

Abstract
Allosteric modulators of G protein-coupled receptors (GPCRs) have a number of potential advantages compared to agonists or antagonists that bind to the orthosteric site of the receptor. These include the potential or receptor selectivity, maintenance of the temporal and spatial fidelity of signaling in vivo, the ceiling effect of the allosteric cooperativity which may prevent overdose issues, and engendering bias by differentially modulating distinct signaling pathways. Here we describe the discovery, synthesis, and molecular pharmacology of δ-opioid receptor-selective positive allosteric modulators (δ PAMs). These δ PAMs increase the affinity and/or efficacy of the orthosteric agonists leu-enkephalin, SNC80 and TAN67, as measured by receptor binding, G protein activation, β-arrestin recruitment, adenylyl cyclase inhibition, and extracellular signal-regulated kinases (ERK) activation. As such, these compounds are useful pharmacological tools to probe the molecular pharmacology of the δ receptor and to explore the therapeutic potential of δ PAMs in diseases such as chronic pain and depression.
Graphical abstract

Introduction
The δ-opioid receptor is a seven transmembrane domain (7TMD) receptor that belongs to the class A family of G protein-coupled receptors (GPCRs). Agonists of the δ receptor have been shown to be antinociceptive especially in chronic pain models1 and to have potential as antidepressant agents.2 The possible dual effects of δ receptor agonists to alleviate chronic pain and mitigate emotional disorders provide a particularly attractive therapeutic strategy because of the high level of comorbidity between chronic pain and depression. However, agonists acting directly at the δ receptor can show proconvulsant effects in animal models, including non-human primates. Indeed, it has been proposed that these seizurogenic properties of δ receptor agonists may be responsible for their antidepressant-like activity analogous to electroconvulsive therapy.3 On the other hand, slowing the rate of administration of the δ receptor agonist SNC80 reduces seizurogenic activity but has no effect on anti-depressant-like effects.4 Also, some δ receptor agonists (e.g., ADL5859) show no seizures in rat or mouse models.5 These and other findings suggest that the convulsive properties of δ receptor agonists can be separated from their antidepressant-like effects.6–8
Allosteric modulators for GPCRs bind to a site on the receptor that is topographically distinct from the site that binds the orthosteric (or endogenous) agonist. Positive allosteric modulators (PAMs) increase the affinity and/or efficacy of bound orthosteric agonist ligands. The operational model of allosterism allows the quantification of allosteric effects, and as such, it can estimate the binding affinity of the allosteric ligand to the free receptor (pKB), the allosteric cooperativity factor (αβ), as well as any intrinsic agonist efficacy (τB) of the allosteric ligand. PAMs that have little or no intrinsic efficacy (τB) but modulate the orthosteric agonist response have a number of advantages over orthosteric ligands.9–11 In particular, these PAMs can theoretically maintain the temporal and spatial fidelity of endogenous receptor activation in vivo. The allosteric modulator binds to the target receptor but remains effectively silent until the endogenous orthosteric agonist is presented to the receptor. Therefore, PAMs can amplify the effect of endogenous signaling molecules without disrupting normal physiological regulation of receptor activation and might therefore be expected to exhibit superior efficacy and side effect profiles compared to traditional orthosteric agonists. Studies with δ receptor selective ligands, or utilizing a genetic deletion of the δ receptor,1 suggest that native opioid peptide signaling at the δ receptor mediates an increase in pain threshold in models of chronic pain and modulates mood states in rodent models.12 Therefore, positive allosteric modulation of the δ receptor should enhance responses to the endogenous agonist peptides and thereby be therapeutically efficacious. In addition, the finite nature of the agonist potency shift (defined by the allosteric cooperativity factor), which saturates when the allosteric site is fully occupied, may increase the safety margin between therapeutic effect and possible side effects associated with overactivation of the target receptor. Finally, and pertinent to the δ-receptor system which is known to exhibit ligand-biased signaling,13 PAMs can modulate the signaling bias of receptor activation toward desired pathways or engender bias from previously unbiased ligands. 14, 15 Thus, δ PAMs may provide a greater therapeutic window between pain relieving and antidepressant-like effects and proconvulsive activity, compared with traditional δ receptor orthosteric agonists.
In this study we report the synthesis and structure–activity relationships (SAR) of the first described δ PAMs. One of the most potent compounds identified, 3,3,6,6-tetramethyl-9-(4-((2-methylbenzyl)oxy)phenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (2, BMS-986187), was further characterized in radioligand binding assays and using a range of cellular functional assays. 2 was shown to positively modulate orthosteric agonist binding affinity and functional potency at the δ receptor and enhance the efficacy of the partial agonist TAN67.
Results
Discovery and Structure–Activity Relationship (SAR) of δ Receptor PAMs
The δ PAM chemotype was identified from a high throughput screen (HTS) using a β-arrestin recruitment assay in a PathHunter U2OS cell line coexpressing μ and δ receptors (U2OS-OPRM1D1) (DiscoveRx, Fremont, CA).16, 17 The screen was executed in PAM mode by measuring activity in the presence of an EC10 concentration of both endomorphin I (a μ-receptor-selective agonist) and leu-enkephalin which in this assay and cell line was a relatively selective agonist for the δ receptor.18 Typically, when using HTS approaches to identify PAMs, an EC20–40 concentration of orthosteric agonist is used.19 However, in this HTS the sum of the two EC10 concentrations of agonists offered a compromise between the detection of both μ and δ receptor PAMs and the ability to maintain the overall signal window so that lower efficacy partial agonists could also be detected. Follow-up in vitro testing to determine structural features necessary for PAM activity was performed utilizing CHO-PathHunter cell lines (CHO-OPRD1 and CHO-OPRM1) obtained from DiscoveRx. Concentration-response curves (CRCs) for HTS hits were determined both in agonist mode (in the absence of orthosteric agonist) to determine agonist activity of the test compounds, and in PAM mode (in the presence of an EC20 concentration of orthosteric agonist) to determine allosteric modulator activity using the β-arrestin recruitment assays. Compound 7 (Table 1) was identified as a δ PAM, producing a robust potentiation of the response to an EC20 concentration of leu-enkephalin.
Table 1. Structure-Activity Relationship of the δ-PAM Chemotype in PathHunter CHO-OPRD1 and CHO-OPRM1 Cells in a β-Arrestin Recruitment Assaya.
