


Abstract
Transcription factor E2F1 is a key regulator of cell proliferation and apoptosis. Recently, it has been shown that aberrant E2F1 expression often detectable in advanced cancers contributes essentially to cancer cell propagation and characterizes the aggressive potential of a tumor. Conceptually, this requires a subset of malignant cells capable of evading apoptotic death through anticancer drugs. The molecular mechanism by which the pro-apoptotic activity of E2F1 is antagonized is widely unclear. Here we report a novel function for EPC1 (enhancer of polycomb homolog 1) in DNA damage protection. Depletion of EPC1 potentiates E2F1-mediated apoptosis in response to genotoxic treatment and abolishes tumor cell motility. We found that E2F1 directly binds to the EPC1 promoter and EPC1 vice versa physically interacts with bifunctional E2F1 to modulate its transcriptional activity in a target gene-specific manner. Remarkably, nuclear-colocalized EPC1 activates E2F1 to upregulate the expression of anti-apoptotic survival genes such as BCL-2 or Survivin/BIRC5 and inhibits death-inducing targets. The uncovered cooperativity between EPC1 and E2F1 triggers a metastasis-related gene signature in advanced cancers that predicts poor patient survival. These findings unveil a novel oncogenic function of EPC1 for inducing the switch into tumor progression-relevant gene expression that may help to set novel therapies.
INTRODUCTION
Members of the E2F transcription factor family play an important role in regulating multiple cellular functions including proliferation, differentiation, and apoptosis (1). Among them, E2F1 is best known to promote apoptosis in response to DNA damage by inducing pro-apoptotic genes such as p73, Bcl-2 homology region 3 (BH3)-only proteins, Apaf-1 and caspases as part of a cellular safeguard mechanism to counteract cancer development (2,3). Therefore, it was intriguing that E2F1 is frequently and predominantly enriched in high-grade tumors and metastases of various human cancers (4,5) associated with therapy resistance and unfavorable patient survival prognosis (6,7). Existing functional evidence indicated that E2F1 directly contributes to several stages of malignant progression by promoting EMT (8), angiogenesis (9), extravasation of CTCs (10) and tumor metastasis (11). In addition, E2F1 regulates matrix metalloproteinase (MMP) genes and collagen degradation (12–14), demonstrating its involvement in extracellular matrix remodeling. H-RAS stimulation of E2F activity leads to the upregulation of cell adhesion molecule β4 integrin and formation of α6β4 integrin complexes that mediate E2F-dependent carcinoma migration and invasion of breast cancer cells (15). The importance of this transcription factor to tumor progression was also shown in a genetic model by interbreeding Neu transgenics with E2F1 knockout mice and in HER2+ breast cancer patients, in which the E2F activation status predicts relapse and metastatic potential of MMTV-Neu-induced tumors (16). In fact, E2F-responsive genes define a novel molecular subset of high-grade human tumors of the breast, ovary and prostate, termed ERGO (E2F-responsive gene overexpressing) cancers (17). We demonstrated that VEGF-C and its cognate receptor VEGFR-3, which are highly upregulated in malignant cells abundantly expressing E2F1, are direct targets of the transcription factor (9). Coregulation of VEGF-C/VEGFR-3 by E2F1 promotes formation of new blood vessels in vivo. Interestingly, E2F1 is activated by VEGFR-3 signaling in a positive feedback loop and both proteins cooperate in the nucleus to coregulate transactivation of the proangiogenic cytokine PDGF-B. Likewise, the hyaluronan receptor RHAMM and EGFR (epidermal growth factor receptor), both directly upregulated by E2F1, also interact with nuclear E2F1 to coactivate expression of fibronectin (10) or other E2F1-dependent genes relevant for metastasis (11). Clinically important is that malignant progression through E2F1 is intimately linked with increased chemoresistance (5). Thus, pro-apoptotic activity of E2F1, in those tumor cells, must be restrained to allow aggressive behavior. However, mechanisms by which its tumor surveillance function is blocked in late cancer stages remains largely unexplored.
Polycomb group proteins (PcGs) are well known for their role in silencing homeotic genes during embryonic development. At the molecular level, PcG proteins are classified into two groups on the basis of their association with distinct classes of multimeric complexes, termed Polycomb repressive complexes (PRC1 and PRC2) (18). A signature activity of PRC2 is methylation of histone H3 on K27 (H3K27me3) (19–21). PRC1 family complexes execute an enzymatic function by ubiquitylation of histone H2A on K119 (22), as well as a nonenzymatic function through compaction of polynucleosomes (23). In recent years, increased levels of several PcG members have been detected in a variety of human tumors, such as lymphoma, prostate and colon cancers (24–26). Even higher PcG level is indicative of metastatic clinical behaviour (27–29). These findings implicate PcG genes in cancer development and progression. However, the mechanism by which PcG regulate tumorigenesis awaits further clarification.
There are several lines of evidence functionally linking PcG-mediated epigenetic events to E2F1-mediated cell function in vertebrates. For example, Enhancer of zeste homolog 2 (EZH2), a core subunit of PRC2 complex, was shown to be activated by E2F1 and required for human cancer cell proliferation (30). Meanwhile, it was found that EZH2 is able to antagonize the induction of E2F1-mediated apoptosis by directly inhibiting the expression of Bim to facilitate cell transformation (31). Moreover, BMI1, a member of the PRC1 complex and target gene of E2F1, is strongly expressed in primary neuroblastomas (32). BMI1 together with other PcGs suppresses the INK4A-ARF locus encoding an upstream regulator of the pRB/E2F pathway, thereby increasing E2F1 expression (33). Finally, a recent study in Drosophila demonstrated that PcG complexes may affect cell proliferation by repressing expression of dE2f1 and certain target genes (34).
In this study, gene expression profiling of conditionally E2F1 activated and knockdown cancer cells has uncovered the epigenetic modifier EPC1 (enhancer of polycomb homolog 1), a component of the NuA4 histone acetyltransferase (HAT) complex with upregulated expression during DNA damage as direct E2F1 target gene. The data reported here indicate a novel mechanism by which chromatin-regulatory protein EPC1 promotes reversible phenotypic alterations toward DNA damage protection and cancer aggressiveness by modifying E2F1 transcriptional activity. We found that EPC1 activates E2F1 to upregulate the expression of antiapoptotic survival genes and inhibits its death-inducing targets via direct interaction. The uncovered cooperativity between EPC1 and E2F1 triggers a metastasis-related gene signature in advanced cancers that predicts poor patient survival.
MATERIALS AND METHODS
Cell culture
SK-Mel-29 and SK-Mel-147 melanoma cells were cultured as described (11). RT4, UMUC3 and T24 bladder cancer, MCF7 and MDA-MB-231 breast cancer, H1299 human non-small cell lung carcinoma and human embryonic kidney HEK293 and HEK293T cells were purchased from American Type Culture Collection (ATCC). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS, Biochrom), 1% penicillin/streptomycin (PAA) and 0.5% amphotericin B (PAA) in a humidified atmosphere of 5% CO2 at 37°C. Stable SK-Mel-29.ER-E2F1, Saos-2.ER-E2F1 and H1299.ER-E2F1 cells, constitutively expressing E2F1 fused to the binding domain of the murine ER receptor, were maintained in medium containing puromycin (Sigma-Aldrich) at a concentration of 0.25 or 1 μg/ml respectively. E2F1 activity in ER-E2F1 stable cell lines was induced by 4-hydroxytamoxifen (4-OHT) at a final concentration of 1 μM. The epirubicin-resistant MCF7-EPIR breast cancer cell line was described before (35) and maintained in DMEM containing 1 μM epirubicin.
Microarray data processing and analysis
SK-Mel-147 melanoma cells were harvested 96 h after infection with Lenti-sh.control or Lenti-sh.EPC1 for RNA extraction. Equal amounts of RNA were analyzed using Affymetrix GeneChip Human Genome U133 Plus 2.0 Arrays (Affymetrix). Analysis was done in duplicate for each sample. Background-corrected signal intensities were determined and processed using MAS5 function of the R/Bioconductor affy package. Normalization of expression data, statistical tests, and clustering was accomplished by GeneSpring GX 9.0 (Agilent). Gene transcripts not detected in any samples were excluded from statistical analysis. To determine differentially expressed genes, expression profiles were grouped and statistically analyzed using t-test and multiple testing correction (Benjamini and Hochberg False Discovery Rate). Only targets displaying a minimum 1.5 fold increase or decrease (P < 0.05) by EPC1 knockdown were included for clustering.
Viral vectors
The cDNA encoding full-length EPC1 was amplified by PCR based on the information available from NCBI database (accession number NM_025209.3), cloned into pcDNA3.1/V5-His TOPO (Invitrogen), and verified by sequencing. Adenoviral plasmid encoding EPC1 was constructed by cutting cDNA from pcDNA3.1/EPC1 usingPmeI and HindIII restriction sites and cloning into pAdTrack-CMV. Recombinant adenovirus was generated by cotransfection of pAdTrack-CMV-EPC1 and pAdEasy-1 as previously described (36). Lentiviral plasmids (pLKO.1-puro) encoding sh.E2F1 (clone ID: TRCN250, TRCN253), sh.EPC1 (clone ID: TRCN263, TRCN264) and sh.control (SHC002) were purchased from Sigma–Aldrich. VSV-G enveloped pseudotyped lentiviral vectors were generated in HEK293T packaging cells by cotransfection with pLKO.1 plasmid containing shRNA sequences, pAX2 and VSV-G/pMD2.G (Addgene) using calcium phosphate method. Media were replaced 8 h after transfection, and 48 h later conditioned media were harvested and filtered.
Promoter reporter constructs and expression plasmids
The human EPC1 promoter (−1576 to +1) was amplified by PCR from human genomic DNA and inserted into pcDNA3.1/V5-His vector, digested with KpnI and HindIII and ligated into pGL3-basic. Bcl-2 promoter reporter plasmid was purchased from Addgene and p73-luciferase reporter was generated before (37). BIRC5 and p27 promoter constructs are described previously (38,39). E2F1, E2F2, E2F3 and E132 plasmids including the pCMV-FLAG vectors containing full-length E2F1 or truncated mutants have been described before (9,40). All plasmids were confirmed by sequencing.
Semi-quantitative and qRT-PCR
Total RNA was isolated with NucleoSpin® RNA II (Macherey-Nagel) and reverse transcribed using Omni-script RT (Qiagen). PCR amplification was performed as previously described (41). For qPCR cDNA samples were mixed with iQTM SYBR® Green Supermix and analyzed on iQTM5 Multicolor Real-Time PCR Detection System (Bio-Rad). Relative gene expression was calculated using iQTM5 Optical System Software. All specific primer pairs are listed in Supplementary Table S1.
ChIP assay
Chromatin immunoprecipitation (ChIP) was performed essentially as described (11). Briefly, SK-Mel-29 either infected with Ad.ER-E2F1 or stably expressing ER-E2F1 were treated with 4-OHT, and SK-Mel-147 cells depleted for E2F1 or EPC1 using gene specific shRNA. Proteins bound to DNA were cross-linked using formaldehyde at a final concentration of 1.42% for 15 min at room temperature. The reaction was stopped by adding glycine to a final concentration of 125 mM. Following sonication, protein-DNA complexes were immunoprecipitated using antibody for E2F1 (KH95, Santa Cruz Biotechnology), EPC1 (ab112043, Abcam), Tip60 (N-17, Santa Cruz Biotechnology), EZH2 (AC22, Cell Signaling), Anti-acetyl-Histone (06-599, Millipore) and Anti-trimethyl-Histone-Lys27 (07-449, Millipore) or appropriate control IgG and placed on a rotating platform at 4°C overnight. Immunoprecipitated chromatin was eluted from the beads in 10% Chelex100 and boiled for 10 min. The resulting DNAs were subjected to semi-quantitative PCRs. For primer sequences see Supplementary Table S1.
