

Abstract
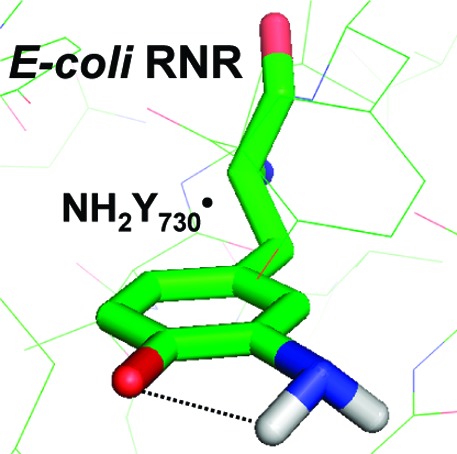
E. coli ribonucleotide reductase (RNR) catalyzes the conversion of nucleotides to deoxynucleotides, a process that requires long-range radical transfer over 35 Å from a tyrosyl radical (Y122•) within the β2 subunit to a cysteine residue (C439) within the α2 subunit. The radical transfer step is proposed to occur by proton-coupled electron transfer via a specific pathway consisting of Y122 → W48 → Y356 in β2, across the subunit interface to Y731 → Y730 → C439 in α2. Using the suppressor tRNA/aminoacyl-tRNA synthetase (RS) methodology, 3-aminotyrosine has been incorporated into position 730 in α2. Incubation of this mutant with β2, substrate, and allosteric effector resulted in loss of the Y122• and formation of a new radical, previously proposed to be a 3-aminotyrosyl radical (NH2Y•). In the current study [15N]- and [14N]-NH2Y730• have been generated in H2O and D2O and characterized by continuous wave 9 GHz EPR and pulsed EPR spectroscopies at 9, 94, and 180 GHz. The data give insight into the electronic and molecular structure of NH2Y730•. The g tensor (gx = 2.0052, gy = 2.0042, gz = 2.0022), the orientation of the β-protons, the hybridization of the amine nitrogen, and the orientation of the amino protons relative to the plane of the aromatic ring were determined. The hyperfine coupling constants and geometry of the NH2 moiety are consistent with an intramolecular hydrogen bond within NH2Y730•. This analysis is an essential first step in using the detailed structure of NH2Y730• to formulate a model for a PCET mechanism within α2 and for use of NH2Y in other systems where transient Y•s participate in catalysis.
Introduction
Ribonucleotide reductases (RNRs) provide the monomeric precursors required for DNA replication and repair by catalyzing the conversion of nucleotides to deoxynucleotides.1,2 The class Ia RNRs are found in eukaryotes and prokaryotes with the RNR from E. coli serving as a model. It consists of a 1:1 complex of two homodimeric subunits: α2 and β2.3,4 α2 contains the active site, including residue C439, which is oxidized to a transient thiyl radical to initiate nucleotide reduction,5−8 and binding sites for allosteric regulators that control the rate and specificity of nucleotide reduction.9,10 β2 harbors a di-iron tyrosyl radical (Y122•) cofactor essential for initiating nucleotide reduction.11−13 A structure of the active RNR complex has not been solved. However, on the basis of in silico docking of the individual subunits, Uhlin and Eklund made the provocative proposal that a pathway consisting of aromatic amino acids is involved in transport of the radical from Y122• in β2 over 35 Å to C439 in the active site of α2.14−17 We have expanded this model to include the fate of the protons as well as the electrons, as oxidation of Ys within the pathway requires proton-coupled electron transfer (PCET).18,19 Within β2, the reaction is proposed to occur by orthogonal PCET,20 while within α2 it is proposed to involve collinear PCET (Figure 1).21,22
Figure 1.

Proposed radical initiation pathway in E. coli class Ia RNR. Radical transfer occurs by orthogonal PCET within β2, where long-distance ET within the pathway is coupled to short-distance off-pathway PT, and by collinear PCET (or hydrogen atom transfer) within α2. Note that the position of Y356 is structurally unknown.19
Recently we have developed new probes to examine the pathway and the involvement of redox-active tyrosines (Y356 in β2 and Y730 and Y731 in α2) and to study the mechanism of this unprecedented radical propagation process.18,23 Incorporation of 3,4-dihydroxyphenylalanine (DOPA), which has a lower reduction potential than Y (260 mV lower at pH 7), into residue 356 of β2 led to trapping of the DOPA radical (DOPA356•).24,25 Formation of DOPA356• occurred concomitantly with disappearance of Y122• and was dependent on the presence of substrate and allosteric effector. These studies support direct participation of residue 356 in radical transfer. We have also inserted a series of fluorotyrosine analogues (FnY, n = 1−4),20,26−28 3-nitrotyrosine,29 and 4-aminophenylalanine30 in order examine the protonation state and the effect of the driving force for radical transfer through residue 356.18
To investigate the roles of residues Y730/Y731 in radical propagation, we recently used the suppressor tRNA/RS method, pioneered by Schultz and co-workers,31,32 to replace site-specifically each residue with 3-aminotyrosine (NH2Y).22 This analogue is 190 mV easier to oxidize than Y (at pH 7), but the pKa of the hydroxyl group is unchanged.33,34 Examination of NH2Y-α2s in the presence of β2, substrate, and allosteric effector demonstrated radical propagation across the subunit interface in a kinetically competent fashion. Concomitant with loss of the Y122•, a new radical species was detected, which we proposed was a NH2Y• on the basis of UV−visible and X-band EPR spectroscopies. Surprisingly, we also showed that these NH2Y-α2s were capable of making deoxynucleotides, an observation that has important implications for the mechanism of radical propagation.22,35
Key to using NH2Y as a probe of PCET reactions in general and the radical propagation step specifically in RNR is a detailed characterization of the electronic properties of the putative NH2Y• by EPR spectroscopy. Prior to our studies, no spectroscopy of this unnatural amino acid radical had been reported. However, studies on the o-aminophenol radical36−41 and the Y•1,42−44 in many different proteins have been examined and provide a foundation for expectations with NH2Y•.
9 GHz EPR studies on the o-aminophenol radical in different solvent matrixes reveal two major coupling constants assigned to the protons and nitrogen of the amino group (Table S1 and Figure S1). The strong temperature dependence of the EPR spectra in aprotic solvents indicated that the two protons may be involved in an intramolecular dynamic process.41 An INDO calculation suggested that of the two possible tautomeric structures of the radical the H is bonded to the N, not the O, and that the amino group is not planar, but at least partially sp3-hydridized. These early calculations further suggested that the •O---H-N hydrogen bond does not lie in the ring plane and that an out-of-plane deformation caused by bending of the C−N bond 5° relative to the plane of the ring stabilizes the radical.41 A more recent computational study about substituent effects on the hybridization state of the nitrogen in anilines showed that pyramidalization of the amino group can occur, but the out-of-plane bending takes place at the protons and not at the nitrogen.45
Transient and stable Y•s have been characterized in detail in many proteins. However, despite the extensive experimental effort in this area, the variability of EPR line shapes associated with different rotational conformations of the phenoxy ring and different patterns of hyperfine interactions have made simulation of Y• spectra challenging. High-field EPR and ENDOR spectroscopies and computational studies have revealed a number of interesting generalizations about the structures of these radicals. High-field EPR studies give rise to accurate g values and to insight about the electrostatic environment and H-bonding to the radical. The gx values for the Y• in class I RNRs range from 2.0077 to 2.0094.46−52 A shift of gx to a lower value indicates the possibility of a H-bond.53,54 Also, variability in line shape and width of gx observed suggests there may be variability of H-bonding within the Y• population.55−57 The EPR spectrum is largely defined by the hyperfine interactions with the β-protons of the methylene group of Y and its four ring protons. The former are primarily responsible for the different patterns in the hyperfine structure. Lessons learned from EPR spectra of the o-aminophenol radical and for many Y•s in different environments provide the foundation for understanding the structure of the NH2Y• described herein.
We now report the characterization of the new radical generated with Y730NH2Y-α2 using multifrequency EPR spectroscopy. Our results demonstrate that this signal is associated with the NH2Y• and provide insight into its conformation within the active RNR complex (Figure 2). The results establish the existence of an intramolecular H-bond between one of the amino protons and the phenol oxygen of the NH2Y730•. This detailed spectroscopic analysis is an essential step in identifying the importance of H-bonding between residues in the radical propagation pathway (Figure 1), which in turn is required for assessing the proposed model for collinear PCET.
Figure 2.

Structure of NH2Y730•. (Top) Numbering scheme (inside numbers), axis system, and the intramolecular H-bond represented by dashed lines. (Bottom) NH2Y730• viewed along the C3-N and the phenol C2−C3 bond within the aromatic plane. Dihedral angles for the amino protons (left) and the Cβ-protons (middle) of NH2Y730• with respect to the phenol plane, which is indicated by dashed lines. (Right) Tetrahedral nature of the NH2 moiety and the 5° tilt of the C3−N bond away from the aromatic plane, which is indicated by a bold line.
