

Summary
Background
Therapies for chronic hepatitis delta virus (HDV) infection are unsatisfactory. Prenylation is essential for HDV and inhibition abrogates HDV production in experimental models. In a proof-of-concept study, we aimed to assess the effect on HDV RNA levels, safety, and tolerability of the prenylation inhibitor lonafarnib in patients with chronic delta hepatitis.
Methods
In this phase 2A double-blind, randomised, placebo-controlled study, patients aged 18 years or older with chronic HDV infection were randomly assigned (3:1 in group 1 and 2:1 in group 2) to receive lonafarnib 100 mg (group 1) or lonafarnib 200 mg (group 2) twice daily for 28 days with 6 months’ follow-up. Participants were randomised by random-number tables blocked in groups of four without stratification. Both groups enrolled six treatment participants and two placebo participants. Group 1 placebo patients received open-label lonafarnib as group 2 participants. The primary therapeutic endpoint was a decrease in HDV RNA viral titre in serum and the primary safety endpoint was the ability to tolerate the drug at the prescribed dose for the full 4-week duration, defined as drug discontinuation due to intolerance or grade 3/4 adverse events. This trial is registered with ClinicalTrials.gov, number NCT01495585.
Findings
Between Jan 19, 2012, and April 28, 2014, 14 patients were enrolled, of whom eight were assigned to group 1 and six were assigned to group 2. At day 28, compared with placebo, mean log HDV RNA declines from baseline were −0.73 log IU/mL in group 1 (95% CI 0.17–1.31; p=0.03) and −1.54 log IU/mL in group 2 (1.21–1.93; p<0.0001). Lonafarnib serum concentrations correlated with HDV RNA change (r2=0.78, p<0.0001). Model fits show that hepatitis B surface antigen (HBsAg) remained stable after a short pharmacological delay (0.75 days [SE 0.24]), lonafarnib effectiveness in blocking HDV production was greater in group 2 than in group 1 (0.952 [SE 0.06] vs 0.739 [0.05], p<0.001), and the HDV half-life was 1.62 days (0.07). There was no evidence of virological resistance. Adverse events were mainly mild to moderate with group 1 patients experiencing diarrhoea in three patients (50%) and nausea in two patients (33%) and in group 2 with all patients (100%) experiencing nausea, diarrhoea, abdominal bloating, and weight loss greater than 2 kg (mean of 4 kg). No treatment discontinuations occurred in any treatment groups.
Interpretation
Treatment of chronic HDV with lonafarnib significantly reduces virus levels. The decline in virus levels significantly correlated with serum drug levels, providing further evidence for the efficacy of prenylation inhibition in chronic HDV.
Funding
National Institute of Diabetes and Digestive and Kidney Diseases and National Cancer Institute, National Institutes of Health, and Eiger Biopharmaceuticals Inc.
Introduction
The hepatitis delta virus (HDV) is an incomplete RNA virus composed of a 1.7 kb single-stranded circular genomic RNA, virally encoded small and large delta antigens, and a surrounding lipid envelope.1,2 It is the smallest pathogenic animal virus known to infect human beings, and it is estimated that up to 20 million people are chronically infected worldwide.3 HDV infection has been described to be endemic across most of the world. Although HDV shares epidemiological patterns with hepatitis B virus (HBV), HDV in its own right continues to be a major global health problem.4,5 The virus was first identified in 1977 and is known only to propagate in individuals infected with HBV, either via superinfection or co-infection.4,6 Eight HDV genotypes have been described and genotype 1, which is most prevalent in North America, Europe, north Africa, and the Middle East, has been associated with more severe disease.7,8 Similar to other forms of viral hepatitis, HDV progresses to cirrhosis, but is the most severe form of human viral hepatitis.5,9–14
At present, therapy for HDV is unsatisfactory and no US Food and Drug Administration approved therapy exists for HDV infection. The American Association for the Study of Liver Diseases and the European Association for the Study of the Liver have recommended treatment with interferon-alfa for chronic HDV infection.15,16 However, therapy with interferon-alfa is generally unsatisfactory with high relapse rates even when therapy is extended out to 5 years.17,18
Prenylation is a post-translational lipid modification that involves the covalent addition of prenyl lipids to proteins resulting in promotion of membrane association and protein–protein interactions.1,19 Prenylation plays a vital part in the life cycle of HDV, and disruption of prenylation of the large delta hepatitis antigen (LDHAg) prevents its ability to interact with, and form secreted particles with, the hepatitis B surface antigen (HBsAg).19,20 Use of prenylation inhibitors has been assessed with success in vitro as well as in vivo in a mouse model of HDV replication.1,19–21 In this proof-of-concept study, we aimed to assess the effect on HDV RNA, safety, and tolerability of the prenylation inhibitor lonafarnib in patients with chronic HDV infection.
