

ABSTRACT
Once transported to the replication sites, human adenoviruses (HAdVs) need to ensure decondensation and transcriptional activation of their viral genomes to synthesize viral proteins and initiate steps to reprogram the host cell for viral replication. These early stages during adenoviral infection are poorly characterized but represent a decisive moment in the establishment of a productive infection. Here, we identify a novel host viral restriction factor, KAP1. This heterochromatin-associated transcription factor regulates the dynamic organization of the host chromatin structure via its ability to influence epigenetic marks and chromatin compaction. In response to DNA damage, KAP1 is phosphorylated and functionally inactive, resulting in chromatin relaxation. We discovered that KAP1 posttranslational modification is dramatically altered during HAdV infection to limit the antiviral capacity of this host restriction factor, which represents an essential step required for efficient viral replication. Conversely, we also observed during infection an HAdV-mediated decrease of KAP1 SUMO moieties, known to promote chromatin decondensation events. Based on our findings, we provide evidence that HAdV induces KAP1 deSUMOylation to minimize epigenetic gene silencing and to promote SUMO modification of E1B-55K by a so far unknown mechanism.
IMPORTANCE Here we describe a novel cellular restriction factor for human adenovirus (HAdV) that sheds light on very early modulation processes in viral infection. We reported that chromatin formation and cellular SWI/SNF chromatin remodeling play key roles in HAdV transcriptional regulation. We observed that the cellular chromatin-associated factor and epigenetic reader SPOC1 represses HAdV infection and gene expression. Here, we illustrate the role of the SPOC1-interacting factor KAP1 during productive HAdV growth. KAP1 binds to the viral E1B-55K protein, promoting its SUMO modification, therefore illustrating a crucial step for efficient viral replication. Simultaneously, KAP1 posttranslational modification is dramatically altered during infection. We observed an HAdV-mediated decrease in KAP1 SUMOylation, known to promote chromatin decondensation events. These findings indicate that HAdV induces the loss of KAP1 SUMOylation to minimize epigenetic gene silencing and to promote the SUMO modification of E1B-55K by a so far unknown mechanism.
INTRODUCTION
Before efficient replication can occur, various DNA virus genomes must be transported into the nucleus. Simultaneously, host cells perceive the introduction of noncellular nucleic acids or unscheduled replication as danger signals and activate a DNA damage response (DDR) that leads to cell cycle arrest and/or apoptosis. To counteract this, human adenovirus (HAdV) expresses early viral genes to degrade or displace key regulators of cellular antiviral measures. In turn, to repress viral expression, cells mobilize a network of transcriptional repressors and activators that normally control cellular homeostasis (1, 2).
The nuclear domains thought to be responsible for repressing viral genomes are promyelocytic nuclear bodies (PML-NBs) (3, 4) combining host proteins with transcriptional repressive functions via covalent posttranslational SUMO modification. The cellular transcription factor Daxx, found in these PML-NBs in a complex with the ATRX protein, induces histone deacetylation. Together these factors negatively regulate HAdV gene expression (5, 6). This Daxx/ATRX-mediated restriction imposed upon virus growth is counteracted by the early viral protein E1B-55K alone and in combination with E4orf6, leading to the reduction of Daxx/ATRX via a proteasome-dependent pathway (5–7).
We have also shown that HAdV inhibits the SPOC1 restriction factor, which is dynamically associated with chromatin and induces chromosome condensation to regulate proper cell division (8). SPOC1 is proposed to increase histone H3 lysine 9 (H3K9) lysine methyltransferases (KMTs) and trimethylated H3K9 (H3K9me3) histone marks to promote chromatin condensation by recruiting histone methyltransferases (HMTs). We found that SPOC1 protein levels were decreased in HAdV-infected cells, which we could attribute to proteasomal degradation mediated by the E1B-55K/E4orf6 E3 ligase complex (8).
Another well-studied SPOC1-interacting protein is the heterochromatin-associated transcription factor KAP1 (Kruppel-associated box [KRAB]-associated protein 1)/transcriptional intermediary factor 1β (TIF1β)/KRAB-interacting protein 1 (KRIP1)/tripartite motif containing 28 (TRIM28) (9, 10). Recruitment of this protein to genetic loci increases H3K9me2/3 repressive histone marks, induces the formation of heterochromatin, and blocks gene expression (8). Upon DNA damage, KAP1 is rapidly phosphorylated at serine 824 (S824) by the nuclear phosphatidylinositol 3 kinase-like (PIKK) family members ataxia telangiectasia mutated (ATM), ataxia telangiectasia and Rad3 related (ATR), and the DNA-dependent protein kinase catalytic subunit (DNA-PKcs). It was shown that the ATM-mediated phosphorylation of KAP1 at S824, in concert with the chromatin remodeler chromodomain helicase DNA binding protein 3 (CHD3), which is essential for DDR in heterochromatin, induces functionally inactive protein and relaxed chromatin (11, 12). This is followed by the activation of proteins involved in cell cycle control, apoptosis, and the interferon response (13, 14). Conversely, inhibition of KAP1 phosphorylation prevents decondensation of heterochromatin repair foci, which renders cells hypersensitive to double-strand break (DSB)-inducing agents.
Besides phosphorylation, KAP1-mediated recruitment of repressive components can also be regulated by SUMOylation of this host factor itself. Upon dephosphorylation by protein phosphatase 1α (PP1α), protein phosphatase 1β (PP1β), or protein phosphatase 4 (PP4) (15–17), the bromodomain of KAP1 can be SUMOylated by its adjacent PHD domain via a PHD/Ubc9 interaction (13, 18); SUMO1 as well as SUMO2 can be conjugated to the C terminus of the KAP1 protein (14, 19). SENP1 and SENP7 SUMO proteases were shown to reverse KAP1 SUMOylation (14).
KAP1 contains an N-terminal RING, B-box, and coiled-coil domain (RBCC)/tripartite interaction motif (TRIM) domain, which is responsible for the interaction with KRAB domain-containing zinc finger (KRAB ZNF) transcription factors (20) and SPOC1 (10). The heterochromatin protein 1 (HP1) box, a central PXVXL pentapeptide region within the KAP1 protein, mediates the interaction with HP1 (21), and the C-terminal PHD domain facilitates the recruitment of the Mi2α subunit within the NuRD histone deacetylase complex (22) and the H3K9-specific HMT SETDB1 (23).
KAP1 plays a role during infection with different human-pathogenic viruses. For example, it was shown that KAP1 inhibits human immunodeficiency virus type 1 (HIV-1) integration by recruiting the viral integrase, followed by its deacetylation through histone deacetylases (HDACs) (24). KAP1 was also identified to be a latency regulator for Kaposi's sarcoma-associated herpesvirus (KSHV), i.e., switching viral latency to lytic replication (25). Clearly, cellular chromatin remodeling mechanisms and DDR impairment play key roles in regulating adenoviral gene expression and illustrate the importance of epigenetic regulation at the chromatin level. Recent studies of Forrester et al. showed evidence that during HAdV infection KAP1 interacts with E1B-55K and is phosphorylated at S824 (26).
In our study, we demonstrate that KAP1 represses HAdV gene expression. This novel host restriction factor is efficiently deSUMOylated upon infection via an SUMO E1B-55K-dependent process. Additionally, we provide evidence that KAP1 induces SUMOylation of the viral factor E1B-55K and therefore plays a major role during productive HAdV infection.
MATERIALS AND METHODS
Cell culture and generation of knockdown cell lines.
H1299 cells (27), A549 cells (DSMZ ACC 107; Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany), and HeLa cells stably expressing 6His-SUMO1 or 6His-SUMO2 (kindly provided by Ron Hay, University of Dundee) (28) were grown as monolayer cultures in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U of penicillin, and 100 μg of streptomycin per ml in a 5% CO2 atmosphere at 37°C.
To generate KAP1-knockdown cell lines, H1299 and A549 cells were transduced with lentiviral vectors expressing short hairpin RNA (shRNA) targeted to the coding strand sequence 5′-GGAGTTGGATCTCTCAGAA-3′ located at nucleotides 626 to 643 in kap1. Transduced cells were selected and maintained in medium containing puromycin (1 μg/ml).
Plasmids and transient transfections.
The HAdV proteins examined in this study were expressed from their respective complementary DNAs under the control of the cytomegalovirus immediate early promoter derived from the pcDNA3 vector (Invitrogen) to express wild-type (wt) adenovirus type 5 (AdV5) E1B-55K (29, 30). pcDNA3.1-Flag-derived plasmids expressing either the KAP1 wild-type construct, the M2 mutant, a mutant lacking the RBCC domain (the ΔRBCC mutant), a mutant lacking the PHD/bromo (PB) domain (the ΔPB mutant), or a mutant lacking both the RBCC and PB domains [the Δ(RBCC+PB) mutant] were kindly provided by Peggy Farnham (University of Southern California, Los Angeles, CA, USA). TIF1β/KAP1 tagged with enhanced yellow fluorescent protein (EYFP) and yellow fluorescent protein (EYFP-YFP-TIF1β/KAP1), YFP-TIF1α, and YFP-TIF1γ were kindly provided by Hans Will (University Hospital Eppendorf, Hamburg, Germany). KAP1 point mutations were introduced by site-directed mutagenesis using the primers shown in Table 1. For transient transfection, subconfluent cells were treated with a transfection mixture of DNA and 25-kDa linear polyethylenimine as described recently (31).
TABLE 1.
Primers used in this study
| Primer no. | Primer descriptiona | Primer sequence |
|---|---|---|
| 2542 | KAP1S824A fwd | 5′-CTGGCCTGAGTGCCCAGGAGCTG-3′ |
| 2543 | KAP1S824A rev | 5′-CAGCTCCTGGGCACTCAGGCCAG-3′ |
| 2548 | KAP1K554A fwd | 5′-GTCTCCTCCTCCGCGACAATGG-3′ |
| 2549 | KAP1K554A rev | 5′-CCATTGTCGCGGAGGAGGAGAC-3′ |
| 2550 | KAP1K779A fwd | 5′-TGCACGTCTGCCGCGTCCTCAG-3′ |
| 2551 | KAP1K779A rev | 5′-CTGAGGACGCGGCAGACGTGCA-3′ |
| 2552 | KAP1K804A fwd | 5′-ACAGCAGAGAACGCGGTGTCACC-3′ |
| 2553 | KAP1K804A rev | 5′-GGTGACACCGCGTTCTCTGCTGT-3′ |
| 2645 | KAP1K676A fwd | 5′-CATCCTCCTCCGCCAGGTCAGG-3′ |
| 2646 | KAP1K676A rev | 5′-CCTGACCTGGCGGAGGAGGATG-3′ |
fwd, forward; rev, reverse.
