

Abstract
Perturbations of astrocytes trigger neurodegeneration in several diseases, but the glial cell–intrinsic mechanisms that induce neurodegeneration remain poorly understood. We found that a protein complex of α2-Na/K ATPase and α-adducin was enriched in astrocytes expressing mutant superoxide dismutase 1 (SOD1), which causes familial amyotrophic lateral sclerosis (ALS). Knockdown of α2-Na/K ATPase or α-adducin in mutant SOD1 astrocytes protected motor neurons from degeneration, including in mutant SOD1 mice in vivo. Heterozygous disruption of the α2-Na/K ATPase gene suppressed degeneration in vivo and increased the lifespan of mutant SOD1 mice. The pharmacological agent digoxin, which inhibits Na/K ATPase activity, protected motor neurons from mutant SOD1 astrocyte–induced degeneration. Notably, α2-Na/K ATPase and α-adducin were upregulated in spinal cord of sporadic and familial ALS patients. Collectively, our findings define chronic activation of the α2-Na/K ATPase/α-adducin complex as a critical glial cell–intrinsic mechanism of non–cell autonomous neurodegeneration, with implications for potential therapies for neurodegenerative diseases.
Astrocytes represent the most abundant cell type in the CNS and have diverse functions in the developing and mature CNS1,2. Astrocytes and neurons share a common lineage during development, and both cell types often express disease genes that trigger neurodegeneration in the CNS. Notably, astrocytes are beginning to emerge as critical targets of CNS disorders that were once thought to selectively afflict neurons. In particular, mounting evidence suggests that astrocytes have a fundamental role in the progression of diverse neurodegenerative diseases3,4. Expression of mutant proteins in astrocytes in ALS, Huntington’s disease and spinocerebellar ataxias induce non–cell autonomous neurodegeneration5–11. However, with few exceptions12, the cell-intrinsic mechanisms operating in mutant astrocytes that trigger non–cell autonomous neurodegeneration remain largely unknown.
The cellular basis of non–cell autonomous neurodegeneration has been best characterized in ALS9,13–15. ALS is the most common motor neuron disease in adults and is characteristically fatal within 5 years of onset. Approximately 5–10% of patients with ALS are familial with an autosomal dominant pattern of inheritance16. Mutations in the gene encoding SOD1 account for 20% of familial ALS with over 140 distinct mutations identified to date16,17.
Transgenic mice expressing the G93A mutation in SOD1 (SOD1G93A) have been an invaluable model for studies of neurodegeneration, as these mice recapitulate the pathological features of ALS, including reactive gliosis, ubiquitin aggregates, loss of motor neurons and lethality13,18. The degeneration of motor neurons in SOD1G93A mice is thought to result in part from cell-autonomous mechanisms19. In addition, expression of mutant SOD1 in astrocytes induces the degeneration of motor neurons in a non–cell autonomous fashion9,14,15,20. Notably, astrocytes from mutant SOD1 mice and astrocytes derived from post-mortem spinal cords of patients with either SOD1 mutations or sporadic ALS induce toxicity in primary motor neurons20. Thus, mutant SOD1 mice provide an excellent model for elucidation of the glial cell–intrinsic mechanisms of non–cell autonomous neurodegeneration.
We found that a complex composed of the ion pump α2-Na/K ATPase and the protein α-adducin in SOD1G93A astrocytes triggers the non–cell autonomous degeneration of motor neurons. Knockdown of α2-Na/K ATPase or α-adducin in SOD1G93A astrocytes markedly inhibited their ability to induce degeneration in co-cultured primary motor neurons. In addition, in vivo knockdown of the α2-Na/K ATPase/α-adducin complex by lentiviral-mediated RNAi in the spinal cord of SOD1G93A mice protected motor neurons from degeneration in vivo. Notably, inactivating one allele of Atp1a2 (the gene encoding α2-Na/K ATPase) in SOD1G93A mice suppressed motor neuron degeneration and substantially increased mouse lifespan. In mechanistic studies, we found that mitochondrial respiration and inflammatory gene expression were induced in SOD1G93A astrocytes, and removal of one allele of Atp1a2 reversed these effects, suggesting that the upregulation of α2-Na/K ATPase stimulates mitochondrial respiration and expression of secreted inflammatory factors in SOD1G93A astrocytes. The Na/K ATPase small molecule inhibitor digoxin, which has been widely used in the treatment of congestive heart failure21, blocked the degeneration of co-cultured primary motor neurons. Finally, α2-Na/K ATPase and α-adducin were substantially upregulated in the spinal cord in individuals with familial ALS bearing distinct SOD1 mutations as well as in sporadic ALS. Together, our findings suggest that the α2-Na/K ATPase/α-adducin complex is critical role for the pathology of non–cell autonomous neurodegeneration and provides a potential drugable target in the treatment of neurodegenerative diseases.
RESULTS
α-adducin induces non–cell autonomous motor neuron degeneration
Using an antibody that recognizes phosphorylation events in cells following exposure to oxidative stress22, we unexpectedly identified a 105-kDa immunoreactive protein band enriched in lysates of spinal cord from symptomatic SOD1G93A mice at 120 d of age as compared with age-matched wild-type littermate mice and non-symptomatic 60-d-old SOD1G93A mice (Fig. 1a). Following incubation of symptomatic SOD1G93A spinal cord lysates with λ-phosphatase, the immunoreactive 105-kDa protein band was eliminated (Fig. 1a). Mass spectrometry analysis following immunoprecipitation assays led to the identification of α-adducin as the putative phosphorylated protein in SOD1G93A spinal cords (data not shown). We validated our mass spectrometry analysis by immunoprecipitating α-adducin and immunoblotting with our phospo-antibody, confirming the identity of α-adducin in symptomatic SOD1G93A mice (Fig. 1b). Notably, immunoblotting for total α-adducin protein levels in symptomatic SOD1G93A mice at 120 d and control age-matched wild-type littermate mice revealed that α-adducin protein was upregulated in spinal cord of symptomatic SOD1G93A mice (Fig. 1c). The upregulation of α-adducin in the spinal cord of SOD1G93A mice was evident as early as 90 d of age, which represents the time of disease onset (Supplementary Fig. 1a). In other experiments, we identified Ser436 as the site of α-adducin phosphorylation in lysates of SOD1G93A spinal cords (data not shown).
Figure 1.

Upregulation of α-adducin in SOD1G93A astrocytes mediates non–cell autonomous degeneration of motor neurons. (a) Lysates of spinal cord from symptomatic SOD1G93A transgenic mice and control non-transgenic mice were subjected to immunoblotting using an antibody that recognizes phosphorylation events in cells following exposure to oxidative stress. Lysates in lanes 5–8 were incubated with λ-phosphatase, which largely eliminated the 105-kDa immunoreactive band. (b) Lysates of spinal cords from symptomatic SOD1G93A and control mice were subjected to immunoprecipitation using the α-adducin antibody followed by immunoblotting with our phospho-antibody. (c) Immunoblots from spinal cord lysates showed an increase in α-adducin and phosphorylated α-adducin relative to the internal control proteins ERK and 14-3-3β in symptomatic SOD1G93A mice as compared with control wild-type littermates (120 d). (d) Immunoblots revealed that α-adducin and phosphorylated α-adducin were predominately expressed in primary glial cultures enriched with the astrocyte marker GFAP relative to primary motor neuron cultures enriched with the neuron marker β-tubulin. HSP60 was used as an internal control (lower panel). Blots shown in a–d are cropped. Full-length blots are presented in Supplementary Figure 11. (e) Immunohistochemistry of symptomatic SOD1G93A lumbar spinal cord sections revealed that phosphorylated-Ser436 α-adducin (phospho-α-adducin) colocalized with the astrocyte protein GFAP. Dashed line indicates ventral horn. The boxed areas are shown at high magnification; scale bars represent 50 μm. (f) Co-cultured astrocytes and motor neurons were subjected to immunocytochemistry with antibodies recognizing the motor neuron nuclear protein Islet1 (red) and the dendrite protein Map2 (green); scale bar represents 50 μm. Wild-type astrocytes transfected with the control U6 or α-adducin RNAi plasmid had little or no effect on motor neuron morphology or survival (upper and lower left panels). Control U6 SOD1G93A astrocytes induced non–cell autonomous motor neuron cell death and dendrite abnormalities (upper right panel). α-adducin knockdown in SOD1G93A astrocytes protected motor neurons against the non–cell autonomous cell death and dendrite abnormality (lower right panel). (g,h) Quantification of motor neuron survival was derived from n ≥ 900 cells per condition and values presented are the average of three independent experiments; two-tailed unpaired t test, P = 0.0095. Quantification of dendrite length was derived from n ≥ 240 images per condition and values presented are the average of three independent experiments; two-tailed unpaired t test, P = 0.0001. Data are presented as mean ± s.e.m. **P < 0.01, ***P < 0.001.
