

Abstract
N1-methyl adenine (1-MeA) is formed in DNA by reaction with alkylating agents and naturally occurring methyl halides. The 1-MeA lesion impairs Watson-Crick base pairing and blocks normal DNA replication. Here we identify the translesion synthesis (TLS) DNA polymerases (Pols) required for replicating through 1-MeA in human cells and show that TLS through this lesion is mediated via three different pathways in which Pols ι and θ function in one pathway and Pols η and ζ, respectively, function in the other two pathways. Our biochemical studies indicate that in the Polι/Polθ pathway, Polι would carry out nucleotide insertion opposite 1-MeA from which Polθ would extend synthesis. In the Polη pathway, this Pol alone would function at both the nucleotide insertion and extension steps of TLS, and in the third pathway, Polζ would extend from the nucleotide inserted opposite 1-MeA by an as yet unidentified Pol. Whereas by pushing 1-MeA into the syn conformation and by forming Hoogsteen base pair with the T residue, Polι would carry out TLS opposite 1-MeA, the ability of Polη to replicate through 1-MeA suggests that despite its need for Watson-Crick hydrogen bonding, Polη can stabilize the adduct in its active site. Remarkably, even though Pols η and ι are quite error-prone at inserting nucleotides opposite 1-MeA, TLS opposite this lesion in human cells occurs in a highly error-free fashion. This suggests that the in vivo fidelity of TLS Pols is regulated by factors such as post-translational modifications, protein-protein interactions, and possibly others.
Keywords: DNA, DNA damage, DNA polymerase, DNA replication, DNA synthesis, DNA polymerase eta, DNA polymerase iota, DNA polymerase theta, N1-methyladenine, translesion synthesis in human cells
Introduction
N1-methyladenine (1-MeA)3 is formed in DNA by reaction with Sn2 methylating agents such as methyl methanesulfonate and naturally occurring methyl halides (1–3). The Sn2 methyl halides are among the most abundant environmental methylating agents released from biomass burning and from decaying vegetation. Exposure to alkylating agents could also occur from food, occupational hazards, and chemotherapeutic treatments (1).
1-MeA is highly cytotoxic because the N1 atom is engaged in Watson-Crick (W-C) base pairing and its modification by a methyl group impairs W-C base pairing and blocks normal DNA replication. In Escherichia coli, AlkB repairs 1-MeA by oxidative demethylation, which liberates formaldehyde from the methylated base and results in complete reversal of the damage (4, 5). In humans, there are nine potential AlkB homologs, two of which, ABH2 and ABH3, can repair the same spectrum of DNA lesions as AlkB (6, 7); ABH2, however, is the primary housekeeping enzyme in humans for repairing 1-MeA (8). Mouse embryonic fibroblast lines derived from ABH2 null mice are highly defective in repairing 1-MeA residues generated in response to methyl methanesulfonate treatment. Because in the absence of any exposure to alkylating agents, 1-MeA residues accumulate over time in the genomic DNA of livers from ABH2 null mice, endogenous DNA methylation contributes to their generation (8).
Previously, we reported on the genetic control of translesion synthesis (TLS) opposite UV-induced cyclobutane pyrimidine dimers and (6-4) pyrimidine-pyrimidone photoproducts and opposite thymine glycol (Tg), which is the most common oxidation product of thymine (9–12). Of the two UV lesions, cyclobutane pyrimidine dimer does not significantly affect the ability of two pyrimidines to form a correct W-C base pair with the purine bases, and it has only a modest effect on DNA structure (13); by contrast, a (6-4) pyrimidine-pyrimidone photoproduct induces a large structural distortion in DNA. It confers a 44º bend in the DNA helix and the 3′T is oriented perpendicular to the 5′T in the (6-4) TT photoproduct (14–16). The Tg lesion also has no significant effect on the ability of oxidized T to form a correct base pair with an A; however, because of the addition of hydroxyl groups at C5 and C6 on Tg, the damaged base becomes non-planar and that prevents the base 5′ to Tg from stacking above it (17–20). Consequently, Tg presents a strong block to extension of synthesis from the Tg:A base pair. Despite the fact that these DNA lesions differ vastly in their effects on DNA structure and on base pairing, they generate only ∼2% mutagenic TLS products in human cells (10–12). This is rather surprising in view of the fact that the various TLS DNA polymerases (Pols) synthesize DNA with a low fidelity (21).
Here we identify the TLS Pols that promote replication through the 1-MeA lesion in human cells and show that TLS opposite this lesion is mediated by three independent pathways, involving Pols ι and θ in one pathway and Pols η and ζ, respectively, in the other two pathways. The observation that similar to that opposite UV and Tg lesions, TLS opposite this W-C blocking lesion also occurs in a highly error-free manner reinforces the notion that the fidelity of TLS Pols is actively regulated in human cells.
Experimental Procedures
Construction of Plasmid Vectors Containing 1-MeA
The 16-mer oligonucleotides containing an N1-methyl deoxyadenosine (Fig. 1A) were purchased from Trilink Biotechnologies (Santa Cruz, CA). The methods used by this company for the synthesis of 1-MeA-derived phosphoramidite and for its incorporation into oligonucleotide employ N6-chloroacetyl protection and controlled anhydrous deprotection conditions to prevent the formation of N6-methyl-2′-deoxyadenosine via a Dimroth rearrangement (22). The in-frame target sequence of lacZ′ gene containing 1-MeA is shown in Fig. 1A. In the lacZ′ gene, the 1-MeA-containing DNA strand harbors an MfeI restriction site, and it encodes functional β-galactosidase (β-gal), whereas the opposite strand harbors an SpeI site containing a +1 frameshift, making it non-functional for β-gal. The 1-MeA containing strand carries the kanamycin gene (Kan+), whereas the other DNA strand has the kan− gene (Fig. 1B). The detailed method for construction of 1-MeA-containing plasmids was as described previously (11).