Viruses.
H5pg4100 served as the wild-type virus in these studies (32). The mutant viruses H5pm4149 and H5pm4154 were generated as described recently (33, 34). Both viruses carry point mutations in the E1B-55K (H5pm4149) or E4orf6 (H5pm4154) open reading frame and do not express the respective viral protein. Viruses were propagated and titrated in HEK293 cell monolayer cultures, and infections were performed as described previously (34).
Antibodies and protein analysis.
Primary antibodies specific for the viral proteins used in this study included E1A mouse monoclonal antibody (MAb) M73 (35), E1B-55K mouse MAb 2A6 (36), E2A-72K mouse MAb B6-8 (37), E4orf6 mouse MAb RSA3 (38), L4-100K rat MAb 6B-10 (39), E4orf3 rat MAb 6A11 (40), and HAdV type 5 (HAdV5) rabbit polyclonal serum L133 (34). Primary antibodies specific for cellular proteins included KAP1 rabbit polyclonal antibody H-300 (Santa Cruz Biotechnology), phospho-KAP1 (S824) rabbit polyclonal antibody (Bethyl Laboratories, Inc.), SPOC1 rat monoclonal antibody 6F6 (8, 10), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) antibody (Ab; catalog number sc-32233; Santa Cruz), H3 antibody (catalog number 1326-1; Epitomics), Mre11 rabbit polyclonal antibody pNB 100-142 (Novus Biologicals, Inc.), anti-Flag mouse MAb M2 (Sigma-Aldrich, Inc.), rabbit polyclonal green fluorescent protein (GFP)/YFP antibody (catalog number ab290; Abcam), and β-actin mouse MAb AC-15 (Sigma-Aldrich, Inc.). Secondary antibodies conjugated to horseradish peroxidase (HRP) for detection of proteins by immunoblotting were anti-rabbit IgG, anti-mouse IgG, and anti-rat IgG (Jackson/Dianova). Fluorescent secondary antibodies were affinity-purified fluorescein isothiocyanate (FITC)-conjugated donkey anti-mouse IgG and Cy3-conjugated donkey anti-rat IgG (Invitrogen). These were used at a 1:100 dilution in all immunofluorescence experiments.
All protein extracts were prepared in radioimmunoprecipitation assay lysis buffer as published recently (41). For immunoprecipitation, Flag-M2-coupled protein A-Sepharose beads (Sigma-Aldrich Inc.) were used or protein A-Sepharose (3 mg/immunoprecipitation) was coupled with 1 μg of MAb for 1 h at 4°C. The Ab-coupled protein A-Sepharose was added to pansorbin-Sepharose (50 μl/lysate; Calbiochem)-precleared extracts and rotated for 2 h at 4°C. Proteins bound to the Ab-coupled protein A-Sepharose were precipitated by centrifugation, washed three times, boiled for 5 min at 95°C in 5× Laemmli buffer, and analyzed by immunoblotting. Cell fractionation was performed on the basis of a modified protocol described by Leppard and Shenk (42), which we reported previously (43). Denaturing purification and analysis of SUMO conjugates were performed as described recently (44). After denaturation, proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and visualized by immunoblotting. Autoradiograms were scanned and cropped using Adobe Photoshop CS6 software, and figures were prepared using Adobe Illustrator CS6 software.
Indirect immunofluorescence.
For indirect immunofluorescence, cells were grown on glass coverslips as described recently (34). At the times indicated below, cells were fixed in ice-cold methanol at −20°C for 15 min. After 1 h of blocking in Tris-buffered saline–BSA–glycine buffer, they were treated for 1 h with the primary antibody diluted in phosphate-buffered saline (PBS) and washed three times in PBS, followed by 1 h of incubation with the corresponding FITC- or Cy3-conjugated secondary antibodies (Invitrogen). Coverslips were washed three times in PBS and mounted in Glow medium (Energene), and digital images were acquired on a DMRB fluorescence microscope (Leica) with a charge-coupled-device camera (Diagnostic Instruments). Images were cropped using Adobe Photoshop CS6 software and assembled with Adobe Illustrator CS6 software.
RESULTS
HAdV induces KAP1 phosphorylation in a virus concentration-dependent manner.
Emerging evidence suggests that KAP1 is subjected to multiple posttranslational modifications (PTMs), including serine/tyrosine phosphorylation, SUMOylation, and acetylation, which coordinately regulate the KAP1 gene repressive function and KAP1 protein abundance. Previous studies by Forrester et al. have shown that HAdV activates DNA damage response (DDR) mechanisms, inducing KAP1 phosphorylation (26). To ascertain whether KAP1 protein levels are modulated, H1299 cells were infected with wt HAdV and total cell lysate was analyzed at various time points. We detected a significant reduction in unphosphorylated KAP1 accompanied by the upregulation of phosphorylated KAP1 (pKAP1) moieties (Fig. 1A, lanes 7 to 9). Consistent with these results, KAP1 was also phosphorylated in infected A549 cells, another independent cell line that we tested (Fig. 1B, lanes 6 to 9). Moreover, infections with different titers of an HAdV suspension revealed the virus concentration-dependent induction of KAP1 phosphorylation (Fig. 1C).
FIG 1.

HAdV induces KAP1 phosphorylation in a virus concentration-dependent manner. (A and B) H1299 cells (A) and A549 cells (B) were infected with wt virus (H5pg4100) at a multiplicity of infection of 50 focus-forming units per cell and harvested after the indicated time points postinfection. Total cell extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting using mouse MAb 2A6 (anti-E1B-55K), rabbit polyclonal Ab H-300 (anti-KAP1), rabbit polyclonal Ab against KAP1 phosphorylated at S824 (pKAP1S824), and mouse MAb AC-15 (β-actin) as a loading control. (C) H1299 cells were infected with wt virus (H5pg4100) at the indicated multiplicity of infection (moi). Cells were harvested at 24 h p.i., and total cell extracts were separated by SDS-PAGE and subjected to immunoblotting using mouse MAb B6-8 (anti-E2A), rabbit polyclonal Ab H-300 (anti-KAP1), rabbit polyclonal Ab against KAP1 phosphorylated at S824, and mouse MAb AC-15 (anti-β-actin) as a loading control. (D) H1299 cells were infected with wt virus (H5pg4100) and mutant viruses (H5pm4149, H5pm4154) at a multiplicity of infection of 50 focus-forming units per cell. Cells were harvested at 24 h p.i., and total cell extracts were separated by SDS-PAGE and subjected to immunoblotting using the Ab indicated in the legend to panel C and mouse MAb RSA3 (E4orf6). (E) H1299 cells were infected and harvested as described in the legend to panel D. Cell extracts were fractionated into soluble and chromatin-bound proteins, separated by SDS-PAGE, and subjected to immunoblotting using the Ab indicated in the legend to panel C plus rabbit MAb H3 (anti-histone 3) and mouse MAb 6C5 (anti-GAPDH).
Since the majority of KAP1 is phosphorylated by ATM and ATR, there might be a strong link with HAdV-induced DDR. Previous studies have shown that viral E4 proteins play an essential role in opposing key DDR factors to support efficient virus growth. Therefore, we next determined KAP1 protein concentrations in wt virus (H5pg4100)-infected cells and cells infected with mutant virus lacking either E1B-55K (H5pm4149) or E4orf6 (H5pm4154) (Fig. 1D). As anticipated, KAP1 was phosphorylated in cells infected with the wt virus (H5pg4100) (Fig. 1D, lane 2). In parallel, this modified cellular protein accumulated to high levels in infected cells lacking E1B-55K (H5pm4149; Fig. 1D, lane 3). However, in cells infected with the E4orf6-minus virus mutant H5pm4154, KAP1 phosphorylation was similar to that in wt virus-infected cells (Fig. 1C, lane 4).
KAP1 resides in distinct cellular compartments, including the pericentric and centromeric heterochromatin, euchromatin, and cytoplasm. Earlier observations also revealed that KAP1 phosphorylation regulates intracellular localization, maintaining the protein functions. Therefore, we examined KAP1 protein levels in H1299 cells infected with wt virus (H5pg4100) and viruses lacking E1B-55K (H5pm4149) or E4orf6 (H5pm4154) and separated the soluble fraction from the chromatin fraction. Again, we observed virus-induced KAP1 phosphorylation and, more intriguingly, that pKAP1 was mainly detected in the soluble fraction and was detected less so in the chromatin fraction (Fig. 1E, left, lanes 2 to 4). Taken together, HAdV-mediated phosphorylation and subsequent relocalization from chromatin might impact KAP1 repressive functions and multiple aspects of HAdV biology and its replication cycle.
KAP1 overexpression reduces viral gene expression.
