

ABSTRACT
Us3 protein kinases encoded by herpes simplex virus 1 (HSV-1) and 2 (HSV-2) play important roles in viral replication and pathogenicity. To investigate type-specific differences between HSV-1 Us3 and HSV-2 Us3 in cells infected by viruses with all the same viral gene products except for their Us3 kinases, we constructed and characterized a recombinant HSV-1 in which its Us3 gene was replaced with the HSV-2 Us3 gene. Replacement of HSV-1 Us3 with HSV-2 Us3 had no apparent effect on viral growth in cell cultures or on the range of proteins phosphorylated by Us3. HSV-2 Us3 efficiently compensated for HSV-1 Us3 functions, including blocking apoptosis, controlling infected cell morphology, and downregulating cell surface expression of viral envelope glycoprotein B. In contrast, replacement of HSV-1 Us3 by HSV-2 Us3 changed the phosphorylation status of UL31 and UL34, which are critical viral regulators of nuclear egress. It also caused aberrant localization of these viral proteins and aberrant accumulation of primary enveloped virions in membranous vesicle structures adjacent to the nuclear membrane, and it reduced viral cell-cell spread in cell cultures and pathogenesis in mice. These results clearly demonstrated biological differences between HSV-1 Us3 and HSV-2 Us3, especially in regulation of viral nuclear egress and phosphorylation of viral regulators critical for this process. Our study also suggested that the regulatory role(s) of HSV-1 Us3, which was not carried out by HSV-2 Us3, was important for HSV-1 cell-cell spread and pathogenesis in vivo.
IMPORTANCE A previous study comparing the phenotypes of HSV-1 and HSV-2 suggested that the HSV-2 Us3 kinase lacked some of the functions of HSV-1 Us3 kinase. The difference between HSV-1 and HSV-2 Us3 kinases appeared to be due to the fact that some Us3 phosphorylation sites in HSV-1 proteins are not conserved in the corresponding HSV-2 proteins. Therefore, we generated recombinant HSV-1 strains YK781 (Us3-chimera) with HSV-2 Us3 and its repaired virus YK783 (Us3-repair) with HSV-1 Us3, to compare the activities of HSV-1 Us3 and HSV-2 Us3 in cells infected by viruses with the same HSV-1 gene products except for their Us3 kinases. We report here that some processes in viral nuclear egress and pathogenesis in vivo that have been attributed to HSV-1 Us3 could not be carried out by HSV-2 Us3. Therefore, our study clarified the biological differences between HSV-1 Us3 and HSV-2 Us3, which may be relevant to viral pathogenesis in vivo.
INTRODUCTION
Protein kinases are key regulators of various cell functions and constitute one of the largest and most functionally diverse gene families (1, 2). By phosphorylating their target (substrate) proteins, kinases regulate protein enzymatic activity, subcellular localization, and/or association with other proteins (1, 2). Herpesviruses commonly encode virus-specific protein kinases (3–5) and utilize them both to regulate viral replication processes and to modify cellular processes by phosphorylating specific viral and cellular proteins (3–5). The Us3 protein kinases encoded by herpes simplex virus 1 (HSV-1) and 2 (HSV-2) are serine/threonine protein kinases with amino acid sequences that are conserved in viruses in the subfamily Alphaherpesvirinae of the family Herpesviridae (6–8).
In vitro biochemical studies identified the consensus target sequence of an HSV Us3 homologue encoded by a porcine alphaherpesvirus, pseudorabies virus (PRV), as RnX(S/T)YY, where n is greater than or equal to 2, X can be Arg, Ala, Val, Pro, or Ser, and Y can be any amino acid except an acidic residue (9–11). The phosphorylation target site specificity of the PRV Us3 homologue has been reported to be similar to that of other alphaherpesvirus Us3 homologues, including those of HSV-1, HSV-2, and varicella-zoster virus (12–15).
It has been reported that HSV-1 Us3, the best-studied alphaherpesvirus Us3 homologue, blocked apoptosis (16–19), promoted vesicle-mediated nucleocytoplasmic transport of nucleocapsids through nuclear membranes (20–23), promoted gene expression by blocking histone deacetylation (24–26), controlled infected-cell morphology (15, 18, 27), modulated host immune systems (28–35), stimulated mRNA translation by activating mTORC1 (36), regulated intracellular trafficking of the abundant virion component UL47 (37) and the essential envelope glycoprotein B (gB) (38, 39), and upregulated the enzymatic activity of viral dUTPase (vdUTPase) (40). These observations suggested that HSV-1 Us3 is a multifunctional protein that regulates various cellular and viral functions by phosphorylating a number of cellular and viral protein substrates.
Vesicle-mediated nucleocytoplasmic transport of nucleocapsids through the host cell nuclear membrane is a unique mechanism by which herpesvirus nucleocapsids traverse the inner nuclear membrane (INM) and outer nuclear membrane (ONM): progeny nucleocapsids acquire primary envelopes by budding through the INM into the perinuclear space between the INM and ONM (primary envelopment), and the enveloped nucleocapsids then fuse with the ONM to release de-enveloped nucleocapsids into the cytoplasm (de-envelopment) (41, 42). HSV-1 proteins UL31 and UL34, which form a complex designated the nuclear egress complex (NEC), play a crucial role in this process (3, 41–45). Us3 has also been reported to regulate viral nuclear egress. Thus, mutations that abrogate either the expression or catalytic activity of HSV-1 Us3, Us3 phosphorylation of UL31, or both Us3 phosphorylation of gB and expression of gH induced membranous structures in infected cells that were adjacent to the nuclear membrane and contained many primary enveloped virions (20–23, 46). These membranous structures have been thought to indicate that the rate of virion egress from the perinuclear space (de-envelopment) may have decreased, while the rate of virion delivery into the perinuclear space (primary envelopment) may have not changed or not decreased as much. Us3 was also shown to phosphorylate lamins A and C; phosphorylation of these lamins leads to dissociation of the nuclear lamina, which may facilitate virion access to the INM (47–51). Furthermore, it has been reported that mutations that mimic constitutive phosphorylation at Us3 phosphorylation sites in UL31 impaired primary envelopment (22).
Similar phosphorylation site specificity of alphaherpesvirus Us3 homologues, as described above, suggested that HSV-1 Us3 functions may be conserved in HSV-2 Us3. In fact, it has been reported that HSV-2 Us3 regulated apoptosis and cell morphology in HSV-2-infected cells similarly to HSV-1 Us3 (27, 52). However, HSV-2 Us3 did not appear to be involved in regulation of intracellular trafficking of HSV-2 gB or in vesicle-mediated nucleocytoplasmic transport of nucleocapsids through the nuclear membrane (27). The kinase-dead mutation in HSV-2 Us3 has been reported to have no effect on vesicle-mediated nucleocytoplasmic transport of nucleocapsids or on cell surface expression of gB, but the kinase-dead mutation in HSV-1 Us3 induced formation of membranous structures adjacent to the nuclear membrane with aberrant accumulations of primary enveloped virions, as described above, and increased cell surface expression of gB (21, 39). In addition, the null mutation in HSV-2 Us3 was reported to significantly reduce accumulation of UL46 protein in HSV-2-infected cells, but the null mutation in HSV-1 Us3 had no effect on UL46 accumulation in HSV-1-infected cells (53, 54). These observations suggested that HSV Us3 kinases may have both conserved and type-specific functions in infected cells. Differences in Us3 type-specific functions may be due to differences in phosphorylation of the HSV-1 Us3 and HSV-2 Us3 protein substrates in infected cells, because of differences between HSV-1 Us3 and HSV-2 Us3 kinases in their catalytic activity, protein substrate specificity, and/or phosphorylation site specificity. In agreement with the possibility of phosphorylation site differences, the Us3 phosphorylation site in HSV-1 gB (gB threonine at the position 887 [Thr-887]), which was reported to be critical for regulation of viral intracellular trafficking, is not conserved in HSV-2 gB, and HSV-2 Us3 has been shown to be unable to regulate gB intracellular trafficking, as described above (27, 39). In contrast, no direct data for differences in HSV-1 Us3 and HSV-2 Us3 kinases have been reported thus far. Previous studies compared the phenotypes of similar mutations in HSV-1 Us3 and HSV-2 Us3 kinases in HSV-1- and HSV-2-infected cells in which the cellular substrates of the kinases were identical, but the viral substrates were not identical. In addition, at least some of the HSV-1 Us3 and HSV-2 Us3 phosphorylation sites in the viral substrates in infected cells were not conserved, as discussed above for regulation of HSV gB. To investigate this issue in the present study, we generated a recombinant HSV-1 in which its Us3 gene was replaced with the HSV-2 Us3 gene. Using this chimera virus together with an HSV-1 Us3 kinase-dead mutant virus, we investigated whether the phenotype of the HSV-1 Us3 kinase-dead mutant was changed by replacement of HSV-1 Us3 with HSV-2 Us3. This strategy enabled us to directly compare HSV-1 Us3 and HSV-2 Us3 kinases in cells infected by HSV-1 strains with all the same viral gene products except for their Us3 kinases. The results of this study indicated that HSV-2 Us3 could compensate only in part for the HSV-1 Us3 kinase, indicating that there is a biological difference between HSV-1 Us3 and HSV-2 Us3.
MATERIALS AND METHODS
Cells and viruses.
Simian kidney epithelial Vero, human neuroblastoma SK-N-SH, and rabbit skin cells were described previously (55, 56). Human keratinocyte HaCaT cells were purchased (CLS Cell Lines Service) (57) and maintained in Dulbecco's modified Eagle's medium containing 10% fetal calf serum. The HSV-1 wild-type strain HSV-1(F), HSV-2 wild-type strain 186 [HSV-2 (186)], recombinant virus YK511 (encoding an enzymatically inactive Us3 mutant in which lysine at Us3 residue 220 was replaced with methionine [Us3K220M]), and recombinant virus YK513 (in which the Us3K220M mutation in YK511 was repaired [Us3K220M-repair]) were described previously (15, 52, 55, 58).
Plasmids.
To generate pBS-HSV-2-Us3, the entire HSV-2 Us3 coding sequence was amplified by PCR from pYEbac356 (27), a full-length infectious HSV-2 (186) clone, and cloned into the NotI and EcoRI sites of pBluescript II KS(+) (Stratagene). To generate pBS-HSV-2-Us3-KanS, for generating recombinant HSV-1 YK781 (Us3-chimera) in which the Us3 gene was replaced with HSV-2 Us3, an I-SceI site, a kanamycin resistance gene, and 60 bp of the HSV-2 Us3 sequence were amplified by PCR from pEP-KanS (15, 59) with primers 5′-GCCTGCAGCTGACACAGGCGCGCAGCCTAGGATGACGACGATAAGTAGGG-3′ and 5′-GCCTGCAGGAGATCTTGGCCCAGATGCACAACCAATTAACCAATTCTGATTAG-3′ and cloned into the PstI site of pBS-HSV-2-Us3.
Mutagenesis of viral genomes and generation of recombinant viruses.
To generate YK780, in which the HSV-1 Us3 gene was replaced by an MEF tag with myc and Flag epitopes and a tobacco etch virus (TEV) protease cleavage site (i.e., myc-TEV-flag) (60) (HSV-1ΔUs3/MEF-1) (Fig. 1), a two-step red-mediated mutagenesis procedure was carried out using Escherichia coli GS1783 containing pYEbac102 (55), a full-length infectious HSV-1(F) clone, as described previously (15, 59), except using primers 5′-CGGGGCCCGTCGTTCGGGGTGCTCGTTGGTTGGCACTCACGGTGCGGCGAATGGAGCAAAAGCTCATTTC-3′ and 5′-TCGGGGTCTTTTTGTGCCAACCCGCAAACAGCACCGCCCCCAGGGGGCGGTCATTTGTCATCGTCGTCCT-3′ and pGEM-MEF (61). Recombinant virus YK781(Us3-chimera), in which the MEF tag in YK780 (HSV-1ΔUs3/MEF-1) was replaced by the HSV-2 (186) Us3 gene, was constructed by the two-step red-mediated mutagenesis procedure described above, except using primers 5′-GGCCCGTCGTTCGGGGTGCTCGTTGGTTGGCACTCACGGTGCGGCGAATGGCCTGTCGTAAGTTCTGTGG-3′ and 5′-TCGGGGTCTTTTTGTGCCAACCCGCAAACAGCACCGCCCCCTGGGGGCGGTCACTTAGGGTGAAATAGCG-3′, pBS-HSV-2-Us3-KanS, and E. coli GS1783 containing the YK780 genome (Fig. 1). Recombinant virus YK782, in which the HSV-2 (186) Us3 gene in YK781 (Us3-chimera) was replaced by a foreign cassette (HSV-1ΔUs3/MEF-2), was constructed by the procedure used to generate YK780 (HSV-1ΔUs3/MEF-1) except using E. coli GS1783 containing the YK781 genome (Fig. 1). Recombinant virus YK783, in which the foreign cassette in YK782 (HSV-1ΔUs3/MEF-2) was replaced by the HSV-1(F) Us3 gene (Us3-repair), was constructed by the same procedure used to generate YK781 (Us3-chimera) except using primers 5′-GGCCCGTCGTTCGGGGTGCTCGTTGGTTGGCACTCACGGTGCGGCGAATGGCCTGTCGTAAGTTTTGTCG-3′ and 5′-TCGGGGTCTTTTTGTGCCAACCCGCAAACAGCACCGCCCCCTGGGGGCGGTCATTTCTGTTGAAACAGCG-3′, pYEbac512 (15), and E. coli GS1783 containing the YK782 genome (Fig. 1). Recombinant virus YK823 encoding Us3 fused to the MEF tag (MEF-Us3) was generated as described previously (15, 59) except using primers 5′-CGGGGCCCGTCGTTCGGGGTGCTCGTTGGTTGGCACTCACGGTGCGGCGAATGGAGCAAAAGCTCATTTC-3′ and 5′-TCCTTCCTCCTGCCCTGTCCCCCGTAAACGCGACAAAACTTACGACAGGCATCTTTGTCATCGTCGTCCT-3′ and pGEM-MEF (Fig. 1). Recombinant virus YK825, in which HSV-2 Us3 in YK781 (Us3-chimera) was fused to the MEF tag, was generated by the same procedure as used to generate YK823 (MEF-Us3) except using the primer 5′-TCCTGTCTCTTGTCGGGTCTACGGTAGACCCCACAGAACTTACGACAGGCATCTTTGTCATCGTCGTCCT-3′ (Fig. 1).