Materials and Methods
Materials
l-Tyrosine, [15N]-HNO3 (conc ∼10 N, 98% 15N enrichment), palladium catalyst on carbon (10 wt % loading), hydroxyurea (HU), Sephadex G-25, 3-nitrotyrosine (NO2Y), 3-aminotyrosine (NH2Y), cytidine-5′-diphosphate (CDP), and adenosine-5′-triphosphate (ATP) were obtained from Sigma-Aldrich. D2O (99.8 atom % in D) was from VWR. β2 was purified as previously described with a specific activity of 7200 nmol/min mg and 1.2 Y122•/dimer. 1H and 13C NMR spectra were recorded on a Varian 300 MHz NMR spectrometer at the MIT Department of Chemistry Instrumentation Facility. Aqueous samples for 1H NMR and 13C (1H-decoupled) NMR were acquired in D2O with 3-(trimethylsilyl)propionic acid-d4 (TSP) as a standard. Absorption spectra were recorded on an Agilent 8453 diode array or a Cary 3 UV−vis spectrophotometer.
Synthesis of 3-[15N]-Nitrotyrosine, [15N]-NO2Y
[15N]-HNO3 (10 N) contained 98% 15N as confirmed by mass spectrometry. Synthesis of [15N]-NO2Y was carried out as previously described with minor modifications.34 L−Y (7.0 mmol) was added to a 25 mL pear-shaped flask, equipped with a stir bar, and dissolved in 5 mL of H2O at room temperature. The mixture was supplemented with 1.55 mL of [15N]-HNO3 (11.6 mmol, 7.5 N) by dropwise addition over 1 min and stirred for 30 min at room temperature to fully dissolve L−Y. The mixture was then cooled by stirring for 15 min in an ice−water bath. Then 4 mL of chilled [15N]-HNO3 (30 mmol) was added to the mixture at 4 °C over 2 h. The solution was then stirred for an additional 5 h at 4 °C and refrigerated overnight, which caused precipitation of [15N]-NO2Y. The precipitate was isolated and dried by vacuum filtration on a coarse Buchner funnel, providing product in 80% yield (5.6 mmol). The product was analyzed by 1H and 13C NMR, and by UV−vis spectroscopy.
1H NMR (300 MHz, D2O, 25 °C): δ 3.18 (dd, 1H, Cβ-H1, 7.4 Hz, 14.8 Hz), 3.30 (dd, 1H, Cβ-H2, 5.8 Hz, 14.8 Hz), 4.24 (dd, 1H, Cα-H, 5.8 Hz, 7.4 Hz), 7.14 (dd, 1H, arom. C−H, 1.1 Hz, 8.7 Hz), 7.54 (dd, 1H, arom. C−H, 2.2 Hz, 8.7 Hz), 8.01 (t, 1H, arom. C−H, 2.2 Hz). 13C NMR (300 MHz, D2O, 25 °C): δ 34.7 (s, Cβ), 54.2 (s, Cα), 120.4 (d, arom. C, 2.1 Hz), 126.1 (d, arom. C, 1.6 Hz), 126.8 (d, arom. C, 2.4 Hz), 134.2 (d, arom. C, 15.2 Hz), 138.2 (s, arom. C), 153.1 (d, arom. C, 1.2 Hz), 172.1 (s, COOH). UV−vis (H2O/HCl, pH 2): λmax ∼277 nm (ε ∼5500 M−1 cm−1), λmax ∼358 nm (ε ∼2600 M−1 cm−1); (H2O/NaOH, pH 12) λmax ∼286 nm (ε ∼4100 M−1 cm−1), λmax ∼431 nm (ε ∼4100 M−1 cm−1).
Synthesis of 3-[15N]-Aminotyrosine, [15N]-NH2Y
[15N]-NO2Y (5.2 mmol) was combined with 18 mL of H2O and 70 mL of MeOH in a 250 mL round-bottom flask, equipped with a stir bar, at room temperature. The solution was supplemented with 0.52 mmol (550 mg) of Pd/C catalyst. The mixture was evacuated gently and filled with H2(g) several times and then stirred for 2 h under H2 using a H2(g) balloon. After the incubation, the H2 was replaced with N2 and the catalyst was removed by filtration through a fine Buchner funnel. The solvent was removed in vacuo, providing product in quantitative yield. The product was exchanged into D2O, lyophilized to dryness, and assessed by 1H and 13C NMR and UV−vis spectroscopic methods. It was pure on the basis of NMR spectroscopy.
1H NMR (300 MHz, D2O, 25 °C): δ 3.12 (m, 2H, Cβ-Hs), 3.90 (t, 1H, Cα-H, 6.3 Hz), 6.99 (m, 1H, arom. C−H), 7.19 (m, 2H, arom. C−H). 13C NMR (300 MHz, D2O, 25 °C): δ 35.2 (s, Cβ), 55.5 (s, Cα), 116.9 (s, arom. C), 118.2 (d, arom. C, 9.4 Hz), 124.8 (d, arom. C, 1.7 Hz), 126.9 (d, arom. C, 1.9 Hz), 131.4 (s, arom. C), 149.5 (s, arom. C), 173.3 (s, COOH). UV−vis (H2O/HCl, pH 1): λmax ∼275 nm (ε ∼1900 M−1 cm−1); (H2O, pH 7) λmax ∼289 nm (ε ∼3200 M−1 cm−1); (H2O/NaOH, pH 12) λmax ∼303 nm (ε ∼4700 M−1 cm−1).
Growth, Expression, and Purification of [15N]-NH2Y730-α2
All procedures were carried out as previously detailed and gave 6 mg of pure [15N]-NH2Y730-α2 per gram of wet cell paste.22
Activity Assays
The activity of [15N]-NH2Y730-α2 was determined by the spectrophotometric assay as described.20
Reaction of [15N]-Y730NH2Y-α2 with β2, CDP/ATP Monitored by EPR Spectroscopy
[15N]-NH2Y730-α2 was prereduced as previously described.22 Prereduced [15N]-Y730NH2Y-α2 and ATP were mixed with β2 and CDP in a final volume of 250 μL to yield final concentrations of 24 μM [15N]-Y730NH2Y-α2/β2, 1 mM CDP, and 3 mM ATP. The reaction was quenched after 20 s by hand-freezing in liquid N2 and its EPR spectrum recorded (see below).
Reaction of Y730NH2Y-α2 or [15N]-Y730NH2Y-α2 with β2, CDP/ATP in Deuterated Buffer Monitored by EPR Spectroscopy
Assay buffer (50 mM Hepes, 15 mM MgSO4, 1 mM EDTA) was prepared in D2O and its pD adjusted to 8.0. Nucleotide stock solutions were prepared in deuterated assay buffer. Y730NH2Y-α2, [15N]-Y730NH2Y-α2, and β2 were exchanged into deuterated assay buffer by multiple concentration/dilution cycles at 4 °C using a Centriprep concentration device and a YM-30 membrane until the solution consisted of >99% deuterated buffer. The reaction in D2O was carried out as described above.
Preparation of High-Field EPR Samples
Prereduced Y730NH2Y-α2 and β2 were combined in an equimolar ratio and concentrated at 4 °C in a Minicon concentration device (YM-30 membrane) to a final complex concentration of 70−100 μM (ε280 nm (α2+β2) = 320 mM−1 cm−1). The concentrated sample was divided into 100 μL aliquots, placed in 1.5 mL Eppendorf tubes, and flash-frozen in liquid N2. High-field EPR samples were prepared by thawing each aliquot on ice and adding CDP and ATP to final concentrations of 2 and 6 mM, respectively. Each reaction was allowed to proceed for 30 s at room temperature, at which point 15 μL of glycerol buffer (100 mM Hepes, 30 mM MgSO4, 2 mM EDTA, 50% glycerol, pH 7.6) was added to yield a final glycerol concentration of ∼6%. The reaction was then quenched by hand-freezing in liquid N2, and the EPR spectra were recorded.
Continuous Wave (CW) X-Band EPR Spectroscopy
EPR spectra were recorded at 77 K in the Department of Chemistry Instrumentation Facility (MIT) on a Bruker ESP-300 X-band spectrometer equipped with a quartz finger dewar filled with liquid N2. EPR parameters were as follows: microwave frequency = 9.34 GHz, power = 30 μW, modulation amplitude = 1.5 G, modulation frequency = 100 kHz, time constant = 5.12 ms, scan time = 41.9 s. The composite spectra obtained were analyzed as described previously.22
Simulation of CW X-Band EPR Spectra
The X-band EPR powder spectra were analyzed using SimFonia software (Bruker). The g values used were determined by 180 GHz EPR spectroscopy and held fixed during the simulations. The spectrum of [15N]-NH2Y730• in D2O was simulated first, followed by that of [14N]-NH2Y730•. Then simulation of spectra in H2O was attempted. The strength and number of hyperfine couplings from I = 1/2 (1H, 15N) and I = 1 (2H, 14N) nuclei were varied systematically to yield the best fit to the experimental data. Since SimFonia employs perturbation theory and forbidden transitions might not be taken into account accurately when the hyperfine couplings exceed the nuclear Zeeman frequency, we checked the 9 GHz simulations using the EasySpin (3.1.0) EPR simulation package and used the option to perform matrix diagonalization.58 The global solutions obtained with SimFonia were also tested in EasySpin using least-squares fitting with the choice of the so-called “genetic” algorithm.
