

ABSTRACT
Epstein-Barr virus (EBV) nuclear antigen 1 (EBNA1) is the EBV-encoded nuclear antigen and sequence-specific DNA binding protein required for viral origin binding and episome maintenance during latency. EBNA1 can also bind to numerous sites in the cellular genome and can provide a host cell survival function, but it is not yet known how EBNA1 sequence-specific binding is responsible for host cell survival. Here, we integrate EBNA1 chromatin immunoprecipitation sequencing (ChIP-Seq) with transcriptome sequencing (RNA-Seq) after EBNA1 depletion to identify cellular genes directly regulated by EBNA1 that are also essential for B-cell survival. We first compared EBNA1 ChIP-Seq patterns in four different EBV-positive cell types, including Burkitt lymphoma (BL) cells, nasopharyngeal carcinoma (NPC) cells, and lymphoblastoid cell lines (LCLs). EBNA1 binds to ∼1,000 sites that are mostly invariant among cell types and share a consensus recognition motif. We found that a large subset of EBNA1 binding sites are located proximal to transcription start sites and correlate genome-wide with transcription activity. EBNA1 bound to genes of high significance for B-cell growth and function, including MEF2B, IL6R, and EBF1. EBNA1 depletion from latently infected LCLs results in the loss of cell proliferation and the loss of gene expression for some EBNA1-bound genes, including MEF2B, EBF1, and IL6R. Depletion of MEF2B, EBF1, or IL6R partially phenocopies EBNA1 depletion by decreasing the cell growth and viability of cells latently infected with EBV. These findings suggest that EBNA1 binds to a large cohort of cellular genes important for cell viability and implicates EBNA1 as a critical regulator of transcription of host cell genes important for enhanced survival of latently infected cells.
IMPORTANCE Epstein-Barr virus (EBV) latent infection is responsible for a variety of lymphoid and epithelial cell malignancies. EBNA1 is the EBV-encoded nuclear antigen that is consistently expressed in all EBV-associated cancers. EBNA1 is known to provide a host cell survival function, but the mechanism is not known. EBNA1 is a sequence-specific binding protein important for viral genome maintenance during latency. Here, by integrating ChIP-Seq and RNA-Seq, we demonstrate that EBNA1 binds directly to the promoter regulatory regions and upregulates the transcription of host genes that are important for the survival of EBV-infected cells. Identification of EBNA1 target genes provides potential new targets for therapeutic intervention in EBV-associated disease.
INTRODUCTION
Epstein-Barr virus (EBV) is clinically associated with a diverse set of lymphoid and epithelial cell malignancies, including Burkitt lymphoma (BL), nasopharyngeal carcinoma (NPC), and lymphoproliferative diseases of immunosuppressed individuals (1–3). In almost all EBV-associated cancers, the viral genome persists as a nuclear, chromatin-associated episome that expresses only a small subset of viral genes and rarely reactivates to produce infectious viral particles (3, 4). This form of persistent infection, referred to as EBV latency, is capable of transforming the growth, differentiation, and survival properties of the host cell. Several EBV latency genes have been implicated in the carcinogenic transformation of host cells, suggesting that the mechanisms of EBV-induced cancer are diverse and complex (5–7).
EBV encodes several nuclear antigens that have well-established roles in transcription regulation and B-cell proliferation. EBV nuclear antigen 2 (EBNA2) binds to the host cell transcription factors (TFs) RBPJ/CBF1 and PU.1 to activate the transcription of viral and host target genes involved in B-cell proliferation (8). EBNA3A and EBNA3C can also bind to RBPJ/CBF1 to recruit epigenetic modifiers, including members of the polycomb family of transcriptional repressors (9–11). EBNA3C has been implicated in the epigenetic silencing of proapoptotic and tumor suppressor genes and thus promotes host cell survival and proliferation. Genome-wide chromatin immunoprecipitation (ChIP) studies have revealed the EBNA2 and EBNA-LP cobind to many promoters and enhancers of host genes important for B-cell activation (12). Similarly, EBNA3C can colocalize with BATF/IRF4 or SPI1/IRF4 composite sites to recruit the corepressor SIN3A (13). While these EBV-encoded nuclear antigens account for much of the transcriptional reprogramming of host cells by EBV during B-cell primary infection of naive B cells, they are often not expressed in EBV-associated cancers such as Burkitt lymphoma, Hodgkin's lymphoma, or epithelial tumors like NPC.
In contrast to these viral nuclear antigens, EBNA1 has a primary role in maintaining the EBV genome during latent infection (14, 15). EBNA1 is also unique among the nuclear antigens for its consistent expression in cells of all EBV-associated tumors and latency types. EBNA1 is a sequence-specific DNA binding protein that binds with high affinity to three well-characterized sites in the viral genome that are important for DNA replication, episome maintenance, and viral gene regulation. EBNA1 tethers viral episomes to host chromosomes through its amino-terminal AT hook domain, which makes nonspecific contacts with host DNA and RNA (16, 17). EBNA1 can also interact with numerous sequence-specific host chromosome sites through its C-terminal DNA binding domain (18–21). To date, studies of these sequence-specific EBNA1 binding sites have failed to identify host gene targets essential for EBV infection or B-cell immortalization.
EBNA1 is also known to provide a host cell survival function that is distinct from its control of viral episome maintenance or viral gene regulation (22). EBNA1 can interact with cellular proteins, including HAUSP7, EBP2, CKII, and PML-associated proteins, that may contribute to its host cell survival function (23). The EBNA1 AT hook chromatin binding domain can alter host global chromatin structure and gene expression, which may also promote host cell survival (24). However, the sequence-specific DNA binding domain has been implicated in promoting host cell survival (22). While EBNA1 is known to bind with sequence specificity to many host genome locations, it is not yet known whether any of these sites regulate cellular genes important for host cell survival. Here, we integrate ChIP sequencing (ChIP-Seq) and transcriptome sequencing (RNA-Seq) with functional studies to identify EBNA1 binding sites that function in the regulation of host genes important for B-cell growth and survival.
