

Abstract
Cardiovascular and metabolic disorders, such as hypertension, insulin resistance, dyslipidemia or obesity are linked with chronic low-grade inflammation and dysregulation of the renin–angiotensin system (RAS). Consequently, RAS inhibition by ACE inhibitors or angiotensin AT1 receptor (AT1R) blockers is the evidence-based standard for cardiovascular risk reduction in high-risk patients, including diabetics with albuminuria. In addition, RAS inhibition reduces the new onset of diabetes mellitus. Yet, the high and increasing prevalence of metabolic disorders, and the high residual risk even in properly treated patients, calls for additional means of pharmacological intervention. In the past decade, the stimulation of the angiotensin AT2 receptor (AT2R) has been shown to reduce inflammation, improve cardiac and vascular remodeling, enhance insulin sensitivity and increase adiponectin production. Therefore, a concept of dual AT1R/AT2R modulation emerges as a putative means for risk reduction in cardio-metabolic diseases. The approach employing simultaneous RAS blockade (AT1R) and RAS stimulation (AT2R) is distinct from previous attempts of double intervention in the RAS by dual blockade. Dual blockade abolishes the AT1R-linked RAS almost completely with subsequent risk of hypotension and hypotension-related events, i.e. syncope or renal dysfunction. Such complications might be especially prominent in patients with renal impairment or patients with isolated systolic hypertension and normal-to-low diastolic blood pressure values. In contrast to dual RAS blockade, the add-on of AT2R stimulation does not exert significant blood pressure effects, but it may complement and enhance the anti-inflammatory and antifibrotic/de-stiffening effects of the AT1R blockade and improve the metabolic profile. Further studies will have to investigate these putative effects in particular for settings in which blood pressure reduction is not primarily desired.
Key Points
| The combination of simultaneous RAS blockade (AT1R) and RAS stimulation (AT2R) represents a novel promising therapeutic concept, which is distinct from previous attempts of double intervention in the RAS by dual blockade. The add-on of AT2R stimulation complements and augments the anti-inflammatory and anti-fibrotic effects of the AT1R blockade. This combined intervention may address the need for further risk reduction in patients with well-controlled blood pressure or where further blood pressure reduction is not desired, such as in patients with isolated systolic hypertension, with renal dysfunction or in the elderly. |
Introduction
Cardiovascular mortality remains high world-wide. In Europe, cardiovascular events remain a major cause of premature deaths, being responsible for 42 % of all deaths in women and for 38 % of all deaths in men aged <75 years [1].
Cardiovascular morbidity and mortality are strongly determined by the presence of metabolic alterations such as different pro-thrombotic states, dyslipidemia, obesity and insulin resistance (with or without its overt clinical manifestation), which are clinically grouped in the metabolic syndrome. The cardio-metabolic syndrome, depending on the definition, usually features at least three of the following dysregulations: obesity (or central obesity), dyslipidemia [hypertriglyceridemia or low high-density lipoprotein (HDL) levels], hypertension or impaired glucose metabolism (impaired fasting glucose, insulin resistance, hyperinsulinemia or diabetes mellitus), while the World Health Organization adds microalbuminuria as a criterion as well [2]. Despite the advances in treatment and prevention of cardiovascular diseases, the continuous rise of metabolic disorders and abnormalities might render the fight for cardiovascular mortality reduction ineffective. The prevalence of overweight and obesity is rising in both developing and developed countries, with 300 million people being obese and 1 billion being overweight world-wide [3]. In the overweight and obese population there is around 40 % prevalence of some type of altered glucose metabolism [4].
Cardiac and metabolic dysregulations represent two sides of the cardio-metabolic coin and are interlinked in both directions. In hypertension, vasoconstriction or vascular rarefication was hypothesized to reduce glucose uptake and thus contribute to hyperglycemia per se [5]. The consequences are hyperinsulinemia followed by insulin resistance. On the other hand, there are several mechanisms by which impaired insulin sensitivity contributes to the development of cardiovascular pathologies. Insulin resistance blunts the vasodilator effects of insulin, and chronic hyperinsulinemia increases sympathetic nervous activity, reduces natriuresis, promotes vascular smooth muscle growth, endothelial dysfunction and hypertriglyceridemia with subsequent atherosclerosis, hypertension and vascular events such as myocardial infarction, stroke or peripheral arterial occlusion disease [6].
Behind the Scenes: Pathophysiological Considerations
Inflammation in Cardio-Metabolic Diseases
Below the surface of the overt cardio-metabolic presentations, there is a complex dysregulation of extracellular and intracellular signaling pathways with chronic low-grade inflammation at its core. This “tonic” inflammation is, besides insulin resistance, now recognized as a main etiological factor of obesity-related metabolic disorders [7, 8]. The adipose tissue is on one hand, the target of the inflammatory process; on the other hand, it can also act as its important modulator. Clinically, the blood levels of pro-inflammatory cytokines such as C-reactive protein or interleukin-6 (IL-6) are not only correlated with excess adipose mass [9, 10], but they also decrease along with weight loss [11]. The levels of pro-inflammatory and anti-inflammatory cytokines depend on the number of adipocytes and even more on the secretory profile of the adipose tissue. In the adipose tissue of healthy, lean subjects, CD4+ Treg cells prevail, producing type 2 T-helper cell (Th2) anti-inflammatory cytokines such as the IL-4 and IL-13 and thereby stimulating the differentiation of the residing macrophages towards the alternatively activated M2 (F4/80+, CD11b+) CD11c− phenotype [12]. This phenotype is characterized by IL-10 production that stimulates the adipocytes to adiponectin production and enhances insulin sensitivity. Adiponectin inhibits the nuclear factor (NF)-κB pathway as well as tumor necrosis factor (TNF)-α signaling and promotes the expression of the peroxisome proliferator-activated receptor (PPAR)-γ co-activator in macrophages, further promoting their M2 phenotype [13]. Adiponectin via 5′ adenosine monophosphate-activated protein kinase (AMPK) and subsequent desensitization of the toll-like receptor (TLR) signaling exhibits anti-inflammatory effects and through the endothelial NO-synthase pathway also cardio-protective effects [14].
High concentrations of saturated free fatty acids via TLR-4, advanced-glycation end-products, or Th1 type signals [such as interferon-gamma (INF-γ)] secreted by CD8+ T cells down-regulate PPAR-γ in the macrophages and stimulate their differentiation towards the classically activated M1 (F4/80+, CD11b+) CD11c+ phenotype [15, 16]. These macrophages produce pro-inflammatory molecules such as TNF-α, IL-6 and the major chemoattractant protein (MCP)-1. Especially MCP-1 via the chemokine receptor type 2 (CCR2) attracts further classically activated macrophages into the adipose tissue thereby increasing their absolute number [17]. Under these conditions, the pro-inflammatory molecules are also generated by the adipocytes, which also produce leptin. Although leptin improves insulin sensitivity and attenuates hyperlipidemia, it shifts macrophage differentiation towards the M1 phenotype, promotes TNF-α and IL-6 production [18, 19] and is thus considered a pro-inflammatory adipokine [14]. Moreover, due to the development of leptin resistance, the beneficial effects of leptin are blunted, despite the high leptin levels in subjects with increased adipose mass [20].
In cells, the pro-inflammatory cytokines release the repression of the nuclear receptor corepressor and subsequently also NF-κB, c-Jun N-terminal kinase (JNK) and phosphoinositide 3-kinase (PI3K) pathways. In macrophages and adipocytes, these pathways lead to the transactivation of overlapping sets of pro-inflammatory molecules [21, 22] and promote cellular insulin resistance as well as leptin secretion in adipocytes. In both macrophages and adipocytes, the loss of nuclear receptor corepressor activity can be prevented by PPAR-γ activation (e.g. by thiazolidinediones) resulting in reduced inflammation and improved insulin sensitivity. As insulin resistance evolves, the adipose tissue undergoes adipocyte hypertrophy and infiltration by inflammatory cells [7].
Renin–Angiotensin-System Activation in Cardio-Metabolic Diseases
The components of the local renin–angiotensin system (RAS), such as angiotensin I (Ang I), angiotensin-converting enzyme (ACE), angiotensin II (Ang II), angiotensin AT1 receptor (AT1R), angiotensin AT2 receptor (AT2R) as well as alternative enzymes such as cathepsin and chymase, are abundantly expressed in adipocytes and adipose tissue of various organs [23, 24]. Overfeeding in rodents results in increased angiotensinogen formation and increased local Ang II levels, alterations which were observed in human hypertensive obese patients as well [25].