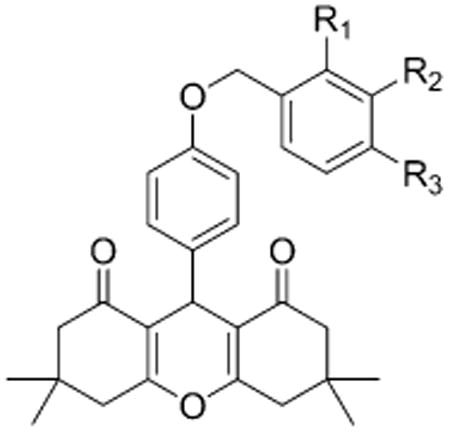
| ||||||
|---|---|---|---|---|---|---|
|
| ||||||
| EC50 (μM) (% Ymax) | ||||||
|
|
||||||
| compd | R1 | R2 | R3 | δ | μ | selectivity μ/δ |
| 1 | H | H | H | 0.2 (126) | 6 (130) | 30 |
| 2 (BMS-986187) | CH3 | H | H | 0.03 (124) | 3 (121) | 100 |
| 3 | H | CH3 | H | 0.2 (136) | 3 (120) | 15 |
| 4 | H | H | CH3 | 0.3 (92) | 4 (65) | 13 |
| 5 | F | H | H | 0.1 (120) | 5 (107) | 50 |
| 6 | H | F | H | 1 (136) | 7 (182) | 7 |
| 7 | H | H | F | 0.1 (95) | 2 (72) | 20 |
| 8 | Cl | H | H | 0.3 (68) | >10 (>100) | >33 |
| 9 | Cl | Cl | H | 0.1 (116) | >10 (>87) | >100 |
| 10 (BMS-986188) | Br | H | H | 0.05 (58) | >10 (>20) | >200 |
| 11 | OCHF2 | H | H | 0.2 (109) | 3 (80) | 15 |
| 12 | OCF3 | H | H | 0.3 (114) | 2 (111) | 7 |
| 13 | S02CH3 | H | H | 0.9 (102) | 5 (42) | 6 |
| 14 | CH2OH | H | H | 1 (92) | 10 (112) | 10 |
| 15 | CF3 | H | H | >10 (>40) | >10 (>20) | |
No activity was observed in agonist mode (in the absence of orthosteric agonist (data not shown)). In PAM mode (in the presence of an EC20 of leu-enkephalin for OPRD1 cells or an EC20 of endomorphin I for OPRM1 cells), robust responses were observed. The mean EC50 values, Ymax values, and potency ratio of δ receptor activity/μ receptor activity in PAM mode are reported in the table (n = 3).
As shown in Scheme 1, we synthesized a series of close analogs of 7 to optimize δ PAM potency and selectivity. None of the compounds exhibited significant agonist activity in a β-arrestin recruitment assay, but all of the compounds produced measurable PAM activity at the δ receptor. 1 with an unsubstituted benzyl ring acted as a δ PAM with an EC50 value of 0.2 μM and showed 30-fold selectivity in the β-arrestin recruitment assay compared with PAM activity at the μ receptor. Introduction of a methyl group in various positions around the phenyl ring (2–4) suggested that ortho substitution increased δ receptor PAM activity by an order of magnitude, with minimal effect on μ receptor PAM activity, while meta and para substitution did not significantly affect δ or μ receptor PAM activity. The corresponding ortho-F analog 5 was not significantly more active than 1, suggesting that the increased δ receptor activity with the o-methyl was due to a steric rather than an electronic effect. Similarly, the meta- and para-F analogs 6 and 7 or the ortho-Cl analog 8 did not afford an increase in δ receptor activity. Introduction of a second Cl group in the meta position (9) provided a modest improvement in δ receptor activity while maintaining selectivity. A more pronounced effect was observed with the ortho-Br analog 10 which produced equipotent PAM activity to 2 at the δ receptor but no observable PAM activity at the μ receptor, suggesting that 9 - (4-((2-bromobenzyl)oxy)phenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (10, BMS-986188) is the most δ receptor-selective analog we have identified to date. The effect of ortho substitution on δ receptor PAM potency and selectivity appears to be restricted to small substituents. As shown with analogs 11–15, larger ortho sub-stituents did not improve δ PAM activity and had no effect on selectivity. Similarly, more drastic changes to the chemotype, such as increasing the chain length between the ether oxygen and the phenyl ring, or replacement of the benzyl ether with a phenyl amide, yielded a significant loss in δ receptor PAM activity (data not shown). The most potent δ PAM identified was 2, which in the presence of an EC20 of leu-enkephalin produced a β-arrestin response with an average EC50 of 33 nM in CHO-OPRD1 cells (Table 1). Representative agonist and PAM mode CRCs for 2 at the μ and δ receptor are shown in Figure 1. In this example, 2 produced little or no activity in agonist mode, but in PAM mode (in the presence of an EC20 of leu-enkephalin (in CHO-OPRD1 cells) or endomorphin 1 (in CHO-OPRM1 cells)) produced a response with an EC50 of 48 nM in CHO-OPRD1 cells and 2 μM in CHO-OPRM1 cells.
Scheme 1.

Figure 1.
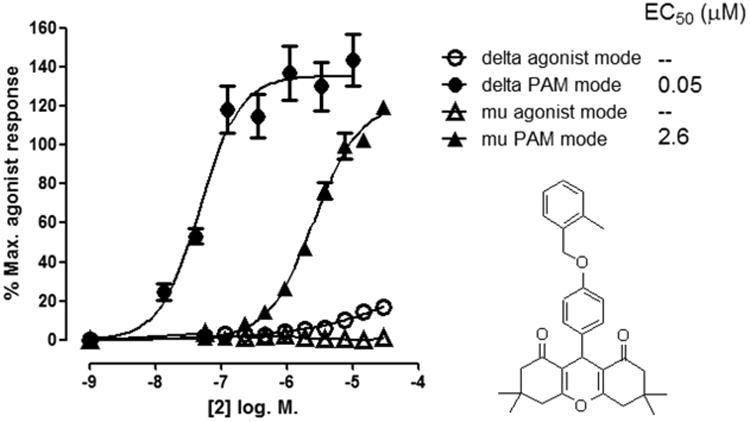
β-Arrestin recruitment response to 2 in agonist mode (in the absence of orthosteric agonist) and in PAM mode (in the presence of an EC20 of orthosteric agonist) in PathHunter cells expressing δ receptors (CHO-OPRD1) and μ receptors (CHO-OPRM1). For CHO-OPRD1 cells the orthosteric agonist was leu-enkephalin, and for CHO-OPRM1 cells the orthosteric agonist was endomorphin I. In PAM mode, the EC20 response of orthosteric agonist was normalized to 0%. 100% represents the response to a maximally effective concentration of orthosteric agonist. Data are presented as the mean ± SEM, n = 4.
Multivariate statistical analysis of a relatively large number of physicochemical properties calculated for the 15 compounds listed in Table 1 (see Methods and Materials for details) revealed specific properties of the 15 compounds that were most likely to predict EC50 values at either δ (Table S3) or μ (Table S4) opioid receptors in line with experimental data. Specifically, the total solvent accessible surface area (SASA, in square angstroms using a probe with a 1.4 Å radius), the π (carbon and attached hydrogen) component of the SASA (PISA), and the parameterized model number 3 (PM3) calculated ionization potential (IP.ev.) allow fitting of the predicted EC50 values at the δ opioid receptor (Figure S1). Similarly, the molecular weight of the molecule (mol.MW), the van der Waals surface area of polar nitrogen and oxygen atoms and carbonyl carbon atoms (PSA), and the number of nitrogen and oxygen atoms (X.N and O) were identified as the best predictors for EC50 values at the μ opioid receptor (Figure S2). An estimation of the δ/μ selectivity based on the ratio of the predicted EC50 values at the δ and μ opioid receptors revealed a definite separation between compounds with a selectivity above or below a 33-fold cutoff (Figure S3).
Binding Characterization of 2
2 (at concentrations up to 30 μM) does not inhibit binding of the orthosteric antagonist 3H-diprenorphine (DPN) to CHO-hDOPr cell membranes, suggesting that 2 is acting at an allosteric site to produce agonist and PAM activity (Figure 2A). However, in competition binding experiments 10 μM 2 increased the affinity of the orthosteric agonists, leu-enkephalin (Figure 2B), SNC80 (Figure 2C), and TAN67 (Figure 2D) to displace 3H-DPN. This suggests that 2 is an affinity modulator (the α component of the cooperativity factor) in the system tested (Table 2). The affinity shift with the partial agonist TAN67 is less than that seen with the full agonists leu-enkephalin and SNC80 (Table 2).