To assess the effect of KAP1 on the production of adenoviral progeny, the total virus yield upon KAP1 overexpression was determined (Fig. 2A). KAP1 overexpression reduced progeny production in H1299 cells 3-fold (24 h postinfection [p.i.]) to 2-fold (48 h p.i.) compared to that in nontreated control cells (Fig. 2A, empty vector). These results suggest that KAP1 mediates repressive effects during the HAdV infectious cycle and thus represents a novel host restriction factor. To further test this idea, expression of viral early and late proteins was monitored after infection (Fig. 2B). In agreement with our results presented above, protein synthesis was partially reduced in KAP1-overexpressing cells. In particular, E1B-55K and E4orf6 expression levels were significantly reduced compared to the levels in nontreated cells infected with wt virus (Fig. 2B, lane 2, and C). To substantiate these results and show that the KAP1-mediated repression of adenoviral gene expression is specific, we overexpressed TIF1α and TIF1γ, two other TIF1 family members. After wt infection, we observed that only KAP1-expressing cells (cells expressing Flag-KAP1 or TIF1β) were able to repress E1B-55K and E4orf6 protein levels (Fig. 3B, lanes 2 and 4), as well as progeny production (Fig. 3C). However, overexpression of KAP1 did not affect cell proliferation within 48 h postinfection (Fig. 3A). To further exclude the possibility of nonspecific effects of the overexpression studies, we stably knocked down KAP1 expression using shRNA (Fig. 4) and validated efficient KAP1 depletion by Western blotting (Fig. 4A) and immunofluorescence analyses (data not shown). We also monitored cell growth behavior and again observed no differences between KAP1-depleted and parental cells (Fig. 4B). Virus growth was analyzed by virus yield experiments (Fig. 4C). Intriguingly, our results showed no significant effect on virus growth in KAP1-depleted cells compared to that in parental cells. Additionally, we monitored the expression of early and late viral proteins and observed no significant differences. Since PML-NBs were recently found to repress HAdV infection, we monitored PML expression in our KAP1-depleted cell line. As anticipated, PML protein concentrations were elevated in cells expressing KAP1 shRNA (Fig. 4D); thus, PML-NB-mediated repression of HAdV transcription might efficiently eliminate any significant increase due to KAP1 depletion (Fig. 4C and D).
FIG 2.

KAP1 overexpression reduces viral gene expression. H1299 cells were transfected with an empty vector control or a plasmid carrying KAP1 and superinfected with wt virus (H5pg4100) at a multiplicity of infection of 20 focus-forming units per cell. (A) Viral particles were harvested at 24 and 48 h p.i., and the virus yield was determined by quantitative immunofluorescence staining of E2A on HEK293 cells. The means and standard deviations from three independent experiments are presented. Two-tailed t tests were applied to calculate significance (*, P < 0.05; **, P < 0.002). Values are shown as the level of progeny production as a percentage of that for cells transfected with the empty vector. (B) Cells were harvested at 24 h p.i., and total cell extracts were separated by SDS-PAGE and subjected to immunoblotting using mouse MAb M73 (anti-E1A), mouse MAb 2A6 (anti-E1B-55K), mouse MAb B6-8 (anti-E2A), mouse MAb RSA3 (anti-E4orf6), rat MAb 6B10 (anti-L4-100K), mouse MAb M2 (anti-Flag), and MAb AC-15 (anti-β-actin) as a loading control. (C) Densitometric analysis of the protein levels in the blot shown in panel B, quantified with ImageJ (version 1.45s) software, normalized to the respective β-actin levels, which were set equal to 100. Values are shown as protein levels as a percentage of those for cells transfected with the empty vector.
FIG 3.

KAP1 overexpression reduces viral gene expression. H1299 cells were transfected with an empty vector control, EYFP-tagged TIF1α, TIF1β (KAP1), TIF1γ, or a plasmid carrying Flag-tagged KAP1 (TIF1β). (A) Absolute cell numbers were determined at 1, 2, and 3 days posttransfection. The means and standard deviations from three independent experiments are presented. The experimental setup for panels B and C is presented as a timeline below the graph. (B) Transfected H1299 cells were superinfected with wt virus (H5pg4100) at a multiplicity of infection of 20 focus-forming units per cell. Cells were harvested at 24 h p.i., and total cell extracts were separated by SDS-PAGE and subjected to immunoblotting using mouse MAb M73 (anti-E1A), mouse MAb 2A6 (anti-E1B-55K), mouse MAb B6-8 (anti-E2A), mouse MAb RSA3 (anti-E4orf6), rabbit polyclonal Ab GFP/YFP (catalog number ab290; Abcam), mouse MAb M2 (anti-Flag), and MAb AC-15 (anti-β-actin) as a loading control. (C) Transfected H1299 cells were superinfected as described in the legend to panel B, viral particles were harvested at 24 and 48 h p.i., and virus yield was determined by quantitative immunofluorescence staining of E2A on HEK293 cells. The means and standard deviations from three independent experiments are presented. Two-tailed t tests were applied to calculate significance (*, P < 0.05). Values are shown as the level of progeny production as a percentage of that for cells transfected with the empty vector.
FIG 4.

Characterization of KAP1-depleted human cells. (A) Endogenous KAP1 levels in H1299 cells transfected with scrambled shRNA (shscrambled) and H1299 cells transfected with shRNA against KAP1 (shKAP1) were determined by preparing total cell extracts, followed by SDS-PAGE and immunoblotting using rabbit polyclonal Ab H-300 (anti-KAP1) and mouse MAb AC-15 (anti-β-actin) as a loading control. (B) H1299 cells (1 × 105) transfected with scrambled shRNA and shRNA against KAP1 were cultivated, and absolute cell numbers were determined after the indicated time points. The means and standard deviations from three independent experiments are presented. (C) H1299 cells transfected with scrambled shRNA and shRNA against KAP1 were infected with wt virus (H5pg4100) at a multiplicity of infection of 20 focus-forming units per cell. Viral particles were harvested at 24, 48, and 72 h p.i., and virus yield was determined by quantitative immunofluorescence staining of E2A on HEK293 cells. The means and standard deviations from three independent experiments are presented. Two-tailed t tests were applied to calculate the significance (n.s., not significant). Values are shown as the level of progeny production as a percentage of that for the cells transfected with scrambled shRNA. (D) H1299 cells transfected with scrambled shRNA and shRNA against KAP1 were infected with wt virus (H5pg4100) at a multiplicity of infection of 50 focus-forming units per cell and harvested after the indicated time points postinfection. Total cell extracts were prepared, separated by SDS-PAGE, and subjected to immunoblotting using rabbit polyclonal Ab H-300 (anti-KAP1), rabbit Ab NB 100-59787 (anti-PML), mouse MAb M73 (anti-E1A), mouse MAb 2A6 (anti-E1B-55K), mouse MAb B6-8 (anti-E2A), rabbit antiserum L133 specific for the HAdV5 capsid, and mouse MAb AC-15 (anti-β-actin) as a loading control.
HAdV E1B-55K interacts with KAP1.
Since we observed enhanced KAP1 phosphorylation in cells infected with HAdV lacking E1B-55K, we next tested whether E1B-55K interacts with the endogenous KAP1 protein in infected cells. As anticipated, in wt virus-infected cells, E1B-55K coimmunoprecipitated with KAP1-specific Ab, revealing an interaction between these factors (Fig. 5A, lane 2). No E1B-55K signal was observed in the corresponding negative controls (Fig. 5A, lane 1). Consistent with the data obtained with infected cells, we also detected KAP1 binding to E1B-55K in the absence of other viral proteins (Fig. 5B, lane 2). The next question was whether E1B-55K interferes with the intracellular localization of KAP1 and vice versa. Immunofluorescence analysis of infected human cells revealed that both proteins show diffuse localization in the host cell nucleus (Fig. 5C, panel h).
FIG 5.

HAdV5 E1B-55K interacts with KAP1. (A) H1299 cells were infected with wt virus (H5pg4100) at a multiplicity of infection of 50 focus-forming units per cell, cells were harvested at 24 h p.i., and total cell extracts were prepared. Immunoprecipitation (IP) of endogenous KAP1 was performed using rabbit polyclonal antibody H-300 (anti-KAP1), and proteins were separated by SDS-PAGE and subjected to immunoblotting. Input levels of total cell lysates and coprecipitated proteins were detected using mouse MAb 2A6 (anti-E1B-55K), rabbit polyclonal Ab H-300 (anti-KAP1), and mouse MAb AC-15 (anti-β-actin) as a loading control. Note that heavy chains (IgH) are detected at 55 kDa. (B) H1299 cells were transfected with an empty vector control or a plasmid carrying E1B-55K and harvested at 48 h posttransfection, and total cell extracts were prepared. Immunoprecipitation of endogenous KAP1 was performed and analyzed as described in the legend to panel A. (C) H1299 cells were infected with wt virus (H5pg4100), fixed with methanol at 24 h p.i., and double labeled with rabbit polyclonal Ab H-300 (anti-KAP1) and mouse MAb 2A6 (anti-E1B-55K). Primary Abs were detected using Cy3-conjugated secondary antibodies (against KAP1; red) and FITC-conjugated secondary antibodies (against E1B-55K; green). Nuclei were labeled with DAPI (4′,6-diamidino-2-phenylindole). Overlays of single images (merge) are shown in panels d and h. Magnifications, ×7,600.
To narrow down the binding domain required for the E1B-55K interaction, we performed coprecipitation experiments with KAP1 truncation mutants (Fig. 6A). KAP1-depleted H1299 cells were cotransfected with plasmids carrying wt E1B-55K and KAP1, the HP1 binding mutant M2, the deletion mutant ΔRBCC or ΔPB, or the double deletion mutant Δ(RBCC+PB) and harvested at 48 h after transfection. These experiments revealed that the deletion mutant lacking the PHD/bromo domain as well as the double mutant additionally lacking the RBCC domain exhibited impaired E1B-55K binding (Fig. 6B, lanes 6 and 7).
FIG 6.

HAdV5 E1B-55K interacts with the PHD/bromo domain region of KAP1. (A) Schematic representation of the KAP1 wt and the truncation constructs M2 (which contains a 2-amino-acid mutation in the HP1 binding domain), ΔRBCC (dRBCC; which contains a deletion of the RBCC domain), ΔPB (dPB; which contains a deletion of the PHD/bromo domain), and Δ(RBCC+PB) [d(RBCC+PB); comprising the region from amino acids 376 to 625]. TSS, transcription start site; aa, amino acid. (B) KAP1-depleted H1299 cells were transfected with plasmids carrying the E1B-55K and KAP1 wt or the KAP1 M2, ΔRBCC, ΔPB, or Δ(RBCC+PB) mutant. Cells were harvested at 48 h posttransfection, and total cell extracts were prepared. Immunoprecipitation of Flag-tagged KAP1 was performed using Flag-M2-coupled agarose beads, and proteins were separated by SDS-PAGE and subjected to immunoblotting. Input levels of total cell lysates and coprecipitated proteins were detected using mouse MAb 2A6 (anti-E1B-55K), mouse MAb M2 (anti-Flag), rabbit polyclonal Ab against KAP1 phosphorylated at S824 (pKAP1S824), and mouse MAb AC-15 (anti-β-actin) as a loading control.