FIGURE 1.

Strategy for examining TLS opposite 1-MeA. A, the target 16-mer sequence containing 1-MeA. The sequence of the N-terminal part of the lacZ′ gene in pBS vector (leading strand) including the 1-MeA site, indicated by mA, is shown. B, selection strategy for TLS assays. The top DNA strand containing the 1-MeA lesion carries a wild type kanamycin gene (Kan+) so that TLS through the 1-MeA lesion will produce a blue colony on LB/Kan plates with isopropyl-1-thio-β-d-galactopyranoside and X-Gal. The other DNA strand harbors an Spe1 sequence opposite 1-MeA and carries the kan− gene. Because the Spe1 sequence puts the lacZ sequence out of frame (see Fig. 1A), replication of this strand will produce kan− colonies. C, assays for TLS and mutation analysis opposite 1-MeA in human cells. The purified cDNA and siRNAs are cotransfected into siRNA-treated human cells. The target sites of 1-MeA and for kanamycin gene selection are shown in the vector. After 30 h of incubation, the rescued plasmid DNA is treated with DpnI to remove any unreplicated plasmid and then transformed into XL-1 blue E. coli cells. TLS frequency is determined by phenotypic selection of transformed bacterial cells grown on LB/kan/X-Gal plates. The mutations induced by TLS opposite 1-MeA are analyzed by colony PCR, MfeI digestion, and by sequencing of the target site. HF, human fibroblast.
Translesion Synthesis Assays and Mutation Analyses ofTLS Products in Human Cells
Normal (MRC5) and XPV (XP30RO) human fibroblast cells were grown in DMEM media (GenDEPOT) with 10% FBS (GenDEPOT) and plated in 6-well plates at 70% confluence (approximately 3 × 105 cells per well). Cells were transfected with 100 pmol of siRNAs with Lipofectamine 2000 (Invitrogen). For the simultaneous siRNA knockdown of two genes, 100 pmol of siRNAs for each gene were mixed and transfected. After 48 h of incubation, the heteroduplex target vector DNA (1 μg) and 50 pmol of siRNA (second transfection) were cotransfected with Lipofectamine 2000 (Invitrogen) (Fig. 1C). After 30 h of incubation, plasmid DNA was rescued from cells by the alkaline lysis method and digested with DpnI to remove unreplicated plasmid DNA. The plasmid DNA was then transformed into E. coli XL1-Blue super competent cells (Stratagene). Transformed bacterial cells were diluted in 1 ml of SOC media (Thermo Fisher Scientific) and plated on LB/kan (25 μg/ml kanamycin, Sigma) plates containing 1 μm isopropyl-1-thio-β-d-galactopyranoside (GenDEPOT) and 100 μg/ml X-Gal (GenDEPOT). After 16 h of incubation at 37 °C, blue and white colonies were counted from kanamycin plates (Fig. 1C). The actual TLS frequency was determined from the number of blue colonies out of the total colonies growing on LB/kan plates. Plasmid DNA obtained from blue colonies was analyzed to determine the mutation frequency, and the mutational changes were incorporated during TLS. For details of these methods, see Yoon et al. (11).
DNA Polymerase Assays
DNA substrates consisted of a radiolabeled oligonucleotide primer annealed to a 75-nucleotide (nt) oligonucleotide DNA template by heating a mixture of primer/template at a 1:1.5 molar ratio to 95 °C and allowing it to cool to room temperature for several hours. The template 75-mer oligonucleotide contained the sequence 5′-AGC AAG TCA CCA ATG TCT AAG AGT TCG TAT AAT GCC TAC ACT GGA GTA CCG GAG CAT CGT CGT GAC TGG GAA AAC-3′, and it harbored an undamaged A or a 1-MeA at the underlined position. For examining the incorporation of dATP, dTTP, dCTP, or dGTP nucleotides individually or of all 4 dNTPs, a 44-mer primer 5′-GTT TTC CCA GTC ACG ACG ATG CTC CGG TAC TCC AGT GTA GGC AT-3′ was annealed to the above-mentioned 75-mer template.
The standard DNA polymerase reaction (5 μl) contained 25 mm Tris·HCl (pH 7.5), 5 mm MgCl2, 1 mm dithiothreitol, 100 μg/ml BSA, 10% glycerol, 10 nm DNA substrate, and 0.2 nm Polι or 0.25 nm Polη. For nucleotide incorporation assays with Polι, 50 μm dATP, dTTP, dCTP, or dGTP (Roche Applied Science) was used, and for examining synthesis through the 1-MeA lesion all 4 dNTPs (50 μm each) were used. For nucleotide incorporation assays with Polη, 25 μm dATP, dTTP, dCTP, or dGTP (Roche Applied Science) was used, and for examining synthesis through the 1-MeA lesion by Polη all 4 dNTPs (25 μm each) were used. Reactions were carried out for 10 min at 37 °C.
For extension studies, a 75-mer oligonucleotide template 5′-AGC AAG TCA CCA ATG TCT AAG AGT TCG TAT AAT GCC TAC ACT GGA GTA CCG GAG CAT CGT CGT GAC TGG GAA AAC-3′ containing an undamaged A or a 1-MeA at the underlined position was annealed to a 23-mer primer 5′ TCC GGT ACT CCA GTG TAG GCA TX-3′, which contained a T at the position indicated by X.