Pulsed EPR Spectroscopy at 9, 94, and 180 GHz
Nine and 94 GHz pulsed EPR spectra were recorded on Bruker Elexsys spectrometers of the series E580 and E680 using a π/2−π spin echo sequence with typical π/2 pulse lengths of 32 ns; 180 GHz pulsed EPR spectra were recorded on a home-built spectrometer previously reported with typical π/2 pulse lengths of 32 ns.59 Other experimental details are given in the figure captions. The pulsed EPR spectra were recorded at 70 K, where the Y122• is not detectable, as it relaxes more rapidly than an NH2Y• due to its vicinity to the di-iron cluster.60 At this temperature, the electron spin echo (ESE) spectra at all three frequencies contain contributions only from the NH2Y730• signal.
Simulation of Pulsed EPR Spectra
The EPR powder spectra were analyzed using SimFonia software (Bruker). To determine independently the optimal number of parameters, g values were obtained from the 180 GHz EPR spectrum, and the majority of the hyperfine couplings were fixed by iterative simulations of the 15N and 14N spectra in D2O at two different frequencies (9 and 94 GHz). Finally, the optimized parameters obtained from the 15N and 14N simulations in D2O were used as input for the simulations of the spectra in H2O. The latter spectra contained additionally the contribution of the two exchangeable protons of the NH2 group. The simulation of the amino proton contribution was complex because of their large hyperfine anisotropy and the noncollinearity of the g and A axes for these protons. The simulation thus required the introduction of the three Euler angles between the g and A axes as additional parameters. The definition of the three Euler rotations around the three axes z, y′, and z′ is illustrated in Figure S3, and the explicit rotation matrix is given in Bennati and Murphy.61
Results
Isotopic Labeling to Identify the NH2Y•
To assess the involvement of residues Y730 and Y731 in radical propagation catalyzed by RNR, we recently reported efficient site-specific incorporation of NH2Y into α2 using the amber suppressor tRNA/RS method. We demonstrated formation of a putative NH2Y• in the presence of CDP/ATP based on UV−visible and EPR spectroscopies and by power saturation studies.22 To further support our assignment of the new EPR species as an NH2Y730• and to obtain additional information regarding its electronic structure, we have performed isotopic replacement studies combined with multifrequency EPR spectroscopic analysis.
Preparation of [15N]-NH2Y
[15N]-NH2Y was prepared from [15N]-NO2Y according to the synthetic route in Scheme S1 in 80% yield with 98% [15N] isotopic enrichment. As expected, the nuclear coupling from 15N to 13C was observed in the 1H-decoupled 13C NMR and in 1H NMR spectra of [15N]-NH2Y.62 To further confirm the identity of the product, UV−vis spectra were acquired as a function of pH. The λmax and extinction coefficients for 3-ammoniumtyrosine, 3-aminotyrosine, and 3-aminotyrosinate were determined with authentic NH2Y (Scheme S2), revealing values that are identical within error to our synthetically prepared [15Ν]-ΝΗ2Y (Table S2).
Preparation of [15N]-NH2Y-α2
[15N]-NH2Y was incorporated into α2 as described for [14N]-NH2Y. Purification by dATP chromatography gave 6 mg of [15N]-Y730NH2Y-α2 with 98% 15N enrichment per gram of wet cell paste. The product was >95% pure by SDS PAGE (Figure S2). Spectrophotometric assays revealed a specific activity of 115 ± 12 nmol/min·mg. This activity is similar to that observed with Y730NH2Y-α2 (100 ± 8 nmol/min·mg) and will be discussed subsequently.22
CW X-Band EPR Spectroscopy of [14N]-NH2Y730• and [15N]-NH2Y730•
To characterize the new radical, [14N]- or [15N]-Y730NH2Y-α2 in H2O or D2O was mixed with β2, ATP, and CDP. The reaction was quenched after 20 s and the 9 GHz EPR spectrum recorded. CDP/ATP were chosen as substrate and effector due to the amount of radical produced versus other substrate/effector pairs (data not shown). The observed spectra were composites of Y122• and NH2Y730• signals. The NH2Y• signal was obtained by subtracting the Y122• contribution as described previously.22 The resulting spectra are shown in Figure 3. The spectra of [14N]-NH2Y730• and [15N]-NH2Y730• in D2O show a marked perturbation relative to each other and relative to those in H2O, as expected for substitution of 15N with 14N and substitution of amine protons for deuterons. The spectrum of [14N]-NH2Y730• in D2O appears as a 1:2:2:1 quartet, consistent with a doublet of triplets with similar hyperfine couplings. The triplet hyperfine coupling arises from the 14N nucleus (I = 1) and the doublet from one of the β-methylene protons (I = 1/2) of NH2Y. The spectrum of [15N]-NH2Y730• in D2O has collapsed to an apparent anisotropic triplet, demonstrating the effect of 15N incorporation (I = 1/2). These isotope-dependent spectral shifts validate our hypothesis that the new signal is associated with the NH2Y probe.
Figure 3.
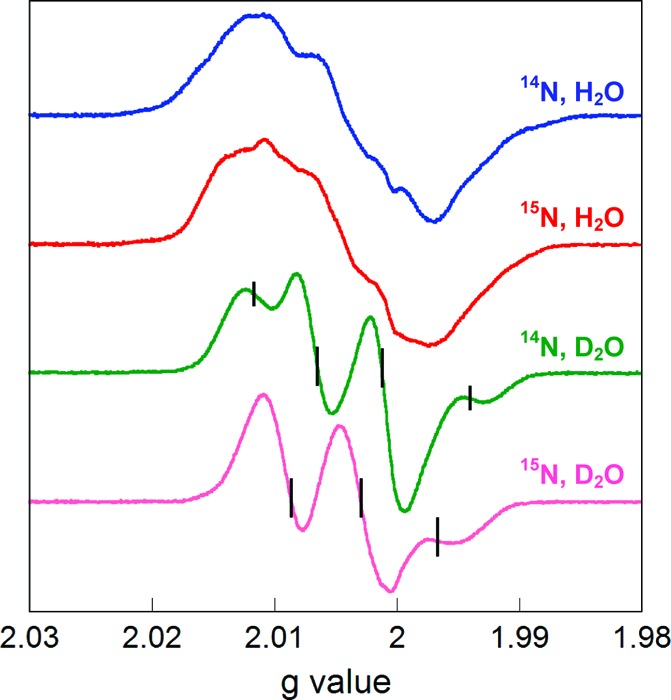
Normalized EPR spectra of isotopically substituted NH2Y730•. Spectra were acquired with Y730NH2Y-α2/β2 containing [14N]-NH2Y or [15N]-NH2Y in H2O or D2O assay buffer in the presence of CDP/ATP. The ticks on the deuterated traces indicate the positions of the quartet and triplet peaks with [14N]- and [15N]-NH2Y730•, respectively. See text for details.
Analysis of NH2Y730• by Pulsed High-Field EPR Spectroscopy
To obtain more detailed information on the anisotropy of the hyperfine interactions and g values, pulsed EPR spectra at different frequencies were acquired. We previously reported that the pulsed EPR method in class I RNRs filters the signal of any new radical in α2 from that of Y122• in β2, when the electron spin echo (ESE) spectrum is recorded at temperatures of 70 K or higher.60 The method is illustrated in Figure 4A with the 180 GHz pulsed EPR spectrum of NH2Y•. At 6 K, the spectrum is a composite of Y122• and NH2Y•. However, at 70 K, the signal associated with Y122•, adjacent to the diferric cluster, is not observed and only the NH2Y• is apparent.
Figure 4.

Multifrequency EPR spectra of NH2Y730• in the presence of CDP/ATP in H2O assay buffer. (A) 180 GHz spectrum at 6 K consisting of Y122• and NH2Y730• signals. Inset: 180 GHz spectrum at 70 K containing only the NH2Y730• signal. (B) Spectrum of NH2Y730• at 9, 94, and 180 GHz, demonstrating the higher g resolution with increasing frequency. The spectrum at 180 GHz yields g values of gx = 2.00520, gy = 2.00420, and gz = 2.00220. The error in these measurements was ±0.0001.
Pulsed EPR spectra were also recorded at the intermediate frequency of 94 GHz and at 9 GHz for comparison. The frequency dependence of the [14N]-NH2Y• ESE spectrum in H2O at 70 K is shown in Figure 4B. The absorption line shape at 9 GHz is quite symmetric and almost without structure, in agreement with the spectra observed in the CW mode (Figure 3). At 94 GHz, an asymmetry caused by the g tensor appears and anisotropic hyperfine interactions are resolved.