MATERIALS AND METHODS
Cells.
Mutu I is an EBV-positive BL cell line with type I latency (gift of J. Sample, Penn State Hershey Medical School). Raji is an EBV-positive BL cell line with irregular type III latency (obtained from the ATCC). C666-1 is an EBV-positive NPC cell line with type II latency (gift of K. K. W. Lo, Chinese University of Hong Kong). The lymphoblastoid cell line (LCL) is an EBV-immortalized lymphoblastoid cell line with the Mutu I virus strain (also referred to as Mutu-LCL). HONE-1 and HK1 are EBV-negative NPC cell lines (obtained from the ATCC). BJAB and DG75 are EBV-negative B-cell lymphoma lines (obtained from the ATCC). All B-cell lines and C666-1 were grown in RPMI medium with 10% fetal bovine serum (FBS) and Glutamax (Life Technologies). All NPC lines were grown in Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and Glutamax.
ChIP assays.
ChIP-Seq and ChIP-quantitative PCR (qPCR) were described previously (19).
Antibodies.
EBNA1 antibody for ChIP-Seq was generated in rabbits immunized with bacterially expressed EBNA1ΔGA (Pocono Rabbit Farms) and then affinity purified with the same antigen. Alternatively, EBNA1 monoclonal antibodies 0211 (AbD Serotec) and MAB8173 (Millipore) were used for Western blot and some ChIP-qPCR analyses, respectively. Interleukin-6 (IL-6) receptor (IL-6R) (catalog number 041581; Millipore), EBF1 (catalog number 041581; Millipore), EBNA2 (MABE8; Millipore), LMP1 (catalog number ETU001; Kerafast), FLAG (catalog number A8592; Sigma), actin (catalog number A3854; Sigma), and in-house-generated anti-rabbit Zta and anti-rabbit BALF2 antibodies were used for Western blot analysis.
ChIP-Seq data analysis. (i) EBNA1 ChIP-Seq.
Data from EBNA1 ChIP-Seq and IgG experiments for 4 cell lines were aligned to the hg19 human genome by using the Bowtie algorithm (25), and all of the redundant tags were removed before downstream analysis. Peak calling was performed by using the Homer algorithm (26) with combined IgG as a reference set. All peaks with false discovery rates (FDRs) of <1% for 4 cell lines were overlapped to create a list of unique binding sites, and only sites with an EBNA1 peak signal of at least 15 RPM (reads per million) in one cell line and at least 5-fold over the IgG and local background signals were considered significant for further analysis. De novo motif discovery on 200-bp sites with significant EBNA1 peaks was performed by using the Homer algorithm (26).
(ii) ENCODE.
The ENCODE database (ENCODE Project Consortium; 46) was used to download ChIP-Seq data available for all transcription factors and histone modification marks in an LCL (GM12878). A list of significant peaks and BigWig tracks provided by the ENCODE database were used.
(a) ChIP-Seq heat maps. All ChIP-Seq heat maps were plotted, with signals of 0 RPM shown in gray, 5 RPM shown in white, and ≥15 RPM shown in bright colors and with intermediate signals shown as gradients of these colors.
(b) Epigenetic heat map. BigWig tracks were used to cluster and visualize the signal distributions of histone modification marks and other epigenetically important factors by using a 10-bp size window spanning positions −1 kb to 1 kb from the EBNA1 site center. Hierarchical clustering of the data was done by using correlation distances and average linkages for both sites and histone modification marks/transcription factors. Genes associated with 3 cohorts were assigned based on 100 kb from the transcription start site (TSS) cutoff. The RNA-Seq expression distribution is shown as medians, 25th/75th percentiles are shown as boxes, and 5th/95th percentiles are shown as whiskers. DAVID (27) was used for enrichment analysis of genes from 2 cohorts.
(c) TF heat map. An EBNA1 site that had a significant transcription factor within 100 bp from its center was called as cooccupied by the TF. Hierarchical clustering of cooccupied transcription factors and sites was done by using the Jaccard distance (1 minus the percentage of nonzero coordinates that differ) with complete linkage.
RNA-Seq data analysis.
RNA-Seq data were aligned by using the Bowtie algorithm, and RSEM v1.2.12 software (28) was used to estimate read counts and reads per kilobase per million (RPKM) values on the gene and exon levels by using the gene table for the hg19 genome downloaded from the UCSC browser (hg9.knownGene and hg9.knownIsoforms tables [http://genome.ucsc.edu/]). EdgeR (29) was used to estimate the significance of differential expression differences between two experimental groups. Overall gene expression level changes were considered significant if they passed thresholds of an FDR of <10% and a fold change of >2. Genes that had an EBNA1 peak within 3 kb from the TSS and that had an FDR of <10% were considered significantly directly changed by EBNA1. The significance of EBNA1 binding and changes in gene expression levels was tested by using a Fisher exact test comparing numbers/percentages of EBNA1-occupied genes that showed expression changes to those of EBNA1-occupied genes that did not show expression changes. Gene set enrichment analysis was done by using Ingenuity Pathway Analysis (IPA) software (Qiagen, Redwood City, CA), using the “Functions” and “Upstream Regulators” options. Functions that passed a threshold of a P value of <10−5 and upstream regulators that passed a threshold of a P value of <10−5 and that had a significantly predicted activation state (|Z| of >2, where |Z| is the absolute value of the Z-score calculated by IPA for prediction of the activation state) were considered.
Lentivirus transduction.
Short hairpin RNAs (shRNA) were expressed in the pLKO.1 vector obtained from Open Biosystems (catalog number TRCN0000013831). The control shRNA (shControl) was generated in the pLKO.1 vector with the target sequence 5′-TTATCGCGCATATCACGCG-3′. All shRNA targeting sequences are provided in Table S1 in the supplemental material. Lentiviruses were produced by cotransfecting the pLKO.1 shRNA expression plasmid packaging vectors pMD2.G and pSPAX2 as described previously (30). Cells in suspension were infected with lentiviruses carrying pLKO.1-puro vectors by spin infection at 450 × g for 90 min at room temperature. The cell pellets were resuspended and incubated in fresh RPMI medium and then treated with 2.5 μg/ml puromycin 48 h after infection.