Primer extension by human Polθ (1 nm) or yeast Polζ (1 nm) was assayed in the presence of 10 μm each dATP, dGTP, dTTP, and dCTP (Roche Applied Science) on DNA containing an A/T or 1-MeA/T primer terminal base pair; the standard DNA polymerase reaction (5 μl) contained 25 mm Tris·HCl (pH 7.5), 5 mm MgCl2, 1 mm dithiothreitol, 100 μg/ml BSA, 10% glycerol, and 10 nm DNA substrate. Reactions containing human Polθ were carried out at 37 °C and yeast Polζ at 30 °C for 10 min.
Results
TLS Pols Required for Replicating through the 1-MeA Lesion
To identify the TLS Pols required for replicating through the 1-MeA lesion, we determined the effects of siRNA depletions of TLS Pols individually and in combinations on TLS frequency opposite this lesion carried on the leading strand template of the SV40-based plasmid. To ascertain that the observed defects result from the deficiency of the particular Pol and not from any off-target effects, we confirmed that the defect in TLS engendered by the siRNA depletion of a Pol was complemented by the respective wild type Pol.
TLS in normal human fibroblasts treated with control (NC) siRNA occurred with a frequency of ∼65% (Table 1). In Polη-depleted cells, TLS frequency was reduced to ∼52%. TLS frequency was reduced to ∼45% in Polι-depleted cells, and a similar reduction in TLS frequency was observed in cells depleted for the Rev3 or the Rev7 subunits of Polζ or for Polθ. To determine whether these Pols function independently of one another or whether some of them function together in replicating through the 1-MeA lesion, we examined the effects of simultaneous depletion of two Pols on TLS frequencies. Our observation that simultaneous depletion of Polι and Polθ confers no further reduction in TLS frequency than that seen upon their individual depletion indicates that Pols ι and θ function together in conducting TLS opposite 1-MeA. By contrast, the observation that simultaneous depletion of Polη with Polι or of Polη with Polθ results in a greater reduction in TLS frequency, to ∼30%, than that observed upon their individual depletion implies that Polη functions in TLS opposite 1-MeA independently of Pols ι and θ. And, because the simultaneous depletion of Polη with the Rev3 or Rev7 subunit of Polζ reduces TLS frequency to a greater extent (∼30%) than that observed upon their individual depletion (∼45–52%), Pols η and ζ act independently of one another. Additionally, because the simultaneous depletion of Polι with Polζ confers a greater reduction in TLS frequency than that observed upon their individual depletion, these Pols function in TLS independently of one another.
TABLE 1.
Effects of siRNA knockdowns of TLS polymerases on the replicative bypass of 1-MeA lesion carried on the leading strand template in human fibroblasts
| siRNA | No. of Kan+ colonies | No. blue colonies among Kan+ | TLS |
|---|---|---|---|
| % | |||
| NC | 423 | 276 | 65.2 |
| Polη | 356 | 187 | 52.5 |
| Polι | 428 | 193 | 45.1 |
| Rev3 | 524 | 228 | 43.5 |
| Rev7 | 458 | 205 | 44.8 |
| Polθ | 415 | 163 | 39.3 |
| Polι + Polθ | 372 | 161 | 43.3 |
| Polη + Polι | 302 | 93 | 30.8 |
| Polη + Polθ | 408 | 122 | 29.9 |
| Polη + Rev3 | 350 | 100 | 28.6 |
| Polη + Rev7 | 296 | 92 | 31.1 |
| Polι + Rev3 | 228 | 63 | 27.6 |
| Polι + Rev7 | 295 | 80 | 27.1 |
As deduced from these genetic observations, TLS opposite 1-MeA occurs via three independent pathways mediated by the combined action of Polι and Polθ or by Polη or Polζ, respectively. To provide confirmatory evidence for this deduction, we analyzed the effects of siRNA depletion of TLS Pols on the frequency of TLS opposite 1-MeA in human XPV fibroblasts. Because of the mutational inactivation of the XPV gene, these cells lack Polη, and they exhibit a reduction in TLS frequency similar to that which occurs upon Polη depletion in wild type human fibroblasts (Table 2). TLS in XPV cells occurs with a frequency of ∼47%. Depletion of Polι, Polθ, or Polζ in XPV cells led to a reduction in TLS frequency to ∼30%, confirming that these Pols function independently of Polη (Table 2), and as expected from the involvement of Pols ι and θ in the same TLS pathway, the simultaneous depletion of Polι and Polθ causes no further reduction in TLS frequency than that seen upon their individual depletion. Because only the Polι/Polθ- and Polζ-dependent TLS pathways would remain functional in XPV cells, we expect the simultaneous depletion of Polι and Polζ or of Polθ and Polζ will confer a drastic reduction in TLS frequency. In accord with this, we find that simultaneous depletion of Polι and Rev3 or of Polθ and Rev3 reduces TLS frequency to ∼4–5% (Table 2). These results provide confirmatory evidence that TLS opposite 1-MeA occurs via three independent pathways that require Pols ι and θ in one pathway and Polη and Polζ, respectively, in the other two pathways.
TABLE 2.
