

Abstract
Radiation-induced genomic instability is a well-studied phenomenon that is measured as mitotically heritable genetic alterations observed in the progeny of an irradiated cell. The mechanisms that perpetuate this instability are unclear; however, a role for chronic oxidative stress has consistently been demonstrated. In the chromosomally unstable LS12 cell line, oxidative stress and genomic instability were correlated with mitochondrial dysfunction. To clarify this mitochondrial dysfunction and gain insight into the mechanisms underlying radiation-induced genomic instability we have evaluated the mitochondrial subproteome and performed quantitative mass spectrometry analysis of LS12 cells. Of 98 quantified mitochondrial proteins, 17 met criteria for fold changes and reproducibility; and 11 were statistically significant in comparison with the stable parental GM10115 cell line. Previous observations implicated defects in the electron transport chain (ETC) in the LS12 cell mitochondrial dysfunction. Proteomic analysis supports these observations, demonstrating significantly reduced levels of mitochondrial cytochrome c, the intermediary between complexes III and IV of the ETC. Results also suggest that LS12 cells compensate for ETC dysfunction and oxidative stress through increased levels of tricarboxylic acid cycle enzymes and upregulation of proteins that protect against oxidative stress and apoptosis. More than one cellular defect is likely to contribute to the genomic instability phenotype, and evaluation of gene and microRNA expression suggests that epigenetics play a role in the phenotype. These data suggest that LS12 cells have adapted mechanisms that allow survival under suboptimal conditions of oxidative stress and compromised mitochondrial function to perpetuate genomic instability.
Keywords: Mitochondria, Quantitative mass spectrometry, Proteomics, Genomic instability, Epigenetics, miR, Free radicals
Historically, the central dogma of radiation biology stated that the effects of ionizing radiation exposure were caused by DNA damage in directly hit cells. However, evidence has shown that deleterious effects of irradiation can be observed as nontargeted effects in the progeny of the irradiated cell at delayed times after exposure [1,2]. Delayed effects of irradiation are mitotically heritable and occur at higher frequencies than expected from Poisson statistics of particle hits to surviving cell nuclei or from conventional gene mutation frequencies [3]. Among these nontargeted phenomena are genomic instability and bystander effects. Genomic instability is measured as chromosomal alterations, changes in ploidy, micronucleus formation, gene mutations and amplifications, microsatellite instability, and/or decreased plating efficiency. This topic has been the subject of numerous reviews [1,2,4,5].
Although studies have successfully characterized the phenomena, the mechanisms driving persistent delayed genomic instability have remained elusive [4]. A common mechanistic thread has emerged, however. Oxidative stress contributes to radiation-induced DNA damage and chronic oxidative stress has been implicated in the perpetuation of delayed radiation-induced genomic instability [6–10]. A series of studies using the GM10115 cell line has characterized the link among increased genomic instability, oxidative stress, and mitochondrial dysfunction. The GM10115 cell line is a human– hamster hybrid, with a single copy of human chromosome 4 in a background of 20–24 Chinese hamster chromosomes. After exposure to radiation, isogenic groups of clones were derived that exhibited chromosomal instability for human chromosome 4 [11–13]. Although numerous studies have been conducted using these unstable clones [6,7,9,11,14–21], arguably the best characterized among these clones is one designated LS12. In comparison to the parental GM10115 cell line, LS12 cells have a distinctive phenotype with many of the classic characteristics of genomic instability (Table 1). The oxidative stress that was shown to correlate with the genomic instability phenotype in general, and observed in LS12 cells specifically, led to the evaluation of mitochondrial function in these cells (Table 2).
Table 1.
LS12 cellular phenotype relative to the parental GM10115 cells in response to radiation exposure or oxidative stress.
| Endpoint | Change | Reference |
|---|---|---|
| Chromosomal instability | ↑ | [11,31] |
| Plating efficiency | ↓ | [11] |
| CAD gene amplification | ↑ | [11] |
| Sister-chromatid exchange | ↑ | [11] |
| HPRT mutation frequency | ↑ | [11] |
| Persistent apoptosis (annexin V, TUNEL) | NC | [9,31] |
| Radiation-induced apoptosis | NC | [31] |
| Endogenous ROS | ↑ | [8] |
| Cytosolic cytochrome c | ↑ | [31] |
| Radiation-induced cytosolic cytochrome c release | NC | [31] |
| IAP, Bcl-2, Apaf-1 protein levels | NC | [31] |
| Radiation-induced IAP, Bcl-2, Apaf-1 protein levels | NC | [31] |
| H2O2-induced NF-κB transcription | ↑ | [44] |
| H2O2-induced Bcl-2 transcription | ↑ | [44] |
| IL-8 secretion | ↑ | [32] |
CAD, carbamyl-P synthetase/aspartate transcarbamylase/dihydroorotase; HPRT, hypoxanthine–guanine phosphoribosyl transferase; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; ROS, reactive oxygen species; NC, no change.
Table 2.
LS12 mitochondrial phenotype relative to the parental GM10115 cells.
| Endpoint | Change | Reference |
|---|---|---|
| Reactive oxygen species | ↑ | [8,15,16] |
| Mitochondrial contribution to cellular H2O2 | ↑ | [15] |
| Number of mitochondria (MitoTracker) | NC | [15] |
| ↑ | [7] | |
| Amount of mitochondrial DNA | NC | [15] |
| State 3 respiration | ↓ | [15] |
| State 4 respiration | NC | [15] |
| Complex IV (cytochrome oxidase) activity | ↓ | [15] |
| MnSOD protein level | NC | [15] |
| MnSOD activity | ↓ | [15] |
| NC | [6] | |
| Intracellular H2O2 | ↑ | [6] |
| Extracellular H2O2 | ↑ | [6] |
| Intracellular superoxide | ↑ | [6] |
| Catalase protein level and activity | NC | [6] |
| GPx protein level and activity | NC | [6] |
| CuZnSOD activity | NC | [6] |
| Mitochondrial membrane potential | NC | [7] |
| ATP levels | NC | [7] |
| SQR activity | ↑ | [7] |
| Soluble and bound SDHB protein levels | ↑ | [7] |
MnSOD, manganese superoxide dismutase; GPx, glutathione peroxidase; CuZn-SOD, copper–zinc superoxide dismutase; SQR, succinate quinone oxidoreductase; SDHB, succinate dehydrogenase B; NC, no change.