Cell viability assay.
Forty-eight hours after lentivirus infection, a 96-well assay plate was set up with 104 cells in 100 μl complete RPMI medium with 2.5 μg/ml puromycin in each well. The transduced cells were cultured for 5 days, 10 μl of a 0.5 mM resazurin (Sigma) solution was mixed in each well, and the wells were incubated at 37°C for 3 to 4 h before the fluorescence at 560 nm excitation and 590 nm emission (560/590 nm) was read by using an Envision plate reader.
EdU incorporation assay.
C666-1 cells were infected with shRNA lentivirus for knockdown of EBNA1 for 2 days and then subjected to puromycin selection for an additional 3 days. Following selection, 1 × 103 cells were seeded into a 96-well culture plate in 100 μl RPMI medium with 10% FBS and appropriate antibiotics, and culturing was then continued for 24 h. On the next day, a final concentration of 10 μM ethynyl deoxyuridine (EdU) was added to the cells, and the cells were maintained in EdU-containing medium and cultured for an additional 24 h before harvest. Following treatment, the cells were fixed and then stained with Oregon Green*488 azide by using a Click-iT EdU microplate assay kit according to the manufacturer's instructions (catalog number C10214; Invitrogen). The images were acquired by using an Operetta high-content imaging system and analyzed with Harmony high-content analysis software according to the manufacturer's instructions (PerkinElmer).
RESULTS
Invariant EBNA1 binding sites in virus and host genomes.
We performed ChIP-Seq studies of EBNA1 in four different cell lines with distinct EBV latency types. We compared the ChIP-Seq profiles for LCLs (latency type III), Mutu I cells (BL) (latency type I), Raji cells (BL) (latency type III with Wp initiation), and C666-1 cells (NPC) (latency type II). We found that EBNA1 binds to the three major binding sites in the viral genome, namely, FR (family of repeats), DS (dyad symmetry), and Qp (Q promoter), indistinguishably in cells of all latency types tested (Fig. 1A; see also Fig. S1 in the supplemental material). Although EBNA1 is thought to function as a transcriptional repressor of Qp in type III latency, we found that EBNA1 is equally enriched at Qp in cells of all latency types. This suggests that EBNA1 has other functions at Qp and that additional factors modulate transcription initiation at Qp. We also observed some differences in the relative ratios of EBNA1 binding to the FR and DS regions of OriP (see Fig. S1 in the supplemental material), but the significance of this is not yet clear.
FIG 1.

EBNA1 binding sites at host cell gene promoters and upstream regulatory regions. (A) EBNA1 ChIP-Seq peaks for Raji cells, Mutu I cells, LCLs, and C666-1 cells mapped to the EBV genome. (B and C) EBNA1 ChIP-Seq peaks for Raji cells, Mutu I cells, LCLs, and C666-1 cells mapped near TSSs of the IL6R and MEF2B genes of the human genome. (D) Distribution of EBNA1 peak signals around the human genome transcription start sites (left) and transcription end sites (TES) (right). (E) Overlap of significant EBNA1 peaks among 4 cell lines. (F) Heat map of EBNA1 peak signals and the combined IgG controls from ChIP-Seq experiments for C666-1 cells (C), LCLs (L), Mutu I cells (M), and Raji cells (R). Each column represents 1,000 bp (10-bp steps) surrounding the EBNA1 binding site in 4 cell lines and the combined IgG signal. The R/M/L/C groups on the left indicate the presence of an EBNA1 peak in the corresponding cell line. (G) Percentage of EBNA1 peaks shared between pairs of cell lines. (H) Hierarchical clustering of cell lines based on the percentage of differentially occupied EBNA1 sites between them. (I) EBNA1 DNA binding motifs discovered by de novo motif analysis of EBNA1 binding sites in 4 cell lines. Numbers indicate percentages of sites with a significant EBNA1 peak that had the binding motif. (J) Western blot analysis of EBNA1 (top) or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (bottom) in BJAB, Mutu I, C666-1, LCL, and Raji cell extracts.
Global analysis of genome-wide binding sites for EBNA1 revealed an average of ∼1,000 significant cellular EBNA1 binding sites in each cell line tested. Many of these sites were common to all four cell types, but some cell type-specific enrichments were observed. Specific examples of EBNA1 ChIP-Seq peaks are shown for the IL6R (Fig. 1B) and MEF2B (Fig. 1C) genes as well as for the RNF145/EBF1, CDC7, KDM4c, and LAIR1 genes (see Fig. S2 in the supplemental material). Genome-wide analysis revealed that EBNA1 binding sites tend to localize to transcription start sites of annotated genes, with the average signal being similar to the background signal at a distance ∼3 kb from the TSS (Fig. 1D). The overlap between the ChIP-Seq data sets (Fig. 1E) suggests that the majority of sites are invariant for all cell types but that there are some cell-specific variations, as further demonstrated by the heat map of the EBNA1 binding signal across the 4 cell types (Fig. 1F). Mutu I cells showed the largest number of strongly occupied sites, with at least 93 to 95% of EBNA1 binding peaks in other cell lines also being present in Mutu I cells (Fig. 1G). Clustering analysis of cell types based on differences in EBNA1 binding shows a common pattern among the BL cell lines Mutu I and Raji, with a more distant overlap for LCLs and even further differences with the NPC cell line C666-1 (Fig. 1H). A de novo motif analysis revealed nearly identical EBNA1 consensus sites in all cell types for the overwhelming majority of binding sites (Fig. 1I). This consensus closely matches that for known EBNA1 binding sites in the EBV genome, suggesting that the majority of cellular sites are bound directly through the EBNA1 DNA binding domain. Western blot analysis of the four cell types indicated that EBNA1 proteins have some variation in molecular mass due to different numbers of Gly-Ala repeats in the central domain as well as some variation in expression levels possibly correlating with different virus copy numbers (Fig. 1J). As expected, LCLs and Raji cells express the type III proteins EBNA2 and LMP1. Taken together, these findings indicate that EBNA1 interacts with host chromosomes near gene start sites through highly specific sequence recognition similar to that on the EBV genome but that cell type variations may limit the access of EBNA1 to some of these potential binding sites.
