

Abstract
A new type of interstrand cross-link resulting from the reaction of a DNA abasic site with a guanine residue on the opposing strand of the double helix was recently identified, but the chemical connectivity of the cross-link was not rigorously established. The work described here was designed to characterize the chemical structure and properties of dG–AP cross-links generated in duplex DNA. The approach involved characterization of the nucleoside cross-link “remnant” released by enzymatic digestion of DNA duplexes containing the dG–AP cross-link. We first carried out a chemical synthesis and complete spectroscopic structure determination of the putative cross-link remnant 9b composed of a 2-deoxyribose adduct attached to the exocyclic N2-amino group of dG. A reduced analogue of the cross-link remnant was also prepared (11b). Liquid chromatography–tandem mass spectrometric (LC-MS/MS) analysis revealed that the retention times and mass spectral properties of synthetic standards 9b and 11b matched those of the authentic cross-link remnants released by enzymatic digestion of duplexes containing the native and reduced dG–AP cross-link, respectively. These results establish the chemical connectivity of the dG–AP cross-link released from duplex DNA and provide a foundation for detection of this lesion in biological samples. The dG–AP cross-link in duplex DNA was remarkably stable, decomposing with a half-life of 22 days at pH 7 and 23 °C. The intrinsic chemical stability of the dG–AP cross-link suggests that this lesion in duplex DNA may have the power to block DNA-processing enzymes involved in transcription and replication.
INTRODUCTION
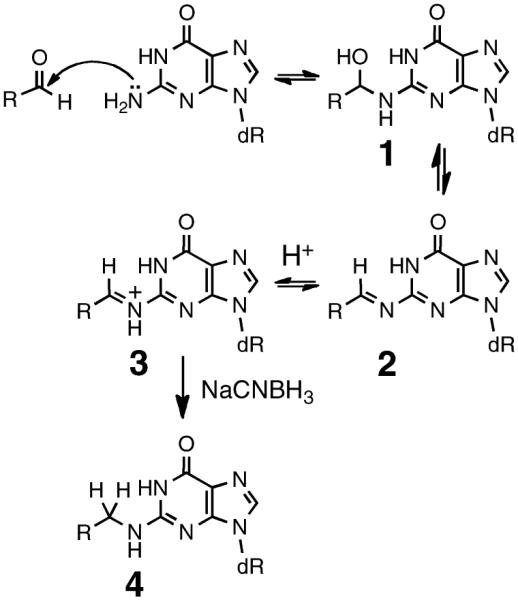
Many molecules that contain an aldehyde functional group display mutagenic or cytotoxic properties that stem from their ability to covalently modify DNA.1–17 Reversible attack of DNA nucleophiles on the electrophilic aldehyde carbon typically yields an equilibrating mixture of hemiaminal and imine adducts (1 and 2, Scheme 1).1,4–14 In some cases, aldehyde–DNA adducts have been stabilized for analysis via irreversible reduction of iminium ion 3 to amine 4 by use of reagents such as NaCNBH3 under mildly acidic conditions (Scheme 1).4,18–20
Scheme 1.
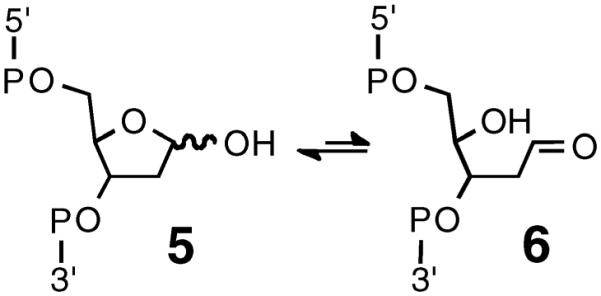
We recently identified a special class of aldehyde–DNA adducts derived from the reaction of DNA abasic (AP) sites with nucleobases on the opposing strand of the double helix.21–23 AP sites are prevalent lesions in genomic DNA that are generated by a wide variety of endogenous cellular processes,24–28 drugs,25 bioactive natural products,29–34 and environmental carcinogens.25,26 AP sites exist as an equilibrium mixture of the ring-closed hemiacetal 5 and the ring-opened aldehyde 6 (Scheme 2).35 We showed that the AP-aldehyde can react reversibly with guanine or adenine residues on the opposing strand of the double helix to generate DNA–DNA interstrand cross-links (Scheme 3).21–23 In the case of the dG–AP cross-link, treatment with NaCNBH3 generated a reduced, chemically stable analogue of the cross-link.21 Interstrand cross-links are extremely deleterious because they block replication and transcription and present serious challenges to cellular DNA repair systems.36–39 Accordingly, AP-derived cross-links have the potential to contribute to aging, sporadic cancers, and biological efiects of the various xenobiotics that induce AP sites in cellular DNA.36–40
Scheme 2.
Scheme 3.
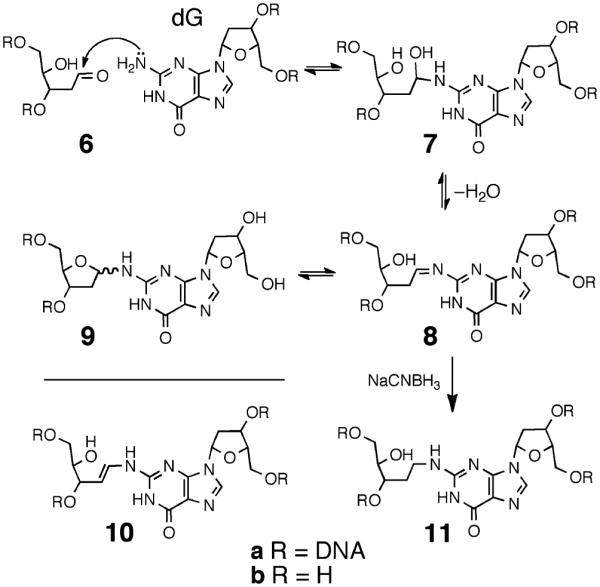
In earlier work, we proposed that dG–AP cross-linking involves attachment of the exocyclic N2-amino group of dG to the anomeric carbon of the AP residue (Scheme 3), but the chemical connectivity of the cross-link was not rigorously determined.21,22 For example, we recognized that the biochemical and mass spectrometric data could not rule out cross-link structures involving attachment of the AP-aldehyde at an endocyclic nitrogen of dG or conversion of an initial imine adduct to the enamine 10a.
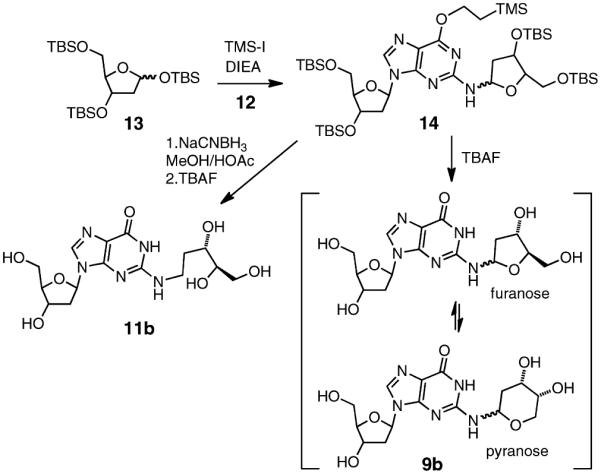
Complete understanding of the molecular events underlying the biological consequences of any given DNA lesion requires precise knowledge of chemical structure. Chemical structure determination of a DNA lesion, in turn, requires full spectroscopic characterizations of the lesion.41 The results described here were designed to shed light on the chemical structure and properties of dG–AP cross-links generated in duplex DNA. Toward this end, we characterized the nucleoside cross-link “remnant” released by enzymatic digestion of a DNA duplex containing the dG–AP cross-link.21 We first carried out a chemical synthesis and complete spectroscopic structure determination of the putative nucleoside cross-link remnant 9b. Liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis was then used to demonstrate that the properties of the authentic cross-link remnant released by enzymatic digestion of a DNA duplex containing the dG–AP cross-link matched those of the synthetic material 9b. The reduced cross-link 11 (Scheme 3) was similarly characterized. The results establish the chemical connectivity of the dG–AP cross-link released from duplex DNA and provide a foundation for detection of this lesion in biological samples. The cross-link remnant 9b was quite stable, decomposing to release dG with a half-life of approximately 17 days at pH 7 and 23 °C. NMR spectroscopic data suggests that the stability of this material is due to the fact that the 2-deoxyribose adduct connected at the N2-position of dG exists as a mixture of the ring-closed α-pyranose, β-pyranose, α-furanose, and β-furanose isomers, with no detectable amounts of the hydrolytically labile imine present (9b, Scheme 5). The intrinsic stability of the cross-link attachment observed in the nucleoside remnant was mirrored in the stability of the actual cross-link in duplex DNA, which dissociated with a half-life of 22 days at pH 7 and 23 °C. This suggests that the dG–AP cross-link may have the power to block critical DNA-processing enzymes.
Scheme 5.
EXPERIMENTAL PROCEDURES
Materials and Methods
Oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). Uracil-DNA glycosylase (UDG, 5000 units/mL) and UDG buffer were purchased from New England Biolabs (Ipswich, MA). 2′-Deoxyguanosine (dG) monohydrate was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). 2-Deoxy-d-ribose (99%), N,N-diisopropylethylamine (redistilled, 99.5%), and tetrabutylammonium fluoride (1 M in tetrahydrofuran, THF) were purchased from Sigma–Aldrich (St. Louis, MO). Iodotrimethylsilane (97%) was purchased from Acros Organics (Thermo Fisher) in a 5 g vial and stored at −15 °C in a desiccator; a small amount of metallic copper was added to the vial as a stabilizer. All other reagents used were purchased from Acros Organics in reagent grade and used without further purification unless otherwise noted. Bulk solvents (hexanes, ethyl acetate, and CH2Cl2) were obtained from Sigma–Aldrich. Methanol, THF, and H2O were purchased from Fisher in HPLC grade. When used as reaction solvents, CH2Cl2 and methanol were dried over 4 and 3 Å molecular sieves, respectively. THF was distilled prior to use. Anhydrous N,N-dimethylformamide (DMF) and dioxane were purchased in AcroSeal bottles from Acros Organics. Column chromatography was performed on silica gel 60 (Sigma–Aldrich) under positive pressure. Glass-backed thin-layer chromatography (TLC) plates with a 254 nm fluorescent indicator were purchased from Sigma–Aldrich and stored in a desiccator. Compounds on developed TLC plates were visualized by use of a 254 nm UV lamp or by dipping in a solution of 10% phosphomolybdic acid hydrate (PMA) in ethanol solution, followed by charring with a heat gun. Unless otherwise specified, reactions were conducted under an atmosphere of dry N2 gas. 1H and 13C NMR spectra were recorded on a Bruker DRX500 at 298 K in CDCl3, deuterated dimethyl sulfoxide (DMSO-d6), or D2O (Cambridge Isotope Laboratories, Inc.).
