

Abstract
The mechanism of ω-6 polyunsaturated fatty acid oxidation by wild-type cyclooxygenase 2 and the Y334F variant, lacking a conserved hydrogen bond to the catalytic tyrosyl radical/tyrosine, was examined for the first time under physiologically relevant conditions. The enzymes show apparent bimolecular rate constants and deuterium kinetic isotope effects that increase in proportion to co-substrate concentrations before converging to limiting values. The trends exclude multiple dioxygenase mechanisms as well as the proposal that initial hydrogen atom abstraction from the fatty acid is the first irreversible step in catalysis. Temperature dependent kinetic studies reinforce the novel finding that hydrogen transfer from the reduced catalytic tyrosine to a terminal peroxyl radical is the first irreversible step that controls regio- and stereospecific product formation.
Keywords: dioxygenase, enzyme mechanism, fatty acid, isotope effect, prostaglandin
Introduction
Cyclooxygenases 1 and 2 (COX-1 and COX-2)2 are tyrosyl radical-utilizing hemoproteins with dioxygenase and peroxidase activities (1–3). Also known as prostaglandin H synthases, these structurally homologous enzymes are expressed by distinct genes, resulting in differences in cellular localization and regulation. COX-1 and COX-2 use O2 to oxidize arachidonic acid (AA) as well as linoleic acid (LA), its dietary precursor, by Equations 1 and 2. COX-1 has a smaller active site, resulting in greater specificity for AA over LA than seen in the larger, more promiscuous COX-2 (4).


Each dioxygenase reaction starts with initial hydrogen abstraction (from AA or LA) by a catalytic tyrosyl radical. Antarafacial O2 trapping (of AA• or LA•) leads to a peroxyl radical that reoxidizes the catalytic tyrosine in a second hydrogen transfer step, thus propagating catalysis. The prostaglandin G2 (PGG2) and acyclic hydroperoxide compounds (HPETEs and HPODEs) formed are processed by the enzyme's peroxidase activity, which consumes two reducing equivalents to generate prostaglandin H2 (PGH2) and acyclic hydroxylated compounds (hydroxyeicosatetraenoic acids (HETEs) and hydroxyoctadecadienoic acids (HODEs)) together with H2O. In vitro studies commonly use phenol as the reductant. Earlier work demonstrated simple, saturating behavior when phenol reacts with COX-2 (5) but not COX-1 where acceleration and inhibition of dioxygenase catalysis are observed upon raising the phenol concentration (6).
AA and LA bind to COX-2 in similar L-shaped conformations (Fig. 1). The reactive bisallylic C–H bond is thereby positioned close to the catalytic tyrosyl radical (Tyr-371•) (7, 8). Tyr-371• forms rapidly upon exposure of the enzyme containing Fe(III)(protoporphyrin IX (Por)) to hydroperoxide compounds present at trace levels in AA and LA preparations (9). A Fe(IV)=O(Por+•) intermediate, typical in heme peroxidases, has been implicated as the oxidant (10). This species resides approximately 5 Å away from Tyr-371, which hydrogen bonds to Tyr-334 (11, 12). By analogy, Mn(III)(Por)-reconstituted COX-2 likely reacts with hydroperoxide compounds via a Mn(V)=O(Por) intermediate to produce Tyr-371• (1).
FIGURE 1.

Overlaid structures of human COX-2 with bound AA (yellow) and LA (cyan). The Fe(III)(Por) was replaced by Co(III)(Por) in both proteins (7, 8).
Prior to findings outlined here, small substrate kinetic isotope effects (KIEs) were reported to characterize dioxygenase catalysis by COX-1 and COX-2 (13–15). Single turnover experiments with COX-1 indicated a deuterium KIE of ∼2 upon AA consumption that was indistinguishable from the deuterium KIE upon the steady-state bimolecular rate constant, Dkcat/Km(AA). This KIE is determined by steps beginning with AA association proceeding up to and including the first irreversible step. The results above, suggesting that initial hydrogen abstraction is the first irreversible step in COX-1, have been extended to COX-2 because of a similar Dkcat/Km(AA) of ∼2 extracted from state-state rate constants (4, 13, 15). This interpretation of the KIE makes it a challenge to explain the regio- and stereospecific reactivity of O2 during dioxygenase catalysis, leading to the hypothesis that distortion of the substrate radical causes accumulation of unpaired spin at a position accessible to O2 via a channel in the protein (16).
Since the works cited above, COX-1 and COX-2 have been reported to exhibit inverse deuterium KIEs and anomalous activation energies associated with Dkcat/Km(AA) as well as Dkcat. The latter has been proposed to reflect an inverse temperature dependence of the deuterium KIEs upon enzyme turnover at saturating concentrations of O2 (15). Such behavior might also arise from kinetic complexity due to competing steps with different temperature dependences instead of the reportedly lower thermal activation barrier to deuterium transfer than protium transfer (17).
In the present work, new data collected under physiologic conditions argue that a second hydrogen transfer, downstream of AA• or LA• formation and O2 trapping of the substrate radical, is the first irreversible step. In this reaction, the terminal peroxyl radical is reduced to the hydroperoxide product, and Tyr-371 is reoxidized to Tyr-371•. In the proposed mechanism, kinetic factors dictate regio- and stereospecific product formation during dioxygenase catalysis, a finding with implications for designing mechanism-based COX-2-specific inhibitors (18).
Experimental Procedures
General Methods
Chemicals were procured in the highest purity available and used as received unless noted. Sodium phosphate, sodium pyrophosphate, sodium chloride, sucrose, polyethylene glycol, and H2O2 were obtained from Fischer. Mn(III)(Por)Cl was obtained from Frontier Scientific. Hematin, soybean lipoxygenase, horseradish peroxidase, phenylmethanesulfonyl fluoride (PMSF), 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt, trimethyl phosphite, glycerol, Tween 20, 2-methoxyphenol, imidazole, phenol, 5(Z),8(Z),11(Z),14(Z)-eicosatetraenoic acid (AA), and 9(Z),12(Z)-octadecadienoic acid (LA) were obtained from Sigma-Aldrich. Perdeuterated linoleic acid (d31-LA) was obtained from Sigma-Aldrich Isotec in >98% purity or isolated as described (5) from an algal mixture of perdeuterated fatty acid methyl esters supplied by Cambridge Isotope Laboratories. Selectively labeled 13,13-d2-AA was a generous gift from Prof. Wilfred van der Donk (University of Illinois at Urbana-Champaign). Products of dioxygenase catalysis, PGG2, 11R-HPETE, 15S-HPETE, 9R-HPODE, and 13S-HPODE (see Equations 1 and 2), and subsequent peroxidase catalysis, prostaglandin H2 (PGH2), 11R-HETE, 15S-HETE, 9R-HODE, and 13S-HODE, were supplied by Cayman Chemicals. PGH2 is unstable in solution with respect to forming secondary prostaglandins PGE2, PGD2, and PGF2α, which were identified by comparison with authentic samples obtained from the same supplier. The oxidation products (above) were stored at −80 °C. LA and AA as well as proteins when necessary were stored at −30 °C inside an N2-filled glove box (MBRAUN LABmaster) containing <1 ppm O2. Ethanolic stock solutions of LA and AA were prepared prior to use in kinetic experiments. Importantly, no differences were discernible with different preparations of the substrates containing variable levels of HPETE or HPODE impurities. Such hydroperoxide species are required to generate Tyr-371• and make COX-2 fully active on the experimental time scale.
Protein Preparation and Characterization
N-terminal His6-tagged proteins were expressed recombinantly in baculovirus-transfected Sf9 insect cells following published protocols (19). Wild-type (WT), Y334F, and Y371F constructs (20) were sequenced and deposited at the Baylor College of Medicine Baculovirus/Monoclonal Antibody facility where proteins were expressed following amplification, titration, and Western blotting characterization (5).