Effects of siRNA knockdowns of TLS polymerases on the replicative bypass of 1-MeA lesion carried on the leading strand template in XPV human fibroblasts
| siRNA | No. of Kan+ colonies | No. blue colonies among Kan+ | TLS |
|---|---|---|---|
| % | |||
| NC | 254 | 120 | 47.2 |
| Polι | 292 | 90 | 30.8 |
| Polθ | 260 | 78 | 30.0 |
| Rev3 | 224 | 71 | 31.7 |
| Rev7 | 212 | 60 | 28.3 |
| Polι + Polθ | 312 | 86 | 27.6 |
| Polι + Rev3 | 238 | 10 | 4.2 |
| Polθ + Rev3 | 308 | 16 | 5.2 |
Mutagenicity of TLS Opposite 1-MeA
As determined by sequence analyses of TLS products, TLS opposite the 1-MeA lesion occurs in a predominantly error-free fashion, as in NC siRNA-treated cells only ∼1% of TLS products harbor mutations. Among the ∼400 TLS products that were analyzed, we observed mutations in only four of the TLS products in which an A or G was inserted opposite 1-MeA instead of the correct nucleotide, T (Table 3). Even though the overall TLS-mediated via three different pathways opposite 1-MeA generates only ∼1% mutations, among the TLS Pols involved in 1-MeA bypass some may function in an error-free manner whereas others may promote a more mutagenic mode of TLS. Thus, for example, even though the overall TLS opposite a Tg lesion generates only ∼2% mutations, the Polκ/Polζ-dependent pathway mediates a predominantly error-free mode of TLS, whereas the Polθ-dependent pathway functions in a more error-prone manner (9, 10). For this reason we examined the frequency and types of mutations generated by the action of different TLS Pols opposite 1-MeA. Interestingly, among the ∼200–300 TLS products analyzed from cells depleted for Polη, Polι, Polθ, or Polζ, we observed mutation frequencies ranging from 0% in Polη-depleted cells to ∼1.5% in Polζ-depleted cells (Table 3). These differences are not statistically significant. We conclude from these results that opposite 1-MeA, all three pathways function in a predominantly error-free manner, generating only ∼1% mutagenic TLS products where an A, or a G, is inserted opposite 1-MeA rather than a T.
TABLE 3.
Effects of siRNA knockdowns of TLS polymerases on the frequencies of nucleotides inserted opposite 1-MeA carried on the leading strand template in human fibroblasts
| siRNA | No. of kan+ blue colonies sequenceda | Nucleotide inserted |
Mutation frequency | |||
|---|---|---|---|---|---|---|
| A | G | C | T | |||
| NC | 384 (4) | 2 | 2 | 0 | 380 | 1.0 |
| Polη | 308 (0) | 0 | 0 | 0 | 308 | 0 |
| Polι | 190 (3) | 2 | 1 | 0 | 187 | 1.6 |
| Polθ | 176 (2) | 2 | 0 | 0 | 174 | 1.1 |
| Rev3 | 288 (4) | 3 | 1 | 0 | 284 | 1.4 |
| Rev7 | 192 (3) | 3 | 0 | 0 | 189 | 1.6 |
a Numbers of mutational TLS events are shown in parentheses.
Biochemical Studies for Roles of TLS Pols in DNA Synthesis Opposite 1-MeA
TLS through a DNA lesion can occur by the action of one Pol, wherein the TLS Pol inserts one nt opposite the DNA lesion and then extends synthesis from the inserted nt (21). Alternatively, TLS through a DNA lesion may occur by the sequential action of two Pols wherein one Pol inserts the nt opposite the lesion and then another Pol extends synthesis (21, 23). To determine the potential of TLS Pols for DNA synthesis opposite 1-MeA, we examined their ability to insert dATP, dTTP, dGTP, or dCTP opposite 1-MeA and to synthesize DNA through the 1-MeA lesion in the presence of all 4 dNTPs. As shown in Fig. 2, Polι inserts dTTP opposite undamaged A, and in the presence of all 4 dNTPs, it extends synthesis by incorporating 1–2 additional nts. Opposite 1-MeA, Polι inserts dTTP, but it also inserts a dATP or dCTP, and in the presence of 4 dNTPs, Polι inserts one nt opposite 1-MeA, but it fails to extend synthesis any further. Thus, Polι is more error-prone opposite 1-MeA than opposite an undamaged A, and because of its failure to extend synthesis from the nt opposite 1-MeA, replication through the lesion would require the action of another TLS Pol. The inability of Polι to extend synthesis opposite from 1-MeA and the requirement of Polθ for Polι-dependent TLS indicated from genetic studies suggested that Polθ would extend synthesis from the nt inserted by Polι opposite 1-MeA. Hence, we examined if in the presence of four dNTPs, Polθ could extend synthesis from a T inserted opposite 1-MeA. As shown in Fig. 3, Polθ exhibits the same propensity for extending synthesis from the T nt opposite 1-MeA as from opposite undamaged A. These biochemical observations support the inference that in the Polι/Polθ pathway, TLS through the 1-MeA lesion would occur by the sequential action of Polι and Polθ, wherein after nt insertion by Polι opposite 1-MeA, Polθ performs the subsequent extension step of TLS.
FIGURE 2.
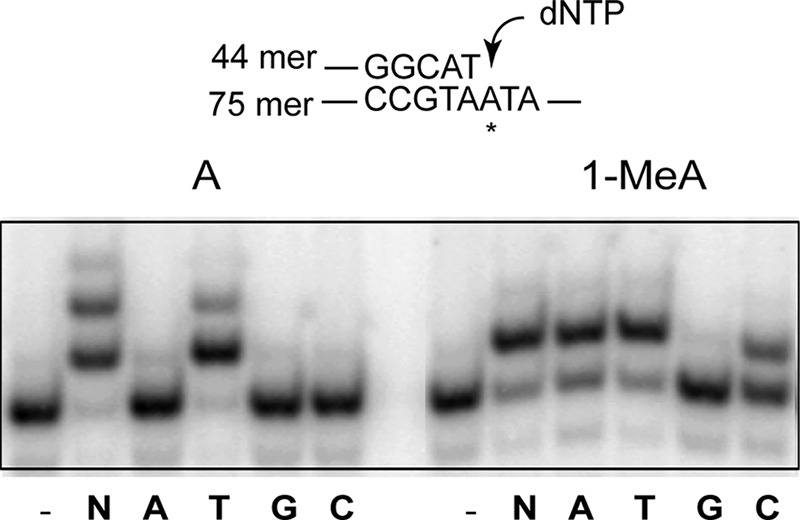
Nucleotide incorporation by Polι opposite an undamaged A and 1-MeA. Polι (0.2 nm) was incubated with DNA (10 nm) and with 50 μm of one of the four dNTPs (A, T, G, or C) or with 50 μm of each of four dNTPs (N) for 10 min at 37 °C. DNA substrate is shown above the gel; the asterisk indicates the site of either an A or a 1-MeA residue.