Methods
Study design and participants
In this proof-of-concept phase 2A double-blinded randomised, placebo-controlled study, patients aged 18 years or older with chronic HDV infection as evidenced by the presence of quantifiable HDV RNA by quantitative PCR (qPCR) in serum and with compensated liver disease were enrolled at the National Institutes of Health (NIH) Clinical Center. Patients were excluded if there was evidence of other forms of liver disease, hepatocellular carcinoma, HIV co-infection, active drug or alcohol abuse, any contraindication to lonafarnib, experimental therapy within 6 months before enrolment, or pregnancy or refusal to use adequate contraception during therapy. Full eligibility criteria are provided in the appendix (p 1). All patients provided written informed consent. This investigator-initiated clinical trial was done at the NIH Clinical Center (and was designed and conducted by the principal and associate investigators who collected the data, monitored the study conduct, performed the statistical analysis, and prepared the report). All study materials were approved by the institutional review board of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) at the NIH Clinical Center. Lonafarnib was provided by Eiger Bio-pharmaceuticals, Inc (San Carlos, CA, USA), under a clinical trial agreement with the NIDDK. Administration of lonafarnib was conducted under an Investigational New Drug Application (IND# 113 137) held by the NIDDK.
Randomisation and masking
14 enrolled patients were sequentially assigned (3:1 in group 1 and 2:1 in group 2) into one of two dosing groups, which consisted of lonafarnib 100 mg twice daily in group 1 and lonafarnib 200 mg twice daily in group 2, with placebo controls in both groups (figure 1). Group 1 participation was completed before the start of group 2. Patients were randomised by the NIH Pharmaceutical Development Services (PDS) with random-number tables blocked in groups of four without stratification. Patients received treatment for 28 days, followed by post-treatment monitoring for 6 months. Each group consisted of eight patients (six treatment and two placebo), and the two patients who received placebo in group 1 were offered treatment as group 2 participants with open-label lonafarnib (200 mg twice daily). All participants and physicians were blinded to treatment or placebo while on therapy except the two participants who received open-label lonafarnib in group 2. Additional details of the study design are described in the appendix (p 1).
Figure 1. Trial profile.
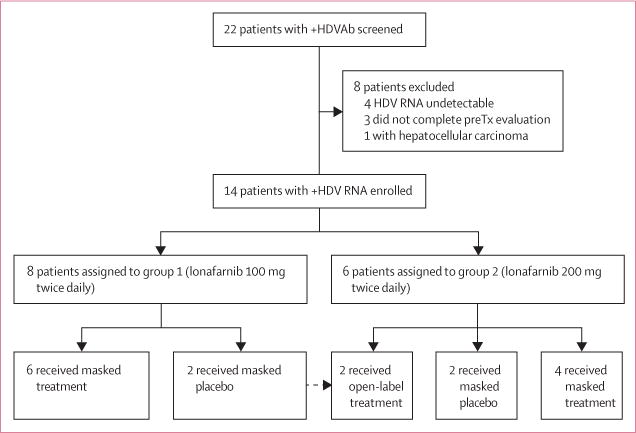
Of the 22 patients with positive hepatitis delta virus antibodies (HDVAb) screened, 14 were enrolled into this study. After the first eight patients completed group 1, the two group 1 placebo patients received open-label lonafarnib 200 mg twice daily as group 2 participants in addition to the six new patients that were randomised to receive either treatment or placebo.
Procedure
Patients received oral lonafarnib (100 mg or 200 mg) or placebo twice daily. Assessments occurred at baseline, 0–72 h, weekly during therapy, weeks 1, 2, and 4 off therapy, and then monthly for a total of 6 months. Assessments included standard laboratory testing (complete blood counts, routine chemistries, and liver function tests), measurement of HDV RNA and HBV DNA by qPCR and HBsAg by ELISA, HDV viral mutations associated with lonafarnib non-response (at baseline, end of therapy, and end of follow-up; appendix pp 2–4), vital signs, electrocardiography (while on therapy and at weeks 1 and 2 post-therapy), and symptom-directed physical examinations. All adverse events were recorded and graded according to the Common Terminology Criteria for Adverse Events version 3.0, with modifications for leucocytes, platelets, prothrombin time, partial thrombo plastin time, alanine amino-transferase (ALT), aspartate aminotransferase (AST), and bilirubin (see study protocol). qPCR was designed to correctly evaluate HDV viral load values by targeting highly conserved regions of the viral genome.
Safety was assessed by the study investigators at each timepoint based on laboratory analysis and study participant reports. Throughout the study period, patients also underwent monitoring for electroretinographic and electrocardiographic changes and reproductive toxicity (appendix pp 4–6).
Characteristion of HDV kinetics during lonafarnib therapy was assessed with a segmented linear regression approach. To study HDV dynamics and estimate lonafarnib effectiveness, a dual HDV and HBsAg model (appendix pp 13, 21) that was recently developed for understanding HDV-host dynamics during peginterferon (pegIFN) therapy was used.22 The model was fitted to the log-scaled HDV viral loads and HBsAg levels with a nonlinear mixed effect modelling approach (appendix pp 14).
Outcomes
The primary therapeutic endpoint of the study was defined as a decrease in HDV RNA viral titre in serum as measured by qPCR. The primary safety endpoint of the study was the ability to tolerate the drug at the prescribed dose for the full 4-week duration. The secondary endpoints included changes in ALT concentration and symptom scales compared with baseline pretreatment values with a visual analogue scale evaluating symptoms including nausea, diarrhoea, indigestion, loss of appetite, and fatigue.
