

Abstract
IsdGs are heme monooxygenases that break open the tetrapyrrole, releasing the iron, and thereby allowing bacteria expressing this protein to use heme as a nutritional iron source. Little is currently known about the mechanism by which IsdGs degrade heme, although the products differ from those generated by canonical heme oxygenases. A synthesis of time-resolved techniques, including in proteo mass spectrometry and conventional and stopped-flow UV/visible spectroscopy, was used in conjunction with analytical methods to define the reaction steps mediated by IsdG from Staphylococcus aureus and their time scales. An apparent meso-hydroxyheme (forming with k = 0.6 min−1, pH 7.4, 10 mm ascorbate, 10 μm IsdG-heme, 22 °C) was identified as a likely common intermediate with the canonical heme oxygenases. Unlike heme oxygenases, this intermediate does not form with added H2O2 nor does it convert to verdoheme and CO. Rather, the next observable intermediates (k = 0.16 min−1) were a set of formyloxobilin isomers, similar to the mycobilin products of the IsdG homolog from Mycobacterium tuberculosis (MhuD). These converted in separate fast and slow phases to β-/δ-staphylobilin isomers and formaldehyde (CH2O). Controlled release of this unusual C1 product may support IsdG's dual role as both an oxygenase and a sensor of heme availability in S. aureus.
Keywords: carbon monoxide, heme, heme oxygenase, iron, Staphylococcus aureus (S. aureus), tetrapyrrole
Introduction
Heme oxygenases (HOs)3 are enzymes that oxidatively liberate iron from the heme tetrapyrrole (1–3). In the well characterized HOs from animals and many bacteria, the same heme molecule acts as both the O2-activating cofactor and substrate. Three successive monooxygenation steps yield Fe(II), CO, and biliverdin IXα as the end products of the reaction (Fig. 1) (2, 3). Animals use HOs to maintain cellular heme homeostasis as part of a constant cycle of heme synthesis and breakdown. The products report on the status of this cycle and serve as antioxidants and signaling agents (4, 5). Many bacteria also use HO homologs, both to control heme homeostasis and to liberate iron from host-derived heme (6, 7). Heme, found primarily in hemoglobin, can therefore be used as a rich nutritional source of iron. Because of the intriguing nature of the reaction, which uses heme as both cofactor and substrate (8–10), as well as the acute biological importance of HO-mediated processes, HOs from several species have been exceptionally well characterized (2, 3).
FIGURE 1.
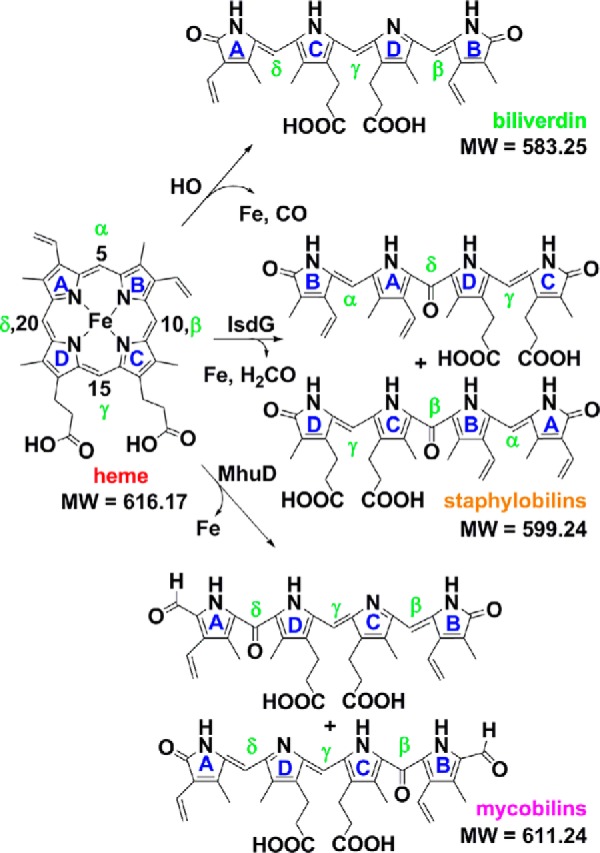
Known enzymatic breakdown products of chemically unfunctionalized heme (heme b). Heme carbons are numbered according to the standard IUPAC convention. The reactive meso-carbons are labeled α, β, γ, and δ. Mycobilin exists as a pair of isomers with the formyl appended at either the heme C4 or C6. Staphylobilins are a pair of isomers with ring cleavage at, and loss of, either the β-meso (heme C10) or δ-meso (heme C20) carbon as formaldehyde. Expected exact masses are given. Exact masses measured by MS for heme and isomers of staphylobilin and mycobilin were within 5 ppm of the expected values for each compound reported.
By the early 2000s, however, it was apparent that many important Gram-positive pathogens that degrade host heme did not possess an HO-encoding gene in their genomes. A new family of heme-degrading proteins known as IsdGs was subsequently discovered, with representatives found in bacteria from both Gram-positive and Gram-negative phyla (11). IsdG family proteins are evolutionarily and structurally distinct from the well studied HOs (12, 13), and they yield different end products. Instead of biliverdin IXα and CO, the IsdG protein from Mycobacterium tuberculosis (known as MhuD) generates triply-oxygenated linear tetrapyrroles called mycobilins (Fig. 1) (14, 15). A formyl group remains appended to pyrrole ring A or B at the site of macrocycle cleavage, and an oxo group is generated on the pyrrole ring on the opposite side. Notably, no C1 product is released (16).
Although homologous to MhuD, the IsdG from Staphylococcus aureus degrades heme to yet a third set of products. The macrocycle is not cleaved at the α-meso- but rather at either the β- or δ-meso-carbon. Oxo groups are generated on both the carbon backbone and the pyrrole rings at the cleavage site, generating tetrapyrrole products known as staphylobilins (Fig. 1) (17). It was recently shown that a C1 product is indeed released by the S. aureus IsdG; however, quite unexpectedly, the major C1 product was determined to be formaldehyde (CH2O) instead of CO (18). Unlike CO, formaldehyde may be undetectable by animal immune systems, offering a potential selective advantage for heme-feeding pathogens that use IsdG-type enzymes (5, 19, 20). Mechanistically, the observation that CH2O instead of CO implies that verdoheme, the green intermediate coproduced with CO in HO-mediated and uncatalyzed heme degradation reactions (21), is not on IsdG's catalytic pathway. IsdG must therefore proceed via a different mechanism from the well studied HOs, in which the identity of any of its reaction intermediates, the relative timing of formaldehyde release and ring opening, and the origins of its distinctions with MhuD are not known.
Previous studies of IsdG reactivity have relied almost exclusively on UV/visible (UV/vis) spectroscopy for monitoring heme decomposition in the presence of a reductant (ascorbate) and O2 (13). Here, using the IsdG enzyme from S. aureus, the time course of the reaction of the IsdG-heme complex was probed using a combination of orthogonal analytical approaches designed to identify intermediates and to define the kinetics of the reaction steps. These included both conventional and stopped-flow UV-vis spectroscopy as well as high resolution in proteo mass spectrometry (MS). The latter method discontinuously measures changes in the mass of the protein-bound substrate in real time (22). We report for the first time the presence of intermediates appearing during the reaction of IsdG with ascorbate and O2, and we propose a likely pathway leading to the formation of staphylobilin that has features in common with both the MhuD and HO mechanisms.
Experimental Procedures
IsdG Purification, Expression, and Reconstitution with Heme
Recombinant IsdG with an N-terminal His6 tag (pET-15b) was expressed in Escherichia coli Tuner (DE3) cells (Merck/Novagen) grown in Terrific Broth supplemented with ampicillin (100 μg/ml). Protein was expressed and purified, as described previously (12), with some modifications. Briefly, cultures were grown at 37 °C in a shaker incubator (250 rpm) to mid-logarithmic phase (absorbance at 600 nm = 0.4). 1 mm isopropyl β-d-thiogalactopyranoside was added to induce protein expression, and the temperature was lowered to 20 °C. After 16 h, cell pellets were collected by centrifugation and stored at −80 °C.