Increased circulatory or local RAS activity is clearly associated with a variety of pathophysiologic processes, including blood pressure elevation, vasoconstriction, vascular and cardiac remodeling, and renal fibrosis [26]. Moreover, there is a distinct pathophysiological connection linking increased RAS activity and the metabolic alterations.
In addition to the hemodynamic link between the vasoconstrictive effects of Ang II and insulin resistance, Ang II opposes the effects of insulin on glucose metabolism in the liver and inhibits glycogen synthetase thereby contributing to insulin resistance [6]. Stimulation of AT1R by Ang II engenders vasoconstriction and hypokalemia, both impairing insulin sensitivity [5, 27]. AT1R activation also inhibits the glucose transporter type 4 (GLUT4) in skeletal muscle and thus reduces insulin sensitivity [28]. Similar effects might be attributed to ACE activity due to increased degradation of bradykinin [29]. Ang II further increases lipogenesis and triacylglyceride content in the adipocytes by increasing fatty acid synthase and glycerol-3-phosphate dehydrogenase [30–32]. Stimulation of the AT1R can also block adipogenesis and adipocyte differentiation thereby reducing insulin sensitivity [33].
Even of higher importance might be the modulatory effects of RAS on inflammatory activity. As described above, chronic inflammation plays a key role in the pathophysiology of the metabolic syndrome. The pro-inflammatory activity of Ang II via its AT1R is well recognized [34]. Among others, AT1R activation stimulates NF-κB, IL-6 and MCP-1 upregulation [35]. In obese mice on high-fat diet, AT1R blockade or AT1R-knock out (AT1R-KO) reduced renal macrophage infiltration, induced a shift from the deleterious M1 to the M2 macrophage phenotype, and prevented renal injury. Finally, AT1R-KO mice displayed lower body fat and cholesterol levels compared to their wild-type controls [36].
RAS Modulation in Cardio-Metabolic Disease
Angiotensin AT1 Receptor Blockers
From a clinical point of view, there is a large body of evidence for the reduction of cardiovascular morbidity and mortality by RAS inhibition, in particular in heart failure, hypertensive, diabetic, or generally, in high-risk patients. Large clinical trials have been performed with ACE inhibitors, for example, SOLVD [37], CAPP [38] STOP-2 [39] or HOPE [40] and AT1R blockers (ARBs), for example, CHARM [41], LIFE [42], VALUE [43] or ONTARGET [44].
The cardio-protective effects of RAS blockade observed in these and other trials are consistent with the effects of RAS blockade in subgroups of diabetic patients. In the HOPE trial, the ACE inhibitor ramipril achieved numerically an even higher relative reduction of the primary end-point (cardiovascular death, myocardial infarction or stroke) in the diabetic compared to the non-diabetic subpopulation [40]. Similarly, the ARB telmisartan was non-inferior compared to ramipril with regard to the reduction of the composite of cardiovascular death, myocardial infarction and stroke with/without hospitalization (primary and secondary efficacy outcomes) in the subgroup of diabetic patients in the ONTARGET trial [44]. Moreover ACE inhibitors and ARBs were not only shown to reduce cardiovascular events in diabetics, but also to reduce the new onset of diabetes [40, 41, 43, 45, 46]. In addition, there are several smaller trials conducted specifically in diabetic patients that have demonstrated the efficacy of ACE inhibitors and ARBs on various surrogate end-points, most notably albuminuria. The benefit was seen in particular in patients with proteinuria [47–52].
Yet, despite these successful trials providing new options for cardio-protection, the high and increasing prevalence of metabolic disorders calls for further reduction of the cardiovascular risk. For example the RENAAL study showed that even the appropriately treated diabetic patients were still at high cardiovascular and renal risk, which could be predicted by the residual albuminuria after RAS blockade [53, 54].
One of the approaches to address this need was the development of ARBs with enhanced PPAR-γ activity [55]. Some data indicate that the activation of PPARs might contribute to the beneficial effects of certain ARBs, such as telmisartan and irbesartan or the losartan metabolite EXP3179 [56–60]. It was demonstrated, that chronic treatment with losartan [61] or with high-dose (160 mg/day) telmisartan [62] are able to activate PPAR-γ in circulating monocytes of patients with hypertension and metabolic syndrome, respectively. The activation of PPAR-γ, on one hand, exerts lipogenic effects, but on the other hand it stimulates adiponectin production along with its insulin-sensitizing effect [63]. A partial PPAR-γ agonism might play a positive role, especially in the context of AT1R blockade, which (via NF-κB inhibition) prevents PPAR-γ down-regulation [64]. However, antidiabetic actions were also observed under ARB treatment with low PPAR-γ activating potential such as valsartan in both preclinical [28] and clinical [43] settings. Thus, the extent of the contribution of PPAR-γ activating properties of the individual ARBs to their clinical antidiabetic outcomes has not been clearly defined [65]. The development of the previously reported metabolically targeted ARBs with PPAR-γ activity, such as K-868 or PF-03838135 did not advance to a clinical phase and no novel data have been reported for these agents [66]. Another putative approach to augment the cardio-protective actions of the ARBs is to complement the AT1R blockade with the activation of the “protective RAS”, i.e. by AT2R stimulation.
Angiotensin AT2 Receptor Agonists
While the current therapeutic strategies aim to prevent an undesired activation of the AT1R, stimulation of the AT2R might provide a mean to oppose many of the AT1R-mediated actions [26, 67, 68]. There is accumulating evidence for the cardio-protective action of the AT2R, which is based not only on indirect evidence, but also on the investigation of the effects of a direct AT2R stimulation. Treatment with the first non-peptide highly selective AT2R agonist, compound 21, prevented or reverted associated vascular remodeling in large and middle-sized arteries in either l-NG-Nitroarginine methyl ester (l-NAME) or spontaneously hypertensive rats (SHR) [69, 70]. Compound 21 also significantly reduced the ischemic area after middle cerebral artery occlusion in rats, along with anti-oxidant and anti-inflammatory effects (such as MCP-1 and TNF-α down-regulation) [71], and in rats with permanent coronary artery ligation it improved cardiac function and myocardial architecture early [72] and later after myocardial infarction [73]. Notably, the cardio-protective effects of AT2R stimulation were not associated with a reduction in blood pressure in these or other experiments when compound 21 was administered peripherally. Compound 21 is under physiological situation not able to cross the blood–brain barrier. However, when compound 21 was administered intracerebroventricularly it reduced blood pressure and exerted sympatholytic action (improved baroreflex sensitivity) in Wistar Kyoto rats and SHR. These effects were not affected by AT1R blockade, but were sensitive to NO-synthase blockade [74]. A similar sympatholytic action was observed after central AT2R stimulation in rats with ligation-induced heart failure [75].
The putative mechanisms behind the beneficial effects of AT2R stimulation include the activation of the NO/cyclic guanosine monophosphate (cGMP) pathway [76], down-regulation and inhibition of mitogen-activated protein kinases (MAPKs) by protein phosphatases [77, 78], or direct disruption of AT1R function by AT1R–AT2R heterodimerization [79]. With regard to cardio-metabolic disease, the anti-inflammatory effects of AT2R stimulation appear to be the most relevant. These include the inhibition of NF-κB activity as a result of 11,12-epoxyeicosatrienoic acid epoxidation [80], augmented IL-10 production and T-cell differentiation to the Treg phenotype [81]. Indeed, the stimulation of the AT2R dose-dependently attenuated TLR-4 lipopolysaccharide (LPS)-induced TNF-α and IL-6 production but it increased IL-10 production via sustained selective extracellular-signal-regulated kinase 1/2 (ERK1/2) phosphorylation [82]. Similar anti-inflammatory effects were observed in human monocytic cells [83] or in high-fat diet-induced vascular inflammation [84].
Concerning AT1R–AT2R interaction, it has to be noted that it might not only take place on the cell membrane by heterodimerization, as previously reported [79], but it might be more related to a negative cytoplasmatic cross-talk [85]. Moreover, the angiotensin receptor function might be modulated by receptor-associated proteins such as AT1 receptor-associated protein (ATRAP), AT1 receptor associated protein 1 (ARAP1) for the AT1R and AT2 receptor interacting protein (ATIP) or AT2 receptor binding protein of 50 kDa for the AT2R (the topic is discussed extensively elsewhere [86]).