Syntheses
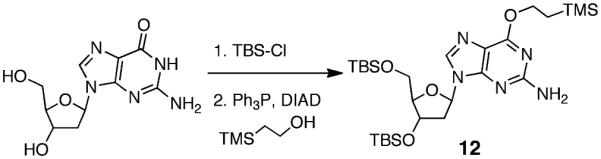
O6-(Trimethylsilylethyl)-3′,5′-bis[O-(tert-butyldimethylsilyl)]-2′-deoxyguanosine (12).
To a solution containing triphenylphosphine (1.91 g, 7.28 mmol) and 2-(trimethylsilyl)ethanol (1.04 mL, 7.28 mmol) in dry dioxane (12 mL) was added 3′,5′-bis-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (1.44 g, 2.91 mmol), which was prepared as described previously.42 Diisopropylazodicarboxylate (1.43 mL, 7.28 mmol) was added slowly by syringe, causing the white slurry to turn translucent yellow upon complete addition. After stirring for 2 h at 23 °C, the solvent was evaporated and the crude oil was redissolved in diethyl ether (12 mL). Triphenylphosphine oxide was crystallized from the mixture by submersion of the flask in liquid N2 and then removed by vacuum filtration. The filtrate was concentrated to a yellow oil and column chromatography on silica gel, eluted with 5:1 hexane/ethyl acetate, gave 12 (1.18 g, 68%, Rf = 0.21 5:1 hexane/ethyl acetate) as a sticky yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.86 (1H, s, H8), 6.30 (1H, t, J = 6.5 Hz, H1′), 4.92 (2H, br s, NH2), 4.59–4.56 (1H, m, H3′), 4.56–4.52 (2H, m, ROCH2CH2TMS), 3.96 (1H, q, J = 3.5 Hz, H4′), 3.79 (1H, dd, J = 4.5, 11 Hz, H5a′), 3.73 (1H, dd, J = 3.3, 11.3 Hz, H5b′), 2.55 (1H, dt, J = 6.5, 13.3 Hz, H2a′), 2.33 (1H, ddd, J = 3.5, 6, 13 Hz, H2b′), 1.24–1.19 (2H, m, ROCH2CH2TMS), 1.00–0.77 [18H, m, SiC(CH3)3], 0.08 [6H, s, Si(CH3)2], 0.06 [6H, s, ROEtSi(CH3)3], 0.06 [3H, s, ROEtSi(CH3)3], 0.05 [6H, s, Si(CH3)2]; 13C NMR (126 MHz, CDCl3) δ 161.3 (C6), 159.2 (C2), 153.3 (C4), 137.3 (C8), 115.9 (C5), 87.6 (C4′), 83.5 (C1′), 71.9 (C3′), 64.8 (ROCH2CH2TMS), 62.8 (C5′), 40.8 (C2′), 25.9, 25.7 [SiC(CH3)3], 18.4, 18.0 [SiC(CH3)3], 17.5 (ROCH2CH2TMS), −1.5 [ROEtSi(CH3)3], −4.7, −4.8, −5.4, −5.6 [Si(CH3)2].
1,3,5-Tris[O-(tert-butyldimethylsilyl)]-2-deoxy-d-ribofuranose (13).
2-Deoxy-d-ribose (1.50 g, 11.18 mmol), tert-butyldimethylsilyl chloride (5.60 g, 37.16 mmol), and imidazole (3.00 g, 44.07 mmol) were dissolved in anhydrous DMF (15 mL) and stirred at 23 °C. After 19 h, the reaction mixture was diluted with hexane (100 mL), washed with water (3 × 50 mL) and brine (1 × 50 mL), and dried over Na2SO4, and the solvent was evaporated under reduced pressure to give a light yellow oil. Column chromatography on silica gel, eluted with 3% ethyl acetate in hexane, gave 13 (3.83 g, 72%) as a colorless gel (Rf = 0.37 in 3% ethyl acetate–hexane): 1H NMR (500 MHz, CDCl3, α and β = anomeric isomers, p and f = pyranose and furanose isomers) δ 5.60 (0.40H, t, J = 4.3 Hz, H1 β-f), 5.46 (0.37H, dd, J = 2.5, 5 Hz, H1 α-f), 5.20 (0.21H, dd, J = 2.3, 5 Hz, H1 β-p), 4.75 (0.02H, dd, J = 2.5, 7.5 Hz, H1 α-p), 4.36 (0.40H, dt, J = 3.2, 5.1 Hz, H3 β-f), 4.18 (0.37H, dt, J = 4.8, 7.8 Hz, H3 α-f), 4.14–4.10 (0.02H, m, H3 α-p), 4.09–4.05 (0.21H, m, H3 β-p), 3.97 (0.37H, q, J = 4.3 Hz, H4 α-f), 3.89–3.86 (0.02H, m, H4 α-p), 3.84 (0.40H, ddd, J = 3, 5, 7.5 Hz, H4 β-f), 3.75–3.69 (0.42H, m, H4 and H5a β-p), 3.67–3.59 (1.37H, m, H5a β-f, H5 α-f, H5b β-p, and H5a α-p), 3.57 (0.40H, dd, J = 7.5, 10.5 Hz, H5b β-f), 3.31 (0.02H, dd, J = 1.5, 12 Hz, H5b α-p), 2.22 (0.37H, ddd, J = 5.4, 7.6, 13.1 Hz, H2a α-f), 2.05–1.97 (1H, m, H2 β-f and H2a β-p), 1.97–1.92 (0.02H, m, H2a α-p), 1.82 (0.37H, ddd, J = 2.4, 4.4, 13.1 Hz, H2b α-f), 1.72–1.68 (0.02H, m, H2b α-p), 1.58 (0.21H, ddd, J = 3.5, 5, 12.5 Hz, H2b β-p), 0.93–0.85 [27H, m, SiC(CH3)3], 0.11–0.03 [18H, m, Si(CH3)2]; 13C NMR (126 MHz, CDCl3) δ 99.1 (C1 β-f), 98.5 (C1 α-f), 94.7 (C1 α-p), 93.0 (C1 β-p), 87.0 (C4 β-f), 85.4 (C4 α-f), 73.3 (C3 β-f), 71.9 (C3 α-f), 70.0 (C4 β-p), 69.9 (C4 α-p), 69.4 (C3 α-p), 68.1 (C3 β-p), 65.8 (C5 α-p), 64.8 (C5 β-f), 64.7 (C5 α-f), 63.0 (C5 β-p), 44.6 (C2 β-f), 44.4 (C2 α-f), 38.7 (C2 β-p), 38.5 (C2 α-p), 26.0, 25.9, 25.8, 25.8, 25.7, 25.7 [SiC(CH3)3], 18.4, 18.3, 18.1, 18.0, 18.0, 18.0, 17.9, 17.9 [SiC(CH3)3], −4.1, −4.2, −4.2, −4.4, −4.5, −4.6, −4.7, −4.7, −4.7, −4.8, −4.8 [Si(CH3)2].
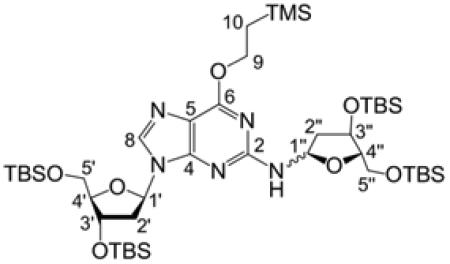
N2-{3,5-Bis[O-(tert-butyldimethylsilyl)]-2-deoxy-d-ribofuranos-1-yl}- O6-benzyl-3′,5′-bis[O-(tert-butyldimethylsilyl)]-2′-deoxyguanosine (14).
Compound 13 (248 mg, 0.520 mmol) was dissolved in dry CH2Cl2 (2 mL) in an oven-dried round-bottom flask. The flask was purged with dry N2 and cooled in a −78 °C dry ice–acetone bath. Trimethylsilyl iodide (TMS-I; 71 μL, 0.499 mmol) was added via syringe to generate the glycosyl iodide, and the resulting light yellow liquid was stirred for 10 min. A solution of 12 (50 mg, 0.084 mmol) and diisopropylethylamine (DIPEA, 120 μL, 0.689 mmol) in dry CH2Cl2 (250 μL) was added by syringe, and the mixture was stirred for 15 min at −78 °C and then for an additional 15 min while warming to room temperature. Unreacted glycosyl iodide was quenched by the addition of dry MeOH (4 mL). The mixture was evaporated to dryness under reduced pressure, redissolved in 7:1 hexane–ethyl acetate, and washed with H2O to remove the diisopropylethylammonium hydroiodide salt. The organic layer was dried over Na2SO4 and evaporated under reduced pressure to yield a yellow oil. Column chromatography on silica gel, eluted with 7:1 hexane–ethyl acetate (v/v), gave 14 (43 mg, 54%, Rf = 0.25) as a white foam: TOF-MS/ES+ 940.5687 M+; 1H NMR (500 MHz, CDCl3) δ 7.86 (0.2H, s, H8 β-f), 7.84 (0.8H, s, H8 α-f), 6.49 (0.8H, d, J = 10.5 Hz, NH α-f), 6.37 (0.2H, m, H1′), 6.31 (0.8H, t, J = 6.5 Hz, H1′), 6.29–6.26 (0.2H, m, H1″ β-f), 6.21 (0.8H, dd, J = 6.5, 10.5 Hz, H1″ α-f), 5.39 (0.2H, d, J = 10 Hz, NH β-f), 4.60–4.55 (1H, m, H3′), 4.55–4.47 (2H, m, OCH2CH2TMS), 4.45 (0.8H, d, J = 4.5 Hz, H3″ α-f), 4.44–4.43 (0.2H, m, H3″ β-f), 4.11 (0.8H, dd, J = 3.8, 7.3 Hz, H4″ α-f), 3.98–3.91 (1H, m, H4′), 3.89 (0.2H, ddd, J = 2.4, 2.4, 4.6 Hz, H4″ β-f), 3.79 (1H, dd, J = 5, 11 Hz, H5a′), 3.75 (1H, dd, J = 3.8, 11 Hz, H5b′), 3.72–3.67 (0.2H, m, H5a″ β-f), 3.67 (0.8H, dd, J = 3.8, 10.8 Hz, H5a″ α-f), 3.58 (0.2H, dd, J = 5, 10.5 Hz, H5b″ β-f), 3.36 (0.8H, dd, J = 7.5, 10.5 Hz, H5b″ α-f), 2.65–2.49 (1H, m, H2a′), 2.39–2.35 (0.2H, m, H2b′ β-f), 2.33 (0.8H, ddd, J = 3.9, 6.1, 13.1 Hz, H2b′ α-f), 2.27–2.20 (0.8H, m, H2a″ α-f), 2.20–2.16 (0.2H, m, H2a″ β-f), 1.99–1.95 (0.2H, m, H2b″ β-f), 1.93 (0.8H, d, J = 13 Hz, H2b″ α-f), 1.28–1.18 (2H, m, OCH2CH2TMS), 0.98–0.88 [36H, m, SiC(CH3)3], 0.15–0.04 [33H, m, Si(CH3)2 and OEtSi(CH3)3];13 C NMR (126 MHz, CDCl3) δ 161.1 (C6), 157.9, 157.6 (C2), 153.3, 153.0 (C4), 137.5, 137.3 (C8), 116.3, 116.2 (C5), 87.7, 87.5 (C4′), 87.0 (C4″α-f), 86.7 (C4″ β-f), 84.1 (C1′), 83.6 (C1″), 83.4 (C1′), 74.5 (C3″ α-f), 73.1 (C3″ β-f), 72.1 (C3′), 64.4 (OCH2CH2TMS), 63.9 (C5″ β-f), 63.7 (C5″ α-f), 63.0 (C5′), 26.0, 26.0, 25.9, 25.8, 25.8 [SiC(CH3)3], 18.4, 18.4, 18.3, 18.2, 18.0, 18.0, 18.0, 17.8, 17.5 [SiC(CH3)3], 17.5 (OCH2CH2TMS), −1.4 [OEtSi(CH3)3], −4.7, −4.7, −4.7, −4.8, −4.9, −5.3, −5.4, −5.5, −5.5, −5.5 [Si(CH3)2]. NMR analysis indicated the presence of a small amount (<5%) of an inseparable isomer whose spectral data are consistent with the N9-α form of 14. Under some conditions, native N9-β-2′-deoxyguanosine can isomerize to N9-α and N7-α/β forms of dG that are not separable by silica gel chromatography.43 We suspected that the electrophilic TMS-I reagent used here may induce small amounts of such isomerization. This was tested by treating 12 with TMS-I and DIPEA, in the absence of 13. After 48 h, 1H NMR of the crude mixture revealed a new singlet downfield of the H8 signal for 12. Similarly, small singlet peaks downfield of H8 were observed in the 1H NMR spectra for 14 and 9b (Figures S5, S10, and S11, Supporting Information). Additionally, the 1H NMR spectrum for 11b contained weak signals whose chemical shifts matched those reported for H8, H3′ and H4′ of the N9-α isomer of dG.44
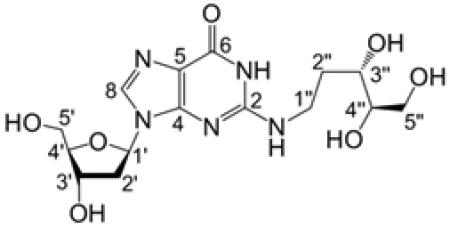
N2-[(3S,4R)-3,4,5-Trihydroxypentyl]-2′-deoxyguanosine (11b).