The pulsed 180 GHz EPR spectrum is displayed in absorption mode and shows a typical EPR line shape dominated by g anisotropy (Figure 4), making it possible to extract g values from the experimental spectrum at the canonical points that characterize singularities in the powder pattern. These points are marked in Figure 4B (bottom trace). The g values were obtained from analysis of spectra at 6 and 70 K using the spectrum of Y122• as a g-factor reference and yield gx = 2.00520, gy = 2.00420, gz = 2.00220 with a precision of ±0.0001. The intrinsic EPR line width is on the order of 22 MHz, and only very large hyperfine couplings can be resolved at 180 GHz.
Pulsed 94 GHz Spectra of [14N]-NH2Y730• in H2O and D2O Assay Buffer
To resolve and disentangle the hyperfine couplings associated with the NH2 protons, we recorded pulsed 94 GHz spectra at 70 K for [14N]-NH2Y730• in H2O and D2O and computed the point-by-point first derivative spectrum (Figure 5). Both spectra show highly resolved hyperfine interactions and indicate the expected anisotropy of some of the nuclei involved. The spectrum in D2O is dominated by two large hyperfine couplings, one almost isotropic and a second largely anisotropic. At the high-field edge (B∥gz), both hyperfine couplings are large with a similar size, giving rise to a quartet with 1:2:2:1 intensity, as observed in the 9 GHz CW spectra. In the center and at the low-field side (B∥gy and B∥gx, respectively) of the 94 GHz spectrum only one large hyperfine interaction is detected. A large anisotropic interaction is typical for the 14N nucleus, which has major spin density in the pz orbital (see below).63,64 Conversely, a large isotropic coupling is typical for β-methylene protons that have a C−H bond oriented almost parallel to the pz orbital of C1 in the aromatic ring (Figure 2), as has been well documented for Y122• of E.coli RNR and other Y•s.65−67 The spectrum in H2O shows a substantially different line shape, which arises from hyperfine contributions of the NH2 protons. The complexity of this spectrum is such that the visible features cannot be interpreted with simple splitting schemes.
Figure 5.
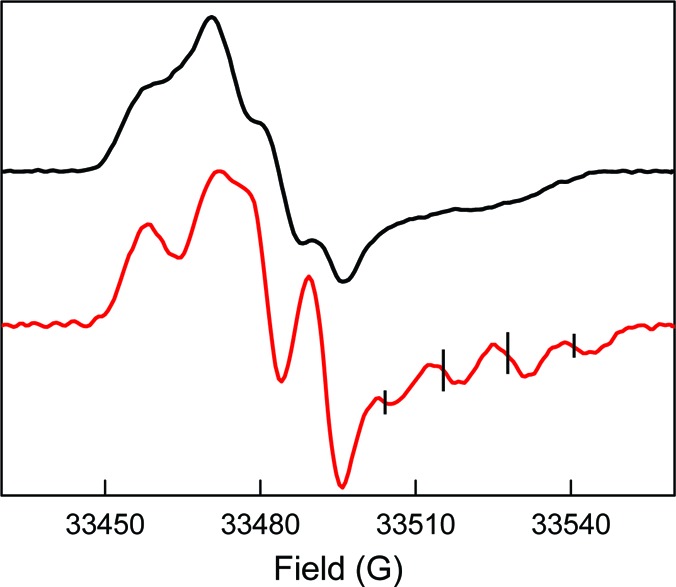
94 GHz pulsed derivative EPR spectra of NH2Y730• in H2O (black trace) and D2O (red trace). Experimental parameters: echo sequence: π/2 = 32 ns, τ = 248 ns (in H2O), τ = 260 ns (in D2O), π = 64 ns; trep = 5 ms; shots/point = 50; scans = 700; T = 70 K. The ticks on the deuterated trace indicate the positions of the quartet. See text for details.
Spectral Simulations
Analysis of the spectra at different frequencies required a simulation strategy in which the constraints posed by the spectra at the different frequencies were determined independently, as far as possible, and then combined to find a global solution. The simulation of the 9 GHz CW-EPR spectra for [15N] and [14N]-NH2Y730• acquired in D2O was attempted first. In this case, hyperfine coupling to the exchangeable amine deuterons is very small, thus facilitating the simulations by lowering the number of unknown hyperfine coupling constants. Consequently, the hyperfine couplings to two nuclei were included in the simulations, as seen in the spectra of Figure 6. One large hyperfine interaction was assumed to be isotropic, as is typical for β-methylene protons, whereas the second one, which generates the triplet pattern and arises from the 14N nucleus, was set anisotropic with the largest tensor component at Az. In this initial set of simulations, for simplicity, all hyperfine tensor axes were assumed collinear with the g tensor axes. The g values determined by EPR spectroscopy at 180 GHz were held as fixed parameters. The best simulation for the 9 GHz spectra could be obtained with the two assumed hyperfine tensors, indicating the couplings to the three ring protons cannot be resolved but contributed to a line width of ∼11 MHz (see Table 1). A simulation with an additional proton that accounted for a coupling at C5 with the parameters of Table S1 did not show any further improvement. For the [15N]-NH2Y730• simulation the parameters were identical to those of [14N]-NH2Y730•, except that the N-nucleus hyperfine constants were divided by 1.4, based on the difference in the gyromagnetic ratios of 14N and 15N. The fact that a single substitution in the 15N simulation with 14N nuclear parameters generates an excellent fit to the [14N]-NH2Y730•/D2O data provides further strong support that the new signal is associated with a neutral NH2Y730•.
Figure 6.
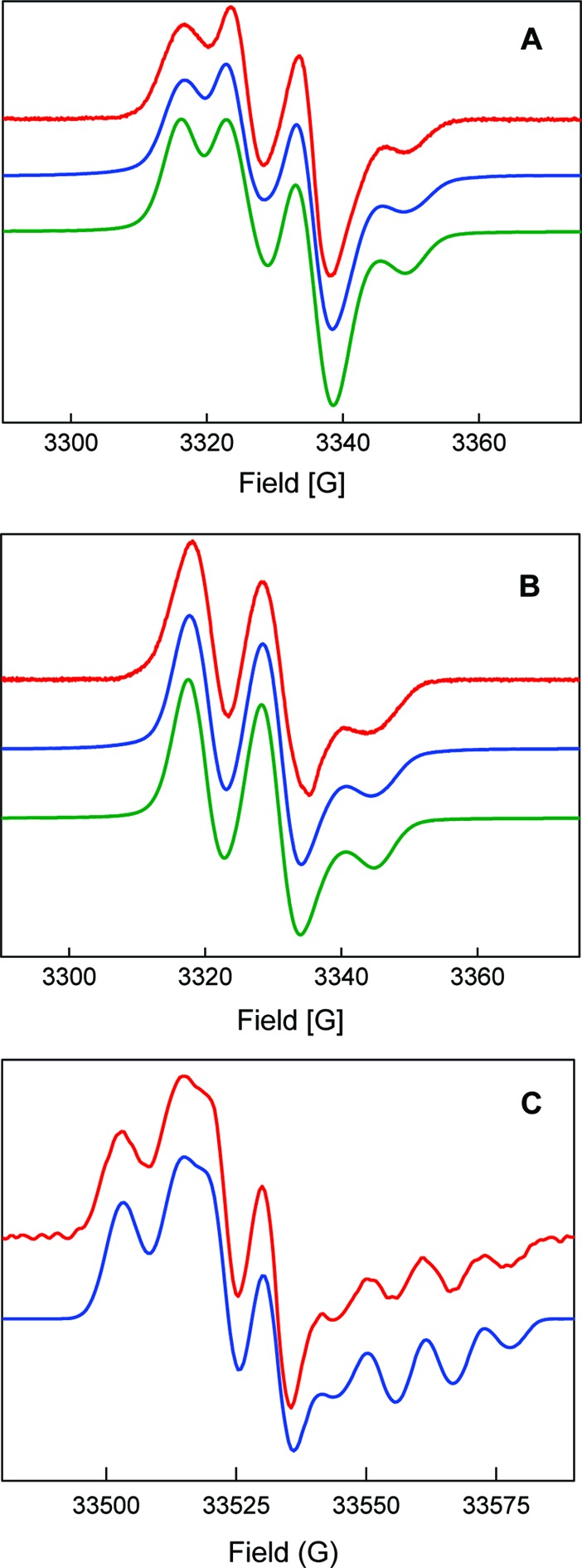
Simulations (blue = SimFonia; green = EasySpin) of the first derivative EPR spectra (red) of [14N]-NH2Y730• and [15N]-NH2Y730• in D2O assay buffer. (A) [14N]-NH2Y730• at 9 GHz. (B) [15N]-NH2Y730• at 9 GHz. (C) [14N]-NH2Y730• at 94 GHz. See Table 1 for parameters. The simulations obtained with EasySpin and the parameters of Table 1 are in agreement with those from SimFonia.