There are several experimental data confirming the beneficial effects of AT2R stimulation in vivo in animal models of metabolic disorders. In type 2 diabetic KKAy mice, compound 21 improved insulin sensitivity, increased adiponectin and reduced TNF-α levels, while protecting the pancreatic β-cells. These effects resulted in reduced body weight and fat mass within two weeks of treatment. Interestingly, the beneficial effects of AT2R activation were blocked by PPAR-γ inhibition [87]. In high-fructose/high-fat fed rats, compound 21 improved insulin sensitivity and glucose tolerance comparably to the ARB, losartan. Moreover, compound 21 lowered triacylglyceride levels as well, an effect not seen by losartan treatment [88]. Very similar results were obtained in high-fat diet mice. In this model, compound 21 improved insulin sensitivity, reduced TNF-α, increased adiponectin and IL-10 levels, and reduced serum triacylglyceride levels. Again, the effects of AT2R stimulation were broader compared to the effects of the ARB (valsartan in this study), which improved glucose tolerance only. Notably, the effects of both compound 21 and valsartan seemed to be mediated via the AT2R as they were not observed in AT2R-KO mice [89]. These results are complemented by the in vitro data from 3T3-L1 adipocytes. In this cell line, compound 21 reduced TNF-α-induced IL-6 and MCP-1 expression, yet without activating PPAR-γ and without inducing adipocyte differentiation [90]. Therefore, it remains controversial, whether PPAR-γ activation is involved in the signaling cascade of the AT2R, at least in the cardio-metabolic area. The AT2R is not only abundantly expressed in pancreatic β-cell of adult rats, but its stimulation significantly improved insulin synthesis and secretion [91]. Moreover, compound 21 up-regulated superoxide dismutase expression, reduced oxidative load and caspase-3 expression and consequently prevented cell death of pancreatic β-cell in streptozotocin-induced diabetic rats [92].
Several further data indicate that the attenuation of inflammation and improvement of the metabolic profile by AT2R stimulation translates into nephroprotective effects. Such nephroprotective effects have previously been reported in hypertensive models such as 2-kidney-1-clip renovascular hypertension [93] or in stroke-prone SHR [94]. The results on the protection against kidney damage observed in diabetic animals are in line with these findings in hypertensive models. In streptozotocin-induced diabetic rats, compound 21 normalized TNF-α and IL-6 levels and increased NO and cGMP with consequent attenuation of urinary albumin to creatinine ratio, yet without affecting blood pressure [95]. Similar protection against diabetic nephropathy was recently reported, in type 1 diabetic mice, in which compound 21 attenuated cystatin C levels, albuminuria, mesangial expansion, and glomerulosclerosis [96]. Therefore, it seems that compound 21 prevents albuminuria via normalization of the NO/cGMP pathway but mainly via reduction of renal inflammatory infiltration and subsequently the prevention structural alterations of the glomerular filter.
Finally, AT2R stimulation exerted anti-inflammatory and nephroprotective effects also in obese Zucker rats. In these rats, the peptide AT2R agonist CGP-42112A reduced systemic inflammation and oxidative load [97], and compound 21 reduced TNF-α expression, renal macrophage infiltration, fibrosis and albuminuria [98]. Although the effects in Zucker rats were not associated with blood pressure-lowering effect by AT2R stimulation, compound 21 prevented salt-sensitive hypertension in these rats. This hemodynamic effect, not generally observed in other animal models, might be related to reduced renal Ang II and AT1R and enhanced ACE2 and Ang (1–7) levels and AT2R expression [99] or to the natriuretic effect by compound 21, which might be unmasked under certain conditions [100–102].
It has to be noted, that the AT2R pathway might be stimulated by the Ang (1–7) produced by ACE2 as well. Congruently, ACE2 overexpression improved fasting glucose in diabetic mice [103] or vice-versa, the loss of ACE2 exaggerated high-calorie diet-induced insulin resistance [104]. However, due to receptor hetero-dimerization, the blockade of either AT2R or Mas receptor seems to block the effects of the other receptor [105]. Thus, we need to be cautious when attributing the effects of ACE2/Ang (1–7) stimulation to the activation of a particular receptor (AT2R or the Mas).
Combined Angiotensin Receptor Modulation
As described above, the RAS blockade currently represents the gold standard in cardiovascular therapy, including diabetic nephropathy. The mechanisms involved include direct blockade of the deleterious effects of Ang II but also indirect immune-modulatory effects. Similar mechanisms are involved in the beneficial (e.g. anti-inflammatory, insulin sensitizing, nephroprotective) effects of the stimulation of the “protective RAS” in various animal models of diabetes or obesity. Yet, because the mechanisms of AT2R stimulation and AT1R blockade are only partly overlapping as described above and display a certain complementarity, there might be a potential for the combination of the AT1R blockade and AT2R stimulation (Fig. 1).
Fig. 1.
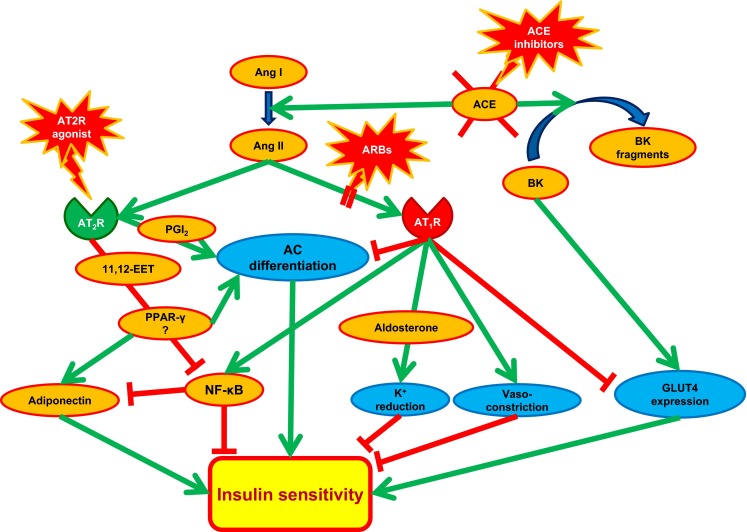
Renin–angiotensin system (RAS) and insulin sensitivity. Scheme showing the modulation [by angiotensin AT2 receptor (AT2R) agonist, angiotensin AT1 receptor AT1R blocker (ARB) or angiotensin-converting enzyme (ACE) inhibition] of insulin sensitivity by the RAS and the involvement of pro-/anti-inflammatory signals. The ACE converts angiotensin (Ang) I to Ang II. It also catalyses the degradation of bradykinin (BK), which enhances the expression of glucose transporter type 4 (GLUT4) [29], an effect opposed by AT1R activation [28]. AT1R also disrupts insulin sensitivity by aldosterone-mediated hypokalemia [65], vasoconstriction [5] and inhibition of adipocyte (AC) differentiation [33]. On the other hand, the AT2R promotes AC differentiation via prostacyclin (PGI2) production [106] and inhibits NF-κB signaling via 11,12-epoxyeicosatrienoic acid (EET) [80], with possible involvement of the peroxisome proliferator-activated receptor-γ (PPAR-γ) [87] vs. [90]. PPAR-γ activation promotes adipocyte differentiation [88] the release of adiponectin with its insulin sensitizing effects [63]
The idea of dual hit in the RAS is not novel. Some early trials with surrogate end-points have investigated the potential benefit of dual RAS blockade with variable outcomes. In the CALM study in diabetic patients with microalbuminuria, the ACE inhibitor + ARB combination led to more pronounced reduction of microalbuminuria compared to either monotherapy [107], while no additive effect on albuminuria was observed in high-risk patients in the IMPROVE trial [108]. The combination of renin inhibition and losartan significantly reduced proteinuria in the AVOID study in diabetic patients with nephropathy [109, 110]. However, the results of the later studies with hard end-points: ONTARGET [44, 111] and ALTITUDE [112] advise against the use of dual RAS blockade. The ONTARGET trial in high-risk patients displayed more adverse events (such as hypotension, syncope, kidney dysfunction, hyperkalemia) in the combined arm (the ACE inhibitor ramipril + the ARB telmisartan). The ALTITUDE trial, which tested the direct renin inhibitor, aliskiren, in diabetic patients with renal disease needed to be halted prematurely due to increased incidence of adverse events (slight numeric increase in stroke rates and significant increase in hyperkalemia and hypotension) in the aliskiren arm (on the background of ACE inhibition or ARB treatment). As such, the concept of dual RAS blockade failed primarily due to safety concerns. A recent meta-analysis demonstrated that dual therapy was associated with 66 % higher risk of hypotension [113]. It was hypothesized that lower blood pressure in the combination arm might have contributed to the increased incidence of renal end-points in the combination arm of the ONTARGET trial [114] and that the low blood pressure at baseline might be responsible for the renal dysfunction observed in the aliskiren arm of the ALTITUDE trial [115]. These considerations suggest that the second hit in the RAS should not “knock-out” the RAS completely in order to minimize the potential for undesired hypotension along with other unwarranted effects. With this regard, the activation of the AT2R represents a putatively better suited candidate for an add-on to the AT1R blocking strategies.
