

Summary
We investigated the EGL-30 (Gqα) pathway in C. elegans by using genetic screens to identify genes that confer phenotypes similar to egl-30 mutants. One such gene, egl-8, encodes a phospholipase Cβ that is present throughout the nervous system and near intestinal cell junctions. EGL-30 and EGL-8 appear to positively regulate synaptic transmission because reducing their function results in strong aldicarb resistance and slow locomotion rates. In contrast, GOA-1 (Goα) and DGK-1 (diacylglycerol kinase) appear to negatively regulate synaptic transmission, because reducing their function results in strong aldicarb hypersensitivity and hyperactive locomotion. A genetic analysis suggests that GOA-1 negatively regulates the EGL-30 pathway and that DGK-1 antagonizes the EGL-30 pathway.
Introduction
To better understand how synaptic transmission is regulated, we are investigating the signaling pathways that regulate neurotransmitter secretion, the presynaptic component of synaptic transmission. Biochemical studies have revealed that one common class of signaling pathways in neurons is built around heterotrimeric G proteins of the Gq/G11 class (Watson and Arkinstall, 1994). The binding of neurotransmitter to a Gq-coupled receptor leads to activation of the G protein, which in turn activates phospholipase Cβ (PLCβ) (Singer et al., 1997). PLCβ cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into the small signaling molecules diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). These molecules, or their analogs, lead to the stimulation of neurotransmitter secretion (Malenka et al., 1986; Shapira et al., 1987; Berridge, 1998). One of the end-points of the Gq pathway in neurons, therefore, appears to be the presynaptic machinery that regulates neurotransmitter secretion.
There are still major questions about the Gq pathway. For example, are there other downstream effectors of Gq besides PLCβ? Is DAG an essential, or merely modulatory, effector of synaptic transmission? Does the Gq pathway interact with other G protein pathways, and, if so, how? Since synaptic transmission is a highly conserved process, genetic studies in model systems provide a way to address these questions. In C. elegans, the egl-30 gene encodes a highly conserved homolog of Gqα (82% identical to vertebrate forms) that is essential for life (Brundage et al., 1996). Hypomorphic egl-30 mutations result in strongly reduced rates of locomotion and egg laying (Trent et al., 1983; Brundage et al., 1996).
Our previous identification of egl-30 alleles in selections for aldicarb resistant mutants suggests that egl-30 regulates synaptic transmission (Miller et al., 1996). Aldicarb, an inhibitor of acetylcholinesterase, is thought to cause a toxic accumulation of secreted acetylcholine (ACh) at synapses (Cambon et al., 1979; Risher et al., 1987). Since the accumulation of secreted ACh can be reduced by mutations that decrease neurotransmitter release, aldicarb selection of mutants is a powerful tool for investigating both the mechanics of synaptic transmission and its regulation by signaling pathways. Of the 13 other aldicarb resistance genes that have been cloned, all encode proteins that either function in the synaptic vesicle cycle, are homologous to synaptic vesicle cycle proteins, or are localized to presynaptic regions (Miller et al., 1996; Iwasaki et al., 1997; Nonet et al., 1997, 1998; Saifee et al., 1998; J. B. R. and K. Grundahl, data not shown). It seems unlikely that the EGL-30 G protein directly functions in the synaptic vesicle cycle, but we hypothesize that it is involved in regulating this machinery.
We observed that egl-30 (Gqα) mutants, along with mutants in at least two other aldicarb resistance genes, egl-10 and ric-8, have phenotypes that distinguish them from other aldicarb resistant mutants. The finding that egl-10 encodes a regulator of G protein signaling (Koelle and Horvitz, 1996) suggests that aldicarb resistant mutants in this subclass may encode signaling proteins that regulate synaptic transmission. To identify additional genes in this subclass, we screened for aldicarb resistant mutants with phenotypes similar to egl-30 mutants. The first part of this study describes one of these genes, egl-8, which we found encodes a homolog of PLCβ, a known downstream effector of Gqα.
To identify proteins that negatively regulate, or act antagonistically to, EGL-30 and EGL-8, we sought mutants with phenotypes that are opposite of egl-30 and egl-8 mutants. In so doing, we identified goa-1, which encodes a highly conserved homolog of Goα (Lochrie et al., 1991; Mendel et al., 1995; Segalat et al., 1995), and dgk-1, also known as sag-1 (Hajdu-Cronin et al., 1999), which encodes a diacylglycerol kinase (Nurrish et al., 1999). GOA-1 and DGK-1 appear to negatively regulate synaptic transmission because loss-of-function mutations in either gene result in hyperactive locomotion, hyperactive egg laying, and, as we show in this study, strong hypersensitivity to aldicarb (Mendel et al., 1995; Segalat et al., 1995; Hajdu-Cronin et al., 1999; Nurrish et al., 1999). We used a genetic analysis to investigate the relationship of EGL-30 and EGL-8 to GOA-1 and DGK-1. The results suggest that GOA-1 negatively regulates the EGL-30 pathway and that DGK-1 acts antagonistically to the EGL-30 pathway.
Results
Reduction-of-Function Mutations in egl-30 (Gqα) and egl-8 Lead to Similar Phenotypes
Many aldicarb resistant mutants have phenotypes roughly similar to snt-1 mutants (Figure 1). These mutants frequently define genes that encode components of the synaptic vesicle cycle (Miller et al., 1996). egl-30 mutants can be distinguished from the snt-1-like class of aldicarb resistant mutants by their larger size, reduced body flexion, and by the accumulation of eggs in their uteri (Figure 1). To identify proteins that are important for EGL-30 (Gqα) signaling, we screened for aldicarb resistant mutants with phenotypes similar to egl-30 mutants. In so doing, we identified alleles of egl-30, egl-8, egl-10, ric-8, and four genes that had not been previously identified. Here, we provide a genetic and molecular characterization of egl-8, which has been previously identified in genetic screens for defective egg laying (Trent et al., 1983) and defecation (Thomas, 1990). The similar appearance of egl-30 and egl-8 mutants is shown in Figure 1.
Figure 1. egl-8 and egl-30 (Gqα) Mutants Share Similar Phenotypes.

Photographs comparing wild-type C. elegans (N2) with strains containing mutations in snt-1 (a synaptotagmin homolog), egl-30, or egl-8. Although all three mutants were isolated based on aldicarb resistance, note the striking phenotypic differences between the snt-1 mutant (smaller in size, few eggs in uterus, kinked or coiled posture) and egl-30 or egl-8 mutants (equal in size or larger than wild-type, bloated with eggs due to defective egg laying, decreased body flexion).
Through a series of aldicarb selections and subsequent noncomplementation screens, we identified 18 egl-8 alleles. As an initial assessment of the amount of egl-8 function remaining in these alleles, we measured their locomotion rates on an agar surface. Wild-type worms exhibit a stereotyped, spontaneous locomotion behavior that is easily quantified by counting body bends. Table 1 compares the mean locomotion rates of wild-type worms and a representative group of egl-8 mutants that form an allelic series with respect to locomotion rate. The locomotion rates of the mildest alleles are similar to wild-type, while those of the strongest are reduced to about 25% of wild-type. The md1971 allele must drastically reduce or eliminate the function of EGL-8, since md1971 in trans to a deficiency had a locomotion rate not significantly different from md1971 homozygotes (Table 1). Molecular analysis of md1971 (described below) supports this conclusion.
Table 1.