The H2O2-scavenging enzyme polyethylene glycol-conjugated catalase was shown to suppress the elevated mutation frequency and rate observed in unstable clones, and inhibition of catalase increased mutation frequency and rate in stable clones [6]. These findings suggest that elevated H2O2 plays a mechanistic role in the genomic instability phenotype, but do not identify the defect that allows this oxidative stress to persist in unstable clones. It has been hypothesized that electron transport chain (ETC) complexes may contribute to radiation-induced genomic instability [8,14]. This hypothesis is supported by the work of Kim et al. [15], which showed decreased respiration and complex IV activity in the LS12 unstable clone. Work by Dayal et al. [7] suggested that complex II defects might have occurred, contributing to increased H2O2. The increased levels of soluble succinate dehydrogenase B suggested that misassembly or weak interactions contribute to complex II defects.
In an effort to clarify the mitochondrial dysfunction observed in LS12 cells and gain insight into the potential mitochondrial mechanisms underlying radiation-induced genomic instability, we have evaluated the mitochondrial subproteome and performed quantitative mass spectrometry analysis. In contrast to the previous proteomic study [16], stable isotope labeling was utilized to increase the robustness of peptide and protein quantitation. Additionally, in the current study, the use of free-flow electrophoresis purification of mitochondria resulted in 44% of the quantified proteins being mitochondrial, as opposed to 25% in the previous proteomic study in which mitochondria were isolated using a commercially available kit [16]. The results of our experiments demonstrate changes in protein levels for proteins that protect against oxidative stress and apoptosis and proteins that are involved in the tricarboxylic acid (TCA) cycle. Further, evaluation of mRNA and microRNA (miR) expression levels showed changes in expression for mitochondrial and cellular redox homeostasis pathways, suggesting that epigenetic regulatory mechanisms are involved. Together, these proteomic, gene expression, and miR data validate the phenotypic observations of others and provide new information regarding the molecular mechanisms underlying genomic instability.
Materials and methods
Cell culture
GM10115 human–hamster hybrid cells were obtained from the Coriell Cell Repositories (Camden, NJ, USA). The LS12 unstable cell line was generated after irradiation of parental GM10115 cells and characterized as unstable cytogenetically [11]. Cells were cultured in Dulbecco's modified Eagle's medium (Invitrogen; Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Hyclone; Logan, UT, USA) and 0.2 mM l-proline (Invitrogen) and maintained in exponential growth as monolayers in a humidified atmosphere of 5% CO2 in air at 34 °C. The LS12 cell line is monitored regularly for instability through metaphase analysis [17]. All cell lines are screened monthly to exclude the presence of mycoplasma (Bionique Testing Laboratories, Saranac Lake, NY, USA).
Isolation of crude mitochondria
Crude mitochondria were isolated from LS12 and GM10115 cells by differential centrifugation according to the method of Attardi and Ching [18]. Briefly, 5 × 108 cells were homogenized in cell homogenization medium (150 mM MgCl2,10 mM KCl, 10 mM Tris-HCl, pH 7.6) with 15–30 strokes of a Dounce homogenizer with a tight-fitting pestle. Nuclei were pelleted by centrifuging at 4 °C for 5 min at 1000g. The supernatant was decanted and centrifuged at 4 °C for 10 min at 5000g. The pellet was resuspended in sucrose/Mg2+ medium (150 mM MgCl2, 250 mM sucrose, 10 mM Tris-HCl, pH 6.7) using 10 strokes of a tightfitting pestle in a Dounce homogenizer followed by centrifugation at 4 °C for 10 min at 5000g. This pellet is referred to as the crude mitochondrial pellet. To this pellet, 3.0 ml of the free-flow electrophoresis (FFE) separation buffer was added before zone electrophoresis (ZE)–FFE. The protein concentration of the crude mitochondria separated by ZE–FFE was ∼1.0 mg/ml.
Free-flow electrophoresis
FFE was performed using the BD FFE system (Becton-Dickinson Biosciences, Sparks, MD, USA) operated horizontally in the ZE mode. All reagents and chemicals used for the ZE–FFE media and buffers were purchased from either Sigma-Aldrich (St. Louis, MO, USA) or Fisher Scientific (Pittsburgh, PA, USA). A separation chamber height of 0.5 mm was used. Separation and counterflow media (10 mM acetic acid, 10 mM triethanolamine, 280 mM sucrose, pH 7.4; medium inlets 2–6 and counterflow inlets 1–3) as well as electrode stabilization medium (100 mM acetic acid, 100 mM triethanolamine, 200 mM sucrose; medium inlets 1 and 7) were injected into the separation chamber at a speed of 200 ml/h. Medium for anodic and cathodic circuit electrolytes consisted of 100 mM acetic acid and 100 mM triethanolamine adjusted to pH 7.4 with 1 M KOH. A voltage of 750 V was applied, which resulted in a current of ∼135 mA The separation chamber was cooled to 5 °C. P-sulfophenylazo-1,8-dihydroxy-3,6-naphthalene disulfate (SPADNS; 1% (v/v); Becton-Dickinson) was added to each sample before FFE separation for use as a tracking dye and the sample injection speed was 2.0–2.5 ml/h. Fractions were collected in 2 ml 96-well plates and the distribution of separated particles was monitored at a wavelength of 450 nm using an Emax plate reader (Molecular Devices, Sunnyvale, CA, USA). Fractions of interest selected on the basis of their optical density (OD) values were pooled and pelleted by centrifugation at 4 °C for 10 min at 5000g.