Pellets were thawed/resuspended in Buffer A (50 mm potassium phosphate (KPi) buffer, pH 7.4, 600 μg/ml lysozyme, and 1 mg of DNase) and lysed by pulsed sonication on ice. The lysates were clarified by centrifugation, and supernatants were loaded onto a 20-ml PerfectPro nickel-nitrilotriacetic acid-agarose (5 PRIME) column. Pure protein was eluted in 3-ml fractions using an AKTA protein purification system (GE Healthcare) (20–500 mm linear gradient of imidazole in Buffer A, flow rate 2 ml/min). Eluted proteins were screened by SDS-PAGE, and pure fractions pooled/buffer-exchanged into Buffer A using SnakeSkin dialysis tubing (10,000 molecular weight cutoff (MWCO), ThermoScientific). For His tag cleavage, the dialyzed protein was incubated with His6-tagged tobacco etch virus protease (50:1 w/w) for 16 h at 4 °C with stirring. Tag cleavage was monitored via SDS-PAGE and MS (see below). Cleaved protein was loaded onto a 10-ml PerfectPro nickel-nitrilotriacetic acid-agarose column to remove the tobacco etch virus protease. The flow-through and a column wash (20 mm imidazole in Buffer A) were pooled and dialyzed into Buffer B (0.1 m KPi, pH 6.8).
The dialyzed IsdG was incubated with 1 eq of hemin chloride (Calbiochem) from a dimethyl sulfoxide (DMSO) stock and incubated overnight at 4 °C with stirring. Excess hemin was removed by centrifugation followed by further purification of the protein by size exclusion chromatography (Sephacryl S200 HR, GE Healthcare). Pure protein was dialyzed into Buffer A, concentrated to 10 mg/ml using an Amicon stirred cell concentrator (10,000 MWCO), frozen in liquid N2, and stored at −80 °C. All IsdG-heme concentrations are given as heme-bound monomer, where [heme] and [protein] were determined by the pyridine hemochrome and Bradford assays, respectively.
IsdG-Heme Reaction with O2 and Reducing Equivalents Monitored by UV/Vis Spectroscopy
To initiate the heme decomposition reaction, 10 μm IsdG-heme was treated in air with 10 mm sodium ascorbate (i.e. 1000 eq) in the presence of 100 units of catalase (50 mm KPi, pH 7.4, 22 °C). These conditions were known from prior work to lead to readily observable loss of the IsdG-heme chromophore. Ascorbate stocks were prepared immediately prior to use and maintained under N2. Spectral changes were monitored over time with a Cary600i spectrophotometer (Agilent Technologies). The absorbance at the heme Soret band maximum (411 nm) plotted versus time was fit by linear least squares regression analysis to exponential decay curves to obtain first-order rate constants (kobs).
All kinetic data plotting and fitting were carried out using KaleidaGraph 4.0 except where noted. All reported kobs values are averages of ≥3 measurements (error = ±1 S.D.).
IsdG-Heme Reaction with O2/Ascorbate Monitored by Time Resolved in Proteo Mass Spectrometry
Reactions were prepared exactly as above but at volumes allowing for multiple successive injections. Alternatively, reactions were prepared at elevated concentrations (50 μm IsdG-heme) to increase the concentrations and thereby the intensity of the signals emitted by intermediates. Reactions and automated analysis used a 1290 ultrahigh pressure (UPLC) series chromatography stack (Agilent Technologies) coupled directly to an electrospray-time of flight mass spectrometer (Bruker Micro-TOF). Rapid reverse-phase chromatography (5 min) was carried out with a PLRP-S 100 Å column (50 mm, 3 μm, flow rate 600 μl/min, autosampler temperature 20 °C). The gradient was as follows: 0.8 min, 20% B; 0.8–4.3 min, 20–95% B; 4.8 min, 20% B at 37 °C column compartment temperature; solvent A = 0.1% formic acid (FA, Sigma) in water (Burdick &Jackson) and solvent B = 0.1% FA in acetonitrile (ACN, Burdick & Jackson). No carryover of heme-associated products or protein was observed in blank runs carried out as controls. Electrospray conditions were as follows: drying gas 8.0 liters/min, dry temperature 180 °C, and capillary exit 150 V. Scan range was 200–3000 m/z. Data processing and analysis were performed using Bruker Data Analysis package 4.0. Mass spectra for the protein and heme products were concurrently obtained and used to verify identity/mass. Measured m/z values for all reported compounds and isomers were <5 ppm from calculated monoisotopic values.
IsdG-Heme Reaction with O2/Ascorbate Monitored by UV/Vis Stopped-flow Spectroscopy
Reactions were carried out at 20 °C on a Hi-Tech stopped-flow instrument (1.5-ms mixing time) with diode array detection (250–700 nm). Aerobic IsdG-heme (20 μm) was mixed 1:1 volume:volume with ascorbate in 50 mm KPi, pH 7.4, prepared immediately prior to use at varying concentrations under N2 (gas) from a 2 m stock. Additionally, the concentration of O2 was varied by equilibrating KPi buffer with O2/N2 gas mixtures and measuring the resulting [O2] with a Clark-type O2 electrode. The variable [O2] buffer was mixed with IsdG-heme equilibrated to air to reach the final [O2]. Spectra were measured over time at 1–100-ms intervals. Absorbance at selected wavelengths near the Soret and visible maxima was fit to one or more exponential curves using both the Hi-Tech data analysis software and KaleidaGraph 4.0, yielding values of kobs. Full spectra (300–700 nm) were fit in parallel to a two-step model via singular value decomposition using the SpecFit software. Reported rate constants are averages of three measurements (error = ±1 S.D.).
Shunt Reactions with H2O2 and Peracetic Acid
5 μm IsdG-heme in 50 mm KPi, pH 7.4 (2 ml), was rendered anaerobic by multiple cycles of headspace gas exchanges with argon carried out on a vacuum manifold and then brought into an anaerobic glove box (Coy). Reactions were initiated by addition of 1–5 eq of H2O2 or peracetic acid. Spectra were recorded every 2 min at 22 °C until they ceased changing. The anaerobic reactions were exposed to air, and changes to UV/vis spectra were subsequently measured. Samples for MS analyses were frozen in liquid N2.
Extraction of Tetrapyrrole Products
Three methods were applied to remove bound products from IsdG at the end of the heme-decomposing reaction. First, concentrated HCl was used to adjust the pH to 2 followed by addition of 2 volumes of 2-butanone. The mixture was vortexed and centrifuged, and the organic layer containing the extracted products was removed and washed with 150 mm NaCl. Second, 1 volume of a 1:1 H2O/ACN mixture with 0.1% trifluoroacetic acid (TFA) was added to the IsdG/product mixtures, followed by centrifugation to pellet the precipitated proteins. The supernatants were subsequently filtered through Centricon Plus spin filters (5 kDa, Millipore) at 4000 rpm. Finally, reactions were applied to a superclean C-18 solid phase extraction column (Supelco), and tetrapyrrole products were eluted two times with 200 μl of 50 mm KPi, pH 7.4 (flow rate 1 ml/min). Extracted products were lyophilized in a mini-SpeedVac.
Tetrapyrrole Product Analysis by Liquid Chromatography/Tandem Mass Spectrometry (LC-MS/MS)
Analyses were performed on an Agilent 1290 UPLC series chromatography instrument coupled to a 6538 UHD Q-TOF mass spectrometer (Agilent Technologies) operated in both positive and negative ion modes. Samples (15 μl) were separated on a Zorbax RRHD Eclipse Plus C-18 column (150 × 2.1 mm; 1.8 μm, Agilent Technologies) at 700 μl/min using the following conditions: 2 min, 5% B; 2–15 min, 5–90% B; 15–17 min, 90% B (50 °C), where solvent A = 0.1% FA in water and solvent B = 0.1% FA in ACN. Electrospray conditions in both targeted and auto-MS/MS modes were as follows: drying gas flow 12.0 liters/min at 350 °C, nebulizer 55 pounds per square inch, and capillary 3500 V, fragmentor 120 V, and skimmer 45 V. Scan range was 50–1300 m/z (auto-MS/MS) and 50–800 m/z (targeted MS/MS) with isolation width 4 m/z and an acquisition rate of 1 spectrum/s. The collision energy was fixed at 35 V in targeted MS/MS mode, whereas a linear voltage gradient was applied for molecules fragmented in auto-MS/MS experiments. Data acquisition and spectral analysis were performed using MassHunter (Qualitative Analysis version B.04.00, Agilent Technologies).
Quantification of C1 Products
IsdG-heme (200 μl) was incubated with ascorbate/air under the same conditions used for time-resolved UV/visible spectroscopy and in proteo MS (see above) and then analyzed for formaldehyde (the major C1 product previously detected) using Nash's reagent (see below and see Ref. 18). Reactions were filtered in a 3-kDa centrifuge concentrator to remove the IsdG protein and any tightly bound products. The filtrate was reacted 1:1 (v/v) with Nash's reagent (0.05 mm acetic acid, 0.02 m acetyl acetone, 2 m ammonium acetate). The retained protein (retentate) was applied to a superclean C-18 solid phase extraction column (Supelco) to remove any bound tetrapyrrole products (extracted retentate). The retentate, extracted retentate, and a whole reaction sample in which the protein had not been removed by filtration were all independently reacted with Nash's reagent, in addition to no-enzyme and no-ascorbate controls.