Locomotion Rates of egl-8 and egl-30 Mutants
| Allele | Locomotion Rate-Body Bends/Minutea |
|---|---|
| N2 (wild-type) | 15.6 ± 1.7 |
| egl-8 (md153) | 15.9 ± 1.3 |
| egl-8 (md1933) | 12 ± 0.77 |
| egl-8 (md2243) | 6.7 ± 0.42 |
| egl-8 (n488) | 4.3 ± 0.56 |
| egl-8 (md1971) | 4.13 ± 0.35 |
| egl-8 (md1971)/sDf32 | 3.4 ± 0.6b |
| egl-30 (ad805) | 0.13 ± 0.07 |
| egl-30 (md186) | 0.20 ± 0.13 |
Mean ± standard error, N = 10 young adults.
Not significantly different from egl-8 (md1971) homozygotes (t-test, p = 0.24)
Although egl-8 and egl-30 mutants share similar phenotypes, we observed that egl-30 reduction-of-function mutants have substantially lower locomotion rates than the egl-8 loss-of-function mutants (Table 1). Taken together with the previous finding that loss of EGL-30 function results in lethality (Brundage et al., 1996), this suggests that EGL-30 has one or more additional functions that are not shared with EGL-8.
egl-8 Encodes a Phospholipase Cβ Homolog
To investigate the molecular basis of the similarities between egl-8 and egl-30 mutants, we cloned the egl-8 gene by transposon tagging (see Experimental Procedures). A sequence comparison identified EGL-8 as a member of the phospholipase Cβ (PLCβ) family of proteins, most closely related to vertebrate PLCβ4. Since PLCβ is a known downstream effector of Gqα, this result is consistent with our finding that EGL-30 and EGL-8 appear to act in the same pathway.
Figure 2 shows a schematic comparison of EGL-8 and PLCβ4. The major domains of PLCβ, all of which are conserved in EGL-8, include an N-terminal pleckstrin homology (PH) domain that mediates activation by Gβ γ for some PLCβs, and X and Y catalytic domains, wherein resides the phospholipase activity (Singer et al., 1997). The most highly conserved regions are the X and Y domains (68% and 75% identical, respectively), which are also identical in size between the two proteins. All other regions of EGL-8 are larger than PLCβ4, although the ends of the two proteins can be precisely aligned.
Figure 2. EGL-8 Encodes a PLCβ Homolog.

Schematic alignment of EGL-8 and rat PLCβ4 (GenBank A48047) amino acid sequences. The X and Y domains are shown as gray shaded boxes. Patterned boxes indicate regions present in EGL-8 but not rat PLCβ4. Inverted V’s delimit regions encoded by alternatively spliced exons. The percentage of rat PLCβ4 residues that are identical to EGL-8 in various regions of the protein are indicated. A BLAST search of C. elegans genomic sequence (99% complete at the time of the search) identified five other genes related to EGL-8 (C. elegans predicted gene numbers: T01E8.3, F31B12.1, Y75B12B.g, R05G6.8, and K10F12.3). Four of these genes appear to be homologs of phospholipases that are not part of the PLCβ family. The fifth gene (Y75B12B.g) appears distantly related to vertebrate PLCβ2, but it is considerably less related to PLCβ2 than EGL-8.
Through sequence analysis of multiple cDNAs, we identified four exons that are alternatively spliced (Figure 3, hatched regions). Due to the transcript size (~5 kb) and the distant spacing of exons 4 and 16, we were unable to obtain cDNAs that spanned all four regions where we identified alternative splicing. However, the major splice variants contained exon 4 (8 of 9 cDNAs) and exon 10 (9 of 11 cDNAs), while exons 9 and 16 seemed to be more rarely included (Figure 3). Since most of the sequences included in these exons cannot be aligned with PLCβ4, the alternatively spliced sequences are not likely to be critical for PLC activity; they may, however, be important for regulating EGL-8 in a cell-, sex-, or stage-specific manner.
Figure 3. The egl-8 Gene, Alternative Transcripts, and Mutations.

Schematic of egl-8 gene structure and mutations. Boxed regions are coding sequence, lines are noncoding. Positions at which introns interrupt the sequence are indicated by inverted arrowheads. Hatched boxes represent alternatively spliced exons, each of which is labeled with the exon number. The major splice variants are as follows: 8 of 9 cDNAs contained exon 4, and 9 of 11 cDNAs contained exon 10. Both cDNAs that were missing exon 10 were also missing exon 9, which was present in only 4 out of 11 cDNAs. Exon 16 was present in 1 of 5 cDNAs. The positions and descriptions of five egl-8 mutations are indicated. The premature stop codon in md1971 results from a C to T change of nucleotide 3138 of the cDNA sequence (GenBank AF179426). The md153 lesion deletes cDNA nucleotides 4411–4825. The lower diagram depicts the region that is deleted in n488, with only the relevant exons shown. The deletion corresponds to nucleotides 23253–25630 on the genomic cosmid clone B0348.
egl-8 Mutations
To investigate how egl-8 mutations affect the EGL-8 protein, we analyzed genomic DNA from five egl-8 mutants. For reference, these are the same five mutants whose locomotion rates are quantified in Table 1. The alleles that most strongly reduce EGL-8 function have lesions that disrupt the highly conserved Y domain. n488 has a deletion that removes exons 10 and 11 and disrupts reading frame before the Y domain, md1933 has a Tc1 transposon insertion in the middle of the Y domain, and md2143 has a 2.1 kb insertion in an adjacent location (Figure 3). Consistent with md1971 being a strong reduction-or loss-of-function allele, we found that this mutant has an early stop codon that should prevent formation of part of the catalytic Y domain and the C-terminal region of the protein (Figure 3).
EGL-8 Protein Is Localized to the Nervous System and Intestinal Cells
We produced a polyclonal antibody against the C-terminal region of EGL-8 and used it to visualize EGL-8 protein in wild-type and mutant strains. In wild-type worms, we observed EGL-8 staining primarily in the nervous system and the intestine (Figure 4A). The absence of this staining in egl-8 (md1971) (Figure 4D) and egl-8 (n488) (data not shown) demonstrates its specificity.
Figure 4. The EGL-8 Protein Is Localized to the Nervous System and near Intestinal Cell Junctions.

(A) Indirect immunofluorescence staining of EGL-8 protein in a wild-type C. elegans adult. Note that the nervous system staining is stronger in the head ganglia than in the ventral cord. Confocal cross sections revealed that the lines of staining running along the body are near the luminal membrane of the intestine, and the loop-like structures emanate from the lumen of the intestine. At the end of the intestine, EGL-8 staining becomes brighter and the pattern more complex as the intestinal cells connect to the epithelial rectal cells and the anus is formed (White, 1988). The vulval muscle staining is present in a mutant control (see [D] in this figure).
(B) Enlarged view of EGL-8 staining in head ganglia. Note staining of neuronal cell somas as well as the axon processes of the nerve ring.
(C) EGL-8 intestinal staining is localized near cell junctions. Shown is a confocal image of EGL-8 staining (green) and MH27 staining (red) in the mid-body region of a wild-type L2 stage larvae. Regions of overlap show up as yellow. The centrally located horizontal yellow and orange line is near the junctions formed as pairs of intestinal cells wrap around the lumen (junctions on the other side of the lumen also stain but are not included in this projection). Short vertical yellow and red lines mark the anterior-posterior ends of adjacent intestinal cells. Loops of EGL-8 staining (white arrowheads; see also [A] in this figure) may correspond to the entire region of cell apposition, whereas MH27 only stains the septate junctions.
(D) The region of EGL-8 used for antibody production is not made in egl-8 (md1971), which serves as a control for specificity of the EGL-8 antibody. Nonspecific staining of the EGL-8 antibody can be seen in the pharynx, germ cells, vulval region, and cuticle. Double staining with a monoclonal antibody to the neuronally localized CHA-1 protein (data not shown) was used as a control for permeabilization.