Figure 2.

Effect of 2 on 3H-diprenorphine (DPN) binding (A) and the effect of 10 μM 2 on leu-enkephalin (B), SNC80 (C), and TAN67 (D) competition binding curves in CHO-hDOPr membranes. Ki values are shown in Table 2. Data are presented as the mean ± SEM of three experiments.
Table 2. Effect of 2 (10 μM) on Orthosteric Agonist Competition Binding Ki Values in CHO-hDOPr Cell Membranesa.
| ligand | Ki (nM) (95% CI (nM)) | affinity ratio (vehicle Ki)/(2 Ki) | |
|---|---|---|---|
|
| |||
| with vehicle | with 2 (10 μM) | ||
| leu-enkephalin | 221 (119–324) | 7(3–12) | 32 |
| SNC80 | 71 (20–122) | 5(3–7) | 14 |
| TAN67 | 10 (7–14) | 3 (0.2–5.8) | 3 |
2 had no effect on 3H-DPN binding (see Figure 2) but increased the affinity of orthosteric agonist competition binding curves. Data are presented as the mean ± SEM of three experiments.
Functional Characterization of 2
The PAM activity of 2 was further characterized in four different functional assays. In the CHO-OPRD1 PathHunter cells, 2 effects on leu-enkephalin potency and efficacy were studied in both β-arrestin recruitment assays and inhibition of forskolin-stimulated cAMP accumulation assays. Unlike the U2OS cell lines used in the HTS, where forskolin was relatively ineffective at stimulating adenylyl cyclase activity, the recombinant CHO PathHunter cell lines allowed us to investigate both β-arrestin recruitment and inhibition of forskolin-stimulated cAMP accumulation in the same cell line. In the β-arrestin recruitment assay, 2 alone (up to 10 μM) produced only marginal agonist activity (∼10% of a maximal response to leu-enkephalin) but produced a robust 18-fold increase in the potency of leu-enkephalin (Figure 3A). A small increase in the maximal response to leu-enkephalin with 2, relative to leu-enkephalin alone, was also observed. This suggests that 2 is a PAM with little or no intrinsic efficacy in this system. In contrast, in the inhibition of forskolin-stimulated cAMP assay, 2 alone produced robust activity resulting in full inhibition of cAMP accumulation at concentrations above 3 μM (Figure 3B). At lower concentrations, 2 increased the potency of leu-enkephalin. At a 370 nM concentration of 2 (the highest concentration at which a potency for leu-enkephalin could be determined) the potency of leu-enkephalin was increased by 56-fold. Similar findings were observed using the small molecule orthosteric agonist SNC80 in these two assays (Table 3).
Figure 3.
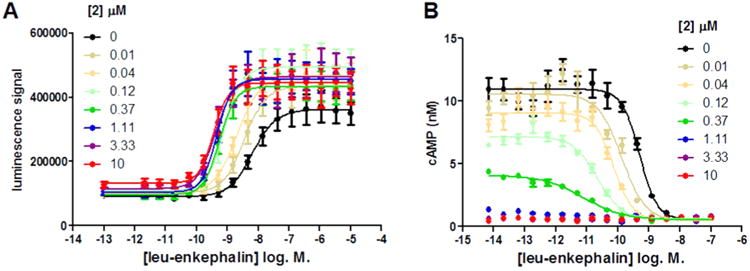
Effect of increasing concentrations of 2 on leu-enkephalin concentration–response curves in β-arrestin recruitment (A) and in inhibition of forskolin-stimulated cAMP accumulation (B) in CHO-OPRD1 cells. Data are presented as the mean ± SEM of four experiments. Data were fitted to the operational model of allosterism (see Table 3).
Table 3. Allosteric Parameters for 2 at the δ Receptora.
| leu-enkephalin | SNC80 | TAN67 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||
| β-arr | GTPγS | cAMP | pERK | β-arr | GTPγS | cAMP | pERK | GTPγS | pERK | |
| logτA | 028 ± 0.04 | 094 ± 006 | 2.8 ± 0.03 | 025 ± 016 | 0.66 ± 0.08 | 0.56 ± 0.10 | 0.86 ± 0.03 | 1.06 ± 0.15 | 0.67 ± 0.09 | 0.22 ± 0.07 |
| logτB | −0.73 ± 0.15 | 0.36 ± 0.17 | 1.45 ± 0.27 | −0.16 ± 0.15 | −1.06 ± 0.11 | 0.29 ± 0.20 | 1.01 ± 0.02 | −0.04 ± 015 | 0.45 ± 0.07 | 0.11 ± 0.08 |
| pKA | 7.90** | 6.6** | 6.7* | 9.13** | 6.38 ± 0.11 | 7.7* | 7.7* | 7.7* | 7.6* | 8.4** |
| pKB | 7.06 ±0.11 | 5.85 ± 0.23 | 5.45 ± 0.02 | 6.13 ± 0.30 | 6.45 ± 0.05 | 6.23 ± 0.32 | 5.52 ± 0.02 | 5.66 ± 0.30 | 6.0*** | 5.80 ± 0.32 |
| log αβ (αβ) | 1.18 ± 0.07 (15) | 1.67 ± 0.21 (47) | 2.80 ± 0.08 (631) | 0.99 ± 0.28 (10) | 1.33 ± 0.05 (21) | 1.00 ± 0.30 (10) | 2.11 ± 0.07 (129) | 0.89 ± 0.29 (8) | 1.11 ± 0.19 (13) | 1.41 ± 0.29 (26) |
Values for affinity, efficacy, and allosteric cooperativity for orthosteric ligands and 2 are derived from the operational model of allosterism. Three different orthosteric agonists were used (leu-enkephalin, SNC80, and TAN67), across up to four functional assays (β-arrestin recruitment, [35S]GTPγS binding, cAMP inhibition, and pERK). In the model τA and τB represent the efficacy of the orthosteric agonist and allosteric modulator, respectively; pKA and pKB represent the binding affinity of the orthosteric agonist and the allosteric modulator, respectively, to the free receptor; and αβ represents the composite allosteric cooperativity factor. Data are presented as the mean ± SEM of three to seven experiments.
pKA is fixed to its equilibrium binding affinity, as ligand is a full agonist in all end points tested.
pKA of leu-enkephalin and TAN67 in end points where they are partial agonists was obtained from fitting their CRC to the operational model of agonism to obtain a functional affinity in each end point tested.
the pKB for TAN67 in [35S]GTPγS binding had to be fixed to the average of the pKB obtained from Leu-enk and SNC80 as neither allosteric agonism or potentiation reached a limit.
Similar to the findings in the cAMP functional assay, 2 was also shown to be a PAM in [35S]GTPγS binding (Figure 4A) and in ERK1/2 phosphorylation (Figure 4B) in CHO-hDOPr cells, showing agonist activity at higher concentrations and increases in the potency of orthosteric agonist at lower concentrations. No agonist activity to 2 was observed in the parental CHO cells (lacking the δ receptor) in ERK1/2 phosphorylation or in parental CHO cells in inhibition of cAMP accumulation assays (data not shown). 2 increased the potency of leu-enkephalin by 16-fold in the [35S]GTPγS binding assay in CHO-hDOPr membranes and by 8-fold in the ERK1/2 phosphorylation assay in CHO-hDOPr cells. Similar experiments were performed replacing leu-enkephalin with the orthosteric agonists SNC80 and the partial agonist TAN67. By use of an operational model of allosterism20 (see Methods and Materials), composite cooperativity (αβ) values and pKB values (denoting the equilibrium dissociation binding constant for 2 at the δ receptor in the absence of orthosteric agonist, i.e., at the free receptor) were determined for 2 across these different assays and with different orthosteric agonists (Table 3).