Diverse environments of EBNA1 binding sites.
EBNA1 binding sites can be clustered into three major categories based on the site's epigenetic context, as determined from the ENCODE ChIP-Seq data for EBV-positive LCLs (Fig. 2A). The first cohort of EBNA1 sites colocalizes with histone modifications associated with active transcription (Fig. 2A, purple), including H3K4me3, H3K9ac, and the histone variant H2AZ. A second cohort of EBNA1-bound sites is enriched with H3K27me3 and H3K9me3 (Fig. 2A, yellow), usually associated with repressed transcription. A third, smaller category of EBNA1 sites colocalizes with H3K36me3 (Fig. 2A, gray), also a marker of active transcription. Indeed, genes near the sites from cohorts 1 and 3 showed a tendency to be expressed in LCLs at higher levels than genes from cohort 2 (Fig. 2B). DAVID analysis of genes associated with cohorts 1 and 2 revealed enrichments of diverse cellular functions (Fig. 2C) (27). Transcriptionally active genes were associated with the functions “response to unfolded protein” and “response to nutrient,” while repressed genes associated with cohort 2 have functions linked to “organ morphogenesis” and “system development.” These findings indicate that EBNA1 binding sites colocalize with histone modifications associated primarily with active transcription and genes associated with the environmental stress response.
FIG 2.

EBNA1 binding sites cluster with epigenetic marks for active or repressed gene transcription in LCLs. (A) Heat map showing correlations between EBNA1 peaks and various histone modification marks and other factors that affect transcription. Clustering of the epigenetic profiles revealed that EBNA1 peaks fell into three distinct cluster groups associated with active and repressed transcription. (B) Averages and distributions of expression values for genes linked to EBNA1 peaks from the 3 epigenetic clusters. FPKM, fragments per kilobase per million. (C) Biological processes enriched among genes linked to EBNA1 peaks from activation and repression epigenetic clusters. (D) List of 23 transcription factors (TF) with peaks that overlapped EBNA1 peaks in at least 10% of EBNA1 sites from cluster 1 associated with active transcription. Columns represent EBNA1 sites, and each row represents a TF, with orange indicating sites cobound by EBNA1 and the TF.
We also assessed whether EBNA1 binding sites tend to colocalize with known transcription factor binding sites, based on available ChIP-Seq data sets from the ENCODE database. We found that 42% of all EBNA1 LCL binding sites were cooccupied by at least 1 of 85 available transcription factors, while the same was true for 73% of sites from epigenetic cohort 1. There were 23 transcription factors that cobound EBNA1 in at least 10% of sites from cohort 1, including PAX5, NRSF, and RXRA as the most frequent binding partners of EBNA1 (Fig. 2C).
EBNA1 depletion leads to loss of viability of EBV-positive cells.
To investigate the functional role of EBNA1 at cellular binding sites, we first developed several shRNA lentivirus expression vectors and assayed these vectors for their ability to efficiently deplete EBNA1 protein in LCLs (Fig. 3A). At least three different shRNAs (shEBNA1.1, -1.2, and -1.5) could deplete EBNA1 to levels below 30% of the levels in control lentivirus-infected LCLs. We assayed these shRNAs for their effects on the viability of cell lines likely to be dependent upon EBV latent infection (LCLs and C666-1 cells) and compared these effects to those on EBV-negative cell lines with comparable growth properties (BJAB and HONE1 cells, respectively) (Fig. 3B). We found that all three shEBNA1 lentiviruses reduced the viability of EBV-positive cells but had little effect on EBV-negative cells (Fig. 3B). The loss of cell viability correlated with an increase in the number of annexin V-positive apoptotic cells (Fig. 3C) and an increase in the population of sub-G1 cells by fluorescence-activated cell sorter (FACS) analyses (Fig. 3D). We also assayed the effect of EBNA1 depletion on cellular DNA replication by assaying the incorporation of ethynyl deoxyuridine (EdU) followed by Click-iT chemistry for high-content imaging (Fig. 3E and F). We found that shEBNA1 led to a large (∼75%) reduction in cellular proliferation relative to that in shControl-treated or untreated cells (Fig. 3E and F). These findings demonstrate that EBNA1 depletion leads to losses of cellular DNA replication and survival in both LCLs and C666-1 cells.
FIG 3.

EBNA1 depletion leads to loss of cell viability and proliferation. (A) LCLs were transduced with lentivirus containing shControl or various shEBNA1 targeting vectors. Cells were selected for puromycin resistance and assayed at 5 days postselection by Western blotting for EBNA1 or actin. (B) C666-1, HONE-1, LCL, or BJAB cell viability was measured by a resazurin assay after selection with lentivirus expressing control shRNA or various EBNA1 shRNAs, as indicated. (C) LCLs transduced with shEBNA1.1 or shControl were assayed for apoptosis by FACS analysis with propidium iodide (PI) (y axis) and annexin V (x axis). (D) LCLs or C666-1 cells were untreated or selected after transduction with shControl or shEBNA1.1 and then assayed at 5 days postselection for cell cycle distribution by PI incorporation. Sub-G1 cells are highlighted in yellow. (E) High-content imaging for incorporation of EdU in C666-1 cells that were untreated or were transduced and selected with shControl or shEBNA1.1. The percentage of cells incorporating EdU was determined by using high-content imaging. * indicates P values of <0.05, as determined by using the Student t test.
Identification of cellular genes regulated by EBNA1.