Table 1. Summary of Parameters (in MHz) Obtained from Simulation of [14N]-NH2Y730• and [15N]-NH2Y730• EPR Spectra in Protonated or Deuterated Buffera.
| nucleus | |Axx| | |Ayy| | |Azz| | α | β | γ |
|---|---|---|---|---|---|---|
| 14N | 2.4 | 1.6 | 30.5 | 60° | |5°| | |
| 15N | 3.4 | 2.2 | 42.7 | 60° | |5°| | |
| Cβ-H | 30.8 | 28.0 | 30.5 | |||
| NH2 (H2) | 13.0 | 4.4 | 27.6 | |126°| ± 10° | −60° ± 5° | |
| NH2 (H1) | 6.7 | 8.0 | 18.0 | 0° to (−40°) | |50°| |
Hyperfine tensor principal values (Aii) and Euler angles (α, β, γ) are indicated. The intrinsic EPR line width was 11−13 MHz at 9 and 94 GHz. In some cases, a range of values is indicated, for which the simulation does not change, as discussed in the text.
The parameters obtained from the simulations of the 9 GHz CW spectra in deuterated buffer were subsequently employed to simulate the 94 GHz spectrum of [14N]-NH2Y730• in D2O. The simulation immediately led to satisfactory results. Nevertheless, it could be improved with the introduction of a small anisotropy in the hyperfine tensor of the Cβ-proton, as previously found for Y122• at 140 GHz,46 and by introducing a 5° tilt angle for the Az14N-hyperfine tensor axis away from the gz axis toward the gx−gy plane.41 The best solution at 94 GHz was then tested for consistency with the 9 GHz spectra, and finally a global solution was found, which is shown in Figure 6 with the parameters in Table 1.
The analysis of the spectra was next attempted in H2O. Simulations first focused on the 94 GHz spectra, as the 9 GHz simulations did not provide sufficient constraints. Two additional couplings associated with the amino protons were added to the parameters for simulation of the spectra in D2O. Assuming that the hyperfine tensor axes of the amino protons were not collinear with the g tensor axes, simulations were performed by varying systematically all hyperfine tensor components as well as the Euler angles between hyperfine and g tensor axes for each nucleus. Our starting parameters were based on the studies on the o-aminophenol radical (Table S1). As a starting point two equivalent tensors for the amino protons with an isotropic coupling constant of 15 MHz were employed (Table S1, entry 1), as were a set of defined Euler rotations that accounted for the directions of the N−H bonds within the molecular plane. After several iterative steps, however, it became clear that the simulation required nonequivalent dipolar tensor components for the amino protons and N−H bond orientations noncollinear with the ring plane. The parameters were further optimized with the simulations of the 9 GHz spectra, and the global solution is displayed in Figure 7 with the parameters shown in Table 1. It is important to note that the simulations do not provide the absolute sign of the hyperfine interaction matrix, which requires ENDOR studies.
Figure 7.
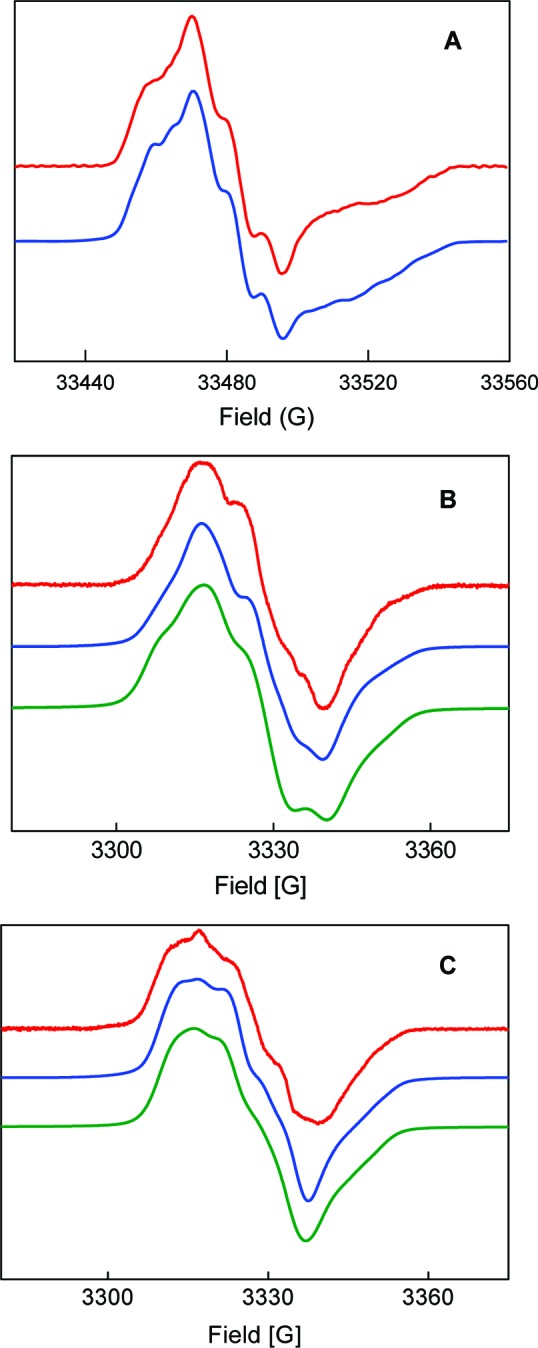
Simulations (blue = SimFonia; green = EasySpin) of the first derivative EPR spectra (red) of [14N]-NH2Y730• and [15N]-NH2Y730• in H2O assay buffer. (A) [14N]-NH2Y730• at 94 GHz. (B) [14N]-NH2Y730• at 9 GHz. (C) [15N]-NH2Y730• at 9 GHz. See Table 1 for parameters. The simulations with EasySpin appear slightly broader due to the effect of forbidden transitions.
Structure of NH2Y•
In order to rationalize the results obtained in Table 1, it is first necessary to establish the relationship between the obtained Euler angles (rotation of the A into the g tensor) and the molecular axes (Figure S3). The detailed location of the g and hyperfine tensors, the latter specifically for the nuclei of the NH2 group, is not known a priori. However, we found that our simulations were very sensitive, within <5°, to the γ angle of the N-(H2) proton (rotation about H(2)-Az′ collinear with gz, Figure S3), which is 60° (see Table 1). With the H(2)-Ax tensor axis along the N−(H2) bond and following the Euler rotations, the angle γ defines the orientation, with respect to gx, of the N−H(2) bond projection in the gx,y plane, i.e., the aromatic plane (Figure S3). Assuming that the gx axis lies along the C−O bond, then the projection of the N−H(2) bond in the gx,y plane forms an angle of about 120° with the C−N bond, a fact that is reasonably consistent with a C-NH2 group forming a trigonal pyramid. We note that the error associated with the uncertainty of the simulated γ angle leads to some error in the location of gx and, in turn, of the g tensor, used to deduce the location of the protons and the nitrogen group, which is difficult to quantify. Nevertheless, the results for the N-H(1) proton (Table 1) show that the simulation is not sensitive to the α angle (rotation about H(1)-Az, Figure S3), as long as it is between 0° and 40°. An α value larger than 0° represents a distortion of the trigonal pyramidal structure of the NH2 group, i.e., a tilt of the N−H(1) bond toward the oxygen atom.
The Euler angle β for the two amino protons is related to the dihedral of each N−H bond with respect to the plane of the aromatic ring (rotation about Ay, Figure S3). The β values in Table 1 lead to an out-of-plane angle between 30° for the N−H(1) and 54° for the N−H(2) bond. Owing to some uncertainty in the direction of gx (see above paragraph), the substantial values of the β angles required in the simulations still infer that the bond direction of the amino protons is considerably out of the molecular ring plane. Similar to the γ angle discussed above, a slight change of the β angle of N−H(2) (for instance by 5°) readily lowers the quality of the simulation. Altogether, the results in Table 1 show that the orientation of N−H(2) within the amino group seems well established, whereas the location of the N−H(1) is less defined. We also note that the hyperfine tensors of these two protons are substantially different. This is likely caused by an additional dipolar interaction of N−H(1) with the oxygen of the C−O group. We conclude that the differences in the hyperfine tensors of the amino protons combined with an α value consistent with a tilt of N−H(1) toward the oxygen strongly suggest the formation of an intramolecular hydrogen bond. A structure of the 3-aminotyrosyl radical is illustrated in Figure 2.
The 94 GHz simulation is not sensitive to the sign of the Euler angles β, in contrast to the 9 GHz simulations. A physical explanation for this observation arises from the largely anisotropic hyperfine couplings of the amino protons and their noncollinearity with the g tensor. Accordingly, the Euler rotations produce large off-diagonal hyperfine tensor elements in the laboratory frame that contribute more strongly to the low-frequency rather than to the high-frequency spectra.61 In Figure 7, the 9 GHz simulations appear slightly narrower than the experimental data. We suggest that some dynamics of the NH2 group could lead to an interconversion of the N−H bond directions (i.e., change in the sign of β) and might account for broadening of the experimental spectra. However, a more quantitative model is not possible due to the low-resolution of the 9 GHz spectra and missing information about detailed conformational restraints of NH2Y730• in the protein environment. To make this point, a 9 GHz EPR simulation of the [15N]-NH2Y730• spectrum with different signs of the N−H proton β angles is displayed in Figure S4.