Compound 14 (48 mg, 0.05 mmol) and NaCNBH3 (32 mg, 0.51 mmol) were dissolved in a mixture of methanol (2 mL) and acetic acid (6 μL, 0.11 mmol). The solution was stirred at room temperature for 4 h, after which the solvent was removed by rotary evaporation under reduced pressure and the resulting residue redissolved in 10 mL of 7:1 hexane–ethyl acetate (v/v). The organic layer was washed with NaHCO3 (2 × 10 mL), H2O (2 × 10 mL), and brine (1 × 10 mL), dried over Na2SO4, and evaporated to give 44 mg of a colorless, sticky oil. This material was dissolved in distilled THF (3 mL) and treated with tetrabutylammonium fluoride (TBAF; 240 μL of a 1 M solution in THF). After 3 h, the mixture was evaporated to give a yellow oil that was redissolved in H2O (5 mL). Addition of NH4PF6 (50 mg, 0.31 mmol) caused precipitation of tetrabutylammonium·PF6 as a white solid that was removed by extraction with CH2Cl2 (4 × 1.5 mL). This step facilitated purification of 11b because tetrabutylammonium salts could not otherwise be removed effectively by column chromatography on silica gel. The aqueous layer was evaporated to a white residue that was subjected to column chromatography on silica gel eluted with 15:5:1 CH2Cl2/MeOH/H2O (Rf = 0.21) to give 11b (9 mg, 46%) as a white solid: LC-LTQ-Orbitrap 386.1678 [M + H]+; 1H NMR (500 MHz, D2O) δ 7.88 (1H, s, H8), 6.25 (1H, t, J = 6.8 Hz, H1′), 4.61 (1H, dt, J = 6.5, 4 Hz, H3′), 4.06 (1H, dt, J = 5.5, 4 Hz, H4′), 3.79 (1H, dd, J = 12.5, 4 Hz, H5a′), 3.74 (1H, dd, J = 12.5, 6.5 Hz, H5b′), 3.73 (1H, dd, J = 18.8, 8.8 Hz, H5a″), 3.71–3.67 (1H, m, H3″), 3.64-.3.61 (1H, m, H4″), 3.59 (1H, dd, J = 19.3, 6.8 Hz, H5b″), 3.49–3.44 (2H, m, H1″), 2.86 (1H, dt, J = 13.9, 6.9 Hz, H2a′), 2.47 (1H, ddd, J = 14, 6.5, 4 Hz, H2b′), 2.00–1.91 (1H, m, H2a″), 1.71–1.61 (1H, m, H2b″); 13C NMR (126 MHz, D2O) δ 159.6 (C6), 153.3 (C2), 152.1 (C4), 116.6 (C5), 138.4 (C8), 87.4 (C4′), 84.5 (C1′), 75.2 (C4″), 71.7 (C3′), 70.1 (C3″), 63.1 (C5″), 62.2 (C5′), 38.7 (C2′), 38.5 (C1″), 32.0 (C2″).
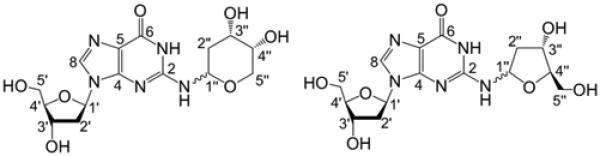
N2-(2-Deoxy-d-ribos-1-yl)-2′-deoxyguanosine (9b)
Compound 14 (88 mg, 0.09 mmol) was dissolved in dry THF (6 mL) and tetrabutylammonium fluoride (562 μL of a 1 M solution in THF) was added by syringe. After the mixture was stirred for 2 h, the solvent was evaporated to a give a yellow oil, which was redissolved in H2O (15 mL). Addition of NH4PF6 (120 mg, 0.74 mmol) gave a white precipitate that was removed by extraction with CH2Cl2 (4 × 5 mL). The aqueous layer was evaporated and the resulting white residue was subjected to column chromatography on silica gel, eluted with 15:5:1 MeOH/ CH2Cl2/H2O (Rf = 0.19), to give 9b (21 mg, 59%) as an off-white solid: LC-LTQ-Orbitrap 384.1521 [M + H]+; 1H NMR (500 MHz, D2O) δ 7.94 (1H, br s, H8), 6.35–6.26 (1H, m, H1′), 6.02–6.00 (0.1H, m, H1″ β-f), 6.00–5.98 (0.1H, m, H1″ α-f), 5.54 (0.3H, dd, J = 8, 2.5 Hz, H1″ β-p), 5.26 (0.6H, dd, J = 9.5, 2.5 Hz, H1″ α-p), 4.67–4.62 (1H, m, H3′), 4.43–4.40 (0.1H, m, H3″ β-f), 4.40–4.37 (0.1H, m, H3″ α-f), 4.22–4.18 (0.3H, m, H3″ β-p), 4.12–4.09 (0.1H, m, H4″ α-f), 4.06 (1H, dd, J = 9, 4 Hz, H4′), 4.05–4.01 (0.6H, m, H3″ α-p), 3.99–3.96 (0.1H, m, H4″ β-f), 3.94 (0.6H, dd, J = 12.5, 3 Hz, H5a″ α-p), 3.87 (0.6H, br s, H4″ α-p), 3.85–3.82 (0.3H, m, H4″ β-p), 3.82–3.76 (2H, m, H5′), 3.76–3.73 (0.6H, m, H5″ β-p), 3.70 (0.6H, d, J = 12.5 Hz, H5b″ α-p), 3.63–3.59 (0.1H, m, H5b″ α-f), 2.93 (0.3H, ddd, J = 13.9, 6.9, 6.9 Hz, H2a′), 2.85 (0.7H, ddd, J = 13.8, 6.8, 6.8 Hz, H2a′), 2.57–2.53 (0.1H, m, H2a″ α-f), 2.53–2.46 (1H, m, H2b′), 2.32 (0.1H, ddd, J = 13.8, 6, 2.3 Hz, H2a″ β-f), 2.22–2.19 (0.1H, m, H2b″ β-f), 2.16 (0.3H, ddd, J = 14, 6.3, 2.8 Hz, H2a″ β-p), 2.09–2.02 (0.6H, m, H2a″ α-p), 2.02–1.99 (0.1H, m, H2b″ α-f), 1.98–1.93 (0.3H, m, H2b″ β-p), 1.93–1.88 (0.6H, m, H2b″ α-p); 13C NMR (126 MHz, D2O) δ 159.2 (C6), 151.8, 151.7 (C2), 151.3 (C4), 139.4, 139.2 (C8), 117.9 (C5), 87.6, 87.5, 87.4 (C4′), 86.5 (C4″ α-f), 86.3 (C4″ β-f), 84.8, 84.7, 84.6, 84.4 (C1′), 82.9 (C1″ β-f), 82.8 (C1″ α-f), 78.7 (C1″ α-p), 76.5 (C1″ β-p), 72.2 (C3″ α-f), 72.1 (C3″ β-f), 71.8, 71.8, 71.5, 71.5 (C3′), 68.2 (C3″ α-p), 67.2 (C4″ β-p), 67.2 (C5″ α-p), 67.1 (C4″ α-p), 66.4 (C3″ β-p), 64.2 (C5″ β-p), 62.6 (C5″ β-f), 62.4 (C5′), 62.3 (C5″ α-f), 62.1 (C5′), 39.5 (C2″ α-f), 39.4 (C2″ β-f), 38.9, 38.8, 38.7 (C2′), 34.9 (C5″ β-p), 33.3 (C5″ α-p).