Yet, because these considerations represent a translational approach with a very specific aim, only few experimental studies have investigated the effects of the combination of AT1R blockade with AT2R stimulation. Experiments in two animal models of hypertension (L-NAME and SHR) indicate that there is no significant additive blood pressure effect by AT2R stimulation when added to the ARB (olmesartan or losartan, respectively) [69, 70]. Nevertheless, a superior effect by the combination treatment on some aspects of vascular remodeling (most notably on hydroxyprolin/collagen content and arterial stiffness) over monotherapy was observed [69, 70]. Therefore a clinical rationale for combined angiotensin receptor modulation includes the AT1R blockade aimed at achieving target blood pressure values and the AT2R stimulation aimed to reduce arterial stiffness in the long term. Arterial stiffness is an independent predictor of cardiovascular events and all-cause mortality [116]. The reduction of arterial stiffness prevents the increase in the systolic blood pressure, without compromising diastolic blood pressure values (Fig. 2). Such a combined effect might be of particular significance in end-stage renal patients in which aortic stiffness attenuation has been indeed shown to improve survival [117]. The mechanisms responsible for these beneficial effects of the combined angiotensin receptor blockade were largely attributed to the anti-inflammatory effects of compound 21. In fact, while the ARBs may also block the NF-κB activation, this pathway might still be triggered independently from Ang II. In contrast, stimulation of the AT2R might prevent such NF-κB activation, with subsequent release of adiponectin and the following insulin sensitizing effects [80, 118].
Fig. 2.
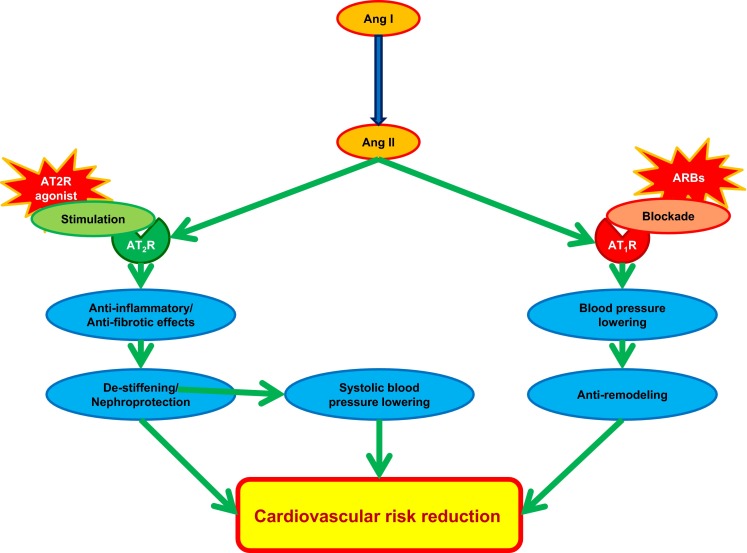
Combined angiotensin receptor modulation. Scheme showing the rationale for combining the stimulation of the AT2 receptor (AT2R) with the blockade of the angiotensin AT1 receptor AT1R by the AT1R blockers (ARBs). The employment of the AT1R blockade is aimed at achieving target blood pressure values and the AT2R stimulation is aimed to reduce arterial stiffness in the long term. The reduction of arterial stiffness prevents the increase in the systolic blood pressure without compromising diastolic blood pressure values. Such a combined effect might be of particular significance in the elderly, in patients with isolated systolic hypertension or in end-stage renal patients
At present, there is only one study which has investigated the effects of combined AT2R stimulation + AT1R blockade specifically in the setting of a metabolic disorder. In this study, compound 21 was tested in comparison and in combination with losartan in Zucker diabetic fatty rats. Add-on of compound 21 extended the reduction of albuminuria from 15 weeks of age (in the losartan group) to 20 weeks of age (the end of the experimental protocol). Moreover, while losartan reduced renal macrophage infiltration, TNF-α expression and glomerular and perivascular fibrosis, compound 21 reduced tubulointerstitial fibrosis (the best correlate for renal disease progression) as well. In the combination arm, there was also a slight numeric tendency for larger effect on glomerular and perivascular fibrosis compared to losartan alone. Consistent with previous studies, reduced triacylglyceride levels were observed only in groups treated with compound 21. Finally, compound 21 neither alone, nor in combination with losartan altered blood pressure, confirming that its beneficial effects are largely blood pressure-independent [98]. Due to these additional beneficial effects of the add-on AT2R stimulation to AT1R blockade, such combination represents an interesting perspective for future clinical investigation.
Conclusion
The combined modulation of angiotensin receptors by AT1R blockade and simultaneous AT2R stimulation provides an intriguing novel therapeutic concept. This approach might be suitable in particular for patient populations with well-controlled blood pressure or where further blood pressure reduction is not desired. In addition, patients with chronic low-grade inflammation and/or isolated systolic hypertension might benefit from the anti-inflammatory and de-stiffening effects of the AT2R stimulation additive to the AT1R blockade. Yet, there are no clinical and only sparse pre-clinical in vivo data available for this approach. Thus, future studies on the combined angiotensin receptor modulation in the field of cardio-metabolic diseases represent an attractive and highly sought endeavor.
Compliance with Ethical Standards
Funding
The work was supported by the Grants of the Scientific Grant Agency VEGA 1-0380-14 and Agency for Science and Research Support APVV 0205-11.
Conflicts of interest
Ludovit Paulis, Sebastien Foulquier, Pawel Namsolleck, Chaira Ricarti and Thomas Unger have no potential conflicts of interest that might be relevant to the contents of this review. Ulrike Steckelings has received short term student fellowship Grant and free drug supply of compound 21 (for research work) from Vicore Pharma.
References
- 1.Perk J, De Backer G, Gohlke H, Graham I, Reiner Z, Verschuren M, Albus C, Benlian P, Boysen G, Cifkova R, Deaton C, Ebrahim S, Fisher M, Germano G, Hobbs R, Hoes A, Karadeniz S, Mezzani A, Prescott E, Ryden L, Scherer M, Syvänne M, Scholte op Reimer WJ, Vrints C, Wood D, Zamorano JL, Zannad F, European Association for Cardiovascular Prevention and Rehabilitation (EACPR) ESC Committee for Practice Guidelines (CPG) European guidelines on cardiovascular disease prevention in clinical practice (version 2012). The Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts) Eur Heart J. 2012;33(13):1635–1701. doi: 10.1093/eurheartj/ehs092. [DOI] [PubMed] [Google Scholar]
- 2.Magliano DJ, Shaw JE, Zimmet PZ. How to best define the metabolic syndrome. Ann Med. 2006;38:34–41. doi: 10.1080/07853890500300311. [DOI] [PubMed] [Google Scholar]
- 3.World Heath Organization. Fact sheet: Obesity and overweight. 2007. http://www.who.int/dietphysical-activity/publications/facts/obesity/en/. Accessed 8 June 2007.