Finally, the largest hyperfine coupling occurs at the β-methylene proton and is due to hyperconjugation with the C1 of the aromatic ring (Table 1, Figure 2). Its strength is modulated by θ, the dihedral angle between the Cβ−H bond and the pz orbital at C1, and by the spin density at C1 according to AH ≈ (B1 × ρC1 × cos2 θ).68,69 Using the isotropic coupling value of 29.8 MHz for one of the β-protons (Table 1), a value of AH ≪ 13 MHz for the second β-proton (the coupling is not visible in the EPR spectra and must be smaller than the intrinsic line width), and a B1 of 162 MHz,70 one calculates θ1 = (16 ± 1)° with θ2 = (104 ± 1)° and a spin density of 0.2 at C1 (Figure 2). With knowledge of θ1 and θ2 we can extract the dihedral angle between Cα and Cβ of the protein chain and pz at C1, which is (46 ± 1)°. The information about the dihedral angle allows us to generate a structure of the radical in α2 as illustrated in the discussion.71
Discussion
We have previously shown that site-specific insertion of NH2Y into the PCET pathway in α2 results in production of a new radical, concomitant with loss of the Y122•. The surprising observation that this mutant can support deoxynucleotide production suggests this new radical is actually an intermediate on the PCET pathway.22 In the present work, by 15N substitution of the amino group of Y and by acquiring data in H2O and D2O, we provide direct evidence that this new species is NH2Y730• and obtain insight into its molecular and electronic structure.
Our experiments and analysis have allowed us to determine the g values and the largest hyperfine tensors of NH2Y730• (Figure 2). The g values, gx = 2.00520, gy = 2.00420, gz = 2.00220, have been resolved using 180 GHz pulsed EPR spectroscopy and are in the range of those previously reported for phenoxy and ortho-substituted phenoxy radicals.72−77 Comparison of the g values of NH2Y730• with those for Y122• (gx = 2.0091, gy = 2.0046, gz = 2.0023 for Y122•)46 reveal that gy and gz are similar and that the gx shift is significantly suppressed.
Two mechanisms have been suggested for gx shifts in phenoxy radicals and their derivatives. The first is ortho substitution of the phenoxy ring with a heteroatom such as O or S, and the second is H-bonding to the phenoxy radical oxygen.54,72−75,77 High-frequency EPR studies on phenoxy radicals substituted at the ortho position with a thio-ether group such as that found in galactose oxidase have revealed a gx value of 2.007 with axial g tensor symmetry.75,77 For o-hydroxyphenoxyl radicals coordinated to a metal, the gx values varied between 2.0045 and 2.006,72 and in our recent studies with Y356DOPA-β2,24,78 a gx value of 2.0056 with an axial g tensor was observed upon formation of the DOPA• (M. Bennati and J. Stubbe, unpublished results).
On the other hand, suppression of gx has been found experimentally and validated by several theoretical studies to be associated with intermolecular hydrogen bonding to the phenoxy radical oxygen.54,79 Indeed, the gx value of Y•s in various RNRs has been used as a diagnostic for the presence of a H-bond. Owing to the complexity of the electronic structure of the NH2Y730•, which contains a heteroatom at the ortho position as well as an intramolecular H-bond (Figure 2), its low gx value (2.0052) could be associated with either or both of these mechanisms. Furthermore, the putative intermolecular H-bonding network at NH2Y730• (Figure 1) may also contribute to suppression of gx and is the subject of current ENDOR studies. Nevertheless, the form of the rhombic g tensor is characteristic for the NH2Y730• radical and is visible not only at 180 but also at 94 GHz.80 With this information at hand, it will be possible to distinguish an NH2Y• from other Y•s within enzymes using high-frequency EPR, if NH2Y is inserted as a probe to trap radical intermediates.
There are several important features of the electronic structure of NH2Y730• that have resulted from our study. First, the requirement for two amino protons in the simulations, along with their inequivalent hyperfine interactions with the nitrogen, is more consistent with an sp3- and not an sp2-hybridized nitrogen. Second, our model suggests the presence of an intramolecular H-bond between the N-H(1) and the oxygen. Our observations are in remarkable agreement with earlier experimental and theoretical studies by Loth et al. on the o-aminophenoxyl radical.41 Their studies suggested an out-of-plane angle of 30° (related to our β angle; see above and Figure S3) for the amino protons in an sp3-hybridized nitrogen, a conformation proposed to stabilize the internal H-bond with the phenoxyl oxygen. Our simulations is consistent with a 5° tilt angle of the C−N bond relative to the ring plane (above or below). Loth et al. found a similar tilt angle stabilized the aminophenol radical by ≥5 kcal/mol and increased the isotropic hyperfine coupling constant of 14N to match their experimental value of about 8.4 MHz. This hyperfine coupling is similar to our isotropic value of about 11 MHz assuming all tensor elements either positive or negative. On the other hand, the hyperfine couplings of their amino protons (Table S1) are different from those determined herein, although only average coupling values, and not full hyperfine tensors, were reported. Finally, recent DFT calculations predicted formation of an H-bond upon oxidation of o-aminophenol,76 in agreement with our results and those of Loth et al.81
Additional insight into the structure of the NH2Y• is provided by a recent report of Carter et al.82 on the synthesis and structural characterization of a neutral imino-semiquinone radical, 4,6-di-tert-butyl-2-tert-butyliminosemiquinone radical. The structure showed bond lengths for C−N and C−O of 1.34 and 1.26 Å, respectively, values intermediate between the reduced and oxidized forms of this molecule. They reported an EPR spectrum at 9 GHz recorded at room temperature in DMSO; however, no simulations of the spectrum were presented. Their protonated imino structure in which the N is sp2-hybridized is inconsistent with our EPR data (see Figure S5 for simulations using their model). Interestingly these authors also reported that their observed radical has an absorption feature at 734 nm with an ε734 that varied between 700 and 900 M−1 cm−1 depending on solvent. These studies provoked a re-examination of the visible spectrum of our NH2Y• in α, where we detected a broad absorption feature between 700 and 760 nm with an ε735 of 800 M−1 cm−1 (Figure S6).
NH2Y was chosen as a probe, as it is 190 mV easier to oxidize than Y at pH 7 and can function as a radical trap.33 It was hoped that trapping the NH2Y• in conjunction with high-field EPR and ENDOR spectroscopies could provide evidence for or against the proposed collinear PCET process to oxidize C439 in α2. The strength of the probe design is also its weakness: the intramolecular H-bonding within NH2Y• makes it a more efficient radical trap, but potentially complicates spectral analysis, as it is not present in Y•. A complete understanding of the electronic structure is thus essential, and computational studies are in progress to understand the unpaired spin density distribution. However, based on our observation that the NH2Y• can function as a catalytically competent radical in nucleotide reduction and on thermodynamic arguments, it is likely that oxidation of C439 occurs through hydrogen atom abstraction.22
Our EPR analysis has provided constraints to model NH2Y at residue 730 in α in order to obtain insight into its position relative to Y731 and C439. The hyperfine coupling of the β methylene proton of the NH2Y• allows calculation of the dihedral angle between Cα and Cβ of the protein chain and pz at C1 (θ = 46° ± 1°). It is similar to the one determined for Y730 in the crystal structure of wt α2 (θ = 36°).14 Thus, replacement of Y730 with NH2Y730 does not appear to significantly alter the orientation of this residue. Four orientations of the amino group are possible. Two of these orientations cause steric clashes with the protein. Recent structural characterization of NO2Y-α2 and NH2Y-α2s (Uhlin, Yokoyama, Minnihan, and Stubbe, unpublished results) suggest that ortho substitution requires the ring to have the orientation shown in Figure 8, thus eliminating a third possible orientation (Figure S7). In this model, the amino protons of NH2Y730• cannot form any hydrogen bonds except the internal one. The distances to Y731 and C439 are 3.3 and 3.5 Å, respectively. Thus, this modeling suggests that the amino group does not affect the distances between the essential amino acids in the proposed PCET pathway, indicating that the NH2Y probe can be used to measure the distance to the protons on Y731 (OH) and C439 (SH). The coupling to these two protons might be visible in high-field ENDOR experiments using D2O buffer, a protocol that permits observation of exchangeable protons and their orientation. The experiment will be aggravated by the concomitant observation of the amino protons, which are also exchangeable. Therefore, the independent determination of the tensors for the amino protons reported herein is a prerequisite for future ENDOR studies with NH2Y. These ENDOR studies as well as studies with C439S-α2 variants are currently underway.
Figure 8.