Statistical analysis
The primary therapeutic endpoint was the change in serum HDV RNA after 28 days of therapy with lonafarnib. Based on a 1 log decline of quantitative HDV RNA with lonafarnib therapy, with a sigma of 0.5 log of normal HDV viral fluctuations over time and an alpha of 0.5 with a power of 0.80, the sample size required to show a difference is three participants. In view of the fact that six patients received therapy in each group and that there were four placebo participants, we feel that this study was adequately powered to show a difference in viral decline and the presence of significant safety signals. Comparison of HDV RNA and HBV DNA changes (after logarithmic transformation) between groups was done via Student’s t test. Other continuous variables (ALT, AST, total bilirubin, and HBsAg) were analysed with the Mann-Whitney U test. Association between HDV RNA change (after logarithmic trans formation) and serum lonafarnib concentration was analysed with linear regression. Viral-host parameters estimated by fitting the dual HDV and HBsAg model (appendix p 13) with the experimental data were tested with a likelihood ratio test. All statistical tests and CIs were two-sided and p<0.05 was considered to be statistically significant. All statistical analyses were done with JMP 11.0 (SAS Inc, Cary, NC, USA) and SAS 9.3 (SAS).
This study is registered with ClinicalTrials.gov, number NCT01495585).
Role of the funding source
The funder of the study had no role in the study design, data collection, data analysis, data interpretation, decision to publish, or writing of the report. The corresponding authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Results
Between Jan 19, 2012, and April 28, 2014, of 22 patients screened, 14 were enrolled, of whom eight were assigned to group 1 and six were assigned to group 2. After completion of the trial for group 1, the two patients receiving placebo in that group were subsequently included in group 2 and received lonafarnib 200 mg twice daily as an open-label treatment. Of the eight patients not enrolled, four had undetectable HDV RNA, three did not complete the pre-enrolment evaluations, and one was diagnosed with hepatocellular carcinoma (figure 1).
Baseline demographics and disease characteristics of the enrolled patients were balanced between the treatment groups (table 1).
Table 1.
Baseline demographic and clinical characteristics
| All participants (n=14) | Placebo group (n=4)* | Group 1 (n=6) | Group 2 (n=6)* | |
|---|---|---|---|---|
| Demographics | ||||
|
| ||||
| Age (years) | 38 (33–48) | 34 (33–39) | 36 (30–42) | 45 (38–55) |
| Sex | ||||
| Male | 10 (71%) | 2 (50%) | 5 (83%) | 4 (67%) |
| Female | 4 (29%) | 2 (50%) | 1 (17%) | 2 (33%) |
| Body-mass index | 24.4 (21.9–27.3) | 26.5 (24.1–28.9) | 22.8 (21.9–23.5) | 26.1 (25.5–27.3) |
| Pre-treatment nucleoside analogues | 5 (36%) | 1 (25%) | 2 (33%) | 2 (33%) |
| Ethnic origin | ||||
| Asian | 7 (50%) | 2 (50%) | 3 (50%) | 3 (50%) |
| White | 6 (43%) | 1 (25%) | 3 (50%) | 3 (50%) |
| African | 1 (7%) | 1 (25%) | 0 | 0 |
|
| ||||
| Disease characteristics | ||||
|
| ||||
| Ishak fibrosis | 3 (3–4) | 3 (3–4) | 3 (3–4) | 3 (3–4) |
| HAI necroinflammation | 10 (10–13) | 13 (12–13) | 10 (10–14) | 11 (10–11) |
| ALT (U/L) | 89 (67–176) | 101 (80–162) | 125 (86–168) | 63 (52–162) |
| AST (U/L) | 61 (42–80) | 72 (56–88) | 61 (50–75) | 42 (41–60) |
| Platelet (109 per L) | 190 (175–242) | 231 (136–218) | 174 (170–183) | 197 (180–229) |
| Total bilirubin (mg/dL) | 0.38 (0.30–0.55) | 0.45 (0.30–0.55) | 0.35 (0.30–0.48) | 0.45 (0.40–0.60) |
| HDV RNA (IU/mL) | 9.27 × 105 (3.20 × 105–2.85 × 106) |
1.72 × 106 (4.28 × 105–4.43 × 106) |
1.29 × 106 (4.74 × 105–4.84 × 106) |
8.68 × 105 (4.60 × 105–1.94 × 106) |
| Log HDV RNA (IU/mL) | 5.94 (5.50–6.43) | 6.06 (5.60–6.51) | 6.06 (5.56–6.60) | 5.92 (5.62–6.26) |
| HBV DNA (IU/mL)† | 22 (<21–684) | <21 (<21–26) | 463 (<21–1239) | 635 (<21–947) |
| HBsAg (ng/mL) | 13 216 (8966–21 792) | 15 718 (10 520–23 534) | 17 707 (11 268–32 085) | 11 417 (8970–13 103) |
Data are n (%) or median (IQR). HAI=histological activity index. ALT=alanine aminotransferase. AST=aspartate aminotransferase. HDV=chronic hepatitis delta virus. HBV=hepatitis B virus. HBsAg=hepatitis B virus surface antigen.
Two of the placebo patients who were group 1 participants were treated with open-label lonafarnib as group 2 participants.