Mitochondria lysis, protein extraction, and trypsin-catalyzed 18O labeling
Mitochondria lysis, protein extraction, and on-pellet digestion were carried out according to a method developed by Duan et al. [19]. Briefly, pelleted material collected from ZE-FFE fractions was solubilized in extraction buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 2% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and Complete protease inhibitors; Roche, Indianapolis, IN, USA). Mitochondria were lysed with three freeze/thaw cycles—one cycle consisted of 5 min in liquid nitrogen, 10 min incubation at 37 °C, and 1 min vortexing. The solution was clarified by centrifuging at 4 °C for 40 min at 22,000 g. Protein was precipitated overnight at −20 °C by the addition of 6 volumes of cold acetone followed by centrifugation at 4 °C for 20 min at 12,000 g. Protein extracted from the LS12 mitochondria was reconstituted in 50 mM ammonium bicarbonate (Sigma-Aldrich) in (Sigma-Aldrich; 97% atom purity), whereas protein extracted from the GM10115 mitochondria was reconstituted in 50 mM ammonium bicarbonate in . The acid-labile surfactant RapiGest (Waters, Milford, MA, USA) was added to each sample to 0.2% final concentration to enhance protein solubility. Protein concentration was measured using the BCA protein assay (Pierce/Thermo Fisher Scientific, Rockford, IL, USA) and 100 μg of protein from each sample was used for the subsequent steps. To further increase protein solubility, trypsin (Promega, Madison, WI, USA) was added at an enzyme:substrate ratio of 1:30 (w/w) and the samples were incubated at 37 °C for 4 h with shaking (600 rpm). Proteins were reduced with 5 mM TCEP (Pierce/Thermo Fisher Scientific) for 5 min at 95 °C followed by alkylation with 20 mM iodoacetamide (Sigma-Aldrich) for 30 min at 37 °C in the dark. A second aliquot of trypsin was added at an enzyme:substrate ratio of 1:25 (w/w) and the mixture was incubated at 37 °C overnight to achieve complete digestion. Formic acid (Sigma-Aldrich) was added to 5% (v/v) followed by a 1 h incubation at 37 °C to stop the digestion and hydrolyze the RapiGest. Digests were then centrifuged at 14,000 rpm for 10 min to pellet RapiGest by-products. The unlabeled (16O; GM10115) and labeled (18O; LS12) samples were combined 1:1 and were desalted using PepClean C18 desalting spin columns (Pierce/Thermo Fisher Scientific) before drying in a vacuum centrifuge. Digests were reconstituted in 0.1% formic acid before 2D liquid chromatography-tandem mass spectrometry (LC–MS/MS). The 18O-labeling efficiency of the LS12 sample was 98.2% as determined by 1D LC–MS/MS.
Liquid chromatography–tandem mass spectrometry
Water (JT Baker), formic acid (Fluka), and acetonitrile (JT Baker) used for LC solvents were LC–MS grade. Ammonium formate was purchased from Sigma-Aldrich. Chromatographic separation of peptides was performed using a Nanoflow Xtreme Simple four-pump, two-dimensional LC system (CVC Micro-Tech, Fontana, CA, USA) equipped with a strong cation-exchange (SCX) column (0.3 × 100 mm, 5 μm particles with 200 Å pores; CVC Micro-Tech) upstream of two C18 peptide traps (0.5 × 2.0 mm; Michrom Bioresources, Auburn, CA, USA) and a C18 reversed-phase column (0.1 × 150 mm, 3 μm particles with 200 Å pores; Michrom Bioresources). The solvents used were 2% acetonitrile, 0.1% formic acid in water (solvents A and C); 95% acetonitrile, 0.1% formic acid (solvent B); and 500 mM ammonium formate, 2% acetonitrile, pH 2.7 (solvent D). Pumps 1 (solvent A) and 2 (solvent B) were operated at a combined flow rate of 0.4 μl/min. The combined flow rate for pumps 3 (solvent A) and 4 (solvent C) was 5 μl/min. Peptides were manually injected into the sample loop and were loaded onto the SCX column with solvent A. Each sample (10 μg) was analyzed in triplicate. A nine-step method was used to separate the peptides. The gradient profile for pumps 1 and 2 was as follows: 25 min 100% solvent A, 90 min 0–35% solvent B, 5 min 90% solvent B, and 15 min 100% solvent A. During each step, the gradient profile for pumps 3 and 4 was as follows: 50 min 100% solvent C, 20 min gradient from % solvent D from previous salt step to x% solvent D, 20 min x% solvent D, and 35 min 100% solvent C, where x=0, 2.5, 7.5, 12.5, 17.5, 22.5, 30, 40, 60, or 100.
Peptides were eluted into an LTQ linear ion trap mass spectrometer (Thermo Electron, San Jose, CA, USA) via an Advance electrospray ionization source (Michrom Bioresources). The spray voltage was 1.4 kV and the heated capillary temperature was 150 °C. Mass spectra were acquired using Xcalibur version 2.0 SR2 software. The five most abundant ions in each MS1 scan were selected for MS/MS. Other mass spectrometric data generation parameters were full-scan MS mass range 400–1800 m/z, collision energy 35%, isolation width 6.0 m/z, activation time 30 ms, and activation q 0.25. Ions selected for fragmentation four times within 60 s were dynamically excluded for 60 s (exclusion list size 250).
Protein identification and quantitation
Xcalibur.RAW files were converted to MGF files using Mascot Daemon 2.2 (Matrix Science, London, UK). The.MGF files were then searched using Mascot 2.2 (Matrix Science) against a UniProt mouse database (66,089 sequences). The following parameters were used—digestion method trypsin, peptide mass tolerance 8 amu, fragment mass tolerance 0.5 amu, max missed cleavages 2, static amino acid residue modification cysteine +57.05 (carba-midomethylation); differential amino acid residue modifications methionine +15.99 (oxidation), and peptide C-terminus+2.0 (18O1) and 4.0 (18O2). Data were filtered using ions score >35. Protein quantitation was conducted using the Mascot 18O-corrected multiplex protocol developed according to the method described by Zhang and Neubert [20]. Quantifiable peptides were those with a minimum of four y-ion pairs (from the 16O and 18O-labeled peptides) that had a minimum ion intensity of 10% of the most abundant ion in the spectrum. Ratios were then manually calculated for those proteins with at least two quantified peptides. The median peptide ratio for each protein was calculated. Peptides with ratios below the first quartile or above the third quartile were classified as outliers and were removed. The protein ratio was then calculated based on the arithmetic mean of the remaining peptide ratios. Protein ratios with CVs ≥ 30% were removed. Lists of protein ratios from all three replicates are provided as supplementary information. Mitochondrial proteins were classified on the basis of Gene Ontology analysis or subcellular localization according to the UniProt knowledgebase.
Pyruvate dehydrogenase activity
Pyruvate dehydrogenase (PDH) activity in GM10115 and LS12 cells was determined using a PDH enzyme activity microplate assay kit (Abcam, Cambridge, MA, USA) according to the protocol provided by the manufacturer. Briefly, mitochondrial protein at a concentration of 2.5 μg/μl isolated from each cell line was loaded in triplicate on the capture antibody precoated microplate and incubated for 3 h at room temperature. After the samples were washed, the reaction mixture was added and the optical density of the reaction product of the reporter dye-coupled reduction of NAD+ to NADH was measured at 450 nm using an Emax plate reader (Molecular Devices) controlled by SoftMax Pro (Molecular Devices) data acquisition and analysis software. Measurements were taken for 50 min at 0.5 min intervals. PDH activity was calculated from the slope of the mOD vs time (min) curve (ΔmOD/min).