FIGURE 3.

Primer extension from a T annealed to an A or a 1-MeA template by Polθ and Polζ. The DNA substrate is shown above the gel where the asterisk indicates the site of either an undamaged A or a 1-MeA residue on a 75-mer template. DNA synthesis by human Polθ (1 nm) was assayed at 37 °C, and DNA synthesis by yeast Polζ (1 nm) was assayed at 30 °C for 10 min in the presence of 10 μm each of dATP, dGTP, dTTP and dCTP on DNA containing an A/T (lane 2) or 1-MeA/T (lane 3) primer terminal base pair; lane 1 contains DNA substrate with no enzyme.
Biochemical studies show that opposite 1-MeA, Polη incorporated dTTP better than the incorrect nts, and the pattern of correct and incorrect nt incorporation opposite 1-MeA resembled that opposite undamaged A (Fig. 4). Thus, in addition to dTTP, Polη inserted the other three nts dATP, dGTP, or dCTP almost equally well opposite 1-MeA and undamaged A. In the presence of four dNTPs, Polη synthesized full-length products on 1-MeA containing DNA, but its ability to extend synthesis from the nt inserted opposite 1-MeA was reduced, as indicated by the presence of a prominent stall site opposite 1-MeA but not opposite undamaged A.
FIGURE 4.
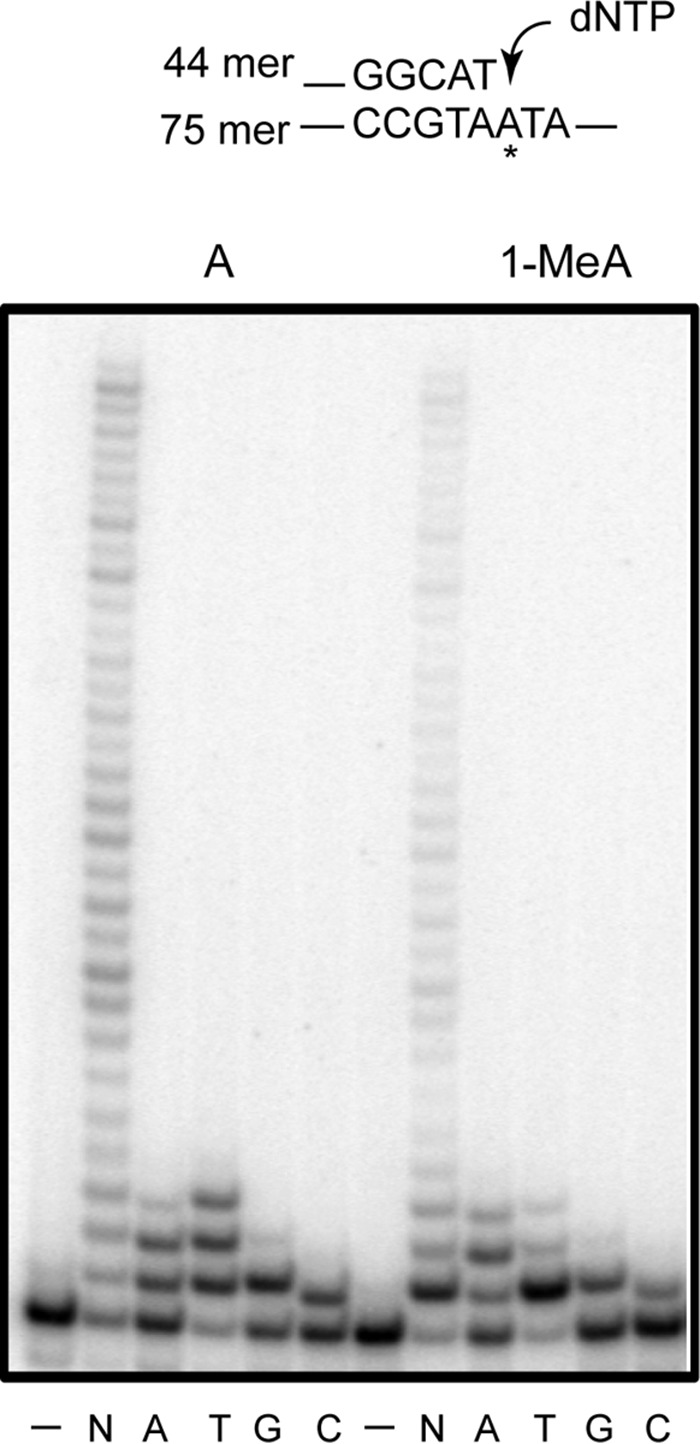
Nucleotide incorporation by Polη opposite an undamaged A and 1-MeA. Polη (0.25 nm) was incubated with DNA (10 nm) and with 25 μm of a single nucleotide (A, T, G, or C) or 25 μm of each of four dNTPs (N) for 10 min at 37 °C. DNA substrate is shown above the gel; the asterisk indicates the site of either an A or a 1-MeA residue.