We next determined the cellular origin of α-adducin in SOD1G93A mice. In immunoblotting analysis of primary SOD1G93A glial cells and motor neurons, α-adducin and Ser436-phosphorylated α-adducin were predominantly expressed in astrocytes rather than motor neurons (Fig. 1d). In complementary immunohistochemical analyses, Ser436-phosphorylated α-adducin colocalized with the astrocyte marker glia fibrillary acidic protein (GFAP) in spinal cord of symptomatic SOD1G93A mice (Fig. 1e). The Ser436-phosphorylated α-adducin immunoreactivity was specifically blocked by a phosphorylated-Ser436 α-adducin peptide, validating the specificity of the phosphorylated-Ser436 α-adducin immunoreactivity in astrocytes (data not shown). In control analyses, Ser436-phosphorylated α-adducin did not colocalize with the motor neuron marker SMi32 in the spinal cord of symptomatic SOD1G93A. In addition, Ser436-phosphorylated α-adducin colocalized with GFAP, but not SMi32, in the spinal cord of wild-type mice (Supplementary Fig. 1b,c). α-adducin forms heterodimers or heterotetramers with β-adducin or γ-adducin23. The adducins function as barbed-end actin-capping proteins that regulate actin filaments length23. Notably, the absence of α-adducin leads to the downregulation of the other subunits, suggesting that α-adducin is the limiting subunit in the formation of adducin heteromers24. The phosphorylation of α-adducin has been described in mutant SOD1 mice and sporadic ALS patients25,26, but the role of α-adducin in neurodegeneration is not yet known. The localization and abundance of α-adducin in astrocytes in symptomatic SOD1G93A mice raises the question of whether α-adducin might be involved in the toxic gain of function in SOD1G93A astrocytes.
To characterize α-adducin function in neurodegeneration, we first employed a cell culture model in which SOD1G93A astrocytes are co-cultured with primary spinal cord motor neurons, which recapitulates the non–cell autonomous degeneration of motor neurons in vivo14,15,20. Using a plasmid-based method of RNA interference (RNAi), we induced efficient knockdown of α-adducin in SOD1G93A astrocytes (Supplementary Fig. 2a). We assessed the effect of α-adducin knockdown in SOD1G93A astrocytes on measures of degeneration in co-cultured motor neurons. SOD1G93A astrocytes, but not wild-type astrocytes, transfected with the control U6 RNAi plasmid induced cell death and a substantial reduction in total dendrite length in motor neurons (Fig. 1f–h), confirming that mutant SOD1 astrocytes trigger non–cell autonomous degeneration of motor neurons. Notably, we found that knockdown of α-adducin in SOD1G93A astrocytes protected motor neurons against the non–cell autonomous induction of motor neuron cell death (Fig. 1f–h). Although control SOD1G93A astrocytes induced cell death in 50% of co-cultured motor neurons, α-adducin knockdown SOD1G93A astrocytes induced cell death in only 23% of co-cultured motor neurons (Fig. 1g). Likewise, knockdown of α-adducin in SOD1G93A astrocytes prevented the ability of SOD1G93A astrocytes to induce abnormalities in motor neuron dendrite morphology (Fig. 1f,h). In control analyses, knockdown of α-adducin in non-transgenic astrocytes had little or no effect on the survival or morphology of co-cultured motor neurons (Fig. 1f–h).
To determine the specificity of the α-adducin RNAi–induced neuroprotective phenotype in SOD1G93A astrocytes, we performed a rescue experiment. We expressed an RNAi-resistant form of α-adducin (Add-Res) in the background of α-adducin RNAi in SOD1G93A astrocytes. Expression of Add-Res in SOD1G93A astrocytes reversed the ability of α-adducin RNAi to protect co-cultured motor neurons from cell death and impairment of dendrite morphology (Supplementary Fig. 2). These data indicate that the α-adducin RNAi–induced neuroprotective effect was the result of specific knockdown of α-adducin in SOD1G93A astrocytes rather than off-target effects of RNAi. Together, our data suggest that α-adducin in SOD1G93A astrocytes is critical for the non–cell autonomous degeneration of motor neurons.
We next assessed the effect of α-adducin knockdown on neurodegeneration in the spinal cord of SOD1G93A mice in vivo. We injected lentivirus encoding α-adducin short hairpin RNAs and GFP (LV-Addi) or the corresponding control lentivirus (LV-U6) unilaterally in the lumbar spinal cord in SOD1G93A mice (Fig. 2a). The in vivo RNAi approach allowed comparison of surviving motor neurons in the injected ventral horn with the non-injected contralateral ventral horn in the same spinal cord sections. We injected lentivirus in the spinal cord of SOD1G93A mice at 90 d, a time at which gliosis had set in, thereby maximizing the targeting of reactive astrocytes. Following injection of the lentivirus encoding GFP into the spinal cord of SOD1G93A mice, 88% of the GFP-positive cells expressed the astrocyte marker GFAP, 8% expressed the motor neuron marker SMi32 and 3% expressed the microglia marker Iba1 (Supplementary Fig. 3a,b). Injection of lentivirus expressing GFP into wild-type mice at 90 d revealed 6% colocalization of GFP with SMi32, 3% with Iba1 and 91% with GFAP (Supplementary Fig. 3c,d). Thus, injected lentivirus into the spinal cord of SOD1G93A and control mice largely targeted astrocytes in vivo.
Figure 2.

Knockdown of α-adducin in SOD1G93A mice suppresses motor neuron degeneration in vivo. (a,c) Spinal cords from SOD1G93A mice injected intraspinally were subjected to immunohistochemistry at end stage. End stage was defined as a time point at which the animal was unable to upright itself within 30 s of placement on its side. Immunohistochemistry with GFP in sections of the spinal cord in SOD1G93A mice lumbar revealed delivery of control virus (LV-U6; top, a) or α-adducin RNAi virus (LV-Addi; top, c) into the ventral horn; scale bars represent 100 μm. Alternating GFP-positive sections were subjected to immunohistochemistry using the GFP antibody and the neurofilament-SMi32 antibody (red), a motor neuron marker, or Nissl stained (bottom) for quantification of surviving motor neurons in GFP-labeled injected ventral horn and contralateral non-injected ventral horn (n ≥ 20 sections per animal); scale bars represent 50 μm. (b) Control LV-U6 SOD1G93A mice (n = 5) displayed equivalent degeneration of motor neurons in injected GFP-labeled ventral horn and non-injected contralateral ventral horn. (d) α-adducin knockdown in SOD1G93A mice (LV-Addi SOD1G93A; n = 5) increased motor neuron survival in GFP-labeled injected ventral horn as compared with non-injected contralateral ventral horn. Two-tailed unpaired t test: LV-U6, P = 0.340; LV-Addi, P = 0.0100. Data are presented as mean ± s.e.m. **P < 0.01.
Injection of control lentivirus in SOD1G93A mice (LV-U6 SOD1G93A) had little or no effect on the survival of motor neurons in the GFP-labeled injected ventral horn when compared with the contralateral non-injected ventral horn in vivo (Fig. 2b,c). By contrast, α-adducin kncockdown in SOD1G93A mice (LV-Addi SOD1G93A) strongly suppressed motor neuron degeneration in vivo (Fig. 2d,e). The α-adducin knockdown mice harbored 7.07 ± 0.98 motor neurons in the GFP-labeled ventral horn injected with α-adducin RNAi virus, whereas the contralateral non-injected ventral horn contained only 3.26 ± 0.56 motor neurons (Fig. 2d,e). In control analyses, we confirmed that α-adducin RNAi induced the knockdown of α-adducin in the GFP-labeled injected ventral horn (Supplementary Fig. 4). Knockdown of α-adducin had little or no effect on the presence of gliosis in the ventral horn (Supplementary Fig. 5). Likewise, α-adducin knockdown did not alter the migration or presence of microglia in the ventral horn in vivo (Supplementary Fig. 6). Collectively, our data suggest that α-adducin mediates non–cell autonomous degeneration of motor neurons in the spinal cord of SOD1G93A mice in vivo.
α2-Na/K ATPase triggers motor neuron degeneration
The identification of a critical role for α-adducin in non–cell autonomous motor neuron degeneration led us to determine the mechanism underlying the function of α-adducin in neurodegeneration. We performed immunoprecipitation of α-adducin followed by mass spectrometry in lysates of spinal cord from symptomatic SOD1G93A mice. These analyses revealed that the ion pump α2-Na/K ATPase interacted with α-adducin in symptomatic SOD1G93A spinal cord lysates (data not shown). The Na/K ATPases are composed of two subunits, a catalytic α subunit and a non-catalytic β subunit27. The four catalytic isoforms, α1–α4, display a unique tissue expression pattern27,28. Notably, the α2 catalytic subunit is selectively expressed in astrocytes in the CNS28. These observations led us to characterize the role of α2-Na/K ATPase in non–cell autonomous degeneration of motor neurons in SOD1G93A mice.
We first validated the interaction of α-adducin with α2-Na/K ATPase by immunoprecipitating α-adducin followed by immunoblotting with an α2-Na/K ATPase antibody in spinal cord lysates of SOD1G93A and littermate control mice (Supplementary Fig. 7a). In immunoblotting analyses of primary astrocytes and motor neurons, α2-Na/K ATPase was predominantly expressed in astrocytes rather than motor neurons (Supplementary Fig. 7a). In immunohistochemical analyses, α2-Na/K ATPase was predominately expressed in astrocytes in the spinal cord of both presymptomatic and symptomatic SOD1G93A mice (Supplementary Fig. 7b,c). Notably, immunoblotting analyses revealed that the levels of α2-Na/K ATPase were upregulated in symptomatic SOD1G93A mice at 120 d (Fig. 3a). Consistent with these results, the increase in α2-Na/K ATPase protein levels was also evident in primary SOD1G93A astrocytes (Fig. 3b). Knockdown of α-adducin in SOD1G93A astrocytes reduced the levels of α2-Na/K ATPase in these cells (Fig. 3b). These data suggest that upregulation of α2-Na/K ATPase might act in concert with upregulated α-adducin to promote the toxic gain of function in SOD1G93A astrocytes.