The following procedure was used to purify COX-2 proteins with all manipulations conducted at 4 °C unless noted. Cell pellets were suspended (1 g/4 ml) in a pH 7.4 buffer consisting of 25 mm NaH2PO4, 100 mm NaCl, 20 mm imidazole, and 1 mm PMSF prior to lysis. Next, sonication was performed using 50% amplitude and cycles of 10 s on and 60 s off. The lysate was isolated and centrifuged for 1 h at 43,000 × g. The supernatant was discarded, and the membrane fraction resuspended in the above lysis buffer supplemented with a 0.1% (v/v) protease inhibitor mixture (Sigma) and Tween 20. The suspension was homogenized (Dounce), stirred on ice for 1.5 h, and then centrifuged for 1 h at 36,000 × g. The cell pellet was discarded, and the supernatant was incubated with nickel-nitrilotriacetic acid resin pretreated with the lysis buffer for 90 min before loading onto a nickel-nitrilotriacetic acid affinity column (2-cm diameter, 10-ml bed volume). Chromatography utilized a pH 7.4 buffer containing 25 mm NaH2PO4, 20 mm imidazole, 0.1% (w/v) Tween 20, and 100 mm NaCl at a flow rate of approximately 0.65 ml/min. The ionic strength (μ) was raised using 300 mm NaCl prior to eluting the protein with 25 mm NaH2PO4, 100 mm NaCl, 0.1% (w/v) Tween 20, and 200 mm imidazole. Aliquots were removed and assayed for peroxidase activity. This procedure involved adding hematin, H2O2, and 2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt to samples followed by colorimetric analysis of COX-2 peroxidase activity. Fractions with peroxidase activity were pooled, loaded onto a PD-10 desalting column (Bio-Rad), and eluted with 100 mm NaH2PO4 at pH 7.0 or 16 mm Na2H2P2O7 at pH 8.0 in the presence of 10% (v/v) glycerol and 0.1% (w/v) Tween 20, allowed isolation of imidazole-free solutions of COX-2 apoproteins lacking the Fe(III)(Por). Alternatively, apoproteins were purified by dialysis for >12 h against the pH 8.0 buffer above.
COX-2 apoproteins were reconstituted at 20 °C. Two equivalents of hematin or Mn(III)(Por)Cl were incubated with solutions of homodimeric forms of COX-2 for 15–20 min. Hydrated DE53 resin was then added, and after 5 min, the mixture was loaded onto a second PD-10 column to remove any unbound metalloporphyrin. The holoprotein was eluted with 100 mm NaH2PO4 buffered to pH 7.0 or 16 mm Na2H2P2O7 buffered to pH 8.0, each containing 10% (v/v) glycerol and 0.1% (w/v) Tween 20. Solutions of holoprotein were concentrated to ≤5 ml using an Amicon stirred cell equipped with a 30-kDa ultrafiltration membrane (Millipore), apportioned into 50–100-μl aliquots, and stored at −80 or −30 °C inside the freezer of the N2-filled glove box described above.
Protein samples were characterized by amino acid analysis (Texas A&M), polyacrylamide gel electrophoresis, and electronic absorption using a diode array spectrophotometer (HP8453a). The extinction coefficient for apo COX-2 (ϵ278 nm = 116,000 m−1 cm−1) (21) was used to compute extinction coefficients associated with the Soret bands of Fe(III)(Por)- and Mn(III)(Por)-bound COX-2 proteins, which ranged from ϵ407 nm = 129,000 m−1 cm− to ϵ375 nm = 66,000 m−1 cm− and ϵ470 nm = 42,000 m−1 cm−, respectively.
Assessing Active Enzyme Concentration
The concentration of active COX-2 was analyzed using extinction coefficients calculated as outlined above and a standard assay conducted at 30.0 ± 0.2 °C. Standard assay buffers contained kinetically saturating concentrations (>6 Km) of the following dioxygenase and peroxidase substrates: [AA] or [LA] = 50–100 μm, [O2] = 237 μm (from air), and [phenol] = >1.5 mm. Hematin or Mn(III)(Por) was added (1 μm) to prevent significant dissociation of the prosthetic group from the holoprotein. Initial rates were measured with an O2 electrode (YSI model 5300A) and compared with predetermined kcat values. As expected, Y371F COX-2 showed no activity due to the absence of Tyr-371•, whereas WT and Y334F COX-2 showed comparable kcat values (22).
Ascertaining the concentration of active enzyme is critical to the investigation of rate constants and deuterium KIEs at variable temperatures. In such experiments, changes in protein structure might result in loss of activity. Therefore, enzyme concentrations were checked by preincubating aliquots of stock solution at reaction temperatures between 5.0 and 50.0 °C for 3 min before performing standard assays. Under these conditions, no decrease in enzyme activity was detectable, and no correction for enzyme concentration was applied.
Steady-state Kinetics
Initial rates were measured at 30.0 ± 0.2 °C unless noted using the O2 electrode (above). All experiments were conducted at pH 8.0 in 0.016 mm sodium pyrophosphate (μ = 0.1 m) using known concentrations of AA or LA and O2 along with optimized concentrations of phenol, hydroperoxide initiator, and 1 μm hematin or Mn(III)(Por)Cl.
Kinetic measurements were initiated by injecting 1–10 μl of enzyme stock solution into 1.0–1.5 ml of stirring reaction solution contained inside a chamber with no headspace. Standard assays were used to check for changes in enzyme concentration over the course of experiments. Diminution in dioxygenase activity is clearly detected as COX-2 undergoes turnover. However, initial rates extracted from the most linear portion of the reaction progress curve after allowing ∼5 s for mixing varied in proportion to the enzyme concentration. This behavior is consistent with no significant loss of enzyme activity on the experimental time scale. In some cases, correction to the O2 uptake rate for background drift of the YSI electrode was required. In no instance was this correction >30% of the measured rate.
Kinetic parameters were obtained by fitting data to the hyperbolic Michaelis-Menten equation: V/[E] = kcat[S]/(Km + [S]) using Kaleidagraph 4.0 (Synergy Software). The concentration of fatty acid or O2, [S], was varied, whereas the co-substrate concentration was held fixed at kinetic saturation; i.e. >6 Km. The apparent maximal rate (Vmax) divided by the active enzyme concentration, [E], plotted versus [S] gave a hyperbolic trend that was fitted to obtain the unimolecular rate constant kcat and bimolecular rate constants kcat/Km(LA) or kcat/Km(AA) and kcat/Km(O2). Bimolecular rate constants were also extracted by linear fitting of data at concentrations below ½Km(S).
Studies of temperature-dependent kinetics at high and low [O2] were performed using protiated and deuterated substrates. The resulting substrate deuterium KIEs upon kcat and kcat/Km(O2) were determined over a 5.0 or 10.0 to 50.0 °C range. Data were analyzed by linear regression of natural log versus inverse temperature plots assuming the phenomenological Arrhenius expression: kcat or kcat/Km(O2) = A exp(−Ea/RT).
Viscosity Effects
Experiments were carried out to probe for diffusion-limited contributions to kcat/Km(O2) in accord with the Stokes-Einstein relation (23). Initial rates were examined at varying [O2] while [AA] or [LA] was held constant at 50–100 μm (>6 Km) in the presence of sucrose or polyethylene glycol, which served as micro- and macroviscosogens, respectively. An Ostwald viscometer allowed the relative viscosity, η/η0, to be measured and correlated to kcat/Km(O2) as a means of probing for diffusion-controlled steps (24, 25). The effect of added micro- or macroviscosogen was examined up to η/η0 = 4 to test whether O2-trapping of AA• or LA• and hydroperoxide product release steps contribute to kcat/Km(O2).
Product Analysis
Products of AA and LA oxidation were quantified in relation to the O2 consumed to confirm the reaction stoichiometries of Equations 1 and 2. Experiments were carried out by adding enzymes to 3–10-ml reaction solutions. After monitoring O2 uptake, 1 ml of acetic acid was added to quench the reaction. Acidification also neutralized the fatty acid oxidation products, allowing for extraction into CH2Cl2. Although COX-2 possesses peroxidase activity, 5% (v/v) trimethyl phosphite was added to ensure reduction of hydroperoxide products; i.e. PGG2 was converted to PGH2, HPETEs were converted to HETEs, and HPODEs were converted to HODEs (26). Solutions of these compounds were evaporated to a residue under a stream of N2 and then redissolved in a known volume of methanol. Samples were analyzed by ultra-high performance liquid chromatography (UPLC) on a Waters Acquity/Xevo-G2 system equipped with a reverse phase C18 column (HSS T3 1.8 μm, 2.1 × 100 mm) and quadrupole time-of-flight mass spectrometer operating in negative ion mode. The mobile phase (60% acetonitrile and 40% water), flowing at a rate of 0.5 ml/min, allowed separation of products, which were identified based on comparisons with authentic samples.