- 4.Benjamin SM, Valdez R, Geiss LS, Rolka DB, Narayan KM. Estimated number of adults with prediabetes in the US in 2000: opportunities for prevention. Diabetes Care. 2003;26:645–649. doi: 10.2337/diacare.26.3.645. [DOI] [PubMed] [Google Scholar]
- 5.Julius S, Gudbrandsson T, Jamerson K, Tariq Shahab S, Andersson O. The hemodynamic link between insulin resistance and hypertension. J Hypertens. 1991;9(11):983–986. doi: 10.1097/00004872-199111000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Katovich MJ, Pachori A. Effects of inhibition of the renin–angiotensin system on the cardiovascular actions of insulin. Diabetes Obes Metab. 2000;2(1):3–14. doi: 10.1046/j.1463-1326.2000.00044.x. [DOI] [PubMed] [Google Scholar]
- 7.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 8.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444(7121):860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 9.Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282(22):2131–2135. doi: 10.1001/jama.282.22.2131. [DOI] [PubMed] [Google Scholar]
- 10.Fried SK, Bunkin DA, Greenberg AS. Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: depot difference and regulation by glucocorticoid. J Clin Endocrinol Metab. 1998;83(3):847–850. doi: 10.1210/jcem.83.3.4660. [DOI] [PubMed] [Google Scholar]
- 11.Esposito K, Pontillo A, Di Palo C, Giugliano G, Masella M, Marfella R, Giugliano D. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: a randomized trial. JAMA. 2003;289(14):1799–1804. doi: 10.1001/jama.289.14.1799. [DOI] [PubMed] [Google Scholar]
- 12.Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, Mathis D. Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med. 2009;15(8):930–939. doi: 10.1038/nm.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, Chawla A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7(6):496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97. doi: 10.1038/nri2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huttunen HJ, Fages C, Rauvala H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J Biol Chem. 1999;274(28):19919–19924. doi: 10.1074/jbc.274.28.19919. [DOI] [PubMed] [Google Scholar]
- 16.Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, Yoshimura K, Kadowaki T, Nagai R. CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med. 2009;15(8):914–920. doi: 10.1038/nm.1964. [DOI] [PubMed] [Google Scholar]
- 17.Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, Ferrante AW., Jr CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lord GM, Matarese G, Howard JK, Baker RJ, Bloom SR, Lechler RI. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature. 1998;394(6696):897–901. doi: 10.1038/29795. [DOI] [PubMed] [Google Scholar]
- 19.Santos-Alvarez J, Goberna R, Sánchez-Margalet V. Human leptin stimulates proliferation and activation of human circulating monocytes. Cell Immunol. 1999;194(1):6–11. doi: 10.1006/cimm.1999.1490. [DOI] [PubMed] [Google Scholar]
- 20.Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395(6704):763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa S, Lozach J, Jepsen K, Sawka-Verhelle D, Perissi V, Sasik R, Rose DW, Johnson RS, Rosenfeld MG, Glass CK. A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc Natl Acad Sci USA. 2004;101(40):14461–14466. doi: 10.1073/pnas.0405786101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, Glass CK. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR depression checkpoint. Mol Cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karlsson C, Lindell K, Ottosson M, Sjöström L, Carlsson B, Carlsson LM. Human adipose tissue expresses angiotensinogen and enzymes required for its conversion to angiotensin II. J Clin Endocrinol Metab. 1998;83(11):3925–3929. doi: 10.1210/jcem.83.11.5276. [DOI] [PubMed] [Google Scholar]
- 24.Crandall DL, Herzlinger HE, Saunders BD, Armellino DC, Kral JG. Distribution of angiotensin II receptors in rat and human adipocytes. J Lipid Res. 1994;35(8):1378–1385. [PubMed] [Google Scholar]
- 25.Engeli S, Schling P, Gorzelniak K, Boschmann M, Janke J, Ailhaud G, Teboul M, Massiéra F, Sharma AM. The adipose-tissue renin–angiotensin-aldosterone system: role in the metabolic syndrome? Int J Biochem Cell Biol. 2003;35(6):807–825. doi: 10.1016/S1357-2725(02)00311-4. [DOI] [PubMed] [Google Scholar]
- 26.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52(3):415–472. [PubMed] [Google Scholar]
- 27.Zillich AJ, Garg J, Basu S, Bakris GL, Carter BL. Thiazide diuretics, potassium, and the development of diabetes: a quantitative review. Hypertension. 2006;48(2):219–224. doi: 10.1161/01.HYP.0000231552.10054.aa. [DOI] [PubMed] [Google Scholar]
- 28.Shiuchi T, Iwai M, Li HS, Wu L, Min LJ, Li JM, Okumura M, Cui TX, Horiuchi M. Angiotensin II type-1 receptor blocker valsartan enhances insulin sensitivity in skeletal muscles of diabetic mice. Hypertension. 2004;43(5):1003–1010. doi: 10.1161/01.HYP.0000125142.41703.64. [DOI] [PubMed] [Google Scholar]
- 29.Shiuchi T, Cui TX, Wu L, Nakagami H, Takeda-Matsubara Y, Iwai M, Horiuchi M. ACE inhibitor improves insulin resistance in diabetic mouse via bradykinin and NO. Hypertension. 2002;40(3):329–334. doi: 10.1161/01.HYP.0000028979.98877.0C. [DOI] [PubMed] [Google Scholar]
- 30.Jones BH, Standridge MK, Moustaid N. Angiotensin II increases lipogenesis in 3T3-L1 and human adipose cells. Endocrinology. 1997;138(4):1512–1519. doi: 10.1210/endo.138.4.5038. [DOI] [PubMed] [Google Scholar]
- 31.Wakil SJ, Stoops JK, Joshi VC. Fatty acid synthesis and its regulation. Annu Rev Biochem. 1983;52:537–579. doi: 10.1146/annurev.bi.52.070183.002541. [DOI] [PubMed] [Google Scholar]
- 32.Weisinger RS, Begg DP, Chen N, Jois M, Mathai ML, Sinclair AJ. The problem of obesity: is there a role for antagonists of the renin–angiotensin system? Asia Pac J Clin Nutr. 2007;16(Suppl 1):359–367. [PubMed] [Google Scholar]
- 33.Janke J, Engeli S, Gorzelniak K, Luft FC, Sharma AM. Mature adipocytes inhibit in vitro differentiation of human preadipocytes via angiotensin type 1 receptors. Diabetes. 2002;51(6):1699–1707. doi: 10.2337/diabetes.51.6.1699. [DOI] [PubMed] [Google Scholar]
- 34.Dagenais NJ, Jamali F. Protective effects of angiotensin II interruption: evidence for antiinflammatory actions Pharmacotherapy. 2005;25:1213–1229. doi: 10.1592/phco.2005.25.9.1213. [DOI] [PubMed] [Google Scholar]
- 35.Phillips MI, Kagiyama S. Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs. 2002;3(4):569–577. [PubMed] [Google Scholar]
- 36.Ma LJ, Corsa BA, Zhou J, Yang H, Li H, Tang YW, Babaev VR, Major AS, Linton MF, Fazio S, Hunley TE, Kon V, Fogo AB. Angiotensin type 1 receptor modulates macrophage polarization and renal injury in obesity. Am J Physiol Renal Physiol. 2011;300(5):F1203–F1213. doi: 10.1152/ajprenal.00468.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.The SOLVD Investigators Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325(5):293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 38.Hansson L, Lindholm LH, Niskanen L, Lanke J, Hedner T, Niklason A, Luomanmäki K, Dahlöf B, de Faire U, Mörlin C, Karlberg BE, Wester PO, Björck JE. Effect of angiotensin-converting-enzyme inhibition compared with conventional therapy on cardiovascular morbidity and mortality in hypertension: the Captopril Prevention Project (CAPP) randomised trial. Lancet. 1999;353:611–616. doi: 10.1016/S0140-6736(98)05012-0. [DOI] [PubMed] [Google Scholar]
- 39.Hansson L, Lindholm LH, Ekbom T, Dahlöf B, Lanke J, Scherstén B, Wester PO, Hedner T, de Faire U. Randomised trial of old and new antihypertensive drugs in elderly patients: cardiovascular mortality and morbidity the Swedish Trial in Old Patients with Hypertension-2 study. Lancet. 1999;354:1751–1756. doi: 10.1016/S0140-6736(99)10327-1. [DOI] [PubMed] [Google Scholar]