Figure 3.

Enrichment of the α2-Na/K ATPase/α-adducin complex in SOD1G93A astrocytes triggers motor neuron degeneration. (a) Immunoblots from spinal cord lysates showed an increase in the protein levels of α2-Na/K ATPase relative to the internal control proteins β2-ATPase and 14-3-3β in symptomatic SOD1G93A mice as compared with control wild-type littermates (120 d). (b) Immunoblots revealed that α2-Na/K ATPase was upregulated in SOD1G93A astrocytes as compared with non-transgenic controls. Knockdown of α-adducin in SOD1G93A astrocytes attenuated α2-Na/K ATPase protein levels. Protein levels are relative to ERK and 14-3-3β. Blots shown in a and b are cropped. Full-length blots are presented in Supplementary Figure 11. (c) Co-cultured astrocytes and motor neurons were subjected to immunocytochemistry with the motor neuron nuclear protein Islet1 (red) and the dendrite protein Map2 (green); scale bar represents 50 μm. Wild-type astrocytes transfected with the control U6 or α2-Na/K ATPase RNAi plasmid had little or no effect on motor neuron morphology or survival (upper and lower left panels). Control U6 SOD1G93A astrocytes induced non–cell autonomous motor neuron cell death and dendrite abnormalities (upper right panel). α2-Na/K ATPase knockdown in SOD1G93A astrocytes protected motor neurons against the non–cell autonomous cell death and dendrite abnormalities (lower right panel). (d,e) Quantification of motor neuron survival was derived from n ≥ 900 cells per condition and values presented are the average of three independent experiments; two-tailed unpaired t test, P = 0.0080. Quantification of dendrite length was derived from n ≥ 240 images per condition and values presented are the average of three independent experiments; two-tailed unpaired t test, P = 0.0001. Data are presented as mean ± s.e.m. **P < 0.01, ***P < 0.001. (f,g) Alternating GFP-positive sections from SOD1G93A mice injected intraspinally with lentivirus expressing α2-Na/K ATPase RNAi were subjected to immunohistochemistry using the GFP and neurofilament-SMi32 (red) antibodies or Nissl stained (bottom panels) for quantification of surviving motor neurons in GFP-labeled injected ventral horn and contralateral non-injected ventral horn at end stage (n ≥ 20 sections per animal); scale bars represent 100 (top) and 50 (middle, bottom) μm. End stage was defined as a time point at which the animal was unable to upright itself within 30 s of placement on its side. α2-Na/K ATPase knockdown in SOD1G93A mice (LV-ATPi SOD1G93A, n = 5) increased motor neuron survival in GFP-labeled injected ventral horn as compared with non-injected contralateral ventral horn. Arrowheads indicate surviving motor neurons (quantified in g). Two-tailed unpaired t test, P = 0.0005. Data are presented as mean ± s.e.m. ***P < 0.001.
We determined the role of α2-Na/K ATPase in the toxic gain of function of SOD1G93A astrocytes. Knockdown of α2-Na/K ATPase in non-transgenic control glia had little or no effect on motor neuron survival or dendrite morphology (Fig. 3c–e). Notably, knockdown of α2-Na/K ATPase in SOD1G93A astrocytes protected co-cultured primary motor neurons against non–cell autonomous cell death and impairment of dendrite morphology (Fig. 3c–e). These data suggest that knockdown of α2-Na/K ATPase phenocopies the neuroprotective effects of α-adducin knockdown in SOD1G93A astrocytes.
We next assessed the role of α2-Na/K ATPase in SOD1G93A-dependent neurodegeneration in vivo. We used a lentiviral approach to induce knockdown of α2-Na/K ATPase in the lumbar spinal cord in SOD1G93A mice. Just as in the α-adducin experiments in vivo, injection of control lentivirus in SOD1G93A mice (LV-U6 SOD1G93A) had no effect on motor neuron survival (Supplementary Fig. 8a,b). By contrast, knockdown of α2-Na/K ATPase in SOD1G93A mice by lentivirus (LV-ATPi SOD1G93A) suppressed the degeneration of spinal cord motor neurons in vivo. The GFP-labeled injected ventral horn in the α2-Na/K ATPase knockdown mice harbored 6.7 ± 0.28 motor neurons, whereas only 4.33 ± 0.31 motor neurons were present in the non-injected contralateral ventral horn in these mice (Fig. 3f,g). Notably, α2-Na/K ATPase knockdown had little or no effect on the presence or abundance of astrocytes or microglia in the ventral horns of SOD1G93A mice (Supplementary Figs. 5 and 6). These data indicate that α2-Na/K ATPase knockdown in SOD1G93A mice suppresses motor neuron degeneration in vivo.
Heterozygous α2-Na/K ATPase suppresses motor neuron degeneration
We also used a genetic knockout approach to define the role of α2-Na/K ATPase in neurodegeneration in SOD1G93A mice. Although the complete absence of α2-Na/K ATPase leads to embryonic lethality, heterozygous-null mice expressing approximately 50% of normal levels of α2-Na/K ATPase protein display no gross abnormalities29. We measured the ability of astrocytes from heterozygous-null Atp1a2+/−; SOD1G93A mice (Atp1a2+/−; SOD1G93A) to induce cell death of co-cultured motor neurons. Control SOD1G93A astrocytes (Atp1a2+/+; SOD1G93A) induced non–cell autonomous cell death in 53% of co-cultured motor neurons (Fig. 4a,b). In contrast, heterozygous-null Atp1a2+/−; SOD1G93A astrocytes (Atp1a2+/−; SOD1G93A) induced non–cell autonomous cell death in only 14% of motor neurons (Fig. 4b). Likewise, Atp1a2+/−; SOD1G93A astrocytes failed to induce dendrite abnormalities in motor neurons as compared to control SOD1G93A astrocytes (Fig. 4a,c). These data corroborate the results of our knockdown analyses and buttress the conclusion that α2-Na/K ATPase is critical for non–cell autonomous degeneration of motor neurons.
Figure 4.

Heterozygous disruption of Atp1a2 in SOD1G93A mice suppresses motor neuron degeneration and enhances mouse lifespan. (a) Downregulation of α2-Na/K ATPase in SOD1G93A astrocytes by crossing SOD1G93A mice with Atp1a2+/− (right) protected motor neurons from non–cell autonomous cell death and dendrite abnormalities induced by control SOD1G93A astrocytes (left). Scale bar represents 50 μm. (b,c) Quantification of motor neuron survival is derived from n ≥ 900 cells per condition and values presented are the average of three independent experiments; unpaired t test, P = 0.0003. Quantification of dendrite length is derived from n ≥ 240 images per condition and values presented are the average of three independent experiments performed in duplicates; two-tailed unpaired t test, P = 0.0001. Data are presented as mean ± s.e.m. ***P < 0.001. (d) Disease onset, that is, initial day of weight loss, was significantly delayed in α Atp1a2+/−; SOD1G93A mice (Atp1a2+/−, n = 14) as compared with control SOD1G93A littermates (Atp1a2+/+, n = 14); two-tailed unpaired t test, P = 0.0009. (e) Early disease process, that is, age at which 10% of weight loss is reached, was significantly delayed in Atp1a2+/−; SOD1G93A mice (Atp1a2+/−; SOD1G93A, red circles, n = 14) as compared with control SOD1G93A littermates (Atp1a2+/+; SOD1G93A, black squares, n = 14) (P = 0.0001). (f) Early phase disease progression, that is, days from onset to 10% weight loss, displayed no change between α Atp1a2+/−; SOD1G93A mice and control SOD1G93A littermates; two-tailed unpaired t test, P = 0.2023. (g) Late-phase disease progression, that is, from 10% weight loss to end stage, displayed significant delay in Atp1a2+/−; SOD1G93A mice (Atp1a2+/−, n = 14) as compared with control SOD1G93A littermates (Atp1a2+/+, n = 14); two-tailed unpaired t test, P = 0.0005. (h) Kaplan-Meier survival plots showed substantial and significant increase in lifespan for Atp1a2+/−; SOD1G93A mice (Atp1a2+/−; SOD1G93A, red circles, n = 14) as compared with control SOD1G93A littermates (Atp1a2+/+; SOD1G93A, black squares, n = 14) (P = 0.0001). (i) Control SOD1G93A mice displayed morbidity of reduced mobility and the inability to upright at endstage (Atp1a2+/+; SOD1G93A), whereas age-matched SOD1G93A littermates that harbor a heterozygous null Atp1a2 allele displayed increased mobility and health (Supplementary Movies 1 and 2). (j) Immunohistochemistry of NMJs from gastrocnemius muscle using the presynaptic marker synapsin (green) and postsynaptic marker α-bungarotoxin (red, Bu Tx) at 120 d showed that control SOD1G93A mice had denervated NMJs (left), whereas Atp1a2+/− displayed an increased NMJ integrity (right); scale bar represents 20 μm. (k) Quantification of NMJs from n = ~300 NMJs from three animals per group (two-tailed unpaired t test: complete denervated, P = 0.0196; partial denervated, P = 0.0203; completely innervated, P = 0.0370). Data are presented as mean ± s.e.m. *P < 0.05.