Competitive Deuterium KIEs
Apparent deuterium KIE upon LA oxidation, ap Dkcat/Km(LA), was measured competitively in 100 μm solutions of perprotiated (h31-LA) and perdeuterated (d31-LA) fatty acid combined in a 1:3 ratio. Experiments were performed with WT and Y334F COX-2 over a range of O2 concentrations by analyzing the ratio of deuterated to protiated products at low conversion (≤10%) relative to 100% conversion. The latter measurements used excess COX-2 or soybean lipoxygenase (26). Following the protocol above, UPLC-MS exposed two baseline resolved signals corresponding to the monodeprotonated isomers of h31-HODE and d31-HODE. This ratio was corroborated by analysis of electronic absorption at 235 nm, the wavelength of maximum absorption associated with the conjugated diene in the product.
Solvent Isotope Exchange
Experiments were undertaken to test whether the reactive hydrogen of the substrate undergoes exchange with the solvent in the presence and absence of Tyr-371•-containing COX-2. WT, Y334F, and Y371F proteins were incubated anaerobically in 3–10 ml of N2-saturated pH 8 buffered H2O containing 50 μm d31-LA. HPODEs in this substrate were present at trace levels sufficient to ensure full enzyme activation. 2 mm phenol was also added to protect enzymes from overoxidation by HPODEs. Samples were incubated at 4 °C under N2 for 12 h before reisolation of unreacted d31-LA. The procedure used was analogous to that described in under “Product Analysis.” Acid-quenched samples were extracted with CH2Cl2, evaporated to a residue, redissolved in methanol, and then analyzed by UPLC-MS. A sample containing all components but no protein was analyzed as a second type of control for detecting spurious isotope exchange.
Results
Recently, Danish et al. (5) demonstrated that the dioxygenase activity of COX-2 could be analyzed independently of the peroxidase activity of the enzyme at sufficiently high concentrations of phenol. Such investigations are not possible with COX-1 where rate acceleration is followed by inhibition upon adding phenol (6). However, rates of dioxygenase catalysis were found to saturate hyperbolically upon increasing the phenol concentration to 1.0–3.0 mm with Fe(III)(Por)-containing WT COX-2 and Y334F COX-2. With these enzymes, trace hydroperoxide impurities present in AA or LA were sufficient to observe optimal rates of turnover. The lower peroxidase activity of Mn(III)(Por)-WT COX-2 eliminates the requirement of added phenol. Even in the presence of 3–10 μm HPODE or HPETE needed to optimize initial rates, no phenol dependence was detected. Furthermore, the inhibition of dioxygenase activity by phenol was undetected in all forms of COX-2 examined in this study.
LA Oxidation Kinetics
Steady-state rate constants for LA oxidation were determined for WT COX-2 and the Y334F variant, which lacks a conserved hydrogen bond to Tyr-371• or Tyr-371 (7, 8, 11, 12). In Figs. 2 and 3, the apparent rate at kinetically saturating co-substrate concentration (>6 Km) is given as V/[E] and plotted versus varied substrate to determine kcat/Km and kcat as outlined under “Experimental Procedures.”
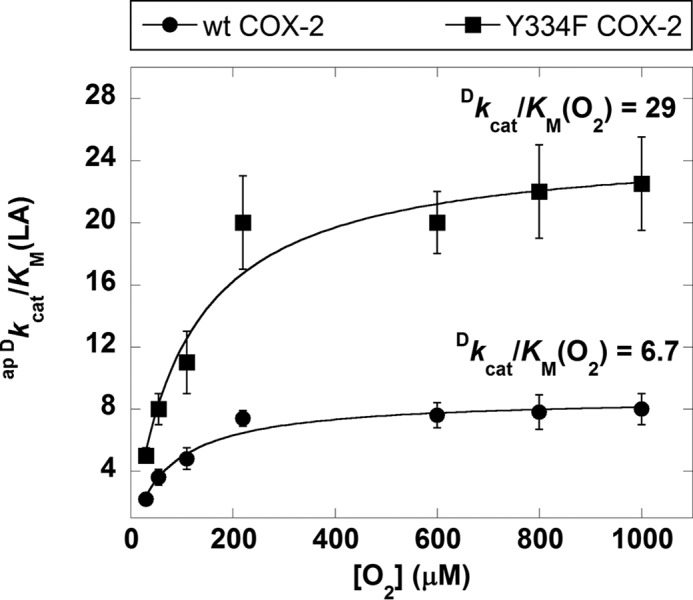
FIGURE 2.

Initial rates of O2 consumption used to determine Dkcat/Km(O2) for dioxygenase catalysis by WT (circles) and Y334F (squares) COX-2. The KIE was extracted from data independently fitted to Michaelis-Menten expressions. The [O2] was varied, and [h31-LA] or [d31-LA] was maintained at 100 μm (i.e. >6Km; see Table 2) in sodium pyrophosphate (0.016 m) at 30 °C, pH 8.0, and μ = 0.1 m as detailed under “Experimental Procedures.”
FIGURE 3.

Initial rates of O2 consumption used to determine the non-competitive Dkcat/Km(LA) for WT (circles) and Y334F (squares) COX-2. The KIE was extracted from data independently fitted to Michaelis-Menten expressions. The [h31-LA] or [d31-LA] was varied, and [O2] was maintained at 1.0 mm (i.e. >6Km; see Table 2) in sodium pyrophosphate (0.016 m) at 30 °C, pH 8.0, and μ = 0.1 m as detailed under “Experimental Procedures.”
Rate constants summarized in Table 1 show that WT and Y334F COX-2 exhibit similar kcat, kcat/Km(AA), and kcat/Km(LA). Comparisons of AA with LA require correction by a factor of 2 for the difference in O2 equivalents consumed in Equations 1 and 2. Quantification of dioxygenase products in tandem with measurements of O2 uptake confirmed the stoichiometry for formation of PGH2 as the major product of AA oxidation and HODEs as the products of LA oxidation. The kcat/Km(O2) was, however, an order of magnitude greater for AA than LA due to a reduction in Km(O2).
TABLE 1.
Rate constants for oxidations of AA and LA at 30 °C
Limiting rate constants are quoted with ±1 S.E. in parentheses.
| COX-2 |
||
|---|---|---|
| WT | Y334F | |
| h31-LA | ||
| kcat (s−1) | 9.4 (0.8) | 8.0 (0.8) |
| kcat/Km(h31-LA) (μm−1 s−1) | 1.6 (0.2) | 1.4 (0.2) |
| kcat/Km(O2) (μm−1 s−1) | 0.050 (0.010) | 0.047 (0.008) |
| d31-LA | ||
| kcat (s−1) | 0.44 (0.03) | 0.27 (0.02) |
| kcat/Km(d31-LA) (μm−1 s−1) | 0.15 (0.02) | 0.050 (0.006) |
| kcat/Km(O2) (μm−1s−1) | 0.0075 (0.0007) | 0.0016 (0.0002) |
| h39-AA | ||
| kcat (s−1) | 16.7 (2.0) | 8.5 (1.0) |
| kcat/Km(AA) (μm−1 s−1) | 5.9 (0.5)a | 3.0 (0.3) |
| kcat/Km(O2) (μm−1 s−1) | 1.1 (0.4) | 0.7 (0.2) |
a Km(h39-AA) is close to the detection limit; thus, kcat/Km(h39-AA) should be viewed as an upper limit.
Deuterium KIEs on LA Oxidation
The apparent rate constants for consumption of h31-LA versus d31-LA afford deuterium KIEs that increase with [O2] until a limiting Dkcat/Km(LA) is attained. Most measurements were made competitively by mixing the substrate isotopologues and analyzing the h31-HODE to d31-HODE product ratios after reduction of h31- and d31-HPODEs following a published procedure (26). Apparent competitive deuterium KIEs upon LA oxidation, ap Dkcat/Km(LA), were found to increase from subsaturating to saturating [O2] in Fig. 4. This behavior is in line with the apparent deuterium KIEs determined non-competitively by measuring O2 uptake rates with h31-LA or d31-LA (see Table 1).