FIG 1.
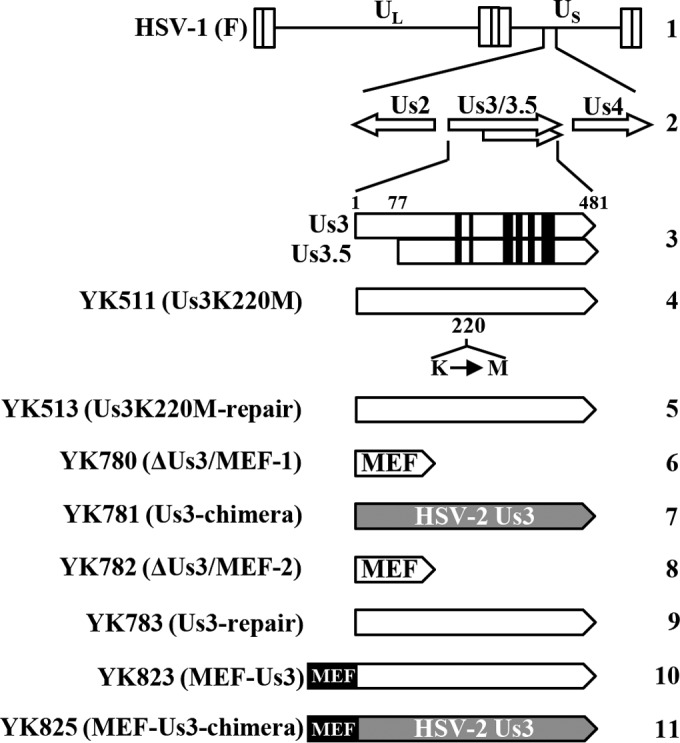
Schematic diagrams of the genome structure of wild-type HSV-1(F) and the relevant domains of the recombinant viruses used in this study. Line 1, wild-type HSV-1(F) genome; line 2, domains encoding the Us3 to Us4 open reading frames; line 3, domains of Us3 and Us3.5; lines 4 to 11, domains in recombinant virus genomes with mutations in Us3.
Immunoblotting, immunofluorescence, and determination of plaque size.
Immunoblotting, immunofluorescence, and determination of plaque size were performed as described previously (62–64).
Antibodies.
Rabbit polyclonal antibodies to Us2, Us3, UL12, UL31, UL34, and vdUTPase were described previously (15, 40, 65–68). Mouse monoclonal antibody that specifically recognizes gB with phosphorylated Thr-887 (gB-T887P) was described previously (69). Mouse monoclonal antibodies to VP5 (3B6), Us4 (7F5), and gB (H1817) were purchased from Virusys. Mouse monoclonal antibodies to gH (52-S), β-actin (AC15), lamin A/C (636), and Myc tag were purchased from the American Type Culture Collection, Sigma, Santa Cruz Biotechnology, and MBL, respectively. Rabbit phospho-protein kinase A (PKA) substrate (100G7) monoclonal antibody was purchased from Cell Signaling Technology. Rabbit polyclonal antibody to lamin B1 was purchased from Abcam.
Induction of apoptosis and measurement of caspase 3/7 activity.
Induction of apoptosis in HSV-1-infected cells was performed as described previously (56). Caspase 3/7 activity was assayed using a Caspase-Glo 3/7 assay kit (Promega) as described previously (56).
Detection of gB on infected-cell surfaces by flow cytometry.
Cell surface expression of gB in infected cells was detected by flow cytometry as described previously (38, 39, 69), except that FACSVerse (BD Bioscience) was used in these experiments.
Electron microscopic analysis.
Vero and HaCaT cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3KM-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 for 18 h were examined by ultrathin-section electron microscopy as described previously (70).
Phos tag SDS-PAGE analysis.
Phos tag (phosphate affinity) SDS-PAGE analyses were performed as described previously (40, 71) except using denaturing gels containing 100 μM MnCl2 and 50 μM Phos tag acrylamide.
Phosphatase treatment.
Lysates of Vero cells that had been mock infected or infected with either wild-type HSV-1(F) or YK781 (Us3-chimera) at a multiplicity of infection (MOI) of 3 for 18 h were treated with alkaline phosphatase, calf intestinal (CIP) (New England BioLabs) as described previously (72).
Animal studies.
Five-week-old female ICR mice were purchased from Charles River. For ocular infections, the mice were infected with 1 × 106 PFU of YK781 (Us3-chimera) or YK783 (Us3-repair) per eye as described previously (37, 69, 70). The scoring scales for the severity of herpes stromal keratitis (HSK) and periocular skin disease were described previously (37, 69, 70). Virus titers in the tear films of infected mice were determined as described previously (37, 69, 70). All animal experiments were carried out in accordance with the guidelines for proper conduct of animal experiments of the Science Council of Japan. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the Institute of Medical Science, The University of Tokyo (IACUC protocol approval numbers PA11-81 and PH12-10).
RESULTS
Characterization of the recombinant viruses generated in this study.
To compare the regulatory effects of HSV-1 Us3 and HSV-2 Us3 kinases in cells infected by HSV-1 strains with the same viral gene products except for their Us3 kinases, recombinant virus YK781 (Us3-chimera), in which the HSV-1 Us3 gene was replaced with the HSV-2 Us3 gene, and its repaired virus YK783 (Us3-repair) were generated (Fig. 1).
To examine whether cells infected with YK781 (Us3-chimera) produced HSV-2 Us3, Vero cells infected with wild-type HSV-1(F), YK781 (Us3-chimera), YK783 (Us3-repair), or wild-type HSV-2 (186) at an MOI of 3 for 18 h were analyzed by immunoblotting (Fig. 2). As described previously (15, 27), anti-Us3 antibody reacted with HSV-1 Us3 and HSV-2 Us3 in the lysates of cells infected with wild-type HSV-1(F) and wild-type HSV-2 (186), respectively (Fig. 2A). However, the amount of HSV-2 Us3 in the band from lysates of cells infected with wild-type HSV-2 (186) was much smaller than that of the HSV-1 Us3 bands from lysates of cells infected with wild-type HSV-1(F), and HSV-2 Us3 migrated faster than HSV-1 Us3 in the denaturing gel (Fig. 2A). The result with cells infected with wild-type HSV-2 (186) was consistent with that for cells infected with YK781 (Us3-chimera), and the wild-type HSV-1 Us3 band was restored in cells infected with YK783 (Us3-repair) (Fig. 2A). These results showed that cells infected with YK781 (Us3-chimera) expressed HSV-2 Us3.
FIG 2.

Immunoblots of electrophoretically separated lysates of Vero cells mock infected or infected with wild-type HSV-1(F), YK781 (Us3-chimera), YK783 (Us3-repair), or wild-type HSV-2 (186) at an MOI of 3. The infected cells were harvested at 18 h (A) and 12 h (B) postinfection and analyzed by immunoblotting with antibodies to Us3, VP5, and β-actin (A) or to Us2, Us4, and UL12 (B).
Since the anti-Us3 antibody used in this study was raised against HSV-1 Us3 protein (15), the reactivity of this anti-Us3 antibody to HSV-2 Us3 could be lower than that to HSV-1 Us3, causing cells infected with wild-type HSV-2 (186) or YK781 (Us3-chimera) to appear to produce less Us3 protein than cells infected with wild-type HSV-1(F) or YK783 (Us3-repair). To address this issue, two additional recombinant viruses, YK823 (MEF-Us3) and YK825 (MEF-Us3-chimera), were generated in which HSV-1 Us3 and the replacement HSV-2 Us3, respectively, were tagged with MEF at their N termini (Fig. 1). YK823 (MEF-Us3) replicated as efficiently as YK825 (MEF-Us3-chimera) in Vero cells infected at MOIs of 3 and 0.01 (data not shown). Anti-Myc antibody should react similarly with MEF-tagged HSV-1 Us3 (MEF-HSV-1 Us3) and HSV-2 Us3 (MEF-HSV-2 Us3) in lysates of cells infected with YK823 (MEF-Us3) and YK825 (MEF-Us3-chimera), respectively. Therefore, the amounts of MEF-HSV-1 Us3 and MEF-HSV-2 Us3 in Vero cells infected with YK823 (MEF-Us3) and YK825 (MEF-Us3-chimera), respectively, were analyzed by immunoblotting with the anti-Myc antibody. As shown in Fig. 3, the density of the MEF-HSV-1 Us3 band in lysates of cells infected with YK823 (MEF-Us3) was comparable to that of the MEF-HSV-2 Us3 band in lysates of cells infected with YK825 (MEF-Us3-chimera), indicating that these two types of infected cells produced MEF-tagged Us3 proteins at similar levels. These results suggested that like cells infected with YK823 (MEF-Us3) or YK825 (MEF-Us3-chimera), cells infected with YK781 (Us3-chimera) produced Us3 protein at a level similar to that of cells infected with wild-type HSV-1(F) or YK783 (Us3-repair). However, we cannot eliminate completely the possibility that the level of expression of the untagged HSV-2 Us3 in cells infected with YK781 (Us3-chimera) was not similar to that of the untagged HSV-1 Us3 in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair), since the tagging of HSV-1 and HSV-2 Us3 with the MEF tag appeared to change their properties. Thus, untagged HSV-1 Us3 migrated slightly more slowly in the denaturing gel than untagged HSV-2 Us3 (Fig. 2A), but MEF-HSV-1 Us3 migrated apparently more rapidly than MEF-HSV-2 Us3 (Fig. 3). Furthermore, untagged HSV-1 Us3 was detected as doublet bands in the denaturing gel (Fig. 2A), but MEF-HSV-1 Us3 was detected as a single band (Fig. 3).
FIG 3.
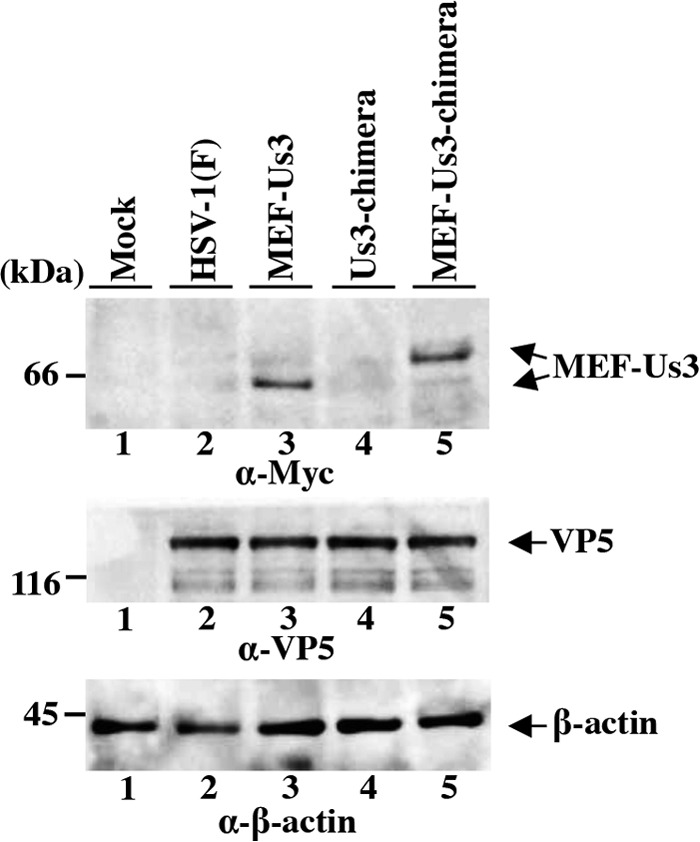
Immunoblots of electrophoretically separated lysates of Vero cells mock infected or infected with wild-type HSV-1(F), YK823 (MEF-Us3), YK781 (Us3-chimera), or YK825 (MEF-Us3-chimera) at an MOI of 3. The infected cells were harvested at 18 h postinfection and analyzed by immunoblotting with antibodies to Myc tag, VP5, and β-actin.
Taken together, these experiments confirmed that HSV-1 Us3 was properly replaced with HSV-2 Us3 in YK781 (Us3-chimera). In addition, the level of expression of the genes on both sides of Us3, Us2 and Us4 (Fig. 1), in Vero cells infected with YK781 (Us3-chimera) was comparable to that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair) (Fig. 2B), indicating that replacement of HSV-1 Us3 with HSV-2 Us3 had little, if any, effect on expression of the Us3-neighboring genes Us2 and Us4.