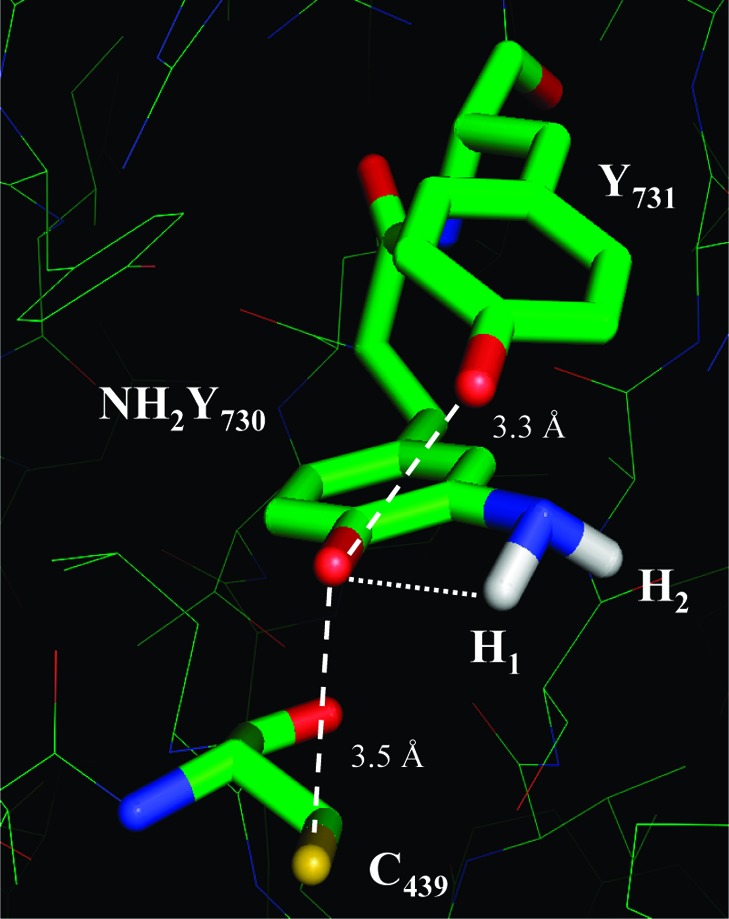
Model of the Y731−NH2Y730−C439 pathway in NH2Y730-α2. NH2Y has been modeled into residue 730 of α2 using the coordinates from ref (14). The pz orientation and Cβ- and amino proton dihedral angles are based on results herein. The distance between the NH1 and the phenol O is indicated. The distances between the essential residues around NH2Y730 are illustrated as discussed in the text.
Finally, understanding PCET within proteins is a major focus of interest of many laboratories, and transient Y•s have been identified in many of these processes.83−87 The availability of the evolved tRNA/tRNA synthetase pair to incorporate a NH2Y site specifically into any protein and our characterization of the NH2Y• reported herein should make this a useful probe to study PCET processes in general.
Acknowledgments
We thank Prof. T. Prisner (Univ. of Frankfurt) for allowing us to record the 180 GHz spectra and Dr. J. Fritscher for valuable discussions. We are indebted to Prof. Frank Neese and Christoph Riplinger (Univ. of Bonn) for starting quantum chemical calculations on NH2Y730• incorporated into α2 in support of this work. We also thank one of the anonymous reviewers for the constructive feedback. This work was supported by the NIH grant 29595 to J.S., the DFG-IRTG 1422 (T.A. and M.B.), and the Max Planck Society.
Supporting Information Available
Summary of EPR parameters previously reported for the o-aminophenol radical; UV spectral properties of authentic [14N]-NH2Y and synthetic [15N]-NH2Y determined in H2O at 25 °C; numbering scheme for the spectral data on the o-aminophenol radical data in Table 1; SDS PAGE analysis of purified [15N]-NH2Y730-α2; Euler rotations for the N-(H1) and N-(H2) protons; simulations of the 9 GHz CW-EPR spectrum of [15N]-NH2Y730• for the four possible combinations of the β angle signs; simulations of the 9 and 94 GHz spectra using a planar NH2 arrangement; spectrum of the 730 nm feature associated with NH2Y•; models of the Y731−NH2Y730−C439 pathway; synthetic scheme used for preparation of 15N-NH2Y; different protonation states and pKas of NH2Y. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Stubbe J.; van der Donk W. A. Chem. Rev. 1998, 98, 705. [DOI] [PubMed] [Google Scholar]
- Jordan A.; Reichard P. Annu. Rev. Biochem. 1998, 67, 71. [DOI] [PubMed] [Google Scholar]
- Brown N. C.; Canellakis Z. N.; Lundin B.; Reichard P.; Thelander L. Eur. J. Biochem. 1969, 9, 561. [DOI] [PubMed] [Google Scholar]
- Thelander L. J. Biol. Chem. 1973, 248, 4591. [PubMed] [Google Scholar]
- Stubbe J. J. Biol. Chem. 1990, 265, 5329. [PubMed] [Google Scholar]
- Stubbe J. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht S.; Gerfen G. G.; Stubbe J. Science 1996, 271, 477. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Riggs-Gelasco P. Trends Biochem. Sci. 1998, 23, 438. [DOI] [PubMed] [Google Scholar]
- Brown N. C.; Reichard P. J. Mol. Biol. 1969, 46, 39. [DOI] [PubMed] [Google Scholar]
- Brown N. C.; Reichard P. J. Mol. Biol. 1969, 46, 25. [DOI] [PubMed] [Google Scholar]
- Ehrenberg A.; Reichard P. J. Biol. Chem. 1972, 247, 3485. [PubMed] [Google Scholar]
- Sjöberg B.-M.; Reichard P.; Gräslund A.; Ehrenberg A. J. Biol. Chem. 1978, 253, 6863. [PubMed] [Google Scholar]
- Reichard P.; Ehrenberg A. Science 1983, 221, 514. [DOI] [PubMed] [Google Scholar]
- Uhlin U.; Eklund H. Nature 1994, 370, 533. [DOI] [PubMed] [Google Scholar]
- Nordlund P.; Sjöberg B.-M.; Eklund H. Nature 1990, 345, 593. [DOI] [PubMed] [Google Scholar]
- Bennati M.; Robblee J. H.; Mugnaini V.; Stubbe J.; Freed J. H.; Borbat P. J. Am. Chem. Soc. 2005, 127, 15014. [DOI] [PubMed] [Google Scholar]
- Uhlin U.; Eklund H. J. Mol. Biol. 1996, 262, 358. [DOI] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G.; Yee C. S.; Chang M. C. Y. Chem. Rev. 2003, 103, 2167. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Hodgkiss J. M.; Stubbe J.; Nocera D. G. Philos. Trans. R Soc. London B Biol. Sci. 2006, 361, 1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Yee C. S.; Reece S. Y.; Nocera D. G.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 1562. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Seyedsayamdost M. R.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2007, 129, 8500. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Xie J.; Chan C. T.; Schultz P. G.; Stubbe J. J. Am. Chem. Soc. 2007, 129, 15060. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Yee C. S.; Stubbe J. Nat. Protoc. 2007, 2, 1225. [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 2522. [DOI] [PubMed] [Google Scholar]
- Jovanovic S. J.; Steenken S.; Tosic M.; Marjanovic B.; Simic M. G. J. Am. Chem. Soc. 1994, 116, 4846. [Google Scholar]
- Seyedsayamdost M. R.; Reece S. Y.; Nocera D. G.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 1569. [DOI] [PubMed] [Google Scholar]
- Reece S. Y.; Seyedsayamdost M. R.; Stubbe J.; Nocera D. G. J. Am. Chem. Soc. 2006, 128, 13654. [DOI] [PubMed] [Google Scholar]
- Yee C. S.; Chang M. C. Y.; Ge J.; Nocera D. G.; Stubbe J. J. Am. Chem. Soc. 2003, 125, 10506. [DOI] [PubMed] [Google Scholar]
- Yee C. S.; Seyedsayamdost M. R.; Chang M. C. Y.; Nocera D. G.; Stubbe J. Biochemistry 2003, 42, 14541. [DOI] [PubMed] [Google Scholar]
- Chang M. C. Y.; Yee C. S.; Nocera D. G.; Stubbe J. J. Am. Chem. Soc. 2004, 126, 16702. [DOI] [PubMed] [Google Scholar]
- Wang L.; Schultz P. G. Angew. Chem., Int. Ed. 2004, 44, 34. [DOI] [PubMed] [Google Scholar]
- Xie J.; Schultz P. G. Methods 2005, 36, 227. [DOI] [PubMed] [Google Scholar]
- DeFelippis M. R.; Murthy C. P.; Broitman F.; Weinraub D.; Faraggi M.; Klapper M. H. J. Phys. Chem. 1991, 95, 3416. [Google Scholar]
- Skawinski W.; Flisak J.; Chung A. C.; Jordan F.; Mendelsohn R. J. Labelled Comp. Radiopharm. 1990, 28, 1179. [Google Scholar]
- Seyedsayamdost M. R.; Chan C. T.; Mugnaini V.; Stubbe J.; Bennati M. J. Am. Chem. Soc. 2007, 129, 15748. [DOI] [PubMed] [Google Scholar]
- Neta P.; Fessenden R. W. J. Phys. Chem. 1974, 78, 523. [Google Scholar]
- Dixon W. T.; M M.; Murphy D. J. Chem. Soc., Faraday Trans. 2 1974, 70, 1713. [Google Scholar]
- Dixon W. T.; Hoyle P. M.; Murphy D. J. Chem. Soc., Faraday Trans. 2 1978, 74, 2027. [Google Scholar]
- Simandi L. I.; Barna T. M.; Korecz L.; Rockenbauer A. Tetrahedron Lett. 1993, 34, 717. [Google Scholar]
- Sur S. K.; Colpa J. P. Organometallics 1989, 8, 2749. [Google Scholar]
- Loth K.; Graf F. Helv. Chim. Acta 1981, 64, 1910. [Google Scholar]