Immunoprecipitation, pull-down assay, and Western blotting
Cells prepared for immunoprecipitation were lysed in lysis buffer (50 mM Tris–HCl, pH 8.0, 5 mM EDTA, 150 mM NaCl, 0.5% Nonidet P-40) with protease inhibitor cocktail (PAA) and centrifuged for 10 min at 13200 rpm, and insoluble debris was discarded. 1 mg of cell lysate was precipitated with 2 μg of anti-E2F1 antibody (KH95), anti-E2F5 (MH-5), anti-E2F6 (TFE61), or anti-mouse IgG antibody from Santa Cruz Biotechnology. Protein A/G PLUS-Agarose beads (sc-2003, Santa Cruz Biotechnology) were added and the immune complexes were pulled down over 2 h at 4°C under rotation. Beads were washed extensively with lysis buffer to remove unbound proteins, boiled in SDS sample buffer, fractionated by SDS–PAGE and immunoblotted using EPC1 and E2F1 antibodies.
Full-length E2F1 was cloned into the pGEX-4T-1 plasmid (GE Healthcare Life Sciences) and used to transform BL21 cells. When cells reached OD 0.6–0.8, Glutathione S-transferase (GST) fusion protein expression was induced by 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 37°C for 1 h. Bacteria were lysed in PBS with protease inhibitors by freeze and thaw following sonification. GST-fusion proteins were purified using Glutathione Sepharose 4B (GE Healthcare Life Sciences) and purified GST-fused proteins were confirmed by Coomassie staining or GST-antibody (B-14, Santa Cruz Biotechnology). GST pull-down assays were performed overnight using beads coated with GST or GST-E2F1 fusion proteins and equivalent amounts of cell lysates from SK-Mel-29 transfected with pcDNA3.1-EPC1. Beads were washed six times with Washing Buffer (1× PBS, pH 7.4, 1% Triton and protease inhibitors). EPC1 proteins retained on the beads were separated by SDS-PAGE and detected by immunoblotting with EPC1 antibody.
Cells prepared for direct protein analysis were lysed in RIPA buffer (50 mM Tris–Cl, 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors. Total protein concentration was quantified by Bradford assay. Equal amounts of total protein were separated by SDS-PAGE, transferred to nitrocellulose membrane (Amersham), and probed with appropriate primary antibodies anti-EPC1 (Santa Cruz Biotechnology/Abcam), anti-E2F1 (Santa Cruz Biotechnology/BD Pharmingen), anti-ß-actin (Sigma–Aldrich), anti-EZH2 (AC22, Cell Signaling), anti-BIRC5 (Abcam), anti-p27 (Santa Cruz Biotechnology), anti-Bim (BD Pharmingen), and anti-Bcl-2 (Calbiochem). The corresponding peroxidase-labeled secondary antibodies were detected using ECL Western blot reagents (Amersham Biosciences).
Luciferase reporter assay
Cells were seeded in 6-well plates and transfected with 0.5 μg luciferase reporter construct and indicated amounts of plasmids expressing E2F1, E2F2 and E2F3. The total amount of transfected plasmids was kept constant (2 μg for a 6-cm plate) by addition of an appropriate empty vector. Luciferase activity was measured 24 h after transfection using luciferase reporter assay system (Promega) and normalized to total protein concentration.
Immunofluorescence
Cells grown on coverslips were fixed in 4% paraformaldehyde, permeabilized, and stained with anti-EPC1 (Santa Cruz Biotechnology) or anti-E2F1 antibody (KH95, Santa Cruz Biotechnology). The secondary antibodies conjugated to Alexa Fluor 488 or Alexa Fluor 633 (Molecular Probes) were used for visualization with a LSM 780 confocal laser-scanning microscope (Carl Zeiss). Cell nuclei were stained with DAPI (5 mg/ml).
Flow cytometry analysis and Hoechst 33342 staining
For FACS analysis, cells were collected at indicated times after infection followed by cisplatin treatment and fixed in 70% ethanol. Fixed cells were stained with propidium iodide (50 mg/ml) after treatment with RNase A (100 mg/ml) and analyzed for DNA content in a FACS Calibur (Becton Dickinson Instrument). Hoechst 33342 was added to cell culture medium at 1 mg/ml. Cells were incubated at 37°C/5% CO2 and subsequently subjected to fluorescence microscopy.
Cell migration assay
Cells growing in 6-cm dishes were infected with indicated viruses and then scratched with a 200 μl pipette tip. Next, plates were washed three times with PBS to remove detached cells and cultured for 24 h in DMEM containing mitomycin C (20 μg/ml). Wound healing was assessed in microscopic visualization of cells that have migrated into the gap.
In silico protein–protein interaction studies
To perform computational protein–protein interaction studies, we used a previously designed model of E2F1 (10) and generated an EPC1 model. To model the structure of EPC1 protein using homology modeling approach, we first retrieved the sequence of EPC1 (Genbank ID: AAH36529.1) and performed the sequence alignment with the protein sequences available on Protein Data Bank (PDB). We applied the iterative threading assembly refinement (I-TASSER) server that uses composite modeling approaches combining various techniques such as threading, ab initio modeling and atomic-level structure refinement approaches (42–44). Template modeling score (TM-score) was used to assess the structural similarity of the model and template protein structure, where TM score >0.5 indicates a model of correct topology. Confidence score (C-score), in the range of [−5, 2], was used to determine the accuracy of the predicted structure by I-TASSER server, where higher score indicate a better structural topology of the model. To further optimize the theoretical model of EPC1 generated by I-TASSER, we first minimized the structure using Smart Minimizer algorithms for 5000 steps by applying CHARMm force field and RMS gradient cut off 0.1 and used Looper and ChiRotor tools available in Accelrys Discovery Studio 3.5. The protein–protein interaction between E2F1 and EPC1 was performed using Zdock protocol (45) available in Accelrys Discovery Studio 3.5. Top 100 interaction poses were analysed and clustered based on all-against-all RMSD matrix by considering clustering algorithm proposed by Lorenzen and Zhang (46). Top poses based on ZDOCK score from each cluster were refined using RDOCK protocols. All the poses were re-ranked based on RDOCK energy (47).
Statistical analysis
Statistical significance was calculated by two-tailed Student's t-test. A P-value below 0.05 was defined as statistically significant. Overall survival curves were calculated by SigmaPlot (Systat Software) according to the Kaplan-Meier method using log-rank test. P < 0.05 was considered to be statistical significant. Bladder and breast cancer patients were grouped according to high and low EPC-1 mRNA levels based on an expression signature generated by using the cancer browser (https://genome-cancer.ucsc.edu).
RESULTS
EPC1 is a direct transcriptional target of E2F1
The critical role of polycomb group (PcG) proteins in regulation of cell fate decisions and how their deregulation potentially contributes to cancer development (48) motivated us to search for PcG genes that are directly controlled by E2F1. We first compared the gene expression profiles of sh.control and sh.E2F1 SK-Mel-147 cells (11). Parental SK-Mel-147 are highly metastatic and this is chiefly due to E2F1 expression in these cells. A previous study showed that two of the core subunits of PRC2 complex, EZH2 and EED, are regulated by E2F1 in fibroblast cells (30). Among the genes that are differentially regulated in dependence on E2F1 expression, we found EZH2, and EPC1 (Figure 1A). These PcG genes were significantly downregulated after knockdown of E2F1 in SK-Mel-147 cells. The array data were further validated by semiquantitative RT-PCR, which showed a marked decrease of EPC1 mRNA in shE2F1 treated cells comparable to the level of the known E2F1 target EZH2 (30), but no obvious effect on EED and SUZ12, other members of PRC2, was detected (Figure 1B). Western blot analysis confirmed that E2F1 depletion using two different shRNA constructs led to reduced expression of endogenous EPC1 and EZH2, indicating a causative role for endogenous E2F1 in regulating EPC1 (Figure 1C). Moreover, activation of E2F1 in stable Saos-2.ER-E2F1 and SK-Mel-29.ER-E2F1 through 4-OHT addition caused a strong increase of EPC1 mRNA and protein levels (Figure 1D and E). Enhanced expression of EPC1 after 4-OHT treatment was also visible by immunofluorescence microscopy in activated SK-Mel-29.ER-E2F1 cells (Figure 1F). These observations led us to conclude that EPC1 is a target of E2F1 transcriptional regulation.
Figure 1.
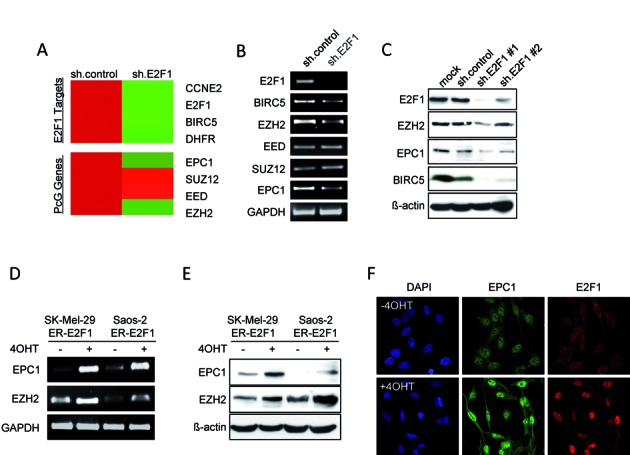
Screening for Polycomb group proteins (PcGs) regulated by E2F1. (A) Gene expression heatmap of representative E2F1 targets and PcG genes that are differentially regulated in SK-Mel-147 upon E2F1 knockdown (11). Red and green shading indicates high and low gene expression relative to sh.control set to 1. (B and C) Analysis of PcGs at mRNA (B) and protein levels (C) following knockdown of E2F1 in SK-Mel-147 cell line. E2F1 target gene BIRC5 is shown as positive control. ß-actin served as loading control. (D and E) mRNA (D) and protein levels (E) of EPC1 and EZH2 in indicated cell lines after induction of ER.E2F1. Saos-2 or SK-Mel-29 cells stable expressing ER-E2F1 were treated with 4-hydroxytamoxifen (4-OHT, 1 μM) or solvent for 24 hrs. Actin was used for loading control. (F) Expression of EPC1 (green) and E2F1 (red) in SK-Mel-29.ERE2F1 treated with 4-OHT for 24 h. Nuclei were counter-stained with DAPI (blue) and fluorescence was visualized by laser scanning microscopy.
To investigate the mechanism of E2F1-dependent EPC1 induction, we performed chromatin immunoprecipitation analysis to determine whether E2F1 can directly bind to the EPC1 gene promoter in vivo. In silico analysis revealed four putative binding motifs for E2F proteins in the promoter region of EPC1 and three different pairs of primers (P1 to P3) were designed to encompass these sites (Figure 2A top). Endogenous E2F1 was immunoprecipitated with a specific E2F1 monoclonal antibody in SK-Mel-29.ERE2F1 cells treated with 4-OHT for 24 hrs followed by genomic PCR analysis of the three regions. The results showed a strong binding of E2F1 to the proximal promoter regions (−825/−507, P2; −230/+42, P3), while there is no significant enrichment at the more distant element (−1292/−1148, P1) (Figure 2A bottom). This finding clearly demonstrates direct binding of E2F1 to the EPC1 promoter.
Figure 2.