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral growth in cell cultures.
To examine the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral growth in cell cultures, Vero and HaCaT cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 or 0.01 were harvested at several times postinfection, and total virus titers in the infected cells and cell culture supernatants were assayed. In agreement with a previous report (15), the Us3 kinase-dead mutant virus YK511 (Us3K220M) grew as well as its repaired virus YK513 (Us3K220M-repair) and wild-type HSV-1(F) in Vero cells infected at an MOI of 3 or 0.01 (Fig. 4A and B). In contrast, YK511 (Us3K220M) grew in HaCaT cells infected at an MOI of 3 or 0.01 much less efficiently than wild-type HSV-1(F) and YK513 (Us3K220M-repair) (Fig. 4C and D), indicating that the kinase activity of HSV-1 Us3 was required for efficient viral replication in HaCaT cells. As shown in Fig. 4A to D, the growth curves of YK781 (Us3-chimera) were comparable to those of wild-type HSV-1(F), YK513 (Us3K220M-repair), and YK783 (Us3-repair) in Vero and HaCaT cells infected at an MOI of 3 or 0.01. These results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 had little effect on viral growth in Vero and HaCaT cells.
FIG 4.
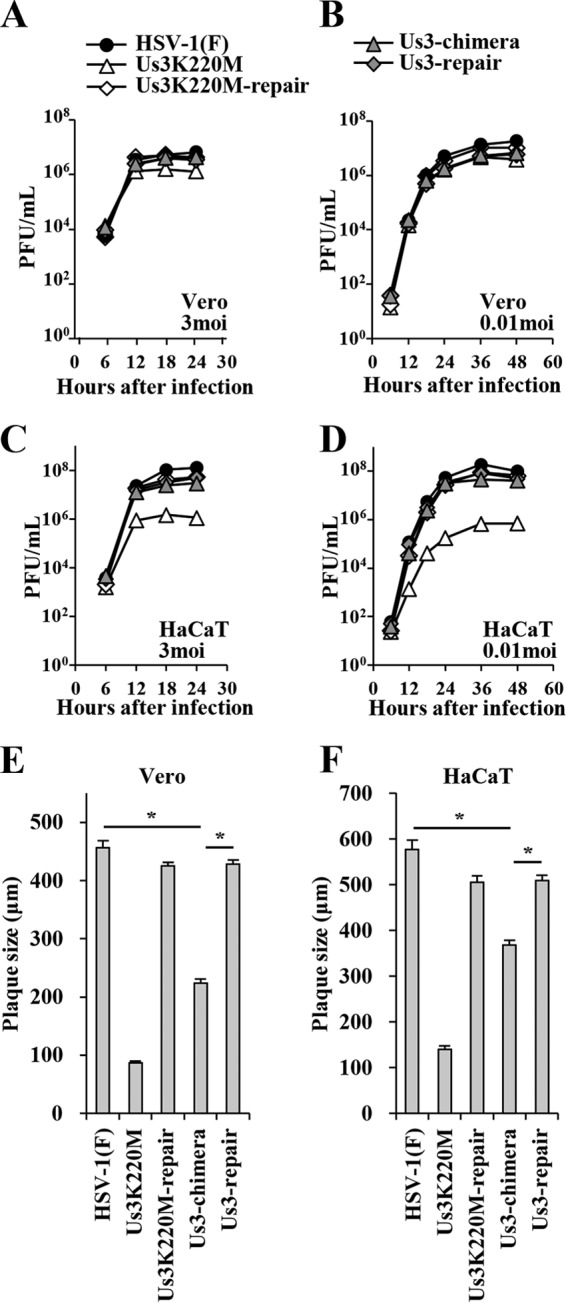
Growth curves and plaque sizes of the recombinant viruses generated in this study. Vero (A and B) and HaCaT (C and D) cells were infected at an MOI of 3 (A and C) or 0.01 (B and D) with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair). Total virus from the cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells. Vero (E) and HaCaT (F) cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 0.0001 under plaque assay conditions. The diameters of 30 single plaques for each of the indicated viruses were measured 48 h postinfection. Each data point is the mean ± standard error of the measured plaque sizes. Statistical analysis was performed by one-way analysis of variance (ANOVA) with the Tukey test. Asterisks indicate statistically significant values (*, P < 0.001).
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on cell-cell spread in cell cultures.
To examine the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral cell-cell spread in cell cultures, we analyzed plaque sizes in Vero and HaCaT cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) under plaque assay conditions. As shown in Fig. 4E and F, YK781 (Us3-chimera) produced significantly smaller plaques than wild-type HSV-1(F) and YK783 (Us3-repair) in Vero and HaCaT cells, although this chimera virus produced larger plaques than YK511 (Us3K220M). These results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 impaired viral cell-cell spread in Vero and HaCaT cells.
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on the profile of phosphorylated proteins in infected cells.
We previously reported that anti-phospho-PKA substrate antibody 100G7, which reacts with proteins containing a phosphorylated serine or threonine residue and arginine at residues −3 and −2 (RRXS or RRXT) (15, 37), was able to detect Us3 phosphorylation of some Us3 substrates, including gB, UL47, and Us3 itself, in infected cells (15, 37, 39). These observations together with a previous study using another anti-phospho-PKA substrate antibody (12, 73) indicated that anti-phospho-PKA antibodies can be used to detect the protein substrates phosphorylated by Us3. To examine the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on the profile of Us3-mediated phosphorylated proteins in infected cells, Vero cells mock infected or infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 for 18 h were analyzed by immunoblotting with anti-phospho-PKA substrate antibody 100G7. As described elsewhere (12), antibody 100G7 reacted with a number of bands in the lysates of cells infected with wild-type HSV-1(F), YK511 (Us3K220M), or YK513 (Us3K220M-repair), but the amount of protein in most of the bands that reacted with the 100G7 antibody in lysates of cells infected with YK511 (Us3K220M) was smaller than in lysates of cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair) (Fig. 5). In contrast, the pattern of protein bands that reacted with the 100G7 antibody in lysates of cells infected with YK781 (Us3-chimera) was almost identical to that in lysates of cells infected with wild-type HSV-1(F) or YK783 (Us3-repair) (Fig. 5). These results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 had little effect on the profile of the proteins in infected cells phosphorylated by Us3 that were detected by the anti-phospho-PKA substrate antibody.
FIG 5.
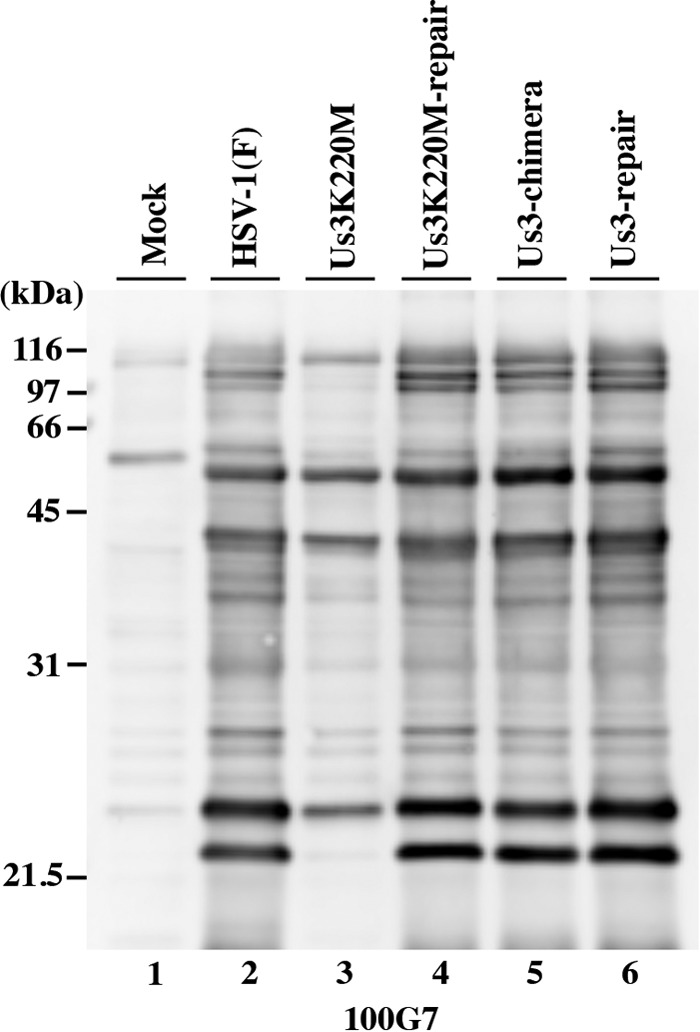
Detection of phosphorylated proteins in Vero cells mock infected or infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3. The infected cells were harvested at 18 h postinfection and analyzed by immunoblotting with anti-PKA substrate antibody 100G7.
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on Us3-mediated regulation of apoptosis, cell surface expression of gB, and infected-cell morphology.
Three series of experiments were carried out to investigate whether replacement of HSV-1 Us3 with HSV-2 Us3 affected some HSV-1 Us3 functions in infected cells, i.e., regulation of apoptosis, cell surface expression of gB, and infected-cell morphology (15, 39). In the first series of experiments, SK-N-SH cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 5, and at 18 h postinfection, apoptosis was induced by osmotic shock for 2 h. The infected cells then were incubated for an additional 5 h, harvested, and assayed for caspase 3/7 activity. In agreement with a previous report (70), caspase 3/7 activity in cells infected with YK511 (Us3K220M) was significantly higher than in cells infected with HSV-1(F) or YK513 (Us3K220M-repair), probably due to the lack of HSV-1 Us3-mediated antiapoptotic activity (Fig. 6A). In contrast, caspase 3/7 activity in YK781 (Us3-chimera)-infected cells was similar to that in wild-type HSV-1(F)- and YK783 (Us3-repair)-infected cells, indicating that replacement of HSV-1 Us3 with HSV-2 Us3 had little effect on caspase 3/7 activity in infected SK-N-SH cells in which apoptosis had been induced. Since upregulation of caspase 3/7 activity is a typical indicator of apoptosis, these results suggested that HSV-2 Us3 in YK781 (Us3-chimera)-infected cells had antiapoptotic activity at a level similar to that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair).
FIG 6.
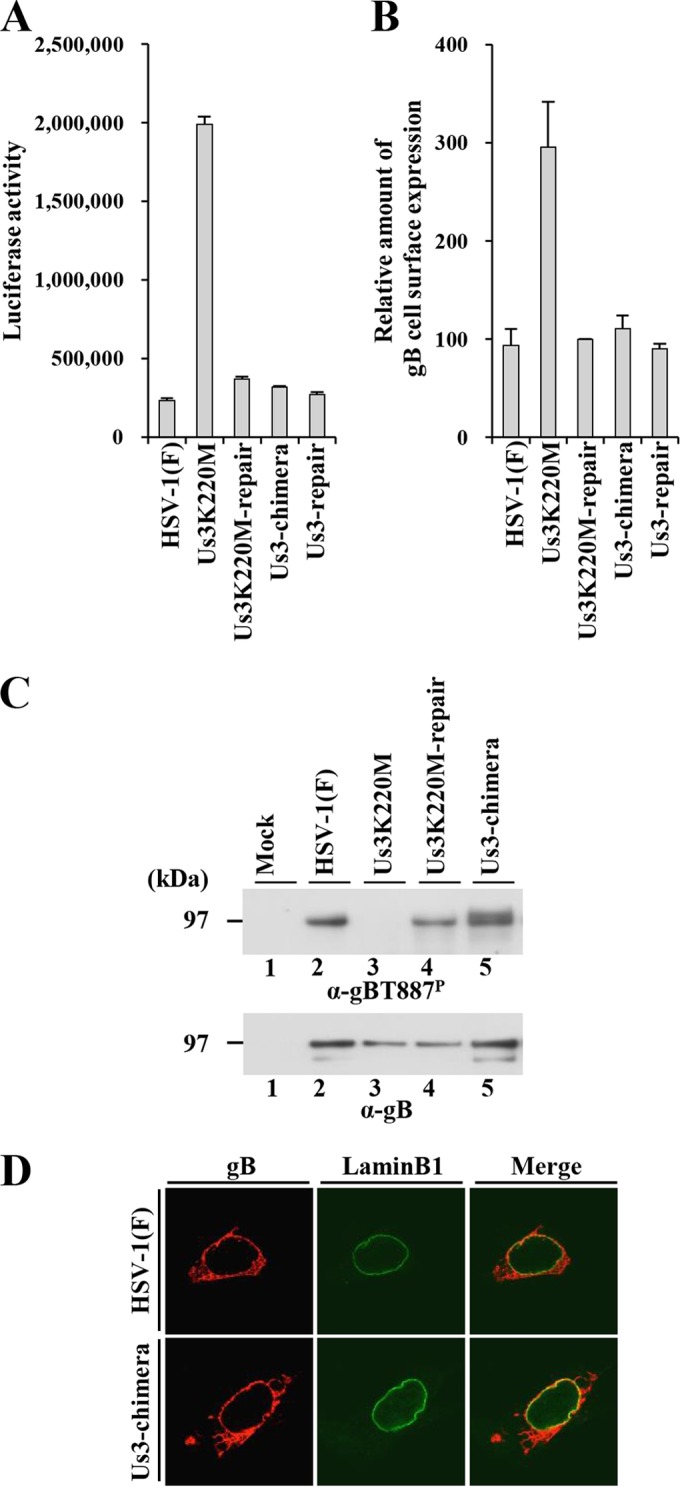
(A) Caspase 3/7 activity of infected SK-N-SH cells after induction of apoptosis by osmotic shock. SK-N-SH cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 5. At 18 h postinfection, the infected cells were exposed to sorbitol for 2 h, incubated for an additional 5 h, harvested, and assayed for caspase 3/7 activity. Each value is the mean ± standard error of the results of triplicate experiments. Data are representative of three independent experiments. (B) Cell surface expression of gB in Vero cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3. At 6 h postinfection, the infected cells were harvested and analyzed by flow cytometry. The relative mean fluorescence intensity for gB expression on the surface of cells infected with the indicated virus is shown as the fluorescence intensity of virus-infected cells relative to that of YK513 (Us3K220M-repair)-infected cells. Each value is the mean ± standard error of the results of three independent experiments. (C) Phosphorylation of gB at Thr-887 in Vero cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), or YK781 (Us3-chimera) at an MOI of 3. At 18 h postinfection, the infected cells were harvested and analyzed by immunoblotting with the monoclonal antibody to gB-T887P and gB. (D) Localization of gB in Vero cells infected with wild-type HSV-1(F) or YK781 (Us3-chimera) at an MOI of 3. At 18 h postinfection, the infected cells were permeabilized, stained with anti-gB and anti-lamin B1 antibodies, and examined by confocal microscopy.