SUMO2 moieties of KAP1 are reduced during HAdV infection.
Li and coworkers demonstrated that dephosphorylation results in increased SUMOylation of KAP1 and vice versa (14). Since the SUMOylation of KAP1 is known to result in the recruitment of repressive components, followed by transcriptional repression (14, 18), we investigated the SUMOylation of KAP1 upon HAdV infection. Cells stably expressing His-SUMO1 or His-SUMO2 were transfected with a Flag-tagged KAP1 expression vector and then superinfected with HAdV (Fig. 7A). Immunoblotting of Ni-nitrilotriacetic acid (NTA)-purified His-SUMO1 and His-SUMO2 conjugates (Fig. 7A, left, lane 6) and input crude lysates (Fig. 7A, right, lane 6) revealed that the KAP1 SUMO2 modification is significantly reduced during HAdV infection. However, SUMO1 conjugation on KAP1 could not be detected in this assay (Fig. 7A, left, lanes 3 and 4).
FIG 7.

SUMO2 chains of KAP1 are diminished during HAdV5 infection. (A) Parental HeLa cells and HeLa cells stably expressing 6His-SUMO1/2 were transfected with a plasmid carrying KAP1 and superinfected with wt virus (H5pg4100) at a multiplicity of infection of 50 focus-forming units per cell. At 48 h p.i., total cell lysates were prepared with guanidinium chloride buffer, subjected to Ni-NTA purification of 6His-SUMO conjugates, and analyzed via SDS-PAGE and immunoblotting. Input levels of total cell lysates and Ni-NTA-purified proteins were detected using rabbit polyclonal Ab H-300 (anti-KAP1), mouse MAb 6His (anti-His), mouse MAb B6-8 (anti-E2A), and mouse MAb AC-15 (anti-β-actin) as a loading control. (B) HeLa cells stably expressing 6His-SUMO2 were transfected with plasmids carrying KAP1 and increasing amounts of E1B-55K. At 48 h posttransfection, total cell lysates were prepared and analyzed as described in the legend to panel A using mouse MAbs Flag-M2 [flag(KAP1)], 6His, 2A6 (E1B-55K), and AC-15 (β-actin) as a loading control. (C) HeLa cells stably expressing 6His-SUMO2 were transfected with plasmids carrying KAP1 and E1B-55K or the SUMO mutant E1B-55K-K104R. At 48 h posttransfection, total cell lysates were prepared and analyzed as described in the legend to panel B. (D) Parental HeLa cells and HeLa cells stably expressing 6His-SUMO2 were transfected with a plasmid carrying KAP1 and superinfected with wt virus (H5pg4100) or the E1B-55K-K104R mutant (H5pm4102) at a multiplicity of infection of 50 focus-forming units per cell. At 48 h p.i., total cell lysates were prepared and analyzed as described in the legend to panel B. Molecular masses are indicated on the left, while the relevant proteins are labeled on the right.
To investigate whether the reduction of high-molecular-mass forms of SUMO2-modified KAP1 depends on the interaction with the viral early protein E1B-55K, we cotransfected SUMO2-expressing cells with plasmids carrying KAP1 and E1B-55K. Intriguingly, we observed a decrease in KAP1 SUMOylation upon cotransfection with a plasmid carrying E1B-55K, although this viral protein has been reported to maintain SUMO ligase-like functions (Fig. 7B). When we cotransfected an E1B-55K variant containing a mutation in the SUMO conjugation motif (SCM) at Lys104 (E1B-55K-K104R), the decreased KAP1 SUMOylation was completely abolished (Fig. 7C, left, lane 3).
To substantiate our novel finding that SUMOylated E1B-55K plays a role in reducing the KAP1 SUMO2 modification, we performed infection assays. HeLa cells expressing His-SUMO2 were transfected with the KAP1 wt and superinfected with wt virus (H5pg4100) or the E1B-55K-K104R mutant virus (H5pm4102) (Fig. 7D). As anticipated, immunoblotting of Ni-NTA-purified His-SUMO conjugates and crude lysates revealed that the reduction of high-molecular-mass forms of SUMO2-modified KAP1 depends on the SUMOylation of E1B-55K at Lys104 during HAdV infection (Fig. 7D, left, lanes 5 and 6). Taken together, these results show that the efficient SUMOylation of E1B-55K significantly reduces KAP1-associated SUMO moieties.
KAP1 promotes E1B-55K SUMOylation, while E1B-55K–SUMO conjugation reduces KAP1-associated SUMO moieties.
To further explore the cross talk between SUMO, KAP1, and E1B-55K, we performed additional Ni-NTA assays in transiently transfected human cells. When expressing KAP1 alone, we detected SUMOylated forms of KAP1, the levels of which were significantly reduced after coexpression of E1B-55K (Fig. 8B, left, lanes 1 and 3). As anticipated, this was not detected when we coexpressed E1B-55K with a nonfunctional SCM (E1B-55K-SCM) (Fig. 8C, left, lanes 1 and 3). These findings provide evidence that the capacity of E1B-55K and SUMO to interact represents a prerequisite for efficient KAP1-SUMO deconjugation.
FIG 8.

KAP1 regulates E1B-55K SUMOylation. (A) Schematic representation of the KAP1 mutants, their positions, and the amino acids changed in the KAP1 mutants. Mutated amino acids are indicated in red. (B and C) HeLa cells stably expressing 6His-SUMO2 were cotransfected with plasmids carrying KAP1, SUMO double mutant K554/676A or K779/804A, or the construct with all four mutated SUMO sites, as well as E1B-55K or the E1B-55K–SCM (E1B-55K-K104R) SUMO mutant, as indicated. At 48 h posttransfection, total cell lysates were prepared with guanidinium chloride buffer, subjected to Ni-NTA purification of 6His-SUMO conjugates, and analyzed via SDS-PAGE and immunoblotting. Input levels of total cell lysates and Ni-NTA-purified proteins were detected using mouse MAb 2A6 (anti-E1B-55K), mouse MAb M2 (anti-flag), mouse MAb 6His (anti-His), and mouse MAb AC-15 (anti-β-actin) as a loading control. (D) Densitometric analysis of KAP1 SUMOylation levels in panels B and C (lanes 1 and 3), quantified with ImageJ (version 1.45s) software, normalized to the respective steady-state KAP1 levels. Values are shown as the level as a percentage of that for cells transfected with the empty vector. (E) Schematic representation of the cross talk between KAP1 deSUMOylation and the increase in E1B-55K SUMOylation in HAdV5-infected cells.
Several studies have revealed that the majority of KAP1 SUMOylation is balanced by the phosphorylation status of KAP1 and that the SUMOylation of KAP1 is required for its repressive function (14). Within or adjacent to the PB domain of KAP1, six Lys residues have been validated to be SUMOylation sites (45). To investigate the effect of KAP1 SUMOylation on KAP1-mediated E1B-55K SUMOylation, we generated KAP1-SUMO mutants with mutations at Lys676, Lys554, Lys779, and Lys804 (Fig. 8A). We also created the double mutants K554/676A and K779/804A as well as one null mutant, the K554/676/779/804A mutant.
When HeLa cells expressing His-SUMO2 were cotransfected with plasmids carrying E1B-55K and the KAP1 wt or KAP1 SUMOylation-deficient mutants (Fig. 8A and B), the mutations resulted in decreased levels of SUMO2-modified proteins (Fig. 8B, left; compare lane 1 with lanes 4 to 6). However, all cells expressing KAP1 mutants showed similar E1B-55K SUMOylation patterns (Fig. 8B, left, lanes 4 to 6), indicating that KAP1 SUMOylation per se does not influence its ability to affect SUMOylation of the viral protein, although the level of SUMO2-modified E1B-55K was the highest in cells cotransfected with the KAP1 mutant harboring all four Lys changes (Fig. 8B, left, lane 6).
Since the viral SUMO mutant E1B-55K–SCM (E1B-55K-K104R) did not reduce the level of SUMO2-modified KAP1, we performed cotransfection experiments with E1B-55K–SCM and the KAP1 wt or the KAP1 SUMOylation mutants (Fig. 8C). As before, cotransfection with the E1B-55K–SCM mutant did not reduce the level of SUMO2-modified KAP1 (Fig. 8C, left [compare lane 3 to lane 1], and E), again pointing to cross talk between KAP1 deSUMOylation and E1B-55K SUMOylation (Fig. 8D and E).
KAP1 bears a SUMO ligase-like capacity toward E1B-55K.
Recent studies have shown that KAP1 recruits the SUMO-conjugating enzyme Ubc9 to act as a SUMO E3 ligase either to auto-SUMOylate its own bromodomain and generate a repressive form or to SUMOylate other cellular substrates, such as interferon regulatory factor 7 (IRF7) or Vps34 (18, 19, 46). To investigate whether KAP1 is able to directly modulate E1B-55K SUMOylation, cells expressing His-SUMO2 were cotransfected with plasmids carrying E1B-55K and increasing amounts of KAP1 (Fig. 9A). After purification of His-SUMO2 conjugates (Ni-NTA), immunoblotting revealed that KAP1 overexpression promotes E1B-55K SUMOylation (Fig. 9A, left, lanes 1 to 3).
FIG 9.