Figure 4.
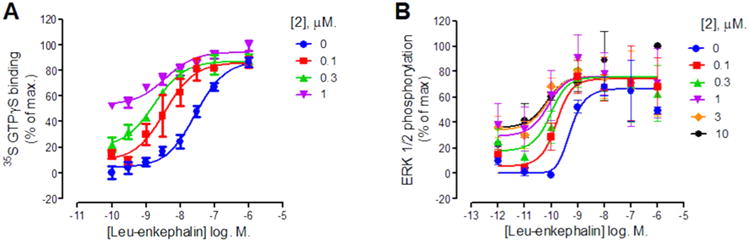
Effect of increasing concentrations of 2 on leu-enkephalin concentration–response curves in [35S]GTPγS binding in CHO-hDOPr membranes (A) and in pERK in CHO-hDOPr cells (B). In the [35S]GTPγS binding assay, 0% and 100% represent the basal response and the maximal response produced, respectively. In the pERK assay, 0% represents basal activity in serum-free media and 100% represents the pERK response in the presence of 10% serum. Data are presented as the mean ± SEM, n = 3–7. Data were fitted to the operational model of allosterism (see Table 3).
The mean ± SEM pKB across all the assays for 2 was 6.02 ± 0.16 (∼1 μM). One would expect that the pKB values should be the same across all the cell lines, functional assays, and orthosteric agonists used, since the pKB represents the binding affinity of 2 to the free receptor. Two way ANOVA with multiple comparison test of the pKB values in Table 3 showed no significant difference between the different orthosteric agonist ligands used in the same functional assay. For SNC80 and TAN67 there were also no significant differences in pKB values across the different functional pathways tested. However, for leu-enkephalin there were significant differences in the pKB values between β-arrestin recruitment and [35S]GTPγS binding (p < 0.01) and between β-arrestin recruitment and cAMP inhibition (p < 0.001).
Discussion
By use of a β-arrestin recruitment assay, the SAR of a δ PAM chemotype identified from HTS was explored, resulting in identification of compounds (1–15) with little or no agonist activity but which produced PAM activity at the δ and μ receptor. To compare the allosteric activity of the compounds, we used increasing concentrations with a single (EC20) concentration of orthosteric agonist and analyzed the EC50 and Ymax values of the functional curves produced. Although the compounds exhibited a range of Ymax values in PAM mode (Table 1) which can correlate with the allosteric cooperativity, the large proportion of the analogs tested exhibited efficacy close to or above 100% limiting the usefulness of the Ymax parameter for selecting compounds for further study. Instead, potency of the PAM response was used and selectivity was determined using potency ratios between the PAM responses at the δ receptor compared to the μ receptor. While this procedure is useful for selecting δ receptor selective PAM candidates to pursue, one must bear in mind that different orthosteric agonist ligands were used in the PAM mode assays: leu-enkephalin for the δ receptor, and endomorphin I for the μ receptor. Since we currently know little about the possible probe dependence of these PAM compounds at the δ and μ receptor, we cannot necessarily assume that the reported selectivity will be the same with different orthosteric probe ligands.
The selected data set used for multivariate statistical analysis do not allow for thorough cross-validation of the presented linear models, but our results suggest initial physicochemical properties that can be used as searching criteria for additional compounds with potential PAM activity at δ and μ opioid receptors. 2 was selected for further characterization, since it had the highest PAM mode potency at the δ receptor and showed 100-fold selectivity compared to the μ receptor.
The multivariate statistical analysis initially suggested that the compounds may not be readily soluble in aqueous buffer at concentrations in the micromolar range. Also, nephelometry data (not shown) suggest that 2 and 10 show particulate matter in phosphate buffered saline solution at concentrations above 1 μM. When nephelometry was repeated using the specific buffer used for the β-arrestin recruitment assays (HBSS + 25 mM HEPES and 10% FBS) in Table 1, 2 and 10 produced particulate matter above 3 μM. While the majority of responses to 2 in cells expressing the δ receptor were maximal at 1 μM (and therefore, within the solubility window predicted for 2), the μ receptor responses (e.g., see Figure 1) also showed sigmoidal responses (i.e., the responses were not biphasic) up to 30 μM 2, suggesting that solubility was not an issue in these assays in the specific buffers used. However, compound solubility should be an important consideration in further studies and optimization of this chemical series.
From competition binding studies, 2 did not affect 3H-DPN binding to the δ receptor but increased the affinity of orthosteric agonists, suggesting that 2 does not bind to the orthosteric site of the δ receptor but can increase the affinity of orthosteric agonists binding to the receptor (α cooperativity). The precise mechanism for this cooperativity remains unknown. However, in this context it is tempting to make comparisons to recently discovered PAMs of the μ opioid receptor.21 The μ receptor PAM 2-(3-bromo-4-methoxyphenyl)-3-((4-chlorophenyl)sulfonyl)thiazolidine (16, BMS-986122) has been found to differentially increase the affinity of various orthosteric agonists, and the magnitude of the affinity increase (α value) produced by 16 correlates with the intrinsic activity of the orthosteric ligand used.22 The mechanism by which 16 induces this affinity modulation is suggested to be via reducing the affinity of Na+ for its binding site on the μ receptor. The precise binding site for 16 on the μ receptor has not been clearly established, and it is unknown whether the δ receptor PAMs described here bind to an analogous binding site on the δ receptor or act via a similar mechanism. However, several analogs of 16 were found to exhibit weak activity at δ receptors, and most of the δ receptor PAMs described here also exhibit some degree of activity at μ receptors. Therefore, it is possible that these δ receptor PAMs may be binding to a site on the δ receptor that is analogous to the 16 binding site on the μ receptor and may work through a similar mechanism. The reduced affinity shift observed with 2 for the partial agonist TAN67 compared with the agonists with higher intrinsic activity, leu-enkephalin and SNC80 (Figure 2, Table 2), is consistent with this hypothesis. It will be interesting to determine whether these δ PAMs reduce the affinity of Na+ for its binding site on the δ receptor. Sodium ions are known to stabilize a lower affinity state of the δ receptor, and the molecular basis for allosteric Na+ control of opioid receptor signaling has been elucidated recently.23, 24
While TAN67 was a partial agonist in the CHO-hDOPr cell line for [35S]GTPγS binding giving 84% of maximal SNC80 response, it had even less intrinsic activity in a C6-DOPr cell line at 41% of maximal SNC80 response (data not shown). In the presence of 2 (300 nM), the maximal stimulation by TAN67 was increased to 67% of maximal SNC80 response. This suggests that 2 has some allosteric efficacy cooperativity (β), as well as the affinity cooperativity (α) observed above.