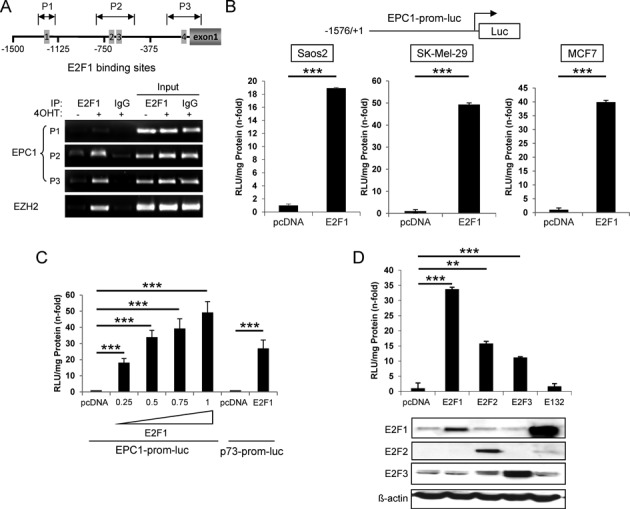
E2F1 directly binds and activates EPC1 promoter. (A) Schematic representation of EPC1 gene promoter region (upper). Putative E2F1-binding sites were predicted using JASPAR database. Arrows indicate position of PCR amplicons in chromatin immunoprecipitation (ChIP) experiments relative to ATG (P1, −1292/−1148; P2, −825/−507; P3, −230/+42). ChIP assay shows the binding of E2F1 on EPC1 promoter in SK-Mel-29.ER-E2F1 treated with or without 4-OHT (1 μM) for 24 h (lower). Input represents 10% of sheared chromatin prior to immunoprecipitation. DNA was analyzed by semi-quantitative PCR with primers corresponding to the amplicons shown in upper panel. Primers amplifying the E2F1 sites in the EZH2 promoter were used as a positive control. (B) Schematic representation of EPC1 full-length promoter region (upper). Luciferase reporter assays in Saos-2, SK-Mel-29 and MCF7 cells upon E2F1 activation (lower). Relative light units (RLU) were measured after co-transfection of EPC1 promoter construct and E2F1. Bars indicate mean values ± SD of three independent experiments. (C) Dose-dependent regulation of EPC1 reporter by E2F1. SK-Mel-29 cells were co-transfected with EPC1 promoter construct and increasing amounts of E2F1 expression plasmid (0.25, 0.5, 0.75 and 1 μg). RLU were measured 24 h post-transfection. p73-prom-luc was used as positive control. (D) Luciferase reporter activities after co-transfection of SK-Mel-29 cells with EPC1 promoter construct and different E2F family members or DNA binding-deficient mutant E132 (1 μg). pcDNA was transfected as control in all reporter assays. E2F1, E2F2, E2F3 and E132 expression was verified by Western blot. Actin serves as loading control. Error bars indicate one SD of the mean. All experiments were performed three times independently. Statistical significance was determined by two-sided Student's t-test. **P ≤ 0.01, ***P ≤ 0.001.
In order to determine the potential activity of E2F1 on EPC1 expression, we cloned the entire promoter region −1576/+1 into the pGL3-luciferase reporter construct. Cells were co-transfected with EPC1-prom-luc and expression plasmids encoding E2F1. Luciferase activity was measured 24 hrs after transfection and normalized to total protein concentration in the cell extract. Consistent with the predictions, a strong upregulation of promoter activity was observed after E2F1 overexpression in each cell line tested (Figure 2B). Our data revealed that stimulation of the EPC1 promoter by E2F1 is dose-dependent (Figure 2C) with the highest 50-fold increase in SK-Mel-29 at 1 μg E2F1, which is significantly higher than for the p73 promoter used as positive control. This clearly indicates that E2F1 controls EPC1 expression directly. In addition, forced expression of E2F2 and E2F3 in these cells, to a lesser extent, can also enhance EPC1 promoter activity but not the DNA binding deficient E2F1 mutant E132 (Figure 2D).
EPC1 expression is elevated in invasive/metastatic and chemoresistant cancer cells
Since little is known about human EPC1, we examined its expression levels in various patient-derived cancer cell lines, including bladder (RT4, T24 and UMUC3), breast (MCF7 and MDA-MB231), and malignant melanoma (SK-Mel-29 and SK-Mel-147). Of these, T24, UMUC3, MDA-MB231, and SK-Mel-147 have been shown to own high invasive potential, while RT4, MCF7 and SK-Mel-29 are non-invasive cells (11,49–51). As demonstrated in Figure 3A, endogenous EPC1 is expressed at large amounts in exact accordance with the abundance of E2F1 in these tumor cell lines. Moreover, we found that EPC1 expression correlates with their aggressive phenotype as its expression is much higher in invasive than non-invasive cells, suggesting that EPC1 might be relevant to E2F1 in facilitating cancer cell propagation.
Figure 3.
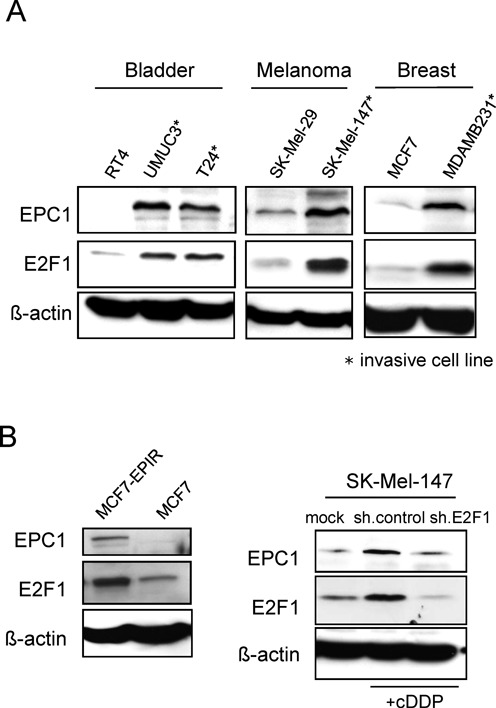
Elevated EPC1 expression in invasive and chemoresistant tumor cells. (A) Endogenous EPC1 and E2F1 expression levels in different cancer cell lines as assessed by Western blot. Actin was used for loading control. (B) Protein levels of endogenous E2F1 and EPC1 in epirubicin-resistant MCF7 human breast cancer cells (MCF7-EPIR) and parental cells (left). Protein levels of E2F1 and EPC1 in cisplatin (cDDP, 30 μM) treated SK-Mel-147 cells expressing sh.E2F1 or sh.control compared with untreated cells (right). Actin was used for loading control.
After DNA damage triggered by genotoxic agents elevated E2F1 values can be detected (52,53), which is involved in increased survival and chemoresistance (5,35,54). As expression of EPC1 is higher in advanced cancer stages and significantly correlates with E2F1 levels, we therefore asked whether EPC1 is also induced through drug treatment. To this end, we evaluated the expression levels of EPC1 and E2F1 in epirubicin-sensitive and -resistant breast cancer cell lines. As shown in Figure 3B, both EPC1 and E2F1 were strongly upregulated in epirubicin-resistant MCF7 (MCF7-EPIR) compared to parental cells. In line with this, treatment of SK-Mel-147 cells with cisplatin leads to a marked upregulation of EPC1 expression that is associated with E2F1 accumulation. Notably, EPC1 induction was abolished after E2F1 knockdown (Figure 3B right). These data clearly suggest that EPC1 is induced in response to chemotherapy in an E2F1-dependent manner enabling cancer cells to survive by antagonizing drug-induced apoptosis.
Loss of EPC1 promotes DNA damage-induced apoptosis and inhibits migration
Having established that EPC1 is an E2F1 target induced by genotoxic agents, we next sought to determine how EPC1 is functionally involved in advanced cancer development. SK-Mel-147 or UMUC3 cells, expressing high EPC1, were infected with sh.EPC1 or sh.control and then exposed to cisplatin (cDDP). FACS analysis showed that ablation of EPC1 in both cell lines leads to a substantial increase of apoptotic cells (Figure 4A). Additionally, we observed elevated chromatin condensation, a classical marker of cell death, in sh.EPC1 expressing SK-Mel-147 cells treated with cDDP (Supplementary Figure S1). In contrast, forced expression of EPC1 in damaged SK-Mel-29 or Saos-2 cells decreased the apoptotic rate, indicating that EPC1 overexpression confers cDDP resistance (Figure 4B). This also demonstrates that suppression of cell death by EPC1 does not require interference with p53 as Saos-2 are p53-negative. Taking into account that high levels of E2F1 can be responsible for the failure of a tumor to respond to anti-cancer drugs (55), we observed that E2F1-induced apoptosis is enhanced in EPC1-depleted cells (Figure 4C). Increased apoptotic rates are also detectable after knockdown of E2F1 itself in cDDP treated metastatic SK-Mel-147 (Figure 4D, left) as drug induced upregulation of normally pro-apoptotic E2F1, leading to increased anti-apoptotic EPC1 (Figure 4D right, lane 5), is abrogated through shE2F1 (right panel, lane 7). Accordingly, apoptosis in response to chemotherapy was partially ablated by concomitant overexpression of EPC1 in these cells (Figure 4D).
Figure 4.
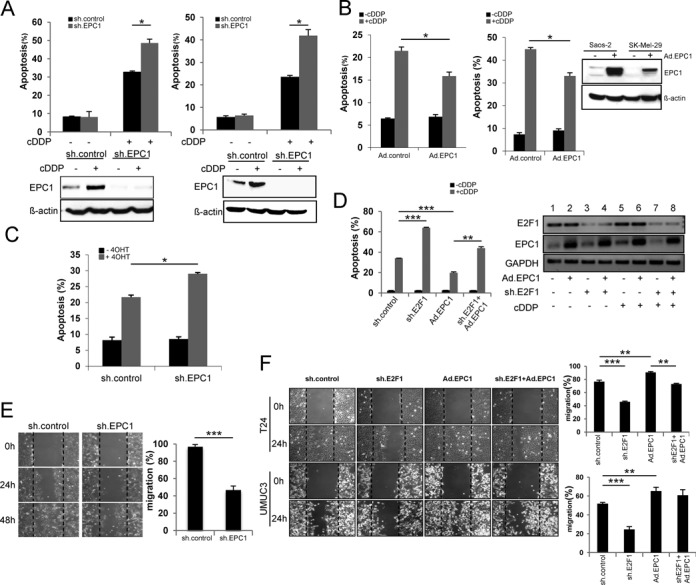
EPC1 inhibits DNA damage-induced apoptosis and promotes cell motility. (A) Percentage apoptotic SK-Mel-147 (left) and UMUC3 (right) cells treated with cisplatin (cDDP, 30 μM) for 48 h upon EPC1 knockdown. Cells were infected with lentivirus expressing sh.EPC1 or scrambled RNA (sh.control) and then treated with cDDP. Apoptosis was measured by PI staining and analyzed by FACS. (B) Percentage cell death of Saos-2 (left) and SK-Mel-29 (right) cells treated with cDDP for 48 h after overexpression of EPC1. Western blot confirmed EPC1 expression. (C) Saos2.ER-E2F1 cells were treated with 4OHT or solvent and apoptosis was measured with and without ablation of EPC1. (D) E2F1 or scrambled shRNA were expressed in SK-Mel-147 cells. Simultaneously, EPC1 was overexpressed by Ad.EPC1 infection for 24 h. Cells were treated with cDDP for 36 h and analyzed by FACS. (E) SK-Mel-147 cells were infected with lentivirus expressing scrambled or EPC1 shRNA for 96 h and analyzed in a scratch migration assay. Representative phase contrast microscopy images of the migration assays at 0, 24 and 48 h after gap creation are shown (left). Quantitative data are shown (right). (F) E2F1 or scrambled shRNA was expressed in metastatic bladder cancer cells T24 (upper) and UMUC3 (lower) followed by EPC1 overexpression by Ad.EPC1 infection. The migratory potential of tumor cells was analyzed by cell migration assays. Representative images at 0 and 24 h after gap creation are shown. Quantitative data are presented as bar graphs. Bars represent the mean ± SD of three independent experiments. Statistical significance was determined by two-sided Student's t-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Anti-cancer drug resistance caused by continuous cycles of chemotherapy promotes enhanced cell migration and cancer aggressiveness (56). Due to the obvious involvement of EPC1 in the repression of drug responsiveness, we analyzed the effect of endogenous EPC1 on cell motility. Knockdown of EPC1 in SK-Mel-147 cells significantly inhibits their migratory potential (Figure 4E). Likewise, depletion of E2F1 in invasive T24 and UMUC3 bladder cancer cell lines reduced cell motility which was, however, completely recovered after re-expression of EPC1, suggesting that EPC1 facilitates migration of cancer cells with elevated E2F1 levels (Figure 4F).