Samples were incubated at 37 °C for 30 min to allow acetyl acetone to couple to any formaldehyde that might be present. A set of formaldehyde standards (0–30 μm) was generated using incubation conditions identical to those above. The samples and standards were analyzed via HPLC on an Agilent 1100 series instrument with UV/vis detection (412 nm). For HPLC, a 150 × 5 mm Phenomenex Luna C-18 column was used at a flow rate of 1.5 ml/min. Elution conditions were as follows: 3 min, 5% B; 3–12 min, 5–95% B; 12–15 min, 95% B at 37 °C; solvent A = 0.1% TFA; solvent B = ACN + 0.1% TFA. A standard curve of the integrated peak area for the formaldehyde/acetyl acetone product (sharp peak, 7.1 min) was constructed from the formaldehyde standard samples, and the curve was used to calculate [formaldehyde] in each sample.
Results and Discussion
Little is known about the steps by which proteins from the IsdG family degrade heme. The reaction can be readily monitored via the disappearance of the intense UV/vis Soret band associated with the aromatic heme macrocycle (λmax (Soret) = 411 nm; ϵ = 91 mm−1 cm−1) (16–18). Heme degradation is typically initiated by adding excess ascorbate in the presence of air and H2O2-scavenging catalase to the IsdG-heme complex. Fig. 2A illustrates the subsequent changes in the heme UV/vis spectrum monitored over time for 30 min, when changes to the UV/vis spectra appear to cease (10 μm IsdG-heme, ascorbate, 50 mm KPi, pH 7.4, 22 °C). Despite the likelihood that the reaction occurs via multiple microscopic steps, the Soret band diminished over time in an apparent single exponential phase. This was fit to give a first-order rate constant (kSoret = 0.13 ± 0.03 min−1; all reported first-order rate constants are averages of ≥3 independent measurements; error = ±1 S.D.). No intermediates were detectable, and the final spectrum resembled those previously published for similar experiments (λmax = 412 nm and 460 nm shoulder) (17). The value for kSoret was insensitive to pH over pH 5–10 (data not shown), a pH range over which the reductant (pKa values of 4.2 and 11.6) is expected to exist predominantly in its singly deprotonated, anionic form. This insensitivity is despite the observation of a transition to an alkaline species with a distinct UV/vis spectrum in this pH range, which was ascribed to formation of the ferric-OH complex at high pH in the related IsdI enzyme (23). However, kSoret depended linearly on the initial ascorbate concentration (k = 10 ± 0.1 m−1 min−1, see Fig. 2B). This suggested that the reaction step that controls kSoret is likewise dependent on reducing equivalents and independent of whether the enzyme is in its acidic or alkaline form.
FIGURE 2.
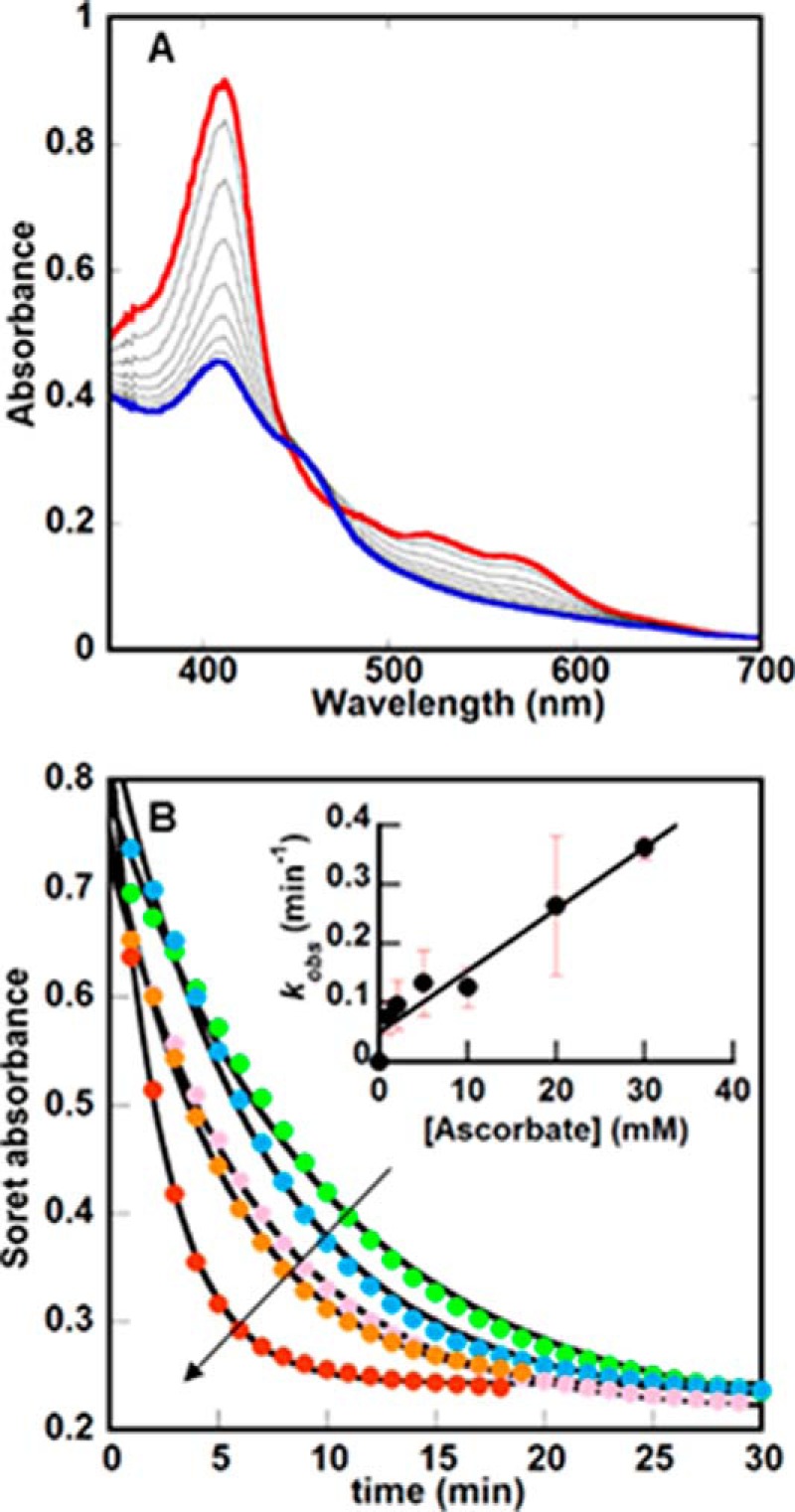
UV/visible spectroscopy illustrates broad changes to the IsdG-heme chromophore during the reaction with O2 and ascorbate. A, changes in the UV/vis spectrum of the heme bound to IsdG (10 μm) over time following the addition of 10 mm ascorbate (50 mm KPi, pH 7.4, 22 °C) are shown. 100 units of catalase are included in the reaction mixture to convert any unwanted H2O2 back to O2 and water. Spectra were measured every minute for 30 min; only spectra measured every 2 min from t = 0 (red) to t = 20 (blue) are shown. Notably, the final spectrum has maxima near 412 and 452 nm, similar to previously measured IsdG reactions but distinct from the UV/vis spectra for pure staphylobilins. (See Fig. 5A, below.) B, changes in the Soret band maximum (411 nm) versus time were plotted as a function of initial [ascorbate] (1, 5, 10, 20, and 30 mm), where the arrow indicates the direction of increasing [ascorbate]. Each curve was fit to a single exponential equation to obtain values for kobs, which in turn were used to determine the second-order rate constant k = 10 ± 0.1 m−1min−1 (inset).
Although a convenient probe of the reaction, conventional UV/visible spectroscopy has several drawbacks as a tool for discerning the mechanism. First, spectra for the starting material, several potential intermediates, and products likely overlap. Second, the acute intensity of the Soret absorbance is attributed to aromaticity in the heme macrocycle; kSoret is therefore expected to reflect predominantly the kinetic step involving loss of aromaticity (for example, scission of the macrocycle). The existence of other steps and intermediates may consequently be masked by monitoring the reaction at the Soret maximum. Finally, conventional UV/visible spectroscopy with manual mixing has relatively poor time resolution. Rapid or early events in the overall reaction, or sequential events with similar time constants, are not expected to be observed by this method.