Analysis of Stability of 9b in Aqueous buffer
Compound 9b (0.4 mg, 1 mM final) was dissolved in 4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid (HEPES) buffer (1 mL, 50 mM, pH 7) containing 100 mM NaCl. Decomposition of 9b to release free dG was monitored by HPLC on an Agilent 1100 series HPLC equipped with autosampler. The reaction was monitored over the course of 34 days at 23 °C, during which time 20 μL aliquots were injected on a Varian Microsorb-MV 100-5 250 × 4.6 mm C18 column that was eluted with 100% H2O from0 to 5 min, 0–10% acetonitrile in H2O from5 to 60 min, and 10–0% acetonitrile from 60 to 65 min. Products were monitored by their absorbance at 254 nm. Peak areas were corrected for the relative molar absorptivities of 9b and dG at 254 nm, which were found to be 13 090 and 12 905 M−1·cm−1, respectively. To obtain the apparent first-order rate constant, data for the disappearance of 9b and the appearance of dG were fit to the appropriate equations for a first-order process.45
Gel Electrophoretic Analysis of Stability of 9a and 11a in Duplex DNA
Cross-links were generated as previously described.21 Single-stranded 2′-deoxyoligonucleotides were 5′-32P-labeled by standard procedures. Labeled DNA was annealed with its complementary strand and treated with the enzyme UDG (50 units/mL, final concentration) to generate AP sites. The enzyme was removed by phenol–chloroform extraction. The AP-containing double-stranded DNA was ethanol-precipitated, resuspended in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM), and incubated at 37 °C for 72 h to generate the dG–AP interstrand cross-link. The reaction mixture was ethanol-precipitated, resuspended in formamide loading buffer, and loaded onto a 20% denaturing polyacrylamide gel. The gel was electrophoresed for 8 h at 200 V to separate cross-linked from uncross-linked DNA. The slower-migrating cross-link band was located by autoradiography and excised from the gel, the gel slice was crushed, and the DNA in the gel was eluted into HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) and incubated at either 37 or 23 °C. Stability of the dG–AP cross-link in duplex DNA was examined by removing aliquots from the assay mixtures at specified time points and freezing them at −20 °C until all the aliquots were collected. The samples were then dissolved in formamide loading buffer and loaded onto a 20% denaturing polyacrylamide gel, and the gel was electrophoresed for 4 h at 1000 V. The amount of radiolabeled DNA in each band on the gel was quantitatively measured by phosphorimager analysis.
Enzymatic Digestion and Mass Spectrometric Analysis
Cross-linked duplex A was prepared as described previously.21 To 500 pmol of the duplex DNA sample was added a cocktail of enzymes including nuclease P1 (1 unit), phosphodiesterase I (0.005 unit), phosphodiesterase II (0.005 unit), and alkaline phosphatase (1 unit), as described previously.46 To inhibit contaminating adenosine deaminase activity during digestion, erythro-9-(2-hydroxy-3-nonyl)adenine was also added to the digestion mixture at a final concentration of 2.5 mM. Enzymes in the digestion mixture were removed by chloroform extraction, the aqueous layer was dried in a Speed-vac, and the residue was reconstituted in water and subjected to mass spectrometric analyses.
The LC-MS/MS and -MS/MS/MS experiments were conducted on an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific). Samples were loaded and separated on a 0.5 × 250 mm Zorbax SB-C18 column (5 μm beads, 80 Å pore size; Agilent Technologies, Santa Clara, CA) at a flow rate of 8.0 μL/min. A solution of 0.1% (v/v) formic acid in water (A) and a solution of 0.1% (v/v) formic acid in methanol (B) were used as mobile phases, and the gradient included 5 min, 0–20% B; 35 min, 20–50% B 10 min, 50% B; and 1 min, 50–0% B.
RESULTS AND DISCUSSION
Synthesis and Spectroscopic Characterizations of Unreduced Cross-Link Remnant 9b
Our initial attempts to prepare 9b involved simply mixing 2-deoxyribose (dR) with 2′-deoxyguanosine (dG). This approach held some promise because, in a number of other cases, the reaction of an aldehyde-containing molecule with unprotected dG yields the corresponding N2-adduct.4,6,14,47,48 Accordingly, we examined the reaction of dG (40 mM) with dR (120 mM) in sodium acetate buffer (200 mM, pH 4) or a mixture of methanol/water/acetic acid (8:1:4).49 Unfortunately, no detectable amounts of product were observed under these conditions as judged by thin-layer chromatographic analysis.
We undertook an alternative approach to the synthesis of 9b involving reaction of an activated 2-deoxyribosyl donor with a protected dG derivative. Our approach was based on the route used by Takamura-Enya et al.50 for the synthesis of N2-(D-ribos-1-yl)-2′-deoxyguanosine. We prepared the protected 2′-deoxyguanosine analogue 12 by reaction of dG with t-butyldimethylsilyl chloride,42 followed by a Mitsunobu reaction with 2-(trimethylsilyl)ethanol to install the (2-trimethylsilyl)- ethyl group at O6 of the guanine residue (Scheme 4).51 The 2-deoxyribosyl donor, 3,5-bis(t-butyldimethylsilyl)-2-deoxyribosyl- 1-iodide was generated in situ via reaction of 1,3,5-tris(t- butyldimethylsilyl)-2-deoxyribose 13 with trimethylsilyl iodide (TMS-I) in CH2Cl2 at −78 °C, and to this mixture was added a solution of 12 and diisopropylethylamine (DIEA) in CH2Cl2 (Scheme 5). Our use of a 2-deoxyribosyl iodide rather than the previously reported ribosyl iodide necessitated some modifications of the literature method,50 including lower reaction temperatures, a larger excess of the iodide, and greater equivalents of DIEA. The reaction generated a major product 14 that eluted faster than 12 on silica gel and was isolated in 54% yield by column chromatography.
Scheme 4.
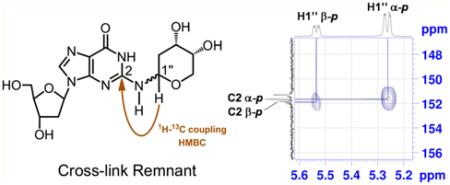
Product 14 was fully characterized by NMR spectroscopy. One-dimensional (1D) 1H NMR and 1H–1H COSY spectra revealed resonances associated with the dG nucleoside52 alongside resonances from the 3,5-bis(tert-butyldimethylsilyl)-2-deoxyribose unit. The 13C resonances associated with each carbon were assigned by use of heteronuclear multiple-quantum correlation (HMQC) and heteronuclear multiple-bond correlation (HMBC) spectra. Two-dimensional (2D) NMR spectra further provided evidence that the 3,5-(tert-butyldimethylsilyl)- 2-deoxyribose adduct was attached to the exocyclic N2-position of the guanine residue as expected. Specifically, the 1H–1H correlation spectroscopy (COSY) experiment showed a correlation between N2-H of the guanine residue and H1″ of the 3,5-bis(tert-butyldimethylsilyl)-2-deoxyribose adduct (Figure 1). In addition, the HMBC experiment showed correlations between C2 of the guanine residue and H1″, along with a correlation between N2-H and C2″.
Figure 1.

Selected regions of 1H–1H COSY and 1H–13C HMBC spectra of 14 in CDCl3 acquired at 500 (1H) and 126 (13C) MHz. Attachment of the 3,5-bis[O-(tert-butyldimethylsilyl)]-2-deoxyribofuranose moiety to N2 of the protected dG was established by (a) homonuclear (COSY) coupling of H1″ to N2-H as well as three-bond HMBC coupling of (b) N2-H to C2″ and (c) H1″ to C2.
A doubling of resonances associated with the 3,5-bis(tert-butyldimethylsilyl)-2-deoxyribose adduct in 14 indicated that this moiety was present as a mixture of isomers. Careful inspection of the NMR spectra suggested that the mixture consisted of a 4:1 diastereomeric mixture of α- and β-anomers of the furanose form of the sugar (Scheme 5). This assessment was based upon the chemical shifts of C4″ in the two isomers of 14 at 86.7 and 87.0 parts per million (ppm), values consistent with a furanose sugar.53 The major isomer showed a strong nuclear Overhauser efiect (NOE) cross-peak between guanine N2-H and H4″, consistent with the α-isomer, while the minor isomer showed a stronger NOE cross-peak between H1″ and H4″, consistent with the β-isomer (Figure 2).44 The high-resolution mass spectrum was consistent with the molecular formula expected for 14.
Figure 2.

1H–1H NOESY spectrum of 14 in CDCl3 acquired at 500 MHz with a mixing time of 800 ms; negative peaks are denoted by red curves. The expanded frame shows the strong through-space correlation between H4″ and N2-H of the major α-furanose isomer. Also shown is a cross-peak between H4″ and H1″ of the minor β-furanose isomer; no correlation between H4″ and N2-H of the minor isomer is evident.
Overall, the spectral data provided evidence that coupling of 12 with 2-deoxyribosyl iodide generated the desired product 14. While this product has the potential to exist as either a ring-opened imine or a cyclic hydroxyalkylhemiaminal, the C1″ chemical shift at 83.6 ppm for this product was consistent with the cyclic structure of 14.53 The corresponding imine 13C-chemical shift would be expected at approximately 160 ppm.20 The observed HMBC correlation between H1″ and C4″ also was consistent with the ring-closed isomer of 14. Table 1 provides a complete list of the chemical shifts and 2D NMR correlations for 14.
Table 1.