EPC1 physically interacts with E2F1 in vivo
As evident from our apoptosis assays, restoration of EPC1 expression in E2F1 ablated cells only partially protected cancer cells from drug-induced apoptosis when compared to cells overexpressing this PcG protein in the presence of E2F1 (Figure 4D). From this, we hypothesized that the pro-survival activity of EPC1 requires, at least in part, co-expression of E2F1. To gain further insight into the mechanism by which EPC1 regulates survival of malignant cells, sequence motif analysis (http://myhits.isb-sib.ch/cgi-bin/motif_scan) was performed for function prediction. EPC1 does not contain a predicted DNA binding domain (data not shown), implying that it most likely requires interaction with transcription factors to be recruited to DNA target sites. Attwool and coauthors previously identified E2F6 as a binding partner for EPC1 and this complex acts as a transcriptional repressor in vivo (57). To investigate whether EPC1 physically interacts with E2F1, we performed computational protein–protein interaction studies. For this purpose, we used our previously designed three-dimensional (3D) model of E2F1 (10). Moreover, in the absence of any X-ray structure of EPC1 protein, the 3D theoretical model was built, minimized and optimized for loops and side chains using the I-TASSER server and Smart Minimizer, Looper as well as ChiRotar tools of the Accelrys DS 3.5 software to obtain the most stable conformation. The optimized structures of E2F1 and EPC1 are shown in Figure 5A (top). Subsequently, protein–protein interactions between E2F1 and EPC1 were predicted by using the ZDOCK, RDOCK and ZRANK algorithms. Interestingly, we observed two distinct binding poses involved in the interactions, suggesting a dual functional role of EPC1 in the regulation of E2F1 activity (Figure 3A, bottom). Residues involved in the hydrogen bond formation between E2F1 and EPC1 are shown in Supplementary Table S2. To validate the predicted interaction between E2F1 and EPC1, we performed co-immunoprecipitation (IP) experiments in SK-Mel-147 cells that displayed the highest endogenous expression of both proteins using antibody specific for E2F1 to precipitate EPC1 from cell lysates. As shown in Figure 5B, EPC1 was precipitated by E2F1 antibody similar to the precipitates obtained from the IP with antibody against E2F6 used as positive control in the same cell lysate, whereas no band was visible in the control IgG lane. To validate the specificity of the IP, we chose E2F5 as a negative control, because it has the lowest sequence identity with E2F6 and further in silico molecular interaction studies between E2F5 and EPC1, performed as described above, predicted very weak interactions in comparison to E2F1 (data not shown). In line with these predictions, complex formation was not observed between EPC1 and E2F5 (Figure 5B). Additional evidence for a direct interaction of EPC1 and E2F1 is provided by in vitro binding assays using purified proteins, which demonstrate that EPC1 binds to GST-E2F1 but not to GST alone (Figure 5C). The interaction between EPC1 and E2F1 is further supported through their sub-cellular localization pattern, indicating that both proteins co-localize in the nucleus of SK-Mel-147 cells (Figure 5D).
Figure 5.

EPC1 directly interacts with E2F1. (A) Optimized protein structure of E2F1 and EPC1 with soft surface added and colored considering functional domains and regions. The model of E2F1 with DNA binding domain in green; dimerization domain in red; transactivation domain in blue; and Cyclin-A/CDK2 binding domain in yellow color. Remaining regions are shown in grey (upper left). The model of EPC1 protein with EPC1-A domain in cyan, EPC1-B domain in purple and EPC1-CQCT region in pink color (upper right). Systematic analyses of all possible interaction poses between E2F1 and EPC1 revealed two distinct sets of binding site patterns (lower). (B) Co-immunoprecipitation (Co-IP) analysis of endogenous EPC1 by anti-E2F1 or anti-E2F6 antibody in SK-Mel-147 cells. Input represents 10% of the protein amount used in the assays (left). UMUC3 cells were co-transfected with plasmids expressing EPC1 and either E2F1 or E2F5 (right). IPs were done by using the indicated antibodies and analyzed by Western blot. (C) GST or GST-E2F1 fusion protein was incubated with lysates from SK-Mel-29 transfected with pcDNA-EPC1 expression plasmid in a pull-down experiment and interaction was analyzed by Western blot. (D) Co-localization of endogenous EPC1 (red) and E2F1 (green) in SK-Mel-147 cells is shown by immunofluorescence. Nuclei were counter-stained with DAPI (blue).
E2F1 target genes involved in apoptosis are differentially regulated by EPC1
Given that EPC1 inhibits apoptosis triggered by anti-cancer drugs and binds to E2F1, we assumed that both proteins share a common set of target genes. We used a microarray-based approach to identify genes whose expression was altered through knockdown of EPC1 in SK-Mel-147 cells with highly metastatic and chemoresistant properties (11,55). Indeed, upon removal of EPC1 we observed the strongest expression changes among genes that are regulated by E2F1, suggesting that EPC1 and E2F1 cooperate in gene transcription (Figure 6A and Supplementary Table S3). In accordance with the two predicted distinct binding poses (Figure 5A), we noted that EPC1 depletion obviously has a differential impact on E2F1-mediated mRNA transcription by either suppressing or promoting the activity of this transcription factor in a target gene-specific manner. While genes implicated in cell survival and DNA repair such as BIRC5, BRCA1, BRCA2 and Bcl-2 were downregulated in sh.EPC1-expressing cells, transcription of E2F targets functionally related to cell cycle arrest and cell death e.g. p27, FAS, Casp3, Apaf-1 and Bim were clearly induced. Several genes were chosen for validation of the microarray results by qPCR analysis. As shown in Figure 6B, mRNA levels of the anti-apoptotic targets Bcl-2 and BIRC5 decreased in EPC1 ablated melanoma cells, whereas transcripts of pro-apoptotic targets Bim and p27 increased after sh.EPC1 treatment (Figure 6B). This differential regulation of E2F1 target genes by EPC1 was also evident on protein level, demonstrating that depletion of EPC1 through two distinct shRNAs facilitates expression of pro-apoptotic and silencing of anti-apoptotic genes (Figure 6C).
Figure 6.
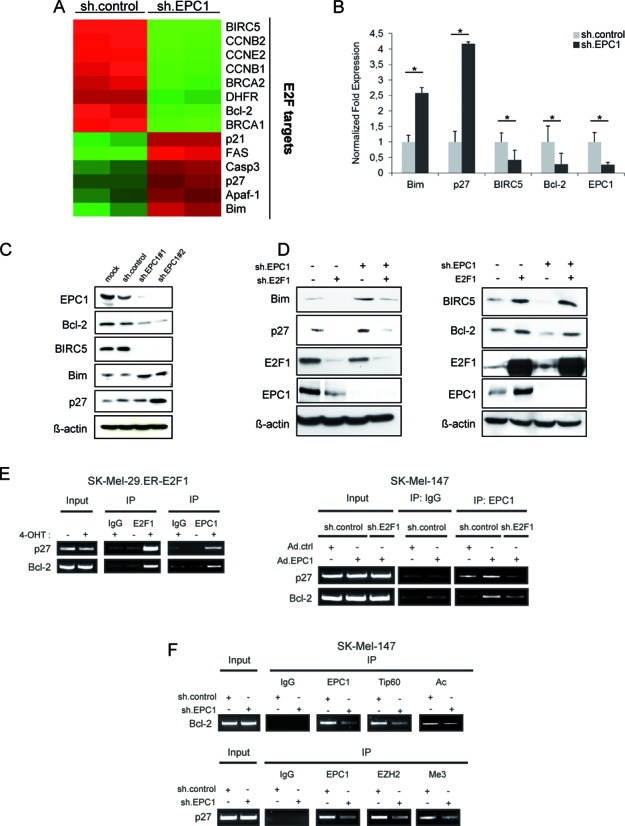
EPC1 regulates E2F1 targets. (A) Equal amounts of RNA from sh.control and sh.EPC1 infected cells were subjected to microarray analysis. Affymetrix gene expression data were analyzed using hierarchical clustering. Gene expression levels for E2F target genes are displayed. Colors represent normalized expression levels. Red column represents high gene expression, while green column indicates low expression. (B and C) mRNA (B) and protein levels (C) of Bcl-2, BIRC5, p27 and Bim were regulated after knockdown of EPC1. SK-Mel-147 cells were infected with Lenti.shRNA against EPC1 or scrambled RNA and harvested 96 h later. GAPDH and actin were used as normalization as well as loading control for qPCR and Western blot, respectively. Bar graphs indicate the mean ± SD of two separate experiments. (D) SK-Mel-147 cells stably expressing sh.EPC1, sh.E2F1 or both were harvested for Western blot analysis of EPC1, E2F1, Bim and p27 expression (left). Actin was used as loading control. SK-Mel-147 stable cell lines expressing EPC1 shRNA or scrambled shRNA were transfected with E2F1 expression vector (right). 48 h after transfection cells were harvested and EPC1, E2F1, BIRC5 and Bcl-2 protein levels were analyzed by Western blot. Actin was used for loading control. (E) Binding of endogenous EPC1 to indicated E2F1-regulated promoters after 4-OHT treatment in SK-Mel-29.ER-E2F1 (left) and E2F1 knockdown in SK-Mel-147 cells (right) was determined by ChIP assays. (F) Recruitment of Tip60 and EZH2 to E2F target gene promoters and associated acetylation (Ac) or tri-methylation (Me3) levels in the presence and absence of EPC1. (E and F) Input represents 10% of sheared chromatin prior to IP. Statistical significance was determined by two-sided Student's t-test. *P ≤ 0.05.