We observed EGL-8 staining in most or all neurons, although the amount of staining varied between individual neurons. In general, neurons in the head and tail ganglia showed stronger staining than the motor neurons of the ventral cord (Figure 4A). Since egl-8 mutants are defective in egg laying, we expected to see staining in the hermaphrodite-specific neurons (which control egg laying) and/or the vulval muscles. However, prominent nonspecific staining around the vulva and weak EGL-8 staining in that region of the ventral cord prevented us from determining if EGL-8 is present at those locations. In the head ganglia, one pair of amphid neurons showed especially strong staining. The amphid neurons relay sensory information, received via the amphid processes, to interneurons in the head ganglia (Bargmann, 1997). Within neurons, we observed EGL-8 staining in both cell somas and in processes (Figure 4B). The presence of EGL-8 in the nerve ring suggests that EGL-8 is present at synapses. Staining was not detected in the sublateral processes or in the dorsal cord.
EGL-8’s intestinal staining was restricted to two lines of staining running down the long axis of the body and to pairs of loop-like structures at regular intervals (Figure 4A). The strongest staining was in the posterior region of the intestine. We used confocal microscopy to determine that the two lines of staining are on the luminal (apical) side of the intestinal cells. In double staining experiments, we observed that the lines of EGL-8 staining colocalize with the staining of MH27, a monoclonal antibody that stains epithelial and intestinal adherens junctions (D. Hall, personal communication) (Figure 4C). MH27 staining also extended part of the way around each of the loops of EGL-8 staining, which may represent sites where the ends of the intestinal cells join together (White, 1988). We conclude that, within intestinal cells, the majority of EGL-8 staining is restricted to regions near adherens junctions.
EGL-30 (Gqα), EGL-8 (PLCβ), and Diacylglycerol Appear to Positively Regulate Synaptic Transmission
Reducing the function of EGL-30 or EGL-8 results in aldicarb resistance. We quantified the aldicarb sensitivity of egl-30 and egl-8 mutants by measuring their population growth rates on various concentrations of aldicarb (Figure 5A). egl-30 and egl-8 mutants are able to grow on concentrations of aldicarb that are about 4-fold higher than the concentration that stops the growth of wild-type. Since aldicarb resistance is most commonly associated with mutations that decrease the rate of neurotransmitter secretion, these results suggest that EGL-30 and EGL-8 normally function to positively regulate synaptic transmission.
Figure 5. Phorbol Esters and Loss-of-Function Mutations in goa-1 and dgk-1 Cause Hypersensitivity to Aldicarb.
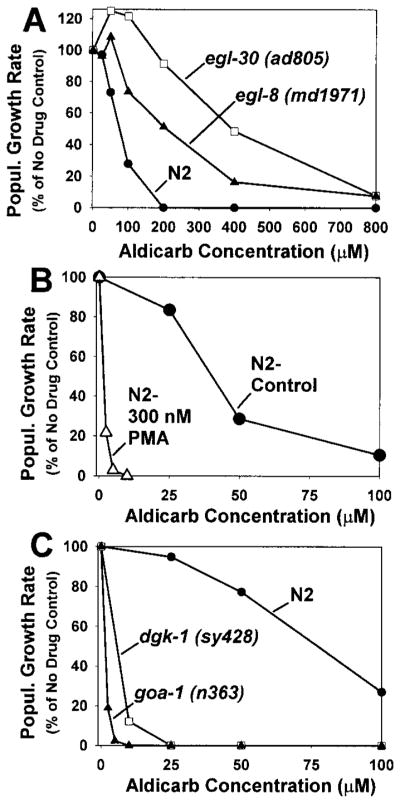
Aldicarb sensitivity is quantified by measuring population growth rates of wild-type and mutant strains on various concentrations of aldicarb. 100% represents the number of progeny produced from a starting population of L1 larva over a 96 hr period in the absence of aldicarb. Curves are representative of duplicate experiments. (A) Reducing the function of egl-30 and egl-8 leads to aldicarb resistance. (B) Phorbol esters cause hypersensitivity to aldicarb. Aldicarb dose-response curves of N2 (wild-type) in the presence and absence of 300 nM phorbol myristate acetate. Note change in scale compared to (A). (C) Loss-of-function mutations in goa-1 and dgk-1 result in hypersensitivity to aldicarb.
Diacylglycerol is a major end product of the Gqα-PLCβ pathway in vertebrates. To address whether or not DAG can positively regulate synaptic transmission in C. elegans, we measured the aldicarb sensitivity of wild-type worms in the presence of phorbol esters, which are molecular analogs of DAG. We observed that, in the presence of 300 nM phorbol myristate acetate, wild-type worms are strikingly hypersensitive to aldicarb. The concentration of aldicarb needed to stop the growth of phorbol ester-treated wild-type is about 35-fold lower than that needed to stop the growth of control-treated wild-type (Figure 5B). This suggests that DAG positively regulates synaptic transmission, consistent with previous studies in vertebrates (Malenka et al., 1986; Shapira et al., 1987).
GOA-1 and DGK-1 Appear to Negatively Regulate Synaptic Transmission
To identify proteins that negatively regulate synaptic transmission, we sought mutants that exhibit hyperactive locomotion and hypersensitivity to aldicarb, phenotypes that are opposite of egl-30 and egl-8 mutants. Loss-of-function mutations in either goa-1 (Goα) or dgk-1 (diacylglycerol kinase) result in hyperactive locomotion and hyperactive egg laying (Mendel et al., 1995; Segalat et al., 1995; Hajdu-Cronin et al., 1999; Nurrish et al., 1999). We observed that these mutants are also very hypersensitive to aldicarb (Figure 5C). A loss-of-function goa-1 allele is greater than 40-fold more hypersensitive than wild-type, while a loss-of-function dgk-1 allele is greater than 10-fold more hypersensitive than wild-type. These data suggest that GOA-1 and DGK-1 normally function to negatively regulate synaptic transmission.
GOA-1 Functions Upstream of EGL-30 or in a Parallel Intersecting Pathway
Given the opposite effects of EGL-30 and GOA-1 mutations, these two G proteins appear to have opposite roles in regulating synaptic transmission. What is the relationship between EGL-30 and GOA-1? Does one negatively regulate the other, or do they act antagonistically? To investigate this question, we analyzed the phenotypes of double mutant animals carrying strong reduction-or loss-of-function mutations in both genes.
Our analysis of goa-1 egl-30 double mutants was complicated by the additive effects of the two mutations. goa-1 egl-30 double mutants exhibited very small brood sizes, and about half of the eggs laid did not hatch (data not shown). As a result, we could not analyze these strains in our standard aldicarb sensitivity assay. We observed, however, that surviving goa-1 egl-30 double mutants are phenotypically similar to egl-30 single mutants. The mean locomotion rate of the double mutant is not significantly different from that of the egl-30 (ad805) single mutant (Figure 6A), and the double mutant also shared the reduced body flexion and egg-laying defective phenotypes of the egl-30 single mutant (data not shown).
Figure 6. GOA-1 Appears to Function Upstream of EGL-30 or in a Parallel Intersecting Pathway.

(A) The locomotion rates of the double mutants egl-30 (ad805); egl-10 [nIs51] and egl-30 (ad805) goa-1 (n363) are indistinguishable from egl-30 (ad805) single mutants. Shown are the mean locomotion rates, expressed as body bends/minute of the double mutants and control strains. Error bars represent the standard error of the mean in a population of 10 young adult animals.
(B) The aldicarb resistance of egl-30 (ad805); egl-10 [nIs51] is similar to egl-30 (ad805) single mutants. Shown are the aldicarb dose-response curves for wild-type, egl-30 (ad805), egl-10 [nIs51], and the double mutant egl-30 (ad805); egl-10 [nIs51]). Curves are representative of duplicate experiments.