In the second series of experiments, Vero cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 were harvested at 6 postinfection, and expression of gB on the surface of the infected cells was analyzed by flow cytometry. In agreement with a previous report (39), cell surface expression of gB in cells infected with HSV-1 YK511 (Us3K220M) was upregulated compared to that in cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair) (Fig. 6B). In contrast, the level of cell surface expression of gB in cells infected with YK781 (Us3-chimera) was similar to that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair). We confirmed that gB was in fact phosphorylated at Thr-887 in cells infected with YK781 (Us3-chimera), like gB in wild-type HSV-1(F)-infected cells, based on the observations that the monoclonal antibody to gB-T887P reacted with gB in Vero cells infected with YK781 (Us3-chimera) (Fig. 6C), whereas this antibody reacted with gB in cells infected with wild-type HSV-1(F) but not that in cells infected with YK511 (Us3K220M) (Fig. 6C) as reported previously (69). Furthermore, the pattern of the localization of gB in Vero cells infected with wild-type HSV-1(F) for 18 h, detected by immunofluorescence with the monoclonal antibody to gB, was similar to that in cells infected with YK781 (Us3-chimera). These results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 had little effect on cell surface expression of gB, phosphorylation of gB at Thr-887, and localization of gB in infected cells and suggested that HSV-2 Us3 in YK781 (Us3-chimera)-infected cells regulated intracellular trafficking of HSV-1 gB as efficiently as HSV-1 Us3 in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair).
In the third series of experiments, Vero cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 and the cytopathic effects (CPEs) in the infected cells were examined at 24 h postinfection. In agreement with a previous report (15), infection of cells with wild-type HSV-1(F) or YK513 (Us3K220M-repair) efficiently induced cell rounding, while YK511 (Us3K220M) infection did so only partially (Fig. 7). In contrast, YK781 (Us3-chimera) infection induced cell rounding as efficiently as infection with wild-type HSV-1(F) or YK783 (Us3-repair). These results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 had little effect on induction of a wild-type CPE and suggested that HSV-2 Us3 in YK781 (Us3-chimera)-infected cells regulated infected-cell morphology as efficiently as HSV-1 Us3 in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair).
FIG 7.
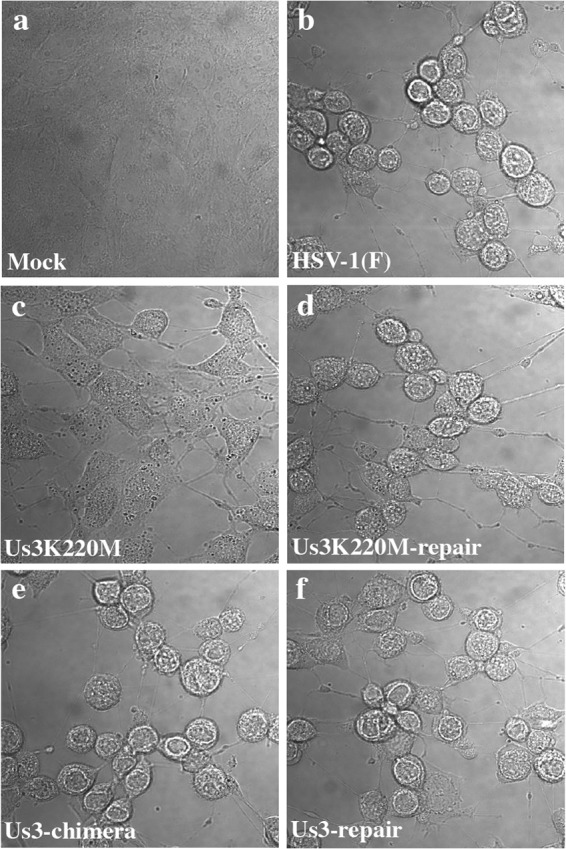
Images of the CPE in Vero cells mock infected or infected at an MOI of 3 with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair). Live cells were examined at 24 h postinfection by confocal microscopy. Digital interference contrast images are shown.
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on intracellular localization of UL31 and UL34.
To investigate whether replacement of HSV-1 Us3 with HSV-2 Us3 affected intracellular localization of UL31 and UL34, Vero and HaCaT cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 for 12 h and localization of UL31 and UL34 in infected cells was analyzed by immunofluorescence using confocal microscopy. It has been reported that localization of HSV-1 UL34 and UL31 in infected cells was regulated by Us3 activity and, in the absence of Us3 protein or its catalytic activity, these viral proteins accumulated aberrantly in punctate structures adjacent to the nuclear membrane in HSV-1-infected cells (15, 21, 43, 56).
In agreement with previous reports (15, 21, 27, 43, 74), in Vero cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair), UL34 and UL31 proteins colocalized smoothly along the nuclear rim, and in Vero cells infected with YK511 (Us3K220M), UL34 and UL31 colocalized in punctate structures adjacent to the nuclear rim (Fig. 8). However, in Vero cells infected with YK781 (Us3-chimera), although UL31 and UL34 colocalized smoothly along the nuclear rim in a majority of cells, they colocalized in punctate structures adjacent to the nuclear rim in a significant fraction (37.7%) of cells (Fig. 8 and 9A). We noted that the sizes of the UL34 and UL31 stained speckles in Vero cells infected with YK781 (Us3-chimera) appeared to be smaller than those in YK511 (Us3K220M)-infected cells (Fig. 8). The UL31 and UL34 punctate structures at the nuclear rim seen in YK781 (Us3-chimera)-infected cells were not found in cells infected with YK783 (Us3-repair), in which UL31 and UL34 colocalized smoothly along the nuclear rim, as found in wild-type HSV-1(F)-infected cells (Fig. 8).
FIG 8.
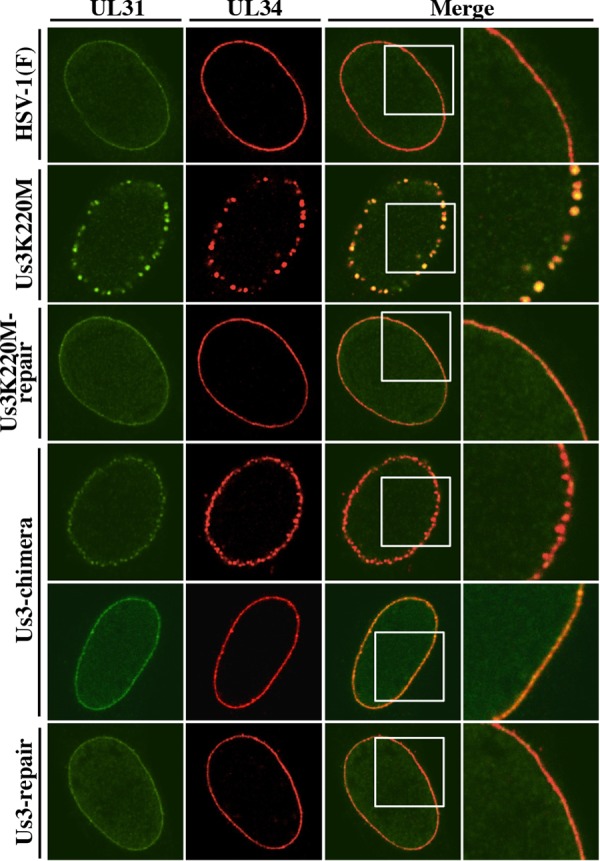
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on localization of UL31 and UL34 in infected Vero cells. Vero cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3, fixed at 12 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy. Each image in the far right column is the magnified image of the boxed area in the image to its left. Since two different patterns of UL31 and UL34 localization were observed in cells infected with YK781 (Us3-chimera), images of two of these infected cells are shown here as examples of UL31 and UL34 localization in these infected cells.
FIG 9.
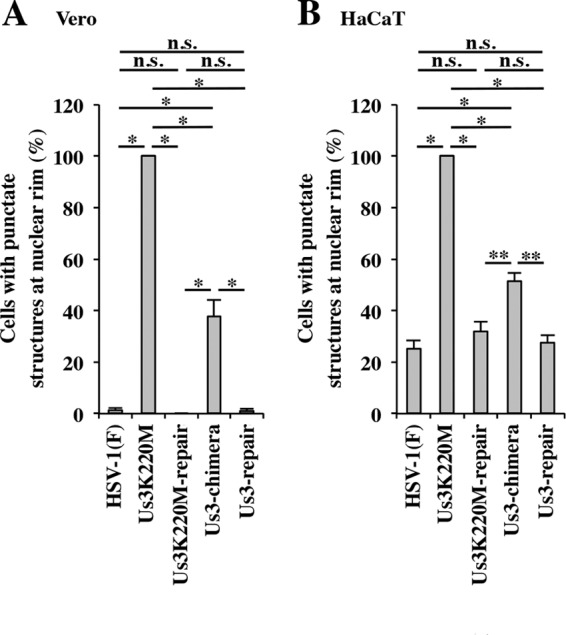
Quantitation of infected Vero (A) and HaCaT (B) cells with aberrant punctate structures of UL31 and UL34 adjacent to the nuclear rim. The experiments were done as described for Fig. 8 and 10, and the percentage of cells with aberrant punctate structures at the nuclear rim was determined. Each value is the mean ± standard error of the results of three independent experiments. Statistical analysis was performed by one-way ANOVA with the Tukey test. Asterisks indicate statistically significant values (*, P < 0.001; **, P < 0.01). n.s., not statistically significant.
Unlike in HSV-1(F)- and YK513 (Us3K220M-repair)-infected Vero cells in which UL34 and UL31 proteins colocalized smoothly along the nuclear rim in almost all cells, in HSV-1(F)- and YK513 (Us3K220M-repair)-infected HaCaT cells, although UL34 and UL31 proteins colocalized smoothly along the nuclear rim in a majority of cells, they also colocalized in punctate structures adjacent to the nuclear rim in a significant fraction of infected cells (25.3 and 32.0%, respectively) (Fig. 9B and 10). In HaCaT cells infected with YK511 (Us3K220M), as in Vero cells infected with YK511 (Us3K220M), UL34 and UL31 colocalized in punctate structures adjacent to the nuclear rim. In YK781 (Us3-chimera)-infected HaCaT cells, as in YK781 (Us3-chimera)-infected Vero cells, UL31 and UL34 colocalized smoothly along the nuclear rim in some cells and in punctate structures adjacent to the nuclear rim in other cells (51.3%): the fraction of YK781 (Us3-chimera)-infected HaCaT cells with punctate structures (51.3%) was greater than the fractions of HSV-1(F)- and YK783 (Us3-repair)-infected HaCaT cells (25.3% and 27.7%, respectively) with punctate structures (Fig. 9B).
FIG 10.