We therefore sought means for monitoring the IsdG-mediated reaction that might be more sensitive to the presence of tetrapyrrole intermediates and therefore more powerful mechanistic probes. Time-resolved in proteo MS was of particular interest because the anticipated changes in mass associated with the reaction, including addition of three oxygen atoms (16 Da each) and loss of CH2O (30 Da) from the tetrapyrrole, are large. Moreover, the reaction is relatively slow (t½ (Soret) ∼5 min; 50 mm KPi, pH 7.4, 22 °C). Time-resolved MS therefore seemed ideally suited for monitoring IsdG intermediates and products, particularly ones that have less intense or overlapping absorbance in their UV/visible spectra relative to heme or which are too unstable to be extracted from the protein in unaltered form.
In proteo MS conditions were adjusted to allow quantitative tracking of tetrapyrroles bound to IsdG (Fig. 3A). The disappearance of a species with an exact mass/charge ratio (m/z) of 616.17 (heme b, Fig. 1) occurred in a single exponential kinetic phase with a rate constant k 616.17 = 0.52 ± 0.09 min−1 (Figs. 3B and 4A), four times the value of kSoret.
FIGURE 3.
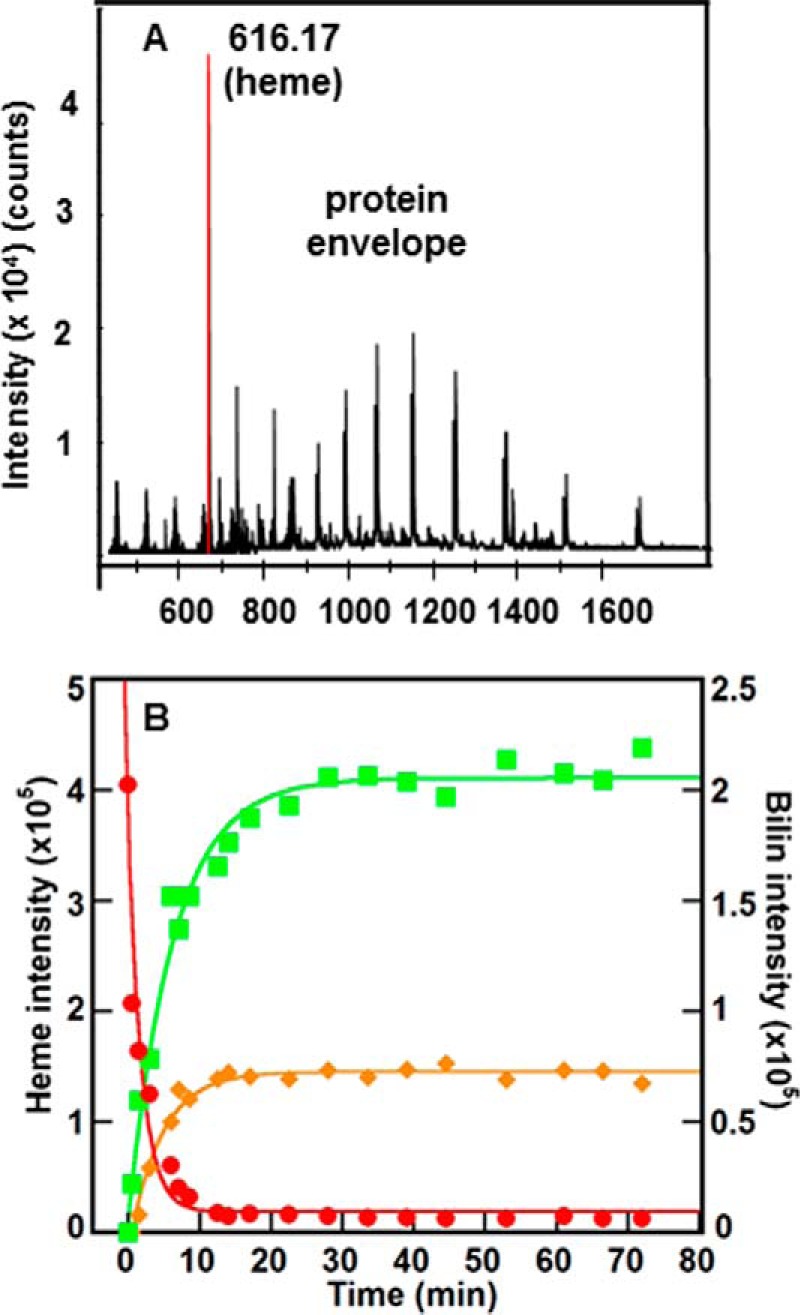
IsdG reaction was monitored using time resolved in proteo MS. A, mass spectrum was measured at t = 0, showing the heme (616.17 Da) and protein envelope. B, integrated intensities of the extracted ion chromatograms (m/z ± 0.01) measured over time for the species with exact masses 616.17 (red), 611.24 (green), and 599.24 (orange) Da. Intensities for the 616.17 Da species (heme) are given on the left y axis; intensities for the other two species are given on the right y axis. The data were fit to single exponential curves as described in the text. Data from a representative experiment are shown; reported rate constants are the average of three values. Conditions were the same as reported in Fig. 2A.
FIGURE 4.
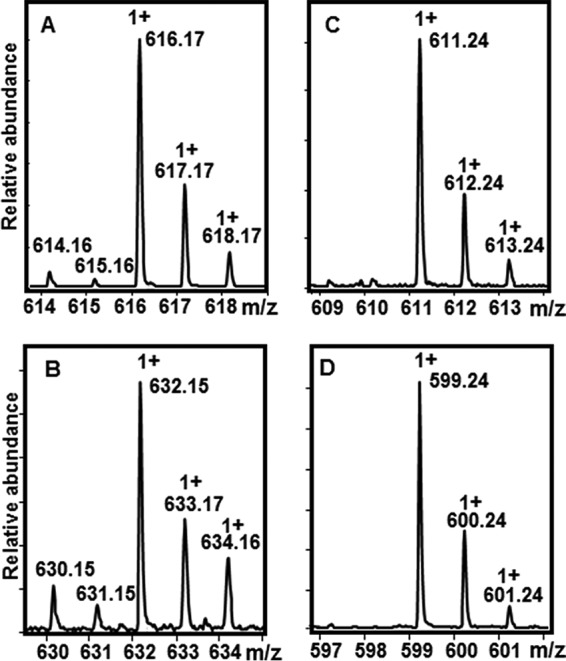
Isotopically resolved mass spectra of heme and its tetrapyrrole breakdown products. Spectra are reported from species isolated in the experiments shown in Fig. 3B. Monocationic species are designated +1. The presence of iron is reflected by the observation of two low intensity, low molecular weight isotopes at m/z = −1 and −2 relative to the most intense peak. The molecular ion for each tetrapyrrole species is (m/z) as follows: A, 616.17 (heme); B, 632.15 (heme + O); C, 611.24 (formyloxobilins); and D, 599.24 (staphylobilins).
In further contrast with the UV/vis data, at least three heme-derived species could be detected by time-resolved MS, starting with a species appearing transiently in positive ion mode at 0.5 min after initiation of the reaction. Reactions measured at 1.5 and 2 min did not contain this species, suggesting that it is relatively short lived. Its exact mass (632.15 m/z, is consistent with the addition of an oxygen atom (16.01 m/z) to the heme, possibly at one of the meso-carbons of the tetrapyrrole (Fig. 1). Even at concentrations nearing the saturation limit of the detector (50 μm IsdG-heme), too little of this species accumulated to allow for further characterization of its structure by collision-induced dissociation (CID) MS/MS fragmentation. However, the exact mass and isotope pattern measured at t = 0.5 min (Fig. 4B) are consistent with the expected formula for a meso-hydroxyheme (C34H32FeN4O5, [M]+). The [M]+ species was previously observed in prior studies of meso-hydroxyheme generated inside H2O2-treated metmyoglobin (24). Furthermore, the presence of iron in this molecule, as in the isotopically resolved mass spectrum for heme (Fig. 4A), is definitively reflected by the observation of two low intensity, low molecular weight isotopes (m/z = 630.15 and 631.15). Hence, this intermediate appears to correspond to heme with one oxygen atom incorporated. Although further in-depth characterization of the intermediate is needed, the available data are consistent with its proposed description as β- or δ-meso-hydroxyheme.