Analysis of patients not on nucleoside analogues.
Overall, the median age of the participants was 38 years (IQR 33–48), ten (71%) were men, all with HBeAg negative status and HDV genotype 1 infection; seven (50%) were Asian, four (43%) were white, and one (7%) was African. 12 (86%) patients originated from endemic regions (northern Asia, eastern Europe, and central Africa) and two (14%) had traditional risk factors for blood and body fluid transmission. Five (36%) patients were on nucleos(t)ide analogues (tenofovir or entecavir) at least 6 months before screening and three (21%) had failed interferon therapy at least 6 months before screening. The median Ishak fibrosis score was 3, histological activity index necroinflammation was 10, and baseline platelets were 190 × 109 per L. The median baseline serum HDV RNA was 9.27 × 105 IU/mL and in patients not taking nucleos(t)ide analogues, the median baseline HBV DNA was 22.5 IU/mL. All but one patient had low level HBV DNA replication less than 2500 IU/mL.
During 28 days of therapy, patients randomised to lonafarnib (100 mg twice daily or 200 mg twice daily) experienced a decline in serum HDV RNA levels at all weeks of therapy compared with baseline and placebo (figure 2). By the end of therapy, the mean HDV RNA decline in serum from baseline was 0.13 log (95% CI −0.14 to 0.34) in the placebo group, 0.73 log (0.17–1.31) in group 1, and 1.54 log (1.21–1.93) in group 2 (table 2). The viral declines seen in group 1 and group 2 were significantly different when compared with placebo (p=0.03 and p<0.0001, respectively). Mean serum lonafarnib concentrations strongly correlated with mean serum log HDV RNA change from baseline to end of therapy (figure 3).
Figure 2.
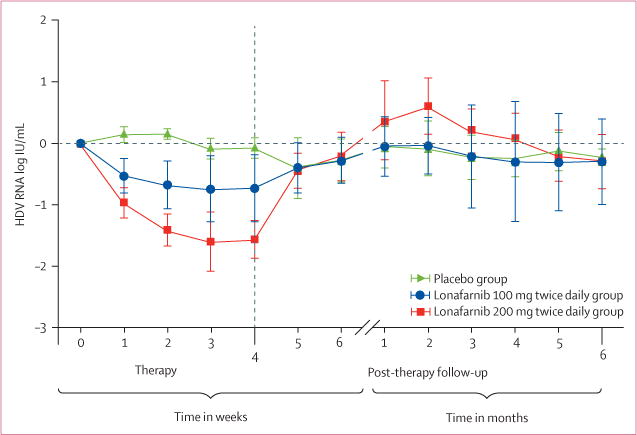
Mean serum hepatitis delta virus RNA (SD) change during therapy with lonafarnib
Table 2.
Laboratory changes at end of lonafarnib therapy (day 28)
| Placebo group | Group 1 | p value (placebo vs group 1) |
Group 2 | p value (placebo vs group 2) |
|
|---|---|---|---|---|---|
| Log HDV RNA (IU/mL) | −0.13 (0.14) | −0.73 (0.54) | 0.03† | −1.54 (0.36) | <0.0001† |
| ALT (U/L) | 18 (13) | 4 (93) | 0.96* | 29 (85) | 0.6* |
| AST (U/L) | 2 (9) | 12 (58) | 0.7* | −3 (14) | 0.6* |
| Total bilirubin (mg/dL) | −0.05 (0.06) | −0.05 (0.18) | 1.0* | −0.08 (0.19) | 0.1* |
| Log HBV DNA (IU/mL) | −0.60 (0.72) | 0.08 (0.13) | 0.1† | 1.12 (1.03) | 0.05† |
| HBsAg (ng/mL) | −1448 (3784) | 3702 (7904) | 0.2* | 3104 (3800) | 0.1* |
Data are mean (SD). HDV=hepatitis delta virus. ALT=alanine aminotransferase. AST=aspartate aminotransferase. HBV=hepatitis B virus. HBsAg=hepatitis B virus surface antigen.
Calculated with Mann-Whitney U test.
Calculated with Student’s t test.
Figure 3. Association between mean serum lonafarnib concentration with mean change in serum HDV RNA from baseline to day 28.
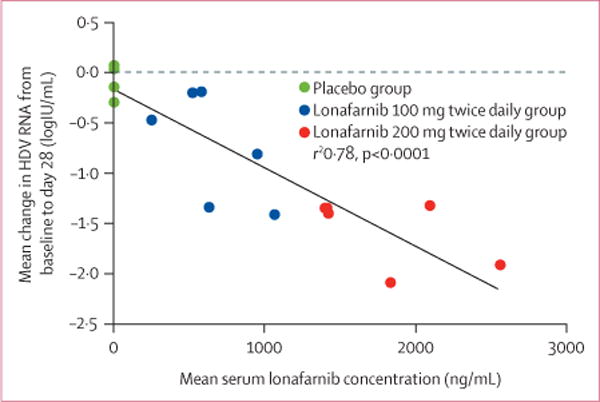
HDV=hepatitis delta virus.
Indeed, population-based sequencing for viral mutations of the large delta antigen (LDHAg) in serum at baseline, end of therapy, and 24 weeks after completion of therapy did not identify viral mutations associated with lonafarnib nonresponse (appendix pp 7–10).