FIGURE 4.
Results of UPLC-MS analyses used to determine competitive deuterium KIEs upon LA consumption in sodium pyrophosphate (0.016 m) at 30 °C, pH 8.0, and μ = 0.1 m as detailed under “Experimental Procedures.” Results for WT (circles) and Y334F (squares) COX-2 are shown at subsaturating [O2] (below ½Km(O2)) and at saturating [O2] (above 6Km(O2)). All solutions initially contained h31-LA and d31-LA mixed in a ratio of 1:3, respectively, at a concentration of 100 μm. The h31-HODE:d31-HODE ratio at <10% reaction conversion was taken relative to the h31-HODE:d31-HODE ratio at 100% conversion to determine the ap Dkcat/Km(LA). These apparent KIEs were fitted to the Michaelis-Menten equation to obtain the limiting competitive Dkcat/Km(LA), which is compared with the corresponding Dkcat/Km(O2) determined in Fig. 2. Error bars are 1 S.D.
Remarkably, the Dkcat/Km(LA) values for WT and Y334F COX-2 are equivalent to the deuterium KIEs upon bimolecular rate constants for O2 uptake, Dkcat/Km(O2), determined under non-competitive conditions. This unusual deuterium KIE upon O2 consumption arises from retention of the hydrogen abstracted from LA at Tyr-371; solvent exchange with this residue is slow on the time scale of enzyme turnover (see below). The observed equivalence of Dkcat/Km(LA) to Dkcat/Km(O2) is consistent with a common irreversible hydrogen transfer in WT and Y334F COX-2 after LA and O2 enter the catalytic cycle.
Table 2 consists of data obtained with two forms of COX-2 containing a Fe(III)(Por) or Mn(III)(Por) prosthetic group. The WT COX-2 exhibits a Dkcat of ∼21, which is 2–3 times larger than Dkcat/Km(LA) and Dkcat/Km(O2). These results, together with reported temperature dependences of Dkcat and Dkcat/Km(O2), expose a change in the irreversible hydrogen transfer step as the [O2] is raised (5). In contrast, Y334F COX-2 exhibits a Dkcat of ∼30 that is indistinguishable from Dkcat/Km(LA) and Dkcat/Km(O2) within the error limits.
TABLE 2.
Deuterium KIEs upon LA oxidation at 30 °C
Limiting deuterium KIEs are quoted with ±1 S.E. in parentheses.
| COX-2 | WT | WT(Mn(III))a | Y334F |
|---|---|---|---|
| Dkcatb | 21.4 (2.3) | 20.0 (3.0) | 29.6 (3.6) |
| Dkcat/Km(LA)b | 10.6 (1.9) | 8.0 (1.0) | 28.0 (3.4) |
| Dkcat/Km(LA)c | 8.7 (0.8) | 7.5 (1.0) | 25.0 (3.2) |
| Dkcat/Km(O2)b | 6.7 (1.4) | 11.0 (2.6) | 29.4 (4.2) |
a Mn(III)(Por)-reconstituted WT COX-2.
b Calculated from non-competitively determined rate constants.
c Determined competitively by UPLC-MS analysis of the products at <10% of the expected [O2] consumed as described under “Experimental Procedures.”
The large [O2]- and [LA]-independent deuterium KIEs implicate a common irreversible and rate-controlling step for Y334F COX-2 turnover at all substrate concentrations. Temperature studies of Dkcat and Dkcat/Km(O2) described below also support a common irreversible step. In addition, competitive oxygen-18 KIEs upon kcat/Km(O2), reflecting steps that begin with O2 encounter and lead up to and include the first irreversible step, are indistinguishable for WT and Y334F COX-2 catalysis with LA (1.0133–1.0156) and AA (1.0194–1.0205). These results, which correlate the magnitude of 18O KIE to the extent of peroxyl radical reduction in the transition state (27, 28), suggest common irreversible steps in WT and Y334F COX-2 at physiological [O2] (see below).
Solvent Isotope Exchange
Under anaerobic conditions, hydroperoxide-activated WT and Y334F COX-2 containing Tyr-371• catalyze exchange of a single protium from H2O into d31-LA. Data obtained as outlined under “Experimental Procedures” reveal the disappearance of the signal at m/z = 310 due to d31-LA deprotonated at the terminal carboxylate, and growth of a signal with m/z = 309, corresponding to h1d30-LA in the same anionic state. Control experiments with hydroperoxide-treated Y371F COX-2 and without added protein show no protium incorporation into d31-LA. In contrast, Tyr-371•-containing forms of COX-2 catalyze solvent isotope exchange into the substrate under anaerobic conditions as shown inFig. 5.
FIGURE 5.

Isotope exchange between d31-LA and H2O is mediated by WT and Y334F COX-2 enzymes (spectra on right) but not the Y371F mutant or in O2-free solutions (spectra on left). Solutions were incubated for equivalent times (∼12 h) at 4 °C, pH 8.0, and μ = 0.1 m as described under “Experimental Procedures.” In the experiments labeled controls 1 and 2, the catalytic Tyr-371• required to abstract hydrogen from the substrate is absent.
Isotope exchange from H2O into d31-LA takes several hours in the presence of WT and Y334F COX-2 because the active site is not accessible to the solvent. No exchange occurs on the same time scale in the presence of O2. Therefore, the results in Fig. 5 indicate that hydrogen transfer between Tyr-371• and LA is thermodynamically reversible but do not address the kinetic reversibility of this reaction in the presence of O2.
Deuterium KIE upon AA Oxidation
In view of the large deuterium KIEs associated with LA-dependent O2 consumption, Dkcat/Km(O2) was examined for AA using unlabeled (h39-AA) and selectively labeled (13,13-d2-AA) substrates. No artificial burst in O2 uptake was detectable with 13,13-d2-AA, and initial rates were identical when this material was used as received and after preincubation with WT COX-2. These results indicate that any unlabeled material present in the 13,13-d2-AA has no impact on the deuterium KIE.
Dkcat/Km(O2) and Dkcat were determined from the data in Fig. 6. Three independent trials were performed by varying [O2], and the resulting rates were analyzed as outlined under “Experimental Procedures.” Linear regression analysis at [O2] < ½Km(O2) gave a Dkcat/Km(O2) of 17.4 ± 3. Data obtained over a range of [O2] and fitted to the Michaelis-Menten equation indicate a Dkcat/Km(O2) of 18.6 ± 3.1 and a Dkcat of 3.1 ± 0. As discussed below, the large Dkcat/Km(O2) is most readily attributed to irreversible hydrogen transfer from Tyr-371 to a peroxyl radical formed after O2 trapping of the terminal carbon radical intermediate (29). Evidently, this rather large deuterium KIE escaped detection in earlier studies because saturating [O2] was used in experiments (13–15, 29–32) rather than physiologic [O2],which is approximately 50 μm (33).
FIGURE 6.

Initial rates of O2 consumption in the presence of 50 μmh39-AA (squares) or 13,13-d2-AA (circles) used to determine Dkcat/Km(O2) for dioxygenase catalysis by WT COX. The KIE was extracted from data independently fitted to Michaelis-Menten expressions and by linear regression (see inset). In each experiment, the [O2] was varied from subsaturating (below ½Km(O2)) to saturating (above 6Km(O2)) in sodium pyrophosphate (0.016 m) at 30 °C, pH 8.0, and μ = 0.1 m as detailed under “Experimental Procedures.”
Probing Viscosity Effects on O2 Consumption
The kinetics of AA and LA oxidation analyzed at variable viscosities failed to provide evidence of diffusion-controlled contributions to kcat/Km(O2). No change in the bimolecular rate constant for O2 uptake was discernible with AA or LA in the presence of micro- and macroviscosogens. The effect of relative viscosity as high as 4 was examined to exclude irreversible O2 trapping and hydroperoxide product release steps (34). In the respective cases, kcat/Km(O2) would be expected to decrease and increase with increasing η/η0.