- 40.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342(3):145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 41.Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Yusuf S. Pocock S; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362(9386):759–766. doi: 10.1016/S0140-6736(03)14282-1. [DOI] [PubMed] [Google Scholar]
- 42.Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H, LIFE Study Group Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2000;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 43.Julius S, Kjeldsen SE, Weber M, Brunner HR, Ekman S, Hansson L, Hua T, Laragh J, McInnes GT, Mitchell L, Plat F, Schork A, Smith B, Zanchetti A, VALUE trial group Outcomes in hypertensive patients at high cardiovascular risk treated with regimens based on valsartan or amlodipine: the VALUE randomised trial. Lancet. 2004;363(9426):2022–2031. doi: 10.1016/S0140-6736(04)16451-9. [DOI] [PubMed] [Google Scholar]
- 44.ONTARGET Investigators. Yusuf S, Teo KK, Pogue J, Dyal L, Copland I, Schumacher H, Dagenais G, Sleight P, Anderson C. Telmisartan, ramipril, or both in patients at high risk for vascular events. N Engl J Med. 2008;358:1547–1559. doi: 10.1056/NEJMoa0801317. [DOI] [PubMed] [Google Scholar]
- 45.Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H, LIFE Study Group Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359(9311):995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 46.Elliott WJ, Meyer PM. Incident diabetes in clinical trials of antihypertensive drugs: a network meta-analysis. Lancet. 2007;369(9557):201–207. doi: 10.1016/S0140-6736(07)60108-1. [DOI] [PubMed] [Google Scholar]
- 47.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329(20):1456–1462. doi: 10.1056/NEJM199311113292004. [DOI] [PubMed] [Google Scholar]
- 48.Parving HH, Lehnert H, Bröchner-Mortensen J, Gomis R, Andersen S, Arner P, Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria Study Group The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N Engl J Med. 2001;345(12):870–878. doi: 10.1056/NEJMoa011489. [DOI] [PubMed] [Google Scholar]
- 49.Brenner BM, Cooper ME, de Zeeuw D, Keane WF, Mitch WE, Parving HH, Remuzzi G, Snapinn SM, Zhang Z, Shahinfar S, RENAAL Study Investigators Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345(12):861–869. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 50.Lewis EJ, Hunsicker LG, Clarke WR, Berl T, Pohl MA, Lewis JB, Ritz E, Atkins RC, Rohde R, Raz I, Collaborative Study Group Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345(12):851–860. doi: 10.1056/NEJMoa011303. [DOI] [PubMed] [Google Scholar]
- 51.Bilous R, Chaturvedi N, Sjølie AK, Fuller J, Klein R, Orchard T, Porta M, Parving HH. Effect of candesartan on microalbuminuria and albumin excretion rate in diabetes: three randomized trials. Ann Intern Med. 2009;151(1):11–20, W3–4. [DOI] [PubMed]
- 52.Haller H, Ito S, Izzo JL, Jr, Januszewicz A, Katayama S, Menne J, Mimran A, Rabelink TJ, Ritz E, Ruilope LM, Rump LC, Viberti G, ROADMAP Trial Investigators Olmesartan for the delay or prevention of microalbuminuria in type 2 diabetes. N Engl J Med. 2011;364(10):907–917. doi: 10.1056/NEJMoa1007994. [DOI] [PubMed] [Google Scholar]
- 53.de Zeeuw D, Remuzzi G, Parving HH, Keane WF, Zhang Z, Shahinfar S, Snapinn S, Cooper ME, Mitch WE, Brenner BM. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004;65(6):2309–2320. doi: 10.1111/j.1523-1755.2004.00653.x. [DOI] [PubMed] [Google Scholar]
- 54.Eijkelkamp WB, Zhang Z, Remuzzi G, Parving HH, Cooper ME, Keane WF, Shahinfar S, Gleim GW, Weir MR, Brenner BM, de Zeeuw D. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007;18(5):1540–1546. doi: 10.1681/ASN.2006050445. [DOI] [PubMed] [Google Scholar]
- 55.Kurtz TW, Klein U. Next generation multifunctional angiotensin receptor blockers. Hypertens Res. 2009;32(10):826–834. doi: 10.1038/hr.2009.135. [DOI] [PubMed] [Google Scholar]
- 56.Schupp M, Janke J, Clasen R, Unger T, Kintscher U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. Circulation. 2004;109(17):2054–2057. doi: 10.1161/01.CIR.0000127955.36250.65. [DOI] [PubMed] [Google Scholar]
- 57.Schupp M, Clemenz M, Gineste R, Witt H, Janke J, Helleboid S, Hennuyer N, Ruiz P, Unger T, Staels B, Kintscher U. Molecular characterization of new selective peroxisome proliferator-activated receptor gamma modulators with angiotensin receptor blocking activity. Diabetes. 2005;54(12):3442–3452. doi: 10.2337/diabetes.54.12.3442. [DOI] [PubMed] [Google Scholar]
- 58.Schupp M, Lee LD, Frost N, Umbreen S, Schmidt B, Unger T, Kintscher U. Regulation of peroxisome proliferator-activated receptor gamma activity by losartan metabolites. Hypertension. 2006;47(3):586–589. doi: 10.1161/01.HYP.0000196946.79674.8b. [DOI] [PubMed] [Google Scholar]
- 59.Clemenz M, Frost N, Schupp M, Caron S, Foryst-Ludwig A, Böhm C, Hartge M, Gust R, Staels B, Unger T, Kintscher U. Liver-specific peroxisome proliferator-activated receptor alpha target gene regulation by the angiotensin type 1 receptor blocker telmisartan. Diabetes. 2008;57(5):1405–1413. doi: 10.2337/db07-0839. [DOI] [PubMed] [Google Scholar]
- 60.Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, Qi N, Wang J, Avery MA, Kurtz TW. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43(5):993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- 61.Kappert K, Tsuprykov O, Kaufmann J, Fritzsche J, Ott I, Goebel M, Bähr IN, Hässle PL, Gust R, Fleck E, Unger T, Stawowy P, Kintscher U. Chronic treatment with losartan results in sufficient serum levels of the metabolite EXP3179 for PPARgamma activation. Hypertension. 2009;54(4):738–743. doi: 10.1161/HYPERTENSIONAHA.109.132886. [DOI] [PubMed] [Google Scholar]
- 62.Bähr IN, Tretter P, Krüger J, Stark RG, Schimkus J, Unger T, Kappert K, Scholze J, Parhofer KG, Kintscher U. High-dose treatment with telmisartan induces monocytic peroxisome proliferator-activated receptor-γ target genes in patients with the metabolic syndrome. Hypertension. 2011;58(4):725–732. doi: 10.1161/HYPERTENSIONAHA.111.173542. [DOI] [PubMed] [Google Scholar]
- 63.Clasen R, Schupp M, Foryst-Ludwig A, Sprang C, Clemenz M, Krikov M, Thöne-Reineke C, Unger T, Kintscher U. PPARgamma-activating angiotensin type-1 receptor blockers induce adiponectin. Hypertension. 2005;46(1):137–143. doi: 10.1161/01.HYP.0000168046.19884.6a. [DOI] [PubMed] [Google Scholar]
- 64.Tham DM, Martin-McNulty B, Wang YX, Wilson DW, Vergona R, Sullivan ME, Dole W, Rutledge JC. Angiotensin II is associated with activation of NF-kappaB-mediated genes and downregulation of PPARs. Physiol Genomics. 2002;11(1):21–30. doi: 10.1152/physiolgenomics.00062.2002. [DOI] [PubMed] [Google Scholar]
- 65.Kintscher U, Foryst-Ludwig A, Unger T. Inhibiting angiotensin type 1 receptors as a target for diabetes. Expert Opin Ther Targets. 2008;12(10):1257–1263. doi: 10.1517/14728222.12.10.1257. [DOI] [PubMed] [Google Scholar]
- 66.Paulis L, Rajkovicova R, Simko F. New developments in the pharmacological treatment of hypertension: dead-end or a glimmer at the horizon? Curr Hypertens Rep. 2015;17(6):557. doi: 10.1007/s11906-015-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Danyel LA, Schmerler P, Paulis L, Unger T, Steckelings UM. Impact of AT2-receptor stimulation on vascular biology, kidney function, and blood pressure. Integr Blood Press Control. 2013;6:153–161. doi: 10.2147/IBPC.S34425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Unger T, Steckelings UM, dos Santos RAS. The protective arm of the renin angiotensin system. Functional aspects and therapeutic implications. ISBN 978-0-12-801364-9 1st ed. Elsevier: Academic Press; 2015.