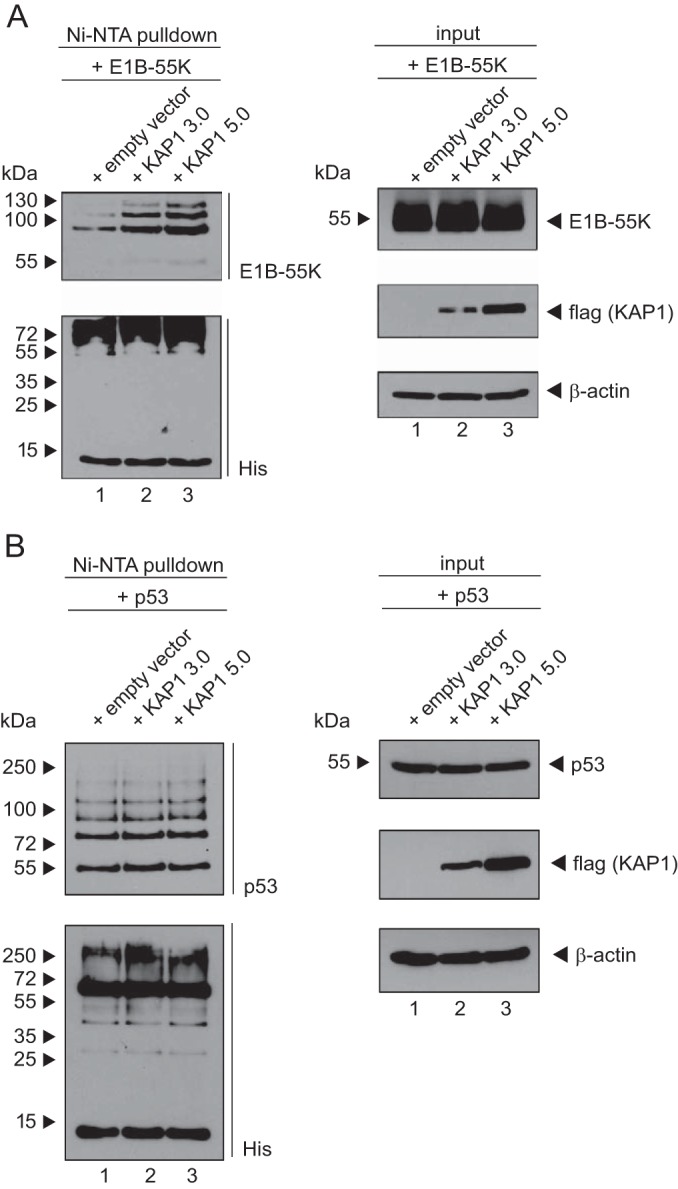
KAP1 SUMOylates E1B-55K. (A and B) HeLa cells stably expressing 6His-SUMO2 were cotransfected with plasmids carrying E1B-55K or p53 and increasing amounts of KAP1. At 48 h posttransfection, total cell lysates were prepared with guanidinium chloride buffer, and 6His-SUMO conjugates were subjected to Ni-NTA purification and analyzed via SDS-PAGE and immunoblotting. Input levels of total cell lysates and Ni-NTA-purified proteins were detected using mouse MAb 2A6 (anti-E1B-55K), mouse MAb M2 (anti-Flag), mouse MAb 6His (anti-His), and mouse MAb AC-15 (anti-β-actin) as a loading control. 3.0 and 5.0, 3.0 μg and 5.0 μg, respectively.
To substantiate our findings that KAP1 specifically induces the SUMO2 modification of E1B-55K, we tested the capacity of KAP1 to SUMOylate the cellular tumor suppressor p53, which is reported to be a host SUMO target. HeLa cells expressing His-SUMO2 were transfected with increasing amounts of the KAP1 wt and human p53 (Fig. 9B). However, it is worth mentioning that HeLa cells express E6 of human papillomavirus 18, and it is not known whether this viral factor influences KAP1/p53 cross talk. Here, KAP1 overexpression did not detectably affect p53 SUMOylation (Fig. 9B, left, lanes 1 to 3). Taken together, KAP1 overexpression had no effect on the SUMO modification of p53, suggesting that the upregulation of viral E1B-55K protein SUMOylation by the cellular KAP1 protein is specific.
DISCUSSION
Here, we illustrate that the SPOC1-interacting factor KAP1 plays the role of a cellular restriction factor during the course of productive HAdV infection. We discovered that KAP1 binds to the HAdV E1B-55K protein, promoting SUMO modification of this viral factor. Conversely, KAP1 posttranslational modification is dramatically altered by an HAdV-mediated decrease in KAP1 SUMO moieties. Our findings point to the possibility that KAP1 deSUMOylation minimizes epigenetic gene silencing. However, KAP1 might also specifically affect the gene regulatory functions of E1B-55K and thus regulate certain E1B-55K-mediated transcriptional events and a subset of adenoviral proteins.
Within the capsids, HAdV genomes are highly condensed by viral core proteins (47–49). Like the herpes simplex virus 1 genome, the HAdV genome was shown to associate with H3.3 (50, 51), suggesting that HAdV DNA is chromatinized. The core/DNA complex enters the nucleus and is decondensed prior to the onset of early viral gene transcription (52). In this context, we recently identified the cellular chromatin remodeling complex Daxx/ATRX to be a negative regulator of HAdV infection. This Daxx/ATRX chromatin remodeling complex recruits HDACs to deacetylate histone tails, resulting in chromatin compaction and transcriptional repression (53). We demonstrated that this complex associates with the HAdV genome, thereby repressing active viral gene transcription (5, 6). Moreover, we recently identified SPOC1 to be a negative regulator of HAdV infection (54). SPOC1 is also involved in regulating the chromatin structure and DDR by modulating the chromatin association of DNA compaction factors (10). SPOC1 was shown to form a chromatin remodeling complex with KAP1, resulting in the recruitment of repressive components, such as HMT and NuRD (10).
Here we show that KAP1 overexpression negatively regulates HAdV progeny production. However, this negative effect is attenuated later during infection, suggesting that HAdV counteracts the KAP1 negative function to ensure proper virus replication. Additionally, we observed reduced levels of HAdV proteins upon KAP1 overexpression. Since chromatin immunoprecipitation analysis recently revealed an interaction of SPOC1 with HAdV5 promoters in transfected cells (54), KAP1-mediated repression might be facilitated via its recruitment to adenoviral promoters by the SPOC1 protein.
Extensive but so far unsuccessful approaches were conducted during this work to generate KAP1-depleted cell lines. Since KAP1 is involved in cell cycle progression and apoptosis, it is not surprising that KAP1 depletion has several downstream effects. Li and coworkers observed that KAP1 depletion increases apoptosis and has severe negative effects on cell proliferation rates (16). Additionally, Kepkay and coworkers observed an increase in PML-NBs upon KAP1 depletion (55). Although our KAP1-knockdown cells showed proliferation rates comparable to those of control cell lines, in agreement with the findings of Kepkay et al. (55), we observed increased PML protein levels upon KAP1 depletion. Since PML is known to play a role in the host cellular antiviral defense (3, 56), its upregulation probably compensates for the loss of KAP1, resulting in unaffected levels of HAdV progeny production. To further clarify the role of KAP1 depletion in HAdV productive infection, how KAP1 correlates with an altered PML-NB composition needs to be fully understood.
SUMOylation of a substrate is known to alter its inter- and/or intramolecular interactions, thereby modulating its stability, localization, or activity. In this context, it is suggested that SUMO modification of a target protein is associated with the recruitment of SUMO-interacting motif-containing effector proteins (57, 58). KAP1 represents a cellular SUMO E3 ligase that not only regulates its own SUMOylation status but also is able to promote SUMO conjugation on other cellular proteins, such as the cellular factor IRF7, hence reducing its transcriptional activity and ultimately resulting in a suppressed interferon-based antiviral response (46).
KAP1's corepressive capacity is flexibly regulated by Ser824 phosphorylation and SUMO modification. KAP1 phosphorylation results in chromatin decondensation, whereas KAP1 SUMOylation increased its repressive function through the recruitment of HMTs and NuRD (16, 59). Here we show that HAdV infection limits the levels of SUMO-KAP1. We speculate that SUMO-depleted KAP1 dissociates from SPOC1 to release the repressive HMTs and HDACs, facilitating H3K9 acetylation and demethylation, which lead to chromatin relaxation, transcriptional activation, and enhanced gene expression. We show that HAdV-mediated KAP1 deSUMOylation depends on the E1B-55K protein, known to contain an SCM and be the substrate for the host cell SUMO modification system (60, 61).
E1B-55K was also reported to interact with the cellular SUMO E2 enzyme Ubc9 (62). Studies from Pennella and coworkers suggest that E1B-55K is also a p53-SUMO1 E3 ligase (63). In line with this, we recently showed that E1B-55K SUMOylation and, hence, PML-NB localization are prerequisites for the SUMO ligase activity of the viral protein (31). However, the detailed mechanism of how E1B-55K mediates the SUMOylation of other proteins is still not fully understood.
For the first time, we have shown here that SUMO modification of E1B-55K is a prerequisite for efficient KAP1 deSUMOylation upon HAdV5 infection. Since the loss of SUMO-KAP1 did not occur in cells expressing the SUMOylation-deficient E1B-55K-K104R mutant, it is tempting to speculate that E1B-55K directly drives KAP1 deSUMOylation through the capture of KAP1-associated SUMO moieties for E1B-55K SUMOylation. Conversely, we also observed that KAP1 induces the SUMO2 modification of E1B-55K in a concentration-dependent manner. However, p53 showed no increase in SUMO modification, eliminating the notion that KAP1 mediates the unspecific SUMOylation of SCM-containing proteins. However, we observed that KAP1 SUMOylation itself is not a prerequisite for this process.
Since high KAP1 expression levels have been linked to various cancer types, including prometastatic cervical cancer, colorectal cancer, gastric cancer, and thyroid carcinoma (64–68), KAP1 has been proposed to be a target for anticancer therapy (69, 70). However, the role of KAP1 in tumorigenesis is still inconclusive and seems to be tissue specific, since KAP1 showed tumor-suppressive functions in early-stage lung cancer by inactivating oncogenic transcription factors (71–73). Furthermore, KAP1 was shown to play a role in the MDM2/p53/HDAC1 complex, thereby promoting deacetylation and MDM2-mediated degradation of p53 (70). This is promoted by the presence of melanoma antigen (MAGE) family proteins (74).
