligomerizes and undergoes dramatic conformational changes to such extents that its N-terminal surface is not available for binding to Fabs (13, 15, 16, 41). Therefore, selected Fabs are expected to be conformation-specific for cytosol- and mitochondrion-associated BAX and inhibit both conformations from proceeding along the BAX activation process. Furthermore, the ability of 3G11 in binding the a9-truncated BAX less potently in comparison with the full-length BAX suggests that 3G11 may be more effective in inhibiting the translocation of BAX than further conformational changes on the membrane.
We found that all of the identified BAX-binding Fabs inhibit BAX activation triggered by pro-apoptotic tBID in liposomal assays. Further investigation of the mechanism, using Fab 3G11, suggested that the Fabs bind to the N-terminal surface of BAX without causing significant conformational changes on BAX. HXMS and mutagenesis showed that 3G11 binds to an extended surface on BAX that includes the N-terminal activation site (helices α1/α6), which BIM, tBID, and p53 upregulated modulator of apoptosis (PUMA) pro-apoptotic BH3 helices bind to trigger BAX activation (10, 11, 37, 39, 42). Therefore, our data are consistent with 3G11 competitively inhibiting tBID-mediated BAX activation by blocking access to the N-terminal trigger site and preventing N-terminal conformational changes associated with BAX activation (11, 37). Indeed, we found that 3G11 binding prevents mitochondrial translocation of BAX, which requires significant conformational changes and integration into the membrane (13, 15, 16, 41). These findings highlight the application of the reported Fabs, as probes, to investigate or modulate structural and functional mechanisms along the BAX activation pathway and its network of protein-protein interactions.
BAX is shown to have two different activation sites depending on its cytosol- or mitochondrion-associated conformations: the N-terminal BH3 pocket (trigger site) and the C-terminal BH3 pocket. Although several proteins have been reported to directly bind BAX and inhibit its activation, only two other studies have reported structural evidence of the binding interaction on the surface of BAX. A stapled helical peptide of the BH4 domain of BCL-2 protein binds to a cleft formed by select residues of the α1-α2 loop: α2, α3, α5, and α6 (20). A helical peptide of the cytomegalovirus protein vMIA binds to a distinct site at the same side of the BAX structure that includes the loops of α3-α4 and α5-α6 (39). In both cases, the peptides bind to a geographically distinct site that has no overlap with either the N-terminal or C-terminal activation site (Fig. 8C). Therefore, these mechanisms of BAX inhibition reflect allosteric mechanisms that suppress conformational changes upon BAX activation. Our data are consistent with 3G11-mediated BAX inhibition through a direct interaction with the N-terminal activation site that has been described previously to be the target of activating BH3 helical peptides or the full-length BH3-only proteins tBID, BIM, and PUMA, representing a novel mechanism of BAX inhibition (10, 11, 36, 37). However, because BAX activation is possible through other mechanisms, the inhibitory activity of Fabs may also be allosteric by suppression of conformational changes in the N-terminal surface of BAX. Nevertheless, it is interesting that at least the Fabs we isolated and tested appear to bind to the N-terminal activation site, which suggests that this N-terminal BH3 binding groove in the full-length BAX structure provides a favorable protein-protein interaction surface for the Fabs to bind.
Pharmacological targeting of BAX to either promote or inhibit its activation has been proposed to be a promising therapeutic strategy. However, discovery of pharmacological modulators of BAX has been challenging because of limited insights or lack of appropriate probes to use for small molecule discovery. Recently, small-molecule BAX activators that bind to the N-terminal activation site have been identified using a competitive binding assay of the stapled BIM BH3 peptide activator that binds to the same site (38). Likewise, the application of synthetic antibodies to BAX provides a novel opportunity to use Fabs as probes for screening small molecule libraries using a competitive binding assay between the identified inhibitory Fabs and BAX. Small-molecule inhibitors that bind directly to the BAX N-terminal activation site will be effective chemical probes for dissecting the function and mechanisms of BAX-mediated cell death and promising starting points for the development of therapeutics. Finally, the success of our screen suggests that synthetic antibodies can also be identified for the structurally homologous anti-apoptotic BCl-2 family proteins, which can potentially reveal novel mechanisms of regulation or binding sites for pharmacological targeting.
Author Contributions
S. S. S. provided the phage library. Z. D. designed and performed the phage selection, ELISA, BI experiments, and data analyses. O. U. designed and performed all other biochemical and structural experiments and data analyses. N. B. assisted with the execution and analysis of the NMR experiments. S. L. and D. L. performed the hydrogen-deuterium exchange mass spectrometry studies. E. G. and J. R. L. conceived and directed the study. E. G. wrote the manuscript, which was edited and reviewed by all authors.