Chemical Shifts and 2D Correlations for 14 in CDCl3
|
|||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| position / isomer | δ C | Ha/b | δH (J in Hz) | COSY | TOCSY | HMBCa | NOESY |
|
| |||||||
| 2 | 157.9, 157.6b | ||||||
| 4 | 153.3, 153.0 | ||||||
| 5 | 116.3, 116.2 | ||||||
| 6 | 161.1 | ||||||
| 8 α | 137.5 | 7.84, s | 4, 5 | 1′, 2a′, 3, 5a′, 5b′ | |||
| 8 β | 137.3 | 7.86, s | 4, 5 | 1′, 2a′, 3′, 5a′, 5b′ | |||
| 1′ α | 84.1 | 6.31, t (6.5) | 2b′ | 2a′, 2b′, 3′ | 4, 8 | 2a′, 2b′, 3′, 4′, 8 | |
| 1′ β | 83.4 | 6.40–6.34, m | 2a′, 2b′ | 2a′, 2b′, 3′ | 8 | 2b′, 3′, 4′, 8 | |
| 2′ α | 41.0 | a | 2.67–2.57, m | 2b′ | 1′, 2b′, 3′ | 1′, 2b′, 3′, 5a′, 5b′, 8 | |
| b | 2.33, ddd (13.1, 6.1, 3.9) | 1′, 2a′, 3′ | 1′, 2a′, 3′, 4′ | 3′, 4′ | 1′, 2a′, 3′, 4′ | ||
| 2′ β | (41.0)c | a | 2.53, ddd (13.4, 6.6, 6.6) | 1′, 2b′, 3′ | 1′, 2b′, 3′ | 1′ | |
| b | 2.39–2.33, m | 1′, 2a′, 3′ | 1′, 2a′, 3′ | 3′, 4′ | |||
| 3 | 72.1, 72.1 | 4.60–4.55, m | 2b′, 4′ | 1′, 2a′, 2b′, 4′, 5′ | 1′, 2a′, 2b′, 4′, 5a′, 5b′, 8 | ||
| 4′ | 87.5 | 3.98–3.91, m | 3′, 5′ | 3′, 5′ | 1′, 2b′, 3′, 5a′, 5b′ | ||
| 5′ | 63.0 | a | 3.79, dd (11.5) | 4′ | 3′, 4′ | 3′ | 2a′, 3′, 4′, 8 |
| b | 3.75, dd (11.3, 3.8) | 4′ | 3′, 4′ | 3′ | 2a′, 3′, 4′, 8 | ||
| 1″ a | 83.6 | 6.21, dd (10.5, 6.5) | 2a″, N2–H | 2a″, 2b″!, 3″, N2–H | 2, 3″, 4″ | 2a″, 2b″, 3″, 4″ | |
| 1″ β | (83.6) | 6.29–6.26, m | 2a″, 2b″, N2–H | 3″ | 2a″, 4″ | ||
| 2″ a | 39.2 | a | 2.27–2.20, m | 1′, 2b″, 3″ | 1″, 2b″, 3″, N2–H | 2, 1″ | 1″, 2b″, 3″, 5b″ |
| b | 1.93, d (13) | 2a″ | 1″, 2a″, 3″, N2–H | 3″, 4″ | 1″, 2a″, 3″, N2–H | ||
| 2″ β | 41.6 | a | 2.20–2.16, m | 1″, 2b″ | 2b″, 3″, N2–H | 3″, 4″ | 1″, 2b″, 3″ |
| b | 1.99–1.95, m | 1″, 2a″ | 2a″, N2–H | 1″ | 2a″, 3″, N2–H | ||
| 3″ α | 74.5 | 4.45, d (4.5) | 2a″ | 1″, 2a″, 2b″, N2–H | 1″, 4″, 5″ | 1″, 2a″, 2b″, 4″, 5a″, 5b″ | |
| 3″ β | 73.1 | 4.43, m | 2b″ | 1″, 2a″, 4″, N2–H | |||
| 4′ α | 87.0 | 4.11, dd (7.3, 3.8) | 5a″, 5b″ | 5a″, 5b″ | 3″ | 3″, 5a″, 5b″, N2–H | |
| 4″ β | 86.7 | 3.89, ddd (4.6, 2.4, 2.4) | 5b″ | 3″, 5a″, 5b″ | |||
| 5″ α | 63.7 | a | 3.67, dd (10.8, 3.8) | 4″, 5b″ | 4″, 5b″ | 3″, 4″ | 3″, 4″, 5b″ |
| b | 3.36, dd (10.5, 7.5) | 4″, 5a″ | 4″, 5a″ | 3″, 4″ | 3″, 4″, 5a″ | ||
| 5″ β | 63.9 | a | 3.72–3.67, m | 5b″ | 4″, 5b″ | 5b″ | |
| b | 3.58, dd (10.5, 5) | 5a″ | 4″, 5a″ | 5a″ | |||
| N2–H α | 6.49, d (10.5) | 1″ | 1″, 2a″, 2b″, 3″ | 2″ | 2b″, 3″, 4″ | ||
| N2–H β | 5.39, d (10) | 1″ | 1″, 2a″, 2b″ | 2″ | 1″, 2b″ | ||
| 9 | 64.7, 64.4 | 4.55–4.47, m | 10 | 10 | 6 | 10 | |
| 10 | 17.5, 17.5 | 1.28–1.18, m | 9 | 9 | 9 | 9 | |
HMBC correlations are from protons in the σH column to the listed carbon(s).
Multiple C peaks represent the two stereoisomers of 14; resonances were assigned to individual isomers where possible.
Parentheses denote 13C signal of the β-isomer is indistinguishable from that of the α-isomer.
The silyl protecting groups were removed from 14 by treatment with tetrabutylammonium fluoride (TBAF) in THF to generate a new product in 59% yield. As expected, the NMR spectrum of the deprotected material 9b was more complex than that of the starting material 14 because removal of the protecting groups from the 2-deoxyribose adduct enables interconversion between pyranose and furanose forms, each of which can exist as a mixture of α- and β-isomers (Scheme 5).53–56 Two-dimensional NMR experiments provided evidence that the 2-deoxyribose adduct in 9b remained connected at the exocyclic N2-amino group of the guanine residue. As described above in the context of 14, the 1H–1H COSY spectrum of 9b in DMSO-d6 showed a correlation between N2-H and H1″ of the 2-deoxyribose adduct (Figure 3). Again, the HMBC spectrum showed a correlation between C2 of the guanine residue and H1″. Observation of the N1-imino proton at 10.9 ppm in the 1H NMR spectrum in DMSO-d6 further provided evidence against attachment of the adduct at N1. Overall, the NMR spectral data provided evidence that the 2-deoxyribose adduct remained attached at the exocyclic N2-amino group of the guanine residue as shown in 9b.
Figure 3.

Selected regions of 1H–13C HMBC and 1H–1H COSY spectra of 9b obtained at 500 (1H) and 126 (13C) MHz, respectively. Correlations between (a) H1″ and C2 in D2O as well as (b) H1″ and N2-H in DMSO-d6 indicate that the 2-deoxyribose moiety remained attached to N2 of dG following deprotection.
All four of the anticipated isomers arising from equilibration of the 2-deoxyribose adduct were observed in the NMR spectra of 9b. Similar to 14, the positions of the 13C shifts of C4″ in the isomers of 9b, along with the results of NOE experiments, allowed us to assign key resonances to each of the four isomers and determine that the 2-deoxyribose adduct connected to N2 of the guanine residue exists as a 6:2:1:1 mixture of α-pyranose, β-pyranose, α-furanose, and β-furanose isomers. This is consistent with the observation that pyranose isomers of unprotected 2-deoxyribose and its arylamino-2-deoxyriboside analogues typically predominate in solution.53–56 The chemical shifts of C1″ in the isomers of 9b at 76.5–82.9 ppm, along with HMBC correlations observed between H1″ and C4″ in the furanose isomers and between H5″ and C1″ in the pyranose isomers, were again consistent with the cyclic hydroxyalkylhemiaminal form. No evidence for a ring-opened imine isomer was seen in the NMR spectra. Predominance of the cyclic isomer is consistent with literature reports for structurally related compounds.14,48,57 The 1H NMR chemical shifts of key positions such as H1″, N2–H, H8, and N1–H in 9b were similar to those observed for related DNA adducts.14,48,57 Table 2 provides a complete list of the chemical shifts and 2D NMR correlations for 9b. High-resolution mass spectrometric analysis was consistent with the molecular formula expected for 9b. When the compound was dissolved in D2O and subjected to ESI-MS analysis with the use of an equivolume mixture of D2O and acetonitrile as the spray solvent, a mass increase of 6 amu was observed, consistent with the presence of six exchangeable hydrogens in 9b (Figure S25, Supporting Information).
Table 2.
Chemical Shifts and 2D Correlations for 9b in D2O
|
|||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| position / isomer | δ c | Ha/b | δH (J in Hz) | COSY | TOCSY | HMBCa | NOESY |
|
| |||||||
| 2 | 152.0, 151.9, 151.8, 151.7b |
||||||
| 4 | 151.3 | ||||||
| 5 | 117.9 | ||||||
| 6 | 159.2 | 4, 5, 6, 1′ | 1′, 2a′, 3′, 5′ | ||||
| 8 | 139.4, 139.2 | 7.94, br s | |||||
| 1′ | 84.8, 84.7, 84.6, 84.4 | 6.35–6.25, m | 2a′, 2b′ | 2a′, 2b′, 3′, 4′ | 4, 8, 3′, 4′ | 8, 2b′, 4′ | |
| 2′ | 38.9, 38.8, 38.7 | a | 2.93, ddd (13.9, 6.9, 6.9); 2.85, ddd (13.8, 6.8, 6.8) |
1′, 2b′, 3′ | 1′, 2b′, 3′, 4′, 5′ | 1′, 3′, 4′ | 8′, 2b′, 3′ |
| b | 2.53–2.46, m | 1′, 2a′, 3′ | 1′, 2a′, 3′, 4′, 5′ | 3′, 4′ | 1′, 2a′, 3′, 4′ | ||
| 3′ | 71.8, 71.7, 71.5, 71.5 | 4.67–4.62, m | 2a′, 2b′, 4′ | 1′, 2a′, 2b′, 4′, 5′ | 1′ | 8, 2a′, 2b′, 4′, 5 |
|
| 4′ | 87.6, 87.5, 87.4 | 4.06, dd (9, 4) | 3′, 5′ | 1′, 2a′, 2b′, 3′, 5′ | 3′ | 3′, 5′ | |
| 5′ | 62.1 | 3.82–3.76, m | 4′ | 2a′, 2b′, 3′, 4′ | 3″, 4″ | 8, 3′, 4′ | |
| 1″ α-p | 78.7 | 5.26, dd (9.5, 2.5) | 2a″, 2b″ | 2a″, 2b″, 3″, 4″ | 2, 3″ | 2a″, 3″, 5b″ | |
| 1″ β-p | 76.5 | 5.54, dd (8, 2.5) | 2a″, 2b″ | 2a″, 2b″, 3″ | 2, 3″ | 2a″, 5″ | |
| 1″ α-f | 82.8 | 6.00–5.98, m | 2a″, 2b″ | 2a″, 2b″, 3″ | 3″, 4″ | 2a″ | |
| 1″ β-f | 82.9 | 6.02–6.00, m | 2a″, 2b″ | 2a″, 2b″, 3″ | 3″, 4″ | 2a″ | |
| 2″ α-p | 33.3 | a | 2.09–2.02, m | 1″, 2b″, 3″ | 1″, 2b″, 3″, 4″ | 1″, 5″ | 1″, 2b″, 3″ |
| b | 1.93-1.88,m | 1″, 2a″, 3″ | 1″, 2a″, 3″, 4″ | 1″, 5″ | 2a″ | ||
| 2″ β-p | 34.9 | a | 2.16, ddd (14, 6.3,2.8) | 1″, 2b″, 3″ | 1″, 2b″, 3″ | 5″ | 1″, 2b″, 3″ |
| b | 1.98–1.93, m | 1″, 2a″, 3″ | 1″, 2a″, 3″ | 2a″, 3″ | |||
| 2″ α-f | 39.5 | a | 2.57–2.53, m | 1″, 2b″, 3″ | 1″, 2b″, 3″ | 1″, 2b″ | |
| b | 2.02–1.99, m | 1″, 2a″, 3″ | 1″, 2a″, 3″ | 2a″ | |||
| 2″ β-f | 39.4 | a | 2.32, ddd (13.8, 6, 2.3) | 1″, 2b″ | 1″, 2b″, 3″ | 1″, 2b″ | |
| b | 2.22–2.19, m | 1″, 2a″, 3″ | 1″, 2a″, 3″ | 1″ | 2a″ | ||
| 3″ α-p | 68.2 | 4.05–4.01, m | 2a″, 2b″, 4″ |
1″, 2a″, 2b″, 4″ | 1″, 2a″, 4″ | ||
| 3″ β-p | 66.4 | 4.22–4.18, m | 2a″, 2b″ | 1″, 2a″, 2b″, 5″ | 2a″, 2b″, 4″ | ||
| 3″ α-f | 72.2 | 4.40–4.37, m | 2a″, 2b″ | 1″, 2a″, 2b″, 4″, 5b″ | 2a″, 5″ | ||
| 3″ β-f | 72.1 | 4.43–4.40, m | 2b″ | 1″, 2a″, 2b″, 4″ | 2b″ | ||
| 4″ α-p | 67.1 | 3.87, br s | 3″ | 1′, 2a″, 2b″, 3″, 5a″, 5b′ |
2′, 3″ | 3″ | |
| 4″ β-p | 67.2 | 3.85–3.82, m | 3″, 5″ | 3″ | |||
| 4″ α-f | 86.5 | 4.12–4.09, m | 5b″ | 3″, 5b″ | 1″ | 5″ | |
| 4″ β-f | 86.3 | 3.99–3.96 | 3″ | ||||
| 5″ α-p | 67.2 | a | 3.94, dd (12.5, 3) | 5b″ | 4″, 5b″ | 1″, 3″ | 5b″ |
| b | 3.70, d (12.5) | 5a″ | 4″, 5a″ | 1″ | 1″, 5a″ | ||
| 5″ β-p | 64.2 | 3.76–3.73, m | 3″ | 1″ | 1″ | ||
| 5″ α-f | 62.3 | b | 3.63–3.59, m | 4″ | 4″ | 3″ | |
| 5″ β-f | 62.6 | ||||||
HMBC correlations are from protons in the δH column to the listed carbon(s).