Quantitation of citrate levels
Citrate levels in GM10115 and LS12 cells were quantified by colorimetric detection using a citrate assay according to instructions provided by the manufacturer (Abcam). Cells (5 × 107) were homogenized in 100 μl of citrate assay buffer by mechanical disruption using a Bullet Blender with 0.5 mm glass beads (Next Advance, Averill Park, NY, USA). Cell debris was removed by centrifugation at 15,000g for 10 min. Samples were deproteinated using a 3-kDa molecular weight cut-off spin column (Millipore, Bedford, MA, USA) and added in triplicate to a 96-well plate. Citrate standards were also added in triplicate to the 96-well plate. Reaction mix (citrate assay buffer, citrate enzyme mix, developer, and citrate probe) was added to the wells containing the citrate standards and the samples. A background control sample containing all components of the reaction mix except the citrate enzyme mix was added in triplicate to the 96-well plate. All samples were incubated for 30 min at room temperature in the dark and the optical density was measured at 540 nm using an Emax plate reader (Molecular Devices) controlled by SoftMax Pro (Molecular Devices) data acquisition and analysis software.
Statistical analysis
Means and standard deviations were calculated for proteins with average ratios <0.45 (1 standard deviation below the overall mean ratio) or > 1.50 (1 standard deviation above the overall mean ratio) that were present in at least two of the three independent biological replicates. The means were compared between parental GM10115 cells and chromosomally unstable LS12 cells by a one-tailed t test; p values of ≤0.05 were considered significant.
Arrays and bioinformatic analysis
Total RNA was extracted using the miRNeasy mini kit (Qiagen) following the kit procedures. MiR microarray analysis of total RNA was performed by LC Sciences (Houston, TX, USA). Quality control for the integrity of total RNA, enrichment of miR from total RNA, labeling, hybridization to μParaflo microfluidics chip, and scanning was performed using miRHuman/Mouse/Chinese Hamster miR array chips, based on Sanger miR-Base release 12.0. Statistical analyses were performed by the service provider using the locally weighted regression (lowess) normalization method on the background-subtracted data, and ANOVA was used to identify differences in miR expression between the LS12 cell line and GM10115. Two technical replicates were performed, so the significance threshold was set at p < 0.10, thereby identifying 13 significant miRNA probes.
Gene expression microarray analysis was performed by LC Sciences using the Affymetrix GeneChip Mouse Genome 430 2.0 array, and three technical replicates were performed. Quality control for the integrity of total RNA was performed and then the Affymetrix's GeneChip IVT Express kit was used for cDNA synthesis and in vitro transcription. Data were normalized using the Robust multiarray analysis, and differentially regulated genes were identified with multiple testing and false discovery rate statistics at p<0.05 using the GeneSpring GX software (Agilent Technologies, Santa Clara, CA, USA).
Gene-set enrichment for Gene Ontology biological process annotation was calculated using the DAVID web portal and functional annotation clustering with p<0.05 [21]. Proteomics data were merged with mRNA array data to identify overlapping members of the mitochondrial processes, and mRNA data were merged with predicted targets for miR significantly altered in LS12 cells relative to GM10115 to identify epigenetic regulatory mechanisms using the Bioinformatics Resource Manager software [22].
Results and discussion
ZE-FFE purification of crude mitochondria
Given the hypothesis that elevated reactive oxygen species levels in cells exhibiting radiation-induced genomic instability result from mitochondrial dysfunction, we adopted a quantitative proteomic approach to determine relative changes in mitochondrial protein expression levels for the LS12 cells in comparison with the unirradiated GM10115 parental cells (Fig. 1). GM10115 and LS12 crude mitochondrial preparations isolated by differential centrifugation were subjected to ZE in a FFE apparatus to increase the purity of the mitochondrial fraction used for proteomic analysis. In the ZE mode of FFE, the separation of charged particles is performed in a constant pH field as a result of the net charge of the compounds to be separated. This enables the rapid separation and purification of organelles, membranes, and whole cells for the analysis of subproteomes such as mitochondria [23]. ZE-FFE is an efficient means of purifying mitochondria to an increased homogeneity compared to differential and isopycnic centrifugation alone [24–26].
Fig. 1.
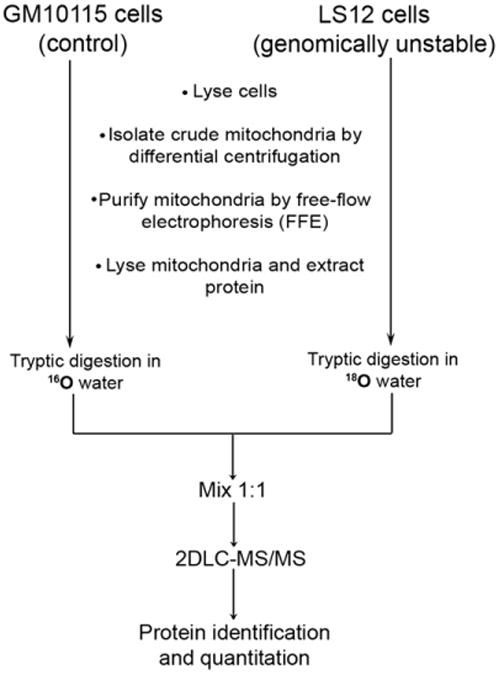
Quantitative proteomic workflow to determine changes in relative protein expression between mitochondrial proteins in chromosomally unstable LS12 cells and the stable unirradiated GM10115 parental cells.