This research was supported by National Institutes of Health Grants R00HL095929 and R01CA178394 (to E. G.); R21CA155472 (to J. R. L.); and R01 GM020501, R56AI110750, and R01AI101436 (to S. L.). This work was partially supported by contributions to the Albert Einstein Center for Experimental Therapeutics by Pamela and Edward S. Pantzer. NMR data were collected at the Einstein NMR Resource with support from National Institutes of Health Awards 1S10OD016305 and P30 CA013330 and at New York Structural Biology Center (NYSBC) with support from a grant from Empire State Development's Division of Science, Technology and Innovation (NYSTAR). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental data.
- MOMP
- Mitochondrial outer membrane permeabilization
- CDR
- complementarity-determining region
- Fab
- antibody fragment
- Ni-NTA
- nickel-nitrilotriacetic acid
- ANTS
- 8-aminonaphthalene-1,3,6-trisulfonic acid
- DPX
- p-xylene-bis-pyridinium bromide
- HXMS
- hydrogen-deuterium exchange mass spectrometry
- tBID
- truncated Bcl2 interacting protein.
References
- 1.Fuchs Y., and Steller H. (2011) Programmed cell death in animal development and disease. Cell 147, 742–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Danial N. N., and Korsmeyer S. J. (2004) Cell death: critical control points. Cell 116, 205–219 [DOI] [PubMed] [Google Scholar]
- 3.Youle R. J., and Strasser A. (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 9, 47–59 [DOI] [PubMed] [Google Scholar]
- 4.Chipuk J. E., Moldoveanu T., Llambi F., Parsons M. J., and Green D. R. (2010) The BCL-2 family reunion. Mol. Cell 37, 299–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsu Y. T., Wolter K. G., and Youle R. J. (1997) Cytosol-to-membrane redistribution of Bax and Bcl-X(L) during apoptosis. Proc. Natl. Acad. Sci. U.S.A. 94, 3668–3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolter K. G., Hsu Y. T., Smith C. L., Nechushtan A., Xi X. G., and Youle R. J. (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139, 1281–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walensky L. D., and Gavathiotis E. (2011) BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem. Sci. 36, 642–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westphal D., Kluck R. M., and Dewson G. (2014) Building blocks of the apoptotic pore: how Bax and Bak are activated and oligomerize during apoptosis. Cell Death Differ. 21, 196–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki M., Youle R. J., and Tjandra N. (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103, 645–654 [DOI] [PubMed] [Google Scholar]
- 10.Gavathiotis E., Suzuki M., Davis M. L., Pitter K., Bird G. H., Katz S. G., Tu H. C., Kim H., Cheng E. H., Tjandra N., and Walensky L. D. (2008) BAX activation is initiated at a novel interaction site. Nature 455, 1076–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H., Tu H. C., Ren D., Takeuchi O., Jeffers J. R., Zambetti G. P., Hsieh J. J., and Cheng E. H. (2009) Stepwise activation of BAX and BAK by tBID, BIM, and PUMA initiates mitochondrial apoptosis. Mol. Cell 36, 487–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavathiotis E., Reyna D. E., Davis M. L., Bird G. H., and Walensky L. D. (2010) BH3-triggered structural reorganization drives the activation of proapoptotic BAX. Mol. Cell 40, 481–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gahl R. F., He Y., Yu S., and Tjandra N. (2014) Conformational rearrangements in the pro-apoptotic protein, Bax, as it inserts into mitochondria: a cellular death switch. J. Biol. Chem. 289, 32871–32882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovell J. F., Billen L. P., Bindner S., Shamas-Din A., Fradin C., Leber B., and Andrews D. W. (2008) Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 135, 1074–1084 [DOI] [PubMed] [Google Scholar]
- 15.Czabotar P. E., Westphal D., Dewson G., Ma S., Hockings C., Fairlie W. D., Lee E. F., Yao S., Robin A. Y., Smith B. J., Huang D. C., Kluck R. M., Adams J. M., and Colman P. M. (2013) Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell 152, 519–531 [DOI] [PubMed] [Google Scholar]
- 16.Bleicken S., Jeschke G., Stegmueller C., Salvador-Gallego R., García-Sáez A. J., and Bordignon E. (2014) Structural model of active Bax at the membrane. Mol. Cell 56, 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sattler M., Liang H., Nettesheim D., Meadows R. P., Harlan J. E., Eberstadt M., Yoon H. S., Shuker S. B., Chang B. S., Minn A. J., Thompson C. B., and Fesik S. W. (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986 [DOI] [PubMed] [Google Scholar]