A second, iron-free species with an m/z of 611.24 (Fig. 4C) accumulated to a more significant level, forming with a first-order rate constant that was the same within error as kSoret: k 611.24 = 0.16 ± 0.03 min−1 (Fig. 3B). This species grew to substantial intensity before decaying very slowly over a period of >12 h. The final species detected by in proteo MS had the exact mass of staphylobilin (599.24 m/z, Fig. 4D), forming with a fitted first-order rate constant k 99.24 = 0.22 ± 0.05 min−1 (Fig. 3B).
Taken together, these results are consistent with the initial rapid conversion of heme to a short lived meso-hydroxyheme (632.15 Da) intermediate and then to a species resembling mycobilin (exact mass 611.24 Da, see Fig. 1). Macrocycle cleavage would presumably occur not at the heme α-meso-carbon but rather at the β- or δ-meso-carbons, with formyl and oxo groups generated on pyrrole rings B/C or A/D to make a formyloxobilin mixture. The similarity in the measured values for kSoret and k 611.24 suggested that loss of heme aromaticity is kinetically coupled to formation of this putative formyloxobilin. In the simplest interpretation of the data, both rate constants could be ascribed to the roughly concomitant opening (kSoret) and second oxygenation (k 611.24) of the heme macrocycle. The final observed tetrapyrrole appeared to be staphylobilin (599.24 Da). The comparable values for k 611.24 and k 599.24 (the same within error) and the failure of 611.24 intensity to diminish as the 599.24 grew argues against a precursor-product relationship between these two species. Instead, although a formyloxobilin would logically form prior to staphylobilin, the kinetic data suggest that the formyloxobilin and staphylobilin appear roughly concurrently.
To test this hypothetical model, the 611.24- and 599.24-Da species, both of which accumulated at the apparent end of the reactions shown in Figs. 2A and 3B (t = 30 min), were further characterized by UPLC-MS/MS. Species with m/z = 599.24 eluted cleanly as two UPLC peaks (at 9.6 and 9.9 min). Each yielded UV/vis (Fig. 5A) spectra and MS/MS fragment patterns (obtainable in both positive and negative ion modes) identical to those previously published for staphylobilin isomers β and δ (data not shown) (17).
FIGURE 5.
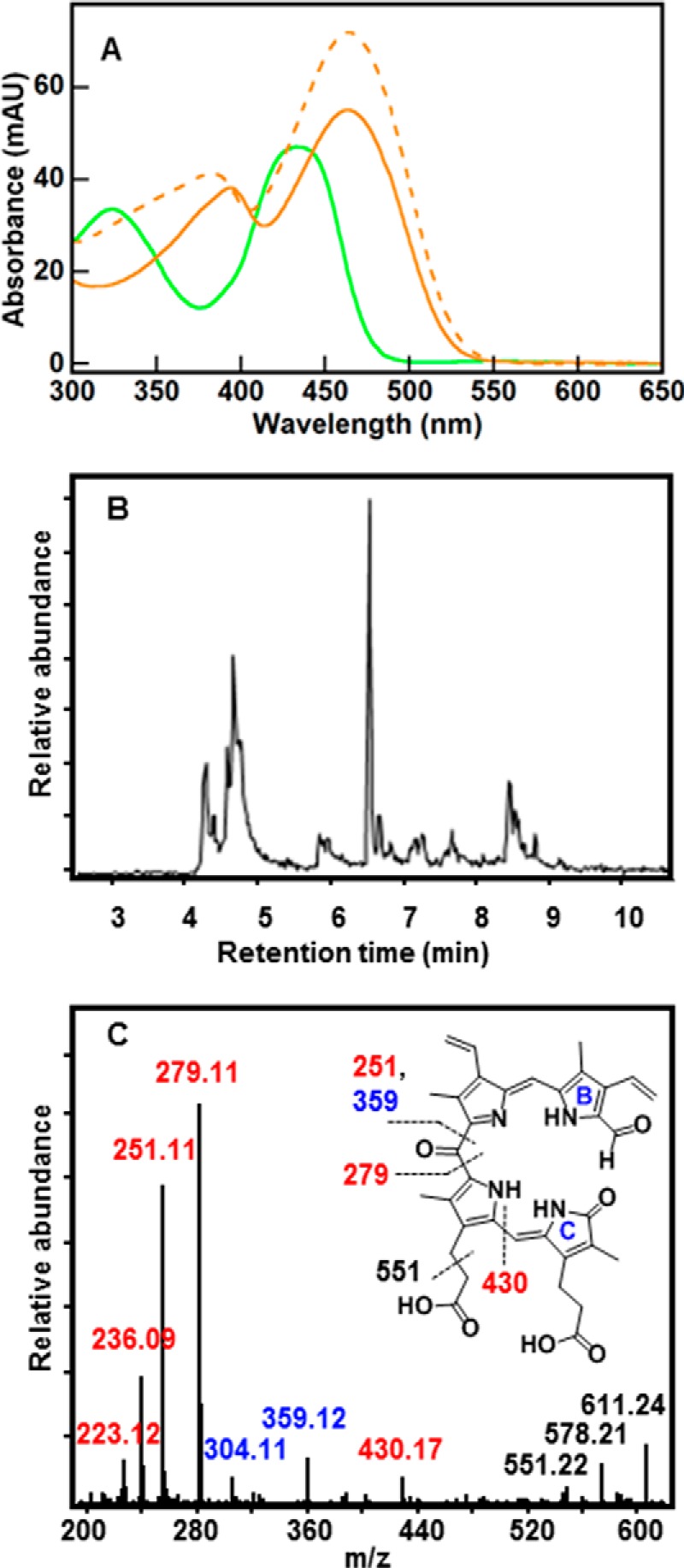
UV/vis and LC-MS reveal a series of formyloxobilin isomers that are distinct from staphylobilins. A, UV/vis spectrum for the formyloxobilin isomer mixture (green) has two broad peaks with maxima at 324 and 435 nm. Spectra for the δ- and β-staphylobilin isomers resolved by UPLC (dashed and solid orange lines, respectively) are shown for comparison. Consistent with prior work (17), these had peak maxima at 383 and 463 nm (δ-staphylobilin, the product of cleavage at the δ-meso-carbon) and 394 and 463 nm (β-staphylobilin). MAU, milliabsorbance units. B, extracted ion chromatogram for m/z 611.24 measured 30 min after initiating the IsdG-mediated heme decomposition reaction (10 μm IsdG-heme, 10 mm ascorbate, 50 mm KPi, pH 7.4, 22 °C, 100 units of catalase). C, CID MS/MS fragmentation pattern of the 611.24 ion eluted at 6.6 min is shown. The presence of signature ions m/z = 279.11, [M]+ and m/z = 251.11, [M]+ indicates a formyl group appended to either ring A or B (cleavage at the β- and δ-meso carbons cannot be distinguished). Ion labels are color-coded to match fragments containing formyl (red), oxo (blue), or both (black). Inset, fragmentation map of 10-formyl,11-oxobilin with indicated cleavage sites generated during CID.
By contrast, the UPLC trace for the species with m/z = 611.24 was far more complicated (Fig. 5B), and it was composed of several temporally resolved peaks. This result is perhaps less surprising in light of the four formyloxobilin isomers that might, in principle, be observed: two cleaved at the δ-meso-carbon, which is converted to a formyl group appended to ring A or D; and the analogous pair of isomers cleaved at the β-meso-carbon with the formyl group on ring B or C.
The most prominent LC peak (retention time = 6.6 min) was extensively characterized by CID MS/MS (Fig. 5C). The intensity of the data in negative ion mode was too low; therefore, the positive ion mode was analyzed. The most abundant fragments were generated by formation of cations containing both pyrrole rings A and B and an appended formyl group, with molecular cleavage occurring on either side of the meso-oxo group (m/z = 279.11 and 251.11, [M]+). If the formyl group had instead been associated with pyrroles C or D, the analogous pair of ions would have had predicted m/z = 267.11 and 241.11, [M]+. These masses are consequently highly diagnostic of the presence and location of the formyl group. Notably, although formylation of rings A/B versus C/D can be distinguished by MS/MS, resolving the highly symmetric β- and δ-formyloxobilin isomers requires parallel methods of characterization. The prominent 6.6-min peak therefore appeared to correspond to either 10-formyl,11-oxo-bilin or 20-formyl,19-oxo-bilin, or a mixture of the two.
Systematic MS/MS analysis of m/z 611.24 peaks eluting between 4 and 8 min confirmed that the fragments diagnostic of formyloxobilin (m/z = 279.11 and 251.11) were ubiquitous. Moreover, several of the same fragments as in Fig. 5C were detected in the peak at 4.8 min, although less prominently, in addition to fragments diagnostic of formylation at the C and/or D rings (m/z = 241.11 and 267.11, [M]+). Together, these results suggest that the m/z 611.24 species is a formyloxobilin that is present as more than one structural isomer (see Fig. 1).