To further investigate the relationship of GOA-1 to EGL-30, we analyzed the aldicarb sensitivity and locomotion rate of egl-30 (ad805) in a double mutant combination with egl-10+ [nIs51], an integrated transgenic array that overexpresses wild-type EGL-10. EGL-10 is an RGS protein that appears to negatively regulate GOA-1 (Koelle and Horvitz, 1996), a finding that is supported by analogous studies in vertebrates (Berman et al., 1996; Hunt et al., 1996; Watson et al., 1996). egl-10+ [nIs51] animals exhibit hyperactive locomotion and hypersensitivity to aldicarb, presumably because the excess EGL-10 protein inhibits GOA-1 activity. However, egl-10+ [nIs51] animals do not exhibit the infertility and small brood size phenotypes of goa-1 loss-of-function mutants. We observed that egl-30 (ad805); egl-10+ [nIs51] double mutants are indistinguishable from egl-30 (ad805) single mutants with respect to locomotion rate (Figure 6A). The aldicarb resistance of the double mutant is also indistinguishable from the egl-30 (ad805) single mutant at high aldicarb concentrations, although the double mutant appears slightly less resistant than egl-30(ad805) at low aldicarb concentrations (Figure 6B). Since the locomotion rate and aldicarb resistance phenotypes of egl-30 (ad805) are not substantially affected by loss of goa-1 function or egl-10 overexpression, EGL-10 and GOA-1 appear to act upstream of EGL-30 or in parallel intersecting pathway.
DGK-1 Acts Antagonistically to EGL-30 and EGL-8
What is the relationship of DGK-1 (diacylglycerol kinase) to the EGL-30 pathway? Since DAG kinases reduce DAG levels by converting DAG to phosphatidic acid (Sakane and Kanoh, 1997), we hypothesized that DGK-1 might act antagonistically to the EGL-30 pathway, which should produce DAG. To test this, we analyzed the phenotype of double mutants carrying a loss-of-function mutation in dgk-1 plus a strong reduction-of-function mutation in egl-30. The egl-30 mutation should result in low levels of DAG, while the dgk-1 mutants should facilitate the accumulation of DAG. The double mutants should tend to restore normal levels of DAG, resulting in intermediate phenotypes that could be close to wild-type.
We found that egl-30; dgk-1 double mutants did indeed exhibit intermediate aldicarb sensitivities that were close to wild-type (Figure 7A). The mean locomotion rates of these strains were also intermediate, falling in between that of egl-30 single mutants and wild-type (Figure 7B). The relationship of DGK-1 to EGL-30, therefore, appears to be distinctly different from the relationship of GOA-1 to EGL-30. Strong reduction-of-function egl-30 mutants are not suppressed by goa-1 (loss of function) or egl-10 (overexpression), but they are suppressed by loss-of-function mutations in dgk-1. Since the egl-30; dgk-1 double mutants exhibited phenotypes that were in between that of either single mutant, these data are most consistent with EGL-30 and DGK-1 acting antagonistically to one another rather than in a linear pathway.
Figure 7. DGK-1 Appears to Act Antagonistically to EGL-30 and EGL-8.

(A) The aldicarb resistance of egl-30 (ad805) is suppressed to near wild-type levels by a dgk-1 loss-of-function mutation. Shown are aldicarb dose-response curves of egl-30; dgk-1 double mutants and control strains. Curves are representative of duplicate experiments. Similar results were observed for egl-30 (md186); dgk-1 (sy428) and egl-30 (md186); dgk-1 (md1777) double mutants.
(B) The mean locomotion rates of egl-30; dgk-1 double mutants are in between those of egl-30 single mutants and wild-type. Error bars represent the standard error of the mean in a population of 10 young adult animals.
(C and D) The aldicarb resistance and sluggish locomotion rate of egl-8 (md1971) is suppressed to near wild-type levels by a dgk-1 loss-of-function mutation. Shown are aldicarb dose-response curves (C) and mean locomotion rates (D) of egl-8; dgk-1 double mutants and control strains. Aldicarb sensitivity curves are representative of duplicate experiments, and locomotion rates are means ±standard error of a population of 10 animals. The mean locomotion of the egl-8; dgk-1 double mutant is not significantly different from wild-type (t-test, p =.149).
DGK-1 also appears to act antagonistically to EGL-8. Double mutants carrying loss-of-function mutations in egl-8 and dgk-1 exhibited aldicarb sensitivities and locomotion rates that were close to wild-type (Figures 7C and 7D).
Discussion
egl-8 Encodes a Phospholipase Cβ Homolog that Is Localized to the Nervous System and near Intestinal Cell Junctions
To investigate the EGL-30 pathway in C. elegans, we screened for aldicarb resistant mutants with phenotypes similar to egl-30. In so doing, we identified, among other genes, alleles of egl-8. egl-30 and egl-8 are thus part of a group of at least 30 genes in C. elegans that can confer resistance to inhibitors of cholinesterase (also known as Ric genes) (Miller et al., 1996; K. G. M. and J. B. R., unpublished data). The aldicarb resistance of egl-30 and egl-8 mutants suggests that they have reduced rates of neurotransmitter secretion. Further studies using electrophysiology, however, will be needed to test this.
Our molecular analysis of the egl-8 gene demonstrates that it encodes a PLCβ homolog that is most closely related to vertebrate PLCβ4. Using immunostaining, we found that the EGL-8 protein is localized to neuronal somas and processes and near intestinal cell junctions. Within the nervous system, we observed EGL-8 staining in most neurons, although the amount of staining varied in different regions. The relatively stronger staining in the head ganglia compared to the ventral cord may indicate that EGL-8 plays a more important role in interneurons and sensory neurons than in the motor neurons that comprise most of the ventral cord. Alternatively, motor neurons might require less EGL-8 to achieve the same level of function. The aldicarb resistance of EGL-8 suggests a defect in the synaptic transmission of acetylcholine; however, EGL-8 is also present in noncholinergic neurons (K. M. and J. Duerr, unpublished data), which suggests that it is involved in their function as well.
The presence of EGL-8 in the adult nervous system suggests that it functions in mature neurons. Although a subtle developmental role cannot be ruled out, a study using the conditional allele egl-8 (sa32) concluded that egl-8 is not required for proper development of the egg laying, defecation, or locomotion behaviors (Thomas, 1990).
EGL-8’s intestinal localization could be related to its role in the posterior body wall muscle contraction (pboc) step of the defecation cycle. The observation that severe synaptic mutants exhibit outwardly normal pbocs (K. G. M., unpublished data) suggests that the signals that generate the pboc originate from a nonneuronal source. Consistent with the intestine being a source of the pboc signal is the recent finding that the flr-1 gene, which plays a role in the pboc and defecation cycle rhythm, is expressed in intestinal cells (Takeuchi et al., 1998). Further investigation using genetic mosaics will be required to determine if EGL-8 functions in the intestine to regulate the pboc. EGL-30 seems not to play a role in the pboc, since strong reduction-of-function egl-30 mutants exhibit apparently normal pbocs (K. G. M., unpublished data).
The EGL-30 (Gqα) Pathway
The phenotypes that result from reducing the function of EGL-30 and EGL-8 suggest that they function in a common pathway to positively regulate synaptic transmission. Since vertebrate studies have shown that Gqα activates PLCβ (Singer et al., 1997), we think it is likely that EGL-30 activates EGL-8. EGL-8, however, appears not to mediate all of the effects of EGL-30, because our comparison of egl-30 and egl-8 mutant phenotypes suggests that substantial EGL-30 signaling occurs independently of EGL-8. One or more of the other genes that we identified in our screens may encode additional EGL-30 effectors.