Temperature Dependence of Deuterium KIEs
The temperature dependences of Dkcat and Dkcat/Km(O2) were measured for AA and LA using WT and Y334F COX-2. Data published for WT COX-2 (5) are compared with those determined for the Y334F variant in Fig. 7. These two enzymes exhibit similar competitive 18O KIEs as described above with each fatty acid substrate, suggesting that the transition state is the same for the step that limits kcat/Km(O2) (28, 35). This result is consistent with a common step giving rise to Dkcat/Km(O2) when the hydrogen initially abstracted from the substrate is retained at Tyr-371 and followed by Tyr-371• formation via irreversible hydrogen transfer after O2 enters the catalytic cycle.
FIGURE 7.

Determination of the temperature dependence of Dkcat/Km(O2) and Dkcat for dioxygenase catalysis by Y334F COX-2 at [LA] = 100 μm (i.e. >6 Km) in sodium pyrophosphate (0.016 m), pH 8.0, and μ = 0.1 m as detailed under “Experimental Procedures.” Data corresponding to kcat/Km(O2) determined with h31-LA (squares) and d31-LA (circles) and those corresponding to kcat determined with h31-LA (diamonds) and d31-LA (crosses) are shown. Error bars are 1 S.D.
Previous findings on LA oxidation by WT COX-2 indicated a change in the first irreversible step upon increasing [O2] from below Km(O2) to >6Km(O2) (5). The activation energy (Ea) associated with kcat/Km(O2) and kcat was determined for h31-LA and d31-LA reacting with Y334F COX-2. In contrast to WT COX-2, the variant shows indistinguishable deuterium KIEs upon kcat, kcat/Km(LA), and kcat/Km(O2) with each approaching 30 at 30 °C. This indication of kinetically simple behavior where the rate constant for dioxygenase catalysis is controlled by a common irreversible step is also consistent with the negligible ΔEa associated with Dkcat = 2.7 ± 0.7 kcal mol−1 and Dkcat/Km(O2) = 2.2 ± 0.6 kcal mol−1 derived from data at saturating and subsaturating [O2], respectively, where Ea(d31-LA) = 9.4 ± 0.7 or 7.9 ± 0.3 and Ea(h31-LA) = 6.7 ± 0.7 or 5.7 ± 0.5. At low [O2], where results reveal irreversible hydrogen transfer from Tyr-371 to an O2-derived peroxyl radical, Y334F COX-2 exhibits a ΔEa that is ∼2 times smaller than that reported for the WT enzyme where ΔEa = 5.0 ± 0.8 kcal mol−1 and AH/AD is much more inverse (5). These results must be reconciled with the proposal of a common irreversible step in WT and Y334F COX-2 under conditions where the enzymes exhibit indistinguishable competitive 18O KIEs and Dkcat/Km(O2) equal to Dkcat/Km(LA).
The availability of 13,13-d2-AA allowed Dkcat and Dkcat/Km(O2) to be examined over a limited temperature range in Fig. 8. Experiments were conducted at a [phenol] of ∼2 mm where optimal dioxygenase activity was observed. The parallel lines in the Arrhenius plots indicate normal temperature dependences of Dkcat and Dkcat/Km(O2).
FIGURE 8.

Determination of the temperature dependence of Dkcat/Km(O2) and Dkcat for dioxygenase catalysis by WT COX-2 at saturating [AA] = 50 μm (i.e. >6 Km) in sodium pyrophosphate (0.016 m), pH 8.0, and μ = 0.1 m as detailed under “Experimental Procedures.” Data corresponding to kcat/Km(O2) determined with h39-AA (squares) and 13,13-d2-AA (circles) and those corresponding to kcat determined with h39-AA (diamonds) and 13,13-d2-AA (crosses) are shown. Error bars are 1 S.D.
Analyzing rate constants associated with WT COX-2 oxidizing AA at subsaturating and saturating [O2], respectively, indicate that Ea(13,13-d2-AA) = 15.2 ± 2.0 and 15.4 ± 0.5 kcal mol−1 and Ea(h39-AA) = 13.2 ± 1.4 and 13.9 ± 0.9 kcal mol−1. Subtracting Ea for labeled AA from Ea for unlabeled AA results in a ΔEa of 2.0 ± 2.4 at low [O2] and 1.5 ± 1.1 kcal mol−1 at high [O2]. These results contradict a recent report by Wu et al. (15) in which deuterium KIEs were found to vary inversely with temperature at pH 7.0, resulting in Ea(h39-AA) > Ea(13,13-d2-AA). Importantly, the same source of 13,13-d2-AA used earlier (13, 15) was used in the present study. In the earlier study, no attention was devoted to optimizing the [phenol] required to observe maximal rates at different temperatures. This oversight might cause kinetic complexity that results in the aberrant ΔEa.
Data reported previously (5) and in this study are compiled in Table 3. Arrhenius analyses suggest a higher thermal barrier to the oxidation of AA than the oxidation of LA. As discussed below, these observations could reflect a greater energy barrier due to reorganization of AA in the active site of COX-2 as well as a mechanism that involves additional pre-equilibrium steps before the first irreversible hydrogen transfer step that forms PGG2 and regenerates Tyr-371•.
TABLE 3.
Activation parameters for AA and LA oxidation by WT and Y334F COX-2
Values in parentheses reflect ±1 S.E.
| Ea(H)a | Ea(D)a | AHb | ADb | |
|---|---|---|---|---|
| WT COX-2 + AA | ||||
| kcatc | 13.9 (0.9) | 15.4 (0.6) | 2.3 (5.5) × 1011 | 9.1 (4.1) × 1011 |
| kcat/Km(O2)d | 13.2 (1.4) | 15.2 (2.0) | 8.6 (30) × 1015 | 8.5 (13) × 1015 |
| WT COX-2 + LAe | ||||
| kcatc | 9.1 (0.2) | 9.1 (0.2) | 3.6 (1.2) × 107 | 1.8 (1.1) × 106 |
| kcat/Km(O2)d | 7.3 (0.6) | 12.3 (0.5) | 6.8 (2.7) × 1010 | 4.9 (2.8) × 1013 |
| Y334F COX-2 + LA | ||||
| kcatc | 6.7 (0.7) | 9.4 (0.7) | 4.6 (1.5) × 105 | 1.8 (0.8) × 106 |
| kcat/Km(O2)d | 5.7 (0.5) | 7.9 (0.3) | 5.6 (2.4) × 108 | 8.1 (1.2) × 108 |
a Units of kcal mol−1.
b The units are the same as those of the rate constants.
c Units of s−1.
d Units of m−1 s−1.
e From Ref. 5.
Discussion
The similar distributions of products formed by COX-1 and COX-2 suggest that the enzymes react by common mechanisms. There are fundamental differences in their substrate specificity profiles, however, because of the larger active site in COX-2 (1). In this study, dioxygenase catalysis with LA to produce HPODEs was examined as a model for HPETE formation from AA, which likely results from the same initial steps as PGG2.
Analyses of Steady-state Kinetic Parameters
In the works cited above (5, 6), COX-1 and COX-2 exhibit apparent second order rate constants that depend upon co-substrate concentrations in a hyperbolic manner. To the best of our knowledge, the present studies are the first to focus upon how substrate deuterium KIEs in COX respond to changes in [O2]. Although COX-1 reacts with high specificity for AA over LA, COX-2 exhibits nearly identical kcat, kcat/Km(AA), and kcat/Km(LA) when a correction is applied for the O2 consumed. In contrast, kcat/Km(O2) is an order of magnitude greater for AA than LA. This difference is due to a diminution in Km(O2), which could indicate a more favorable O2 uptake. The mechanism for LA is less complex because a single equivalent of O2 traps the LA• before irreversible production of 9R- and 13S-HPODEs. Fewer steps contributing to kcat/Km(O2) could also explain why Ea determined with LA is 5–6 kcal mol−1 smaller than Ea determined with AA as discussed below.
Deuterium KIEs upon LA Oxidation
The ap Dkcat/Km(LA) and ap Dkcat/Km(O2) exhibit hyperbolic dependences upon co-substrate concentrations analogous to that seen for apparent rate constants (5, 6). Each increases to a limiting deuterium KIE at sufficiently high co-substrate concentration. The apparent competitive deuterium KIEs upon LA consumption were analyzed at varying [O2] to test the kinetic mechanism and the relationship of Dkcat/Km(LA) to Dkcat/Km(O2) and Dkcat (27, 35).