The genetic knockout approach facilitated analysis of the role of α2-Na/K ATPase in the motor neuron disease phenotype of mutant SOD1 mice. We characterized disease onset, progression and lethality in heterozygous-null Atp1a2; SOD1G93A mice and control SOD1G93A littermates. Disease onset, defined as the first day of weight loss, was significantly delayed in Atp1a2+/−; SOD1G93A mice (Atp1a2+/−) as compared with control Atp1a2+/+; SOD1G93A mice (Atp1a2+/+) (P = 0.0009; Fig. 4d). Accordingly, disruption of Atp1a2 in SOD1G93A mice delayed the age at which 10% weight loss was reached, which is a measurement of early disease (Fig. 4e). Early phase disease progression, as measured from the first day of weight loss to 10% weight loss, was not significantly altered between Atp1a2+/−; SOD1G93A and control SOD1G93A littermates (P = 0.2023; Fig. 4f). In contrast, late-phase disease progression, as measured from 10% weight loss to end stage, was significantly delayed in Atp1a2+/−; SOD1G93A mice compared with control SOD1G93A littermates (P = 0.0005; Fig. 4g). Because expression of SOD1G37R in astrocytes drives late-phase motor neuron degeneration progression in mice9, the suppression of late-phase disease progression in Atp1a2+/−; SOD1G93A mice supports the conclusion that α2-Na/K ATPase mediates astrocyte-dependent mechanism of neurodegeneration. Notably, the overall survival of SOD1G93A mice was increased after reducing the expression of α2-Na/ATPase to an average lifespan of 171.0 ± 2.5 d compared with 151.5 ± 2.7 d in control SOD1G93A mice (Fig. 4h). In addition, the Atp1a2+/−; SOD1G93A mice were substantially more mobile at the time at which the control SOD1G93A mice were at the endstage of the disease (Fig. 4i and Supplementary Movies 1 and 2). Thus, reducing the expression of α2-Na/K ATPase in SOD1G93A mice delays the onset and slows the progressive process of neurodegeneration, thereby substantially increasing lifespan.
We next determined if the improvement in the lifespan of SOD1G93A mice that are heterozygous for Atp1a2 is associated with suppression of motor neuron degeneration in vivo. We quantified motor neurons at the endstage of disease in control SOD1G93A mice and aged-matched SOD1G93A littermates heterozygous-null for Atp1a2. Control SOD1G93A mice harbored 3.39 ± 0.43 motor neurons per ventral horn (Supplementary Fig. 9a,b). By contrast, littermate Atp1a2+/−; SOD1G93A mice had more than twice the number of motor neurons at 7.27 ± 0.63 per ventral horn (Supplementary Fig. 9a,b). These results demonstrate that reducing the expression of α2-Na/K ATPase in SOD1G93A mice delays motor neuron degeneration.
We also determined the effect of disruption of Atp1a2 in SOD1G93A mice on motor neuron innervation of muscles in vivo. We quantified neuromuscular junction (NMJ) innervation of the gastrocnemius muscle in 120-d SOD1G93A mice heterozygous for the null Atp1a2 allele and age-matched control SOD1G93A mice using published parameters30. We categorized NMJs as completely innervated (as reflected by complete overlap of synapsin with α-bungarotoxin staining), completely denervated (displaying no overlap staining) or partially denervated (displaying partial overlap). In control SOD1G93A mice, 75% of NMJs were completely denervated, 23% were partially denervated and less than 1.5% were completely innervated (Fig. 4j,k). By contrast, in Atp1a2+/−; SOD1G93A mice, 47% of NMJs were completely denervated, 45% were partially denervated and 7.6% were completely innervated NMJs (Fig. 4j,k). These analyses demonstrate that knockout of one allele of Atp1a2 substantially improves the integrity of motor neuron innervation of peripheral muscles.
Na/K ATPase stimulates mitochondria and inflammatory genes
The elevated levels of the α2-Na/K ATPase/α-adducin complex in SOD1G93A astrocytes raised the question of whether the activity of α2-Na/K ATPase per se has a pathogenic role in the toxic gain of function of SOD1G93A astrocytes. We used the Na/K ATPase small molecule inhibitors ouabain and digoxin, the latter of which is used widely as a therapeutic drug in treatment of heart failure21, to assess the role of Na/K ATPase activity in the toxic effects of SOD1G93A astrocytes. Following the co-culturing of motor neurons with control and mutant SOD1G93A astrocytes, we added ouabain, digoxin or control vehicle at a final concentration of 1 μm, which is sufficient to inhibit the α2-Na/K ATPase in primary glia cells31. We found that inhibition of Na/K ATPase with ouabain or digoxin substantially reduced motor neuron cell death induced by SOD1G93A astrocytes to 22% and 19%, respectively, as compared with 56% motor neuron cell death in cultures treated with vehicle (Fig. 5a,b). Likewise, both ouabain and digoxin prevented impairment of dendrite morphology in motor neurons induced by SOD1G93A astrocytes (Fig. 5a,c). In control analyses, co-cultures of motor neurons and control astrocytes exposed to ouabain and digoxin did not alter survival or morphology of motor neurons (Fig. 5a–c). These results support the conclusion that the catalytic activity of α2-Na/K ATPase/α-adducin complex in SOD1G93A astrocytes triggers non–cell autonomous degeneration of motor neurons.
Figure 5.

Na/K ATPase stimulates mitochondrial respiration and expression of inflammatory genes in SOD1G93A astrocytes. (a) Control SOD1G93A astrocytes (left) induced non–cell autonomous motor neuron cell death and dendrite abnormalities. Pharmacological inhibition of Na/K ATPase with ouabain (middle) and digoxin (right) was neuroprotective from the non–cell autonomous cell death and dendrite abnormalities induced by SOD1G93A astrocytes (left); scale bar represents 50 μm. Pharmacological inhibition of Na/K ATPase in control co-cultures non-transgenic astrocytes (Non-tg) and motor neurons with ouabain and digoxin did not alter motor neuron survival or morphology. (b,c) Quantification of motor neuron survival was derived from n ≥ 900 cells per condition and values presented are the average of three independent experiments (two-tailed unpaired t test: ouabain, P = 0.0024; digoxin, P = 0.0001). Quantification of dendrite lengths was derived from n ≥ 240 images per condition and values presented are the average of three independent experiments (two-tailed unpaired t test: ouabain, P = 0.0002; digoxin, P = 0.0001). Data are presented as mean ± s.e.m. **P < 0.01, ***P < 0.001. (d) A representative plot of oxygen consumption measured in astrocytes from control non-transgenic, SOD1G93A and heterozygous-null SOD1G93A mice using Seahorse Bioscience XF Analyzer. Arrow indicates time when the mitochondrial uncoupler FCCP was added. (e) Basal and maximum oxygen consumption were quantified in control non-transgenic (n = 3), SOD1G93A (n = 3) and heterozygous-null SOD1G93A mice (n = 3) astrocytes using Seahorse Bioscience XF Analyzer (two-tailed unpaired t test: basal consumption, P = 0.0001; maximum consumption, P = 0.0001). Data are presented as mean ± s.e.m. ***P < 0.001. (f) Total RNA of astrocytes from control non-transgenic (n = 3), SOD1G93A (n = 3) and heterozygous-null SOD1G93A mice (n = 3) were subjected to qRT-PCR analyses using primers to a panel of pro-inflammatory genes. Gene expression was normalized to GAPDH expression. The expression of 18 inflammatory genes was upregulated in SOD1G93A astrocytes. Downregulation of α2-Na/K ATPase in SOD1G93A astrocytes significantly decreased expression of half of the upregulated inflammatory genes (two-tailed unpaired t test: SPP1, P = 0.0253; LCN2, P = 0.0013; CLM1, P = 0.0052; WNT1, P = 0.0198; CCL11, P = 0.0387; CXCL1, P = 0.0246; CCR4, P = 0.0314; Il1b2, P = 0.0421; Itgb2, P = 0.0215; II1r1, P = 0.0092). Data are presented as mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001.
Because Na/K ATPase is the major consumer of ATP in the CNS, we would anticipate that an increase in the levels and activity of Na/K ATPase would alter demand for cellular metabolism and ATP. Mitochondria generate the majority of the cellular demand for ATP. We reasoned that elevated levels and activity of α2-Na/K ATPase in SOD1G93A astrocytes might reduce basal ATP levels and influence mitochondrial respiration. Measurement of oxygen consumption revealed that mutant SOD1G93A astrocytes had significantly higher levels of basal oxygen consumption (P = 0.0001) and significantly increased maximum oxidative capacity (P = 0.0001), the latter of which was induced by the electron transport uncoupler p-triflouromethoxyphenylhydrazon (FCCP) (Fig. 5d,e). By contrast, Atp1a2+/−; SOD1G93A astrocytes had reduced basal and maximum oxidative capacity (Fig. 5d,e). These data suggest that α2-Na/K ATPase in SOD1G93A astrocytes enhance mitochondrial respiration, presumably secondary to increased ATP demand. Following the downregulation of α2-Na/K ATPase in Atp1a2+/−; SOD1G93A, demand for ATP production is attenuated, thereby reducing basal and maximum oxidative capacity.