Polζ is very inefficient in inserting nts opposite DNA lesions but it is highly proficient at extending synthesis from nts opposite from a diverse array of DNA lesions (21, 23–26). To verify that Polζ plays a similar role opposite 1-MeA, we examined the proficiency of Polζ for extending from the 1-MeA:T base pair. As shown in Fig. 3, in the presence of 4 dNTPs, Polζ extends synthesis from the 1-MeA:T base pair as well as from the undamaged A:T base pair.
Discussion
The important findings of this study are as follows. 1) TLS provides a major means for replication of 1-MeA containing DNA. 2) Multiple pathways promote replication through the 1-MeA lesion. 3) TLS opposite 1-MeA occurs in a predominantly error-free manner. We consider the implications of these observations for the prominent role of TLS in the replication of 1-MeA-damaged DNA, for the roles of TLS Pols in 1-MeA bypass as related to their structural and biochemical features, and for the fact that in human cells, TLS opposite 1-MeA occurs in a highly error-free fashion.
A Major Role of TLS in the Replicative Bypass of 1-MeA
Previously, we have determined the extent, genetic control, and mutagenicity of TLS opposite three DNA lesions, cis-syn TT dimer, (6-4) TT photoproduct, and thymine glycol. These studies indicated that in nucleotide excision repair-defective XPA human fibroblasts, TLS opposite a cis-syn TT dimer or a (6-4) TT photoproduct occurs with a frequency of ∼40%, whereas TLS opposite these lesions in nucleotide excision repair-proficient fibroblasts occurs with a frequency of ∼20%. Thus, a very substantial fraction of these UV photoproducts are excised from plasmid DNA before its replication. Similar to that for UV photoproducts, in wild type fibroblasts, TLS opposite the thymine glycol lesion occurs with a frequency of ∼20–25%. In striking contrast to the TLS frequency of ∼20–25% opposite UV photoproducts or thymine glycol in wild type fibroblasts, we find that opposite 1-MeA, TLS contributes to ∼65% of lesion bypass. Because the 1-MeA lesion carried on the duplex plasmid will be subject to removal by base excision repair or nucleotide excision repair, we presume that TLS provides a predominant, if not the sole means for replicating through this lesion.
Multiple Pathways for Replication of 1-MeA-damaged DNA
From siRNA depletion of TLS Pols, we have inferred that replication through the 1-MeA lesion occurs via three different pathways that, respectively, require Polι/Polθ, Polη, or Polζ (Fig. 5). Because biochemical studies have indicated that Polι can insert a nt opposite 1-MeA but lacks the ability to extend synthesis from the inserted nt whereas Polθ can carry out efficient extension of synthesis from the nt opposite 1-MeA, we suggest that Polι/Polθ-dependent TLS would occur by the sequential action of Polι and Polθ at the insertion and extension step of TLS, respectively (Fig. 5). In the Polη-dependent pathway, Polη alone would carry out both the insertion and extension steps of TLS, and because of the proficient ability of Polζ to extend synthesis from the nt opposite 1-MeA, we expect that in the Polζ-dependent TLS pathway, an as yet unidentified TLS Pol would insert a nt opposite 1-MeA from which Polζ would extend (Fig. 5). We are now checking whether any of the Pols not yet examined for their role in TLS opposite 1-MeA, such as Polκ, Polν, or Polλ, could function as an inserter in this pathway.
FIGURE 5.
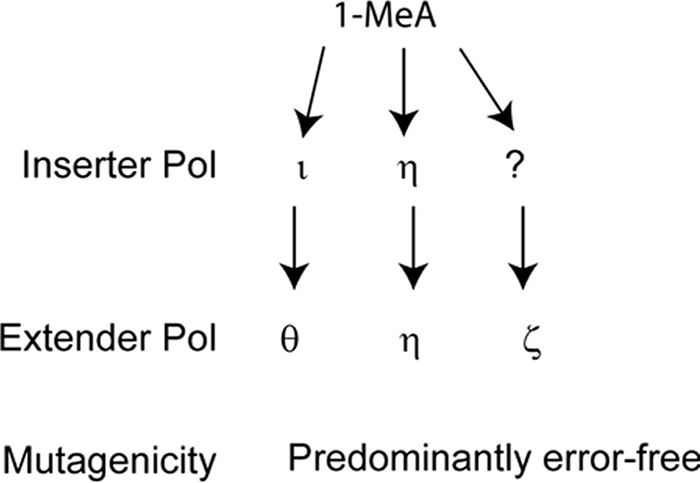
Pathways for replicating through 1-MeA in human cells. Replication through 1-MeA occurs by the combined action of Polι and Polθ in one pathway in which Polι inserts a nt opposite 1-MeA and Polθ extends and by Pols η and ζ, respectively, in the other two pathways. In the Polη pathway, Polη would act alone at both the insertion and extension steps, whereas in the Polζ pathway, after nt insertion by an unidentified Pol, Polζ would extend.
Roles of TLS Pols in the Replication of 1-MeA-damaged DNA: Structural and Biochemical Considerations
In the Polι/Polθ-dependent pathway, the ability of Polι to push 1-MeA into a syn conformation and to form a Hoogsteen base pair with the incoming T residue would allow it to carry out an nt insertion opposite 1-MeA from which Polθ would extend (26, 27). Previously, we have shown that Polθ can replicate through the Tg lesion by both inserting an A opposite Tg and then by extending from there (9). The ability of Polθ to extend synthesis on DNA containing a Tg lesion that blocks the base 5′ to Tg from stacking above it, and the potential ability of Polθ to extend from the 1-MeA:T Hoogsteen base pair handed over to it by Polι would suggest that Polθ, an A-family Pol, can extend synthesis opposite from a diverse array of DNA lesions which distort the DNA helix or which inhibit the W-C base pairing.