The ap Dkcat/Km(LA) increases upon raising the [O2] until an [O2]-independent Dkcat/Km(LA) is reached in Fig. 4. The Dkcat/Km(LA) is indistinguishable from Dkcat/Km(O2) in WT and Y334F COX-2, indicating that the same irreversible hydrogen transfer contributes to the deuterium KIE. Unlike WT COX-2 where a change in the first irreversible step occurs as the [O2] is raised, Y334F COX-2 exhibits similar deuterium KIEs at all substrate concentrations; i.e. Dkcat, Dkcat/Km(LA), and Dkcat/Km(O2) all approach 30. These results indicate a common irreversible and rate-limiting step at all substrate concentrations. On these grounds, the mechanism in Fig. 9 is proposed to involve reversible LA• formation and O2 trapping followed by irreversible peroxyl radical reduction by Tyr-371.
FIGURE 9.

The random kinetic mechanism and microscopic rate constants (bold) proposed to contribute to Dkcat/Km(LA) and Dkcat/Km(AA). At low [O2], the observed Dkcat/Km(O2) indicates irreversible/rate-limiting transfer of hydrogen, initially abstracted from LA or AA and retained at the Tyr-371 during enzyme turnover, and product dissociation after the first irreversible step. For simplicity, the competitive KIE defined by Equation 3 neglects equilibrium isotope effects upon LA binding and initial hydrogen transfer to Tyr-371•. In the analogous pathway for AA (dashed box), 2 eq of O2 are consumed before the first irreversible step.
Certain mechanisms can be excluded based on the [O2] dependence of ap Dkcat/Km(LA) and the [LA] dependence of ap Dkcat/Km(O2). For instance, a “ping-pong” type mechanism where the fatty acid is oxidized and O2 is reduced in kinetically independent steps is inconsistent with the co-substrate dependence of the apparent deuterium KIEs (36). Ordered sequential mechanisms where (i) initial LA or AA binding is followed by O2 uptake and (ii) initial O2 binding is followed by LA or AA uptake can also be ruled out based on the increase in ap Dkcat/Km(LA) upon raising the [O2]. Published derivations (36, 37) indicate that in (i) the ap Dkcat/Km(LA) should decrease upon raising [O2] and in (ii) the ap Dkcat/Km(LA) should be O2-independent.
Findings with WT and Y334F COX-2 expose an increase in ap Dkcat/Km(LA) upon raising the [O2] until a limiting Dkcat/Km(LA) equal to Dkcat/Km(O2) is reached. This behavior is explained by the random kinetic mechanism in Fig. 9 under the simplifying assumptions that reversible LA binding couples to reversible hydrogen transfer and that the reactions exhibit negligible equilibrium isotope effects. These assumptions allow use of the previously derived Equation 3 as a starting point (37). Considering the limit where [O2] approaches 0 results in Equation 4, taking the limit where [O2] approaches ∞ gives Equation 5.



A ternary complex is hypothesized to result from O2 binding to LA• in Fig. 9. This intermediate, consisting of the reduced catalytic Tyr-371 and LA-derived peroxyl radical, is expected to react by irreversible hydrogen transfer to form either 9R- or 13S-HPODE. In competition, the ternary complex could release O2 from the LA-derived peroxyl radical via k−2. Alternatively, O2 could dissociate from the peroxyl radial and partition into an active site pocket in COX-2, and reverse hydrogen transfer from Tyr-371 to LA• could release LA via k−4. When k−4 ≫ k−2, raising [O2] results in an increase in the ap Dkcat/Km(LA) to the intrinsic KIE defined by Hk5/Dk5. This scenario reproduces the trends in Fig. 4 in accord with Equations 3–5.
The mechanism also allows for observations that Dkcat/Km(LA) is equivalent to Dkcat/Km(O2). The random kinetic mechanism in Fig. 9 predicts that the deuterium KIE approaches a minimum as the [O2] is lowered. At the lowest [O2], deviation from unity is expected when the equilibrium isotope effect upon initial hydrogen transfer is significant. A negligible effect is anticipated in COX-2 because of the similar vibrational frequencies of the O–H/D in Tyr-371 and C–H/D of the bisallylic position in the fatty acid.
The Large Deuterium KIE on AA Oxidation
The present results reveal a large Dkcat/Km(O2) when oxidation of h39-AA is compared with 13,13-d2-AA. As with LA, this result requires the initially abstracted hydrogen to be retained at Tyr-371 to explain the presence of a deuterium KIE upon the rate constant for O2 consumption. During catalysis with AA, Tyr-371 is reoxidized by PGG2• in k8′ of Fig. 9. Evidence that initial hydrogen transfer from AA to Tyr-371• might be reversible comes from earlier work in which the AA• was detected at vanishingly low [O2] (12).
A large Dkcat of ∼21 and smaller Dkcat/Km(O2) of ∼7 are observed during LA oxidation by WT COX-2. This behavior has been attributed to a change in the irreversible hydrogen transfer step upon varying [O2]. A similar change in the first irreversible step might also occur during AA oxidation where Dkcat is ∼3 and Dkcat/Km(O2) is ∼18. Alternatively, an ordered sequential mechanism where AA binding and formation of AA• occur before O2 is consumed might account for the difference in deuterium KIEs. Assignment of the mechanism requires examining apparent competitive KIEs at varying [O2].
The results highlighted in this study pertain to physiologically relevant conditions for the oxidation of LA and AA by COX-2. Previously, Dkcat/Km(AA) was estimated to be ∼2 in non-competitive experiments at an [O2] of ∼250 μm; however, the use of non-optimal [phenol] could cause kinetic complexity under these conditions (5). Inhibition of dioxygenase activity by peroxidase turnover could also obscure Dkcat/Km(AA) so that it is smaller than the intrinsic Hk8′/Dk8′ in Fig. 9.
The Dkcat/Km(O2) for AA is proposed to reflect Tyr-371 oxidation by the terminal peroxyl radical. It is unclear, however, if the deuterium KIE arises from pre-equilibrium hydrogen transfer (from AA to Tyr-371•) coupled to reversible 5-exocyclization steps in Fig. 9. Although Dkcat/Km(O2) exposes irreversible hydrogen transfer from Tyr-371 to PGG2•, the possibility that O2 equivalents are consumed independently cannot be rigorously excluded. Such a reaction seems unlikely in view of the greater Ea and larger 18O KIE associated with dioxygenase catalysis. If two irreversible reactions consume O2 during AA oxidation, the observed 18O KIE would be an average reflecting the 18O KIE upon O2 trapping of AA• followed by irreversible 5-exocyclization and the 18O KIE upon O2 trapping at the 15S-position followed by reduction of PGG2•. This averaged 18O KIE should be smaller than that observed with LA where the evidence points to an 18O KIE arising from reversible O2 trapping of LA• followed by irreversible reduction of the terminal peroxyl radical by Tyr-371. Based on earlier experiments and computations (27, 35), the size of the 18O KIE should increase in response to polarization of the hydrogen transfer to a proton/deuteron (H+/D+)-coupled electron transfer-like transition state (38). Such reactivity could explain the moderate to large Dkcat/Km(O2) and competitive 18O KIEs measured with AA and LA as well as the absence of viscosity effects upon kcat/Km(O2), which reveals thermally activated hydrogen transfer rather than diffusion control.
Temperature Studies of Dioxygenase Catalysis
The temperature-dependent rate constants for WT and Y334F COX-2 summarized in Table 3 contradict findings recently reported by Wu et al. (15) that Ea(h39-AA) significantly exceeds Ea(13,13-d2-AA). Such results conflict with expectations that in the normal thermodynamic range tunneling is more probable for protium than deuterium (39) and requires less reorganization to achieve the reactive configuration. These properties make Ea smaller for the lighter isotope of hydrogen. Although there are instances where inversely temperature-dependent deuterium KIEs have been attributed to hydrogen tunneling, this generally requires a highly exergonic “inverted region” in which the reorganization energy is offset by very favorable Gibbs free energy (λ ≤ −ΔG0) (17). This situation is unlikely in COX-2 where oxidation of Tyr-371 by a peroxyl radical is estimated to be thermoneutral, ΔG0 ∼ 0, because of the similar O–H bond strengths in Tyr-371 and the hydroperoxide product (40).