Mitochondrial separation was monitored by OD measurements at 450 nm—a wavelength at which the heme group of cytochrome c, an abundant mitochondrial protein, absorbs visible light. Fig. 2 shows representative plots of the OD of each of the 96 fractions from the ZE-FFE separation of crude mitochondria prepared from GM10115 (Fig. 2A) and LS12 cells (Fig. 2B). Mitochondria are negatively charged at physiological pH [27]; thus they deflect in an electrical field toward the anode. Fraction 1 is the most anodic, and fraction 96 is the most cathodic. Fractions 20–23 from the ZE-FFE-separated GM10115 mitochondria were pooled and fractions 22–27 from the ZE-FFE-separated LS12 mitochondria were pooled. Protein extracted from the mitochondria was enzymatically digested with trypsin. Protein from the LS12 mitochondria was labeled with 18O by trypsin-catalyzed labeling whereby the two C-terminal 16O atoms of each tryptic peptide were exchanged with 18O atoms from the -containing digestion buffer. Protein from the GM10115 mitochondria was not labeled with 18O and is hereafter referred to as the 16O-labeled sample. An equivalent amount of peptides from the 16O- and 18O-labeled samples was combined before 2D LC–MS/MS analysis.
Fig. 2.
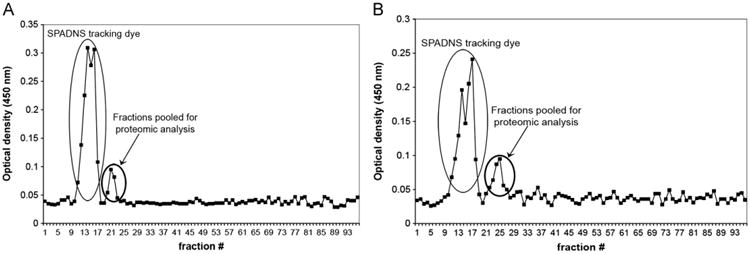
Optical density plots of fractions collected from ZE-FFE separation of crude mitochondria isolated from (A) GM10115 and (B) LS12 cells. The indicated fractions from each sample were pooled and the organelles pelleted by centrifugation at 5000g and used for in-solution tryptic digestion followed by 2D LC–MS/MS.
Differential expression of LS12 and GM10115 mitochondrial proteins
The relative quantitation of LS12 and GM10115 proteins was determined by calculating the relative intensities of the 18O- and 16O-labeled peptides identified by MS. Specifically, the multiplex quantitation method we used is based on the relative intensities of peptide ion fragment peaks within MS/MS spectra [20]. A precursor ion selection window of 6 m/z was selected to permit the cofragmentation of each set of 16O- (GM10115) and 18O-(LS12) labeled peptides.
Representative MS and MS/MS spectra of a quantified voltage-dependent anion-selective 2 (VDAC2) peptide from our dataset are presented in Fig. 3. Shown in the inset of Fig. 3A are the ions from the 16O-, 18O1-, and 18O2-labeled VDAC2 peptide: 1052.4, 1053.4, and 1054.3 m/z, respectively. The 16O-labeled VDAC2 peptide was from the GM10115 mitochondria, whereas the 18O1- and 18O2-labeled VDAC2 peptides were from the LS12 mitochondria. A representative MS/MS spectrum from the cofragmentation of these peptides is presented in Fig. 3B. The 18O (18O1+18O2)/16O peptide ratio was calculated based on the relative intensities of the y-ions in the MS/MS spectra. The y9 ions from the 16O-, 18O1-, and 18O2-labeled peptides are shown in the inset of Fig. 3B. The 18O/16O ratio for the VDAC2 peptide shown in Fig. 3B was 2.7. The mean 18O/16O VDAC2 protein ratio was 2.08±.28. VDAC2 was significantly upregulated in LS12 cells (p=0.011; n=8 peptides).
Fig. 3.
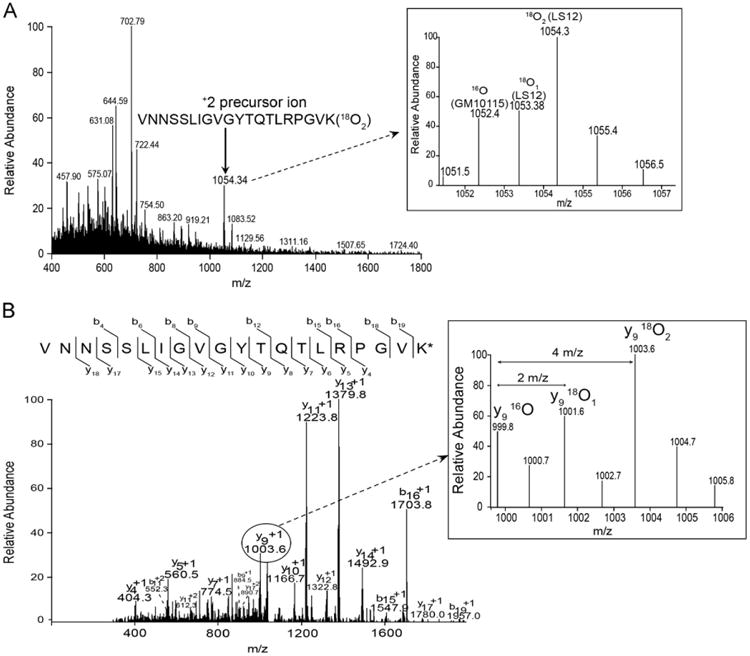
Representative quantified mass spectra of VDAC2 peptide, 249VNNSSLIGVGYTQTLRPGVK268. (A) Full MS1 scan showing +2 precursor ion (1054.34 m/z, indicated by arrow) that was selected for MS2 fragmentation. The inset shows the ions in addition to the precursor ion that were included in the MS2 fragmentation window so as to permit fragmentation of both the labeled (18O1, 1053.4 m/z; 18O2, 1054.3 m/z) and unlabeled (16O, 1052.4 m/z) peptides. (B) MS2 spectrum of 1054.3±3 m/z. The y-ions were used for quantitation. The inset displays the y9 ions from the labeled (18O1, 18O2) and unlabeled (16O) VDAC2 peptides from LS12 and GM10115 mitochondria, respectively. The 18O/16O ratio for this peptide was 2.7. The asterisk indicates 18O2-labeled lysine (K).
Of the 222 proteins that were quantified by 2D LC–MS/MS, 44% were mitochondrial. The distributions of all proteins quantified in at least two replicates are shown in Fig. 4. The mean ratio of all proteins quantified in at least two replicates was 1.07±0.47, whereas the mean ratio of all mitochondrial proteins quantified in at least two replicates was 1.27±0.42. Of the 98 quantified mitochondrial proteins, 17 met our criteria for fold change in at least two replicates: protein ratio either less than the overall mean ratio minus 1 standard deviation (≤0.45) or greater than the overall mean ratio plus 1 standard deviation (≥1.50). The proteins meeting these criteria are listed in Table 3. Of these, 11 were statistically significant (p<0.05) and are indicated in bold.