During therapy, excluding patients on nucleos(t)ide analogues, serum HBV DNA levels did not significantly change in group 1 patients (table 2). Notably, there was a trend towards increasing HBV DNA at the end of therapy in group 2 patients with an increase in HBV DNA of 1.12 log (p=0.05). Baseline HBsAg levels did not significantly differ between groups at baseline (table 1) nor did they change during or after therapy (table 2).
During therapy, no significant changes in liver-associated enzymes were noted on comparison of either dosing group with placebo (table 2). The mean baseline ALT was 150 U/L (SD 91) in group 1 and 120 U/L (106) in group 2, and the mean end of therapy ALT was 146 U/L (95) in group 1 and 85 U/L (80) in group 2. In the placebo group, the mean baseline ALT was 140 U/L (SD 111) and the mean ALT at the end of therapy was 122 U/L (103). No patients in group 1 had normalisation of ALT at the end of therapy. Two patients (not on nucleoside analogues) had normalisation of ALT (35 U/L and 31 U/L); however, their baseline ALT value was 47 U/L.
On post-therapy follow-up, HDV RNA levels returned to baseline in all group 1 patients by week 4 (range week 1–4; appendix p 11). One patient (not on nucleos[t]ide analogue) had a virological flare of HDV RNA greater than 0.5 log over baseline at week 4 of post-therapy follow-up, which was accompanied by an ALT increase of 1.5 times the patient’s baseline level.
In group 2, HDV RNA levels returned to baseline in all patients by week 4 of post-therapy follow-up (range week 1–4). Four patients (two on nucleos[t]ide analogues) had a flare of HDV RNA greater than 0.5 log over baseline between week 4 and 8 (range 0.79–1.48 log IU/mL) of post-therapy follow-up. In these four patients, at the time of virological rebound, ALT was increased no more than 2.5 times (mean 1.81 [SD 0.6]) the patients’ baseline levels.
A biphasic decline of serum HDV RNA during therapy was characterised in all patients given lonafarnib, with a first rapid decline phase followed by a second slower (or plateau) phase (appendix pp 16, 17). The first phase duration was longer in group 2 than in group 1 (12.3 days [SE 2.1] vs 8.7 days [2.5]) and the decline was faster in group 2 than in group 1 (−0.143 log IU/mL per day [0.02] vs −0.098 log IU/mL per day [0.03]), leading to a significantly greater first-phase decline (1.59 log IU/mL vs 0.78 log IU/mL, p=0.015) in the group 2 patients. The second slope phase did not differ significantly between the two groups (−0.06 log IU/mL per day, p=0.94; appendix pp 16, 17).
Fitting the HDV and HBsAg dual model (appendix pp 13, 21) with measured HDV and HBsAg kinetics yielded excellent fit curves in both groups (appendix p 22). The pretreatment serum HDV RNA level was estimated as 5.97 (SE 0.13) log IU/mL and the HBsAg level as 4.17 (0.12) log ng/mL. A short pharmacological delay of t0=0.73 day (SE 0.24), in which HDV remained at baseline levels, was not associated with lonafarnib dose. The HDV clearance rate in blood, c, was estimated as 0.426 (SE 0.04) per day, corresponding to an HDV half-life in blood of 1.63 days (SE 0.15). Lonafarnib effectiveness in blocking HDV production was significantly (p<0.001) higher in group 2 (ε=0.952 [SE 0.057]) than in group 1 (ε=0.739 [0.05]; appendix p 19). Individual parameters are shown in appendix p 18).
No patients prematurely discontinued lonafarnib because of grade 3 or 4 adverse events or a serious adverse event (table 3). No significant changes from baseline on digitised electrocardiography, ophthalmological examination with retinal photography, or reproductive toxicity testing were noted throughout the study (appendix p 6). The most common adverse events in group 1 patients included mild diarrhoea (three [50%] of six patients), nausea (two [33%]), anorexia (one [17%]), fatigue (one [17%]), abdominal bloating/dyspepsia (one [17%]), and weight loss greater than 2 kg (one [17%]; table 3). In group 2 patients, most common adverse events included nausea, diarrhoea, abdominal bloating and weight loss of more than 2 kg (mean of 4 kg) in all patients (six [100%] of six patients) and anorexia in five (83%) of six patients followed by intermittent vomiting (three [50%] of six), headaches and fatigue (one [17%]). In the placebo group, one [25%] of four patients complained of abdominal bloating/dyspepsia. The symptoms experienced by group 1 patients given lonafarnib were indistinguishable from placebo patients, whereas the symptoms experienced by group 2 patients given lonafarnib could be distinguished from placebo patients.
Table 3.