Cellular stresses, including increased metabolic rates, induce the production of mitochondrial reactive oxygen species (ROS) that in turn activate an inflammatory response associated with the secretion of cytokines and other inflammatory factors that promote chronic inflammatory diseases32–34. Notably, both ROS and inflammation have been linked to the pathogenesis of ALS20,35. Consistent with this possibility, whereas conditioned medium from SOD1G93A astrocytes induces degeneration of motor neurons, conditioned medium from Atp1a2+/−; SOD1G93A astrocytes failed to induce motor neuron degeneration (Supplementary Fig. 10a,b). These results suggest that the upregulation of α2-Na/K ATPase in SOD1G93A astrocytes mediates the non–cell autonomous degeneration of motor neurons via secreted factors.
We next assessed whether the upregluation of α2-Na/K ATPase in SOD1G93A astrocytes might stimulate the expression of specific inflammatory genes20,36. In quantitative RT-PCR (qRT-PCR) analyses, we characterized a panel of 20 inflammatory genes reported to be increased in SOD1G93A astrocytes20,36. We found that expression of a set of cytokines, chemokines and related inflammatory astrocytic genes was increased in SOD1G93A astrocytes compared with control wild-type astrocytes (Fig. 5f). Among this panel, more than half were downregulated in Atp1a2+/−; SOD1G93A astrocytes (Fig. 5f). These data support the conclusion that the upregulation of α2-Na/K ATPase in SOD1G93A astrocytes promotes an inflammatory response, which may in turn induce motor neuron degeneration.
α2-Na/K ATPase and α-adducin are upregulated in ALS spinal cord
To determine the clinical relevance of the α2-Na/K ATPase/α-adducin mechanism in non–cell autonomous neurodegeneration, we characterized the expression of the α2-Na/K ATPase/α-adducin complex in spinal cord lysates from both familial ALS expressing distinct mutations in SOD1 and sporadic ALS patients and controls. Familial ALS cases demonstrated autosomal dominant inheritance. All of these patients had definite ALS by the El Escorial criteria37, with a mean age of 42 years (range of 21–65). The mean age of sporadic ALS patients was 60 years (range 46–69). All patients with sporadic ALS met modified El Escorial criteria for probable or definite ALS, with their spinal cords displaying gliosis, demyelination and long-term motor neuron degeneration37. The mean age of control patients was 64 years (range 54–70). The CNS had normal appearance in samples of control patients. Immunoblotting of lysates of familial ALS, sporadic ALS and control patients revealed that the levels of α2-Na/K ATPase were significantly increased in lysates of spinal cord in both familial (P = 0.0302) and sporadic (P = 0.0467) ALS patients as compared with controls (Fig. 6). Likewise, the protein levels of α-adducin were also significantly increased in the spinal cord of ALS patients (familial, P = 0.0336; sporadic, P = 0.0181; Fig. 6a,d). Quantification of α2-Na/K ATPase and α-adducin immunoreactivity as a continuous variable revealed that the levels of these two proteins doubled in both familial and sporadic ALS (Fig. 6b,c,e,f). Together, these data suggest that the abundance of α2-Na/K ATPase and α-adducin in familial and sporadic ALS mimics the elevated levels of the α2-Na/K ATPase/α-adducin complex in SOD1G93A mice and may therefore contribute to neurodegeneration.
Figure 6.
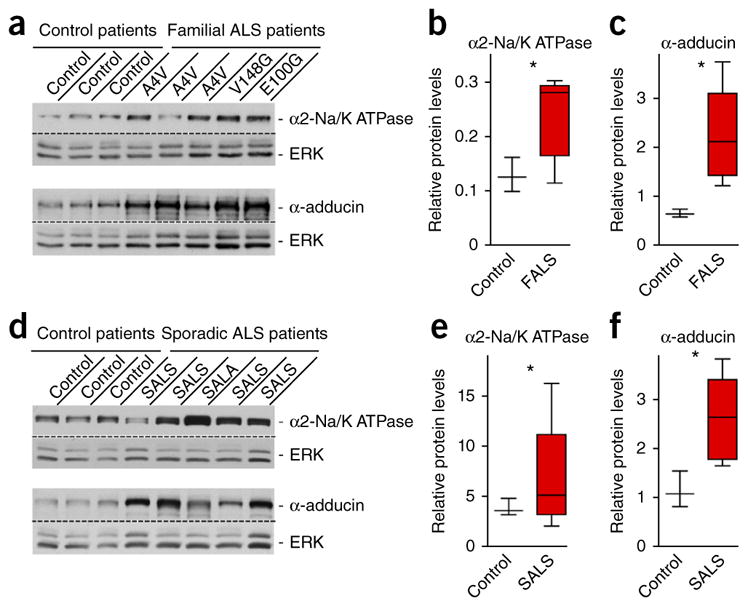
The α2-Na/K ATPase/α-adducin complex is upregulated in spinal cord in familial and sporadic ALS patients. (a) Immunoblots from patients with familial ALS (n = 5) revealed elevated protein levels of α2-Na/K ATPase (top) and α-adducin (bottom) in spinal cord lysates as compared to control patients (n = 3). ERK served as a loading control. (b,c) Quantification of the relative densitometry protein levels for α2-Na/K ATPase and α-adducin relative to the internal control ERK; two-tailed unpaired t test: α2-Na/K ATPase, P = 0.0302; α-adducin, P = 0.0336. Blots shown in a and b are cropped. Full-length blots are presented in Supplementary Figure 11. (d) Immunoblots from patients with sporadic ALS (n = 5) showed elevated protein levels of α2-Na/K ATPase (top) and α-adducin (bottom) in spinal cord lysates as compared with control patients (n = 3). (e,f) Quantification of the relative densitometry protein levels for α2-Na/K ATPase and α-adducin relative to the internal control ERK; two-tailed unpaired t test: α2-Na/K ATPase, P = 0.0467; α-adducin, P = 0.0181. Data are presented as mean ± s.e.m. *P < 0.05.
DISCUSSION
Our results reveal a glial cell–intrinsic mechanism of non–cell autonomous neurodegeneration. We found that α2-Na/K ATPase and α-adducin form a complex that is highly abundant in mutant SOD1 astrocytes. Knockdown of α2-Na/K ATPase or α-adducin by RNAi or by introducing a null allele of Atp1a2 in mutant SOD1 astrocytes protects motor neurons from non–cell autonomous degeneration. Notably, introducing a null allele of Atp1a2 in mutant SOD1 mice suppressed motor neuron degeneration and muscle denervation in vivo and thereby substantially increased mouse lifespan. Mechanistic studies revealed that mitochondrial respiration and inflammatory genes were induced in mutant SOD1 astrocytes and that these alterations were suppressed in mutant SOD1 Atp1a2+/− astrocytes. We have also found that the therapeutic drug digoxin, which inhibits Na/K ATPases and is commonly used to treat heart failure21, protected motor neurons from non–cell autonomous degeneration. Finally, we found that α2-Na/K ATPase and α-adducin were upregulated in both familial ALS patients expressing distinct SOD1 mutations and sporadic ALS patients, suggesting a pathogenic role for this protein complex in human disease. Taken together, our results define the α2-Na/K ATPase/α-adducin complex as a key glial cell–intrinsic regulator of non–cell autonomous neurodegeneration.
Although neurodegenerative diseases were once thought to be selectively neuronal, growing evidence suggests that expression of neurodegenerative disease genes in astrocytes triggers neuronal degeneration non–cell autonomously3,4,7,10. However, the glial cell-intrinsic mechanism that mediate neurodegeneration have remained poorly understood. Our study reveals that upregulation of α2-Na/K ATPase/α-adducin complex stimulates mitochondrial respiration and an inflammatory response in mutant SOD1 astrocytes, leading to the non–cell autonomous degeneration of motor neurons. The finding that the α2-Na/K ATPase/α-adducin complex is upregulated in familial ALS patients expressing distinct mutations of SOD1 and in sporadic ALS supports the conclusion that the α2-Na/K ATPase/α-adducin complex may represent a common mechanism of non–cell autonomous degeneration in motor neuron disease. Identification of a critical pathogenic role for the glial α2-Na/K ATPase/α-adducin complex in non–cell autonomous neurodegeneration in ALS suggests that it will be important to determine whether chronic activation of this complex in astrocytes might contribute to the pathogenesis of other neurodegenerative diseases.