It is unsurprising that hydrogen transfer from Tyr-371 to the terminal peroxyl radical derived from LA is associated with a smaller Ea than the analogous reaction of AA. Following antarafacial O2 trapping of LA• or AA•, rearrangement of the terminal peroxyl radical must occur to remove hydrogen from Tyr-371. The hydrogen transfer that limits kcat/Km(O2) is expected to require less thermal activation to reorganize the smaller 9R or 13S peroxyl radical derived from LA than the larger PGG2• derived from AA, although multiple explanations are possible.
The reduction of PGG2• might take place over a shorter distance than the 9R or 13S peroxyl radical, explaining the larger magnitude of the Arrhenius prefactor (A). Favorable pre-equilibria could also elevate A to a value approaching 1016 m−1 s−1 in the case of AA (see Table 3). Although the temperature dependence of kcat also indicates a large A for AA compared with LA, the value of 1011–1012 s−1 falls within the upper limit defined by transition state theory where A is associated with κkBT/h value of ∼1013 s−1 where κ is a probability factor related to hydrogen tunneling, kB is the Boltzmann constant, T is temperature, and h is the Planck constant.
Catalysis by WT and Y334F COX-2
Oxidation of Tyr-371 by the oxidized prosthetic group is slower in Y334F COX-2 than in the WT enzyme (22); however, there are no major differences in the steady-state rate constants of the enzymes (see Table 1). This behavior is consistent with the observed deuterium and 18O KIEs but raises questions concerning how their magnitudes might depend upon polarization of the hydrogen transfer in transition states with differently sized AA- and LA-derived peroxyl radicals (27, 35).
Mutating Tyr-334 to Phe reduces complexity of the enzyme kinetics and unmasks a single irreversible step controlling LA oxidation. The evidence for this change is inflation of Dkcat, Dkcat/Km(LA), and Dkcat/Km(O2) to values approaching 30. Additional support comes from temperature studies that reveal similar a Ea for kcat and kcat/Km(O2). This behavior is consistent with irreversible hydrogen transfer from Tyr-371 to the LA-derived peroxyl radical.
In contrast to Y334F COX-2, the WT enzyme exhibits a Dkcat of ∼21 along with Dkcat/Km(LA) and Dkcat/Km(O2) values that are 3 times smaller. Complementary temperature studies indicate disparate ΔEa associated with Dkcat and Dkcat/Km(O2), implicating a change in the irreversible hydrogen transfer step. At high [O2], the KIE reflects initial hydrogen transfer from LA to Tyr-371•, whereas at low [O2], initial hydrogen transfer becomes reversible, and the second hydrogen transfer where the peroxyl radical is reduced by Tyr-371 becomes irreversible. This behavior is different in Y334F COX-2 where Ea and ΔEa are invariant to the [O2].
Although the magnitude of kcat/Km(O2) is essentially the same for WT and Y334F COX-2 oxidizing LA, a 4-fold increase in Dkcat/Km(O2) is observed. This is possibly the result of expanding the hydrogen transfer distance upon removal of the conserved hydrogen bond to Tyr-371. The smaller Ea observed for Y334F COX-2 relative to WT COX-2 could be associated with greater protein flexibility accommodating reorganization of enzyme-bound intermediates required for hydrogen transfer from Tyr-371 to the terminal peroxyl radical.
Conclusions
Conditions have been identified where dioxygenase and peroxidase catalysis can be examined independently in recombinant human COX-2. This feature allows for the analyses of apparent rate constants and deuterium KIEs at variable co-substrate concentrations to address the kinetic mechanism of the enzyme. Studies of dioxygenase catalysis by Y334F COX-2, which lacks a conserved hydrogen bond to the catalytic Tyr-371 and Tyr-371•, reveal kinetically uncomplicated behavior. Using the Y334F variant together with the simpler reacting LA substrate exposes a random sequential mechanism and hydrogen transfer from Tyr-371 to a terminal peroxyl radical in the first irreversible step.
Temperature-dependent deuterium KIEs at subsaturating [O2] revealed differences in the behaviors of WT and Y334F COX-2. In both enzymes, the deuterium KIE upon kcat/Km(O2) arises from retention of the hydrogen abstracted from the substrate at Tyr-371 in the absence of solvent isotope exchange. In the first irreversible step, the terminal peroxyl radical is proposed to accept the hydrogen retained at Tyr-371 via a polarized hydrogen transfer or H+/D+-coupled electron transfer-like transition state. The less temperature-dependent Dkcat/Km(O2) observed for Y334F COX-2 relative to the WT enzyme suggests that removal of the conserved hydrogen bond creates greater protein flexibility or lower reorganization energy within the active site.
In this study, the reactivity of WT COX-2 with AA was also examined at physiologically relevant [O2] for the first time, and a large deuterium KIE was observed upon kcat/Km(O2). This result again indicates irreversible hydrogen transfer step after O2 enters the catalytic cycle at physiologically relevant concentrations. Smaller deuterium KIEs at saturating [AA] and [O2], i.e. Dkcat, could arise from a change in hydrogen tunneling distance or in the kinetic mechanism. Comparing the temperature dependences of Dkcat and Dkcat/Km(O2) suggests a higher activation barrier required to attain the distance required for hydrogen tunneling from Tyr-371 to the enzyme-bound PGG2• derived from AA than from Tyr-371 to the smaller acyclic peroxyl radical derived from LA. Future studies of deuterium KIEs upon AA oxidation will address the origin of discrepancies in the temperature-dependent deuterium KIEs and mechanistic inconsistencies in the literature to date.
Author Contributions
Y. L. was responsible for the acquisition of all data in this paper. J. P. R. was responsible for the preparation of the manuscript. Both authors participated in the analysis of data described herein.
Acknowledgment
Prof. Wilfred van der Donk is acknowledged for the generous gift of 13,13-d2-arachidonic acid.
This work was supported by National Science Foundation Grant MCB0919898. The authors declare that they have no conflicts of interest with the contents of this article.
- COX
- cyclooxygenase
- AA
- arachidonic acid
- LA
- linoleic acid
- KIE
- kinetic isotope effect
- HETE
- hydroxyeicosatetraenoic acid
- HPETE
- hydroperoxyeicosatetraenoic acid
- HPODE
- hydroperoxyoctadecadienoic acid
- HODE
- hydroxyoctadecadienoic acid
- PGG2
- prostaglandin G2
- PGH2
- prostaglandin H2
- PGD2
- prostaglandin D2
- PGE2
- prostaglandin E2
- Por
- protoporphyrin IX
- UPLC
- ultra-high performance liquid chromatography.