Our genetic and biochemical observations support a role for Polη in replicating through the 1-MeA lesion by inserting a nt opposite 1-MeA and then by extending synthesis. Such a role for Polη is intriguing in view of the fact that biochemical studies have indicated that Polη uses W-C base pairing for DNA synthesis. For example, synthesis by Polη is severely impaired by a difluorotoluene base, which is virtually identical in shape, size, and conformation to thymine but lacks the ability to form W-C hydrogen bonds with adenine (28). And opposite the UV-induced cis-syn TT dimer, Polη forms W-C base pairs with an A opposite both the 3′-T and 5′-T residues of the dimer (29–31). Curiously, although biochemical studies with yeast Polη have indicated that the incorporation of 1-MeA opposite the 3′T or the 5′T of a TT dimer or opposite the undamaged T residue is reduced by ∼100–200-fold compared with the incorporation of an A opposite the undamaged or UV-damaged T residue (30), human Polη inserts a T opposite 1-MeA almost as well as it inserts a T opposite undamaged A. It remains to be seen how in the absence of W-C base pairing, Polη can manage to insert the correct T nt opposite 1-MeA and then extend synthesis from the 1-MeA:T base pair.
In the Polζ-dependent pathway, the proficient ability of Polζ to extend synthesis from the 1-MeA:T base pair is in accord with the role of this Pol in extending synthesis opposite from a wide variety of DNA lesions that distort the DNA helix or which impair W-C base pairing. It may turn out that the TLS Pol found to be involved in inserting the nt opposite 1-MeA requires W-C hydrogen bonding for synthesizing DNA. The ability of TLS Pols such as Polη and others to bypass the requirement of W-C base-pairing opposite 1-MeA and presumably opposite other such lesions would suggest that at these lesion sites, TLS Pols can stabilize the nascent base pair in their active site by using means other than W-C hydrogen bonding.
Predominantly Error-free TLS Opposite 1-MeA
Rather surprisingly, in human cells, despite the poor fidelity of Pols η and ι for nt insertion opposite 1-MeA, TLS opposite this lesion occurs with a high fidelity, and only ∼1% of TLS products harbor mutations. The high fidelity with which TLS Pols manage to replicate 1-MeA damaged DNA recapitulates the relatively high fidelity TLS that occurs opposite the cis-syn TT dimer, (6-4) TT photoproduct, and thymine glycol, where despite the low fidelity of DNA synthesis by the TLS Pols involved in their replicative bypass, only ∼2% of TLS products harbor mutations (10–12). Thus, opposite these different types of DNA lesions, which confer structural distortions or affect W-C base pairing, human cells have adapted TLS Pols to function in a much more error-free manner than would be predicted from their fidelity for nt incorporation. It remains enigmatic how human cells manage to achieve such a high fidelity for replicating through DNA lesions. A likely possibility is that protein-protein interactions and post-translational modifications regulate the fidelity of TLS Pols in human cells.
Author Contributions
J. C., J.-H. Y., and J. R. C. performed the experiments. J. C., J.-H. Y., J. R. C., L. P., and S. P. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This work was supported by National Institutes of Health Grants ES020833 and ES021452. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- 1-MeA
- N1-methyladenine
- W-C
- Watson-Crick
- TLS
- translesion synthesis
- Tg
- thymine glycol
- Pol
- DNA polymerase
- nt
- nucleotide(s)
- NC
- negative control.
References
- 1.Sedgwick B. (2004) Repairing DNA-methylation damage. Nat. Rev. Mol. Cell Biol. 5, 148–157 [DOI] [PubMed] [Google Scholar]
- 2.Sedgwick B., Bates P. A., Paik J., Jacobs S. C., and Lindahl T. (2007) Repair of alkylated DNA: recent advances. DNA Repair 6, 429–442 [DOI] [PubMed] [Google Scholar]
- 3.Shrivastav N., Li D., and Essigmann J. M. (2010) Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkyaltion. Carcinogenesis 31, 59–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falnes P. Ø., Johansen R. F., and Seeberg E. (2002) AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 419, 178–182 [DOI] [PubMed] [Google Scholar]
- 5.Trewick S. C., Henshaw T. F., Hausinger R. P., Lindahl T., and Sedgwick B. (2002) Oxidative demethylation by Escherichia coli AlkB directly reverts DNA base damage. Nature 419, 174–178 [DOI] [PubMed] [Google Scholar]
- 6.Duncan T., Trewick S. C., Koivisto P., Bates P. A., Lindahl T., and Sedgwick B. (2002) Reversal of DNA alkylations damage by two human dioxygenases. Proc. Natl. Acad. Sci. U.S.A. 99, 16660–16665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang C.-G., Yi C., Duguid E. M., Sullivan C. T., Jian X., Rice P. A., and He C. (2008) Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature 452, 961–965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ringvoll J., Nordstrand L. M., Vågbø C. B., Talstad V., Reite K., Aas P. A., Lauritzen K. H., Liabakk N. B., Bjørk A., Doughty R. W., Falnes P. Ø., Krokan H. E., and Klungland A. (2006) Repair-deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. EMBO J. 25, 2189–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoon J. H., Roy Choudhury J., Park J., Prakash S., and Prakash L. (2014) A role for DNA polymerase θ in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J. Biol. Chem. 289, 13177–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon J.