Our findings shed light on the role of Na/K ATPase activity in motor neuron disease. The levels of Na/K ATPase isoforms have been reported to be downregulated in mutant SOD1 spinal cord38. However, consistent with our findings, the activity of Na/K ATPase has been shown to be elevated in spinal cord lysates from symptomatic mutant SOD1 mice39. We found that α2-Na/K ATPase was upregulated in the spinal cord of symptomatic mutant SOD1 mice and that the upregulation of α2-Na/K ATPase occurred specifically in glia in these mice. Notably, consistent with our findings in mutant SOD1 mice, we also found that the levels of α2-Na/K ATPase were elevated in spinal cord of familial ALS patients harboring distinct SOD1 mutations as well as sporadic ALS. Our distinct genetic approaches of knockdown and knockout analyses both in primary cultures and in mice in vivo suggest a requirement for α2-Na/K ATPase in motor neuron degeneration.
Recent studies have suggested that elevated levels of the α1 isoform of Na/K ATPase may induce hippocampal morphological abnormalities and memory deficits in Angelman syndrome, a neurodevelopmental disorder that features developmental delay, speech impairment and seizures40,41. Attenuation of the α1 Na/K ATPase levels suppresses the hippocampal morphological abnormalities and memory deficits in a mouse model of Angelman syndrome. Whether the α1 isoform of Na/K ATPase acts non–cell autonomously in glial cells or cell autonomously in neurons and whether α1-Na/K ATPase operates in a complex with α-adducin in Angelman syndrome remains unknown. In view of our findings, it will be interesting to determine whether the glial α2-Na/K ATPase/α-adducin complex contributes to the pathogenesis of other neurological diseases beyond neurodegeneration, including developmental disorders of cognition.
Our study also illuminates a mechanism by which the α2-Na/K ATPase/α-adducin complex elicits neurodegeneration non–cell autonomously. The upregulation of α2-Na/K ATPase is anticipated to reduce ATP levels and increase demand for mitochondria function. Accordingly, we found that the upregulation of α2-Na/K ATPase stimulates mitochondrial respiration in mutant SOD1 astrocytes. Mitochondrial respiration may increase ROS in mutant SOD1 astrocytes, which in turn may activate the observed induction of inflammatory factors leading to non–cell autonomous degeneration of motor neurons.
The role of α-adducin and Na/K ATPase activity in the renal epithelium may be instructive for the study of neurodegeneration. Consistent with our findings in mutant SOD1 astrocytes, α-adducin promotes the stability and activity of Na/K ATPase in renal epithelial cells42,43. Notably, polymorphisms in α-adducin may enhance Na/K-ATPase activity in the renal epithelium, leading to abnormal renal ionic handling and hypertension in humans42–44. Furthermore, in models of renal interstitial fibrosis, chronic activation of Na/K ATPase in proximal tubule cells and macrophages leads to the release of pro-inflammatory cytokines that facilitate the development of chronic inflammation, oxidative stress and fibrosis45,46. Thus, our findings that α2-Na/K ATPase/α-adducin complex in astrocytes triggered non–cell autonomous degeneration suggest the exciting idea of parallels in the pathogenesis of hypertension and neurodegenerative diseases.
The unexpected finding that chronic activation of α2-Na/K ATPase in astrocytes is critical for neurodegeneration suggests that α2-Na/K ATPase might represent an attractive target for the identification of new therapies for neurodegenerative diseases. We found that the therapeutic drug digoxin, which has been widely used to treat heart failure21, protects motor neurons against degeneration. Consistent with our findings, cardiac glycosides appear to be neuroprotective in models of ischemic stroke, prevents polyglutamine-induced cell death, and inhibits SOD1 and TDP-43 aggregation in cells47–50. Whether chronic activation of the α2-Na/K ATPase/adducin complex in astrocytes has an active role in other neurodegenerative diseases and in brain injury remains to be determined. The enormous experience with Na/K ATPase inhibitors in the treatment of heart disease should prove useful in the development of inhibitors selective for the α2 isoform of Na/K ATPase in glial-dependent neurodegeneration.
ONLINE METHODS
Human tissue
All human spinal cord tissue samples were acquired by way of an Investigational Review Board and Health Insurance Portability and Accountability Act compliant process. The SOD1 A4V, V148G and E100G nervous system were confirmed by DNA testing. All patients who had been followed during the clinical course of their illness had met El Escorial criteria for definite ALS. After death, autopsies were performed immediately by an on-call tissue acquisition team. Control spinal cord tissue samples were from patients from the hospital’s critical care unit when life support was withdrawn or patients on hospice. Tissue samples were completed within 4–6 h of death and the entire motor system was dissected and elaborately archived for downstream applications by creating two parallel tissue sets from alternating adjacent regions. For biochemical studies, segments were embedded in cutting media, frozen on blocks of dry ice and stored at −80 °C.
Animals
We used transgenic mice overexpressing mutant G93A human superoxide dismutase 1 (Jackson Laboratory stock# 002726), α2-Na/K ATPase null heterozygous (provided by J.B.L.) and C57BL6 (Charles River). Mouse husbandry with timed mating and analysis of age-dependent phenotypes was carried out as described51. Two or three males or two to four females per cage were housed under a 12-h/12-h light-dark cycle. Age-matched littermates of both males and females were used in the experiments. No animals were excluded from analysis. All analysis was done with littermate mice derived from SOD1G93A heterozygous mating with non-transgenic wild-type and heterozygous α2-Na/K ATPase mice. All animal experiments were conducted under the institutional guidelines and were approved by the Institutional Animal Care and Use Committees of Harvard Medical School and Washington University School of Medicine.
Plasmids
RNAi plasmids were designed as described previously52. The U6/α-adducin plasmids were cloned using the following primers: 5′-agt cga aga cta agt gga ctt-3′ and 5′-agt cga aga cta agt gga cta-3′. The U6/α2-Na/K ATPase plasmids were cloned using the following primers: 5′-gca tca tat cag agg gta acc-3′ and 5′-gtg gca aga aga aac aga aac-3′. α-adducin and α2-Na/K ATPase were cloned from C57BL/6J mice cDNA and inserted into the pcDNA3 vector (Invitrogen) at the ECOR1 and Xho1 sites. The primer for site direct mutagenesis for RNAi-resistant form of α-adducin was 5′-aac gga agc agt ccc aaA tcA aaA acA aaA tgg acA aaa gag gat gga cat ag-3.
Antibodies
Antibodies to α-adducin (1:1,000, H-100, Santa Cruz), phospho-Ser436-adducin (1:1,000, Ser436, Santa Cruz), α2-Na/K ATPase (1:1,000, A-16, Santa Cruz), α2-Na/K ATPase (1:5,000, a5, Developmental Studies Hybridoma Bank), α2-Na/K ATPase (1:1,000, ATP1A2, Thermo Fisher Scientific), Islet-1 (1:1,000, 39.4D5, Developmental Studies Hybridoma Bank), MAP2 (1:1,000, ab5622, Fisher), GFAP (1:1,000, AB5804, Millipore), Iba-1 (019-19741, WACO), 14-3-3β (1:2,000, H-8, Santa Cruz), ERK (1:2,000, Molecular Probes), superoxide dismutase (1:1,000, SD-G6, SIGMA), ERK (4685, Cell Signaling), GFP (1:1,000, Molecular Probes) and synapsin (1:500, AB1543, Millipore) were purchased.
Protein biochemistry
To determine the relative amounts of total proteins, whole spinal cords from aged match littermate mice were homogenized in 50 mM Tris-buffer (pH 7.2), 100 mM NaCl, 1 mM EDTA, 1 mM PMSF and a cocktail of protease and phosphatase inhibitors. Equal concentrations from each sample were solubilized in SDS-loading buffer and boiled for 10 min. For analyses of α2-Na/K ATPase protein levels, equal concentrations (1 mg ml−1) from each sample were solubililzed in SDS-loading buffer. Using an ultrasonic water sonicator protein samples in SDS-loading buffer were sonicated in an ice bath to prevent oligomerization of the α2-Na/K ATPase (6 pulses for 30 s with 30-s intervals). Protein quantification was carried out by immunoblotting using Amersham ECL plus and analyzed with Bio-Rad ChemiDoc gel imaging system. Signals were normalized for internal controls run on the same blots. For immunoprecipitation of α-adducin, proteins were extracted under non-denaturing (1% Triton X-100 for 1 h at 4 °C, vol/vol) and denaturing conditions (1% SDS and 0.5% DOC for 1 h at 4 °C, vol/vol) and the insoluble material was removed by centrifugation (18,000 g for 10 min). Following the denaturing conditions the supernatant was diluted to a final concentration of 0.33% SDS and 0.16% Triton. The supernatants were precleared with protein A for 30 min. at 4 °C before the addition of α-adducin antibody for 1 h at 4 °C. For co-immunoprecipitation of α-adducin and α2-Na/K ATPase, immunoprecipitated α-adducin antibody was eluted from protein A with 0.2 M glycine pH 2.5 on ice for 15 min followed by quenching with 1 mM Tris pH 8.8.
Primary astrocyte cultures
We prepared monolayer astrocytes culture from P2 SOD1G93A and non-transgenic littermate mice that were previously described14,15. Astrocytes were plated in DMEM (GIBCO) supplemented with 10% FBS (vol/vol) containing 100 U ml−1 penicillin and 100 mg ml−1 streptomycin for 2 weeks or until confluent. Once confluent, cells were replated onto glass cover slips pre-coated with poly-d-lysine at a density of 40,000 cells per well in a 24-well dish. For RNAi experiment, astrocyte cultures were transfected with RNAi or control U6 plasmid using Lipofectamine 2000 (Invitrogen) according to the guidelines of the manufacturer 4 d before co-culturing with motor neurons.