Fig. 4.
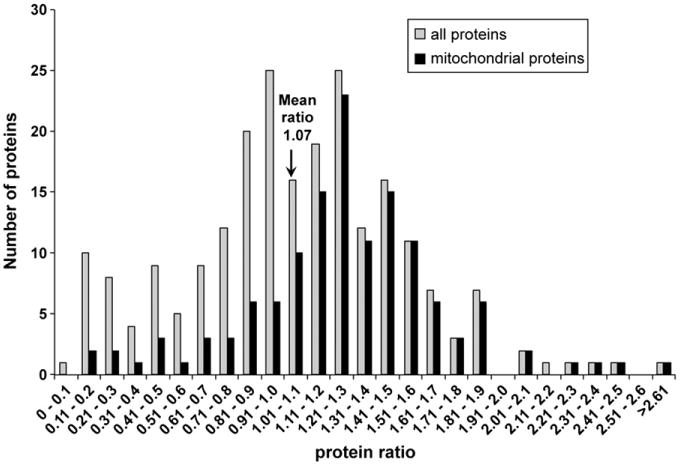
Distributions of the ratios for all proteins and for mitochondrial proteins quantified in at least two replicates.
Table 3.
Differentially regulated mitochondrial proteins in LS12 cells as determined by quantitative 2D LC–MS/MS.
| Accession no. | Description | No. of replicates | Protein ratio±SD | No. of peptides quantified±SD | p valuea |
|---|---|---|---|---|---|
| Q56A15|Q56A15_mouse | Cytochrome c, somatic | 3 | 0.28±0.15 | 19±2 | 0.014 |
| Q5SS40|Q5SS40_mouse | Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein, ε polypeptide | 2 | 0.41±0.08 | 8±1 | 0.032 |
| Q9DB15|RM12_mouse | 39 S ribosomal protein L12, mitochondrial | 2 | 1.50±0.44 | 7±4 | 0.17 |
| Q3UW66|Q3UW66_mouse | Adult male cecum cDNA, RIKEN full-length enriched library, clone 9130405K21, product: mercaptopyruvate sulfur transferase, full insert sequence | 2 | 1.52±0.01 | 4±0 | 0.006 |
| Q3TW93|Q3TW93_mouse | Osteoclast-like cell cDNA, RIKEN full-length enriched library, clone I420031E23, product: heat shock protein A, full insert sequence | 2 | 1.52±0.14 | 27±5 | 0.060 |
| Q99JY0|ECHB_mouse | Trifunctional enzyme subunit β, mitochondrial precursor | 2 | 1.55±0.01 | 11±1 | 0.003 |
| Q3V2D0|Q3V2D0_mouse | Lon protease homolog | 3 | 1.55±0.18 | 4±1 | 0.016 |
| Q99JR1|SFXN1_mouse | Sideroflexin-1 | 3 | 1.57±0.12 | 14±15 | 0.007 |
| Q3UFJ3|Q3UFJ3_mouse | Adult pancreas islet cells cDNA, RIKEN full-length enriched library, clone C820004O15, product: pyruvate dehydrogenase E1 α1, full insert sequence | 3 | 1.58±0.28 | 10±4 | 0.034 |
| Q99LL2|Q99LL2_mouse | Dlat protein (fragment) | 2 | 1.73±0.14 | 9±4 | 0.045 |
| Q80Y14|GLRX5_mouse | Glutaredoxin-related protein 5 | 2 | 1.70±0.20 | 5±1 | 0.063 |
| Q922R9|Q922R9_mouse | Trap1 protein (fragment) | 2 | 1.70±0.01 | 17±13 | 0.003 |
| Q53ZD4|Q53ZD4_mouse | Microsomal glutathione S-transferase 1 | 2 | 1.71±0.18 | 3±0 | 0.058 |
| Q9CZU6|CISY_mouse | Citrate synthase, mitochondrial precursor | 2 | 1.79±0.67 | 3±0 | 0.17 |
| P54071|IDHP_mouse | Isocitrate dehydrogenase [NADP], mitochondrial precursor | 2 | 1.84±0.30 | 3±1 | 0.078 |
| Q60930|VDAC2_mouse | Voltage-dependent anion-selective channel protein 2 | 3 | 2.08±0.28 | 8±4 | 0.011 |
| Q3THU8|Q3THU8_mouse | CRL-1722 L5178Y-R cDNA, RIKEN full-length enriched library, clone I730059K20, product: solute carrier family 25 (mitochondrial carrier; phosphate carrier), member 3, full insert sequence | 2 | 2.55±0.08 | 3±1 | 0.013 |
Statistically significant protein ratios (p ≤ 0.05) are in bold.
One-tailed t test.
Mitochondrial proteins with protective roles against oxidative stress are upregulated in LS12 cells
One of the hallmarks of the LS12 cell line is increased oxidative stress, increased H2O2, in particular (Table 2). Among the proteins that were identified as upregulated were a number that play a role in protecting against oxidative stress. Among these was 3-mercaptopyruvate sulfur transferase (MST; 1.52±0.01, p=0.006). In addition to protecting against oxidative stress, MST contributes to the maintenance of cellular redox homeostasis through metabolic regulation of cysteine degradation [28].
Another of these protective proteins was the Lon protease homolog (1.55±0.18, p=0.016). Aconitase is susceptible to oxidation by H2O2 and damaged aconitase accumulates during oxidative stress. It has been hypothesized that Lon protease degrades damaged aconitase and other proteins, preventing the accumulation of damaged proteins that can compromise mitochondrial function and cell viability [29]. That aconitase is upregulated in the LS12 cells supports this observation, although it did not meet our criteria for 1.5-fold change above the overall mean ratio (1.46±0.01, p=0.002; supplementary table). The third significantly upregulated protector against oxidative stress is tumor necrosis factor-associated protein 1 (TRAP1), a protein with heat shock protein (Hsp) homology (1.70±0.01, p=0.003). Cells that overexpress TRAP1 have been shown to exhibit a phenotype resistant to H2O2-induced DNA damage and apoptosis [30]. Other proteins considered to protect against the adverse effects of oxidative stress that had trends for upregulation in LS12 cells were HspA, glutaredoxin-related protein 5, and microsomal glutathione S-transferase 1 (1.52±0.14, 1.70±0.20, 1.71±70.18, respectively; p=0.06).