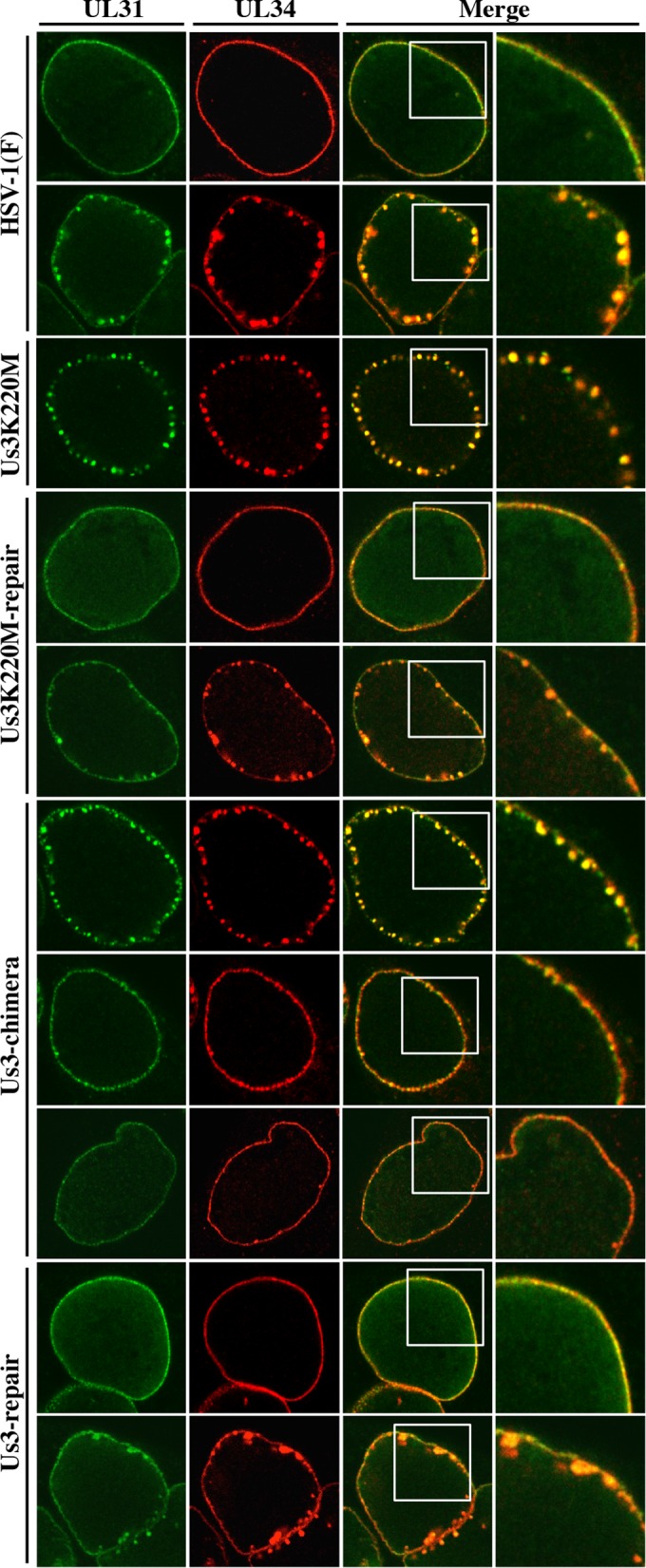
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on localization of UL31 and UL34 in infected HaCaT cells. HaCaT cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3, fixed at 12 h postinfection, permeabilized, stained with anti-UL34 and anti-UL31 antibodies, and examined by confocal microscopy. Each image in the far right column is the magnified image of the boxed area in the image to its left. Since different patterns of UL31 and UL34 localization were observed in cells infected with HSV-1(F), YK513 (Us3K220M-repair), YK781 (Us3-chimera), and YK783 (Us3-repair), images of two of the HSV-1(F)-, YK513 (Us3K220M-repair)- and YK783 (Us3-repair)-infected cells and three of the YK781 (Us3-chimera)-infected cells are shown here as examples of UL31 and UL34 localization in these infected cells.
Collectively, these results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 produced an aberrant localization of UL34 and UL34 in a significant fraction of YK781 (Us3-chimera)-infected cells.
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on HSV-1 nuclear egress.
To investigate the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on HSV-1 nuclear egress, Vero and HaCaT cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3, and viral morphogenesis in the infected cells was investigated by electron microscopy 18 h postinfection. In agreement with a previous report (21), in Vero and HaCaT cells infected with YK511 (Us3K220M), membranous structures containing primary enveloped virions, formed by invaginations of the INM into the nucleoplasm, were readily observed adjacent to the nuclear membrane (Fig. 11 and 12). These membranous invagination structures were barely detectable in Vero cells infected with wild-type HSV-1(F) or YK513 (Us3-K220M-repair) but were observed with low but consistent frequency in HaCaT cells infected with wild-type HSV-1(F) or YK513 (Us3-K220M-repair) (Fig. 11 and 12 and Table 1). In Vero and HaCaT cells infected with YK511 (Us3K220M), 63.8 and 54.6% of enveloped virions, respectively, were in the perinuclear space and invagination structures, but in Vero and HaCaT cells infected with wild-type HSV-1(F), 28.5 and 17.4% of enveloped virions, respectively, were in these compartments (Table 1). Results similar to those with wild-type HSV-1(F) were also obtained with YK513 (Us3K220M-repair)-infected Vero and HaCaT cells (Fig. 12B and Table 1). These results indicated that primary enveloped virions accumulated aberrantly in YK511 (Us3K220M)-infected cells. Similar to the results with YK511 (Us3K220M)-infected HaCaT cells, membranous invagination structures containing primary enveloped virions were readily observed adjacent to the nuclear membrane in HaCaT cells infected with YK781 (Us3-chimera), with 43.0% of the enveloped virions in the perinuclear space and invagination structures. In contrast, in HaCaT cells infected with wild-type HSV-1(F) or YK783 (Us3-repair), 17.4% and 22.6%, respectively, of primary enveloped virions were in the perinuclear space and invagination structures. However, in Vero cells infected with YK781 (Us3-chimera), in addition to the invagination structures observed in Vero cells infected with YK511 (Us3K220M), membranous structures containing numerous primary enveloped virions, formed by evaginations of the ONM into the cytoplasm, were also induced. In YK781 (Us3-chimera)-infected Vero cells, 57.6% of enveloped virions were in the perinuclear space and membranous invaginations, but in Vero cells infected with wild-type HSV-1(F) or YK783 (Us3-repair), 28.5% and 36.1%, respectively, were in the perinuclear space and membranous invaginations. Collectively, these results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 induced membranous invaginations and/or evaginations containing primary enveloped virions adjacent to the nuclear membrane in infected cells and that primary enveloped virions accumulated aberrantly in the perinuclear space and membranous invagination and/or evagination structures.
FIG 11.
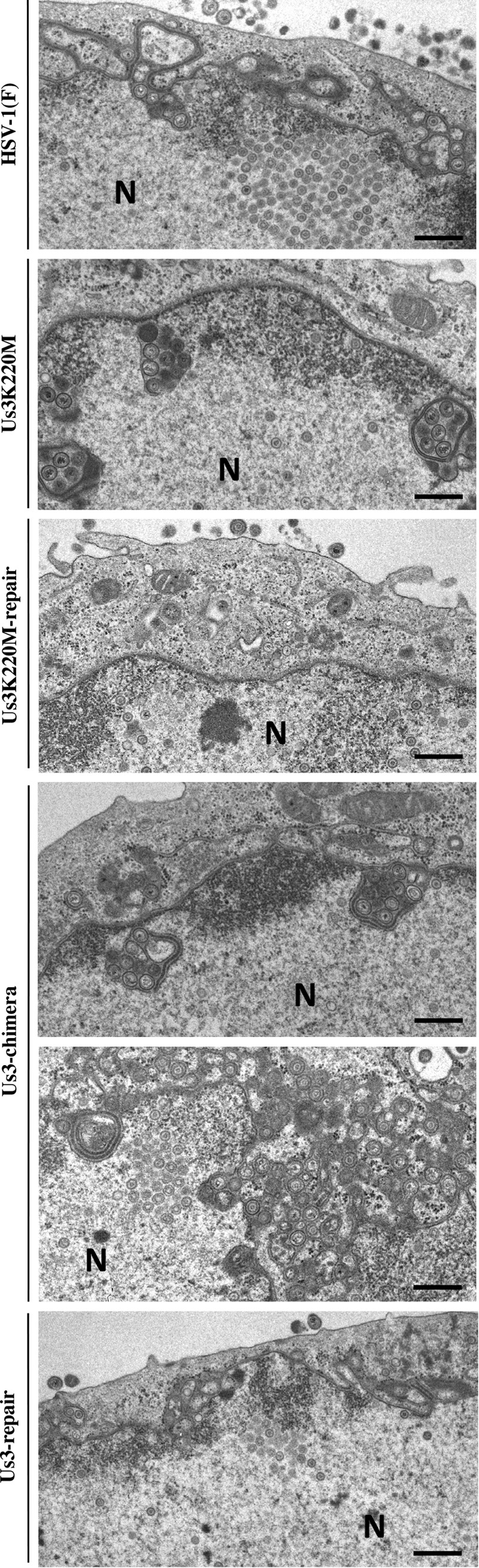
Ultrastructural analysis of the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral nuclear egress in Vero cells. Vero cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 were fixed at 18 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. Bars, 200 nm. N, nucleus.
FIG 12.

Ultrastructural analysis of the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral nuclear egress in HaCaT cells. (A) HaCaT cells infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 were fixed at 18 h postinfection, embedded, sectioned, stained, and examined by transmission electron microscopy. (B) Area of some cells infected with wild-type HSV-1(F), YK513 (Us3K220M-repair), or YK783 (Us3-repair) showing the invagination structures. Bars, 200 nm. N, nucleus.
TABLE 1.
Electron microscopic localization of enveloped virions in infected Vero and HaCaT cells
| Cells | Virus | No. of enveloped virions in compartment (mean ± SE)a |
Total no. of enveloped virions/cells | |||
|---|---|---|---|---|---|---|
| Intranuclear invaginations | Perinuclear space | Cytoplasm | Extracellular | |||
| Vero | HSV-1(F) | 0.8 ± 0.4 (1.3) | 16.4 ± 2.6 (27.2) | 13.6 ± 1.7 (22.6) | 29.3 ± 2.9 (48.6) | 2,716/45 |
| YK511 (Us3K220 M) | 29.6 ± 2.4 (59.9) | 1.9 ± 0.4 (3.9) | 1.3 ± 0.6 (2.7) | 11.5 ± 1.1 (23.2) | 2,222/45 | |
| YK513 (Us3KM-repair) | 1.7 ± 0.7 (4.1) | 9.7 ± 2.1 (23.4) | 7.0 ± 1.4 (16.9) | 22.6 ± 1.9 (54.7) | 1,861/45 | |
| YK781 (Us3-chimera) | 9.7 ± 2.2 (16.9) | 23.4 ± 3.3 (40.7) | 6.9 ± 1.1 (12.1) | 15.6 ± 1.7 (27.1) | 2,586/45 | |
| YK783 (Us3-repair) | 1.0 ± 0.5 (2.3) | 14.6 ± 2.6 (33.8) | 8.0 ± 1.3 (18.5) | 19.3 ± 2.1 (44.8) | 1,936/45 | |
| HaCaT | HSV-1(F) | 12.8 ± 2.2 (9.8) | 10.0 ± 1.0 (7.6) | 18.4 ± 3.1 (14.0) | 87.8 ± 7.1 (67.1) | 5,885/45 |
| YK511 (Us3K220 M) | 46.1 ± 3.5 (52.9) | 1.5 ± 0.3 (1.7) | 2.9 ± 0.6 (3.3) | 28.7 ± 3.1 (33.0) | 3,921/45 | |
| YK513 (Us3KM-repair) | 12.6 ± 2.1 (9.5) | 11.0 ± 1.0 (8.3) | 22.5 ± 3.8 (17.0) | 84.0 ± 7.1 (63.6) | 5,947/45 | |
| YK781 (Us3-chimera) | 42.1 ± 4.9 (35.7) | 8.6 ± 1.0 (7.3) | 7.2 ± 1.3 (6.1) | 54.2 ± 5.1 (46.0) | 5,305/45 | |
| YK783 (Us3-repair) | 18.1 ± 5.0 (13.8) | 11.6 ± 1.7 (8.8) | 21.8 ± 4.2 (16.7) | 77.6 ± 4.8 (59.3) | 5,890/45 | |
Numbers in parentheses are the percentages of enveloped virus particles in compartments.
We noted that the phenotype in viral nuclear egress observed with replacement of HSV-1 Us3 with HSV-2 Us3, as shown above (Fig. 11 and 12), was quite similar to that with mutations that abrogate the expression of both gB and gH, as reported previously (46). Mutations in both gB and gH induced invagination structures containing primary enveloped virions adjacent to the nuclear membrane in infected HaCaT cells, but these mutations induced both invagination and evagination structures in infected Vero cells (46). Therefore, to examine whether replacement of HSV-1 Us3 with HSV-2 Us3 affected expression of gB and gH in infected cells, Vero cells were infected with wild-type HSV-1(F), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 for 18 h and expression of gB and gH was analyzed by immunoblotting. As shown in Fig. 13, cells infected with YK781 (Us3-chimera) produced gB and gH at levels similar to those in Vero cells infected with wild-type HSV-1(F) or YK783 (Us3-repair). Therefore, replacement of HSV-1 Us3 with HSV-2 Us3 did not indirectly affect viral nuclear egress by downregulating expression of gB and gH.
FIG 13.