Real-time PCR reactions were performed using Light cycler 480 SYBR Green 1 Master (Roche). The following primers were used: ATPase forward, tcttagcctatggtatcc; ATPase reverse, gcatcttttctccctctc; GFAP forward, ggaccagcttacggccaacagtgc; GFAP reverse, ccagcgattcaacctttctctcc; SPP1 forward, ccctcccggtgaaagtgactgattc; SPP1 reverse, ggtctccatcgtcatcatcatcgtc; LCN2 forward, ggcccaggactcaactcagaacttg; LCN2 reverse, cctgaccaggatggaggtgacattg; CLM1 forward, attgctggtcccctttctct; CLM1 reverse, ggaactccttggcaccagta; WNT11 forward, caggatcccaagccaataaa; WNT 11 reverse, gacaggtagcgggtcttgag; CCL8 forward, gcttctttgcctgctgctcatag; CCL8 reverse, ccatgtactcactgacccacttc; CCL11 forward, gagctccacagcgcttctattcc; CCL11 reverse, ccacttcttcttggggtcagcac; CXCL1 forward, gcttctttgcctgctgctcatag; CXCL1 reverse, gatctccatgtactcactgaccc; CXCL12 forward, gctctgcatcagtgacggtaaacc; CXCL12 reverse, cgggtcaatgcacacttgtctgttg; AIMP1 forward, gctgttctgaagaggctggagca; AIMP1 reverse, agttcgtctgcagtggagtactc; CCR4 forward, tgaccctcggaggatgtacacaa; CCR4 reverse, accatgtttccgagttctgccgg; CCR10 forward, atctggcagccagacgaactacc; CCR10 reverse, agagctcgtgcgatggccacata; CEBP forward, aaggccaaggccaagaagacggt; CEBP reverse, tcgggcagctgcttgaacaagtt; Il1b2 forward, ggctgcttccaaacctttgacct; Il1b2 reverse, catcatcccatgagtcacagagg; Itgb2 forward, attcaatgtgactttccggcggg; Itgb2 reverse, caggccttctccttgttgggaca; Il10ra forward, gaagtggccctcaaacagtacgg; Il10ra, gatgccgtccattgctttcagag; Il1R1 forward, ggggctcatttgtctcatggtgcc; Il1R1 reverse, gctgatgaatcctggagtcccggtc; Il1R2 forward, cgcatcccactgtgagcaaatgt; Il1R2 reverse, tgtggggtctcctgcactcagaa; TNFr1 forward, acttggtgagtgactgtccgagc; TNFr1 reverse, ggaagtgtgtctcactcaggta; IFNγ forward, ggctttgcagctcttcctcatgg; IFNγ reverse, cgcttatgttgttgctgatggcc.
Primary motor neuron cultures and morphological analyses
We performed spinal motor neuronal cultures from E12.5 wild-type rodents that were described previously53. We plated at 12,000 cells per well onto astrocyte monolayer in Neurobasal-A medium (Gibco) supplemented with 2% B-27 serum free supplement (vol/vol) and 0.5 mM glutamine; DMEM/F12 (3:1, vol/vol ratio) with a cocktail of trophic factors composed of 0.5 ng ml−1 glia-derived neurotrophic factor, 1 ng ml−1 brain-derived neurotrophic factor and 10 ng ml−1 ciliary neurotrophic factor (trophic factor cocktail, R&D Systems).
Motor neurons were fixed after 7 d of co-cultures with astrocytes and immunostained with motor neurons marker Islet1 and dendrite marker MAP2. Cell death of motor neurons was assessed on the basis of the integrity of neuritis and the morphology of the nucleus (condensation or fragmentation) as determined using DNA dye Hoechst 33258 (Sigma) previously described22. Approximately 150 cells were counted per cover slip per condition. For motor neuron morphology, images were taken using a NIKON eclipse TE2000 epifluorescence microscope using a digital CCD camera (DIAGNOSTIC instruments) and imported into the SPOT imaging software. Dendrites length was measured by tracing the dendrites emanating from the soma in a manner that produced the longest continuous path. Total dendrite length was quantified as a sum of all dendrites emanating from the soma. Approximately 40 cells were analyzed per cover slip.
Condition medium studies were performed as previously described14. For pharmacological inhibition of Na/K ATPase in co-cultures, 1.0 μM of ouabian or digoxin was added 4 h following the co-culturing of motor neurons with control or G93A astrocytes (d in vitro (DIV) 0). This was followed by administration of 0.5 μM of ouabain or digoxin every 48 h (DIV2, DIV4 and DIV6).
Seahorse XF-24 metabolic flux analysis
Oxygen consumption rates and extracellular acidification rate were measured at 37 °C using an XF24 extracellular analyzer (Seahorse Bioscience). Astrocytes were plated in 24-well plates for 24 h (50,000 cells per well) in DMEM 10% FBS containing 100 U ml−1 penicillin and 100 mg ml−1 streptomycin. Cells were changed to unbuffered DMEM (DMEM supplemented either 25 mM glucose, 1 mM sodium pyruvate, 31 mM NaCl, 2 mM GlutaMax, pH 7.4) and incubated at 37 °C in a non-CO2 incubator for 60 min. Four baseline measurements were taken before sequential injection of mitochondrial inhibitors. Four measurements were taken following the addition of 1.0 μM oligomycin, 1.0 μM FCCP, 1.0 μM rotenone/antimycin A. Oxygen consumption rate and extracellular acidification rate were automatically calculated by Seahorse XF-24 software. Every point represents an average of n = 3.
Morphological studies
All morphological studies were performed as previously described51. Briefly, mice were deeply anesthetized and perfused with 4% paraformaldehyde (vol/vol) in phosphate buffer. Spinal cords were post-fixed in the same fixative for 4 h and processed for cryoprotective embedding. All immunohistochemistry analyses were carried out on 30-μm cryosections from spinal cords with the indicated antibodies. For Na/K ATPase, immunohistochemical analyses antigen retrieval was performed using 1 mg ml−1 pepsin in 0.2 M HCl for 10 min followed by blocking in 3% BSA (vol/vol) for 30 min. For measurement of motor neurons survival, alternating GFP-positive sections were either immunostained or Nissl stained and images were taken using a NIKON eclipse TE2000 epifluorescence microscope or brightfield using a digital CCD camera (DIAGNOSTIC Instruments) and imported into the SPOT imaging software. For lentiviral-mediated RNAi, a minimum of 20 sections were quantified for surviving motor neurons in the injected and non-injected ventral horns from Nissl-stained and immunofluorescence-labeled sections.
Lentiviral injections
Lentivirus was produced in 293T cells and concentrated by ultracentrifuge. All surgical procedures were performed as described54. Briefly, 90-d-old mice were anesthetized with ketamine/xylazine intraperitoneally (90–200 mg per kg of body weight ketamine/10 mg per kg xylazine). A 2-cm longitudinal skin incision was made above the lumbar region under a dissecting microscope. Using a dental drill, a small (1 mm) hole was made into the spinal cord. The lentiviral solution was injected into the L3–L4 region using a stereotaxic frame (Stoelting) at 2 mm unilaterally. The viral solution (50 nl) was injected 25 times per animal with 45-s intervals using a fine micropipette (Drummond 30-μl microcapillaries pulled with P-9 capillary puller, Sutter Instruments) and a Nanoject II (Drummond). The micropipette was then left for an additional 5 min and gently withdrawn.
Statistics
Statistical analyses were done using GraphPad Prism 4 software. Bar graphs are presented as the mean ± s.e.m. For experiments in which only two groups were analyzed, the two-tailed unpaired t test was used. No statistical methods were used to pre-determine sample sizes, but our sample sizes are similar to those generally employed in the field. Data distribution was assumed to be normal, but this was not formally tested. Blinding and randomization was performed on weight and survival data analyses.
A Supplementary Methods Checklist is available.
Supplementary Material
Acknowledgments
We thank members of the Bonni laboratory for helpful discussions. We thank L. Zinman (University of Toronto) for providing human patient tissue samples. This work was supported by a grant from the Edward R. and Anne G. Lefler Foundation (A.B.) and The Ruth L. Kirschstein National Research Service Awards T32 5T32AG00222 (G.G.). Human spinal cord material provided from Northwestern University autopsy program is partially funded from the Les Turner ALS Foundation. Additional human tissue samples were obtained from the Human Brain and Spinal Fluid Resource Center, which is sponsored by the National Institute of Neurological Disorders and Stroke and the US National Institutes of Health, National Multiple Sclerosis Society, and Department of Veterans Affairs.