Since activation of PLCβ by Gqα leads to the cleavage of PIP2 to form DAG and IP3 (Berridge, 1984), a deficiency of DAG and/or IP3 in egl-30 and egl-8 mutants may cause or contribute to their shared phenotypes. Although we have not yet determined if IP3 plays a role in synaptic transmission in C. elegans, our finding that phorbol esters induce strong hypersensitivity to aldicarb suggests that DAG can positively regulate synaptic transmission.
What are the downstream targets of DAG? Protein kinase C (PKC), which is directly activated by DAG and calcium (Berridge, 1984), could potentially regulate synaptic transmission by phosphorylating synaptic vesicle cycle proteins. Studies have shown that interactions between SNAP-25 and syntaxin, between Munc-18 and syntaxin, and between N-type calcium channels and syntaxin or SNAP-25 are all inhibited by phosphorylation with PKC (Fujita et al., 1996; Shimazaki et al., 1996; Yokoyama et al., 1997). Although these studies are consistent with a role for PKC in regulating synaptic vesicle fusion, their in vivo relevance is not yet clear, and there are many other potential PKC targets in the synapse.
Another likely downstream target of DAG in the synapse is UNC-13, which, like PKC, contains a C1 domain that binds phorbol esters and diacylglycerol (Maruyama and Brenner, 1991). Consistent with a role in regulating neurotransmitter secretion, UNC-13 and its vertebrate homolog are localized to presynapses (Betz et al., 1998; R. Eustance and J. B. R., unpublished data). unc-13 reduction-of-function mutants are also strongly aldicarb resistant and exhibit severely reduced locomotion rates (Nguyen et al., 1995; Miller et al., 1996). A vertebrate homolog of UNC-13 interacts with syntaxin (Betz et al., 1997), an essential component of the synaptic vesicle fusion machinery (Schulze et al., 1995). UNC-13, therefore, could be a crucial link between the EGL-30 (Gqα) pathway and neurotransmitter secretion.
GOA-1 and DGK-1 Negatively Regulate the EGL-30 (Gqα) Pathway
The hyperactive locomotion and hypersensitivity to aldicarb exhibited by loss-of-function mutants in goa-1 and dgk-1 suggest that GOA-1 and DGK-1 negatively regulate synaptic transmission. Our genetic analysis suggests that both proteins interact with the EGL-30 pathway, although they appear to do so in distinctly different ways. Our analysis of egl-30; dgk-1 doubles and egl-8; dgk-1 doubles suggests that EGL-30 and EGL-8 act antagonistically to DGK-1. The knowledge that dgk-1 encodes a diacylglycerol kinase (Nurrish et al., 1999) suggests a simple explanation for these results. Specifically, we suggest that low DAG levels in the neurons of egl-30 and egl-8 mutants are restored to near normal levels by loss-of-function mutations in dgk-1, which should block phosphorylation of DAG (i.e., block its conversion to phosphatidic acid). Although the source of this putative DAG in the egl-30 and egl-8 mutants has not been determined, it could be derived from partial function of egl-30 pathway or from one of the other pathways that are known to produce DAG (Singer et al., 1997).
Our analysis of goa-1 egl-30 double mutants and egl-10+ [nIs51]; egl-30 double mutants suggests that GOA-1 acts upstream of the EGL-30 pathway. However, this kind of genetic analysis cannot determine which component(s) of the EGL-30 pathway are regulated by GOA-1. GOA-1 could act upstream of the EGL-30 G protein itself or, alternatively, it could regulate a downstream effector or product of the EGL-30 pathway. For example, GOA-1 could act to prevent the accumulation of DAG, possibly by activating DGK-1. If this were the case, however, goa-1 mutants, like dgk-1 mutants, should suppress egl-30 mutants, and they do not. We think, therefore, that GOA-1 most likely regulates upstream components of the EGL-30 pathway, such as EGL-30 itself.
GOA-1 could negatively regulate the EGL-30 pathway directly or through intermediate proteins. One candidate for an intermediate protein is EAT-16, an RGS protein that appears to negatively regulate EGL-30 and also appears to function downstream of GOA-1 (Hajdu-Cronin et al., 1999). Consistent with a role in negatively regulating EGL-30, reducing the function of EAT-16 results in hyperactive locomotion, hyperactive egg laying, and hypersensitivity to aldicarb (Hajdu-Cronin et al., 1999; K. G. M., unpublished data).
Gene expression and protein localization data support an important assumption of this model, which is that these proteins function together in the same cells. EGL-30, GOA-1, DGK-1, EGL-10, EAT-16, and EGL-8 appear to be broadly, though not in all cases exclusively, expressed in the nervous system (Mendel et al., 1995; Segalat et al., 1995; Koelle and Horvitz, 1996; Hajdu-Cronin et al., 1999; Lackner et al., 1999 [this issue of Neuron]; Nurrish et al., 1999; this paper). These studies suggest that the GOA-1/EGL-30 signaling network is a core component of most, or all, neurons in C. elegans.
Why Negatively Regulate the EGL-30 (Gqα) Pathway?
In vertebrates, many neurotransmitters/neuropeptides have been shown to activate Gqα via G protein linked receptors (Watson and Arkinstall, 1994). These include acetylcholine, glutamate, and serotonin, all of which are present in C. elegans (Rand and Nonet, 1997). Since the proteins of the GOA-1/EGL-30 signaling network appear to be widely expressed in the nervous system, EGL-30 activation may be coupled to different neurotransmitters in a cell-specific manner.
If the activity of the Gqα pathway can be controlled via its input (i.e., neurotransmitters binding to receptors), why is there a mechanism for its negative regulation? One possibility is that it gives synapses the added flexibility of a gain control mechanism. Depending on the activity of the negative regulators, the output of the synapse (i.e., the amount of neurotransmitter released) could vary in response to the same amount of input. Interestingly, GOA-1 and DGK-1 provide two different mechanisms for negative regulation that could be used in concert. Activation of GOA-1 should decrease DAG production, while concurrent activation of DGK-1 should deplete the existing stores of DAG, an effect that would shorten the time needed to downregulate the pathway.
Experimental Procedures
Strains
Worms were cultured using standard methods (Brenner, 1974). Wild-type worms were C. elegans variety Bristol, strain N2. Variety Bergerac, strain EM1002, was used for STS mapping. TR638 and RM25, derived from TR403, were used as starting strains for isolating spontaneous aldicarb resistant mutants. The following C. elegans mutant strains were used in this work: RM2224 dgk-1 (sy428) X, RM1790 dgk-1 (md1777) X, KP828 dgk-1 (nu199) X, MT1083 egl-8 (n488) V, RM153 egl-8 (md153) V, RM2163 egl-8 (md1933) V, RM2215 egl-8 (md1933 md1934R) V, RM2243 egl-8 (md2243) V, RM2221 egl-8 (md1971) V, RM2246 egl-8 (md1971) V; dgk-1 (sy428) X, RM2158 egl-8 (md1933) V; dpy-11 (e224) V, MT8190 lin-15 (n765) egl-10+ [nIs51], RM2289 egl-30 (ad805); lin-15 (n765) egl-10+ [nIs51], DA823 egl-30 (ad805) I, RM1758 egl-30 (md186) I, RM2217 egl-30 (ad805) I; dgk-1 (sy428) X, RM2183 egl-30 (md186) I; dgk-1 (md1777) X, RM2210 egl-30 (md186) I; dgk-1 (sy428) X, RM2226 goa-1 (n363) I, RM2279 goa-1 (n363) egl-30 (ad805) I, RM2278 goa-1 (n363) egl-30 (md186) I, TR638 mut-3 (r456).