In all of the functional assays, 2 acted as a PAM, increasing the potency of the response to orthosteric agonists. No activity was observed in functional assays when 2 was added alone in CHO-parental cells (lacking the recombinant δ opioid receptor) in either the ERK activation assay or cAMP assay (data not shown). However, in cells expressing the δ receptor, 2 (when added alone) produced significant activity in cAMP inhibition, [35S]GTPγS binding, and ERK activation assays, suggesting that this activity is due to intrinsic efficacy of 2 at the τ receptor. Thus, 2 is a PAM-agonist in these systems. In cells expressing the δ receptor, 2 showed little to no agonist activity in the β-arrestin recruitment assay, which measures an event proximal to receptor activation with limited signal amplification. The difference in observed agonist activity for 2 between the β-arrestin recruitment assay and the cAMP assay (Figure 3) is likely reflective of a higher level of signal amplification and thus a higher receptor reserve in the cAMP assay compared to the β-arrestin recruitment assay in the same cell line.26, 27 Phosphorylation of the receptor by G protein-coupled receptor kinases (GRKs) is thought to be a prerequisite for β-arrestin recruitment.28 It would be interesting to see how 2 impacts δ receptor phosphorylation by GRKs and consequently desensitization and internalization of the receptor.
Calculation of pKB values for 2 across the various functional assays with leu-enkephalin, SNC80, and TAN67, using the allosteric operational model, showed some variability. These differences in pKB values may result from the allosteric effect not reaching a plateau or ceiling. This could reflect that the allosteric effect was submaximal at concentrations below those at which full agonism was observed with 2 or that the highest concentrations of 2 used did not cause the allosteric EC50 shift to reach its ceiling. This can make accurate assessment of the allosteric parameters more difficult to estimate in the model. Other variables, including the use of different cell lines or use of a tagged receptor (in the case of the PathHunter CHO-OPRD1 cell line), may also contribute to the variability of values obtained in the model.
The fact that we observed PAM effects with 2 at concentrations lower than those which produced agonist effects is entirely consistent with the allosteric ternary complex model because the former effects (PAM effects) are observed in the presence of orthosteric agonist, and hence the affinity of the modulator for the receptor is higher, whereas the latter effects (agonist effects) reflect the actions of the modulator at the free receptor and thus require higher concentrations to achieve the same level of fractional occupancy. Therefore, a PAM with a large cooperativity factor (αβ) can exhibit functional activity that is far more potent than its KB value. This has potential implications for PAM drug discovery programs, suggesting that it is important to track functional PAM activity rather than KB values when designing assays to support SAR. Additionally, this suggests that assays assessing target engagement may dramatically underestimate the relevant receptor occupancy of a PAM, since the affinity of the PAM (and therefore its fractional receptor occupancy) will be much higher at sites where orthosteric agonist is present. While such sites may represent only a small fraction of the receptor population in vivo, they nonetheless represent the relevant receptor population, since positive allosteric modulation can only occur when and where orthosteric agonist is bound.
Despite the complexities discussed above, all available data suggest that 2 is a δ PAM or a δ PAM-agonist. In future studies, it will be important to confirm the activity of 2 and its analogs in cells or tissues natively expressing δ receptors. Further, it will be of significant interest to determine whether compounds such as 2 also exhibit direct agonist activity in native systems expressing endogenous levels of δ receptors. PAMs devoid of intrinsic agonist activity could theoretically have therapeutic advantages over PAM-agonists, particularly in the maintenance of the temporal and spatial fidelity of endogenous receptor activation in vivo, as they would effectively be silent when bound to the receptor until orthosteric (endogenous) agonist is presented to the receptor. A key issue will be the determination of these effects in vivo. We intend to evaluate the in vivo activity of 2 and its analogs in models of acute and chronic pain,29 migraine,30 depression,31 and convulsive activity3 which is a known liability of δ opioid receptor agonists that has limited the pursuit of δ receptor agonists as potential therapeutics.
In summary, we have identified and characterized δ receptor-selective PAMs including our lead compound 2. Further studies are planned to assess probe dependence and signaling bias for these PAMs using a variety of orthosteric opioid receptor ligands and functional assays. Additional research is also ongoing to determine if this new class of compounds could represent a viable approach to develop new medicines for chronic pain, depression, and other therapeutic indications.
Methods and Materials
Chemistry
Analogs were purchased from external vendors (1, 3–5, 7) or synthesized according to Scheme 1 (2, 6, 8–15). All purchased and newly synthesized analogs provided analytical data consistent with their assigned structures and were >95% pure based on LCMS.
Synthesis of Intermediate A (Scheme 1)
To a solution of 4-hydroxybenzaldehyde (1.5 g, 12.28 mmol) in 2-propanol (35 mL) were added 5,5-dimethylcyclohexane-1,3-dione (3.44 g, 24.57 mmol) and H2SO4 (98%, 0.098 mL, 1.842 mmol). The reaction mixture was refluxed for 1.5 h in an oil bath and then cooled to room temperature, forming a white precipitate. After filtration, 3 g of 9-(4-hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione was obtained in 65% yield (98% purity by LCMS analysis). 1H NMR (400 MHz, CD3Cl) δ 7.09 (d, J = 8.6 Hz, 2H), 6.56 (d, J = 8.6 Hz, 2H), 4.67 (s, 1H), 2.46 (s, 4H), 2.23 (s, 2H), 2.21 (s, 2H), 1.10 (s, 6H), 1.00 (s, 6H); ESI-MS m/z = 367.08 [M + H]+.
Synthesis of Analogs 1-15
General Procedure
To a solution of 9-(4-hydroxyphenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (100 μmol, 36.6 mg) in DMF (1.2 mL) were added ArCH2Br (200 μmol) and Cs2CO3 (65.2 mg, 200 μmol). The reaction mixture was stirred at room temperature overnight. Then 10 μL of the reaction solution was taken, dissolved in MeOH (0.2 mL), and analyzed by LCMS. The LCMS showed that the reaction was complete and the desired product as a major peak was found. The product was purified via preparative LC/MS with the following conditions. Column: XBridge C18, 19 mm × 200 mm, 5-μm particles. Mobile phase A: 5:95 acetonitrile/water with 10 mM ammonium acetate. Mobile phase B: 95:5 acetonitrile/water with 10 mM ammonium acetate. Gradient: 70–100% B over 15 min, then a 5 min hold at 100% B. Flow: 20 mL/min. Fractions containing the desired product were combined and dried via centrifugal evaporation.
Two analytical LC/MS injections were used to determine the final purity. Injection 1 conditions were the following. Column: Waters BEH C18, 2.0 mm × 50 mm, 1.7 μm particles. Mobile phase A: 5:95 acetonitrile/water with 10 mM ammonium acetate. Mobile phase B: 95:5 acetonitrile/water with 10 mM ammonium acetate. Temperature: 50 °C. Gradient: 0% B, 0–100% B over 3 min, then a 0.5 min hold at 100% B. Flow: 1 mL/min. Detection: UV at 220 nm.
Injection 2 conditions were the following. Column: Waters BEH C18, 2.0 mm × 50 mm, 1.7 μm particles. Mobile phase A: 5:95 methanol/water with 10 mM ammonium acetate. Mobile phase B: 95:5 methanol/water with 10 mM ammonium acetate. Temperature: 50 °C. Gradient: 0% B, 0–100% B over 3 min, then a 0.5 min hold at 100% B. Flow: 0.5 mL/min. Detection: UV at 220 nm.
Proton NMR was acquired in deuterated CDCl3 or DMSO.