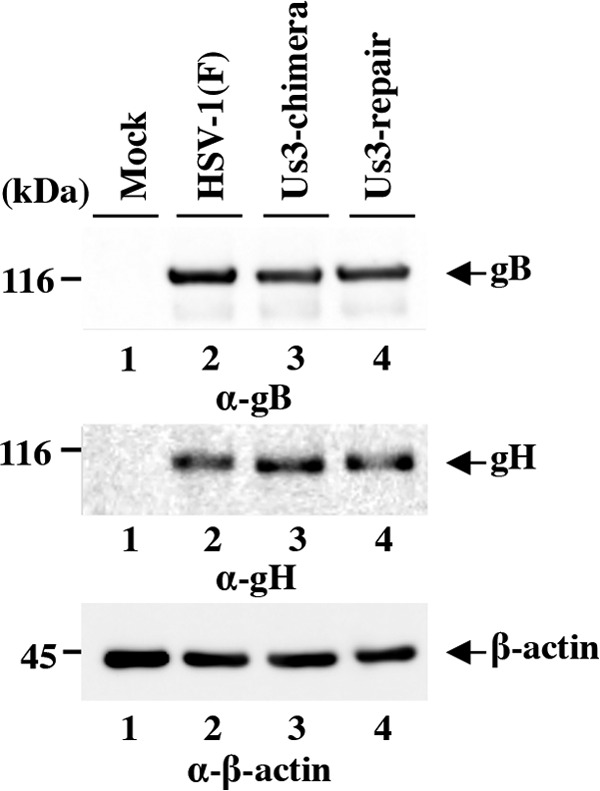
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on expression of gB and gH. Vero cells mock infected or infected with wild-type HSV-1(F), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3 were harvested at 18 h postinfection and analyzed by immunoblotting with antibodies to gB, gH, and β-actin.
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on phosphorylation status of UL31 and UL34.
To investigate whether replacement of HSV-1 Us3 with HSV-2 Us3 affected the phosphorylation status of UL31 and UL34, we used phosphate affinity SDS-polyacrylamide gel electrophoresis (Phos tag SDS-PAGE) analysis, which enables the phosphorylation status of a protein to be visualized as a distinct band shift (40, 71). Vero cells were infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3, harvested at 18 h postinfection, and analyzed on Phos tag(+) SDS-PAGE or Phos tag(−) SDS-PAGE gels. UL31 proteins from Vero cells infected with wild-type HSV-1(F), YK513 (Us3K220M-repair), or YK783 (Us3-repair) were detected as at least eight bands with mobility differences in the Phos tag SDS-PAGE(+) gel (Fig. 14A). After phosphatase treatment of the lysate from wild-type HSV-1(F)-infected cells, at least six of the slower migrating UL31 bands disappeared, two faster-migrating bands remained, and two distinct slower bands appeared in the Phos tag SDS-PAGE(+) gel (Fig. 14B). In YK511 (Us3K220M)-infected Vero cells, the three slowest of the six slower UL31 bands were not present and the amount of protein in the three fastest of the six slower UL31 bands decreased compared to findings with wild-type HSV-1(F)- and YK513 (Us3K220M-repair)-infected cells. These results indicated that the six slower UL31 bands were phosphorylated forms of UL31 with different amounts of phosphorylation; i.e., Us3 regulated phosphorylation of UL31 to produce proteins with different numbers of phosphorylated sites in infected cells. These results were in agreement with previous reports (22, 72) that Us3 phosphorylated UL31 in infected cells and that UL31 contained multiple phosphorylation sites. As shown in Fig. 14A, in cells infected with YK781 (Us3-chimera), the amount of protein in the slowest-migrating UL31 band in the Phos tag SDS-PAGE(+) gel decreased significantly compared to that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair).
FIG 14.
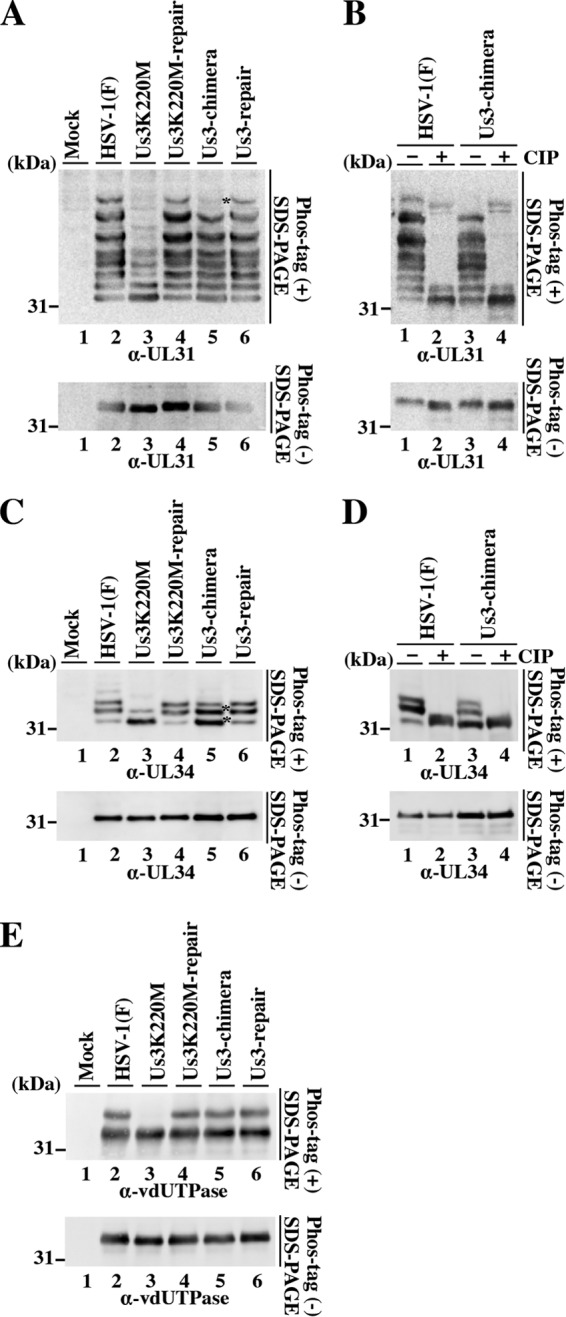
Phosphorylation status of UL31, UL34, and vdUTPase in infected cells. Vero cells were mock infected or infected with wild-type HSV-1(F), YK511 (Us3K220M), YK513 (Us3K220M-repair), YK781 (Us3-chimera), or YK783 (Us3-repair) at an MOI of 3, harvested at 18 h postinfection, and analyzed on a Phos tag(+) SDS-PAGE gel (top) or Pho tag(−) SDS-PAGE gel (bottom) (A to E). The gels were immunoblotted with anti-UL31 (A and B), anti-UL34 (C and D), or anti-vdUTPase (E) antibody. The infected cell lysates were also either left untreated or treated with CIP (B and D). Asterisks indicates bands in which the amount of protein was affected in YK781 (Us3-chimera)-infected cells compared to that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair).
UL34 proteins from Vero cells infected with wild-type HSV-1(F), YK513 (Us3K220M-repair), or YK783 (Us3-repair) were detected as at least three bands with mobility differences in the Phos tag SDS-PAGE(+) gel (Fig. 14C), with the upper and middle bands being the dominant bands (Fig. 14C). After phosphatase treatment, the upper and middle bands were not seen (Fig. 14D). In Vero cells infected with YK511 (Us3K220M), the upper UL34 band was not seen, and relative to the HSV-1(F) Phos tag SDS-PAGE(+) gel pattern, the amount of protein in the middle UL34 band decreased and the amount of protein in the lower UL34 band increased (Fig. 14C, upper image). These results indicated that the two slower-migrating UL34 bands were phosphorylated UL34 proteins, with different amounts of phosphorylation, and the fastest-migrating UL34 band was unphosphorylated UL34. Therefore, Us3 regulated the UL34 phosphorylation status in infected cells. These results were in agreement with previous reports (21, 72, 75) that Us3 phosphorylated UL34 at multiple sites in infected cells. As shown in Fig. 14C, in cells infected with YK781 (Us3-chimera), the amount of protein in the lower and middle UL34 bands in the Phos tag SDS-PAGE(+) gel increased compared to the corresponding UL34 bands from cells infected with wild-type HSV-1(F) or YK783 (Us3-repair). In contrast, the amount of protein in the upper UL34 band from cells infected with YK781 (Us3-chimera) remained relatively constant compared to the corresponding band from cells infected with wild-type HSV-1(F) or YK783 (Us3-repair).
It has been reported that vdUTPase proteins in Vero cells infected with wild-type HSV-1(F) or YK513 (Us3K220M-repair) were detected as two bands with mobility differences in Phos tag SDS-PAGE(+) gels, with the slower-migrating band being vdUTPase phosphorylated by Us3 and the faster-migrating band being unphosphorylated vdUTPase (40). In agreement with these results, the slower-migrating vdUTPase band was not seen in YK511 (Us3K220M)-infected cells (Fig. 14E). In cells infected with YK781 (Us3-chimera), the amount of protein in the two vdUTPase bands in the Phos tag SDS-PAGE(+) gel was similar to that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair) (Fig. 14E), indicating that HSV-2 Us3 efficiently replaced HSV-1 Us3 and phosphorylated vdUTPase in YK781 (Us3-chimera)-infected cells.
Taken together, these results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 specifically changed the phosphorylation status of UL31 and UL34 in infected cells.
Effect of the replacement of HSV-1 Us3 with HSV-2 Us3 on viral growth and pathogenesis in mice.
Us3 has been reported to play critical roles in HSV-1 pathogenesis in experimental animals (27, 70, 76–79). To determine the effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral growth and pathogenesis in vivo, mice were ocularly infected with YK781 (Us3-chimera) and its repaired virus YK783 (Us3-repair) and observed daily for 14 days postinfection for development of herpes stromal keratitis (HSK) and periocular skin disease. In addition, to examine viral growth at the infection site, tear film samples were collected at 1 and 5 days postinfection and viral titers were assayed. As shown in Fig. 15A and B, mice infected with YK781 (Us3-chimera) exhibited significantly reduced severities of HSK and periocular skin disease compared to those of mice infected with YK783 (Us3-repair). In addition, YK781 (Us3-chimera) replicated significantly less efficiently than YK783 (Us3-repair) in the tear films of these infected mice, with titers approximately 3.4- and 16.0-fold lower than those of YK783 (Us3-repair) at 1 and 5 days, respectively (Fig. 15C). These results indicated that replacement of HSV-1 Us3 with HSV-2 Us3 significantly reduced viral growth and pathogenicity in the murine HSK model.
FIG 15.
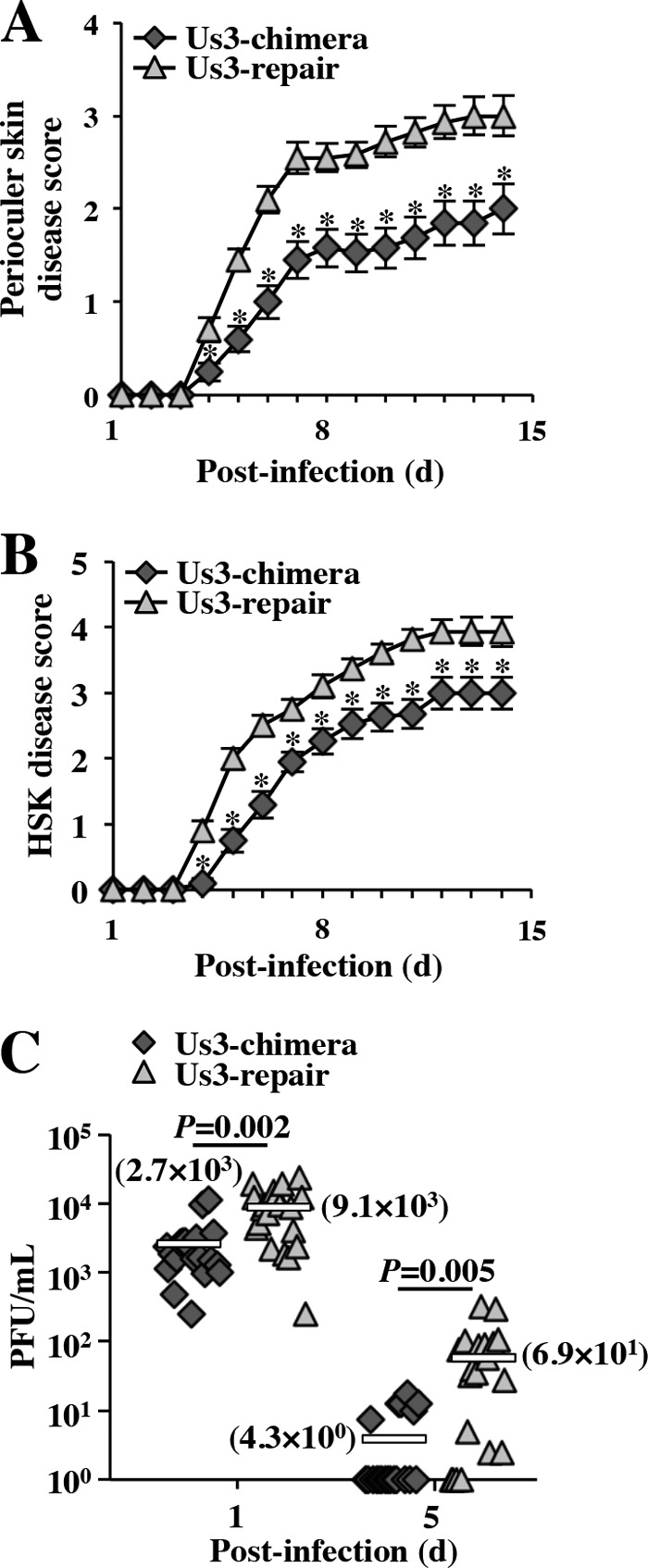
Effect of replacement of HSV-1 Us3 with HSV-2 Us3 on viral pathogenesis and replication in mice following ocular infection. (A and B) Five-week-old female ICR mice were ocularly infected with YK781 (Us3-chimera) or YK783 (Us3-repair) and scored for HSK (A) and periocular skin disease (B) daily for 14 days (d). Each data point is the mean ± standard error of the scores. Asterisks indicate statistically significant values (*, P < 0.05) according to the two-tailed Student t test. (C) Tear films of infected mice at 1 and 5 days postinfection in the experiments shown in panels A and B were harvested, and virus titers were assayed. Each data point is the virus titer in the tear film of one mouse. The horizontal bars mark the means for the groups. P shows statistically significant values according to the two-tailed Student t test.