HAdV early oncoproteins are able to transform primary rodent cells (75–80). For efficient transformation, E1A supports cell cycle progression and induces the immortalization of primary rodent cells by modulating key regulators controlling cell cycle progression in the course of an abortive infection (81, 82). E1B contributes to a completely transformed phenotype (83–90). The tumorigenic functions of E1B-55K are mainly mediated by the functional inhibition of the tumor suppressor p53 (60, 61, 91–95). Here, SUMOylation of the oncoprotein E1B-55K is required for PML-IV/V interaction and p53/Daxx inhibition (5, 31, 62). Since we observed that E1B-55K SUMOylation is a prerequisite for its ability to limit KAP1 SUMO moieties, it is tempting to ask whether the SUMO status of KAP1 plays an important role during transformation of primary rodent cells.
KSHV was recently shown to exploit the KAP1 chromatin remodeling function via phosphorylation of KAP1 S824 by the viral protein kinase, leading to the activation of its lytic genes and subsequent lytic KSHV replication (96). Furthermore, KAP1 phosphorylation is thought to support the chronic inflammatory environment of Kaposi's sarcoma by activating STAT3 (97). Additionally, the latency-associated nuclear antigen (LANA) was shown to functionally interact with KAP1 to repress lytic gene expression during the early stage of KSHV infection, which may help establish KSHV latency after primary infections (25).
HAdV is also thought to establish long-term low-level persistent or even latent infections in nonpermissive cells (98–100). It is proposed that the viral genome is maintained in the cell in an unintegrated episomal state. This model might explain the high prevalence of HAdV infections in immunocompromised individuals. Whether KAP1 contributes to HAdV latency remains unclear. However, KAP1 is involved in regulating episomal gene expression through KRAB/KAP1-mediated histone modifications (101).
Alternatively, KAP1 might be involved in maintaining an HAdV latent state by repressing adenoviral episomal gene expression from specific viral promoters, viral protein stability, or viral splicing processes. Since KAP1 is phosphorylated upon DNA DSBs, in chemotherapy-treated patients, irradiation-mediated KAP1 phosphorylation could result in activation of HAdV lytic gene expression. Hence, KAP1 could play an important role in HAdV reactivation in immunocompromised patients, underlining the importance of considering this factor in cancer therapy.
In summary, our results reveal that adenovirus mediates the complex regulation of KAP1 corepressor and chromatin remodeler functions (Fig. 10). Based on our results, we suggest a model in which early in infection HAdV5 exploits KAP1 phosphorylation to generate a positive environment for virus replication and circumvent host cellular proapoptotic pathways. Consequently, HAdV-induced KAP1 phosphorylation and the loss of SUMO moieties promote decondensation of the cellular and viral genome, resulting in transcriptional activation and enhanced cellular and viral gene expression.
FIG 10.

Model of cross talk between KAP1 and E1B-55K. Schematic representation illustrating a proposed model linking the modulation of KAP1 PTMs by viral factor E1B-55K and alteration of the E1B-55K PTM by KAP1. Upon HAdV5 infection, KAP1 is phosphorylated and simultaneously deSUMOylated by SUMO-modified E1B-55K, resulting in chromatin decondensation and enhanced adenoviral gene expression due to dissociation of the repressive complex.
ACKNOWLEDGMENTS
We thank Ron Hay, Peggy Farnham, and Elisabeth Kremmer for providing reagents and Nicole Fischer, Tobias Schubert, Sarah Kinkley, and Hans Will for scientific discussions.
C.B. was supported by a grant from the Deutsche Forschungsgemeinschaft (DFG) to T.D. (DO 343/7-1). A.M. is supported by a Marie Curie Intra-European Fellowship for Career Development (project no. 627187). T.D. is supported by the DFG and the Wilhelm Sander-Stiftung. S.S. is supported by grants from the Dräger Stiftung, the Else Kröner-Fresenius-Stiftung, and the Deutsche Krebshilfe e.V. Part of this work was supported by the B. Braun Stiftung and the Fonds der Chemischen Industrie. The Heinrich Pette Institute, Leibniz Institute for Experimental Virology, is supported by the Freie und Hansestadt Hamburg and the Bundesministerium für Gesundheit (BMG).
REFERENCES
- 1.Berk AJ. 2005. Recent lessons in gene expression, cell cycle control, and cell biology from adenovirus. Oncogene 24:7673–7685. doi: 10.1038/sj.onc.1209040. [DOI] [PubMed] [Google Scholar]
- 2.Weitzman MD, Lilley CE, Chaurushiya MS. 2010. Genomes in conflict: maintaining genome integrity during virus infection. Annu Rev Microbiol 64:61–81. doi: 10.1146/annurev.micro.112408.134016. [DOI] [PubMed] [Google Scholar]
- 3.Everett RD, Chelbi-Alix MK. 2007. PML and PML nuclear bodies: implications in antiviral defence. Biochimie 89:819–830. doi: 10.1016/j.biochi.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 4.Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J Interferon Cytokine Res 31:145–158. doi: 10.1089/jir.2010.0111. [DOI] [PubMed] [Google Scholar]
- 5.Schreiner S, Wimmer P, Sirma H, Everett RD, Blanchette P, Groitl P, Dobner T. 2010. Proteasome-dependent degradation of Daxx by the viral E1B-55K protein in human adenovirus-infected cells. J Virol 84:7029–7038. doi: 10.1128/JVI.00074-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreiner S, Bürck C, Glass M, Groitl P, Wimmer P, Kinkley S, Mund A, Everett RD, Dobner T. 2013. Control of human adenovirus type 5 (Ad5) gene expression by cellular Daxx/ATRX chromatin-associated complexes. Nucleic Acids Res 41:3532–3550. doi: 10.1093/nar/gkt064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schreiner S, Wimmer P, Dobner T. 2012. Adenovirus degradation of cellular proteins. Future Microbiol 7:211–225. doi: 10.2217/fmb.11.153. [DOI] [PubMed] [Google Scholar]
- 8.Kinkley S, Staege H, Mohrmann G, Rohaly G, Schaub T, Kremmer E, Winterpacht A, Will H. 2009. SPOC1: a novel PHD-containing protein modulating chromatin structure and mitotic chromosome condensation. J Cell Sci 122:2946–2956. doi: 10.1242/jcs.047365. [DOI] [PubMed] [Google Scholar]
- 9.Iyengar S, Farnham PJ. 2011. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem 286:26267–26276. doi: 10.1074/jbc.R111.252569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mund A, Schubert T, Staege H, Kinkley S, Reumann K, Kriegs M, Fritsch L, Battisti V, Ait-Si-Ali S, Hoffbeck A, Soutoglou E, Will H. 2012. SPOC1 modulates DNA repair by regulating key determinants of chromatin compaction and DNA damage response. Nucleic Acids Res 40:11363–11379. doi: 10.1093/nar/gks868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, Shiloh Y. 2006. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol 8:870–876. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 12.Goodarzi AA, Kurka T, Jeggo PA. 2011. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat Struct Mol Biol 18:831–839. doi: 10.1038/nsmb.2077. [DOI] [PubMed] [Google Scholar]
- 13.Lee YK, Thomas SN, Yang AJ, Ann DK. 2007. Doxorubicin down-regulates Kruppel-associated box domain-associated protein 1 sumoylation that relieves its transcription repression on p21WAF1/CIP1 in breast cancer MCF-7 cells. J Biol Chem 282:1595–1606. doi: 10.1074/jbc.M606306200. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Lee YK, Jeng JC, Yen Y, Schultz DC, Shih HM, Ann DK. 2007. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem 282:36177–36189. doi: 10.1074/jbc.M706912200. [DOI] [PubMed] [Google Scholar]
- 15.Lee DH, Goodarzi AA, Adelmant GO, Pan Y, Jeggo PA, Marto JA, Chowdhury D. 2012. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J 31:2403–2415. doi: 10.1038/emboj.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, Lin HH, Chen H, Xu X, Shih HM, Ann DK. 2010. SUMOylation of the transcriptional co-repressor KAP1 is regulated by the serine and threonine phosphatase PP1. Sci Signal 3:ra32. doi: 10.1126/scisignal.2000781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Wang Z, Ghosh S, Zhou Z. 2013. Defective ATM-Kap-1-mediated chromatin remodeling impairs DNA repair and accelerates senescence in progeria mouse model. Aging Cell 12:316–318. doi: 10.1111/acel.12035. [DOI] [PubMed] [Google Scholar]