Treatment discontinuations and adverse events in treatment groups
| Placebo group (n=4) | Group 1 (n=6) | Group 2 (n=6) | |
|---|---|---|---|
| Discontinuation of therapy due to an adverse event | 0 | 0 | 0 |
| Serious adverse events | 0 | 0 | 0 |
| Common adverse events | |||
| Nausea | 1 (25%) | 2 (33%) | 6 (100%) |
| Diarrhoea | 0 | 3 (50%) | 6 (100%) |
| Decreased appetite | 0 | 1 (17%) | 5 (83%) |
| Abdominal bloating/dyspepsia | 1 (25%) | 1 (17%) | 6 (100%) |
| Vomiting | 0 | 0 | 3 (50%) |
| Weight loss >2 kg | 0 | 1 (17%) | 6 (100%) |
| Headache | 1 (25%) | 1 (17%) | 1 (17%) |
| Testicular pain | 0 | 1 (17%) | 0 |
| Lightheadedness | 1 (25%) | 0 | 0 |
| Fatigue | 0 | 1 (17%) | 1 (17%) |
| Data are n (%). |
Discussion
In this first-in-man, proof-of-concept study, we show that lonafarnib can decrease serum HDV levels in a dose-dependent manner during 28 days of therapy. This study has value in that it shows that an oral small molecule prenylation inhibitor can affect HDV and thereby warrants further study in a field with unmet medical needs. Moreover, half of the patients in the higher dosing group achieved a decrease from baseline to nadir of greater than −2 log IU/mL. As further evidence supporting the antiviral efficacy of prenylation inhibition against HDV, two-thirds of patients in the higher dosing group had HDV virological rebound with accompanied ALT flare after stopping therapy. Although this is the first demonstration of virological rebound after stopping prenylation inhibitor therapy, the concept of HDV rebound after treatment cessation has been shown in interferon-based studies for HDV.23 As expected, there was no evidence of HDV viral mutations associated with lonafarnib non-response throughout the study, which is consistent with the mechanism of action of prenylation inhibitors that target host enzymes. Additionally, although this study consisted only of HDV genotype 1 patients, in-vitro studies with farnesyltransferase inhibitors suggest that comparable results would be expected with other HDV genotypes.20 However, a challenge across HDV genotypes could be to adequately monitor and compare viral levels based on the various qPCR techniques available.
Although small in patient numbers and short in duration, this study provides evidence in human beings of a novel mechanism for potential therapeutic eradication of HDV. In the higher dosing group of patients not taking nucleos(t)ide analogues, there was a notable, albeit non-significant, trend of increased HBV DNA levels at the end of therapy (p=0.05). Previous studies where interferon was used for therapy have described HBV reactivation when HDV is suppressed or eliminated.3,24 However, unlike interferon-based studies, which might treat both HDV and HBV, prenylation inhibitors only inhibit HDV, and this allows for the study of the effect of HDV on HBV. With extended therapy, HBV monitoring and nucleos(t)ide therapy should be considered.
In this study, no premature discontinuations or serious adverse events were recorded during 28 days of therapy with lonafarnib. Gastrointestinal side-effects (nausea, diarrhoea, abdominal bloating, and decreased appetite) were the most common, and are similar to studies evaluating lonafarnib for other disease states.25–27 The exploration of antiemetics, antidiarrhoeal, and acid suppressive therapies might improve tolerability in future studies. The average weight loss of 4 kg in group 2 suggests that prolonged dosing could be challenging, especially in higher doses, and would warrant close monitoring. One potential route of further exploration could include the fact that lonafarnib is metabolised by CYP3A4, allowing for enhanced post-absorption hepatic concentrations. By blocking this pathway and with lower doses of lonafarnib, it could be possible to minimise side-effects, while potentiating the relation between HDV decline and drug concentrations. Alternatively, future exploration as a combination therapy with other novel therapies could be considered as they become available.
Analysis of viral kinetics showed additional promising benefits of lonafarnib. The modelling analysis indicated a dose-dependent effect of lonafarnib in blocking HDV release. Moreover, the 95% efficacy of the 200 mg lonafarnib dose was similar to recent estimates in patients given peg-interferon (ε=96%)22 and was achieved much earlier with lonafarnib (median 12 days [IQR 9–17]) than with peg-interferon (25 days [23–58]). Frequent measurements during lonafarnib therapy allowed for a refined estimate of HDV half-life of 1.6 days that was about two-fold shorter than during peg-interferon therapy (2.9 days), and suggests a higher HDV production rate than recently estimated.22 Finally, additional viral kinetic analyses based on these results and future studies should determine the strategy for proper eradication of those infected with HDV by lonafarnib. Unlike interferon-based therapies, in which only a percentage of patients respond, the mechanism of action of lonafarnib and our study results suggest that a treatment response in all patients might be possible.
In conclusion, despite the small numbers, this study shows that treatment of chronic HDV with the prenylation inhibitor lonafarnib significantly reduces virus levels in patients. The drug was well tolerated by all participants. The decline in virus levels significantly correlated with increased serum drug concentrations, providing further evidence for the efficacy of prenylation inhibition in chronic HDV. These results provide a compelling rationale for next studies aimed to achieve eradication of HDV in man.
Supplementary Material
Research in context.
Evidence before this study
We reviewed advances in the treatment of hepatitis delta virus (HDV) to identify therapies that have been studied for chronic HDV infection by consulting systematic reviews and searching PubMed with the terms “HDV treatment” and “Delta hepatitis treatment” published between Jan 1, 1984, and April 15, 2015, in English language only. So far, most therapeutic trials for HDV have assessed the use of interferon, peg-interferon, and nucleos(t)ide analogues either as monotherapy or combination therapies for differing durations. Systematic reviews have also described these studies and their absence of success, but have suggested that prenylation inhibition could be the future of HDV therapy in view of the success seen in two decades worth of preclinical HDV studies. To date, no published studies in man have assessed oral prenylation inhibitors for chronic HDV infection.