Genetic Screens
Four different mutant screens were used to identify the egl-8 mutants described here. In the first, RM25 or the Tc1 mutator strain TR638 was used as a starting strain to isolate spontaneous aldicarb resistant mutants as previously described (Miller et al., 1996). The egl-8 alleles identified in this screen were md153 (described here) and seven other alleles (six derived from RM25, one derived from TR638). In the second screen, TR638 was again used as the starting strain to look for Tc1-induced aldicarb resistant mutants, but a secondary screen was applied to identify mutants with phenotypes similar to egl-30 and/or egl-10 mutants (defective egg laying and/or straightened posture). Four hundred independent lines were analyzed in this screen (~20,000 animals per line), and a single egl-8 allele, md1933, was identified. In a similar screen, 8000 EMS-mutagenized genomes were subjected to aldicarb selection followed again by secondary screening for phenotypes similar to egl-10 and/or egl-30. This screen identified five alleles of egl-8 including md1971 (described here). The fourth screen was a noncomplementation screen in which EMS mutagenized N2 males were crossed to egl-8 (md1933) dpy-11 (e224). Plates were allowed to produce F1 cross progeny for 2 days, at which point progeny were rinsed off and transferred to 0.3 mM aldicarb plates for selection of non-complementing cross progeny (the dpy-11 mutation causes self-progeny to die under these conditions). Five alleles of egl-8 were identified in this way, including md2243 (described here).
Mutants identified in these screens (with the exception of the noncomplementation screen) were mapped to a chromosomal sub-region using STS mapping of EM1002 strain polymorphisms (Williams et al., 1992). Mutants were identified as egl-8 alleles after first mapping them to the left arm of linkage group (LG) V (left of STS marker stP3) followed by complementation testing. Several combinations of egl-8 alleles (n488/md2116, n488/md1971, and md2116/md1971) showed partial complementation for the Egl phenotype but noncomplementation for the aldicarb resistance and locomotion phenotypes.
dgk-1 (md1777) was obtained in a screen for suppressors of the mutant ric-8 (md303) and was mapped left of stP41 on the left arm of LG X by STS mapping. Subsequent complementation tests revealed that its hyperactive locomotion and egg laying are allelic with dgk-1 (nu199) and sag-1 (sy428).
Mutants identified in this study or obtained from other labs were backcrossed at least twice before use in assays or double mutant construction.
Double Mutant Strain Construction and Verification
For double mutant constructions, we chose strong reduction-of-function or loss-of-function alleles. egl-30 (ad805) and egl-30 (md186) are strong reduction-of-function alleles (Brundage et al., 1996; Miller et al., 1996; this paper), while egl-8 (md1971), dgk-1 (sy428), and goa-1 (n363) are loss-of-function or null alleles (this paper; Segalat et al., 1995; Hajdu-Cronin et al., 1999). dgk-1 (sy428) contains an early stop codon in the coding sequences of dgk-1 (S. Nurrish and J. Kaplan, personal communication). The molecular lesion of dgk-1 (md1777) has not been determined, but comparison to dgk-1 (sy428) suggests that it strongly reduces dgk-1 function.
Double mutants were constructed using standard genetic methods, without additional marker mutations. Homozygosity of alleles in each double mutant was confirmed by sequencing amplified genomic DNA of strains containing egl-30 (ad805), egl-30 (md186), or egl-8 (md1971), and by PCR for strains containing goa-1 (n363). Homozygosity of dgk-1 alleles (X-linked) was confirmed by noncomplementation tests. Homozygosity of egl-10+ [nIs51] in double mutants was confirmed by PCR, using primers specific for the ampicillin resistance gene of the integrated array (25/25 progeny of the cloned double mutant contained this marker), and by outcrossing to wild-type, where we observed segregation of the egl-10 overexpressing phenotype in 100% of the F1 generation.
Cloning and Analysis of egl-8 Genomic and cDNA Sequences
Genomic sequence corresponding to egl-8 was identified using a transposon display method developed by Henri van Luenen and Ronald Plasterk. In brief, the egl-8 (md1933) mutant was obtained from the Tc1 mutator strain TR638 as described above. A wild-type revertant of the mutant was subsequently isolated (md1933 md1934R), and Tc1 copy number of the mutant and revertant was reduced by backcrossing. Genomic DNA prepared from these strains and a wild-type control was digested with the 4-cutter Sau 3A followed by heat inactivation. A vectorette oligo cassette (consisting of 55 nucleotide complementary oligos that form a Sau 3A overhang upon annealing) was ligated to the digested genomic DNA. Primers complementary to Tc1 and the vectorette cassette were then used to amplify genomic sequences flanking all of the Tc1s in the genome. After 20 cycles, the PCR reaction was diluted 100-fold, and a second PCR reaction was performed using nested primers. The nested Tc1 primer was end-labeled with 33P γATP before inclusion in the reaction. After 20 cycles, a portion of the reaction was run out on a 6% sequencing gel. The gel was dried and exposed to X-ray film, producing a pattern of bands made up of Tc1-containing genomic Sau 3A fragments. The Tc1 pattern was compared between wild-type, mutant, and revertant strains, and a single band was identified that was present in the md1933 mutant but was missing in wild-type and the revertant. The dried gel was aligned with the autoradiogram, and the band was excised, eluted in water, and amplified for 30 cycles using the same primers as for the second PCR of the transposon display. The amplification product was sequenced, labeled, and used to screen a C. elegans mixed-stage oligo dT-primed cDNA library, which resulted in the isolation of cDNAs derived from the 3′ two thirds of the egl-8 transcript. Genomic sequence was analyzed with Genefinder to predict the 5′ most exon of the gene, and primers designed from this exon were used to amplify a 100 bp fragment from genomic DNA, which was used to probe a C. elegans mixed-stage random-primed cDNA library. A 5002 nucleotide chimeric sequence was assembled from all of the unique sequence in the 5′ and 3′ cDNA clones. In several cDNA clones, this sequence was preceded by a portion of the trans-spliced leader SL1, which is found on the 5′ end of some C. elegans transcripts (Krause and Hirsh, 1987). Clones used for the chimeric sequence were sequenced on both strands. The chimeric sequence was used to search the database of C. elegans genomic sequence, and intron/exon boundaries were determined. The predicted peptide sequence was used to search GenBank databases using BLAST 2.0 with default search parameters (Altschul et al., 1997). Related sequences were compared to each other using the Genetics Computer Group programs Bestfit and Pileup with default parameters.
Molecular Analysis of Mutations
In cases where genomic regions were amplified from egl-8 mutants, populations of mutant animals were processed for PCR using the method of Williams et al. (1992).
Identification of the transposon insertion in md1933 is described above. PCR amplification of the relevant genomic region was used to confirm that the transposon was present in md1933 mutant strains but absent in N2, TR638 (from which md1933 was derived), and the revertant strain.
The md153 deletion was identified by probing a Southern blot of genomic DNA from md153 and control strains. The appropriate genomic region of md153 was amplified and sequenced along with the corresponding region from the parental strain RM25.
Genomic DNA containing all egl-8 exons and intron/exon boundaries was amplified using PCR in md2243, n488, and md1971. The insertion in md2243 and the deletion in n488 were localized using PCR, and the PCR products spanning the lesions were sequenced. All exons and intron/exon boundaries were sequenced for the md1971 mutant.
EGL-8 Antibodies and Immunostaining
Pfu polymerase (Stratagene) was used to amplify codons 1041–1382 from an egl-8 cDNA clone. BamHI and XhoI sites were incorporated into the primers to facilitate directional in-frame cloning into the bacterial expression vector pRSETb (Invitrogen). The construct was transformed into the bacterial expression host BL21(DE3) pLysS, and EGL-8 fusion protein was purified under denaturing conditions using nickel resin.