Altered apoptotic signaling in LS12 cells
Previously, LS12 cells had been evaluated for their basal apoptotic index and ability to undergo radiation-induced apoptosis [9,31]. Despite chromosomal instability and oxidative stress, the background level of apoptosis for LS12 cells was similar to that of the parental GM10115 cell line. Increased secretion of the antiapoptotic cytokine interleukin 9 was thought to contribute to the prosurvival phenotype [32]. LS12 cells were shown to have high background levels of apoptosis-inducing cytosolic cytochrome c (Cyt c) [29]. After irradiation of the LS12 cells, no further increase in cytosolic Cyt c was observed and the cells did not upregulate the downstream apoptotic proteins BAD, BAX, BID, or APAF1, nor did they complete apoptosis [31]. Our data support the observed increase in cytosolic Cyt c and initiation of apoptotic signaling, as well as the hypothesis that defects in apoptotic signaling could prevent cell death (Fig. 5). Proteomic analysis showed that VDAC2 is upregulated in LS12 cells (2.08±0.28, p=0.01). This VDAC upregulation may play a role in apoptosis by mediating Cyt c release from the mitochondria [33]. We observed significantly lower levels of mitochondrial Cyt c in the LS12 cells compared to the parental cell line (0.28±0.15, p=0.01), validating the observations of Nagar et al. that Cyt c is being released into the cytosol [31]. These observations are also supported by Ripple et al., who demonstrated that the mitochondrial concentration of Cyt c is decreased and the cytosolic concentration of Cyt c is increased when Cyt c is effluxed out of the mitochondria [34].
Fig. 5.
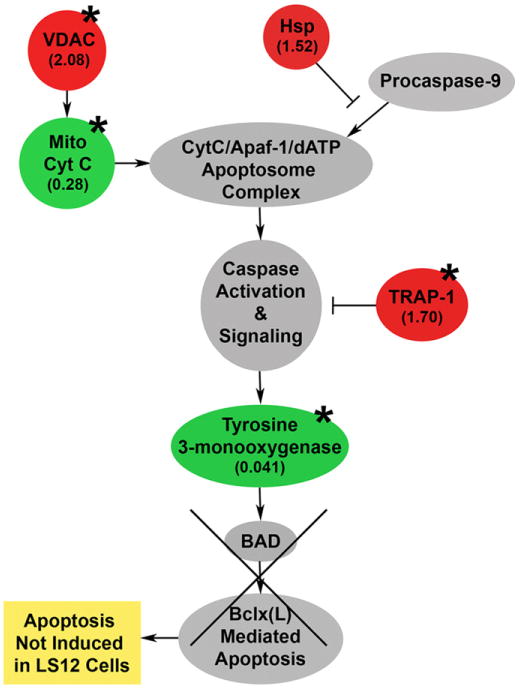
Apoptosis signaling in LS12 cells. LS12 cells have background levels of apoptosis similar to the parental GM10115 cell line and they do not complete radiation-induced apoptosis [31]. Based on proteomic analysis, it is hypothesized that upregulation of VDAC permits efflux of Cyt c from the mitochondria, initiating the apoptosis signaling cascade, but that upregulated antiapoptotic proteins inhibit apoptosis, despite downstream inhibition of the proapoptotic factor tyrosine 3-mono-oxygenase. Red, proteins identified in proteomic and MS analysis as upregulated; green, downregulated; with fold change indicated in parentheses; *p<0.05.
In apoptotic signaling, cytosolic Cyt c then binds to Apaf-1 and dATP to form the apoptosome. The apoptosome recruits and cleaves procaspase-9, activating caspase-9, which leads to the activation of caspase-3, the executor molecule of apoptosis [35]. Hsp70 family proteins have been shown to block recruitment of procaspase-9 to the apoptosome complex, preventing its cleavage and downstream effects [36]. HspA, a member of this Hsp70 family, is increased in LS12 cells and, although not statistically significant, this increase may indicate suboptimal induction of apoptosis (1.52±0.14, p=0.06). Upregulation of TRAP1 may also contribute to the apoptotic-resistant phenotype of LS12 cells. In addition to protecting against H2O2-mediated stress, TRAP1 has been shown to inhibit apoptosis by interfering with caspase-3 activation [30]. This inhibition provides the mechanism for both the low basal levels of apoptotic proteins in LS12 cells relative to the parental cell line and the inability of the LS12 cells to induce these proteins and undergo apoptosis after irradiation [31].
The final player in apoptosis signaling that was identified by proteomic analysis is tyrosine 3-monooxygenase, a member of the 14-3-3 family, also known as 14-3-3ε or YWHAE. Members of this protein family have been shown to play roles in inflammatory responses, mitogenic and cell survival signaling, cell cycle regulation, transcriptional activity, DNA replication, DNA damage, and apoptosis [37]. 14-3-3ε mediates antiapoptotic effects through the sequestering of proapoptotic factors including BAD [38]. Activation of the caspase-mediated apoptotic pathway leads to cleavage of 14-3-3ε and cell death [39]. We see a decrease in expression of this antiapoptotic protein in the LS12 cells that would seem to favor increased cell death (0.41±0.08, p=0.03). It is possible, however, that the other upstream antiapoptotic signals make this decrease in 14-3-3ε irrelevant.
Electron transport chain proteins in LS12 mitochondria
Although ETC dysfunction was proposed to play a significant role in the LS12 phenotype [7,15], the only ETC protein affected in the LS12 cells was mitochondrial Cyt c. For LS12 cells the Cyt c protein level was significantly decreased relative to parental GM10115 cells (0.28±0.15, p=0.01). However, given that the role of Cyt c in the ETC is to shuttle electrons from complex III (cytochrome bc1 complex) to complex IV (cytochrome c oxidase), this downregulation could be sufficient to compromise ATP synthesis and cellular respiration. Downregulation of Cyt c, the intermediary between State 3 respiration and cytochrome oxidase, validates the findings of Kim and colleagues regarding decreased complex IV activity and ETC dysfunction [15].