- 69.Paulis L, Becker ST, Lucht K, Schwengel K, Slavic S, Kaschina E, Thöne-Reineke C, Dahlöf B, Baulmann J, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation in Nω-nitro-l-arginine-methyl ester-induced hypertension: the effect on pulse wave velocity and aortic remodeling. Hypertension. 2012;59(2):485–492. doi: 10.1161/HYPERTENSIONAHA.111.185496. [DOI] [PubMed] [Google Scholar]
- 70.Rehman A, Leibowitz A, Yamamoto N, Rautureau Y, Paradis P, Schiffrin EL. Angiotensin type 2 receptor agonist compound 21 reduces vascular injury and myocardial fibrosis in stroke-prone spontaneously hypertensive rats. Hypertension. 2012;59(2):485–492. doi: 10.1161/HYPERTENSIONAHA.111.180158. [DOI] [PubMed] [Google Scholar]
- 71.Min LJ, Mogi M, Tsukuda K, Jing F, Ohshima K, Nakaoka H, Kan-No H, Wang XL, Chisaka T, Bai HY, Iwanami J, Horiuchi M. Direct stimulation of angiotensin II type 2 receptor initiated after stroke ameliorates ischemic brain damage. Am J Hypertens. 2014;27(8):1036–1044. doi: 10.1093/ajh/hpu015. [DOI] [PubMed] [Google Scholar]
- 72.Kaschina E, Grzesiak A, Li J, Foryst-Ludwig A, Timm M, Rompe F, Sommerfeld M, Kemnitz UR, Curato C, Namsolleck P, Tschöpe C, Hallberg A, Alterman M, Hucko T, Paetsch I, Dietrich T, Schnackenburg B, Graf K, Dahlöf B, Kintscher U, Unger T, Steckelings UM. Angiotensin II type 2 receptor stimulation: a novel option of therapeutic interference with the renin–angiotensin system in myocardial infarction? Circulation. 2008;118(24):2523–2532. doi: 10.1161/CIRCULATIONAHA.108.784868. [DOI] [PubMed] [Google Scholar]
- 73.Lauer D, Slavic S, Sommerfeld M, Thöne-Reineke C, Sharkovska Y, Hallberg A, Dahlöf B, Kintscher U, Unger T, Steckelings UM, Kaschina E. Angiotensin type 2 receptor stimulation ameliorates left ventricular fibrosis and dysfunction via regulation of tissue inhibitor of matrix metalloproteinase 1/matrix metalloproteinase 9 axis and transforming growth factor β1 in the rat heart. Hypertension. 2014;63(3):e60–e67. doi: 10.1161/HYPERTENSIONAHA.113.02522. [DOI] [PubMed] [Google Scholar]
- 74.Brouwers S, Smolders I, Wainford RD, Dupont AG. Hypotensive and sympathoinhibitory responses to selective central AT2 receptor stimulation in spontaneously hypertensive rats. Clin Sci (Lond). 2015;129:81–92. doi: 10.1042/CS20140776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gao J, Zucker IH, Gao L. Activation of central angiotensin type 2 receptors by compound 21 improves arterial baroreflex sensitivity in rats with heart failure. Am J Hypertens. 2014;27(10):1248–1256. doi: 10.1093/ajh/hpu044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gohlke P, Pees C, Unger T. AT2 receptor stimulation increases aortic cyclic GMP in SHRSP by a kinin-dependent mechanism. Hypertension. 1998;31(1 Pt 2):349–355. doi: 10.1161/01.HYP.31.1.349. [DOI] [PubMed] [Google Scholar]
- 77.Dandapat A, Hu CP, Chen J, Liu Y, Khan JA, Remeo F, et al. Over-expression of angiotensin II type 2 receptor (agtr2) decreases collagen accumulation in atherosclerotic plaque. Biochem Biophys Res Commun. 2008;366:871–877. doi: 10.1016/j.bbrc.2007.11.061. [DOI] [PubMed] [Google Scholar]
- 78.Fischer TA, Singh K, O’Hara DS, Kaye DM, Kelly RA. Role of AT1 and AT2 receptors in regulation of MAPKs and MKP-1 by ANG II in adult cardiac myocytes. Am J Physiol. 1998;275(3 Pt 2):H906–H916. doi: 10.1152/ajpheart.1998.275.3.H906. [DOI] [PubMed] [Google Scholar]
- 79.AbdAlla S, Lother H, Abdel-tawab AM, Quitterer U. The angiotensin II AT2 receptor is an AT1 receptor antagonist. J Biol Chem. 2001;276(43):39721–39726. doi: 10.1074/jbc.M105253200. [DOI] [PubMed] [Google Scholar]
- 80.Rompe F, Artuc M, Hallberg A, Alterman M, Ströder K, Thöne-Reineke C, Reichenbach A, Schacherl J, Dahlöf B, Bader M, Alenina N, Schwaninger M, Zuberbier T, Funke-Kaiser H, Schmidt C, Schunck WH, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension. 2010;55(4):924–931. doi: 10.1161/HYPERTENSIONAHA.109.147843. [DOI] [PubMed] [Google Scholar]
- 81.Valero-Esquitino V, Lucht K, Namsolleck P, Monnet-Tschudi F, Stubbe T, Lucht F, Liu M, Ebner F, Brandt C, Danyel LA, Villela DC, Paulis L, Thoene-Reineke C, Dahlöf B, Hallberg A, Unger T, Sumners C, Steckelings UM. Direct angiotensin type 2 receptor (AT2R) stimulation attenuates T-cell and microglia activation and prevents demyelination in experimental autoimmune encephalomyelitis in mice. Clin Sci (Lond). 2015;128(2):95–109. doi: 10.1042/CS20130601. [DOI] [PubMed] [Google Scholar]
- 82.Dhande I, Ma W, Hussain T. Angiotensin AT2 receptor stimulation is anti-inflammatory in lipopolysaccharide-activated THP-1 macrophages via increased interleukin-10 production. Hypertens Res. 2015;38(1):21–29. doi: 10.1038/hr.2014.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Menk M, Graw JA, von Haefen C, Sifringer M, Schwaiberger D, Unger T, Steckelings U, Spies CD. Stimulation of the angiotensin II AT2 receptor is anti-inflammatory in human lipopolysaccharide-activated monocytic cells. Inflammation. 2015 [Epub ahead of print]. [DOI] [PubMed]
- 84.Sampson AK, Irvine JC, Shihata WA, Dragoljevic D, Lumsden N, Huet O, Barnes T, Unger T, Steckelings UM, Jennings GL, Widdop RE, Chin-Dusting JP. Compound 21 prevents endothelial inflammation and leukocyte adhesion in vitro and in vivo. Br J Pharmacol. 2015. doi:10.1111/bph.13063. [DOI] [PMC free article] [PubMed]
- 85.Miura S, Matsuo Y, Kiya Y, Karnik SS, Saku K. Molecular mechanisms of the antagonistic action between AT1 and AT2 receptors. Biochem Biophys Res Commun. 2010;391(1):85–90. doi: 10.1016/j.bbrc.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mogi M, Iwai M, Horiuchi M. New insights into the regulation of angiotensin receptors. Curr Opin Nephrol Hypertens. 2009;18(2):138–143. doi: 10.1097/MNH.0b013e328324f5fa. [DOI] [PubMed] [Google Scholar]
- 87.Oshima K, Mogi M, Jing F, Iwanami J, Tsukuda K, Min LJ, Ogimoto A, Dahlöf B, Steckelings UM, Unger T, Higaki J, Horiuchi M. Direct angiotensin II type 2 receptor stimulation ameliorates insulin resistance in type 2 diabetes mice with PPARγ activation. PLoS One. 2012;7(11):e48387. doi: 10.1371/journal.pone.0048387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shum M, Pinard S, Guimond MO, Labbé SM, Roberge C, Baillargeon JP, Langlois MF, Alterman M, Wallinder C, Hallberg A, Carpentier AC, Gallo-Payet N. Angiotensin II type 2 receptor promotes adipocyte differentiation and restores adipocyte size in high-fat/high-fructose diet-induced insulin resistance in rats. Am J Physiol Endocrinol Metab. 2013;304(2):E197–E210. doi: 10.1152/ajpendo.00149.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wardat S, Iwai M, Horiuchi M, Dahlöf B, Hallberg A, Unger T, Kintscher U, Steckelings UM, Foryst-Ludwig. A glucose tolerance and insulin sensitivity is improved in obese mice by direct stimulation of angiotensin AT2-receptors, 16th annual meeting of the european council for cardiovascular research (ECCR) 30.9-2.10 2011 La Colle sur Loup, Nice, France.