- 18.Ding J., Zhang Z., Roberts G. J., Falcone M., Miao Y., Shao Y., Zhang X. C., Andrews D. W., and Lin J. (2010) Bcl-2 and Bax interact via the BH1–3 groove-BH3 motif interface and a novel interface involving the BH4 motif. J. Biol. Chem. 285, 28749–28763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding J., Mooers B. H., Zhang Z., Kale J., Falcone D., McNichol J., Huang B., Zhang X. C., Xing C., Andrews D. W., and Lin J. (2014) After embedding in membranes antiapoptotic Bcl-XL protein binds both Bcl-2 homology region 3 and helix 1 of proapoptotic Bax protein to inhibit apoptotic mitochondrial permeabilization. J. Biol. Chem. 289, 11873–11896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barclay L. A., Wales T. E., Garner T. P., Wachter F., Lee S., Guerra R. M., Stewart M. L., Braun C. R., Bird G. H., Gavathiotis E., Engen J. R., and Walensky L. D. (2015) Inhibition of Pro-apoptotic BAX by a noncanonical interaction mechanism. Mol. Cell 57, 873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czabotar P. E., Lessene G., Strasser A., and Adams J. M. (2014) Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 15, 49–63 [DOI] [PubMed] [Google Scholar]
- 22.Sidhu S. S., and Fellouse F. A. (2006) Synthetic therapeutic antibodies. Nat. Chem. Biol. 2, 682–688 [DOI] [PubMed] [Google Scholar]
- 23.Paduch M., Koide A., Uysal S., Rizk S. S., Koide S., and Kossiakoff A. A. (2013) Generating conformation-specific synthetic antibodies to trap proteins in selected functional states. Methods 60, 3–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fellouse F. A., Esaki K., Birtalan S., Raptis D., Cancasci V. J., Koide A., Jhurani P., Vasser M., Wiesmann C., Kossiakoff A. A., Koide S., and Sidhu S. S. (2007) High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J. Mol. Biol. 373, 924–940 [DOI] [PubMed] [Google Scholar]
- 25.Koellhoffer J. F., Chen G., Sandesara R. G., Bale S., Saphire E. O., Chandran K., Sidhu S. S., and Lai J. R. (2012) Two synthetic antibodies that recognize and neutralize distinct proteolytic forms of the Ebola virus envelope glycoprotein. ChemBioChem 13, 2549–2557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koerber J. T., Thomsen N. D., Hannigan B. T., Degrado W. F., and Wells J. A. (2013) Nature-inspired design of motif-specific antibody scaffolds. Nat. Biotechnol. 31, 916–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao J., Sidhu S. S., and Wells J. A. (2009) Two-state selection of conformation-specific antibodies. Proc. Natl. Acad. Sci. U.S.A. 106, 3071–3076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsh J. J., Guan H. S., Li S., Chiles P. G., Tran D., and Morris T. A. (2013) Structural insights into fibrinogen dynamics using amide hydrogen/deuterium exchange mass spectrometry. Biochemistry 52, 5491–5502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li S., Tsalkova T., White M. A., Mei F. C., Liu T., Wang D., Woods V. L. Jr., and Cheng X. (2011) Mechanism of intracellular cAMP sensor Epac2 activation: cAMP-induced conformational changes identified by amide hydrogen/deuterium exchange mass spectrometry (DXMS). J. Biol. Chem. 286, 17889–17897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z., and Smith D. L. (1993) Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 2, 522–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Vries S. J., van Dijk M., and Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 32.Marcatili P., Olimpieri P. P., Chailyan A., and Tramontano A. (2014) Antibody structural modeling with prediction of immunoglobulin structure (PIGS). Nat. Protoc. 9, 2771–2783 [DOI] [PubMed] [Google Scholar]
- 33.Morin A., Eisenbraun B., Key J., Sanschagrin P. C., Timony M. A., Ottaviano M., and Sliz P. (2013) Collaboration gets the most out of software. eLife 2, e01456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Persson H., Ye W., Wernimont A., Adams J. J., Koide A., Koide S., Lam R., and Sidhu S. S. (2013) CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J. Mol. Biol. 425, 803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yethon J. A., Epand R. F., Leber B., Epand R. M., and Andrews D. W. (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J. Biol. Chem. 278, 48935–48941 [DOI] [PubMed] [Google Scholar]
- 36.Edwards A. L., Gavathiotis E., LaBelle J. L., Braun C. R., Opoku-Nsiah K. A., Bird G. H., and Walensky L. D. (2013) Multimodal interaction with BCL-2 family proteins underlies the proapoptotic activity of PUMA BH3. Chem. Bio. 20, 888–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leshchiner E. S., Braun C. R., Bird G. H., and Walensky L. D. (2013) Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. U.S.A. 110, E986–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gavathiotis E., Reyna D. E., Bellairs J. A., Leshchiner E. S., and Walensky L. D. (2012) Direct and selective small-molecule activation of proapoptotic BAX. Nat. Chem. Biol. 8, 639–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma J., Edlich F., Bermejo G. A., Norris K. L., Youle R. J., and Tjandra N. (2012) Structural mechanism of Bax inhibition by cytomegalovirus protein vMIA. Proc. Natl. Acad. Sci. U.S.A. 109, 20901–20906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 41.Annis M. G., Soucie E. L., Dlugosz P. J., Cruz-Aguado J. A., Penn L. Z., Leber B., and Andrews D. W. (2005) Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 24, 2096–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai C. J., Liu S., Hung C. L., Jhong S. R., Sung T. C., and Chiang Y. W. (2015) BAX-induced apoptosis can be initiated through a conformational selection mechanism. Structure 23, 139–148 [DOI] [PubMed] [Google Scholar]