To further explore the intermediacy of a meso-hydroxyheme, we attempted to isolate this species using either H2O2 or peracetic acid. Classic experiments with human HO-1 (hHO-1) showed that the ferric heme-HO complex converted to the α-meso-hydroxyheme within 4 min of the anaerobic addition of 1 eq of H2O2 (23 °C) (21). Exposure of this species to air brought about its rapid (within ∼5 s) and quantitative conversion to verdoheme and CO, where verdoheme (visible absorbance ∼690 nm) is an intermediate on the pathway to biliverdin-IXα. Peracids yielded neither verdoheme nor biliverdin-IXα, suggesting that a ferric heme-hydroperoxy species was an obligate hHO-1 intermediate.
Using the ferric IsdG-heme complex, the same experiments were repeated here. Following anaerobic treatment with either H2O2 or peracetic acid, the Soret peak diminished slightly, consistent with the possible formation of meso-hydroxyheme (Fig. 6). However, upon exposure of the oxidant-treated species to air, the UV/vis spectra changed little. In particular, the prominent absorbance band centered near 690 nm and known to be responsible for the characteristic green color of verdoheme (21) was not observed. MS analyses of the air-treated products revealed neither staphylobilin nor biliverdin IXα. Instead, the overall amount of heme appeared to diminish, and a very small amount of 611.24 m/z species formed specifically in the H2O2-treated sample. These results suggest that either the β-/δ-meso-hydroxyheme is not an intermediate in the IsdG-catalyzed reaction or that this intermediate forms and reacts in a manner that is distinct from its hHO counterpart.
FIGURE 6.
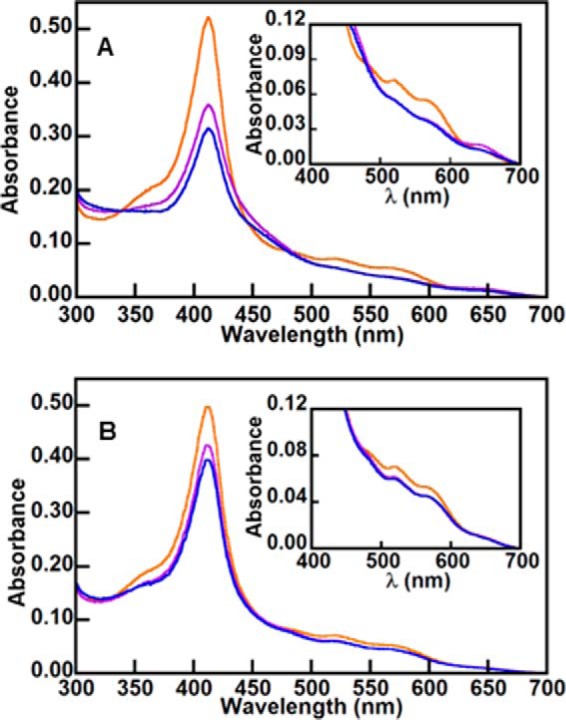
Sequentially added H2O2 and air (O2) do not generate the same products observed in hHO. UV/vis spectra of 5 μm IsdG-heme (red) and following the anaerobic addition of 5 eq of H2O2 (A) or peracetic acid (B) (purple). In each case, the sample was subsequently exposed to air, and the spectra in blue were recorded. (Insets, visible bands are shown on an expanded scale.) The UV/vis spectra indicate that, in contrast to hHO, verdoheme is not formed by the sequential addition of H2O2 and O2. MS analysis of the ultimate products confirmed the absence of verdoheme, biliverdin IXα, and staphylobilin. A small amount of 611.24 species was observed in the sample treated with H2O2.
The latter explanation is plausible because the heme environment in HOs and IsdGs is highly distinct (25, 26). In particular, the IsdG-bound ferric heme, uniquely, is profoundly distorted from planarity (ruffled) (12). The vicinity of the meso-carbons, unlike in HOs, is devoid of water or conserved polar residues. Moreover, although a mechanism leading from this intermediate to either the IsdG/hHO products can be drawn, only the hHO meso-hydroxyheme intermediate is expected to convert to verdoheme plus CO. The IsdG enzyme would therefore have to have some way of blocking the analogous conversion of a β- or δ-meso-hydroxyheme intermediate. Hence, inside the IsdG environment, the meso-hydroxyheme would almost certainly have to be structurally or electronically distinct from its hHO counterpart.
We next asked whether the higher resolution kinetic analysis afforded by stopped-flow UV/visible spectroscopy might provide evidence for early or other reaction intermediates. Indeed, like the time-resolved MS, stopped-flow experiments monitoring the reaction between aerobic solutions of IsdG-heme and ascorbate gave evidence for a multistep heme degradation process. Time traces for absorbances at the Soret (411 nm) (Fig. 7A) and charge transfer band (500–600 nm) (Fig. 7B) regions were both best modeled as sums of single exponential functions, indicating at least two detectable kinetic events.
FIGURE 7.
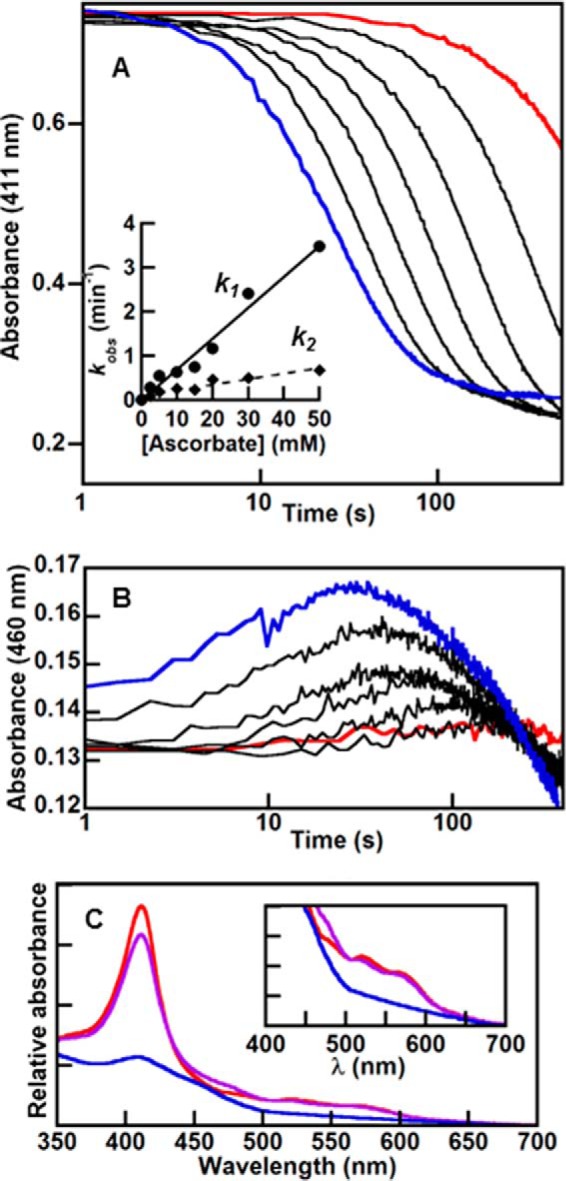
Stopped-flow kinetic traces at selected wavelengths present a detailed spectroscopic view of early events in heme degradation. Stopped-flow kinetic traces were measured at 411 nm (A) and 460 nm (B) for the reaction of IsdG-heme with varying [ascorbate] (10 μm IsdG-heme, 50 mm KPi, pH 7.4, 22 °C, 100 units of catalase, 285 μm O2). Final ascorbate concentrations were as follows: 2.5 (red), 20, 50, 100, 250, and 500 (blue) mm. Inset, each curve in A was fit to the sum of two exponentials to obtain values for k1 (circles) and k2 (diamonds). First-order rate were plotted as a function of [ascorbate] and fit to linear equations. The second-order rate constants determined from the slopes are 69 m−1 min−1 (k1) and 13 m−1 min−1 (k2). C, singular value decomposition of stopped-flow data measured over 300–700 nm were fit to an A → B → C model and used to obtain a simulated spectrum for the intermediate. The starting spectrum is in red, intermediate in purple, and end spectrum in blue. Inset, visible bands on an expanded scale.