Added value of this study
This study reports the results from our first-in-man study testing the hypothesis that a host-targeting small molecule prenylation inhibitor has authentic anti-HDV activity in human beings. This short-term proof-of-concept study shows that HDV viral load decreases on prenylation inhibitor therapy and rebounds upon withdrawal of the drug—thereby showing in-vivo specific antiviral activity in humans of this novel mechanism of action. Additionally, this study describes a clear dose-dependent antiviral effect with dose response curves indicating that there is no inherent limit on the ability to drive viral loads down with increased drug levels. Finally, this study shows that the kinetics of response to prenylation inhibition is superior to those noted with interferon without evidence of viral resistance despite 28 days of monotherapy treatment with lonafarnib.
Implications of all the available evidence
In our study, the prenylation inhibitor lonafarnib significantly reduced virus levels providing the first-in-human evidence for the efficacy of prenylation inhibition in chronic HDV. Although this 28-day study did not result in HDV eradication, it provides the underpinnings that HDV could be susceptible to eradication through an alternative mechanism other than interferon and that this represents the beginning of a new era of therapy free of interferon for HDV.
Acknowledgments
This research was supported by the Intramural Research Programs of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the National Cancer Institute (NCI) of the National Institutes of Health (NIH) and extramural NIH grants R01-AI078881 and P20-GM103451 and the US Department of Energy contract DE-AC52-06NA25396. Lonafarnib was provided by Eiger Biopharmaceuticals Inc under a clinical trial agreement with the NIDDK. Blinded analysis for HDV RNA quantitation (performed by MAW) and lonafarnib pharmacokinetic studies (performed by Quest Pharmaceutical Services, Newark, DE, USA) was contracted through Eiger Biopharmaceuticals Inc under a clinical trial agreement with the NIDDK.
Footnotes
Contributors
CK and TH conceived and designed the study. CK, VH-W, SLC, PP, EFW, RB, and MATH acquired the data. CK, LC, HD, XZ, SLU, MAW, GS, PP, EFW, RB, MATH, RI, DEK, CY, SJC, DEK, JSG, and TH analysed and interpreted the data. CK, LC, HD, XZ, SJC, SLU, JSG, and TH drafted the report. CK, LC, HD, XZ, SLU, VH-W, MAW, GS, SLC, PP, EFW, RB, MATH, SJC, OK, RI, CY, JSG, and TH critically revised the report for important intellectual content. CK, LC, HD, XZ, and EFW did the statistical analysis. TH obtained the funding. VH-W provided administrative support. TH supervised the study.
Declaration of interests
JSG has equity interest in Eiger Biopharmaceuticals Inc. HD has received partial travel support from Eiger Biopharmaceuticals Inc to attend scientific meetings. CY has served on an advisory board for Merck, AbbVie, Janssen, and Gilead Pharma; been on a speaker’s bureau of Roche, Gilead, BMS, and Merck Pharma; and has received a research grant from BMS. All other authors declare no competing interests.
References
- 1.Glenn JS, Marsters JC, Jr, Greenberg HB. Use of a prenylation inhibitor as a novel antiviral agent. J Virol. 1998;72:9303–06. doi: 10.1128/jvi.72.11.9303-9306.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gudima S, Chang J, Moraleda G, Azvolinsky A, Taylor J. Parameters of human hepatitis delta virus genome replication: the quantity, quality, and intracellular distribution of viral proteins and RNA. J Virol. 2002;76:3709–19. doi: 10.1128/JVI.76.8.3709-3719.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hughes SA, Wedemeyer H, Harrison PM. Hepatitis delta virus. Lancet. 2011;378:73–85. doi: 10.1016/S0140-6736(10)61931-9. [DOI] [PubMed] [Google Scholar]
- 4.Rizzetto M, Ponzetto A, Forzani I. Hepatitis delta virus as a global health problem. Vaccine. 1990;8:S10–14. doi: 10.1016/0264-410x(90)90207-3. [DOI] [PubMed] [Google Scholar]
- 5.Wedemeyer H, Manns MP. Epidemiology, pathogenesis and management of hepatitis D: update and challenges ahead. Nat Rev Gastroenterol Hepatol. 2010;7:31–40. doi: 10.1038/nrgastro.2009.205. [DOI] [PubMed] [Google Scholar]
- 6.Rizzetto M, Canese MG, Arico S, et al. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut. 1977;18:997–1003. doi: 10.1136/gut.18.12.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radjef N, Gordien E, Ivaniushina V, et al. Molecular phylogenetic analyses indicate a wide and ancient radiation of African hepatitis delta virus, suggesting a deltavirus genus of at least seven major clades. J Virol. 2004;78:2537–44. doi: 10.1128/JVI.78.5.2537-2544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makuwa M, Caron M, Souquiere S, Malonga-Mouelet G, Mahe A, Kazanji M. Prevalence and genetic diversity of hepatitis B and delta viruses in pregnant women in Gabon: molecular evidence that hepatitis delta virus clade 8 originates from and is endemic in central Africa. J Clin Microbiol. 2008;46:754–56. doi: 10.1128/JCM.02142-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Govindarajan S, Kanel GC, Peters RL. Prevalence of delta-antibody among chronic hepatitis B virus infected patients in the Los Angeles area: its correlation with liver biopsy diagnosis. Gastroenterology. 1983;85:160–62. [PubMed] [Google Scholar]