- 18.Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, Sadofsky MJ, Zhou MM, Rauscher FJ III. 2007. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28:823–837. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang Y, Fiskus W, Yong B, Atadja P, Takahashi Y, Pandita TK, Wang HG, Bhalla KN. 2013. Acetylated hsp70 and KAP1-mediated Vps34 SUMOylation is required for autophagosome creation in autophagy. Proc Natl Acad Sci U S A 110:6841–6846. doi: 10.1073/pnas.1217692110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ III. 1996. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10:2067–2078. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 21.Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, Rauscher FJ III. 1999. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol 19:4366–4378. doi: 10.1128/MCB.19.6.4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz DC, Friedman JR, Rauscher FJ III. 2001. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev 15:428–443. doi: 10.1101/gad.869501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ III. 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev 16:919–932. doi: 10.1101/gad.973302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allouch A, Di Primio C, Alpi E, Lusic M, Arosio D, Giacca M, Cereseto A. 2011. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 9:484–495. doi: 10.1016/j.chom.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Sun R, Liang D, Gao Y, Lan K. 2014. Kaposi's sarcoma-associated herpesvirus-encoded LANA interacts with host KAP1 to facilitate establishment of viral latency. J Virol 88:7331–7344. doi: 10.1128/JVI.00596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forrester NA, Patel RN, Speiseder T, Groitl P, Sedgwick GG, Shimwell NJ, Seed RI, Catnaigh PO, McCabe CJ, Stewart GS, Dobner T, Grand RJ, Martin A, Turnell AS. 2012. Adenovirus E4orf3 targets transcriptional intermediary factor 1gamma for proteasome-dependent degradation during infection. J Virol 86:3167–3179. doi: 10.1128/JVI.06583-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitsudomi T, Steinberg SM, Nau MM, Carbone D, D'Amico D, Bodner HK, Oie HK, Linnoila RI, Mulshine JL, Minna JD, Gazdar AF. 1992. p53 gene mutations in non-small-lung cell cancer cell lines and their correlation with the presence of ras mutations and clinical features. Oncogene 7:171–180. [PubMed] [Google Scholar]
- 28.Tatham MH, Rodriguez MS, Xirodimas DP, Hay RT. 2009. Detection of protein SUMOylation in vivo. Nat Protoc 4:1363–1371. doi: 10.1038/nprot.2009.128. [DOI] [PubMed] [Google Scholar]
- 29.Nevels M, Täuber B, Spruss T, Wolf H, Dobner T. 2001. “Hit-and-run” transformation by adenovirus oncogenes. J Virol 75:3089–3094. doi: 10.1128/JVI.75.7.3089-3094.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rubenwolf S, Schütt H, Nevels M, Wolf H, Dobner T. 1997. Structural analysis of the adenovirus type 5 E1B 55-kilodalton-E4orf6 protein complex. J Virol 71:1115–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wimmer P, Berscheminski J, Blanchette P, Groitl P, Branton PE, Hay RT, Dobner T, Schreiner S. 16 March 2015. PML isoforms IV and V contribute to adenovirus-mediated oncogenic transformation by functionally inhibiting the tumor-suppressor p53. Oncogene. doi: 10.1038/onc.2015.63. [DOI] [PubMed] [Google Scholar]
- 32.Groitl P, Dobner T. 2007. Construction of adenovirus type 5 early region 1 and 4 virus mutants. Methods Mol Med 130:29–39. [DOI] [PubMed] [Google Scholar]
- 33.Blanchette P, Kindsmuller K, Groitl P, Dallaire F, Speiseder T, Branton PE, Dobner T. 2008. Control of mRNA export by adenovirus E4orf6 and E1B55K proteins during productive infection requires E4orf6 ubiquitin ligase activity. J Virol 82:2642–2651. doi: 10.1128/JVI.02309-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kindsmuller K, Groitl P, Hartl B, Blanchette P, Hauber J, Dobner T. 2007. Intranuclear targeting and nuclear export of the adenovirus E1B-55K protein are regulated by SUMO1 conjugation. Proc Natl Acad Sci U S A 104:6684–6689. doi: 10.1073/pnas.0702158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harlow E, Franza BR Jr, Schley C. 1985. Monoclonal antibodies specific for adenovirus early region 1A proteins: extensive heterogeneity in early region 1A products. J Virol 55:533–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sarnow P, Hearing P, Anderson CW, Reich N, Levine AJ. 1982. Identification and characterization of an immunologically conserved adenovirus early region 11,000 Mr protein and its association with the nuclear matrix. J Mol Biol 162:565–583. doi: 10.1016/0022-2836(82)90389-8. [DOI] [PubMed] [Google Scholar]
- 37.Reich NC, Sarnow P, Duprey E, Levine AJ. 1983. Monoclonal antibodies which recognize native and denatured forms of the adenovirus DNA-binding protein. Virology 128:480–484. doi: 10.1016/0042-6822(83)90274-X. [DOI] [PubMed] [Google Scholar]
- 38.Marton MJ, Baim SB, Ornelles DA, Shenk T. 1990. The adenovirus E4 17-kilodalton protein complexes with the cellular transcription factor E2F, altering its DNA-binding properties and stimulating E1A-independent accumulation of E2 mRNA. J Virol 64:2345–2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kzhyshkowska J, Kremmer E, Hofmann M, Wolf H, Dobner T. 2004. Protein arginine methylation during lytic adenovirus infection. Biochem J 383:259–265. doi: 10.1042/BJ20040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nevels M, Täuber B, Kremmer E, Spruss T, Wolf H, Dobner T. 1999. Transforming potential of the adenovirus type 5 E4orf3 protein. J Virol 73:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wimmer P, Schreiner S, Everett RD, Sirma H, Groitl P, Dobner T. 2010. SUMO modification of E1B-55K oncoprotein regulates isoform-specific binding to the tumour suppressor protein PML. Oncogene 29:5511–5522. doi: 10.1038/onc.2010.284. [DOI] [PubMed] [Google Scholar]
- 42.Leppard KN, Shenk T. 1989. The adenovirus E1B 55 kd protein influences mRNA transport via an intranuclear effect on RNA metabolism. EMBO J 8:2329–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gunther T, Schreiner S, Dobner T, Tessmer U, Grundhoff A. 2014. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLoS Pathog 10:e1004274. doi: 10.1371/journal.ppat.1004274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berscheminski J, Wimmer P, Brun J, Ip WH, Groitl P, Horlacher T, Jaffray E, Hay RT, Dobner T, Schreiner S. 2014. Sp100 isoform-specific regulation of human adenovirus type 5 (Ad5) gene expression. J Virol 88:6076–6092. doi: 10.1128/JVI.00469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng CT, Kuo CY, Ann DK. 2014. KAPtain in charge of multiple missions: emerging roles of KAP1. World J Biol Chem 5:308–320. doi: 10.4331/wjbc.v5.i3.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang Q, Deng H, Li X, Wu X, Tang Q, Chang TH, Peng H, Rauscher FJ III, Ozato K, Zhu F. 2011. Tripartite motif-containing protein 28 is a small ubiquitin-related modifier E3 ligase and negative regulator of IFN regulatory factor 7. J Immunol 187:4754–4763. doi: 10.4049/jimmunol.1101704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chatterjee PK, Vayda ME, Flint SJ. 1986. Identification of proteins and protein domains that contact DNA within adenovirus nucleoprotein cores by ultraviolet light crosslinking of oligonucleotides 32P-labelled in vivo. J Mol Biol 188:23–37. doi: 10.1016/0022-2836(86)90477-8. [DOI] [PubMed] [Google Scholar]
- 48.Matsumoto K, Nagata K, Ui M, Hanaoka F. 1993. Template activating factor I, a novel host factor required to stimulate the adenovirus core DNA replication. J Biol Chem 268:10582–10587. [PubMed] [Google Scholar]
- 49.Okuwaki M, Nagata K. 1998. Template activating factor-I remodels the chromatin structure and stimulates transcription from the chromatin template. J Biol Chem 273:34511–34518. doi: 10.1074/jbc.273.51.34511. [DOI] [PubMed] [Google Scholar]
- 50.Placek BJ, Huang J, Kent JR, Dorsey J, Rice L, Fraser NW, Berger SL. 2009. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J Virol 83:1416–1421. doi: 10.1128/JVI.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ross PJ, Kennedy MA, Christou C, Risco Quiroz M, Poulin KL, Parks RJ. 2011. Assembly of helper-dependent adenovirus DNA into chromatin promotes efficient gene expression. J Virol 85:3950–3958. doi: 10.1128/JVI.01787-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Komatsu T, Haruki H, Nagata K. 2011. Cellular and viral chromatin proteins are positive factors in the regulation of adenovirus gene expression. Nucleic Acids Res 39:889–901. doi: 10.1093/nar/gkq783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. 2002. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J Cell Sci 115:3319–3330. [DOI] [PubMed] [Google Scholar]
- 54.Schreiner S, Kinkley S, Bürck C, Mund A, Wimmer P, Schubert T, Groitl P, Will H, Dobner T. 2013. SPOC1-mediated antiviral host cell response is antagonized early in human adenovirus type 5 infection. PLoS Pathog 9:e1003775. doi: 10.1371/journal.ppat.1003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kepkay R, Attwood KM, Ziv Y, Shiloh Y, Dellaire G. 2011. KAP1 depletion increases PML nuclear body number in concert with ultrastructural changes in chromatin. Cell Cycle 10:308–322. doi: 10.4161/cc.10.2.14551. [DOI] [PubMed] [Google Scholar]
- 56.Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim Biophys Acta 1783:2207–2221. doi: 10.1016/j.bbamcr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 57.Kerscher O, Felberbaum R, Hochstrasser M. 2006. Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu Rev Cell Dev Biol 22:159–180. doi: 10.1146/annurev.cellbio.22.010605.093503. [DOI] [PubMed] [Google Scholar]
- 58.Song J, Durrin LK, Wilkinson TA, Krontiris TG, Chen Y. 2004. Identification of a SUMO-binding motif that recognizes SUMO-modified proteins. Proc Natl Acad Sci U S A 101:14373–14378. doi: 10.1073/pnas.0403498101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JH, Paull TT. 2007. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26:7741–7748. doi: 10.1038/sj.onc.1210872. [DOI] [PubMed] [Google Scholar]
- 60.Endter C, Hartl B, Spruss T, Hauber J, Dobner T. 2005. Blockage of CRM1-dependent nuclear export of the adenovirus type 5 early region 1B 55-kDa protein augments oncogenic transformation of primary rat cells. Oncogene 24:55–64. doi: 10.1038/sj.onc.1208170. [DOI] [PubMed] [Google Scholar]