Multiple 13C peaks arise from the four isomers of 9b.
Synthesis and Spectroscopic Characterization of Reduced Cross-Link Remnant 11b
In our previous characterization of the dG–AP cross-link in duplex DNA, we employed conditions of reductive amination18 to generate good yields of a chemically stable cross-linked species, whose structure was tentatively assigned as 11a (Scheme 3).21,22 Reduction of imine and cyclic hydroxyalkylhemiaminal adducts attached at the N2-amino group of guanine residues has been used by others to facilitate the detection and analysis of aldehyde-derived DNA adducts.4,14,19,20,48 To elucidate the structure of the reduced dG–AP cross-link in DNA, we wished to prepare a synthetic standard for the proposed structure of the reduced dG–AP cross-link remnant 11b. Toward this end, we treated the protected dG-dR derivative 14 with NaCNBH3 in methanol–acetic acid. The silyl groups were then removed from the crude product by treatment with TBAF in THF, and the resulting material was purified by column chromatography on silica gel eluted with 15:5:1 CH2Cl2/CH3OH/H2O. As anticipated, the NMR spectra of the product 11b were greatly simplified compared to those of either 14 or 9b, consistent with loss of the potential for pyranose–furanose and anomeric isomerism (Scheme 5). Significant changes were especially evident at the C1″ position of the reduced adduct. The splitting patterns and integrations associated with the H1″ and H2″ protons were diagnostic for the presence of a CH2 group at C1″. The resonances for the H1″ protons appeared at 3.44–3.49 ppm, a range consistent with the published data for an analogous structure.58 The 13C shift of C1″ changed dramatically from 76.5 to 82.9 ppm in 9b to 38.5 ppm in 11b, consistent with the acyclic alkyl adduct in 11b. As described above in the context of 14 and 9b, 1H–1H COSY and HMBC spectra of 11b provided evidence for attachment of the 2-deoxyribose unit at the N2-position of the guanine residue. Observation of the N1-imino proton at 10.6 ppm in the 1H NMR spectrum in DMSO-d6 provided evidence against attachment of the adduct at N1. The 13C chemical shifts of the guanine residue in 14 were similar (±3.5 ppm or less) to those in the native nucleoside dG and in other known N2-alkyl-dG adducts.59,60 Table 3 provides a complete list of the chemical shifts and 2D-NMR correlations for 11b. Plots of NMR and UV–vis spectra for 14, 9b, and 11b are provided in Supporting Information. In addition, ESI-MS analysis of 11b in H2O and D2O revealed the presence of seven exchangeable protons in the [M + Na]+ ion of the compound, which is consistent with the proposed structure (Figure S25, Supporting Information).
Table 3.
Chemical Shifts and 2D Correlations for 11b in D2O
|
|||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| position | δ C | Ha/b | δH (J in Hz) | COSY | TOCSY | HMBCa | NOESY |
|
| |||||||
| 2 | 153.3 | ||||||
| 4 | 152.1 | ||||||
| 5 | 116.6 | ||||||
| 6 | 159.6 | ||||||
| 8 | 138.4 | 7.88, s | 4, 5, 6, 1′ | 1′, 2a′, 3′, 5a′, 5b′ | |||
| 1′ | 84.5 | 6.25, t (6.8) | 2a′, 2b′ | 2a′, 2b′, 3′, 4′ | 4, 8, 2′, 3′, 4′ | 8, 2a′, 2b′, 4′ | |
| 2′ | 38.7 | a | 2.86, ddd (13.9, 6.9, 6.9) | 1′, 2b′, 3′ | 1′, 2b′, 3′, 4′ | 1′, 3′, 4′ | 1′, 2b′, 3′ |
| b | 2.47, ddd (14.0, 6.5, 4) | 1′, 2a′, 3′ | 1′, 2a′, 3′, 4′ | 3′, 4′ | 1′, 2a′, 3′, 4′ | ||
| 3′ | 71.7 | 4.61, dt (6.5, 4) | 2a′, 2b′, 4′ | 1′, 2a′, 2b′, 4′, 5a′, 5b′ | 1′, 5′ | 8, 2a′, 2b′, 5a′, 5b′ | |
| 4′ | 87.4 | 4.06, dt (5.5, 4) | 3′, 5a′, 5b′ | 1′, 2a′, 2b′, 3′, 5a′, 5b′ | 1′, 3′ | 1′, 2b′ | |
| 5′ | 62.2 | a | 3.79, dd (12.5,4) | 5b′ | 3′, 4′, 5b′ | 3′, 4′ | 8, 3′ |
| b | 3.74, dd (12.5, 6.5) | 5a′ | 3′, 4′, 5a′ | 3′, 4′ | 8, 3′ | ||
| 1″ | 38.5 | 3.49–3.44, m | 2a″, 2b″ | 2a″, 2b″, 3″, 4″ | 2, 2″, 3″ | 2a″, 2b″, 3″ | |
| 2″ | 32.0 | a | 2.00–1.91, m | 1″, 2b″, 3 | 1″, 2b″, 3″, 4″ | 1″, 3″, 4″ | 1″, 2b″, 3″, 5b″ |
| b | 1.71–1.61, m | 1″, 2a″, 3″ | 1″, 2a″, 3″, 4″ | 1″, 3″, 4″ | 1″, 2a″, 3″, 5a″ | ||
| 3″ | 70.1 | 3.71–3.67, m | 2a″, 2b″ | 1″, 2a″, 2b″, 4″, 5a″, 5b″ | 1″, 2″, 4″ | 1″, 4″ | |
| 4″ | 75.2 | 3.64–3.61, m | 1″, 2a″, 2b″, 3″, 5a″, 5b″ | 2″ | 3″ | ||
| 5″ | 63.1 | a | 3.73, dd (18.8, 8.8) | 5a″ | 3″, 4″, 5b″ | 2b″ | |
| b | 3.59, dd (19.3, 6.8) | 5b″ | 3″, 4″, 5a″ | 3″, 4″ | 2a″ | ||
HMBC correlations are from protons in the δH column to the listed carbon(s).
Stability of Unreduced Cross-Link Remnant 9b and Native Interstrand DNA–DNA Cross-Link
The formation of N-aryl aminoglycosides is reversible.61,62 Consequently, we recognized that the cross-link remnant 9b had the potential to revert to dG and 2-deoxyribose. Therefore, before employing 9b as an analytical standard, we examined the stability of this material. We used HPLC to monitor the stability of 9b (1 mM) at 23 °C in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM). Our results indicated that 9b was quite stable under these conditions, disappearing with a half-life of approximately 17 days (corresponding to a first-order rate constant of 4 × 10−2 day−1). As expected, decomposition of 9b was accompanied by release of the native nucleoside dG (Figure 4). The stability of 9b was also monitored by thin-layer chromatography (Figure S19, Supporting Information).
Figure 4.

Stability of dG–AP cross-linkage. (A) Synthetic cross-link remnant 9b was incubated at 23 °C in HEPES buffer (pH 7, 50 mM) containing NaCl (100 mM). Disappearance of 9b and concomitant appearance of dG were monitored by HPLC. (B) Gel electrophoretic analysis of purified, unreduced cross-link in duplex A, incubated at 37 °C in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM). The fast-moving fragment at the bottom of the gel corresponds to the 3′-4-hydroxy-2-pentenal-5-phosphate cleavage product resulting from β-elimination at the AP site.
We compared the stability of cross-link remnant 9b to that of the dG–AP cross-link in duplex DNA. Accordingly, we generated the dG–AP cross-link in a 32P-labeled version of the 21 base pair duplex A (5′-32P-labeled on the top strand) and isolated the cross-linked DNA by gel electrophoresis. Dissociation of the cross-linked DNA into the single strand 6a in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 23 °C was monitored by gel electrophoresis. The cross-linked DNA decomposed with apparent first-order kinetics and a half-life of approximately 22 days (k = 3.1 × 10−2 day−1; Figure 4B and Figure S20 in Supporting Information). Overall, the stability of dG–AP cross-link in duplex DNA mirrors that of cross-link remnant 9b.
At pH 5.6 and 37 °C, cross-link remnant 9b decomposed with a half-life of 7.7 days (30 mM sodium acetate, pH 5.6, containing 1 mM ZnCl2; Figure S17, Supporting Information). These solvent conditions resemble those employed during enzymatic digestion of DNA prior to mass spectrometric analysis.
Evidence for Release of Cross-Link Remnants 9b and 11b from DNA Duplexes Containing Native and Reduced dG–AP Cross-Link
With a synthetic standard of the cross-link remnant 9b in hand, we set out to determine whether this was, in fact, the structure of the authentic crosslink remnant released by enzymatic digestion of a DNA duplex containing the dG–AP cross-link. Duplex A, containing the dG–AP cross-link, was prepared as previously described.21 Briefly, an abasic site was site-selectively introduced into the duplex by treatment of the corresponding 2′-deoxyuridine-containing duplex with uracil-DNA glycosylase (UDG). The AP-derived cross-link was generated in duplex DNA by incubation in HEPES buffer (50 mM, pH 7) containing NaCl (100 mM) at 37 °C. The dG–AP cross-link was generated in approximately 2–3% yield in this sequence under these conditions.21 The resulting native (unreduced) cross-link was digested with a four-enzyme cocktail that included nuclease P1, alkaline phosphatase, and phosphodiesterases I and II. This treatment led to the liberation of the fully digested dG–AP cross-link (Figure 5). The digestion mixture was subjected to LC-MS/MS and MS/MS/MS analysis on an LTQ linear ion trap mass spectrometer, under previously reported experimental conditions.63–65 We also analyzed the synthetic dG–AP cross-link remnant (9b) by LC-MS/MS and MS/MS/MS under the same experimental conditions.