3,3,6,6-Tetramethyl-9-(4-((2-methylbenzyl)oxy)phenyl)-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (2, BMS-986187)
1H NMR (400 MHz, chloroform-d) δ 7.51–7.35 (m, 2H), 7.26–7.18 (m, 4H), 6.89 (dd, J = 14.2, 8.6 Hz, 2H), 5.05 (s, 2H), 4.72 (s, 1H), 2.49 (d, J = 5.9 Hz, 4H), 2.38 (d, J = 7.8 Hz, 4H), 2.27–2.21 (m, 3H), 1.16–1.10 (m, 6H), 1.07–1.00 (m, 6H). HRMS: calcd C31H35O4, 471.2530; found, 471.2538
9-(4-((2-Bromobenzyl)oxy)phenyl)-3,3,6,6-tetramethyl-3,4,5,6,7,9-hexahydro-1H-xanthene-1,8(2H)-dione (10, BMS-986188)
The yield of the product was 20.6 mg, and its estimated purity by LCMS analysis was 100%. 1H NMR (500 MHz, DMSO-d6) δ 7.67 (d, J = 7.7 Hz, 1H), 7.56 (d, J = 7.3 Hz, 1H), 7.42 (t, J = 7.5 Hz, 1H), 7.31 (t, J = 7.3 Hz, 1H), 7.10 (d, J = 8.1 Hz, 2H), 6.88 (d, J = 8.4 Hz, 2H), 5.04 (s, 2H), 4.48 (s, 1H), 2.54 (d, J = 11.4 Hz, 4H), 2.26 (d, J = 16.1 Hz, 2H), 2.09 (d, J = 16.1 Hz, 2H), 1.04 (s, 6H), 0.91 (s, 6H). HRMS: calcd C30H32O4Br, 535.1478; found, 535.1478.
Calculation and Analysis of Physicochemical Properties
The Schrodinger Suite 2014-4 was used throughout. The two-dimensional (2D) sketcher tool of Maestro 10.0 was used to draw 2D structures of each compound listed in Table 1. These 15 2D structures were imported as 3D structures into LigPrep, version 3.2, and their protonation state was assigned at physiological pH using Epik, version 3.0.32 Fifty-two physicochemical properties were calculated using QikProp, version 4.2. Among them, 38 properties (see Tables S1 and S2) displayed nonzero variance across the 15 ligands of Table 1 and were therefore used for multivariate statistical analysis.
The bayesglm function in the “arm” R package was used to build a Bayesian linear regression model for the relationship between the calculated physicochemical properties of the 15 compounds in Table 1 and their measured EC50 values at δ- or μ-opioid receptors. Specifically, each linear model was built so that P = ΣCiXi + C0, where P is the EC50 value to be predicted, C0 is the interception, and Xi and Ci are the calculated property and associated regression coefficient, respectively. We arbitrarily assigned a numerical value of 100 to EC50 values listed as “>10” in Table 1. To better discriminate the most potent compounds, we used a logarithmic scale for EC50. All models containing combinations of up to three properties (without interaction terms) were estimated, and the best linear models for δ-opioid (see Table S3 and Figure S1) and μ-opioid (see Table S4 and Figure S2) receptors were selected based on the Akaike information criterion (AIC). Following prediction of −log EC50 values at δ- or μ-opioid receptors, the δ/μ selectivity was estimated as the difference of −Δ log EC50= − log[EC50(δ)/EC50(μ)] (see Figure S3).
Cell Lines
Chinese hamster ovary (CHO) PathHunter cells expressing enzyme acceptor (EA) tagged β-arrestin 2 and either ProLink (PK) tagged δ receptor (CHO-OPRD1) or PK-tagged μ receptor (CHO-OPRM1) were from DiscoveRx (Fremont, CA). PathHunter is a trademark of DiscoveRx. Cells were grown in F-12 media (Invitrogen 11765), containing Hyclone FBS 10%, Hygromycin 300 μg/mL (Invitrogen 10687), G418 800 μg/mL (Invitrogen 10131) and maintained at 37 °C in a humidified incubator containing 5% CO2. These cells were used for β-arrestin recruitment assays and inhibition of forskolin-stimulated cAMP accumulation assays described below.
FlpIn CHO cells (Invitrogen, Carlsbad, CA, USA) were grown in Dulbecco's modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum and maintained at 37 °C in a humidified incubator containing 5% CO2. FlpIn CHO cells were transfected with the pOG44 vector encoding Flp recombinase and the pDEST vector encoding the human δ receptor (hDOPr) at a ratio of 9:1 using polyethylenimine as transfection reagent. At 24 h after transfection the cells (CHO-hDOPr) were subcultured and the medium was supplemented with 700 μg/mL HygroGold as selection agent. Cells were grown and maintained in DMEM containing 20 mM HEPES, 5% fetal bovine serum, and 200 μg/mL Hygromycin-B. Cells were maintained at 37 °C in a humidified incubator containing 5% CO2. These cells were used for ERK phosphorylation assays, and membranes derived from these cells were used for [35S]GTPγS binding and 3H DPN binding studies as described below.
Materials
PathHunter detection reagents were from DiscoveRx (Fremont, CA). Cell culture media and supplements were from Life Technologies (Carlsbad, CA). Lance-Ultra cAMP detection reagents, Surefire ERK assay reagents, [3H]diprenorphine (DPN), and [35S]GTPγS (guanosine-5′-O-(3-thio)triphosphate) were from PerkinElmer Life Sciences (Cambridge, MA). Endomorphin I and TAN67 were obtained from Tocris. All other chemicals, unless otherwise specified, were purchased from Sigma (St. Louis, MO).
PathHunterβ-Arrestin Assay
Confluent flasks of CHO-OPRM1 and CHO-OPRD1 cells were harvested with TrypLE Express and resuspended in F-12 media supplemented with 10% FBS and 25 mM HEPES, at a density of 6.67 × 10 cells/mL and plated (3 μL/well) into white solid TC-treated 1536-well plates (Corning, NY). Plates were incubated overnight at 37 °C in a 5% CO2 humidified incubator. The next day, compounds (40 nL of 100 × final concentration in 100% DMSO) were added to cell plates by acoustic dispense using an Echo-550 (Labcyte, Sunnyvale, CA) from Echo-qualified 1536-well source plates (Labcyte). Next, 1 μL of assay buffer (agonist mode), or assay buffer containing a low concentration (∼4 × EC20) of orthosteric agonist (PAM mode), was added to assay plates. The orthosteric agonists used are described in the Results and Discussion. Plates were covered with a lid and incubated at room temperature for 90 min. Incubations were terminated by the addition of 2 μL of PathHunter Reagent (DiscoveRx). One hour later luminescence was detected using a Viewlux imaging plate reader (PerkinElmer).
Inhibition of Forskolin-Stimulated cAMP Accumulation Assays
CHO-OPRD1 cells were grown to confluence (as described above). Cells were harvested and resuspended at 1 × 10 cells/mL in assay buffer (HBSS + 25 mM HEPES, +0.05% BSA). Compounds (30 nL of 100 × final concentration in 100% DMSO) were added to 1536-well white solid NT plates by acoustic dispense using an Echo-550 (Labcyte, CA) followed by a 1 μL addition of cells (2000 cells/well) to all wells. Next, 1 μL of either assay buffer (for agonist mode) or assay buffer containing a 3 × EC20 concentration of orthosteric agonist (PAM mode) was added. Finally, 1 μL of 3 × forskolin (2 μM final) was added. Plates were lidded and incubated for 45 min at rt. Incubations were terminated by the addition of Lance-Ultra cAMP detection reagent (PerkinElmer) (1.5 μL of Eu-cryptate-labeled cAMP tracer in lysis buffer, followed by 1.5 μL of U-light conjugated anti-cAMP antibody in lysis buffer). After a 1 h incubation at room temperature, time-resolved fluorescence (TRF) was detected on a Viewlux or Envision plate reader (PerkinElmer) with excitation at 337 nm and emission reads at 615 and 665 nm. The ratiometric data (665 nm read/615 nm read) × 10 000 were then converted to cAMP (nM) based on a standard curve for cAMP (replacing the cell addition step) run at the same time and under identical conditions to the assay.