Qualitatively similar results were obtained for both regions of the UV/vis spectrum. For the Soret (Fig. 7A) maximum, a fast phase (k1 = 0.63 ± 0.06 min−1) followed by a second slower decay phase (k2 = 0.26 ± 0.04 min−1; 10 mm ascorbate, 50 mm KPi, pH 7.4, 22 °C) could be readily modeled. The majority of the fast phase (t½ = 1 min) was complete before the Soret band diminished appreciably in intensity (t½ (Soret) = 5 min), consistent with the possible conversion of strongly absorbing heme to a slightly less strongly absorbing meso-hydroxyheme, as expected from work with hHO (21). Instead, most of the Soret intensity was lost in the slower second phase (k2), which dominates the kinetic trace at 411 nm and has a rate constant that is approximately the same as both kSoret and k 611.24. The full spectral data set was subsequently fit using singular value decomposition, yielding values for k1 and k2 and an intermediate component spectrum (Fig. 7C) that were consistent with analysis of the data at the Soret maximum.
Together, these results suggested that k2, kSoret, and k 611.24, measured by three different methods, are each predominantly controlled by the same reaction step. This step is most plausibly associated with the conversion of a meso-hydroxyheme intermediate to a formyloxobilin in which the macrocycle is cleaved and the iron has been released. Notably, neither k1 nor k2 exhibited a strong dependence on [O2], although each had a measurable linear dependence on ascorbate concentration (second-order rate constants for k1 and k2 are 69 and 13 m−1 min−1, respectively, see Fig. 7A, inset). This again indicated that the kinetic events described by these constants are rate-limited by the delivery of reducing equivalents (ascorbate).
In combination, these results suggest a model (Fig. 8) whereby the ferric IsdG-heme complex rapidly (t½ ∼1 min) converts to a (β/δ)-meso-hydroxyheme intermediate, which then more slowly (t½ ∼3 min) goes on to form a set of formyloxobilin isomers. A subpopulation of these converts rapidly to β/δ-staphylobilins. However, a sizable fraction stays unconverted and appears to remain associated with the protein in the formyloxobilin precursor state (∼55% based on integrated extracted ion chromatogram intensities measured by UPLC for the 611.24- and 599.24-Da species and assuming identical ionization efficiencies). Measurements of the product distribution over longer time periods indicated that the formyloxobilins and staphylobilins, which are known to degrade readily under ambient light (17), both decay over a period of ≥12 h.
FIGURE 8.

Summary of the kinetic scheme determined using in proteo MS and conventional time-resolved and stopped-flow UV/vis approaches. Approximate rate constants measured by each of these methods are given. Standard conditions used to obtain all first-order rate constants were 10 μm IsdG-heme, 10 mm ascorbate, 50 mm KPi, pH 7.4, 22 °C, 100 units of catalase.
If both formyloxobilin and staphylobilin species are present at the end of the IsdG reaction (as assessed by monitoring changes at the Soret maximum by UV/vis, Fig. 2A), why have only staphylobilins been isolated in previous analyses of IsdG (tetrapyrrole) products? We hypothesized that the staphylobilins observed in Fig. 3B arose from one, possibly isomer-dependent, group of formyloxobilin precursors that spontaneously released formaldehyde. The other group might be more stable when associated with IsdG. Release from the protein into the buffer medium could induce their spontaneous conversion to staphylobilin isomers and formaldehyde. In reaction mixtures from which the protein was extracted, only staphylobilins would be detected.
To address this hypothesis, IsdG reaction mixtures were fractionated and analyzed quantitatively for their formaldehyde and bilin content (Table 1). When the protein was filtered from the reaction mixture following 40 min of reaction time, much of the characteristic orange color of the tetrapyrrole product remained associated with the protein-containing retentate. MS analysis of this fraction showed that it contained both staphylobilin and formyloxobilin, with a roughly even ratio in their integrated extracted ion chromatogram peaks. By contrast, the filtrate contained no observable bilins.
TABLE 1.
Stoichiometries of formaldehyde produced following the IsdG-mediated breakdown of heme
The following reaction conditions were used: 10 μm IsdG-heme, air, 50 mm KPi, pH 7.4, 22 °C, 100 units of catalase. Reactions were analyzed 40 min following initiation with 10 mm ascorbate, at which time the UV/vis absorbance at the Soret band had appeared to stop changing.
| Sample/fraction | eq of CH2O |
|---|---|
| IsdG-heme (control) | 0 |
| Whole reaction mixture (untreated) | 0.4 ± 0.1 |
| Whole reaction mixture (solid phase extracted) | 0.9 ± 0.1 |
| Filtrate (3000 MWCO) | 0.5 ± 0.1 |
| Retentate (solid phase extracted) | 0.5 ± 0.1 |
Parallel analyses for formaldehyde showed that 0.5 (±0.1) eq could be detected in the filtrate and no CH2O in the washed, protein-containing retentate fraction. Following solid phase (C-18 column) extraction to remove protein-bound tetrapyrroles from the retentate, the protein component became colorless. An additional 0.5 (±0.1) eq of CH2O emerged in the extract. Moreover, the bilin component of the extract shifted strongly in favor of staphylobilins (93:7), according to their integrated extracted ion chromatograms. HPLC analysis further confirmed that, in reaction mixtures in which ACN was used to precipitate the protein or either 2-butanone or a C-18 column to extract the tetrapyrroles, only staphylobilins were detected.
Consistent with these and prior observations (18), a total of 0.9 (± 0.1) eq of CH2O were detected in whole reaction mixtures that had been passed over the solid phase extraction column. Collectively, these results suggest that some formaldehyde remained protein-associated, likely appended to the 611.24-Da species, until the tetrapyrrole was released from the protein. Notably, the addition of further equivalents of ascorbate and/or O2 did not accelerate conversion of this protein-associated species to staphylobilin and CH2O.
Conclusions
The results presented here are consistent with a model for the S. aureus IsdG reaction that has elements in common with both HO and MhuD. An initial intermediate, tentatively assigned as a mixture of β/δ-meso-hydroxyhemes, is analogous to the α-meso-hydroxyheme of HOs, although the two enzyme families carry the intermediate forward toward different fates.
The observation of a formyloxobilin as a second likely staphylobilin intermediate is chemically intuitive and satisfying from a biological perspective. As described above, the formyloxobilin known as mycobilin is the final reaction product of the IsdG homolog, MhuD (Fig. 1). Formyloxobilins are also observed as intermediates in plant chlorophyll degradation, as products of O2-mediated chlorin ring cleavage by the enzyme pheophorbide a oxygenase (27). Interestingly, a cytochrome P450 was recently discovered that oxidatively frees formaldehyde from the chlorophyll derivative (28), converting the formyloxobilin into a staphylobilin-like dioxobilin product (28). There is no obvious candidate P450 to carry out the analogous reaction in S. aureus. However, enzymes in the IsdG family belong to a class of cofactor-independent monooxygenases, where reactions very similar to the proposed oxidative deformylation are known to occur autocatalytically (29). Autocatalytic O2-dependent release of the formyl group would therefore appear to be plausible for members of the IsdG family. However, uncatalyzed conversion of the formyloxobilins to staphylobilins plus CH2O appears to occur when the former are released from the protein and into aerobic aqueous solution.
Why the MhuD product does not spontaneously release formaldehyde, and why the product of the S. aureus IsdG appears to generate formaldehyde from only some of its formyloxobilin population are not clear. Given the biological roles of the latter enzyme as both a catalyst and a sensor of S. aureus cellular heme status (30, 31), it may be that the tendency of IsdG to retain at least some of the formyloxobilin, which is slowly released to yield formaldehyde, has some yet-known biological role. It is likewise possible that the affinity of the formyloxobilin for IsdG may be isomer-dependent or that another protein is involved in facilitating formyloxobilin release. These are questions that will be addressed in future work.
Author Contributions
J. L. D. conceived and coordinated the study and wrote the paper. B. R. S., R. K., M. T.-L., A. I. C., and M. M. M. performed and analyzed experiments designed by B. B. and J. L. D. E. P. S. provided material and technical assistance and contributed to the preparation of the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We acknowledge Dr. Jonathan Hilmer for sharing technical expertise in mass spectrometry. We thank Garrett Moraski for helpful discussions. Dr. Sunil Ojha is thanked for contributing to the initiation of this project. The mass spectrometry facility at Montana State University receives funding from the Murdock Charitable Trust and National Institutes of Health Grant 5P20RR02437 of the CoBRE program.