To analyze whether upregulation of pro-apoptotic genes after loss of EPC1 strictly depends on E2F1 activity, we examined whether the concomitant knockdown of both proteins prevents induction of Bim and p27 gene expression. Bim and p27 protein levels increased upon EPC1 depletion, which was not the case when sh.E2F1 was co-expressed (Figure 6D, left). In addition, overexpression of E2F1 in sh.EPC1 cells completely rescues Bcl-2 and BIRC5 expression (Figure 6D, right) indicating that EPC1 requires E2F1 to trigger the activation of both genes. In vivo binding of endogenous EPC1 to E2F1-regulated promoters was verified by ChIP. As shown in Figure 6E (left), activation of E2F1 through 4-OHT in SK-Mel-29.ER-E2F1 cells led to an enrichment of the p27 and Bcl-2 promoter regions bound by E2F1 using EPC1 antibody. Binding of EPC1 to these promoters decreased sharply upon E2F1 depletion in SK-Mel-147 overexpressing EPC1 (Figure 6E, right), demonstrating that EPC1 association to E2F1-regulated promoters depends on E2F1. Furthermore, we observed that Tip60, a component of the NuA4 acetyltransferase complex and interaction partner of E2F1 and EPC1 (58–60), is recruited to the E2F site of the Bcl-2 promoter and knockdown of EPC1 in SK-Mel-147 cells reduces Tip60 binding and associated histone acetylation levels (Figure 6F, top). In contrast, ablation of EPC1 removes the PRC2 component EZH2 from the p27 promoter resulting in decreased histone methylation (Figure 6F, bottom) for E2F1-mediated gene activation as shown in Figure 6D (left). Together, these results suggest that EPC1 might alter the balance between anti- and pro-apoptotic genes induced by E2F1 in high-grade cancer cells, enabling evasion from drug therapy and promoting malignant progression.
EPC1 acts as activator or repressor of E2F1 transcriptional activity on distinct promoters
EPC1 was initially supposed to act as a transcriptional repressor (57). However, our data support a model where EPC1 as cofactor has dual functions in the regulation of E2F1 activity by either stimulating or repressing target mRNA transcription. Clearly, luciferase reporter assays revealed a strong increase of E2F1-induced Bcl-2 promoter activity upon addition of EPC1 in less aggressive SK-Mel-29 cells, although EPC1 alone has no stimulating effect (Figure 7A, left). A similar activity of EPC1 was observed for the BIRC5 promoter (Figure 7A, right). By contrast, co-expression of E2F1 and EPC1 in these cells resulted in a marked reduction of p27 promoter activation (Figure 7B), suggesting that EPC1 contributes to the inhibition of E2F1 transcriptional activity on this promoter. These results demonstrate that EPC1 is capable of serving as both, transcriptional activator and repressor in a gene-specific manner. This occurs only in conjunction with E2F1 since EPC1 alone has no effect on these promoters. Our in silico docking analyses suggested that interaction of EPC1 involves the transactivation domain of E2F1 in both potential binding poses (Figure 5A and Supplementary Table S2). To test this hypothesis, we cotransfected EPC1 expression vector with Flag-tagged full-length E2F1 (1–437) or mutants lacking the transactivation domain (1–368). Whereas addition of EPC1 led to an increase of E2F1 transcriptional activity on the Bcl-2 promoter, p27 reporter activity decreased, but had no influence on the transactivation-deficient E2F1 mutant (1–368) on both promoters (Figure 7C and D). Co-expression of truncated E2F1 together with EPC1 was verified by Western blot (Figure 7E). In addition, we performed E2F1-driven promoter assays by using two different EPC1 truncated constructs and observed that EPC1-A and EPC1-B domains alone are sufficient to enhance Bcl-2 promoter activity comparable to full length protein (Figure 8A). Likewise, EPC1-A and EPC1-B are also required for inhibiting p27 promoter induction by E2F1 (Figure 8B). These findings validate our in silico interaction model predicting that E2F1 binds EPC1-A and EPC1-B domains. Overall this suggests that EPC1 modulates E2F1-dependent transcription by forcing survival-related and blocking tumor suppressor gene signatures, thereby acting as a driver of disease progression. This is also supported by clinical data from patients with bladder and breast cancer, indicating that EPC1 overexpression significantly correlates with poor survival prognosis as revealed by Kaplan-Meier analysis of RNA-Seq data from the TCGA cohorts (Figure 9A). In addition, we compared gene expression profiles of metastatic SK-Mel-147 cells expressing shRNAs either against E2F1 or EPC1 (Figures 1A and 6A) and observed a concerted downregulation of an E2F1 target cluster previously described to promote drug resistance and tumor cell invasion (10–11,35,61–64). Intriguingly, these genes are highly coexpressed in patients with bladder and breast cancer, suggesting that EPC1 and E2F1 cooperativity causes activation of metastasis-related gene signatures in these late-stage tumors (Figure 9B).
Figure 7.
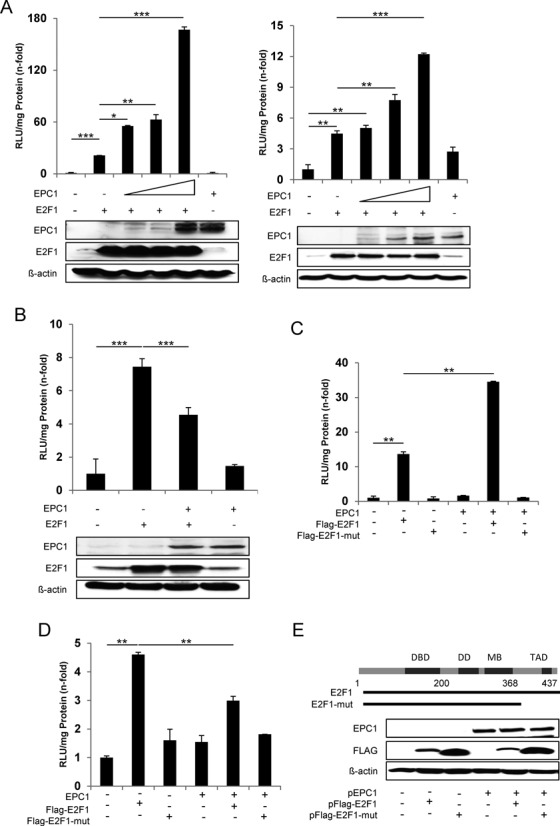
Modulation of E2F1 transcriptional activity by EPC1. (A) Regulation of E2F1 transcriptional activity by EPC1. SK-Mel-29 cells were transfected with 0.5 μg Bcl-2-luc (left) or BIRC5-luc reporter plasmids (right) together with expression vectors for E2F1 (0.5 μg) in the absence or presence of EPC1 expression vector (1, 2, 3 μg). E2F1 and EPC1 expression was confirmed by Western blot. Actin was used as loading control. (B) SK-Mel-29 cells were cotransfected with the p27-luc reporter plasmid (0.5 μg) and expression plasmids for E2F1 (0.5 μg) in the absence or presence of EPC1 expression vectors (3 μg). E2F1, EPC1 and actin expression was confirmed by Western blot. (C and D) H1299 cells were transfected with Bcl-2-luc (C) or p27-luc reporter plasmids (D) together with expression vectors for Flag-tagged full-length E2F1 or Flag-tagged E2F1 mutant in the presence or absence of EPC1 expression vector. Luciferase assays were performed after 24 h using pcDNA plasmid as control that was set to 1. Data are presented as the mean with standard deviation of technical replicates (duplicates or triplicates) in an experiment representative of several independent others. (E) Schematic representation of full-length and truncated E2F1 proteins. Western blot shows expression of Flag-tagged full-length E2F1 or Flag-tagged E2F1 mutant and EPC1 in H1299 cells. Actin was used as loading control. Statistical significance was determined by two-sided Student's t-test. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 8.
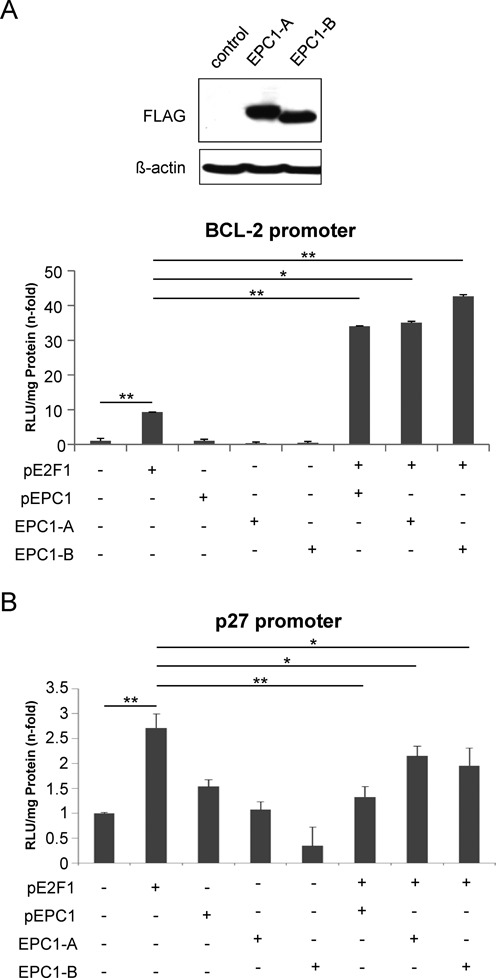
EPC1-A and EPC1-B domains alone are sufficient to modulate transcriptional activity of E2F1. (A and B) H1299 cells were co-transfected with expression plasmids for full-length EPC1, Flag-tagged EPC1-A or EPC1-B together with Bcl-2-luc (A) or p27-luc promoter (B) reporter constructs in the absence or presence of transient E2F1 expression. Expression of truncated EPC1 proteins was confirmed by Western blot by using anti-FLAG antibody. Actin was used as loading control. Promoter assays were performed 24 h post transfection using pcDNA plasmid as control that was set to 1. Data are presented as the mean with standard deviation of technical duplicates in an experiment representative of several independent others. Statistical significance was determined by two-sided Student's t-test. *P ≤ 0.05, **P ≤ 0.01.
Figure 9.
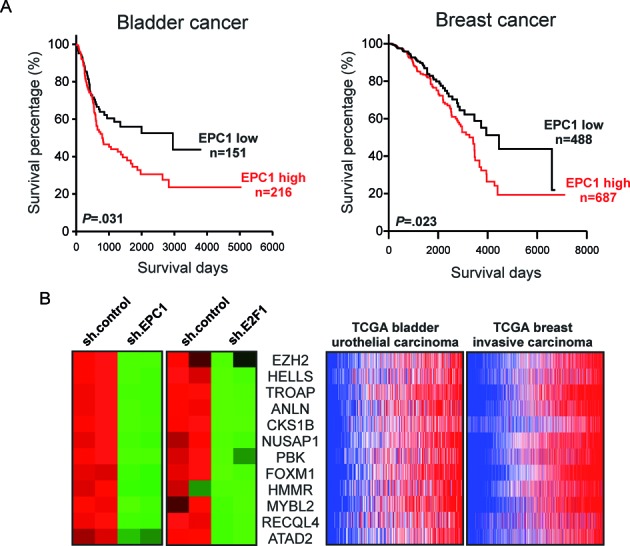
High EPC1 expression coincides with poor overall survival of bladder and breast cancer patients and activation of E2F1 target signatures promoting metastasis. (A) Overall survival curves of Kaplan-Meier analyses indicate that patient with high EPC1 transcript levels had significantly poorer survival than patients with low EPC1 expression. P-values for survival analyses were determined using log-rank test. (B) Heatmaps depicting differential gene expression of a metastasis-related genes in EPC1 or E2F1 knockdown SK-Mel-147 cells (left) and their coexpression in TCGA RNA-Seq datasets by using the cancer browser.
DISCUSSION
In the present study, we identified EPC1, a largely uncharacterized member of the polycomb group protein family, as novel E2F1 target gene that restrains E2F1-mediated cell death in response to anti-cancer drug treatment. This is achieved by direct protein–protein interaction with E2F1, which enhances or inhibits its transactivation potential on distinct target genes. Our data provide a plausible explanation for the aberrant expression of E2F1 in highly chemotherapy-resistant cancers and the loss of its tumor suppressor activity in these cells. As our findings suggest that the established E2F1-EPC1 regulatory circuit is potentiated under genotoxic treatment in order for cancer to progress, targeting EPC1 might be a promising avenue to reactivate the proapoptotic function of E2F1 and avoid cancer propagation.