Characterization of δ-opioid receptor-selective PAMs in the CHO-OPRD1 cAMP assay, using curve-shift assays, were performed as described above using orthosteric agonists described in the Results and Discussion.
Membrane Preparation
Confluent cells were rinsed with phosphate buffered saline and then detached using harvesting buffer (0.68 mM EDTA, 0.15 M NaCl, 20 mM HEPES, pH 7.4). Cells were pelleted by centrifugation at 300g for 3 min, followed by resuspension in cold 50 mM Tris-HCl buffer, pH 7.4. Pellet was rehomogenized using a Tissue Tearor and then centrifuged at 20 000g for 20 min at 4 °C. The supernatant was discarded, and the process was repeated for an additional rehomogenization and centrifugation. The supernatant was discarded, and the final pellet was resuspended in 50 mM Tris-HCl and flash-frozen in aliquots using liquid nitrogen. Aliquots were kept at −80 °C until assays. Protein concentration was determined using BCA protein assay with bovine serum albumin as the standard.
Radioligand Binding Assay
Cell membranes (as prepared above, 10 μg/well) were incubated in the following mixture for 90 min at 25 °C: assay buffer (50 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 10 μM GTPγS), various concentrations of orthosteric and allosteric ligand, and 0.35-0.45 nM [3H]DPN. Nonspecific binding was determined in the presence of 10 μM naloxone. Reactions were terminated by rapid filtration through glass microfiber GF/C filters (Whatman) using a Brandell harvester and washed three times using cold 50 mM Tris-HCl buffer. Filters were dried in a 50 °C oven, and radioactivity was measured by liquid scintillation counting with EcoLume liquid scintillation cocktail (MP Biomedicals) in a Wallac 1450 MicroBeta counter (PerkinElmer).
[35S]GTPγS Assay
CHO-hDOPr cell membranes (as prepared above, 10 μg/well) were incubated for 1 h at 30 °C in buffer comprising 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, 5 mM MgCl2, 100 mM NaCl, 0.1 nM [35S]GTPγS, and 30 μM GDP (guanosine 5-diphosphate) in a final volume of 200 μL. Orthosteric and allosteric ligands were also included, with SNC80 used as the maximal standard and assay buffer used to assess basal [35S]GTPγS binding. The reaction was terminated by filtration through glass microfiber GF/C filters (Whatman) using a Brandell harvester. The filters were rinsed, dried, and radioactivity was counted by liquid scintillation counting using EcoLume liquid scintillation cocktail (MP Biomedicals) in a Wallac 1450 McroBeta counter (PerkinElmer).
ERK1/2 Phosphorylation Assay
hDOPr FlpIn CHO cells (CHO-hDOPr) were seeded into 96-well plates at a density of 50 000 cells/well. After 5–7 h, cells were washed with phosphate buffered saline (PBS) and incubated overnight in serum-free DMEM. Initially, time-course experiments were conducted at least twice for each ligand to determine the time required to maximally promote ERK1/2 phosphorylation via the δ-receptor. Concentration–response experiments were performed for the orthosteric ligands in the absence or presence of increasing concentrations of the allosteric modulator at 37 °C. Stimulation of the cells was terminated by removal of the media and the addition of 100 μL of SureFire lysis buffer (PerkinElmer) to each well. The plate was shaken for 5 min at room temperature before transferring 5 μL of the lysates to a white 384-well Proxiplate (PerkinElmer). Then 8 μL of a 240:1440:7:7 mixture of Surefire activation buffer/Surefire reaction buffer/Alphascreen acceptor beads/Alphascreen donor beads was added to the samples and incubated in the dark at 37 °C for 1.5 h. Plates were read using a Fusion plate reader (PerkinElmer).
Data Analysis
For all experiments data were analyzed and EC50 or K i values determined using nonlinear regression analysis to fit a logistic equation using GraphPad Prism, version 6 (GraphPad, San Diego, CA). pKB and αβ values were determined using the “operational model of allosterism”20 (see eq 1), using Graphpad Prism, version 6.
| (1) |
Within this model, E is the pharmacological effect, KA and KB denote the equilibrium binding constants for the orthosteric ligand A and the allosteric ligand B at the receptor. The binding cooperativity factor α represents the effect of the allosteric ligand on orthosteric agonist binding affinity and vice versa. An activation cooperativity factor β denotes the effect the allosteric ligand has on orthosteric agonist efficacy. Agonism constants τA and τB represent the intrinsic activity of the orthosteric agonist and any intrinsic activity of the allosteric ligand, respectively, which is dependent on the cell context and receptor expression level of the cell system and intrinsic efficacy of the ligands used. The remaining parameters Em and n denote the maximal response of the system and the slope, respectively.
Supplementary Material
Acknowledgments
The authors thank Dr. Davide Provasi for providing the scripts needed for the statistical analyses. M.F. acknowledges support for this work from the National Institutes of Health (NIH) Grants DA026434 and DA034049. J.R.T. also acknowledges support from NIH Grant DA035316. Computations relied on the Extreme Science and Engineering Discovery Environment (XSEDE) under Grant MCB080109N and on the computational resources and staff expertise provided by the Scientific Computing Facility at the Icahn School of Medicine at Mount Sinai.
Footnotes
Supporting Information: Specific properties of the 15 compounds listed in Table 1 that were most likely to predict the experimentally determined EC50 values at either δ or μ opioid receptors are revealed from multivariante statistical analysis. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.5b00007.
Notes: The authors declare no competing financial interest.