- 10.Colombo M, Cambieri R, Rumi MG, Ronchi G, Del Ninno E, De Franchis R. Long-term delta superinfection in hepatitis B surface antigen carriers and its relationship to the course of chronic hepatitis. Gastroenterology. 1983;85:235–39. [PubMed] [Google Scholar]
- 11.Manesis EK, Vourli G, Dalekos G, et al. Prevalence and clinical course of hepatitis delta infection in Greece: a 13-year prospective study. J Hepatol. 2013;59:949–56. doi: 10.1016/j.jhep.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 12.Fattovich G, Giustina G, Christensen E, et al. Influence of hepatitis delta virus infection on morbidity and mortality in compensated cirrhosis type B. The European Concerted Action on Viral Hepatitis (Eurohep) Gut. 2000;46:420–26. doi: 10.1136/gut.46.3.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ji J, Sundquist K, Sundquist J. A population-based study of hepatitis D virus as potential risk factor for hepatocellular carcinoma. J Natl Cancer Inst. 2012;104:790–92. doi: 10.1093/jnci/djs168. [DOI] [PubMed] [Google Scholar]
- 14.Romeo R, Del Ninno E, Rumi M, et al. A 28-year study of the course of hepatitis Delta infection: a risk factor for cirrhosis and hepatocellular carcinoma. Gastroenterology. 2009;136:1629–38. doi: 10.1053/j.gastro.2009.01.052. [DOI] [PubMed] [Google Scholar]
- 15.Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology. 2009;50:661–62. doi: 10.1002/hep.23190. [DOI] [PubMed] [Google Scholar]
- 16.European Association For The Study Of The Liver. EASL clinical practice guidelines: management of chronic hepatitis B virus infection. J Hepatol. 2012;57:167–85. doi: 10.1016/j.jhep.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Wedemeyer H, Yurdaydin C, Ernst S, et al. 96 weeks of pegylated-interferon-alfa-2a plus tenofovir or placebo for the treatment of hepatitis delta: the HIDIT-2 study. Hepatology. 2013;58:222A–23A. [Google Scholar]
- 18.Heller T, Rotman Y, Koh C, et al. Long-term therapy of chronic delta hepatitis with peginterferon alfa. Aliment Pharmacol Ther. 2014;40:93–104. doi: 10.1111/apt.12788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Einav S, Glenn JS. Prenylation inhibitors: a novel class of antiviral agents. J Antimicrob Chemother. 2003;52:883–86. doi: 10.1093/jac/dkg490. [DOI] [PubMed] [Google Scholar]
- 20.Bordier BB, Marion PL, Ohashi K, et al. A prenylation inhibitor prevents production of infectious hepatitis delta virus particles. J Virol. 2002;76:10465–72. doi: 10.1128/JVI.76.20.10465-10472.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bordier BB, Ohkanda J, Liu P, et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J Clin Invest. 2003;112:407–14. doi: 10.1172/JCI17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guedj J, Rotman Y, Cotler SJ, et al. Understanding early serum hepatitis D virus and hepatitis B surface antigen kinetics during pegylated interferon-alpha therapy via mathematical modeling. Hepatology. 2014;60:1902–10. doi: 10.1002/hep.27357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yurdaydin C, Bozkaya H, Onder FO, et al. Treatment of chronic delta hepatitis with lamivudine vs lamivudine + interferon vs interferon. J Viral Hepat. 2008;15:314–21. doi: 10.1111/j.1365-2893.2007.00936.x. [DOI] [PubMed] [Google Scholar]
- 24.Castelnau C, Le Gal F, Ripault MP, et al. Efficacy of peginterferon alpha-2b in chronic hepatitis delta: relevance of quantitative RT-PCR for follow-up. Hepatology. 2006;44:728–35. doi: 10.1002/hep.21325. [DOI] [PubMed] [Google Scholar]
- 25.Feldman EJ, Cortes J, DeAngelo DJ, et al. On the use of lonafarnib in myelodysplastic syndrome and chronic myelomonocytic leukemia. Leukemia. 2008;22:1707–11. doi: 10.1038/leu.2008.156. [DOI] [PubMed] [Google Scholar]
- 26.Ravoet C, Mineur P, Robin V, et al. Farnesyl transferase inhibitor (lonafarnib) in patients with myelodysplastic syndrome or secondary acute myeloid leukaemia: a phase II study. Ann Hematol. 2008;87:881–85. doi: 10.1007/s00277-008-0536-2. [DOI] [PubMed] [Google Scholar]
- 27.Gordon LB, Kleinman ME, Miller DT, et al. Clinical trial of a farnesyltransferase inhibitor in children with Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2012;109:16666–71. doi: 10.1073/pnas.1202529109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.