EPC1 was first identified in Drosophila to enhance the phenotypes of homozygotic mutations of other PcG genes (65,66). The homologues of EPC1 were also found in other eukaryotes and largely conserved from yeast to human (67). EPC1 belongs to the PcG gene family which functions mainly through epigenetic gene silencing and the maintenance of cell type specificity (68). In addition, EPC1 was identified as a component of the NuA4 (or Tip60-EP400) complex, which possesses histone acetyltransferase activity. EPC1 contains different domains and previous studies suggested transcriptional activation and repression capabilities between the EPC1-A region, on one hand, and the EPC1-B region on the other (69). A dual function of PcG proteins is also observed for other family members. For example, EZH2 silences gene expression through its histone methyltransferase activity but was also reported to act as a coactivator inducing a subset of its targets in castration-resistant prostate cancer in a polycomb-independent manner (70,71). The current study indicates that EPC1, in analogy to EZH2, has a unique role amongst the PcG protein family.
In mammalian biology, most reports focused on the function of EPC1 regarding mouse skeletal muscle differentiation (72–74). EPC1 is required for initiating skeletal muscle differentiation and appears to induce relevant genes via the interaction with Homeodomain Only Protein (Hop), serum response factor (SRF), and/or RET finger protein (RFP) in mice (69,74–75). Apart from this, there are only a few hints linking EPC1 to cancer. The gene is mapped to human chromosome 10p11, a genomic region frequently rearranged in various human tumors (76,77). EPC1 deregulation has been observed in Adult T-cell leukemia/lymphoma (ATLL), a malignant tumor caused by latent human T-lymphotropic virus 1 (HTLV-1) infection. In acute-type ATLL, there is a common breakpoint cluster region at the EPC1 gene locus (78). Biankin et al. reported somatic gene mutations in pancreatic adenocarcinoma (76) and another study demonstrated that side population (SP) cells, exhibiting tumor-initiating characteristics, express EPC1 more strongly than non-SP cells (79). Very recently, a targeted knockdown screen of chromatin regulatory genes, identified EPC1 and EPC2 as critical cofactors in acute myeloid leukemia (AML) required to sustain the oncogenic potential of MLL leukemia stem cells (80). These data imply that EPC1 might contribute to solid tumor development although the mechanisms were unclear. Our data are mostly consistent with this idea as high-grade melanoma, bladder, and breast cancer cells express higher EPC1 levels than low-grade tumors. Since E2F1 and thereby EPC1 is induced through chemotherapy of primary tumors, patient treatment may favour drug resistance and metastasis. Establishing a significant link between EPC1 and E2F1 in advanced cancer cells, these findings led us to postulate that their direct interaction contributes to an aggressive tumor phenotype.
Compelling evidence indicates that DNA damage signaling induces stabilization of E2F1, which in turn blocks apoptosis induced by cellular stress. Our group recently demonstrated that E2F1 confers anti-cancer drug resistance by influencing ABC transporter family members and Bcl-2 in melanoma through a DNp73-mediated mechanism, which involves miR-205 downregulation (55). Despite this progress, little is known on how the apoptotic activity of E2F1 is repressed in therapy resistant tumors. Here, we have shown that EPC1 overexpression results in a dramatic decrease in cancer cell death in response to drug treatment, whereas its knockdown increased the sensitivity to DNA damage. Moreover, the effect of EPC1 on E2F1-induced drug resistance was demonstrated by depleting E2F1 and simultaneous EPC1 overexpression. Increased apoptosis after E2F1 knockdown is partially abolished upon EPC1 replacement, suggesting that E2F1-mediated drug resistance is to a large extent dependent on its target EPC1. This apoptosis inhibitory effect of EPC1 in cancer cells is achieved by either preventing expression of pro-apoptotic genes such as Bim or driving accumulation of anti-apoptotic proteins like Bcl-2. In this regard, EPC1 clearly differs from EZH2, which has been shown to inhibit E2F1-induced apoptosis but unlike EPC1, EZH2 is only able to repress E2F1 pro-apoptotic target genes (31).
In accordance with the observed physical interaction between EPC1 and E2F1 in advanced chemoresistant cells, depletion of the latter abrogated differential regulation of gene expression by EPC1, demonstrating that E2F1 is required for EPC1 activity. Previous reports indicated that chemoresistant cancer cells exhibit an increased migratory and invasive potential (81), which was also observed in epirubicin-resistant MCF-7 cells with elevated E2F1 and EPC1 protein levels (data not shown). Together with our observation that the migratory capacity of aggressive tumor cells, which strongly decreased after ablation of E2F1, is rescued by ectopic expression of EPC1, this emphasizes that the oncogenic activity of this PcG protein in transformed cells extends beyond inhibition of apoptosis when connected to E2F1, likely playing also an active role in stimulating cancer progression-relevant genes. Supporting a specific function of the EPC1-E2F1 complex in inducing resistant and aggressive traits, the EPC1-E2F6 repressive complex, shown to interact with EZH2 through binding to EPC1 in a proliferation-specific manner in human fibroblast cells, rather contributes to cell cycle control and transformation (57).
EPC1 has been identified as a component of the PRC2 complex, which includes EZH2, EED and SUZ12, as well as the NuA4 complex, both involved in repression or activation of transcription. EZH2 is thought to confer histone lysine methyltransferase activity (H3K27me3) that is associated with the repressive activity of the PRC2 complex. This complex promotes T-cell leukemogenesis through inhibiting the expression of HOXA9 and HOXA10, two major differentiation factors of the NK-cell lineage (82). Therefore; EPC1 may repress downstream genes by recruiting EZH2 or other PcG proteins for methylation of histone residues. In fact, our results showing EPC1-dependent EZH2 enrichment in conjunction with elevated histone methylation levels at E2F sites of the p27 promoter suggest that EPC1-mediated PRC2 promoter occupancy is an important mechanism to restrict p27 induction by E2F1 in metastatic cancer cells. On the other hand, EPC1 as a part of NuA4 complex is involved in histone H4/H2A aceyltransferase activity (AcH4) (83). Complexes containing histone acetyltransferases induce transactivation of downstream genes by introducing structural modifications in the nucleosome of the promoter region, for example the NuA4 complex is required for transactivation mediated by p53 (83,84). In murine embryonic stem (ES) cells as well as cancer cells, MYC was shown to interact with the NuA4 complex, which has a positive correlation with active histone signatures (58). In concert with these earlier observations we show that ablation of EPC1 prevents binding of Tip60 to the Bcl2 promoter and that this is associated with reduced histone acetylation. These findings indicate that EPC1 is required for the formation of distinct complexes to either repress or activate E2F1 target gene expression by gene-specific epigenetic modifications.
The marked box and flanking region of E2F1 was first found to be important in the interaction of E2F1 with the adenoviral protein E4 (85), but it is also critical for the regulation of transcriptional activity, promoter selectivity and induction of apoptosis by E2F1 (86). Previous studies identified proteins interacting specifically with the marked box domain of E2F1, such as KAP1 (87), Jab1 (88), DP1 (89), prohibitin (90) and ppRB (91). Our in silico analyses implicate that EPC1 directly interacts with different domains of E2F1 including the marked box and the transactivation domain. Moreover, the computational protein–protein interaction studies of EPC1 and E2F1 predicted that only the EPC1-A and EPC1-B domains were involved. This is in accordance with our experimental results where the transcriptional activity of E2F1 was either increased or suppressed in the presence of EPC1-A and EPC1-B domains in a promoter-specific manner. An earlier study described a unique transactivating capacity of the A region and a weak repressive effect of the B region on luciferase reporter constructs harboring the SV40 promoter (69).
Intriguingly, our experiments clearly indicate that both, A and B domains, are able to repress or enhance E2F1 transactivation efficiency suggesting the recruitment of cofactors such as EZH2 or Tip60 by EPC1 which induce gene activation or silencing apparently depending on the promoter-context of the E2F1 target. We performed additional computational protein–protein interaction studies clearly indicating that isolated EPC1-A and B domains can each interact with E2F1 and EZH2 or Tip60 to form stable complexes (data not shown). In this scenario EPC1 might act as a crucial bridging factor to connect E2F1 to essential components of the chromatin remodeling complexes NuA4 or PRC2.
Epigenetic mechanisms like chromatin remodeling play an important role in tumor metastasis since they activate gene programs leading to mesenchymal tumor phenotypes and cell dissemination. In this regard, we uncovered tumor progression-relevant genes that are co-regulated by E2F1 and EPC1 which promote metastasis in different types of cancer predicting poor patient survival. Further studies are needed to reveal the mechanism(s) enabling EPC1 to differentiate between transactivation of oncogenes and inhibition of tumor suppressor genes in order to understand how this epigenetic regulator contributes to the radical switch of E2F1 from a mediator of cell death toward an accelerator of tumor progression.
In conclusion, our study defines a novel regulatory mechanism by which E2F1 pro-apoptotic activity is abolished through EPC1 in human cancer cells, raising the potential to develop inhibitors of EPC1 as a new therapeutic option for cancer. In addition, EPC1 silencing cooperates with DNA damaging agents in re-sensitizing aggressive tumor cells to apoptosis and inhibits cell migration. Thus, an effective combination treatment may set novel standards against metastatic cancer.