Rabbits were injected with 500 μg of fusion protein and boosted four times. To affinity purify EGL-8 antibodies, 1.0 mg of EGL-8 fusion protein was run out on a 9% SDS-PAGE gel and blotted to nitrocellulose. The blot was stained with Ponceau S, and the EGL-8 fusion protein band was excised and washed for 30 min in blocker (10 mM tris [pH 8.0], 0.05% Tween 20, 150 mM NaCl, 3% nonfat dry milk, and 0.05% sodium azide) with rocking. The blocker solution was then removed, and 4.5 ml of serum was incubated with the EGL-8 protein on the nitrocellulose strips for 1 hr. The serum was removed, and the strips were rinsed with TBST (10 mM tris [pH 8.0], 0.05% Tween 20, and 150 mM NaCl), followed by 3 × 5 min washes in TBST. Antibodies were eluted from the strips using glycine elution buffer (5 mM glycine, 0.01% BSA, 0.05% Tween 20, and 500 mM NaCl [pH 2.3]) followed by 50 mM triethylamine (pH 11.5). Secondary antibodies were adsorbed against 4% formaldehyde-fixed worms to remove antibodies to nematode proteins.
Whole mounts of C. elegans for antibody staining were prepared as previously described, using the methanol/acetone fixation protocol (Duerr et al., 1999). Affinity-purified anti-EGL-8 antibodies (KM3B-5.1) were used at a 1/500 dilution. In double staining experiments, the mouse monoclonal antibodies MH27 (1/1000 dilution) or anti-CHA-1 (undiluted supernatant) were included. Secondary antibodies were donkey anti-rabbit antibodies (Jackson ImmunoResearch) coupled to Alexa 488 dye (Molecular Probes). In double staining experiments, donkey anti-mouse antibodies coupled to Cy3 were also added. Slides were viewed and images were collected using a Leica TCS NT Confocal Microscope and accompanying software.
Aldicarb Sensitivity Assays
Aldicarb sensitivity was quantified by placing a fixed number of L1 stage larva on culture plates containing 0, 10, 25, 50, 100, 200, 400, 800, or 1600 μM aldicarb and allowing them to grow at 20°C for 96 hr. Growth was then stopped by putting the plates at 4°C. The number of progeny (eggs and larva) produced during this period were counted on a gridded background, and a percentage of the number of progeny produced on the no drug control plate was calculated for each concentration. The number of L1 larva plated for the assay was chosen so that at least 300 progeny were produced on the no drug control plate. For strains with wild-type fertility and growth rate, 3 L1s were placed on each plate in the series; for strains with decreased fertility or growth rate, correspondingly more L1s were placed on each plate.
Plates used in the assay were prepared fresh for each set of assays. Aldicarb was added after autoclaving, when media had cooled to 60°C. Additions were made from a 105 mM stock of aldicarb prepared fresh in 70% ethanol. The volume of drug +70% ethanol carrier was kept constant for each concentration. Twenty-four hours after the plates had been poured, 15 μl of OP-50 bacterial culture was spread on each plate, leaving a clear border around the edge of the plate to minimize wandering onto the sides of the plate. Plates were incubated for 2 more days at room temperature, placed at 4°C for 1 day, and then immediately used for the assay. To allow comparison between different sets of assays, plates were always prepared as described above, duplicate assays were performed, and an N2 control was included with each set of strains. Over a series of eight independent assays, N2 values (expressed as percentage of the no drug control) ranged from 91% to 104% for 25 μM, 73% to 86% for 50 μM, 16% to 46% for 100 μM, and 0% to 6% for 200 μM.
To test the aldicarb sensitivity of phorbol ester-treated wild-type worms, phorbol myristate acetate (Research Products; 5 mg/ml stock dissolved in 100% ethanol) was added to the 60°C cooled media to a final concentration of 300 nM before addition of aldicarb. The assay then proceeded as described above except that six instead of three wild-type L1s were plated, since the phorbol myristate acetate reduced the growth rate of wild-type by about half.
Locomotion Assays
Locomotion rate was quantified by counting body bends of young adult animals on reproducibly thin lawns of OP-50 bacteria. Locomotion assay plates were produced by spreading standard culture plates with 100 μl of OP-50 culture (grown 16 hr at 30°C from a single colony). The bacterial lawn was grown 16 hr at 37°C and then plates were stored at 4°C until needed. The assay was set up by placing single late L3 or early L4 stage larva on each of 10 locomotion assay plates. Plates were then placed at 20°C for 24 hr, during which time the larva matured to young adults (thus allowing egg-laying defective mutants to be assayed before they became engorged with eggs). To count body bends, a plate was equilibrated to 23–24°C, placed on the scope, and, after a 30 second period, body bends were counted for 3 min. A body bend was counted each time the tip of the tail passed through maximum or minimum amplitude. After all 10 plates had been counted, the process was repeated two more times on the same set of animals. Mean body bends/minute were calculated for each animal, and these means were used to calculate the mean and standard error of the entire set of 10. For strains with strongly reduced locomotion rate, the number of animals in a set was reduced to five due to low variability.
Acknowledgments
We thank Henri van Luenen and Ronald Plasterk for providing protocols and advice concerning their transposon display method; Paul Sternberg and Joshua Kaplan for providing alleles of dgk-1 and for communicating results prior to publication; Lorna Brundage for sharing information on her analysis of the egl-30 (md186) mutation; David Sonneborn for sharing egl-8 cDNA sequence; Janet Duerr for assistance with confocal microscopy, image analysis, and cell identification; Jim Waddle and Ross Francis for providing MH27 antibody; Bob Barstead for providing cDNA libraries; and John McManus for help in analysis of the egl-8 (n488) mutation. Some of the strains used here were provided by the C. elegans Genetics Center. This work was supported by a grant from the National Institute of Neurological Disorders and Stroke to J. B. R. (NS33187).
Footnotes
GenBank Accession Number
The egl-8 sequence is deposited in the GenBank database under accession number AF179426.