References
- 1.Gaveriaux-Ruff C, Kieffer BL. Delta opioid receptor analgesia: recent contributions from pharmacology and molecular approaches. Behav Pharmacol. 2011;22(5-6):405–414. doi: 10.1097/FBP.0b013e32834a1f2c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lutz PE, Kieffer BL. Opioid receptors: distinct roles in mood disorders. Trends Neurosci. 2013;36(3):195–206. doi: 10.1016/j.tins.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broom DC, Nitsche JF, Pintar JE, Rce KC, Woods JH, Traynor JR. Comparison of receptor mechanisms and efficacy requirements for delta-agonist-induced convulsive activity and antinociception in mice. J Pharmacol Exp Ther. 2002;303(2):723–729. doi: 10.1124/jpet.102.036525. [DOI] [PubMed] [Google Scholar]
- 4.Jutkiewicz EM, Rce KC, Traynor JR, Woods JH. Separation of the convulsions and antidepressant-like effects produced by the delta-opioid agonist SNC80 in rats. Psychopharmacology (Berlin, Ger) 2005;182(4):588–596. doi: 10.1007/s00213-005-0138-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Le Bourdonnec B, Windh RT, Ajello CW, Leister LK, Gu M, Chu GH, Tuthill PA, Barker WM, Koblish M, Wiant DD, Graczyk TM, Belanger S, Cassel JA, Feschenko MS, Brogdon BL, Smith SA, Christ DD, Derelanko MJ, Kutz S, Little PJ, DeHaven RN, DeHaven-Hudkins DL, Dolle RE. Potent, orally bioavailable delta opioid receptor agonists for the treatment of pain: discovery of N,N-diethyl-4-(5-hydroxyspiro-[chromene-2,4′-piperidine]-4-yl)benzamide (ADL5859) J Med Chem. 2008;51(19):5893–5896. doi: 10.1021/jm8008986. [DOI] [PubMed] [Google Scholar]
- 6.Jutkiewicz EM, Baladi MG, Folk JE, Rice KC, Woods JH. The convulsive and electroencephalographic changes produced by nonpeptidic delta-opioid agonists in rats: comparison with pentylene-tetrazol. J Pharmacol Exp Ther. 2006;317(3):1337–1348. doi: 10.1124/jpet.105.095810. [DOI] [PubMed] [Google Scholar]
- 7.Chu Sin Chung P, Kieffer BL. Delta opioid receptors in brain function and diseases. Pharmacol Ther. 2013;140(1):112–120. doi: 10.1016/j.pharmthera.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung PCS, Boehrer A, Stephan A, Matifas A, Scherrer G, Darcq E, Befort K, Kieffer BL. Delta opioid receptors expressed in forebrain GABAergic neurons are responsible for SNC80-induced seizures. Behav Brain Res. 2014;278C:429–434. doi: 10.1016/j.bbr.2014.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54(2):323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- 10.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2007;47:1–51. doi: 10.1146/annurev.pharmtox.47.120505.105159. [DOI] [PubMed] [Google Scholar]
- 11.Burford NT, Traynor JR, Alt A. Positive allosteric modulators of the mu-opioid receptor: a novel approach for future pain medications. Br J Pharmacol. 2015;172(2):277–286. doi: 10.1111/bph.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pradhan AA, Befort K, Nozaki C, Gaveriaux-Ruff C, Kieffer BL. The delta opioid receptor: an evolving target for the treatment of brain disorders. Trends Pharmacol Sci. 2011;32(10):581–590. doi: 10.1016/j.tips.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pradhan AA, Smith ML, Kieffer BL, Evans CJ. Ligand-directed signalling within the opioid receptor family. Br J Pharmacol. 2012;167(5):960–969. doi: 10.1111/j.1476-5381.2012.02075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discovery. 2013;12(3):205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 15.Leach K, Loiacono RE, Felder CC, McKinzie DL, Mogg A, Shaw DB, Sexton PM, Christopoulos A. Molecular mechanisms of action and in vivo validation of an M4 muscarinic acetylcholine receptor allosteric modulator with potential antipsychotic properties. Neuropsychopharmacology. 2010;35(4):855–869. doi: 10.1038/npp.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bassoni DL, Raab WJ, Achacoso PL, Loh CY, Wehrman TS. Measurements of beta-arrestin recruitment to activated seven transmembrane receptors using enzyme complementation. Methods Mol Biol. 2012;897:181–203. doi: 10.1007/978-1-61779-909-9_9. [DOI] [PubMed] [Google Scholar]
- 17.Zhao X, Jones A, Olson KR, Peng K, Wehrman T, Park A, Mallari R, Nebalasca D, Young SW, Xiao SH. A homogeneous enzyme fragment complementation-based beta-arrestin translocation assay for high-throughput screening of G-protein-coupled receptors. J Biomol Screening. 2008;13(8):737–747. doi: 10.1177/1087057108321531. [DOI] [PubMed] [Google Scholar]
- 18.Burford NT, Wehrman T, Bassoni D, O'Connell J, Banks M, Zhang L, Alt A. Identification of selective agonists and positive allosteric modulators for mu- and delta-opioid receptors from a single high-throughput screen. J Biomol Screening. 2014;19(9):1255–1265. doi: 10.1177/1087057114542975. [DOI] [PubMed] [Google Scholar]
- 19.Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol. 2011;81(6):691–702. doi: 10.1016/j.bcp.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Leach K, Sexton PM, Christopoulos A. Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci. 2007;28(8):382–389. doi: 10.1016/j.tips.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 21.Burford NT, Clark MJ, Wehrman TS, Gerritz SW, Banks M, O'Connell J, Traynor JR, Alt A. Discovery of positive allosteric modulators and silent allosteric modulators of the mu-opioid receptor. Proc Natl Acad Sci USA. 2013;110(26):10830–10835. doi: 10.1073/pnas.1300393110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livingston KE, Traynor JR. Disruption of the Na+ ion binding site as a mechanism for positive allosteric modulation of the mu-opioid receptor. Proc Natl Acad Sci USA. 2014;111(51):18369–18374. doi: 10.1073/pnas.1415013111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, Roth BL, Stevens RC. Molecular control of delta-opioid receptor signalling. Nature. 2014;506(7487):191–196. doi: 10.1038/nature12944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shang Y, LeRouzic V, Schneider S, Bisignano P, Pasternak GW, Filizola M. Mechanistic insights into the allosteric modulation of opioid receptors by sodium ions. Biochemistry. 2014;53(31):5140–5149. doi: 10.1021/bi5006915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christopoulos A, Changeux JP, Catterall WA, Fabbro D, Burris TP, Cidlowski JA, Olsen RW, Peters JA, Neubig RR, Pin JP, Sexton PM, Kenakin TP, Ehlert FJ, Spedding M, Langmead CJ. International union of basic and clinical pharmacology. XC. Multisite pharmacology: recommendations for the nomenclature of receptor allosterism and allosteric ligands. Pharmacol Rev. 2014;66(4):918–947. doi: 10.1124/pr.114.008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehlert FJ. Analysis of allosterism in functional assays. J Pharmacol Exp Ther. 2005;315(2):740–754. doi: 10.1124/jpet.105.090886. [DOI] [PubMed] [Google Scholar]
- 27.Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick SA. simple method for quantifying functional selectivity and agonist bias. ACS Chem Neurosci. 2012;3(3):193–203. doi: 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierce KL, Lefkowitz RJ. Classical and new roles of beta-arrestins in the regulation of G-protein-coupled receptors. Nat Rev Neurosci. 2001;2(10):727–733. doi: 10.1038/35094577. [DOI] [PubMed] [Google Scholar]
- 29.Vanderah TW. Delta and kappa opioid receptors as suitable drug targets for pain. Clin J Pain. 2010;26(Suppl. 10):S10–S15. doi: 10.1097/AJP.0b013e3181c49e3a. [DOI] [PubMed] [Google Scholar]
- 30.Pradhan AA, Smith ML, Zyuzin J, Charles A. delta-Opioid receptor agonists inhibit migraine-related hyperalgesia, aversive state and cortical spreading depression in mice. Br J Pharmacol. 2014;171(9):2375–2384. doi: 10.1111/bph.12591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jutkiewicz EM. The antidepressant-like effects of delta-opioid receptor agonists. Mol Interventions. 2006;6(3):162–169. doi: 10.1124/mi.6.3.7. [DOI] [PubMed] [Google Scholar]
- 32.(a) Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, Uchimaya M. Epik: a software program for pK(a) prediction and protonation state generation for drug-like molecules. J Comput-Aided Mol Des. 2007;21(12):681–691. doi: 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]; (b) Greenwood JR, Calkins D, Sullivan AP, Shelley JC. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J Comput-Aided Mol Des. 2010;24(6-7):591–604. doi: 10.1007/s10822-010-9349-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.