DISCUSSION
In this study, we generated recombinant viruses YK781 (Us3-chimera) and its repaired virus YK783 (Us3-repair) to compare the activities of HSV-1 Us3 and HSV-2 Us3 in cells infected by viruses with the same viral gene products except for their Us3 kinases. With these recombinant viruses, wild-type HSV-1(F), the HSV-1 Us3 kinase-dead mutant YK511 (Us3K220M), and its repaired virus YK513 (Us3K220M-repair), we investigated whether the phenotypes observed with the HSV-1 Us3 kinase-dead mutant YK511 (Us3K220M) could be compensated for by replacement of HSV-1 Us3 with HSV-2 Us3. The results of this study showed that replacement of HSV-1 Us3 with HSV-2 Us3 was able to almost completely compensate for the protein phosphorylation activity that was lost in the HSV-1 Us3 kinase-dead mutant. This suggested that HSV-2 Us3 in YK781 (Us3-chimera)-infected cells was able to carry out most of the phosphorylation processes catalyzed by HSV-1 Us3 in HSV-1-infected cells. In support of this hypothesis, we showed that replacement of HSV-1 Us3 with HSV-2 Us3 was able to compensate for functional defects due to the HSV-1 Us3 kinase-dead mutation, i.e., defects in (i) viral growth in HaCaT cells, (ii) blockage of apoptosis induced by osmotic shock, (iii) downregulation of cell surface expression of gB, and (iv) induction of infected-cell rounding.
However, we also showed that the defects in viral nuclear egress induced by the HSV-1 Us3 kinase-dead mutation were only partially compensated for by replacement of HSV-1 Us3 with HSV-2 Us3, i.e., defects such as (i) aberrant localization of UL31 and UL34 at the nuclear membrane, (ii) formation of membranous invagination structures containing primary enveloped virions adjacent to the nuclear membrane, and (iii) aberrant accumulation of primary enveloped virions in the perinuclear space and the induced invagination structures. Interestingly, in infected Vero cells, replacement of HSV-1 Us3 with HSV-2 Us3 induced not only invaginations of the INM containing primary enveloped virions into the nucleus but also evaginations of the ONM containing numerous primary enveloped virions into the cytoplasm; these were not observed with the kinase-dead mutation. Furthermore, the effects of replacement of HSV-1 Us3 with HSV-2 Us3 on viral nuclear egress were dependent on cell types. Thus, the evagination structures induced by replacement of HSV-1 Us3 with HSV-2 Us3 in Vero cells were not observed in HaCaT cells, and replacement of HSV-1 Us3 with HSV-2 Us3 tended to accumulate primary enveloped virions in the perinuclear space in Vero cells but in intranuclear invagination structures in HaCaT cells. Theses cell-type-dependent effects on viral nuclear egress might reflect the cell type difference(s) in the structural flexibility of the ONM and/or the INM. These results suggested that HSV-2 Us3 phosphorylated regulatory proteins for HSV-1 nuclear egress differently than those of HSV-1 Us3, resulting in improper functioning of these regulatory proteins. In agreement with this hypothesis, we showed that the pattern of UL31 and UL34 proteins with different numbers of phosphorylated sites in cells infected with YK781 (Us3-chimera) was different from that in cells infected with wild-type HSV-1(F) or YK783 (Us3-repair). We should note that we have no data, at present, on the relationship between the pattern of phosphorylated UL31 and UL34 in YK781 (Us3-chimera)-infected cells and the defects in viral nuclear egress in cells infected with YK781 (Us3-chimera). However, it has been reported that many of the effects of HSV-1 Us3 on viral nuclear egress involved phosphorylation of UL31 and that strict and dynamic regulation of Us3 phosphorylation of UL31 was required for proper HSV-1 nuclear egress (22). Therefore, it seems likely that the changed phosphorylation pattern of UL31, and probably UL34 as well, induced by replacement of HSV-1 Us3 with HSV-2 Us3 may impair the NEC function in HSV-1 nuclear egress. Alternatively, HSV-2 Us3, unlike HSV-1 Us3, may not have been able to mediate proper phosphorylation of other cellular and HSV-1 regulatory proteins for viral nuclear egress. The NEC has recently been reported to form a complex(es) with HSV-1 UL47, Us3, and ICP22 and cellular proteins p32, CD98 heavy chain, and β1 integrin (67, 74, 80, 81), suggesting that the NEC is a high-order complex consisting of these viral and cellular regulatory proteins and probably some not-yet-identified proteins. Among the regulatory proteins, UL47, Us3, and ICP22 are known to be substrates for Us3 (15, 37, 72, 82), and p32 has been reported to contain multiple sites similar to Us3 target consensus sequences (74, 83). Therefore, differences in the phosphorylation of HSV-1 and infected-cell regulatory proteins for viral nuclear egress, due to the replacement of HSV-1 Us3 with HSV-2 Us3, may have impaired the function of the high-order NEC, leading to defects in viral nuclear egress. These observations implied that HSV-1 nuclear egress was tightly and dynamically regulated by the phosphorylated status of the regulatory proteins involved in this process. Thus, although HSV-2 can phosphorylate most of the HSV-1 Us3 substrates as described above, there was a clear biological difference between HSV-1 Us3 and HSV-2 Us3, especially in their effects on specific substrates involved in viral nuclear egress. In agreement with this conclusion, we showed that replacement of HSV-1 Us3 with HSV-2 Us3 significantly reduced viral cell-cell spread in cell cultures and viral growth in the mouse cornea and pathogenic manifestations of HSK and periocular skin disease in mice. These observations not only suggested that HSV-1 Us3 and HSV-2 Us3 phosphorylated Us3 substrates involved in viral cell-cell spread and pathogenesis differently but also indicated that the regulatory role of HSV-1 Us3 in viral nuclear egress, which was missing in HSV-2 Us3, was important for HSV-1 cell-cell spread and pathogenesis. In addition, we cannot exclude the possibility that a currently unidentified HSV-1 Us3 regulatory function(s), other than in viral nuclear egress, is involved in HSV-1 cell-cell spread and pathogenesis but is missing in HSV-2 Us3.
The amino acid sequences of HSV-1 Us3 and HSV-2 Us3 show that their C-terminal domains (amino acids [aa] 152 to 481) containing the kinase domains are well conserved, with 84% sequence identity, but the 151 amino acids of their N-terminal domains are less well conserved, with 54% sequence identity. Therefore, the differences in the activity and/or substrate specificity between HSV-1 Us3 and HSV-2 Us3, shown in this study, may be due to the difference in their N-terminal domains. In support of this hypothesis, it has been reported that the N-terminal domain of HSV-1 Us3 was important for expression of specific HSV-1 Us3 functions (84). It should be noted that the HSV-1 Us3 gene encodes not only full-length Us3 but also Us3.5, a truncated amino-terminal protein that initiates at Us3 codon 77 (Fig. 1) (84). It is interesting that similar to the case with HSV-2 Us3, the range of substrates phosphorylated by HSV-1 Us3.5, detected by the anti-PKA substrate antibody, was reported to be similar to the substrates phosphorylated by HSV-1 Us3, but Us3.5 did not regulate viral nuclear egress and apoptosis (84). These observations suggested that HSV-1 Us3 and Us3.5 phosphorylated regulatory proteins for viral nuclear egress and apoptosis differently, and these differences may have been due to the N-terminal domain of Us3 that is missing in Us3.5.
The results in this study showing that replacement of HSV-1 Us3 with HSV-2 Us3 completely compensated for the defect in downregulation of gB in cells infected by the HSV-1 Us3 kinase-dead mutation indicated that HSV-2 Us3, like HSV-1 Us3, regulated gB intracellular trafficking in HSV-1 (Us3-chimera)-infected cells. However, despite this HSV-2 Us3 activity in HSV-1 (Us3-chimera)-infected cells, HSV-2 Us3 has been reported to have no effect on downregulation of cell surface expression of gB in HSV-2-infected cells, since the Us3 phosphorylation site in HSV-1 gB is critical for gB intracellular trafficking and is not conserved in HSV-2 gB (39). Therefore, evolution of the HSV-1 Us3 phosphorylation site in gB may have been related to regulation of gB intracellular trafficking. These observations raised the interesting possibility that the phosphorylation sites in viral proteins may have evolved to enable viral kinases to acquire new functions in viral protein regulation, and they may provide new insight into the evolution of viral protein kinases and their substrates. In agreement with this possibility, an autophosphorylation site in HSV-1 Us3 which was reported to regulate optimal catalytic activity of HSV-1 Us3 (15, 70) is not conserved in HSV-2 Us3 (15, 27). Since all herpesviruses are known to have at least one protein kinase, this might also be the case in other herpesviruses.
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS), grants for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan, a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases (J-GRID) from MEXT and the Japan Agency for Medical Research and Development (AMED), and grants from the Takeda Science Foundation and the Mitsubishi Foundation.
REFERENCES
- 1.Tuccinardi T, Martinelli A. 2011. Protein kinase homology models: recent developments and results. Curr Med Chem 18:2848–2853. doi: 10.2174/092986711796150441. [DOI] [PubMed] [Google Scholar]
- 2.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002. The protein kinase complement of the human genome. Science 298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 3.Roizman B, Knipe DM, Whitley RJ (ed). 2013. Herpes simplex viruses. Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Jacob T, Van den Broeke C, Favoreel HW. 2011. Viral serine/threonine protein kinases. J Virol 85:1158–1173. doi: 10.1128/JVI.01369-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawaguchi Y, Kato K. 2003. Protein kinases conserved in herpesviruses potentially share a function mimicking the cellular protein kinase cdc2. Rev Med Virol 13:331–340. doi: 10.1002/rmv.402. [DOI] [PubMed] [Google Scholar]
- 6.Frame MC, Purves FC, McGeoch DJ, Marsden HS, Leader DP. 1987. Identification of the herpes simplex virus protein kinase as the product of viral gene US3. J Gen Virol 68(Part 10):2699–2704. [DOI] [PubMed] [Google Scholar]
- 7.McGeoch DJ, Davison AJ. 1986. Alphaherpesviruses possess a gene homologous to the protein kinase gene family of eukaryotes and retroviruses. Nucleic Acids Res 14:1765–1777. doi: 10.1093/nar/14.4.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Purves FC, Longnecker RM, Leader DP, Roizman B. 1987. Herpes simplex virus 1 protein kinase is encoded by open reading frame US3 which is not essential for virus growth in cell culture. J Virol 61:2896–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leader DP. 1993. Viral protein kinases and protein phosphatases. Pharmacol Ther 59:343–389. doi: 10.1016/0163-7258(93)90075-O. [DOI] [PubMed] [Google Scholar]
- 10.Leader DP, Deana AD, Marchiori F, Purves FC, Pinna LA. 1991. Further definition of the substrate specificity of the alpha-herpesvirus protein kinase and comparison with protein kinases A and C. Biochim Biophys Acta 1091:426–431. [DOI] [PubMed] [Google Scholar]
- 11.Purves FC, Deana AD, Marchiori F, Leader DP, Pinna LA. 1986. The substrate specificity of the protein kinase induced in cells infected with herpesviruses: studies with synthetic substrates [corrected] indicate structural requirements distinct from other protein kinases. Biochim Biophys Acta 889:208–215. doi: 10.1016/0167-4889(86)90106-0. [DOI] [PubMed] [Google Scholar]
- 12.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daikoku T, Yamashita Y, Tsurumi T, Maeno K, Nishiyama Y. 1993. Purification and biochemical characterization of the protein kinase encoded by the US3 gene of herpes simplex virus type 2. Virology 197:685–694. doi: 10.1006/viro.1993.1644. [DOI] [PubMed] [Google Scholar]
- 14.Eisfeld AJ, Turse SE, Jackson SA, Lerner EC, Kinchington PR. 2006. Phosphorylation of the varicella-zoster virus (VZV) major transcriptional regulatory protein IE62 by the VZV open reading frame 66 protein kinase. J Virol 80:1710–1723. doi: 10.1128/JVI.80.4.1710-1723.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J Virol 82:6172–6189. doi: 10.1128/JVI.00044-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leopardi R, Van Sant C, Roizman B. 1997. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc Natl Acad Sci U S A 94:7891–7896. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munger J, Chee AV, Roizman B. 2001. The U(S)3 protein kinase blocks apoptosis induced by the d120 mutant of herpes simplex virus 1 at a premitochondrial stage. J Virol 75:5491–5497. doi: 10.1128/JVI.75.12.5491-5497.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munger J, Roizman B. 2001. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc Natl Acad Sci U S A 98:10410–10415. doi: 10.1073/pnas.181344498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogg PD, McDonell PJ, Ryckman BJ, Knudson CM, Roller RJ. 2004. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology 319:212–224. doi: 10.1016/j.virol.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds AE, Wills EG, Roller RJ, Ryckman BJ, Baines JD. 2002. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J Virol 76:8939–8952. doi: 10.1128/JVI.76.17.8939-8952.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryckman BJ, Roller RJ. 2004. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J Virol 78:399–412. doi: 10.1128/JVI.78.1.399-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mou F, Wills E, Baines JD. 2009. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J Virol 83:5181–5191. doi: 10.1128/JVI.00090-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wisner TW, Wright CC, Kato A, Kawaguchi Y, Mou F, Baines JD, Roller RJ, Johnson DC. 2009. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J Virol 83:3115–3126. doi: 10.1128/JVI.01462-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poon AP, Liang Y, Roizman B. 2003. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein no. 0. J Virol 77:12671–12678. doi: 10.1128/JVI.77.23.12671-12678.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Poon AP, Gu H, Roizman B. 2006. ICP0 and the US3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc Natl Acad Sci U S A 103:9993–9998. doi: 10.1073/pnas.0604142103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walters MS, Kinchington PR, Banfield BW, Silverstein S. 2010. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J Virol 84:9666–9676. doi: 10.1128/JVI.00981-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morimoto T, Arii J, Tanaka M, Sata T, Akashi H, Yamada M, Nishiyama Y, Uema M, Kawaguchi Y. 2009. Differences in the regulatory and functional effects of the Us3 protein kinase activities of herpes simplex virus 1 and 2. J Virol 83:11624–11634. doi: 10.1128/JVI.00993-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cartier A, Komai T, Masucci MG. 2003. The Us3 protein kinase of herpes simplex virus 1 blocks apoptosis and induces phosphorylation of the Bcl-2 family member Bad. Exp Cell Res 291:242–250. doi: 10.1016/S0014-4827(03)00375-6. [DOI] [PubMed] [Google Scholar]