To identify cellular genes that are transcriptionally regulated by EBNA1, we first performed a gene expression profile analysis using RNA-Seq for LCLs transduced with shEBNA1.1 or shControl lentivirus. We identified a large number of genes (∼700) that are both positively and negatively regulated >2-fold by EBNA1 shRNA depletion (Fig. 4). We found that these genes were significantly associated with a number of diverse functions, as identified by DAVID and Ingenuity Pathway Analysis (IPA) software. Among the top functions were those associated with cancer, attraction of T lymphocytes, and synthesis of fatty acid (Fig. 4A). Among the genes whose expression levels were most reduced by EBNA1 depletion were glucagon, IL6, CCR4, CXCL10, and defensinβ4A. Among the genes most activated by EBNA1 depletion were PHGDH, SREBF1, and MAG. We attempted to integrate these data with the ChIP-Seq data to determine which EBNA1-bound genes tended to have increased or decreased expression in response to EBNA1 depletion. Overall, we found that genes with EBNA1 bound within 3 kb of the TSS were 2.6-fold more likely (P = 0.007) to be downregulated by EBNA1 depletion, suggesting that EBNA1 functions more frequently as a transcriptional activator of neighboring genes (Fig. 4B). We further analyzed genes with strong EBNA1 peaks located within 3 kb of the TSS and changes in mRNA expression after EBNA1 depletion (Fig. 4C), which may indicate direct regulation by EBNA1. In this group, we found that USP30, MEF2B, and SLC37A4 were among the genes most downregulated by EBNA1 depletion, while EPHA4, SGK1, and MRPL30 were among the genes most upregulated by EBNA1 depletion. Further analysis by using IPA revealed that EBNA1 may regulate several pathways, including the activation of various cytokine pathways (IL-18, IL-6, IL-12, and IL-2) and the p38 mitogen-activated protein (MAP) kinase network and the inhibition of cytokine IL-10 and ADIPOQ networks (Fig. 4D).
FIG 4.

Integration of RNA-Seq with ChIP-Seq to identify genes regulated directly by EBNA1. LCLs were transduced twice with shControl (shC1 and shC2) or shEBNA1 (shE1 and shE2) and assayed by RNA-Seq. The RNA-Seq data were then analyzed to find significantly affected genes. (A) Genes from 3 major functions significantly affected by EBNA1 depletion. Red, significantly upregulated by EBNA1 depletion; blue, significantly downregulated by EBNA1 depletion. CoA, coenzyme A. (B) Significant association of EBNA1 depletion and downregulation of genes occupied by EBNA1 within 3 kb of the TSS. (C) Genes occupied by EBNA1 within 3 kb of the TSS whose expression levels changed at least 1.5-fold upon EBNA1 depletion. (D) Regulators significantly affected by EBNA1 depletion, as indicated by expression level changes of their known targets. GF, growth factor; HGF, human growth factor; MAPK, mitogen-activated protein kinase.
EBNA1 regulates genes essential for B-cell survival.
To identify cellular genes directly and positively regulated by EBNA1, we tested several of the highly significant candidate genes by reverse transcription-PCR (RT-PCR) (Fig. 5A). EBNA1 depletion led to a substantial loss of IL6R, KDM4C, EBF1, and MEF2b mRNA expression. We also confirmed previous observations that EBNA1 depletion leads to increased expression of viral lytic cycle genes, including the immediate early genes BRLF1 and BZLF1 (Fig. 5B). Western blot analysis confirmed these findings for the loss of the cellular proteins IL-6R and EBF1 and increased expression of the viral lytic proteins BALF2 and Zta (BZLF1) (Fig. 5C).
FIG 5.

EBNA1 target genes contribute to cell viability. (A) Cellular mRNA expression in LCLs transduced with shControl or shEBNA1.1. RT-PCR was performed to quantify the expression level changes relative to shControl treatment for each gene indicated. (B) Viral mRNA expression in LCLs treated as described above for panel A. (C) Western blot analysis of LCLs transduced with shControl or shEBNA1.1 and antibodies for EBNA1, IL-6R, EBF1, BALF2, ZTA, or actin. (D) Immunoblot (IB) analysis of EBV-negative cells of the NPC cell line HK1 transduced with lentivirus expressing FLAG-EBNA1 (+) or the control (−) probed for FLAG, IL-6R, or actin. Fold changes in expression levels of IL-6R, MEF2B, and EBF1 as determined by RT-PCR are shown on the right. (E) Same as panel D except that the EBV-negative B-cell line BJAB was transduced with FLAG-EBNA1 lentivirus and assayed by immunoblotting (left) or RT-PCR for IL-6R, MEF2B, and EBF1 (right). (F) Cell viability assay for LCLs transduced with lentivirus vectors for either shControl, shEBF1 (left), or shIL6R (right). Transduced LCLs were analyzed by Western blotting for EBF1 (left) or IL-6R (right). (G) Cell viability assay for EBV-negative BJAB and DG75 cells and EBV-positive Mutu I, LCL352, and Raji cells transduced with lentivirus for shMEF2B. RT-PCR shows that shMEF2B vectors efficiently deplete MEF2B mRNA in all cell types (bottom). * indicates a P value of <0.05, as determined by using the Student t test (n = 6).
We also assayed the effect of ectopic expression of EBNA1 on the EBV-negative NPC cell line HK1 or the B-cell lymphoma line BJAB (Fig. 5D and E). Western blot analysis demonstrated that FLAG-EBNA1 protein was expressed in each cell type, and a modest increase in the IL-6R level was also observed in HK1 cells (Fig. 5D). RT-PCR also revealed that ectopic expression of EBNA1 induced the expression of IL6R mRNA in both cell types, while MEF2B and EBF1 were induced only in BJAB cells (Fig. 5E). Each of these genes has a strong EBNA1 peak at its transcription start site in EBV-positive cell lines. These findings suggest that ectopic expression of EBNA1 may activate the transcription of bound genes in a cell type- and context-dependent manner.