Figure 5.

LC-MS/MS and MS/MS/MS for characterization of the dG–AP cross-link remnant that was obtained synthetically (a and c) or released from purified AP-derived cross-link-containing duplex DNA sequence A (b and d). (a, b) Selected-ion chromatograms for monitoring the loss of 2-deoxyribose (i.e., m/z 384 → 268 transition) from [M + H]+ ion of the unreduced dG–AP cross-link remnant that was (a) obtained synthetically or (b) liberated from the four-enzyme digestion of purified cross-link-containing duplex DNA. (c, d) Corresponding selected-ion chromatograms for monitoring the formation of [M + H]+ ion of guanine (i.e., m/z 384 → 268 → 152 transition) from the ion of m/z 268 observed in MS/MS. Depicted in the insets are the corresponding MS/MS and MS/MS/MS averaged from the retention times of (a, c) 23.7 min and (b, d) 23.0 min. The MS/MS/MS averaged from the two other peaks are displayed in Figure S26. The ion of m/z 250 observed in panel d is attributed to isobaric interference.
The LC-MS/MS results revealed that collisional activation of the [M + H]+ ions of 9b led to formation of a dominant fragment ion of m/z 268. The selected-ion chromatogram (SIC) for the m/z 384 → 268 transition showed three major peaks at 22.3, 23.7, and 25.1 min. The presence of multiple peaks in the SIC is in agreement with the fact that 9b exists as an isomeric mixture of α- and β-anomers of both pyranose and furanose forms (vide supra). Further collisional activation of the ion of m/z 268 resulted in facile formation of the protonated ion of guanine at m/z 152. Similar LC-MS/MS analysis of the aforementioned enzymatic digestion mixture of purified cross-link-containing duplex A also revealed three major peaks at 22.0, 23.0, and 24.8 min in the SIC for the m/z 384 → 268 transition (Figure 5b). Further collisional activation of the ion at m/z 268 again gave rise to formation of the ion at m/z 152 as the major fragment ion in MS/MS/MS (Figure 5 and Figure S26 in Supporting Information). The similar HPLC elution time, as well as the similar fragmentation behaviors observed in MS/MS and MS/MS/MS, suggested strongly that the cross-link remnant released from duplex DNA shares the same structure as synthetic cross-link 9b.
We next generated the reduced dG–AP cross-link in duplex A by incubation of the AP-containing duplexes in sodium acetate buffer (750 mM, pH 5.2) containing NaCNBH3 (250 mM) at 37 °C for 24 h, as previously described.21 Digestion of the duplex containing the reduced cross-link, followed by LC-MS/MS analysis, revealed a major peak eluting at 20.8 min in the SIC for the m/z 386 → 270 transition (Figure 6). The synthetic reduced cross-link remnant 11b displayed very similar retention time and nearly identical fragmentation patterns in MS/MS and MS/MS/MS as that released from duplex DNA (Figure 6). In this regard, the sequential losses of multiple H2O molecules in MS/MS/MS of the reduced dG–AP remnant, which differ from the predominant loss of the 2-deoxyribose remnant observed for the corresponding MS/MS/MS of the unreduced dG–AP remnant, are also consistent with the ring-opened structure of the reduced derivative. Together, these results provided further evidence regarding the structural nature of the dG–AP cross-link in duplex DNA.
Figure 6.

LC-MS/MS and MS/MS/MS for characterization of reduced dG–AP cross-link remnant that was obtained synthetically or released from NaCNBH3-treated cross-linked duplex DNA. Shown are selected-ion chromatograms for monitoring the loss of a 2-deoxyribose (i.e., m/z 386 → 270 transition) from the [M + H]+ ion of reduced dG–AP cross-link remnant that was (a) obtained synthetically or (b) released from the 4-enzyme digestion of reduced product of the purified cross-linked duplex A. (c, d) Corresponding selected-ion chromatograms for monitoring the further loss of H2O from the ion at m/z 270 observed in MS/MS (i.e., m/z 386 → 270 → 252 transition). (Insets) Corresponding MS/MS and MS/MS/MS averaged from retention times of (a, c) 20.5 and (b, d) 20.8 min.
CONCLUSIONS
The goals of this work were to characterize the chemical structure and properties of dG–AP cross-links generated in duplex DNA. Our approach centered upon characterization of the nucleoside cross-link “remnant” released by enzymatic digestion of a DNA duplex containing the dG–AP cross-link.21 Accordingly, our work began with the chemical synthesis of an analytical standard of the putative cross-link remnant 9b, composed of a 2-deoxyribose adduct attached to the exocyclic N2-amino group of dG. NMR spectroscopic analysis revealed that the 2-deoxyribose adduct in 9b exists as an equilibrating mixture of the ring-closed α-pyranose, β-pyranose, α-furanose, and β-furanose isomers (Scheme 5). A synthetic standard for the reduced cross-link remnant 11b was prepared by treatment of 14 with NaCNBH3, followed by removal of protecting groups. Reduction ablates the stereocenter at C1″ of the 2-deoxyribose adduct to give 11b as a single entity lacking the isomeric complexity of the unreduced remnant 9b.
LC-MS/MS analysis revealed that the retention times and mass spectral properties of the synthetic cross-link remnants 9b and 11b matched materials generated by enzymatic digestion of DNA duplexes containing native and reduced dG–AP cross-link, respectively. This provided evidence that the dG–AP cross-link formed at 5′-CAP/AG sequences in duplex DNA21 involves attachment of the exocyclic N2-amino group of dG to the aldehydic carbon of the AP-site on the opposing strand of the duplex. The LC-MS/MS methodology reported here provides a foundation for detection of the dG–AP cross-links 9b and 11b in cellular DNA. Analogous to some other aldehyde-derived DNA adducts,20,66 the native (unreduced) dG–AP cross-link in duplex DNA has the potential to exist in several different forms (Scheme 3), and the exact distribution of these cross-link isomers present in duplex DNA remains to be determined.
We also examined the chemical stability of cross-links 9 and 11 in both the synthetic nucleoside remnants and duplex DNA. As expected, the reduced cross-link 11 was completely stable in the remnant and in duplex DNA (Figure S24). More interestingly, we found that the unreduced cross-link remnant 9b was quite stable in aqueous buffered solution, decomposing with a half-life of 17 days at pH 7 and 23 °C. Cyclization of the C4″–OH group onto the imine residue in 8 may serve to limit the amount of hydrolytically labile imine present in solution, thus stabilizing the cross-link.67–69 Consistent with this notion, it is known that N-aryl aminoglycosides that exist as cyclic hydroxyalkylhemiaminals typically undergo hydrolysis with half-lives in the range of hours to days at pH values near neutral.70–72 In contrast, simple imines that lack this protective “hydroxyl lock” mechanism are hydrolytically labile. For example, the half-life for hydrolysis of N-p-chlorobenzylideneaniline in aqueous buffer at pH 7 and 25 °C is approximately 3.5 min (see Figure 4 in ref 73). Most interestingly, the stability of the cross-link attachment observed in the nucleoside remnant was reflected in the properties of the cross-link in duplex DNA. We found that the cross-link in duplex DNA decomposed with a half-life of 22 days at pH 7 and 23 °C. The intrinsic chemical stability of the dG–AP linkage in duplex DNA suggests that this cross-link may have the power to block cellular DNA-processing enzymes involved in transcription and replication.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health for support of this work (ES021007).
Footnotes
Supporting Information
Twenty-seven figures, showing 1D and 2D NMR spectra of 9b, 11b, 12, 13, and 14; UV absorption spectra; HPLC and TLC chemical kinetics analysis of stability of 9b and 11b; gel electrophoretic analysis of stability of cross-links 9a and 11a in duplex DNA; ESI-MS of 9b and 11b; and MS/MS/MS analysis of 9b released by enzymatic digestion of cross-linked duplex A. This material is available free of charge via the Internet at http://pubs.acs.org/.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Gates KS. In: Comprehensive Natural Products Chemistry. Kool ET, editor. Vol. 7. Pergamon; New York: 1999. pp. 491–552. [Google Scholar]
- (2).Delaney JC, Essigmann JM. Chem. Res. Toxicol. 2008;21:232–252. doi: 10.1021/tx700292a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Marnett LJ. In: DNA Adducts: Identification and Biological Significance. Hemminki K, Dipple A, Shuker DEG, Kadlubar FF, Segerback D, Bartsch H, editors. Vol. 125. IARC Scientific Publications; Lyon, France: 1994. pp. 151–162. [Google Scholar]
- (4).Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Chem. Res. Toxicol. 2000;13:1149–1157. doi: 10.1021/tx000118t. [DOI] [PubMed] [Google Scholar]
- (5).Shapiro R, Cohen BI, Shiuey S-J, Maurer H. Biochemistry. 1969;8:238–245. doi: 10.1021/bi00829a034. [DOI] [PubMed] [Google Scholar]
- (6).Chaw YFM, Crane LE, Lange P, Shapiro R. Biochemistry. 1980;19:5525–5531. doi: 10.1021/bi00565a010. [DOI] [PubMed] [Google Scholar]
- (7).Wang H, Marnett LJ, Harris TM, Rizzo CJ. Chem. Res. Toxicol. 2004;17:144–149. doi: 10.1021/tx034174g. [DOI] [PubMed] [Google Scholar]
- (8).Olsen R, Molander P, Øvrebø S, Ellingsen DG, Thorud S, Thomassen Y, Lundanes E, Greibrokk T, Backman J, Sjöholm R, Kronberg L. Chem. Res. Toxicol. 2005;18:730–739. doi: 10.1021/tx0496688. [DOI] [PubMed] [Google Scholar]