- 61.Endter C, Kzhyshkowska J, Stauber R, Dobner T. 2001. SUMO-1 modification required for transformation by adenovirus type 5 early region 1B 55-kDa oncoprotein. Proc Natl Acad Sci U S A 98:11312–11317. doi: 10.1073/pnas.191361798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wimmer P, Blanchette P, Schreiner S, Ching W, Groitl P, Berscheminski J, Branton PE, Will H, Dobner T. 2013. Cross-talk between phosphorylation and SUMOylation regulates transforming activities of an adenoviral oncoprotein. Oncogene 32:1626–1637. doi: 10.1038/onc.2012.187. [DOI] [PubMed] [Google Scholar]
- 63.Pennella MA, Liu Y, Woo JL, Kim CA, Berk AJ. 2010. Adenovirus E1B 55-kilodalton protein is a p53-SUMO1 E3 ligase that represses p53 and stimulates its nuclear export through interactions with promyelocytic leukemia nuclear bodies. J Virol 84:12210–12225. doi: 10.1128/JVI.01442-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hector S, Chen H, Kijanka G, Murray F, Prehn JH. 2012. A reverse-ELISA for the detection of TRIM28/KAP1 serum autoantibodies in colorectal cancer patients. Acta Oncol 51:394–396. doi: 10.3109/0284186X.2011.652742. [DOI] [PubMed] [Google Scholar]
- 65.Lin LF, Li CF, Wang WJ, Yang WM, Wang DD, Chang WC, Lee WH, Wang JM. 2013. Loss of ZBRK1 contributes to the increase of KAP1 and promotes KAP1-mediated metastasis and invasion in cervical cancer. PLoS One 8:e73033. doi: 10.1371/journal.pone.0073033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang YY, Li L, Zhao ZS, Wang HJ. 2013. Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J Surg Oncol 11:81. doi: 10.1186/1477-7819-11-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yokoe T, Toiyama Y, Okugawa Y, Tanaka K, Ohi M, Inoue Y, Mohri Y, Miki C, Kusunoki M. 2010. KAP1 is associated with peritoneal carcinomatosis in gastric cancer. Ann Surg Oncol 17:821–828. doi: 10.1245/s10434-009-0795-8. [DOI] [PubMed] [Google Scholar]
- 68.Martins MB, Marcello MA, Morari EC, Cunha LL, Soares FA, Vassallo J, Ward LS. 2013. Clinical utility of KAP-1 expression in thyroid lesions. Endocr Pathol 24:77–82. doi: 10.1007/s12022-013-9245-z. [DOI] [PubMed] [Google Scholar]
- 69.Okamoto K, Kitabayashi I, Taya Y. 2006. KAP1 dictates p53 response induced by chemotherapeutic agents via Mdm2 interaction. Biochem Biophys Res Commun 351:216–222. doi: 10.1016/j.bbrc.2006.10.022. [DOI] [PubMed] [Google Scholar]
- 70.Wang C, Ivanov A, Chen L, Fredericks WJ, Seto E, Rauscher FJ III, Chen J. 2005. MDM2 interaction with nuclear corepressor KAP1 contributes to p53 inactivation. EMBO J 24:3279–3290. doi: 10.1038/sj.emboj.7600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen L, Chen DT, Kurtyka C, Rawal B, Fulp WJ, Haura EB, Cress WD. 2012. Tripartite motif containing 28 (Trim28) can regulate cell proliferation by bridging HDAC1/E2F interactions. J Biol Chem 287:40106–40118. doi: 10.1074/jbc.M112.380865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang C, Martin S, Pfleger C, Du J, Buckner JH, Bluestone JA, Riley JL, Ziegler SF. 2013. Cutting edge: a novel, human-specific interacting protein couples FOXP3 to a chromatin-remodeling complex that contains KAP1/TRIM28. J Immunol 190:4470–4473. doi: 10.4049/jimmunol.1203561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsuruma R, Ohbayashi N, Kamitani S, Ikeda O, Sato N, Muromoto R, Sekine Y, Oritani K, Matsuda T. 2008. Physical and functional interactions between STAT3 and KAP1. Oncogene 27:3054–3059. doi: 10.1038/sj.onc.1210952. [DOI] [PubMed] [Google Scholar]
- 74.Yang B, O'Herrin SM, Wu J, Reagan-Shaw S, Ma Y, Bhat KM, Gravekamp C, Setaluri V, Peters N, Hoffmann FM, Peng H, Ivanov AV, Simpson AJ, Longley BJ. 2007. MAGE-A, mMage-b, and MAGE-C proteins form complexes with KAP1 and suppress p53-dependent apoptosis in MAGE-positive cell lines. Cancer Res 67:9954–9962. doi: 10.1158/0008-5472.CAN-07-1478. [DOI] [PubMed] [Google Scholar]
- 75.Branton PE, Bayley ST, Graham FL. 1985. Transformation by human adenoviruses. Biochim Biophys Acta 780:67–94. [DOI] [PubMed] [Google Scholar]
- 76.Gaggar A, Shayakhmetov DM, Lieber A. 2003. CD46 is a cellular receptor for group B adenoviruses. Nat Med 9:1408–1412. doi: 10.1038/nm952. [DOI] [PubMed] [Google Scholar]
- 77.Graham FL, Rowe DT, McKinnon R, Bacchetti S, Ruben M, Branton PE. 1984. Transformation by human adenoviruses. J Cell Physiol Suppl 3:151–163. [DOI] [PubMed] [Google Scholar]
- 78.Täuber B, Dobner T. 2001. Adenovirus early E4 genes in viral oncogenesis. Oncogene 20:7847–7854. doi: 10.1038/sj.onc.1204914. [DOI] [PubMed] [Google Scholar]
- 79.Ricciardi RP. 1995. Transformation and tumorigenesis mediated by the adenovirus E1A and E1B oncogenes, p 195–210. In Barbanti-Brodano G. (ed), DNA tumor viruses: oncogenic mechanisms. Plenum Press, New York, NY. [Google Scholar]
- 80.Nevins JR, Vogt PK. 1996. Cell transformation by viruses, p 301–343. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 3rd ed Lippincott-Raven, New York, NY. [Google Scholar]
- 81.Gallimore PH, Byrd PJ, Grand RJ. 1984. Adenovirus genes involved in transformation. What determines the oncogenic phenotype?, p 125–172. In Rigby PWJ. (ed), Viruses and Cancer Symposium of the Society for General Microbiology. Cambridge University Press, Cambridge, United Kingdom. [Google Scholar]
- 82.Gallimore PH, Byrd PJ, Grand RJ, Whittaker JL, Breiding D, Williams J. 1984. An examination of the transforming and tumor-inducing capacity of a number of adenovirus type 12 early region 1, host-range mutants and cells transformed by subgenomic fragments of Ad12 E1 region. Cancer Cells 2:519–526. [Google Scholar]
- 83.Debbas M, White E. 1993. Wild-type p53 mediates apoptosis by E1A, which is inhibited by E1B. Genes Dev 7:546–554. doi: 10.1101/gad.7.4.546. [DOI] [PubMed] [Google Scholar]
- 84.Grand RJ, Grant ML, Gallimore PH. 1994. Enhanced expression of p53 in human cells infected with mutant adenoviruses. Virology 203:229–240. doi: 10.1006/viro.1994.1480. [DOI] [PubMed] [Google Scholar]
- 85.Lowe SW, Ruley HE. 1993. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 7:535–545. doi: 10.1101/gad.7.4.535. [DOI] [PubMed] [Google Scholar]
- 86.Mymryk JS, Shire K, Bayley ST. 1994. Induction of apoptosis by adenovirus type 5 E1A in rat cells requires a proliferation block. Oncogene 9:1187–1193. [PubMed] [Google Scholar]
- 87.Ruley HE. 1983. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature 304:602–606. doi: 10.1038/304602a0. [DOI] [PubMed] [Google Scholar]
- 88.Sabbatini P, Chiou SK, Rao L, White E. 1995. Modulation of p53-mediated transcriptional repression and apoptosis by the adenovirus E1B 19K protein. Mol Cell Biol 15:1060–1070. doi: 10.1128/MCB.15.2.1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Samuelson AV, Lowe SW. 1997. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc Natl Acad Sci U S A 94:12094–12099. doi: 10.1073/pnas.94.22.12094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Turnell AS, Grand RJ, Gorbea C, Zhang X, Wang W, Mymryk JS, Gallimore PH. 2000. Regulation of the 26S proteasome by adenovirus E1A. EMBO J 19:4759–4773. doi: 10.1093/emboj/19.17.4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kao CC, Yew PR, Berk AJ. 1990. Domains required for in vitro association between the cellular p53 and the adenovirus 2 E1B 55K proteins. Virology 179:806–814. doi: 10.1016/0042-6822(90)90148-K. [DOI] [PubMed] [Google Scholar]
- 92.Martin ME, Berk AJ. 1998. Adenovirus E1B 55K represses p53 activation in vitro. J Virol 72:3146–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martin ME, Berk AJ. 1999. Corepressor required for adenovirus E1B 55,000-molecular-weight protein repression of basal transcription. Mol Cell Biol 19:3403–3414. doi: 10.1128/MCB.19.5.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sarnow P, Ho YS, Williams J, Levine AJ. 1982. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 28:387–394. doi: 10.1016/0092-8674(82)90356-7. [DOI] [PubMed] [Google Scholar]
- 95.Yew PR, Liu X, Berk AJ. 1994. Adenovirus E1B oncoprotein tethers a transcriptional repression domain to p53. Genes Dev 8:190–202. doi: 10.1101/gad.8.2.190. [DOI] [PubMed] [Google Scholar]
- 96.Chang PC, Fitzgerald LD, Van Geelen A, Izumiya Y, Ellison TJ, Wang DH, Ann DK, Luciw PA, Kung HJ. 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res 69:5681–5689. doi: 10.1158/0008-5472.CAN-08-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.King CA. 2013. Kaposi's sarcoma-associated herpesvirus kaposin B induces unique monophosphorylation of STAT3 at serine 727 and MK2-mediated inactivation of the STAT3 transcriptional repressor TRIM28. J Virol 87:8779–8791. doi: 10.1128/JVI.02976-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Garnett CT, Talekar G, Mahr JA, Huang W, Zhang Y, Ornelles DA, Gooding LR. 2009. Latent species C adenoviruses in human tonsil tissues. J Virol 83:2417–2428. doi: 10.1128/JVI.02392-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gustafsson B, Huang W, Bogdanovic G, Gauffin F, Nordgren A, Talekar G, Ornelles DA, Gooding LR. 2007. Adenovirus DNA is detected at increased frequency in Guthrie cards from children who develop acute lymphoblastic leukaemia. Br J Cancer 97:992–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kosulin K, Haberler C, Hainfellner JA, Amann G, Lang S, Lion T. 2007. Investigation of adenovirus occurrence in pediatric tumor entities. J Virol 81:7629–7635. doi: 10.1128/JVI.00355-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barde I, Laurenti E, Verp S, Groner AC, Towne C, Padrun V, Aebischer P, Trumpp A, Trono D. 2009. Regulation of episomal gene expression by KRAB/KAP1-mediated histone modifications. J Virol 83:5574–5580. doi: 10.1128/JVI.00001-09. [DOI] [PMC free article] [PubMed] [Google Scholar]