Reference List
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;17:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI. Chemotaxis and thermotaxis. In: Riddle DH, Blumenthal T, Meyer B, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 717–737. [PubMed] [Google Scholar]
- Berman DM, Wilkie TM, Gilman AG. GAIP and RGS4 are GTPase-activating proteins for the Gi subfamily of G protein α subunits. Cell. 1996;86:445–452. doi: 10.1016/s0092-8674(00)80117-8. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and diacylglycerol as second messengers. Biochem J. 1984;220:345–360. doi: 10.1042/bj2200345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Betz A, Okamoto M, Benseler F, Brose N. Direct interaction of the rat unc-13 homologue Munc13–1 with the N terminus of syntaxin. J Biol Chem. 1997;272(4):2520–2526. doi: 10.1074/jbc.272.4.2520. [DOI] [PubMed] [Google Scholar]
- Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- Brenner S. The genetics of C. elegans. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundage L, Avery L, Katz A, Kim U, Mendel JE, Sternberg PW, Simon MI. Mutations in a C. elegans Gqα gene disrupt movement, egg laying, and viability. Neuron. 1996;16:999–1009. doi: 10.1016/s0896-6273(00)80123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambon C, Declume C, Derache R. Effect of the insecticidal carbamate derivatives (carbofuran, pirimicarb, aldicarb) on the activity of acetylcholinesterase in tissues from pregnant rats and fetuses. Toxicol Appl Pharmacol. 1979;49(2):203–208. doi: 10.1016/0041-008x(79)90242-4. [DOI] [PubMed] [Google Scholar]
- Duerr JS, Frisby DL, Gaskin J, Duke A, Asermely K, Huddleston D, Eiden LE, Rand JB. The cat-1 gene Caenorhabditis elegans encodes a vesicular monoamine transporter required for specific monoamine-dependent behaviors. J Neurosci. 1999;19(1):72–84. doi: 10.1523/JNEUROSCI.19-01-00072.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Sasaki T, Fukui K, Kotani H, Kimura T, Hata Y, Sudhof TC, Scheller RH, Takai Y. Phosphorylation of Munc-18/n-Sec1/rbSec1 by protein kinase C: its implication in regulating the interaction of Munc-18/n-Sec1/rbSec1 with syntaxin. J Biol Chem. 1996;271(13):7265–7268. doi: 10.1074/jbc.271.13.7265. [DOI] [PubMed] [Google Scholar]
- Hajdu-Cronin YM, Chen WJ, Patikoglou G, Koelle MR, Sternberg PW. Antagonism between Goα and Gqa in C. elegans: the RGS protein EAT-16 is necessary for Goα signaling and regulates Gqα activity. Genes Dev. 1999;13:1780–1793. doi: 10.1101/gad.13.14.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt TW, Fields TA, Casey PJ, Peralta EG. RGS10 is a selective activator of Gαi GTPase activity. Nature. 1996;383:175–177. doi: 10.1038/383175a0. [DOI] [PubMed] [Google Scholar]
- Iwasaki K, Staunton JE, Saifee O, Nonet ML, Thomas JH. aex-3 encodes a novel regulator of presynaptic activity in C. elegans. Neuron. 1997;18:613–622. doi: 10.1016/s0896-6273(00)80302-5. [DOI] [PubMed] [Google Scholar]
- Koelle MR, Horvitz HR. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell. 1996;84:112–125. doi: 10.1016/s0092-8674(00)80998-8. [DOI] [PubMed] [Google Scholar]
- Krause M, Hirsh D. A trans-spliced leader sequence on actin mRNA in C. elegans. Cell. 1987;49:753–761. doi: 10.1016/0092-8674(87)90613-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Nurrish S, Kaplan JM. Facilitation of synaptic transmission by EGL-30 Gαq and EGL-8 phospholipase Cβ: DAG-binding to UNC-13 is required to stimulate acetylcholine release. Neuron. 1999;24:335–346. doi: 10.1016/s0896-6273(00)80848-x. this issue. [DOI] [PubMed] [Google Scholar]
- Lochrie MA, Mendel JE, Sternberg PW, Simon MI. Homologous and unique G protein alpha subunits in the nematode C. elegans. Cell Regul. 1991;2(2):135–154. doi: 10.1091/mbc.2.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- Maruyama IN, Brenner SA. Phorbol ester/diacylglycerol-binding protein encoded by the unc-13 gene of Caenorhabditis elegans. Proc Natl Acad Sci USA. 1991;88:5729–5733. doi: 10.1073/pnas.88.13.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, Plasterk RHA, Sternberg PW. Modulation of serotonin-controlled behaviors by Go in Caenhorabditis elegans. Nature. 1995;267:1648–1651. [Google Scholar]
- Miller KG, Alfonso A, Nguyen M, Crowell JA, Johnson CD, Rand JB. A genetic selection for Caenorhabditis elegans synaptic transmission mutants. Proc Natl Acad Sci USA. 1996;93:12593–12598. doi: 10.1073/pnas.93.22.12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen M, Alfonso A, Johnson CD, Rand JB. Caenorhabditis elegans mutants resistant to inhibitors of acetylcho-linesterase. Genetics. 1995;140:527–535. doi: 10.1093/genetics/140.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Staunton JE, Kilgard MP, Fergestad T, Hartwieg E, Horvitz HR, Jorgensen EM, Meyer BJ. Caenorhabditis elegans rab-3 mutant synapses exhibit impaired function and are partially depleted of vesicles. J Neurosci. 1997;17(21):8061–8073. doi: 10.1523/JNEUROSCI.17-21-08061.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Saifee O, Zhao H, Rand JB, Wei L. Synaptic transmission deficits in Caenorhabditis elegans synaptobrevin mutants. J Neurosci. 1998;18(1):70–80. doi: 10.1523/JNEUROSCI.18-01-00070.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurrish S, Segalat L, Kaplan JM. Serotonin inhibition of synaptic transmission: Gαo decreases the abundance of UNC-13 at release sites. Neuron. 1999;24:231–242. doi: 10.1016/s0896-6273(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Rand JB, Nonet ML. Synaptic transmission. In: Riddle DH, Blumenthal T, Meyer BJ, Priess JR, editors. C. elegans II. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. pp. 611–643. [PubMed] [Google Scholar]
- Risher JF, Mink FL, Stara JF. The toxicologic effects of the carbamate insecticide aldicarb in mammals: a review. Environ Health Perspect. 1987;72:267–281. doi: 10.1289/ehp.8772267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifee O, Wei L, Nonet ML. The Caenorhabditis elegans unc-64 locus encodes a syntaxin that interacts genetically with synaptobrevin. Mol Biol Cell. 1998;9(6):1235–1252. doi: 10.1091/mbc.9.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane F, Kanoh H. Molecules in focus: diacylglycerol kinase. Int J Biochem Cell Biol. 1997;29(10):1139–1143. doi: 10.1016/s1357-2725(97)00037-x. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80(2):311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- Segalat L, Elkes DA, Kaplan JM. Participation of the protein Go in multiple aspects of behavior in C. elegans. Nature. 1995;267:1652–1655. [Google Scholar]
- Shapira R, Silberberg SD, Ginsburg S, Rahamimoff R. Activation of protein kinase C augments evoked transmitter release. Nature. 1987;325:58–60. doi: 10.1038/325058a0. [DOI] [PubMed] [Google Scholar]
- Shimazaki Y, Nishiki T, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25-kDa synaptosome-associated protein. Possible involvement in protein kinase C-mediated regulation of neurotransmitter release. J Biol Chem. 1996;271(24):14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- Singer WD, Brown HA, Sternweis PC. Regulation of eukaryotic phosphatidylinositol-specific phospholipase C and phospholipase D. Annu Rev Biochem. 1997;66:475–509. doi: 10.1146/annurev.biochem.66.1.475. [DOI] [PubMed] [Google Scholar]
- Take-uchi M, Kawakami M, Ishihara T, Amano T, Kondo K, Katsura I. An ion channel of the degenerin/epithelial sodium channel superfamily controls the defecation rhythm in Caenorhabditis elegans. Proc Natl Acad Sci USA. 1998;95:11775–11780. doi: 10.1073/pnas.95.20.11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JH. Genetic analysis of defecation in Caenorhabditis elegans. Genetics. 1990;124:855–872. doi: 10.1093/genetics/124.4.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent C, Tsung N, Horvitz HR. Egg-laying defective mutants of the nematode Caenorhabditis elegans. Genetics. 1983;104:619–647. doi: 10.1093/genetics/104.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson S, Arkinstall S. The G-Protein Linked Receptor Factsbook. New York: Academic Press; 1994. [Google Scholar]
- Watson N, Linder ME, Druey KM, Kehrl JH, Blumer KJ. RGS family members: GTPase-activating proteins for hetero-trimeric G-protein α-subunits. Nature. 1996;383:172–175. doi: 10.1038/383172a0. [DOI] [PubMed] [Google Scholar]
- White J. The Anatomy. In: Wood WB, editor. The Nematode Caenorhabditis elegans. Cold Spring Harbor Laboratory Press; 1988. pp. 81–122. [Google Scholar]
- Williams BD, Schrank B, Huynh C, Shownkeen R, Waterston RH. A genetic mapping system in Caenorhabditis elegans based on polymorphic sequence-tagged sites. Genetics. 1992;131:609–624. doi: 10.1093/genetics/131.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama CT, Sheng ZH, Catterall WA. Phosphorylation of the synaptic protein interaction site on N-type calcium channels inhibits interactions with SNARE proteins. J Neurosci. 1997;17(18):6929–6938. doi: 10.1523/JNEUROSCI.17-18-06929.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]