To investigate the potential importance of EBNA1-regulated genes for B-cell survival, we first tested whether shRNA depletion of IL-6R or EBF1 led to a loss of LCL viability (Fig. 5F). Depletion of IL-6R (∼50% reduction) led to an ∼40% decrease in B-cell viability (Fig. 5F, left). Depletion of EBF1 (∼80% reduction) reduced LCL viability by ∼70% (Fig. 5F, right). MEF2B was among the genes most highly enriched for EBNA1 binding and responsive to EBNA1 for mRNA transcription levels. MEF2B is of particular interest because of its frequent deregulation in B-cell lymphomas (31, 32). Depletion of MEF2B resulted in a significant loss of cell viability in EBV-positive cells (Mutu I cells, Raji cells, and LCLs) but not in EBV-negative B cells (DG75 and BJAB cells) (Fig. 5G). Interestingly, MEF2B mRNA levels were depleted to a greater extent in EBV-negative cells than in EBV-positive cells (Fig. 5G, bottom), suggesting that EBV-negative cells are less dependent upon MEF2B for viability. These findings indicate that genes bound and regulated by EBNA1 contribute, at least in part, to host cell viability.
DISCUSSION
We have used ChIP-Seq, RNA-Seq, and targeted gene depletion to identify candidate cellular genes that are directly bound and regulated by EBNA1. We show that EBNA1 binds with high occupancy to ∼1,000 host chromosome sites in various cell types. These sites are mostly invariant between cell and latency types, although a subset of sites has cell-specific enrichments or losses. The majority of these sites are in close proximity to genes transcriptionally active in B lymphocytes and are affected, either positively or negatively, by depletion of EBNA1. We show that EBV-positive cells require EBNA1 for viability and proliferation. We identified a small cohort of genes that have strong EBNA1 binding sites within known promoter regions that were also positively coregulated by EBNA1. Finally, we demonstrated that some of these genes, namely, IL6R, EBF1, and MEF2B, are required for the viability of EBV-positive cells. We suggest that EBNA1 modulates the transcription of numerous genes and gene pathways that contribute to the overall viability and fitness of latently EBV-infected cells (Fig. 6).
FIG 6.
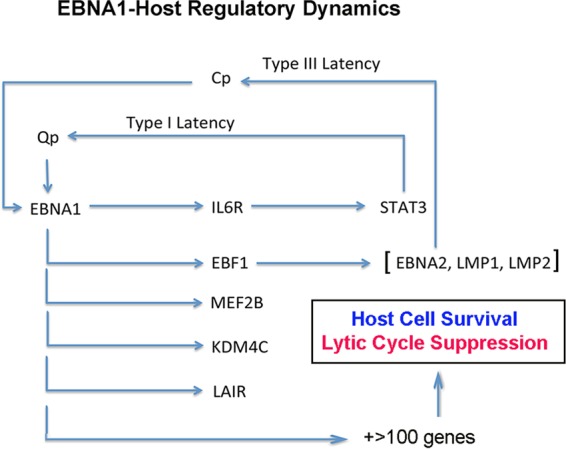
EBNA1 regulatory network. The schematic shows some of the salient cellular and viral target genes regulated directly by EBNA1 DNA binding and how this controls latency type and latent-lytic cycle switch decisions.
EBNA1 may promote host cell survival through multiple mechanisms. EBNA1 can interact physically with several cellular proteins that may affect cellular fitness independent of EBNA1 DNA binding functions (14). On the other hand, the EBNA1 DNA binding domain can function as a dominant negative mutant capable of inhibiting cell growth and survival (33). EBNA1 is also known to have essential functions in the transcription activation of EBV genes, including the activation of EBNA2 during the establishment of type III latency. EBNA1 can also function in the suppression of EBV lytic reactivation (34). EBNA1 has been shown to activate let-7 microRNA (miRNA) and to downregulate Dicer, which in turn prevents lytic cycle gene activation (35). However, we did not find any EBNA1 binding sites close to the transcription start sites of these genes, suggesting that they may be regulated indirectly by EBNA1. EBNA1 has been thought to bind selectively to Qp in type III latency, where it functions to repress transcription initiation (36). However, our ChIP-Seq data indicate that Qp is bound similarly in all latency types tested. The fact that EBNA1 can activate and repress viral promoters is consistent with data from our genome-wide analysis revealing a cohort of EBNA1-bound genes with either active or repressed chromatin marks. This suggests that EBNA1 may function as a scaffold for host factors that can either activate or repress transcription depending on the context. Thus, EBNA1 is unlikely to be a potent activator or repressor of transcription but rather is a cofactor in mediating nucleoprotein complexes at selective sites in the genome.
The majority of cellular EBNA1 binding sites conformed to the consensus site found in the EBV genome, consistent with EBNA1 binding DNA through a single major recognition mechanism defined by structural studies (37). Although we did not find statistically significant alternative consensus sites, as previously reported (18, 19), it is possible that a small amount of EBNA1 binds directly or indirectly to cellular sites that diverge from the canonical consensus. EBNA1 can also binds to sites that do not correspond to known transcriptional regulatory elements. Approximately 8% of EBNA1 binding sites overlap CTCF binding sites, suggesting that EBNA1 may have chromatin architectural functions. EBNA1 binds in close proximity to CTCF at Qp in the viral genome, where CTCF functions to limit DNA methylation and facilitate DNA loop formation with the OriP enhancer. EBNA1 is known to form homotypic interactions through its amino-terminal zinc binding motif (38–42), suggesting that an important function of EBNA1 is the formation of DNA loops and interaction networks, similar to that proposed for CTCF. Thus, EBNA1 sites may regulate transcription by linking regulatory elements through DNA loop formations.