This work was supported in part by National Institutes of Health Grants GM090260 and 5P20RR02437 of the CoBRE Program (to J. L. D.) and Grant RO1 AI069233 (to E. P. S.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HO
- heme oxygenase
- IPTG
- isopropyl β-d-thiogalactopyranoside
- MWCO
- molecular weight cutoff
- UPLC
- ultrahigh pressure liquid chromatography
- FA
- formic acid
- ACN
- acetonitrile
- CID
- collision induced dissociation
- hHO
- human heme oxygenase
- UV/vis
- UV/visible.
References
- 1.Galbraith R. (1999) Heme oxygenase: Who needs it? Proc. Soc. Exp. Biol. Med. 222, 299–305 [DOI] [PubMed] [Google Scholar]
- 2.Ortiz de Montellano P. R., and Wilks A. (2000) Heme oxygenase structure and mechanism. Adv. Inorg. Chem. 51, 359–402 [Google Scholar]
- 3.Wilks A. (2002) Heme oxygenase: evolution, structure, and mechanism. Antioxid. Redox Signal. 4, 603–614 [DOI] [PubMed] [Google Scholar]
- 4.Ryter S. W., Alam J., and Choi A. M. (2006) Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol. Rev. 86, 583–650 [DOI] [PubMed] [Google Scholar]
- 5.Maines M. D. (1997) The heme oxygenase system: a regulator of second messenger gases. Ann. Rev. Pharm. Toxicol. 37, 517–554 [DOI] [PubMed] [Google Scholar]
- 6.Pishchany G., and Skaar E. P. (2012) Taste for blood: hemoglobin as a nutrient source for pathogens. PLoS Pathog. 8, e1002535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pishchany G., McCoy A. L., Torres V. J., Krause J. C., Crowe J. E. Jr., Fabry M. E., and Skaar E. P. (2010) Specificity for human hemoglobin enhances Staphylococcus aureus infection. Cell Host Microbe 8, 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caignan G. A., Deshmukh R., Zeng Y., Wilks A., Bunce R. A., and Rivera M. (2003) The hydroxide complex of Pseudomonas aeruginosa heme oxygenase as a model of the low-spin iron(III) hydroperoxide intermediate in heme catabolism: C-13 NMR spectroscopic studies suggest the active participation of the heme in macrocycle hydroxylation. J. Am. Chem. Soc. 125, 11842–11852 [DOI] [PubMed] [Google Scholar]
- 9.Caignan G. A., Deshmukh R., Wilks A., Zeng Y., Huang H. W., Moënne-Loccoz P., Bunce R. A., Eastman M. A., and Rivera M. (2002) Oxidation of heme to β- and δ-biliverdin by Pseudomonas aeruginosa heme oxygenase as a consequence of an unusual seating of the heme. J. Am. Chem. Soc. 124, 14879–14892 [DOI] [PubMed] [Google Scholar]
- 10.Matsui T., Unno M., and Ikeda-Saito M. (2010) Heme oxygenase reveals its strategy for catalyzing three successive oxygenation reactions. Acc. Chem. Res. 43, 240–247 [DOI] [PubMed] [Google Scholar]
- 11.Skaar E. P., Gaspar A. H., and Schneewind O. (2004) IsdG and IsdI, heme-degrading enzymes in the cytoplasm of Staphylococcus aureus. J. Biol. Chem. 279, 436–443 [DOI] [PubMed] [Google Scholar]
- 12.Lee W. C., Reniere M. L., Skaar E. P., and Murphy M. E. (2008) Ruffling of metalloporphyrins bound to IsdG and IsdI, two heme-degrading enzymes in Staphylococcus aureus. J. Biol. Chem. 283, 30957–30963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu R., Skaar E. P., Zhang R., Joachimiak G., Gornicki P., Schneewind O., and Joachimiak A. (2005) Staphylococcus aureus IsdG and IsdI, heme-degrading enzymes with structural similarity to monooxygenases. J. Biol. Chem. 280, 2840–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chim N., Iniguez A., Nguyen T. Q., and Goulding C. W. (2010) Unusual diheme conformation of the heme-degrading protein from Mycobacterium tuberculosis. J. Mol. Biol. 395, 595–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graves A. B., Morse R. P., Chao A., Iniguez A., Goulding C. W., and Liptak M. D. (2014) Crystallographic and spectroscopic insights into heme degradation by Mycobacterium tuberculosis MhuD. Inorg. Chem. 53, 5931–5940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nambu S., Matsui T., Goulding C. W., Takahashi S., and Ikeda-Saito M. (2013) A new way to degrade heme: the Mycobacterium tuberculosis enzyme MhuD catalyzes heme degradation without generating CO. J. Biol. Chem. 288, 10101–10109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reniere M. L., Ukpabi G. N., Harry S. R., Stec D. F., Krull R., Wright D. W., Bachmann B. O., Murphy M. E., and Skaar E. P. (2010) The IsdG-family of haem oxygenases degrades haem to a novel chromophore. Mol. Microbiol. 75, 1529–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsui T., Nambu S., Ono Y., Goulding C. W., Tsumoto K., and Ikeda-Saito M. (2013) Heme degradation by Staphylococcus aureus IsdG and IsdI liberates formaldehyde rather than carbon monoxide. Biochemistry 52, 3025–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts G. P., Youn H., and Kerby R. (2004) CO-sensing mechanisms. Microbiol. Mol. Biol. Rev. 68, 453–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clark R. W., Lanz N. D., Lee A. J., Kerby R. L., Roberts G. P., and Burstyn J. N. (2006) Unexpected NO-dependent DNA binding by the CooA homolog from Carboxydothermus hydrogenoformans. Proc. Natl. Acad. Sci. U.S.A. 103, 891–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu Y., Moënne-Loccoz P., Loehr T. M., and Ortiz de Montellano P. R. (1997) Heme oxygenase-1, intermediates in verdoheme formation and the requirement for reduction equivalents. J. Biol. Chem. 272, 6909–6917 [DOI] [PubMed] [Google Scholar]
- 22.Bothner B., Chavez R., Wei J., Strupp C., Phung Q., Schneemann A., and Siuzdak G. (2000) Monitoring enzyme catalysis with mass spectrometry. J. Biol. Chem. 275, 13455–13459 [DOI] [PubMed] [Google Scholar]
- 23.Takayama S. J., Ukpabi G., Murphy M. E., and Mauk A. G. (2011) Electronic properties of the highly ruffled heme bound to the heme degrading enzyme IsdI. Proc. Nat. Acad. Sci. U.S.A. 108, 13071–13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sugiyama K., Highet R. J., Woods A., Cotter R. J., and Osawa Y. (1997) Hydrogen peroxide-mediated alteration of the heme prosthetic group of metmyoglobin to an iron chlorin product: evidence for a novel oxidative pathway. Proc. Nat. Acad. Sci. U.S.A. 94, 796–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schuller D. J., Wilks A., Ortiz de Montellano P. R., and Poulos T. L. (1999) Crystal structure of human heme oxygenase-l. Nat. Struct. Biol. 6, 860–867 [DOI] [PubMed] [Google Scholar]
- 26.Sugishima M., Sakamoto H., Higashimoto Y., Omata Y., Hayashi S., Noguchi M., Fukuyama K. (2002) Crystal structure of rat heme oxygenase-1 in complex with heme bound to azide. Implication for regiospecific hydroxylation of heme at the α-meso carbon. J. Biol. Chem. 277, 45086–45090 [DOI] [PubMed] [Google Scholar]
- 27.Hörtensteiner S., Wüthrich K. L., Matile P., Ongania K. H., and Kräutler B. (1998) The key step in chlorophyll breakdown in higher plants–cleavage of pheophorbide α-macrocycle by a monooxygenase. J. Biol. Chem. 273, 15335–15339 [DOI] [PubMed] [Google Scholar]
- 28.Christ B., Süssenbacher I., Moser S., Bichsel N., Egert A., Müller T., Kräutler B., and Hörtensteiner S. (2013) Cytochrome P450 CYP89A9 is involved in the formation of major chlorophyll catabolites during leaf senescence in Arabidopsis. Plant Cell 25, 1868–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fetzner S., and Steiner R. A. (2010) Cofactor-independent oxidases and oxygenases. Appl. Microbiol. Biotechnol. 86, 791–804 [DOI] [PubMed] [Google Scholar]
- 30.Reniere M. L., Haley K. P., and Skaar E. P. (2011) The flexible loop of Staphylococcus aureus IsdG is required for its degradation in the absence of heme. Biochemistry 50, 6730–6737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reniere M. L., and Skaar E. P. (2008) Staphylococcus aureus haem oxygenases are differentially regulated by iron and haem. Mol. Microbiol. 69, 1304–1315 [DOI] [PMC free article] [PubMed] [Google Scholar]