-H., Bhatia G., Prakash S., and Prakash L. (2010) Error-free replicative bypass of thymine glycol by the combined action of DNA polymerases κ and ζ in human cells. Proc. Natl. Acad. Sci. U.S.A. 107, 14116–14121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoon J.-H., Prakash L., and Prakash S. (2009) Highly error-free role of DNA polymerase η in the replicative bypass of UV induced pyrimidine dimers in mouse and human cells. Proc. Natl. Acad. Sci. U.S.A. 106, 18219–18224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoon J.-H., Prakash L., and Prakash S. (2010) Error-free replicative bypass of (6−4) photoproducts by DNA polymerase ζ in mouse and human cells. Genes Dev. 24, 123–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kemmink J., Boelens R., Koning T., van der Marel G. A., van Boom J. H., and Kaptein R. (1987) 1H NMR study of the exchangeable protons of the duplex d(GCGTTGCG).d(CGCAACGC) containing a thymine photodimer. Nucleic Acids Res. 15, 4645–4653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J.-K., and Choi B.-S. (1995) The solution structure of DNA duplex-decamer containing the (6-4) photoproduct of thymidyl(3′ → 5′)thymidine by NMR and relaxation matrix refinement. Eur. J. Biochem. 228, 849–854 [DOI] [PubMed] [Google Scholar]
- 15.Kim J.-K., Patel D., and Choi B.-S. (1995) Contrasting structural impacts induced by cis-syn cyclobutane dimer and (6-4) adduct in DNA duplex decamers: implication in mutagenesis and repair activity. Photochem. Photobiol. 62, 44–50 [DOI] [PubMed] [Google Scholar]
- 16.Lee J.-H., Hwang G.-S., and Choi B.-S. (1999) Solution structure of a DNA decamer duplex containing the stable 3′ T. G base pair of the pyrimidine(6-4)pyrimidone photoproduct [(6-4) adduct]: implications for the highly specific 3′ T → C transition of the (6-4) adduct. Proc. Natl. Acad. Sci. U.S.A. 96, 6632–6636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aller P., Rould M. A., Hogg M., Wallace S. S., and Doublié S. (2007) A structural rationale for stalling of a replicative DNA polymerase at the most common oxidative thymine lesion, thymine glycol. Proc. Natl. Acad. Sci. U.S.A. 104, 814–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kao J. Y., Goljer I., Phan T. A., and Bolton P. H. (1993) Characterization of the effects of a thymine glycol residue on the structure, dynamics, and stability of duplex DNA by NMR. J. Biol. Chem. 268, 17787–17793 [PubMed] [Google Scholar]
- 19.Kung H. C., and Bolton P. H. (1997) Structure of a duplex DNA containing a thymine glycol residue in solution. J. Biol. Chem. 272, 9227–9236 [DOI] [PubMed] [Google Scholar]
- 20.McNulty J. M., Jerkovic B., Bolton P. H., and Basu A. K. (1998) Replication inhibition and miscoding properties of DNA templates containing a site-specific cis-thymine glycol or urea residue. Chem. Res. Toxicol. 11, 666–673 [DOI] [PubMed] [Google Scholar]
- 21.Prakash S., Johnson R. E., and Prakash L. (2005) Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 74, 317–353 [DOI] [PubMed] [Google Scholar]
- 22.Mikhailov S. N., Timofev E. D., Drenichev M. S., Efimtseva E. V., Herdewijn P., Roesch E. B., and Lemaitre M. M. (2009) Oligodeoxynucleotides containing N1-methyl-2′-deoxyadenosine and N6-methyl-2-deoxyadenosine. Curr. Protoc. Nucleic Acid Chem. 10.1002/0471142700.nc0436s38 [DOI] [PubMed] [Google Scholar]
- 23.Johnson R. E., Washington M. T., Haracska L., Prakash S., and Prakash L. (2000) Eukaryotic polymerases ι and ζ act sequentially to bypass DNA lesions. Nature 406, 1015–1019 [DOI] [PubMed] [Google Scholar]
- 24.Haracska L., Unk I., Johnson R. E., Johansson E., Burgers P. M., Prakash S., and Prakash L. (2001) Roles of yeast DNA polymerases δ and ζ and of Rev1 in the bypass of abasic sites. Genes Dev. 15, 945–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson R. E., Haracska L., Prakash S., and Prakash L. (2001) Role of DNA polymerase η in the bypass of a (6-4) TT photoproduct. Mol. Cell. Biol. 21, 3558–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nair D. T., Johnson R. E., Prakash L., Prakash S., and Aggarwal A. K. (2006) Hoogsteen base pair formation promotes synthesis opposite the 1, N6-ethenodeoxyadenosine lesion by human DNA polymerase ι. Nat. Struct. Mol. Biol. 13, 619–625 [DOI] [PubMed] [Google Scholar]
- 27.Nair D. T., Johnson R. E., Prakash S., Prakash L., and Aggarwal A. K. (2004) Replication by human DNA polymerase ι occurs via Hoogsteen base-pairing. Nature 430, 377–380 [DOI] [PubMed] [Google Scholar]
- 28.Washington M. T., Helquist S. A., Kool E. T., Prakash L., and Prakash S. (2003) Requirement of Watson-Crick hydrogen bonding for DNA synthesis by yeast DNA polymerase η. Mol. Cell. Biol. 23, 5107–5112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Biertümpfel C., Zhao Y., Kondo Y., Ramón-Maiques S., Gregory M., Lee J. Y., Masutani C., Lehmann A. R., Hanaoka F., and Yang W. (2010) Structure and mechanism of human DNA polymerase η. Nature 465, 1044–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnson R. E., Prakash L., and Prakash S. (2005) Distinct mechanisms of cis-syn thymine dimer bypass by Dpo4 and DNA polymerase η. Proc. Natl. Acad. Sci. U.S.A. 102, 12359–12364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silverstein T. D., Johnson R. E., Jain R., Prakash L., Prakash S., and Aggarwal A. K. (2010) Structural basis for the suppression of skin cancers by DNA polymerase η. Nature 465, 1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]