- Pesavento R. P.; van der Donk W. A. Adv. Protein Chem. 2001, 58, 317. [DOI] [PubMed] [Google Scholar]
- Lendzian F. Biochim. Biophys. Acta 2005, 1707, 67. [DOI] [PubMed] [Google Scholar]
- Hoganson C. W.; Tommos C. Biochim. Biophys. Acta 2004, 1655, 116. [DOI] [PubMed] [Google Scholar]
- Alabugin I. V.; Manoharan M.; Buck M.; Clark R. J. THEOCHEM 2007, 813, 21. [Google Scholar]
- Gerfen G. J.; Bellew B. F.; Un S.; Bollinger J. M. Jr.; Stubbe J.; Griffin R. G.; Singel D. J. J. Am. Chem. Soc. 1993, 115, 6420. [Google Scholar]
- Allard P.; Barra A. L.; Andersson K. K.; Schmidt P. P.; Atta M.; Gräslund A. J. Am. Chem. Soc. 1996, 118, 895. [Google Scholar]
- Schmidt P. P.; Andersson K. K.; Barra A. L.; Thelander L.; Gräslund A. J. Biol. Chem. 1996, 271, 23615. [DOI] [PubMed] [Google Scholar]
- Sauge-Merle S.; Laulhere J. P.; Coves J.; Le Pape L.; Menage S.; Fontecave M. J. Biol. Inorg. Chem. 1997, 2, 586. [Google Scholar]
- Dam P. J. v.; Willems J.; Schmidt P. P.; Pötsch S.; Barra A. L.; Hagen W. R.; Hoffman B. M.; Andersson K. K.; Gräslund A. J. Am. Chem. Soc. 1998, 120, 5080. [Google Scholar]
- Liu A.; Barra A. L.; Rubin H.; Lu G.; Gräslund A. J. Am. Chem. Soc. 2000, 122, 1974. [Google Scholar]
- Bar G.; Bennati M.; Nguyen H. H.; Ge J.; Stubbe J. A.; Griffin R. G. J. Am. Chem. Soc. 2001, 123, 3569. [DOI] [PubMed] [Google Scholar]
- Un S.; Atta M.; Fontecave M.; Rutherford A. W. J. Am. Chem. Soc. 1995, 117, 10713. [Google Scholar]
- Un S. Magn. Reson. Chem. 2005, 43, S229. [DOI] [PubMed] [Google Scholar]
- Wilson J. C.; Wu G.; Tsai A.-L.; Gerfen G. J. J. Am. Chem. Soc. 2005, 127, 1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svistunenko D. A.; Cooper C. E. Biochem. J. 2004, 87, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warncke K.; Babcock G. T.; McCracken J. J. Phys. Chem. 1996, 100, 4654. [Google Scholar]
- Stoll S.; Schweiger A. J. Magn. Reson. 2006, 178, 42. [DOI] [PubMed] [Google Scholar]
- Hertel M. M.; Denysenkov V. P.; Bennati M.; Prisner T. F. Magn. Reson. Chem. 2005, 43, S248. [DOI] [PubMed] [Google Scholar]
- Lawrence C. C.; Bennati M.; Obias H. V.; Bar G.; Griffin R. G.; Stubbe J. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 8979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennati M.; Murphy D.Electron Paramagnetic Resonance Spectra in the Solid State. In Electron Paramagentic Resonance; Brustolon M., Giamello E., Eds.; Wiley & Sons: New York, 2009. [Google Scholar]
- Interestingly, in the case of [15N]-NO2Y, 1J, 2J, and 3J couplings were observed in the 13C NMR spectrum at 15.2, 2.1−2.4, and 1.2−1.6 Hz, respectively. With [15N]-NH2Y, however, only the 1J and 2J couplings are present in the 13C NMR spectrum, at 9.4 and 1.7−1.9 Hz, respectively. This is consistent with previous NMR studies on aniline, which have shown 1J, 2J, and 3J15N−13C couplings on the order of 13.9−14.8, 2−4, and 0−1.5 Hz, respectively, depending on the solvent and concentration.
- Huyett J. E.; Doan P. E.; Gurbiel R.; Houseman A. L. P.; Sivaraja M.; Goodin D. B.; Hoffman B. M. J. Am. Chem. Soc. 1995, 117, 9033. [Google Scholar]
- Bleifuss G.; Kolberg M.; Pötsch S.; Hofbauer W.; Bittl R.; Lubitz W.; Gräslund A.; Lassmann G.; Lendzian F. Biochemistry 2001, 40, 15362. [DOI] [PubMed] [Google Scholar]
- Hulsebosch R. J.; van den Brink J. S.; Nieuwenhuis S. A. M.; Gast P.; Raap J.; Lugtenburg J.; Hoff A. J. J. Am. Chem. Soc. 1997, 119, 8685. [Google Scholar]
- Bender C. J.; Sahlin M.; Babcock G. T.; Barry B. A.; Chandrashekar T. K.; Salowe S. P.; Stubbe J.; Lindstrom B.; Petersson L.; Ehrenberg A.; Sjöberg B.-M. J. Am. Chem. Soc. 1989, 111, 8076. [Google Scholar]
- Hoganson C. W.; Sahlin M.; Sjöberg B.-M.; Babcock G. T. J. Am. Chem. Soc. 1996, 118, 4672. [Google Scholar]
- McConnell H. M. J. Chem. Phys. 1956, 24, 764. [Google Scholar]
- Heller C.; McConnell H. M. J. Chem. Phys. 1960, 32, 1535. [Google Scholar]
- Fessenden R. W.; Schuler R. H. J. Chem. Phys. 1963, 39, 2147. [Google Scholar]
- We note that the large hyperfine coupling of the amine nitrogen suggests that it bears significant unpaired spin density consistent with recent computational work. Additional studies are in progress, using the structural features obtained herein as input, to delineate the complete distribution of the spin density.
- Brezgunov A. Y.; Dubinskii A. A.; Poluektov O. G.; Prokof ’ev A. I.; Chemerisov S. D.; Lebedev Y. S. J. Struct. Chem. 1992, 33, 69. [Google Scholar]
- Engstrom M.; Himo F.; Agren H. Chem. Phys. Lett. 2000, 319, 191. [Google Scholar]
- Kaupp M.; Gress T.; Reviakine R.; Malkina O. L.; Malkin V. G. J. Phys. Chem. B 2003, 107, 331. [Google Scholar]
- Gerfen G. J.; Bellew B. F.; Griffin R. G. J. Phys. Chem. 1996, 100, 16739. [Google Scholar]
- Himo F.; Eriksson L. A.; Blomberg M. R. A.; Siegbahn P. E. M. Int. J. Quantum Chem. 2000, 76, 714. [Google Scholar]
- Lee Y. K.; Whittaker M. M.; Whittaker J. W. Biochemistry 2008, 47, 6637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M. R.; Stubbe J. J. Am. Chem. Soc. 2007, 129, 2226. [DOI] [PubMed] [Google Scholar]
- Bennati M.; Stubbe J.; Griffin R. G. Appl. Magn. Reson. 2001, 21, 389. [Google Scholar]
- The g values of the NH2Y• radical incorporated at position Y731 in α2 are the same as those for NH2Y730• (M. Bennati and J. Stubbe, unpublished results).
- Preliminary calculations on the isolated NH2Y• lead to a planar amino group. While this is a reasonable result, it is at variance with the experimental data obtained in this work. This strongly suggests a significant impact of the protein environment. Cluster and QM/MM calculations with more than 100 atoms in the quantum region are planned to clarify this point.
- Carter S. M.; Sia A.; Shaw M. J.; Heyduk A. F. J. Am. Chem. Soc. 2008, 130, 5838. [DOI] [PubMed] [Google Scholar]
- Babcock G. T.; Espe M.; Hoganson C.; Lydakis-Simantiris N.; McCracken J.; Shi W. J.; Styring S.; Tommos C.; Warncke K. Acta Chem. Scand. 1997, 51, 533. [DOI] [PubMed] [Google Scholar]
- Pujols-Ayala I.; Barry B. A. Biochim. Biophys. Acta 2004, 1655, 205. [DOI] [PubMed] [Google Scholar]
- Gupta A.; Mukherjee A.; Matsui K.; Roth J. P. J. Am. Chem. Soc. 2008, 130, 11274. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee S.; Deterding L. J.; Jiang J.; Bonini M. G.; Tomer K. B.; Ramirez D. C.; Mason R. P. J. Am. Chem. Soc. 2007, 129, 13493. [DOI] [PubMed] [Google Scholar]
- Rigby S. E. J.; Hynson R. M. G.; Ramsay R. R.; Munro A. W.; Scrutton N. S. J. Biol. Chem. 2005, 280, 4627. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.