- (9).Wang M, McIntee EJ, Cheng G, Shi Y, Villalta PW, Hecht SS. Chem. Res. Toxicol. 2000;13:1065–1074. doi: 10.1021/tx000095i. [DOI] [PubMed] [Google Scholar]
- (10).Lee WH, Rindgen D, Bible RHJ, Hajdu E, Blair IA. Chem. Res. Toxicol. 2000;13:565–574. doi: 10.1021/tx000057z. [DOI] [PubMed] [Google Scholar]
- (11).Garcia CCM, Angeli JPF, Freitas FP, Gomes OF, de Oliveira TF, Loureiro APM, Di Mascio P, Medeiros MHG. J. Am. Chem. Soc. 2011;133:9140–9143. doi: 10.1021/ja2004686. [DOI] [PubMed] [Google Scholar]
- (12).McGhee JD, von Hippel PH. Biochemistry. 1975;14:1281–1296. doi: 10.1021/bi00677a029. [DOI] [PubMed] [Google Scholar]
- (13).McGhee JD, von Hippel PH. Biochemistry. 1975;14:1297–1303. doi: 10.1021/bi00677a030. [DOI] [PubMed] [Google Scholar]
- (14).Young-Sciame R, Wang M, Chung F-L, Hecht SM. Chem. Res. Toxicol. 1995;8:607–616. doi: 10.1021/tx00046a016. [DOI] [PubMed] [Google Scholar]
- (15).Yuan B, Cao H, Jiang Y, Hong H, Wang Y. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8679–8684. doi: 10.1073/pnas.0711546105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang M, Yu N, Chen L, Villalta PW, Hochalter JB, Hecht SM. Chem. Res. Toxicol. 2006;19:319–324. doi: 10.1021/tx0502948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Cho Y-J, Wang H, Kozekov ID, Kurtz AJ, Jacob J, Voehler M, Smith J, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. Chem. Res. Toxicol. 2006;19:195–208. doi: 10.1021/tx050239z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Borch RF, Hassid AI. J. Org. Chem. 1972;37:1673–1674. [Google Scholar]
- (19).Lu K, Moeller B, Doyle-Eisele M, McDonald JP, Swenberg JA. Chem. Res. Toxicol. 2011;24:159–161. doi: 10.1021/tx1003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cho Y-J, Kim H-Y, Huang H, Slutsky A, Minko IG, Wang H, Nechev LV, Kozekov ID, Kozekova A, Tamura P, Jacob J, Voehler M, Harris TM, Lloyd RS, Rizzo CJ, Stone MP. J. Am. Chem. Soc. 2005;127:17686–17696. doi: 10.1021/ja053897e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Johnson KM, Price NE, Wang J, Fekry MI, Dutta S, Seiner DR, Wang Y, Gates KS. J. Am. Chem. Soc. 2013;135:1015–1025. doi: 10.1021/ja308119q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dutta S, Chowdhury G, Gates KS. J. Am. Chem. Soc. 2007;129:1852–1853. doi: 10.1021/ja067294u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Price NE, Johnson KM, Wang J, Fekry MI, Wang Y, Gates KS. J. Am. Chem. Soc. 2014;136:3483–3490. doi: 10.1021/ja410969x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Guillet M, Bioteux S. Mol. Cell. Biol. 2003;23:8386–8394. doi: 10.1128/MCB.23.22.8386-8394.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Gates KS, Nooner T, Dutta S. Chem. Res. Toxicol. 2004;17:839–856. doi: 10.1021/tx049965c. [DOI] [PubMed] [Google Scholar]
- (26).Gates KS. Chem. Res. Toxicol. 2009;22:1747–1760. doi: 10.1021/tx900242k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).De Bont R, van Larebeke N. Mutagenesis. 2004;19:169–185. doi: 10.1093/mutage/geh025. [DOI] [PubMed] [Google Scholar]
- (28).Dong M, Wang C, Deen WM, Dedon PC. Chem. Res. Toxicol. 2003;16:1044–1055. doi: 10.1021/tx034046s. [DOI] [PubMed] [Google Scholar]
- (29).Dutta S, Abe H, Aoyagi S, Kibayashi C, Gates KS. J. Am. Chem. Soc. 2005;127:15004–15005. doi: 10.1021/ja053735i. [DOI] [PubMed] [Google Scholar]
- (30).Fekry MI, Price N, Zang H, Huang C, Harmata M, Brown P, Daniels JS, Gates KS. Chem. Res. Toxicol. 2011;24:217–228. doi: 10.1021/tx100282b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nooner T, Dutta S, Gates KS. Chem. Res. Toxicol. 2004;17:942–949. doi: 10.1021/tx049964k. [DOI] [PubMed] [Google Scholar]
- (32).Shipova K, Gates KS. Bioorg. Med. Chem. Lett. 2005;15:2111–2113. doi: 10.1016/j.bmcl.2005.02.058. [DOI] [PubMed] [Google Scholar]
- (33).Zang H, Gates KS. Chem. Res. Toxicol. 2003;16:1539–1546. doi: 10.1021/tx0341658. [DOI] [PubMed] [Google Scholar]
- (34).Fekry M, Szekely J, Dutta S, Breydo L, Zang H, Gates KS. J. Am. Chem. Soc. 2011;132:17641–17651. doi: 10.1021/ja2046149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wilde JA, Bolton PH, Mazumdar A, Manoharan M, Gerlt JA. J. Am. Chem. Soc. 1989;111:1894–1896. [Google Scholar]
- (36).Muniandy PA, Liu J, Majumdar A, Liu S.-t., Seidman MM. Crit. Rev. Biochem. Mol. Biol. 2010;45:23–49. doi: 10.3109/10409230903501819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Scharer OD. ChemBioChem. 2005;6:27–32. doi: 10.1002/cbic.200400287. [DOI] [PubMed] [Google Scholar]
- (38).Shen X, Li L. Environ. Mol. Mutagen. 2010;51:493–499. doi: 10.1002/em.20558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Noll DM, Mason TM, Miller PS. Chem. Rev. 2006;106:277–301. doi: 10.1021/cr040478b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Clauson C, Scharer OD, Niedernhofer LJ. Cold Spring Harbor Perspect. Biol. 2013;5(a012732) doi: 10.1101/cshperspect.a012732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Sturla SJ. Curr. Opin. Chem. Biol. 2007;11:293–299. doi: 10.1016/j.cbpa.2007.05.021. [DOI] [PubMed] [Google Scholar]
- (42).Woo J, Sigurdsson ST, Hopkins PB. J. Am. Chem. Soc. 1993;115:3407–3415. [Google Scholar]
- (43).Golankiewicz B, Ostrowski T, Leonard P, Seela F. Helv. Chim. Acta. 2002;85:388–398. [Google Scholar]
- (44).Gambino J, Yang T-F, Wright GE. Tetrahedron. 1994;50:11363–11368. [Google Scholar]
- (45).Espenson JH. Chemical Kinetics and Reaction Mechanisms. 2nd McGraw-Hill, Inc.; New York: 1995. [Google Scholar]
- (46).Liu S, Wang J, Su Y, Guerrero C, Zeng Y, Mitra D, Brooks PJ, Fisher DE, Song H, Wang Y. Nucleic Acids Res. 2013;41:6421–6429. doi: 10.1093/nar/gkt360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Riggins JN, Daniels JS, Rouzer CA, Marnett LJ. J. Am. Chem. Soc. 2004;126:8237–8243. doi: 10.1021/ja040009r. [DOI] [PubMed] [Google Scholar]
- (48).Hermida SAS, Possari EPM, Souza DB, de Arruda Campos IP, Gomes OF, Di Mascio P, Medeiros MHG, Loureiro APM. Chem. Res. Toxicol. 2006;19:927–936. doi: 10.1021/tx060033d. [DOI] [PubMed] [Google Scholar]
- (49).Mons S, Fleet GW. J. Org. Biomol. Chem. 2003;1:3685–3691. doi: 10.1039/b307795k. [DOI] [PubMed] [Google Scholar]
- (50).Takamura-Enya T, Watanabe M, Totsuka Y, Kanazawa T, Matsushima-Hibya Y, Koyama K, Sugimura T, Wakabayashi K. Proc. Natl. Acad. Sci. U.S.A. 2001;98:12414–12419. doi: 10.1073/pnas.221444598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Hou X, Wang G, Gaffney BL, Jones RA. Nucleosides Nucleotides. 2009;28:1076–1094. doi: 10.1080/15257770903368385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Narukulla R, Shuker DEG, Ramesh V, Xu Y-Z. Magn. Reson. Chem. 2008;46:1–8. doi: 10.1002/mrc.2111. [DOI] [PubMed] [Google Scholar]
- (53).Christov PP, Brown KL, Kozekov ID, Stone MP, Harris TM, Rizzo C. J. Chem. Res. Toxicol. 2008;21:2324–2333. doi: 10.1021/tx800352a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Tomasz M, Lipman R, Lee MS, Verdine GL, Nakanishi K. Biochemistry. 1987;26:2010–2027. doi: 10.1021/bi00381a034. [DOI] [PubMed] [Google Scholar]
- (55).Berger M, Cadet JZ. Naturforsch. 1985;40B:1519–31. [Google Scholar]
- (56).Cortes SJ, Mega TL, Van Etten RL. J. Org. Chem. 1991;56:943–947. [Google Scholar]
- (57).Amann N, Wagenknecht H-A. Tetrahedron Lett. 2003;44:1685–1690. [Google Scholar]
- (58).Singh MP, Hill GC, Peoc’h D, Rayner B, Imbach J-L, Lown JW. Biochemistry. 1994;33:10271–10285. doi: 10.1021/bi00200a007. [DOI] [PubMed] [Google Scholar]
- (59).Veldhuyzen WF, Lam Y-F, Rokita SE. Chem. Res. Toxicol. 2001;14:1345–1351. doi: 10.1021/tx0101043. [DOI] [PubMed] [Google Scholar]
- (60).Synold T, Xi B, Wuenschell GE, Tamae D, Figarola JL, Rahbar S, Termini J. Chem. Res. Toxicol. 2008;21:2148–2155. doi: 10.1021/tx800224y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Cordes EH, Jencks WP. J. Am. Chem. Soc. 1963;85:2843–2848. [Google Scholar]
- (62).Holton S, Runquist O. J. Org. Chem. 1961;26:5193–5195. [Google Scholar]
- (63).Wang Y, Wang Y. Anal. Chem. 2003;75:6306–6313. doi: 10.1021/ac034683n. [DOI] [PubMed] [Google Scholar]
- (64).Cao H, Hearst JE, Corash L, Wang Y. Anal. Chem. 2008;80:2932–2938. doi: 10.1021/ac7023969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Lai C, Cao H, Hearst JE, Corash L, Luo H, Wang Y. Anal. Chem. 2008;80:8790–8798. doi: 10.1021/ac801520m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6615–6620. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Adrover M, Vilanova B, Mun~oz F, Donoso J. Bioorg. Chem. 2009;37:26–32. doi: 10.1016/j.bioorg.2008.11.002. [DOI] [PubMed] [Google Scholar]
- (68).Caldes C, Vilanova B, Adrover M, Mun~oz F, Donoso J. Chem. Biodiversity. 2011;8:1318–1332. doi: 10.1002/cbdv.201000296. [DOI] [PubMed] [Google Scholar]
- (69).Capon B, Connett B. E. J. Chem. Soc. 1965:4497–4502. doi: 10.1039/jr9650004497. [DOI] [PubMed] [Google Scholar]
- (70).Bridiau N, Benamansour M, Legoy MD, Maugard T. Tetrahedron. 2007;63:4178–4183. [Google Scholar]
- (71).Na Y, Shen H, Byers LD. Bioorg. Chem. 2011;39:111–113. doi: 10.1016/j.bioorg.2011.02.003. [DOI] [PubMed] [Google Scholar]
- (72).Sianipar A, Parkin JE, Sunderland VB. Int. J. Pharm. 1998;176:55–61. [Google Scholar]
- (73).Cordes EH, Jencks WP. J. Am. Chem. Soc. 1962;84:832–837. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.