- 29.Cartier A, Masucci MG. 2004. Differential regulation of MHC class-I-restricted and unrestricted cytotoxicity by the Us3 protein kinase of herpes simplex virus-1. Scand J Immunol 60:592–599. doi: 10.1111/j.0300-9475.2004.01523.x. [DOI] [PubMed] [Google Scholar]
- 30.Sloan DD, Zahariadis G, Posavad CM, Pate NT, Kussick SJ, Jerome KR. 2003. CTL are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J Immunol 171:6733–6741. doi: 10.4049/jimmunol.171.12.6733. [DOI] [PubMed] [Google Scholar]
- 31.Rao P, Pham HT, Kulkarni A, Yang Y, Liu X, Knipe DM, Cresswell P, Yuan W. 2011. Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J Virol 85:8093–8104. doi: 10.1128/JVI.02689-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai T, Koyanagi N, Ogawa R, Shindo K, Suenaga T, Sato A, Arii J, Kato A, Kiyono H, Arase H, Kawaguchi Y. 2013. Us3 kinase encoded by herpes simplex virus 1 mediates downregulation of cell surface major histocompatibility complex class I and evasion of CD8+ T cells. PLoS One 8:e72050. doi: 10.1371/journal.pone.0072050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xiong R, Rao P, Kim S, Li M, Wen X, Yuan W. 2015. Herpes simplex virus 1 US3 phosphorylates cellular KIF3A to downregulate CD1d expression. J Virol 89:6646–6655. doi: 10.1128/JVI.00214-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Wang K, Lin R, Zheng C. 2013. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J Virol 87:12814–12827. doi: 10.1128/JVI.02355-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalamvoki M, Roizman B. 2014. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc Natl Acad Sci U S A 111:E611–E617. doi: 10.1073/pnas.1323414111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85:9599–9613. doi: 10.1128/JVI.00845-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Imai T, Arii J, Minowa A, Kakimoto A, Koyanagi N, Kato A, Kawaguchi Y. 2011. Role of the herpes simplex virus 1 Us3 kinase phosphorylation site and endocytosis motifs in the intracellular transport and neurovirulence of envelope glycoprotein B J Virol 85:5003–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J Virol 83:250–261. doi: 10.1128/JVI.01451-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kato A, Tsuda S, Liu Z, Kozuka-Hata H, Oyama M, Kawaguchi Y. 2014. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral dUTPase and regulates its catalytic activity in infected cells. J Virol 88:655–666. doi: 10.1128/JVI.02710-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mettenleiter TC, Muller F, Granzow H, Klupp BG. 2013. The way out: what we know and do not know about herpesvirus nuclear egress. Cell Microbiol 15:170–178. doi: 10.1111/cmi.12044. [DOI] [PubMed] [Google Scholar]
- 42.Johnson DC, Baines JD. 2011. Herpesviruses remodel host membranes for virus egress. Nat Rev Microbiol 9:382–394. doi: 10.1038/nrmicro2559. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds AE, Ryckman BJ, Baines JD, Zhou Y, Liang L, Roller RJ. 2001. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J Virol 75:8803–8817. doi: 10.1128/JVI.75.18.8803-8817.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roller RJ, Zhou Y, Schnetzer R, Ferguson J, DeSalvo D. 2000. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J Virol 74:117–129. doi: 10.1128/JVI.74.1.117-129.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bigalke JM, Heuser T, Nicastro D, Heldwein EE. 2014. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat Commun 5:4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farnsworth A, Wisner TW, Webb M, Roller R, Cohen G, Eisenberg R, Johnson DC. 2007. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc Natl Acad Sci U S A 104:10187–10192. doi: 10.1073/pnas.0703790104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mou F, Forest T, Baines JD. 2007. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J Virol 81:6459–6470. doi: 10.1128/JVI.00380-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mou F, Wills EG, Park R, Baines JD. 2008. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J Virol 82:8094–8104. doi: 10.1128/JVI.00874-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Park R, Baines JD. 2006. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J Virol 80:494–504. doi: 10.1128/JVI.80.1.494-504.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Muranyi W, Haas J, Wagner M, Krohne G, Koszinowski UH. 2002. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 297:854–857. doi: 10.1126/science.1071506. [DOI] [PubMed] [Google Scholar]
- 51.Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek MC, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog 5:e1000275. doi: 10.1371/journal.ppat.1000275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Asano S, Honda T, Goshima F, Watanabe D, Miyake Y, Sugiura Y, Nishiyama Y. 1999. US3 protein kinase of herpes simplex virus type 2 plays a role in protecting corneal epithelial cells from apoptosis in infected mice. J Gen Virol 80(Part 1):51–56. [DOI] [PubMed] [Google Scholar]
- 53.Matsuzaki A, Yamauchi Y, Kato A, Goshima F, Kawaguchi Y, Yoshikawa T, Nishiyama Y. 2005. US3 protein kinase of herpes simplex virus type 2 is required for the stability of the UL46-encoded tegument protein and its association with virus particles. J Gen Virol 86:1979–1985. doi: 10.1099/vir.0.80949-0. [DOI] [PubMed] [Google Scholar]
- 54.Eaton HE, Saffran HA, Wu FW, Quach K, Smiley JR. 2014. Herpes simplex virus protein kinases US3 and UL13 modulate VP11/12 phosphorylation, virion packaging, and phosphatidylinositol 3-kinase/Akt signaling activity. J Virol 88:7379–7388. doi: 10.1128/JVI.00712-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391. doi: 10.1128/JVI.77.2.1382-1391.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kato A, Yamamoto M, Ohno T, Tanaka M, Sata T, Nishiyama Y, Kawaguchi Y. 2006. Herpes simplex virus 1-encoded protein kinase UL13 phosphorylates viral Us3 protein kinase and regulates nuclear localization of viral envelopment factors UL34 and UL31. J Virol 80:1476–1486. doi: 10.1128/JVI.80.3.1476-1486.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. 1988. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J Cell Biol 106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ejercito PM, Kieff ED, Roizman B. 1968. Characterization of herpes simplex virus strains differing in their effects on social behaviour of infected cells. J Gen Virol 2:357–364. doi: 10.1099/0022-1317-2-3-357. [DOI] [PubMed] [Google Scholar]
- 59.Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 60.Tanaka Y, Kanai F, Ichimura T, Tateishi K, Asaoka Y, Guleng B, Jazag A, Ohta M, Imamura J, Ikenoue T, Ijichi H, Kawabe T, Isobe T, Omata M. 2006. The hepatitis B virus X protein enhances AP-1 activation through interaction with Jab1. Oncogene 25:633–642. [DOI] [PubMed] [Google Scholar]
- 61.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 62.Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J Virol 82:5198–5211. doi: 10.1128/JVI.02681-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J Virol 71:7328–7336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fujii H, Kato A, Mugitani M, Kashima Y, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 2014. The UL12 protein of herpes simplex virus 1 is regulated by tyrosine phosphorylation. J Virol 88:10624–10634. doi: 10.1128/JVI.01634-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang YM, Yamada H, Goshima F, Daikoku T, Oshima S, Wada K, Nishiyama Y. 1998. Characterization of the herpes simplex virus type 2 (HSV-2) US2 gene product and a US2-deficient HSV-2 mutant. J Gen Virol 79(Part 11):2777–2784. [DOI] [PubMed] [Google Scholar]
- 66.Fujii H, Mugitani M, Koyanagi N, Liu Z, Tsuda S, Arii J, Kato A, Kawaguchi Y. 2014. Role of the nuclease activities encoded by herpes simplex virus 1 UL12 in viral replication and neurovirulence. J Virol 88:2359–2364. doi: 10.1128/JVI.03621-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu Z, Kato A, Shindo K, Noda T, Sagara H, Kawaoka Y, Arii J, Kawaguchi Y. 2014. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J Virol 88:4657–4667. doi: 10.1128/JVI.00137-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shiba C, Daikoku T, Goshima F, Takakuwa H, Yamauchi Y, Koiwai O, Nishiyama Y. 2000. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J Gen Virol 81:2397–2405. doi: 10.1099/0022-1317-81-10-2397. [DOI] [PubMed] [Google Scholar]
- 69.Imai T, Sagou K, Arii J, Kawaguchi Y. 2010. Effects of phosphorylation of herpes simplex virus 1 envelope glycoprotein B by Us3 kinase in vivo and in vitro. J Virol 84:153–162. doi: 10.1128/JVI.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sagou K, Imai T, Sagara H, Uema M, Kawaguchi Y. 2009. Regulation of the catalytic activity of herpes simplex virus 1 protein kinase Us3 by autophosphorylation and its role in pathogenesis. J Virol 83:5773–5783. doi: 10.1128/JVI.00103-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. 2006. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 5:749–757. [DOI] [PubMed] [Google Scholar]
- 72.Kato A, Yamamoto M, Ohno T, Kodaira H, Nishiyama Y, Kawaguchi Y. 2005. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J Virol 79:9325–9331. doi: 10.1128/JVI.79.14.9325-9331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Finnen RL, Roy BB, Zhang H, Banfield BW. 2010. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 397:23–33. doi: 10.1016/j.virol.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Z, Kato A, Oyama M, Kozuka-Hata H, Arii J, Kawaguchi Y. 17 June 2015. Role of host cell p32 in herpes simplex virus 1 de-envelopment during viral nuclear egress. J Virol doi: 10.1128/JVI.01220-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Purves FC, Spector D, Roizman B. 1991. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J Virol 65:5757–5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kato A, Shindo K, Maruzuru Y, Kawaguchi Y. 18 December 2013. Phosphorylation of a herpes simplex virus 1 dUTPase by a viral protein kinase Us3 dictates viral pathogenicity in the central nervous system but not at the periphery. J Virol doi: 10.1128/JVI.03300-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koyanagi N, Imai T, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 Us3 in viral neuroinvasiveness. Microbiol Immunol 58:31–37. doi: 10.1111/1348-0421.12108. [DOI] [PubMed] [Google Scholar]
- 78.Nishiyama Y, Yamada Y, Kurachi R, Daikoku T. 1992. Construction of a US3 lacZ insertion mutant of herpes simplex virus type 2 and characterization of its phenotype in vitro and in vivo. Virology 190:256–268. doi: 10.1016/0042-6822(92)91212-D. [DOI] [PubMed] [Google Scholar]
- 79.Meignier B, Longnecker R, Mavromara-Nazos P, Sears AE, Roizman B. 1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251–254. doi: 10.1016/0042-6822(88)90417-5. [DOI] [PubMed] [Google Scholar]
- 80.Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88:7445–7454. doi: 10.1128/JVI.01057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirohata Y, Arii J, Liu Z, Shindo K, Oyama M, Kozuka-Hata H, Sagara H, Kato A, Kawaguchi Y. 2015. Herpes simplex virus 1 recruits CD98 heavy chain and beta1 integrin to the nuclear membrane for viral de-envelopment. J Virol 89:7799–7812. doi: 10.1128/JVI.00741-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Purves FC, Roizman B. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc Natl Acad Sci U S A 89:7310–7314. doi: 10.1073/pnas.89.16.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hall KT, Giles MS, Calderwood MA, Goodwin DJ, Matthews DA, Whitehouse A. 2002. The herpesvirus saimiri open reading frame 73 gene product interacts with the cellular protein p32. J Virol 76:11612–11622. doi: 10.1128/JVI.76.22.11612-11622.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Poon AP, Benetti L, Roizman B. 2006. U(S)3 and U(S)3.5 protein kinases of herpes simplex virus 1 differ with respect to their functions in blocking apoptosis and in virion maturation and egress. J Virol 80:3752–3764. doi: 10.1128/JVI.80.8.3752-3764.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]