The cohort of genes with EBNA1 binding sites near transcription start sites (cluster 1) includes many genes with known functions in cell survival and proliferation. We examined three of these genes, IL6R, EBF1, and MEF2B, for their regulation by EBNA1 and for their contribution to cell viability. The level of IL6R was found to be elevated in EBV-positive lymphoid cells (43) and nasopharyngeal carcinoma cells (44). IL6R was shown to be overexpressed in NPC tumor tissue and important for an enhanced response to IL-6–STAT3 signaling that is important for tumor cell growth and invasiveness (44). The MEF2B level was found to be elevated in several B-cell malignancies, including diffuse large B-cell lymphoma (DLBCL), which may be commonly infected by EBV (31, 32). EBF1 was identified as a B-cell identity factor important for maintaining EBV latency (45). EBF1 was also found to occupy many of the EBNA2 chromosome binding sites (8). EBNA2 is expressed in type III latency, where it activates the transcription of many cellular genes important for B-cell activation, functioning in concert with B-cell transcription factors like EBF1 and RBP-jK (8). EBNA2 binds to many sites in the host chromosome that overlap cellular transcription factor binding sites, especially RBP-jk and EBF1. EBNA1 overlaps ∼7% of EBNA2 binding sites, but it is not yet known if EBNA1 modulates EBNA2 function at these sites. The overwhelming majority of EBNA2 sites are EBNA1 independent, suggesting that these viral factors act through distinct and likely complementary mechanisms on the host chromosome.
In conclusion, we find that EBNA1 binds to an invariant group of sites that tend to localize with transcription start sites of active and euchromatic genes. By combining ChIP-Seq data for multiple EBV-positive cells and RNA-Seq data for EBNA1-depleted cells, we were able to identify a cohort of cellular genes that are direct targets of EBNA1 and contribute to the survival and proliferation functions provided by EBNA1 in latently infected cells.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Wistar Institute Cancer Center Core Facilities for Molecular Screening, Genomics, and Bioinformatics.
A.D.L. was a Pasteur Cenci Fellowship recipient.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.02318-15.
REFERENCES
- 1.Young LS, Rickinson AB. 2004. Epstein-Barr virus: 40 years on. Nat Rev Cancer 4:757–768. doi: 10.1038/nrc1452. [DOI] [PubMed] [Google Scholar]
- 2.Thorley-Lawson DA, Allday MJ. 2008. The curious case of the tumour virus: 50 years of Burkitt's lymphoma. Nat Rev Microbiol 6:913–924. doi: 10.1038/nrmicro2015. [DOI] [PubMed] [Google Scholar]
- 3.Longnecker R, Kieff E, Cohen JI. 2013. Epstein-Barr virus, p 1898–1959. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, vol I Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 4.Lieberman PM. 2013. Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat Rev Microbiol 11:863–875. doi: 10.1038/nrmicro3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3:227–232. doi: 10.1016/j.coviro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seifert M, Scholtysik R, Kuppers R. 2013. Origin and pathogenesis of B cell lymphomas. Methods Mol Biol 971:1–25. doi: 10.1007/978-1-62703-269-8_1. [DOI] [PubMed] [Google Scholar]
- 7.Mesri EA, Feitelson MA, Munger K. 2014. Human viral oncogenesis: a cancer hallmarks analysis. Cell Host Microbe 15:266–282. doi: 10.1016/j.chom.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE, Kieff E. 2011. Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc Natl Acad Sci U S A 108:14902–14907. doi: 10.1073/pnas.1108892108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allday MJ. 2009. How does Epstein-Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt's lymphoma? Semin Cancer Biol 19:366–376. doi: 10.1016/j.semcancer.2009.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paschos K, Parker GA, Watanatanasup E, White RE, Allday MJ. 2012. BIM promoter directly targeted by EBNA3C in polycomb-mediated repression by EBV. Nucleic Acids Res 40:7233–7246. doi: 10.1093/nar/gks391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skalska L, White RE, Franz M, Ruhmann M, Allday MJ. 2010. Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP. PLoS Pathog 6:e1000951. doi: 10.1371/journal.ppat.1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Portal D, Zhao B, Calderwood MA, Sommermann T, Johannsen E, Kieff E. 2011. EBV nuclear antigen EBNALP dismisses transcription repressors NCoR and RBPJ from enhancers and EBNA2 increases NCoR-deficient RBPJ DNA binding. Proc Natl Acad Sci U S A 108:7808–7813. doi: 10.1073/pnas.1104991108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Portal D, Zhou H, Zhao B, Kharchenko PV, Lowry E, Wong L, Quackenbush J, Holloway D, Jiang S, Lu Y, Kieff E. 2013. Epstein-Barr virus nuclear antigen leader protein localizes to promoters and enhancers with cell transcription factors and EBNA2. Proc Natl Acad Sci U S A 110:18537–18542. doi: 10.1073/pnas.1317608110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frappier L. 2012. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 4:1537–1547. doi: 10.3390/v4091537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Westhoff Smith D, Sugden B. 2013. Potential cellular functions of Epstein-Barr nuclear antigen 1 (EBNA1) of Epstein-Barr virus. Viruses 5:226–240. doi: 10.3390/v5010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chakravorty A, Sugden B. 2015. The AT-hook DNA binding ability of the Epstein Barr virus EBNA1 protein is necessary for the maintenance of viral genomes in latently infected cells. Virology 484:251–258. doi: 10.1016/j.virol.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Norseen J, Johnson FB, Lieberman PM. 2009. Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J Virol 83:10336–10346. doi: 10.1128/JVI.00747-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dresang LR, Vereide DT, Sugden B. 2009. Identifying sites bound by Epstein-Barr virus nuclear antigen 1 (EBNA1) in the human genome: defining a position-weighted matrix to predict sites bound by EBNA1 in viral genomes. J Virol 83:2930–2940. doi: 10.1128/JVI.01974-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, Showe L, Davuluri RV, Lieberman PM. 2010. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr virus nuclear antigen 1 (EBNA1). Virol J 7:262. doi: 10.1186/1743-422X-7-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu F, Tempera I, Lee HT, Dewispelaere K, Lieberman PM. 2014. EBNA1 binding and epigenetic regulation of gastrokine tumor suppressor genes in gastric carcinoma cells. Virol J 11:12. doi: 10.1186/1743-422X-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Canaan A, Haviv I, Urban AE, Schulz VP, Hartman S, Zhang Z, Palejev D, Deisseroth AB, Lacy J, Snyder M, Gerstein M, Weissman SM. 2009. EBNA1 regulates cellular gene expression by binding cellular promoters. Proc Natl Acad Sci U S A 106:22421–22426. doi: 10.1073/pnas.0911676106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy G, Komano J, Sugden B. 2003. Epstein-Barr virus provides a survival factor to Burkitt's lymphomas. Proc Natl Acad Sci U S A 100:14269–14274. doi: 10.1073/pnas.2336099100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malik-Soni N, Frappier L. 2012. Proteomic profiling of EBNA1-host protein interactions in latent and lytic Epstein-Barr virus infections. J Virol 86:6999–7002. doi: 10.1128/JVI.00194-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coppotelli G, Mughal N, Callegari S, Sompallae R, Caja L, Luijsterburg MS, Dantuma NP, Moustakas A, Masucci MG. 2013. The Epstein-Barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group A proteins. Nucleic Acids Res 41:2950–2962. doi: 10.1093/nar/gkt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 28.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deng Z, Wang Z, Stong N, Plasschaert R, Moczan A, Chen HS, Hu S, Wikramasinghe P, Davuluri RV, Bartolomei MS, Riethman H, Lieberman PM. 2012. A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J 31:4165–4178. doi: 10.1038/emboj.2012.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, Califano A, Pasqualucci L, Basso K, Dalla-Favera R. 2013. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol 14:1084–1092. doi: 10.1038/ni.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, Johnson NA, Severson TM, Chiu R, Field M, Jackman S, Krzywinski M, Scott DW, Trinh DL, Tamura-Wells J, Li S, Firme MR, Rogic S, Griffith M, Chan S, Yakovenko O, Meyer IM, Zhao EY, Smailus D, Moksa M, Chittaranjan S, Rimsza L, Brooks-Wilson A, Spinelli JJ, Ben-Neriah S, Meissner B, Woolcock B, Boyle M, McDonald H, Tam A, Zhao Y, Delaney A, Zeng T, Tse K, Butterfield Y, Birol I, Holt R, Schein J, Horsman DE, Moore R, Jones SJ, Connors JM, Hirst M, Gascoyne RD, Marra MA. 2011. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 476:298–303. doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mack AA, Sugden B. 2008. EBV is necessary for proliferation of dually infected primary effusion lymphoma cells. Cancer Res 68:6963–6968. doi: 10.1158/0008-5472.CAN-08-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sivachandran N, Wang X, Frappier L. 2012. Functions of the Epstein-Barr virus EBNA1 protein in viral reactivation and lytic infection. J Virol 86:6146–6158. doi: 10.1128/JVI.00013-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mansouri S, Pan Q, Blencowe BJ, Claycomb JM, Frappier L. 2014. Epstein-Barr virus EBNA1 protein regulates viral latency through effects on let-7 microRNA and dicer. J Virol 88:11166–11177. doi: 10.1128/JVI.01785-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshioka M, Crum MM, Sample JT. 2008. Autorepression of Epstein-Barr virus nuclear antigen 1 expression by inhibition of pre-mRNA processing. J Virol 82:1679–1687. doi: 10.1128/JVI.02142-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, Edwards AM. 1996. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84:791–800. doi: 10.1016/S0092-8674(00)81056-9. [DOI] [PubMed] [Google Scholar]
- 38.Aras S, Singh G, Johnston K, Foster T, Aiyar A. 2009. Zinc coordination is required for and regulates transcription activation by Epstein-Barr nuclear antigen 1. PLoS Pathog 5:e1000469. doi: 10.1371/journal.ppat.1000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Su W, Middleton T, Sugden B, Echols H. 1991. DNA looping between the origin of replication of Epstein-Barr virus and its enhancer site: stabilization of an origin complex with Epstein-Barr nuclear antigen 1. Proc Natl Acad Sci U S A 88:10870–10874. doi: 10.1073/pnas.88.23.10870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frappier L, O'Donnell M. 1991. Epstein-Barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of Epstein-Barr virus. Proc Natl Acad Sci U S A 88:10875–10879. doi: 10.1073/pnas.88.23.10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goldsmith K, Bendell L, Frappier L. 1993. Identification of EBNA1 amino acid sequences required for the interaction of the functional elements of the Epstein-Barr virus latent origin of DNA replication. J Virol 67:3418–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Middleton T, Sugden B. 1992. EBNA1 can link the enhancer element to the initiator element of the Epstein-Barr virus plasmid origin of DNA replication. J Virol 66:489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klein SC, Jucker M, Abts H, Tesch H. 1995. IL6 and IL6 receptor expression in Burkitt's lymphoma and lymphoblastoid cell lines: promotion of IL6 receptor expression by EBV. Hematol Oncol 13:121–130. doi: 10.1002/hon.2900130302. [DOI] [PubMed] [Google Scholar]
- 44.Zhang G, Tsang CM, Deng W, Yip YL, Lui VW, Wong SC, Cheung AL, Hau PM, Zeng M, Lung ML, Chen H, Lo KW, Takada K, Tsao SW. 2013. Enhanced IL-6/IL-6R signaling promotes growth and malignant properties in EBV-infected premalignant and cancerous nasopharyngeal epithelial cells. PLoS One 8:e62284. doi: 10.1371/journal.pone.0062284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davies ML, Xu S, Lyons-Weiler J, Rosendorff A, Webber SA, Wasil LR, Metes D, Rowe DT. 2010. Cellular factors associated with latency and spontaneous Epstein-Barr virus reactivation in B-lymphoblastoid cell lines. Virology 400:53–67. doi: 10.1016/j.virol.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 46.ENCODE Project Consortium. The ENCODE (ENcylopedia Of DNA Elements) Project. 2004. Science 306:636–640. doi: 10.1126/science.1105136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.