Supplementary Material
Acknowledgments
The authors thank Dr D. Koczan (Rostock University Medical Center, Germany) for microarray analysis. We are grateful to Dr T. Sakai (Kyoto Prefectural University of Medicine, Japan), Dr P. Obexer (Tyrolean Cancer Research Institute, Austria) and Dr W. Xiao (Institute of Hydrobiology, CAS, China) for providing p27 and BIRC5 promoter constructs as well as E2F1 pCMV-FLAG vectors. We greatly appreciate the kind gift of Dr E.W. Lam (Imperial College London, UK) for providing the MCF7-EPIR cell line.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
German Cancer Aid, Dr Mildred Scheel Stiftung [109801 to B.M.P. and D.E.]; German Federal Ministry of Education and Research (BMBF) as part of the eBio:SysMet project [0316171 to B.M.P.]. Funding for open access charge: German Cancer Aid, Dr Mildred Scheel Stiftung [109801 to B.M.P. and D.E.]; German Federal Ministry of Education and Research (BMBF) as part of the eBio:SysMet project [0316171 to B.M.P.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Polager S., Ginsberg D. E2F - at the crossroads of life and death. Trends Cell Biol. 2008;18:528–535. doi: 10.1016/j.tcb.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 2.Engelmann D., Putzer B.M. Translating DNA damage into cancer cell death-A roadmap for E2F1 apoptotic signalling and opportunities for new drug combinations to overcome chemoresistance. Drug Resist. Updat. 2010;13:119–131. doi: 10.1016/j.drup.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 3.Stanelle J., Putzer B.M. E2F1-induced apoptosis: turning killers into therapeutics. Trends Mol. Med. 2006;12:177–185. doi: 10.1016/j.molmed.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 4.Engelmann D., Putzer B.M. The Dark Side of E2F1: In Transit beyond Apoptosis. Cancer Res. 2012;72:571–575. doi: 10.1158/0008-5472.CAN-11-2575. [DOI] [PubMed] [Google Scholar]
- 5.Putzer B.M., Engelmann D. E2F1 apoptosis counterattacked: evil strikes back. Trends Mol. Med. 2013;19:89–98. doi: 10.1016/j.molmed.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Lee J.S., Leem S.H., Lee S.Y., Kim S.C., Park E.S., Kim S.B., Kim S.K., Kim Y.J., Kim W.J., Chu I.S. Expression signature of E2F1 and its associated genes predict superficial to invasive progression of bladder tumors. J. Clin. Oncol. 2010;28:2660–2667. doi: 10.1200/JCO.2009.25.0977. [DOI] [PubMed] [Google Scholar]
- 7.Sharma A., Yeow W.S., Ertel A., Coleman I., Clegg N., Thangavel C., Morrissey C., Zhang X., Comstock C.E., Witkiewicz A.K., et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. 2010;120:4478–4492. doi: 10.1172/JCI44239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Knoll S., Furst K., Kowtharapu B., Schmitz U., Marquardt S., Wolkenhauer O., Martin H., Putzer B.M. E2F1 induces miR-224/452 expression to drive EMT through TXNIP downregulation. EMBO Rep. 2014;15:1315–1329. doi: 10.15252/embr.201439392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Engelmann D., Mayoli-Nussle D., Mayrhofer C., Furst K., Alla V., Stoll A., Spitschak A., Abshagen K., Vollmar B., Ran S., et al. E2F1 promotes angiogenesis through the VEGF-C/VEGFR-3 axis in a feedback loop for cooperative induction of PDGF-B. J. Mol. Cell Biol. 2013;5:391–403. doi: 10.1093/jmcb/mjt035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Meier C., Spitschak A., Abshagen K., Gupta S., Mor J.M., Wolkenhauer O., Haier J., Vollmar B., Alla V., Putzer B.M. Association of RHAMM with E2F1 promotes tumour cell extravasation by transcriptional up-regulation of fibronectin. J. Pathol. 2014;234:351–364. doi: 10.1002/path.4400. [DOI] [PubMed] [Google Scholar]
- 11.Alla V., Engelmann D., Niemetz A., Pahnke J., Schmidt A., Kunz M., Emmrich S., Steder M., Koczan D., Putzer B.M. E2F1 in melanoma progression and metastasis. J. Natl. Cancer Inst. 2010;102:127–133. doi: 10.1093/jnci/djp458. [DOI] [PubMed] [Google Scholar]
- 12.Johnson J.L., Pillai S., Pernazza D., Sebti S.M., Lawrence N.J., Chellappan S.P. Regulation of Matrix Metalloproteinase Genes by E2F Transcription Factors: Rb-Raf-1 Interaction as a Novel Target for Metastatic Disease. Cancer Res. 2012;72:516–526. doi: 10.1158/0008-5472.CAN-11-2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z., Guo Y., Jiang H., Zhang T., Jin C., Young C.Y., Yuan H. Differential regulation of MMPs by E2F1, Sp1 and NF-kappa B controls the small cell lung cancer invasive phenotype. BMC Cancer. 2014;14:276. doi: 10.1186/1471-2407-14-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanelle J., Stiewe T., Theseling C.C., Peter M., Putzer B.M. Gene expression changes in response to E2F1 activation. Nucleic Acids Res. 2002;30:1859–1867. doi: 10.1093/nar/30.8.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon S.O., Shin S., Mercurio A.M. Ras stimulation of E2F activity and a consequent E2F regulation of integrin alpha6beta4 promote the invasion of breast carcinoma cells. Cancer Res. 2006;66:6288–6295. doi: 10.1158/0008-5472.CAN-06-0826. [DOI] [PubMed] [Google Scholar]
- 16.Andrechek E.R. HER2/Neu tumorigenesis and metastasis is regulated by E2F activator transcription factors. Oncogene. 34:217–225. doi: 10.1038/onc.2013.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shackney S.E., Chowdhury S.A., Schwartz R. A novel subset of human tumors that simultaneously overexpress multiple E2F-responsive genes found in breast, ovarian, and prostate cancers. Cancer Inform. 2014;13:89–100. doi: 10.4137/CIN.S14062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Levine S.S., King I.F., Kingston R.E. Division of labor in polycomb group repression. Trends Biochem. Sci. 2004;29:478–485. doi: 10.1016/j.tibs.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16:2893–2905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller J., Hart C.M., Francis N.J., Vargas M.L., Sengupta A., Wild B., Miller E.L., O'Connor M.B., Kingston R.E., Simon J.A. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell. 2002;111:197–208. doi: 10.1016/s0092-8674(02)00976-5. [DOI] [PubMed] [Google Scholar]
- 21.Simon J.A., Kingston R.E. Occupying chromatin: Polycomb mechanisms for getting to genomic targets, stopping transcriptional traffic, and staying put. Mol. Cell. 2013;49:808–824. doi: 10.1016/j.molcel.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao R., Tsukada Y., Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol. Cell. 2005;20:845–854. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Francis N.J., Kingston R.E., Woodcock C.L. Chromatin compaction by a polycomb group protein complex. Science. 2004;306:1574–1577. doi: 10.1126/science.1100576. [DOI] [PubMed] [Google Scholar]
- 24.Kirmizis A., Bartley S.M., Farnham P.J. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol. Cancer Ther. 2003;2:113–121. [PubMed] [Google Scholar]
- 25.Sawa M., Yamamoto K., Yokozawa T., Kiyoi H., Hishida A., Kajiguchi T., Seto M., Kohno A., Kitamura K., Itoh Y., et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int. J. Hematol. 2005;82:42–47. doi: 10.1532/IJH97.05013. [DOI] [PubMed] [Google Scholar]
- 26.van Kemenade F.J., Raaphorst F.M., Blokzijl T., Fieret E., Hamer K.M., Satijn D.P., Otte A.P., Meijer C.J. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001;97:3896–3901. doi: 10.1182/blood.v97.12.3896. [DOI] [PubMed] [Google Scholar]
- 27.Kleer C.G., Cao Q., Varambally S., Shen R., Ota I., Tomlins S.A., Ghosh D., Sewalt R.G., Otte A.P., Hayes D.F., et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varambally S., Dhanasekaran S.M., Zhou M., Barrette T.R., Kumar-Sinha C., Sanda M.G., Ghosh D., Pienta K.J., Sewalt R.G., Otte A.P., et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419:624–629. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 29.Vekony H., Raaphorst F.M., Otte A.P., van Lohuizen M., Leemans C.R., van der Waal I., Bloemena E. High expression of Polycomb group protein EZH2 predicts poor survival in salivary gland adenoid cystic carcinoma. J. Clin. Pathol. 2008;61:744–749. doi: 10.1136/jcp.2007.054262. [DOI] [PubMed] [Google Scholar]
- 30.Bracken A.P., Pasini D., Capra M., Prosperini E., Colli E., Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z.L., Zheng S.S., Li Z.M., Qiao Y.Y., Aau M.Y., Yu Q. Polycomb protein EZH2 regulates E2F1-dependent apoptosis through epigenetically modulating Bim expression. Cell Death Differ. 2010;17:801–810. doi: 10.1038/cdd.2009.162. [DOI] [PubMed] [Google Scholar]
- 32.Nowak K., Kerl K., Fehr D., Kramps C., Gessner C., Killmer K., Samans B., Berwanger B., Christiansen H., Lutz W. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–1754. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bracken A.P., Kleine-Kohlbrecher D., Dietrich N., Pasini D., Gargiulo G., Beekman C., Theilgaard-Monch K., Minucci S., Porse B.T., Marine J.C., et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007;21:525–530. doi: 10.1101/gad.415507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ji J.Y., Miles W.O., Korenjak M., Zheng Y., Dyson N.J. In vivo regulation of E2F1 by Polycomb group genes in Drosophila. G3 (Bethesda) 2012;2:1651–1660. doi: 10.1534/g3.112.004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Millour J., de Olano N., Horimoto Y., Monteiro L.J., Langer J.K., Aligue R., Hajji N., Lam E.W. ATM and p53 regulate FOXM1 expression via E2F in breast cancer epirubicin treatment and resistance. Mol. Cancer Ther. 2011;10:1046–1058. doi: 10.1158/1535-7163.MCT-11-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Engelmann D., Knoll S., Ewerth D., Steder M., Stoll A., Putzer B.M. Functional interplay between E2F1 and chemotherapeutic drugs defines immediate E2F1 target genes crucial for cancer cell death. Cell. Mol. Life Sci. 2010;67:931–948. doi: 10.1007/s00018-009-0222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stiewe T., Putzer B.M. Role of the p53-homologue p73 in E2F1-induced apoptosis. Nat. Genet. 2000;26:464–469. doi: 10.1038/82617. [DOI] [PubMed] [Google Scholar]
- 38.Obexer P., Hagenbuchner J., Unterkircher T., Sachsenmaier N., Seifarth C., Bock G., Porto V., Geiger K., Ausserlechner M. Repression of BIRC5/survivin by FOXO3/FKHRL1 sensitizes human neuroblastoma cells to DNA damage-induced apoptosis. Mol. Biol. Cell. 2009;20:2041–2048. doi: 10.1091/mbc.E08-07-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murata K., Hattori M., Hirai N., Shinozuka Y., Hirata H., Kageyama R., Sakai T., Minato N. Hes1 directly controls cell proliferation through the transcriptional repression of p27Kip1. Mol. Cell. Biol. 2005;25:4262–4271. doi: 10.1128/MCB.25.10.4262-4271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji W., Zhang W., Xiao W. E2F-1 directly regulates thrombospondin 1 expression. PLoS ONE. 2010;5:e13442. doi: 10.1371/journal.pone.0013442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Racek T., Buhlmann S., Rust F., Knoll S., Alla V., Putzer B.M. Transcriptional repression of the prosurvival endoplasmic reticulum chaperone GRP78/BIP by E2F1. J. Biol. Chem. 2008;283:34305–34314. doi: 10.1074/jbc.M803925200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roy A., Kucukural A., Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins. 2007;69(Suppl. 8):108–117. doi: 10.1002/prot.21702. [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li L., Chen R., Weng Z. RDOCK: refinement of rigid-body protein docking predictions. Proteins. 2003;53:693–707. doi: 10.1002/prot.10460. [DOI] [PubMed] [Google Scholar]
- 46.Lorenzen S., Zhang Y. Identification of near-native structures by clustering protein docking conformations. Proteins. 2007;68:187–194. doi: 10.1002/prot.21442. [DOI] [PubMed] [Google Scholar]