Footnotes
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
A.B. directed and coordinated the project. G.G. designed and performed or participated in all experiments. J.B. performed mouse husbandry and survival studies. J.B.L. provided α2-Na/K ATPase knockout mice. H.S. performed mass spectrometry analysis. J. Ravits, T.S. and J. Robertson provided human tissue samples. The manuscript was written by G.G. and A.B. and commented on by all authors.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Allen NJ, Barres BA. Signaling between glia and neurons: focus on synaptic plasticity. Curr Opin Neurobiol. 2005;15:542–548. doi: 10.1016/j.conb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 2.Fields RD, Stevens-Graham B. New insights into neuron-glia communication. Science. 2002;298:556–562. doi: 10.1126/science.298.5593.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ilieva H, Polymenidou M, Cleveland DW. Non–cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGann JC, Lioy DT, Mandel G. Astrocytes conspire with neurons during progression of neurological disease. Curr Opin Neurobiol. 2012;22:850–858. doi: 10.1016/j.conb.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garden GA, et al. Polyglutamine-expanded ataxin-7 promotes non–cell-autonomous purkinje cell degeneration and displays proteolytic cleavage in ataxic transgenic mice. J Neurosci. 2002;22:4897–4905. doi: 10.1523/JNEUROSCI.22-12-04897.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoo SY, et al. SCA7 knockin mice model human SCA7 and reveal gradual accumulation of mutant ataxin-7 in neurons and abnormalities in short-term plasticity. Neuron. 2003;37:383–401. doi: 10.1016/s0896-6273(02)01190-x. [DOI] [PubMed] [Google Scholar]
- 7.Evert BO, et al. Inflammatory genes are upregulated in expanded ataxin-3–expressing cell lines and spinocerebellar ataxia type 3 brains. J Neurosci. 2001;21:5389–5396. doi: 10.1523/JNEUROSCI.21-15-05389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clement AM, et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 9.Yamanaka K, et al. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Faideau M, et al. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shin JY, et al. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol. 1995;38:73–84. doi: 10.1002/ana.410380114. [DOI] [PubMed] [Google Scholar]
- 13.Boillée S, et al. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 14.Nagai M, et al. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Non–cell autonomous effect of glia on motor neurons in an embryonic stem cell–based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dion PA, Daoud H, Rouleau GA. Genetics of motor neuron disorders: new insights into pathogenic mechanisms. Nat Rev Genet. 2009;10:769–782. doi: 10.1038/nrg2680. [DOI] [PubMed] [Google Scholar]
- 17.Rosen DR, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 18.Gurney ME, et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- 19.Bruijn LI, Miller TM, Cleveland DW. Unraveling the mechanisms involved in motor neuron degeneration in ALS. Annu Rev Neurosci. 2004;27:723–749. doi: 10.1146/annurev.neuro.27.070203.144244. [DOI] [PubMed] [Google Scholar]
- 20.Haidet-Phillips AM, et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodman LS, Brunton LL, Chabner B, Knollmann BC. Goodman & Gilman’s Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 2011. [Google Scholar]
- 22.Lehtinen MK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 23.Matsuoka Y, Li X, Bennett V. Adducin: structure, function and regulation. Cell Mol Life Sci. 2000;57:884–895. doi: 10.1007/PL00000731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robledo RF, et al. Targeted deletion of alpha-adducin results in absent beta- and gamma-adducin, compensated hemolytic anemia, and lethal hydrocephalus in mice. Blood. 2008;112:4298–4307. doi: 10.1182/blood-2008-05-156000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shan X, Hu JH, Cayabyab FS, Krieger C. Increased phospho-adducin immunoreactivity in a murine model of amyotrophic lateral sclerosis. Neuroscience. 2005;134:833–846. doi: 10.1016/j.neuroscience.2005.04.036. [DOI] [PubMed] [Google Scholar]
- 26.Hu JH, Zhang H, Wagey R, Krieger C, Pelech SL. Protein kinase and protein phosphatase expression in amyotrophic lateral sclerosis spinal cord. J Neurochem. 2003;85:432–442. doi: 10.1046/j.1471-4159.2003.01670.x. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem. 2002;71:511–535. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 28.Watts AG, Sanchez-Watts G, Emanuel JR, Levenson R. Cell-specific expression of mRNAs encoding Na+, K(+)-ATPase alpha- and beta-subunit isoforms within the rat central nervous system. Proc Natl Acad Sci USA. 1991;88:7425–7429. doi: 10.1073/pnas.88.16.7425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moseley AE, et al. Deficiency in Na,K-ATPase alpha isoform genes alters spatial learning, motor activity, and anxiety in mice. J Neurosci. 2007;27:616–626. doi: 10.1523/JNEUROSCI.4464-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang G, et al. Death receptor 6 (DR6) antagonist antibody is neuroprotective in the mouse SOD1G93A model of amyotrophic lateral sclerosis. Cell Death Dis. 2013;4:e841. doi: 10.1038/cddis.2013.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartford AK, Messer ML, Moseley AE, Lingrel JB, Delamere NA. Na,K-ATPase alpha 2 inhibition alters calcium responses in optic nerve astrocytes. Glia. 2004;45:229–237. doi: 10.1002/glia.10328. [DOI] [PubMed] [Google Scholar]
- 32.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bulua AC, et al. Mitochondrial reactive oxygen species promote production of pro-inflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS) J Exp Med. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 35.Wu DC, Re DB, Nagai M, Ischiropoulos H, Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci USA. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phatnani HP, et al. Intricate interplay between astrocytes and motor neurons in ALS. Proc Natl Acad Sci USA. 2013;110:E756–E765. doi: 10.1073/pnas.1222361110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 38.Ellis DZ, Rabe J, Sweadner KJ. Global loss of Na,K-ATPase and its nitric oxide–mediated regulation in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurosci. 2003;23:43–51. doi: 10.1523/JNEUROSCI.23-01-00043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin LJ, et al. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J Comp Neurol. 2007;500:20–46. doi: 10.1002/cne.21160. [DOI] [PubMed] [Google Scholar]
- 40.Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E. Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J Neurosci. 2011;31:17637–17648. doi: 10.1523/JNEUROSCI.4162-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaphzan H, et al. Genetic reduction of the alpha1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Reports. 2013;4:405–412. doi: 10.1016/j.celrep.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efendiev R, et al. Hypertension-linked mutation in the adducin alpha-subunit leads to higher AP2-mu2 phosphorylation and impaired Na+,K+-ATPase trafficking in response to GPCR signals and intracellular sodium. Circ Res. 2004;95:1100–1108. doi: 10.1161/01.RES.0000149570.20845.89. [DOI] [PubMed] [Google Scholar]
- 43.Torielli L, et al. alpha-adducin mutations increase Na/K pump activity in renal cells by affecting constitutive endocytosis: implications for tubular Na reabsorption. Am J Physiol Renal Physiol. 2008;295:F478–F487. doi: 10.1152/ajprenal.90226.2008. [DOI] [PubMed] [Google Scholar]
- 44.Cusi D, et al. Polymorphisms of alpha-adducin and salt sensitivity in patients with essential hypertension. Lancet. 1997;349:1353–1357. doi: 10.1016/S0140-6736(97)01029-5. [DOI] [PubMed] [Google Scholar]
- 45.Kennedy DJ, et al. CD36 and Na/K-ATPase-alpha1 form a pro-inflammatory signaling loop in kidney. Hypertension. 2013;61:216–224. doi: 10.1161/HYPERTENSIONAHA.112.198770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu J, Kennedy DJ, Yan Y, Shapiro JI. Reactive oxygen species modulation of Na/K-ATPase regulates fibrosis and renal proximal tubular sodium handling. Int J Nephrol. 2012;2012:381320. doi: 10.1155/2012/381320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang JK, et al. Cardiac glycosides provide neuroprotection against ischemic stroke: discovery by a brain slice-based compound screening platform. Proc Natl Acad Sci USA. 2006;103:10461–10466. doi: 10.1073/pnas.0600930103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Piccioni F, Roman BR, Fischbeck KH, Taylor JP. A screen for drugs that protect against the cytotoxicity of polyglutamine-expanded androgen receptor. Hum Mol Genet. 2004;13:437–446. doi: 10.1093/hmg/ddh045. [DOI] [PubMed] [Google Scholar]
- 49.Corcoran LJ, Mitchison TJ, Liu Q. A novel action of histone deacetylase inhibitors in a protein aggresome disease model. Curr Biol. 2004;14:488–492. doi: 10.1016/j.cub.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 50.Burkhardt MF, et al. A cellular model for sporadic ALS using patient-derived induced pluripotent stem cells. Mol Cell Neurosci. 2013;56:355–364. doi: 10.1016/j.mcn.2013.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandra S, Gallardo G, Fernandez-Chacon R, Schluter OM, Sudhof TC. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell. 2005;123:383–396. doi: 10.1016/j.cell.2005.09.028. [DOI] [PubMed] [Google Scholar]
- 52.Gaudilliere B, Shi Y, Bonni A. RNA interference reveals a requirement for myocyte enhancer factor 2A in activity-dependent neuronal survival. J Biol Chem. 2002;277:46442–46446. doi: 10.1074/jbc.M206653200. [DOI] [PubMed] [Google Scholar]
- 53.Gingras M, Gagnon V, Minotti S, Durham HD, Berthod F. Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J Neurosci Methods. 2007;163:111–118. doi: 10.1016/j.jneumeth.2007.02.024. [DOI] [PubMed] [Google Scholar]
- 54.Raoul C, et al. Lentiviral-mediated silencing of SOD1 through RNA interference retards disease onset and progression in a mouse model of ALS. Nat Med. 2005;11:423–428. doi: 10.1038/nm1207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.