References
- 1.Rouzer C. A., and Marnett L. J. (2003) Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 103, 2239–2304 [DOI] [PubMed] [Google Scholar]
- 2.Rouzer C. A., and Marnett L. J. (2011) Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 111, 5899–5921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith W. L., Urade Y., and Jakobsson P. J. (2011) Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 111, 5821–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsai A. L., and Kulmacz R. J. (2010) Prostaglandin H synthase: resolved and unresolved mechanistic issues. Arch. Biochem. Biophys. 493, 103–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Danish H. H., Doncheva I. S., and Roth J. P. (2011) Hydrogen tunneling steps in cyclooxygenase-2 catalysis. J. Am. Chem. Soc. 133, 15846–15849 [DOI] [PubMed] [Google Scholar]
- 6.Mukherjee A., Brinkley D. W., Chang K. M., and Roth J. P. (2007) Molecular oxygen dependent steps in fatty acid oxidation by cyclooxygenase-1. Biochemistry 46, 3975–3989 [DOI] [PubMed] [Google Scholar]
- 7.Vecchio A. J., Simmons D. M., and Malkowski M. G. (2010) Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 285, 22152–22163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vecchio A. J., Orlando B. J., Nandagiri R., and Malkowski M. G. (2012) Investigating substrate promiscuity in cyclooxygenase-2: the role of Arg-120 and residues lining the hydrophobic groove. J. Biol. Chem. 287, 24619–24630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kulmacz R. J., van der Donk W. A., and Tsai A.-L. (2003) Comparison of the properties of prostaglandin H synthase-1 and -2. Prog. Lipid Res. 42, 377–404 [DOI] [PubMed] [Google Scholar]
- 10.Poulos T. L. (2014) Heme enzyme structure and function. Chem. Rev. 114, 3919–3962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dorlet P., Seibold S. A., Babcock G. T., Gerfen G. J., Smith W. L., Tsai A. L., and Un S. (2002) High-field EPR study of tyrosyl radicals in prostaglandin H2 synthase-1. Biochemistry 41, 6107–6114 [DOI] [PubMed] [Google Scholar]
- 12.Wilson J. C., Wu G., Tsai A. L., and Gerfen G. J. (2005) Determination of the structural environment of the tyrosyl radical in prostaglandin H2 synthase-1: a high frequency ENDOR/EPR study. J. Am. Chem. Soc. 127, 1618–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu G., Lü J. M., van der Donk W. A., Kulmacz R. J., and Tsai A. L. (2011) Cyclooxygenase reaction mechanism of prostaglandin H synthase from deuterium kinetic isotope effects. J. Inorg. Biochem. 105, 382–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffmann I., Hamberg M., Lindh R., and Oliw E. H. (2012) Novel insights into cyclooxygenases, linoleate diol synthases, and lipoxygenases from deuterium kinetic isotope effects and oxidation of substrate analogs. Biochim. Biophys. Acta 1821, 1508–1517 [DOI] [PubMed] [Google Scholar]
- 15.Wu G., Kulmacz R. J., and Tsai A. L. (2014) Kinetic isotope effect of prostaglandin H synthase exhibits inverted temperature dependence. Catalysts 4, 174–185 [Google Scholar]
- 16.Smith W. L., and Song I. (2002) The enzymology of prostaglandin endoperoxide H synthases-1 and -2. Prostaglandins Other Lipid Mediat. 68, 115–128 [DOI] [PubMed] [Google Scholar]
- 17.Ludlow M. K., Soudackov A. V., and Hammes-Schiffer S. (2009) Theoretical analysis of the unusual temperature dependence of the kinetic isotope effect in quinol oxidation. J. Am. Chem. Soc. 131, 7094–7102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marnett L. J. (2009) The COXIB experience: a look in the rearview mirror. Annu. Rev. Pharmacol. Toxicol. 49, 265–290 [DOI] [PubMed] [Google Scholar]
- 19.Wu G., Tsai A. L., and Kulmacz R. J. (2009) Cyclooxygenase competitive inhibitors alter tyrosyl radical dynamics in prostaglandin H synthase-2. Biochemistry 48, 11902–11911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogge C. E., Liu W., Wu G., Wang L. H., Kulmacz R. J., and Tsai A. L. (2004) Identification of Tyr504 as an alternative tyrosyl radical site in human prostaglandin H synthase-2. Biochemistry 43, 1560–1568 [DOI] [PubMed] [Google Scholar]
- 21.Goodwin D. C., Rowlinson S. W., and Marnett L. J. (2000) Substitution of tyrosine for the proximal histidine ligand to the heme of prostaglandin endoperoxide synthase 2: implications for the mechanism of cyclooxygenase activation and catalysis. Biochemistry 39, 5422–5432 [DOI] [PubMed] [Google Scholar]
- 22.Rogge C. E., Ho B., Liu W., Kulmacz R. J., and Tsai A. L. (2006) Role of Tyr348 in Tyr385 radical dynamics and cyclooxygenase inhibitor interactions in prostaglandin H synthase-2. Biochemistry 45, 523–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Edward J. T. (1970) Molecular volumes and the Stokes-Einstein equation. J. Chem. Educ. 47, 261 [Google Scholar]
- 24.Hasinoff B. B., and Chishti S. B. (1982) Viscosity dependence of the kinetics of the diffusion-controlled reaction of carbon monoxide and myoglobin. Biochemistry 21, 4275–4278 [DOI] [PubMed] [Google Scholar]
- 25.Brouwer A. C., and Kirsch J. F. (1982) Investigation of diffusion-limited rates of chymotrypsin reactions by viscosity variation. Biochemistry 21, 1302–1307 [DOI] [PubMed] [Google Scholar]
- 26.Lewis E. R., Johansen E., Holman T. R. (1999) Large competitive kinetic isotope effects in human 15-lipoxygenase catalysis measured by a novel HPLC method. J. Am. Chem. Soc. 121, 1395–1396 [Google Scholar]
- 27.Huff G. S., Doncheva I. S., Brinkley D. W., Angeles-Boza A. M., Mukherjee A., Cramer C. J., and Roth J. P. (2011) Experimental and computational investigations of oxygen reactivity in a heme and tyrosyl radical-containing fatty acid α-(di)oxygenase. Biochemistry. 50, 7375–7389 [DOI] [PubMed] [Google Scholar]
- 28.Roth J. P. (2009) Oxygen isotope effects as probes of electron transfer mechanism and structures of activated O2. Acc. Chem. Res. 42, 399–408 [DOI] [PubMed] [Google Scholar]
- 29.Peng S., and van der Donk W. A. (2003) An unusual isotope effect on substrate inhibition in the oxidation of arachidonic acid by lipoxygenase. J. Am. Chem. Soc. 125, 8988–8989 [DOI] [PubMed] [Google Scholar]
- 30.Knapp M. J., and Klinman J. P. (2003) Kinetic studies of oxygen reactivity in soybean lipoxygenase-1. Biochemistry 42, 11466–11475 [DOI] [PubMed] [Google Scholar]
- 31.Schneider C., and Brash A. R. (2000) Stereospecificity of hydrogen abstraction in the conversion of arachidonic acid to 15R-HETE by aspirin-treated cyclooxygenase-2. Implications for the alignment of substrate in the active site. J. Biol. Chem. 275, 4743–4746 [DOI] [PubMed] [Google Scholar]
- 32.McGinley C. M., and van der Donk W. A. (2003) Enzymatic hydrogen atom abstraction from polyunsaturated fatty acids. Chem. Commun. 23, 2843–2846 [DOI] [PubMed] [Google Scholar]
- 33.Halliwell B., and Gutteridge J. M. C. (2006) Free Radicals in Biology and Medicine, 4th Ed., pp. 3–4, Clarendon Press, Oxford [Google Scholar]
- 34.Roth J. P., and Klinman J. P. (2003) Oxygen isotope effects on electron transfer to O2 probed using chemically modified flavins bound to glucose oxidase. Proc. Natl. Acad. Sci. U.S.A. 100, 62–67 [DOI] [PubMed] [Google Scholar]
- 35.Mukherjee A., Angeles-Boza A. M., Huff G. S., and Roth J. P. (2011) Catalytic mechanism of a heme and tyrosyl radical-containing fatty acid alpha-(di)oxygenase. J. Am. Chem. Soc. 133, 227–238 [DOI] [PubMed] [Google Scholar]
- 36.Glickman M. H., and Klinman J. P. (1995) Nature of rate-limiting steps in the soybean lipoxygenase-1 reaction. Biochemistry 34, 14077–14092 [DOI] [PubMed] [Google Scholar]
- 37.Klinman J. P., Humphries H., and Voet J. G. (1980) (1980) Deduction of kinetic mechanism in multisubstrate enzyme reactions from tritium isotope effects. Application to dopamine β-hydroxylase. J. Biol. Chem. 255, 11648–11651 [PubMed] [Google Scholar]
- 38.Hammes-Schiffer S., and Stuchebrukhov A. A. (2010) Theory of coupled electron and proton transfer reactions. Chem. Rev. 110, 6939–6960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Layfield J. P., and Hammes-Schiffer S. (2014) Hydrogen tunneling in enzymes and biomimetic models. Chem. Rev. 114, 3466–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smirnov V. V., and Roth J. P. (2014) Tyrosine oxidation in heme oxygenase: examination of long-range proton-coupled electron transfer. J. Biol. Inorg. Chem. 19, 1137–1148 [DOI] [PubMed] [Google Scholar]