- 47.Chen R., Li L., Weng Z. ZDOCK: an initial-stage protein-docking algorithm. Proteins. 2003;52:80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- 48.Bracken A.P., Helin K. Polycomb group proteins: navigators of lineage pathways led astray in cancer. Nat. Rev. Cancer. 2009;9:773–784. doi: 10.1038/nrc2736. [DOI] [PubMed] [Google Scholar]
- 49.Lee Y.G., Macoska J.A., Korenchuk S., Pienta K.J. MIM, a potential metastasis suppressor gene in bladder cancer. Neoplasia. 2002;4:291–294. doi: 10.1038/sj.neo.7900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang F., Zhang R., He Y., Zou M., Guo L., Xi T. MicroRNA-125b induces metastasis by targeting STARD13 in MCF-7 and MDA-MB-231 breast cancer cells. PLoS ONE. 2012;7:e35435. doi: 10.1371/journal.pone.0035435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thomas S., Overdevest J.B., Nitz M.D., Williams P.D., Owens C.R., Sanchez-Carbayo M., Frierson H.F., Schwartz M.A., Theodorescu D. Src and caveolin-1 reciprocally regulate metastasis via a common downstream signaling pathway in bladder cancer. Cancer Res. 2011;71:832–841. doi: 10.1158/0008-5472.CAN-10-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin W.C., Lin F.T., Nevins J.R. Selective induction of E2F1 in response to DNA damage, mediated by ATM-dependent phosphorylation. Genes Dev. 2001;15:1833–1844. [PMC free article] [PubMed] [Google Scholar]
- 53.Pediconi N., Ianari A., Costanzo A., Belloni L., Gallo R., Cimino L., Porcellini A., Screpanti I., Balsano C., Alesse E., et al. Differential regulation of E2F1 apoptotic target genes in response to DNA damage. Nat. Cell Biol. 2003;5:552–558. doi: 10.1038/ncb998. [DOI] [PubMed] [Google Scholar]
- 54.de Olano N., Koo C.Y., Monteiro L.J., Pinto P.H., Gomes A.R., Aligue R., Lam E.W. The p38 MAPK-MK2 axis regulates E2F1 and FOXM1 expression in epirubicin treatment. Mol. Cancer Res. 2012;10:1189–1202. doi: 10.1158/1541-7786.MCR-11-0559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alla V., Kowtharapu B.S., Engelmann D., Emmrich S., Schmitz U., Steder M., Putzer B.M. E2F1 confers anticancer drug resistance by targeting ABC transporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry. Cell Cycle. 2012;11:3067–3078. doi: 10.4161/cc.21476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Su Y.C., Lin Y.H., Zeng Z.M., Shao K.N., Chueh P.J. Chemotherapeutic agents enhance cell migration and epithelial-to-mesenchymal transition through transient up-regulation of tNOX (ENOX2) protein. Biochim. Biophys. Acta. 2012;1820:1744–1752. doi: 10.1016/j.bbagen.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 57.Attwooll C., Oddi S., Cartwright P., Prosperini E., Agger K., Steensgaard P., Wagener C., Sardet C., Moroni M.C., Helin K. A novel repressive E2F6 complex containing the polycomb group protein, EPC1, that interacts with EZH2 in a proliferation-specific manner. J. Biol. Chem. 2005;280:1199–1208. doi: 10.1074/jbc.M412509200. [DOI] [PubMed] [Google Scholar]
- 58.Kim J., Woo A.J., Chu J., Snow J.W., Fujiwara Y., Kim C.G., Cantor A.B., Orkin S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell. 2010;143:313–324. doi: 10.1016/j.cell.2010.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taubert S., Gorrini C., Frank S.R., Parisi T., Fuchs M., Chan H.M., Livingston D.M., Amati B. E2F-dependent histone acetylation and recruitment of the Tip60 acetyltransferase complex to chromatin in late G1. Mol. Cell. Biol. 2004;24:4546–4556. doi: 10.1128/MCB.24.10.4546-4556.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Van Den Broeck A., Nissou D., Brambilla E., Eymin B., Gazzeri S. Activation of a Tip60/E2F1/ERCC1 network in human lung adenocarcinoma cells exposed to cisplatin. Carcinogenesis. 2012;33:320–325. doi: 10.1093/carcin/bgr292. [DOI] [PubMed] [Google Scholar]
- 61.Gulzar Z.G., McKenney J.K., Brooks J.D. Increased expression of NuSAP in recurrent prostate cancer is mediated by E2F1. Oncogene. 2013;32:70–77. doi: 10.1038/onc.2012.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim H.E., Symanowski J.T., Samlowski E.E., Gonzales J., Ryu B. Quantitative measurement of circulating lymphoid-specific helicase (HELLS) gene transcript: a potential serum biomarker for melanoma metastasis. Pigment Cell Melanoma Res. 2010;23:845–848. doi: 10.1111/j.1755-148X.2010.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su Y., Meador J.A., Calaf G.M., Proietti De-Santis L., Zhao Y., Bohr V.A., Balajee A.S. Human RecQL4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010;70:9207–9217. doi: 10.1158/0008-5472.CAN-10-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.von Eyss B., Maaskola J., Memczak S., Mollmann K., Schuetz A., Loddenkemper C., Tanh M.D., Otto A., Muegge K., Heinemann U., et al. The SNF2-like helicase HELLS mediates E2F3-dependent transcription and cellular transformation. EMBO J. 2012;31:972–985. doi: 10.1038/emboj.2011.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hayes P.H., Sato T., Denell R.E. Homoeosis in Drosophila: the ultrabithorax larval syndrome. Proc. Natl. Acad. Sci. U.S.A. 1984;81:545–549. doi: 10.1073/pnas.81.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sato T., Denell R.E. Homoeosis in Drosophila: anterior and posterior transformations of Polycomb lethal embryos. Dev. Biol. 1985;110:53–64. doi: 10.1016/0012-1606(85)90063-6. [DOI] [PubMed] [Google Scholar]
- 67.Stankunas K., Berger J., Ruse C., Sinclair D.A., Randazzo F., Brock H.W. The enhancer of polycomb gene of Drosophila encodes a chromatin protein conserved in yeast and mammals. Development. 1998;125:4055–4066. doi: 10.1242/dev.125.20.4055. [DOI] [PubMed] [Google Scholar]
- 68.Orlando V. Polycomb, epigenomes, and control of cell identity. Cell. 2003;112:599–606. doi: 10.1016/s0092-8674(03)00157-0. [DOI] [PubMed] [Google Scholar]
- 69.Shimono Y., Murakami H., Hasegawa Y., Takahashi M. RET finger protein is a transcriptional repressor and interacts with enhancer of polycomb that has dual transcriptional functions. J. Biol. Chem. 2000;275:39411–39419. doi: 10.1074/jbc.M006585200. [DOI] [PubMed] [Google Scholar]
- 70.Cavalli G. Molecular biology. EZH2 goes solo. Science. 2012;338:1430–1431. doi: 10.1126/science.1232332. [DOI] [PubMed] [Google Scholar]
- 71.Xu K., Wu Z.J., Groner A.C., He H.H., Cai C., Lis R.T., Wu X., Stack E.C., Loda M., Liu T., et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–1469. doi: 10.1126/science.1227604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Joung H., Kwon J.S., Kim J.R., Shin S., Kang W., Ahn Y., Kook H., Kee H.J. Enhancer of polycomb1 lessens neointima formation by potentiation of myocardin-induced smooth muscle differentiation. Atherosclerosis. 2012;222:84–91. doi: 10.1016/j.atherosclerosis.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 73.Kee H.J., Kim J.R., Joung H., Choe N., Lee S.E., Eom G.H., Kim J.C., Geyer S.H., Jijiwa M., Kato T., et al. Ret finger protein inhibits muscle differentiation by modulating serum response factor and enhancer of polycomb1. Cell Death Differ. 2012;19:121–131. doi: 10.1038/cdd.2011.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kim J.R., Kee H.J., Kim J.Y., Joung H., Nam K.I., Eom G.H., Choe N., Kim H.S., Kim J.C., Kook H., et al. Enhancer of polycomb1 acts on serum response factor to regulate skeletal muscle differentiation. J. Biol. Chem. 2009;284:16308–16316. doi: 10.1074/jbc.M807725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kee H.J., Kim J.R., Nam K.I., Park H.Y., Shin S., Kim J.C., Shimono Y., Takahashi M., Jeong M.H., Kim N., et al. Enhancer of polycomb1, a novel homeodomain only protein-binding partner, induces skeletal muscle differentiation. J. Biol. Chem. 2007;282:7700–7709. doi: 10.1074/jbc.M611198200. [DOI] [PubMed] [Google Scholar]
- 76.Biankin A.V., Waddell N., Kassahn K.S., Gingras M.C., Muthuswamy L.B., Johns A.L., Miller D.K., Wilson P.J., Patch A.M., Wu J., et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature. 2012;491:399–405. doi: 10.1038/nature11547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Micci F., Panagopoulos I., Bjerkehagen B., Heim S. Consistent rearrangement of chromosomal band 6p21 with generation of fusion genes JAZF1/PHF1 and EPC1/PHF1 in endometrial stromal sarcoma. Cancer Res. 2006;66:107–112. doi: 10.1158/0008-5472.CAN-05-2485. [DOI] [PubMed] [Google Scholar]
- 78.Nakahata S., Saito Y., Hamasaki M., Hidaka T., Arai Y., Taki T., Taniwaki M., Morishita K. Alteration of enhancer of polycomb 1 at 10p11.2 is one of the genetic events leading to development of adult T-cell leukemia/lymphoma. Genes Chromosomes Cancer. 2009;48:768–776. doi: 10.1002/gcc.20681. [DOI] [PubMed] [Google Scholar]
- 79.Nara M., Teshima K., Watanabe A., Ito M., Iwamoto K., Kitabayashi A., Kume M., Hatano Y., Takahashi N., Iida S., et al. Bortezomib reduces the tumorigenicity of multiple myeloma via downregulation of upregulated targets in clonogenic side population cells. PLoS ONE. 2013;8:e56954. doi: 10.1371/journal.pone.0056954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Huang X., Spencer G.J., Lynch J.T., Ciceri F., Somerville T.D., Somervaille T.C. Enhancers of Polycomb EPC1 and EPC2 sustain the oncogenic potential of MLL leukemia stem cells. Leukemia. 2014;28:1081–1091. doi: 10.1038/leu.2013.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yang X.L., Lin F.J., Guo Y.J., Shao Z.M., Ou Z.L. Gemcitabine resistance in breast cancer cells regulated by PI3K/AKT-mediated cellular proliferation exerts negative feedback via the MEK/MAPK and mTOR pathways. Oncol. Targets Ther. 2014;7:1033–1042. doi: 10.2147/OTT.S63145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nagel S., Venturini L., Marquez V.E., Meyer C., Kaufmann M., Scherr M., MacLeod R.A., Drexler H.G. Polycomb repressor complex 2 regulates HOXA9 and HOXA10, activating ID2 in NK/T-cell lines. Mol. Cancer. 2010;9:151. doi: 10.1186/1476-4598-9-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Doyon Y., Selleck W., Lane W.S., Tan S., Cote J. Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol. Cell. Biol. 2004;24:1884–1896. doi: 10.1128/MCB.24.5.1884-1896.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Legube G., Trouche D. Regulating histone acetyltransferases and deacetylases. EMBO Rep. 2003;4:944–947. doi: 10.1038/sj.embor.embor941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jost C.A., Ginsberg D., Kaelin W.G. Jr. A conserved region of unknown function participates in the recognition of E2F family members by the adenovirus E4 ORF 6/7 protein. Virology. 1996;220:78–90. doi: 10.1006/viro.1996.0288. [DOI] [PubMed] [Google Scholar]
- 86.Hallstrom T.C., Nevins J.R. Specificity in the activation and control of transcription factor E2F-dependent apoptosis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:10848–10853. doi: 10.1073/pnas.1831408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang C., Rauscher F.J. 3rd, Cress W.D., Chen J. Regulation of E2F1 function by the nuclear corepressor KAP1. J. Biol. Chem. 2007;282:29902–29909. doi: 10.1074/jbc.M704757200. [DOI] [PubMed] [Google Scholar]
- 88.Hallstrom T.C., Nevins J.R. Jab1 is a specificity factor for E2F1-induced apoptosis. Genes Dev. 2006;20:613–623. doi: 10.1101/gad.1345006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Vidal M., Braun P., Chen E., Boeke J.D., Harlow E. Genetic characterization of a mammalian protein-protein interaction domain by using a yeast reverse two-hybrid system. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10321–10326. doi: 10.1073/pnas.93.19.10321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang S., Nath N., Fusaro G., Chellappan S. Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals. Mol. Cell. Biol. 1999;19:7447–7460. doi: 10.1128/mcb.19.11.7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cecchini M.J., Dick F.A. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J. 2011;434:297–308. doi: 10.1042/BJ20101210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.