- 90.Wardat S, Kintscher U, Steckelings UM, Foryst-Ludwig A. TNF-α-mediated inflammatory response in adipocytes is attenuated by the direct activation of angiotensin II AT2-receptors. Hypertension. 2009;54(5):1179. [Google Scholar]
- 91.Shao C, Zucker IH, Gao L. Angiotensin type 2 receptor in pancreatic islets of adult rats: a novel insulinotropic mediator. Am J Physiol Endocrinol Metab. 2013;305(10):E1281–E1291. doi: 10.1152/ajpendo.00286.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shao C, Yu L, Gao L. Activation of angiotensin type 2 receptors partially ameliorates streptozotocin-induced diabetes in male rats by islet protection. Endocrinology. 2014;155(3):793–804. doi: 10.1210/en.2013-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matavelli LC, Huang J, Siragy HM. Angiotensin AT2 receptor stimulation inhibits early renal inflammation in renovascular hypertension. Hypertension. 2011;57(2):308–313. doi: 10.1161/HYPERTENSIONAHA.110.164202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gelosa P, Pignieri A, Fändriks L, de Gasparo M, Hallberg A, Banfi C, Castiglioni L, Turolo L, Guerrini U, Tremoli E, Sironi L. Stimulation of AT2 receptor exerts beneficial effects in stroke-prone rats: focus on renal damage. J Hypertens. 2009;27(12):2444–2451. doi: 10.1097/HJH.0b013e3283311ba1. [DOI] [PubMed] [Google Scholar]
- 95.Matavelli LC, Zatz R, Siragy HM. A nonpeptide angiotensin II type 2 receptor agonist prevents renal inflammation in early diabetes. J Cardiovasc Pharmacol. 2015;65(4):371–376. doi: 10.1097/FJC.0000000000000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Koulis C, Chow BS, McKelvey M, Steckelings UM, Unger T, Thallas-Bonke V, Thomas MC, Cooper ME, Jandeleit-Dahm KA, Allen TJ. AT2R agonist, compound 21, is reno-protective against type 1 diabetic nephropathy. Hypertension. 2015;65(5):1073–1081. doi: 10.1161/HYPERTENSIONAHA.115.05204. [DOI] [PubMed] [Google Scholar]
- 97.Sabuhi R1, Ali Q, Asghar M, Al-Zamily NR, Hussain T. Role of the angiotensin II AT2 receptor in inflammation and oxidative stress: opposing effects in lean and obese Zucker rats. Am J Physiol Renal Physiol. 2011;300(3):F700–6. [DOI] [PMC free article] [PubMed]
- 98.Castoldi G, di Gioia CR, Bombardi C, Maestroni S, Carletti R, Steckelings UM, Dahlöf B, Unger T, Zerbini G, Stella A. Prevention of diabetic nephropathy by compound 21, selective agonist of angiotensin type 2 receptors, in Zucker diabetic fatty rats. Am J Physiol Renal Physiol. 2014;307:F1123–F1131. doi: 10.1152/ajprenal.00247.2014. [DOI] [PubMed] [Google Scholar]
- 99.Ali Q, Patel SN, Hussain T. Angiotensin AT2 receptor agonist prevents salt-sensitive hypertension in obese Zucker rats. Am J Physiol Renal Physiol. 2015. doi:10.1152/ajprenal.00002.2015. [DOI] [PMC free article] [PubMed]
- 100.Brouwers S, Smolders I, Massie A, Dupont AG. Angiotensin II type 2 receptor-mediated and nitric oxide-dependent renal vasodilator response to compound 21 unmasked by angiotensin-converting enzyme inhibition in spontaneously hypertensive rats in vivo. Hypertension. 2013;62:920–926. doi: 10.1161/HYPERTENSIONAHA.112.00762. [DOI] [PubMed] [Google Scholar]
- 101.Hilliard LM, Chow CL, Mirabito KM, Steckelings UM, Unger T, Widdop RE, Denton KM. Angiotensin type 2 receptor stimulation increases renal function in female, but not male, spontaneously hypertensive rats. Hypertension. 2014;64(2):378–383. doi: 10.1161/HYPERTENSIONAHA.113.02809. [DOI] [PubMed] [Google Scholar]
- 102.Kemp BA, Howell NL, Gildea JJ, Keller SR, Padia SH, Carey RM. AT2 receptor activation induces natriuresis and lowers blood pressure. Circ Res. 2014;115(3):388–399. doi: 10.1161/CIRCRESAHA.115.304110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bindom SM, Hans CP, Xia H, Boulares AH, Lazartigues E. Angiotensin I-converting enzyme type 2 (ACE2) gene therapy improves glycemic control in diabetic mice. Diabetes. 2010;59(10):2540–2548. doi: 10.2337/db09-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Takeda M, Yamamoto K, Takemura Y, Takeshita H, Hongyo K, Kawai T, Hanasaki-Yamamoto H, Oguro R, Takami Y, Tatara Y, Takeya Y, Sugimoto K, Kamide K, Ohishi M, Rakugi H. Loss of ACE2 exaggerates high-calorie diet-induced insulin resistance by reduction of GLUT4 in mice. Diabetes. 2013;62(1):223–233. doi: 10.2337/db12-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Villela D, Leonhardt J, Patel N, Joseph J, Kirsch S, Hallberg A, Unger T, Bader M, Santos RA, Sumners C, Steckelings UM. Angiotensin type 2 receptor (AT2R) and receptor Mas: a complex liaison. Clin Sci (Lond). 2015;128(4):227–234. doi: 10.1042/CS20130515. [DOI] [PubMed] [Google Scholar]
- 106.Négrel R, Gaillard D, Ailhaud G. Prostacyclin as a potent effector of adipose-cell differentiation. Biochem J. 1989;257(2):399–405. doi: 10.1042/bj2570399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mogensen CE, Neldam S, Tikkanen I, Oren S, Viskoper R, Watts RW, Cooper ME. Randomised controlled trial of dual blockade of renin–angiotensin system in patients with hypertension, microalbuminuria, and non-insulin dependent diabetes: the candesartan and lisinopril microalbuminuria (CALM) study. BMJ. 2000;321(7274):1440–1444. doi: 10.1136/bmj.321.7274.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bakris GL, Ruilope L, Locatelli F, Ptaszynska A, Pieske B, de Champlain J, Weber MA, Raz I. Treatment of microalbuminuria in hypertensive subjects with elevated cardiovascular risk: results of the IMPROVE trial. Kidney Int. 2007;72(7):879–885. doi: 10.1038/sj.ki.5002455. [DOI] [PubMed] [Google Scholar]
- 109.Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK, AVOID Study Investigators Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358(23):2433–2446. doi: 10.1056/NEJMoa0708379. [DOI] [PubMed] [Google Scholar]
- 110.Persson F, Lewis JB, Lewis EJ, Rossing P, Hollenberg NK, Parving HH. Aliskiren in combination with losartan reduces albuminuria independent of baseline blood pressure in patients with type 2 diabetes and nephropathy. Clin J Am Soc Nephrol. 2011;6(5):1025–1031. doi: 10.2215/CJN.07590810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mann JF, Schmieder RE, McQueen M, Dyal L, Schumacher H, Pogue J, Wang X, Maggioni A, Budaj A, Chaithiraphan S, Dickstein K, Keltai M, Metsärinne K, Oto A, Parkhomenko A, Piegas LS, Svendsen TL, Teo KK, Yusuf S, ONTARGET investigators Renal outcomes with telmisartan, ramipril, or both, in people at high vascular risk (the ONTARGET study): a multicentre, randomised, double-blind, controlled trial. Lancet. 2008;372(9638):547–553. doi: 10.1016/S0140-6736(08)61236-2. [DOI] [PubMed] [Google Scholar]
- 112.Parving HH, Brenner BM, McMurray JJ, de Zeeuw D, Haffner SM, Solomon SD, Chaturvedi N, Persson F, Desai AS, Nicolaides M, Richard A, Xiang Z, Brunel P, Pfeffer MA, ALTITUDE Investigators Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N Engl J Med. 2012;367(23):2204–2213. doi: 10.1056/NEJMoa1208799. [DOI] [PubMed] [Google Scholar]
- 113.Makani H, Bangalore S, Desouza KA, Shah A, Messerli FH. Efficacy and safety of dual blockade of the renin–angiotensin system: meta-analysis of randomised trials. BMJ. 2013;346:f360. doi: 10.1136/bmj.f360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Volpe M, Danser AH, Menard J, Waeber B, Mueller DN, Maggioni AP, Ruilope LM. Inhibition of the renin–angiotensin-aldosterone system: is there room for dual blockade in the cardiorenal continuum? J Hypertens. 2012;30(4):647–654. doi: 10.1097/HJH.0b013e32834f6e00. [DOI] [PubMed] [Google Scholar]
- 115.de Boer RA, Azizi M, Danser AJ, Nguyen G, Nussberger J, Ruilope LM, Schmieder RE, Volpe M. Dual RAAS suppression: recent developments and implications in light of the ALTITUDE study. J Renin Angiotensin Aldosterone Syst. 2012;13(3):409–412. doi: 10.1177/1470320312455271. [DOI] [PubMed] [Google Scholar]
- 116.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–1327. doi: 10.1016/j.jacc.2009.10.061. [DOI] [PubMed] [Google Scholar]
- 117.Guerin AP, Blacher J, Pannier B, Marchais SJ, Safar ME, London GM. Impact of aortic stiffness attenuation on survival of patients in end-stage renal failure. Circulation. 2001;103:987–992. doi: 10.1161/01.CIR.103.7.987. [DOI] [PubMed] [Google Scholar]
- 118.Sodhi K, Inoue K, Gotlinger KH, Canestraro M, Vanella L, Kim DH, Manthati VL, Koduru SR, Falck JR, Schwartzman ML, Abraham NG. Epoxyeicosatrienoic acid agonist rescues the metabolic syndrome phenotype of HO-2-null mice. J Pharmacol Exp Ther. 2009;331(3):906–916. doi: 10.1124/jpet.109.157545. [DOI] [PMC free article] [PubMed] [Google Scholar]