Upregulation of TCA cycle enzymes in LS12 mitochondria
In addition to ETC defects, Kim et al. hypothesized that TCA cycle enzymes might be responsible for LS12 mitochondrial dysfunction [15]. Proteomic analysis demonstrates that TCA cycle proteins are upregulated in the unstable LS12 cells compared to the parental cells (Fig. 6). These data suggest that rather than being responsible for mitochondrial dysfunction, the LS12 cells upregulate TCA cycle proteins to compensate for ETC defects. Succinate dehydrogenase (complex II of the ETC) is the only enzyme that participates in both the TCA cycle and the ETC [40]. This enzyme is a likely candidate for dysfunction in the LS12 cells given their phenotype; however, no change in succinate dehydrogenase protein level was observed (supplementary table). Barring that, the protein most frequently implicated in mitochondrial dysfunction is PDH. The PDH complex is the link from glycolysis to the TCA cycle, converting pyruvate to acetyl-CoA and CO [41]. Two components of this multienzyme complex, pyruvate dehydrogenase E1 component subunit α and dihydrolipoamide acetyltransferase (Dlat, E2 component), are upregulated in LS12 cells (1.58±0.28, p=0.03, and 1.73±0.14, p=0.05, respectively [42]). These observations are supported by measurement of PDH enzyme activity in the LS12 cells. Although not statistically significant, PDH activity was consistently higher in LS12 than in GM10115 cells (Fig. 7A). After acetyl-CoA synthesis, the first five enzymatic steps in the TCA cycle involve citrate synthase, aconitase, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase. Although not statistically significant, our data suggest trends for upregulation of all of these proteins in the LS12 cells (Fig. 6, Table 3, supplementary table). Measurement of intracellular citrate levels strongly supports these observations for upregulation of TCA cycle proteins. The citrate level for the LS12 cells was ∼3.4-fold higher than in the parental GM10115 cell line (89.8±2.2 and 26.7±2.2 μM, respectively; Fig. 7B; p<0.0001). After the succinyl-CoA synthetase and succinic dehydrogenase steps, the last two major enzymatic steps in the TCA cycle are mediated by fumarase and malate dehydrogenase, both of which also have trends for upregulation in LS12 (Fig. 6, supplementary table). The number of upregulated TCA cycle enzymes identified in the proteomic analysis and the increase in PDH activity and intracellular citrate level indicate that these proteomic changes are functional.
Fig. 6.
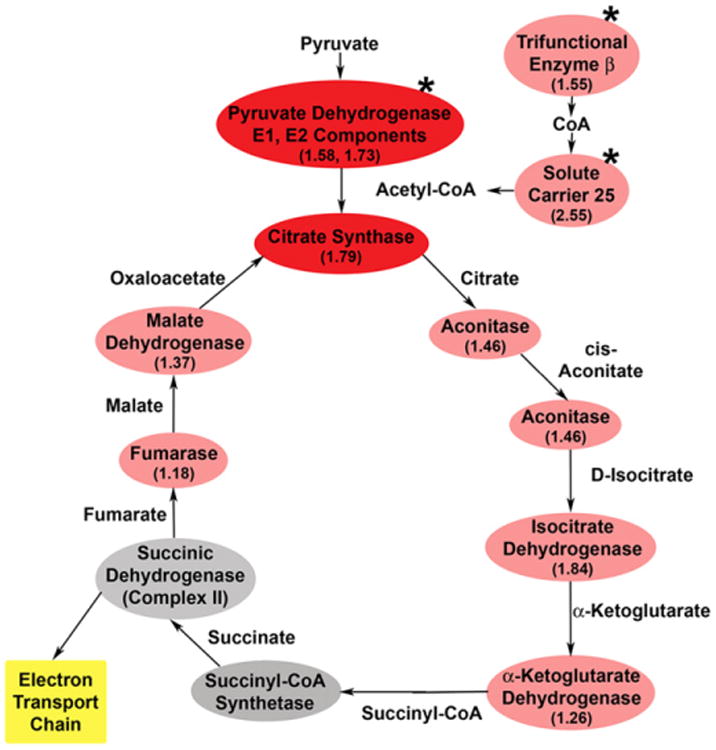
Upregulation of TCA cycle proteins in LS12 cells. Analysis of the mitochondrial subproteome demonstrates that, in addition to proteins involved in β oxidation, seven of nine TCA proteins are upregulated in LS12 cells, suggesting increased TCA cycle activity in compensation for ETC defects. Gray, proteins not identified in proteomic and MS analysis; pink, proteins identified in proteomic and MS analysis as upregulated (*p<0.05); red, proteins validated in subsequent enzyme activity assays.
Fig. 7.
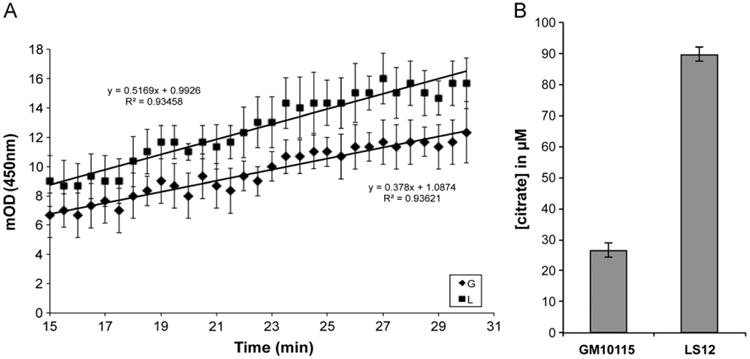
Validation of TCA cycle protein upregulation. (A) Analysis of pyruvate dehydrogenase (PDH) enzyme activity indicates a consistent, although not statistically significant, upregulation of PDH activity in LS12 cells compared to GM10115 cells in three replicate experiments (G, GM10115; L, LS12). (B) Measurement of intracellular citrate levels demonstrates a significant increase in LS12 cells relative to GM10115 cells and supports the observation regarding increased citrate synthase enzyme levels in LS12 cells. Columns represent the means±SD for three replicate experiments (p<0.0001).
Upstream of the TCA cycle is β oxidation, the process by which fatty acids are processed by the mitochondria to generate acetyl-CoA, the starting molecule for the TCA cycle [43]. Two proteins involved in β oxidation, trifunctional enzyme subunit β and solute carrier 25, are upregulated in the LS12 cell line (1.55±0.01, p=0.003, and 2.55±0.08, p=0.01, respectively). Although β oxidation occurs in peroxisomes as well as in mitochondria, distinct groups of enzymes mediate this process in each subcellular compartment.
Altered gene and miR expression in LS12 cells
In an effort to determine the regulatory mechanism for mitochondrial effects measured by proteomics, we used data from a separate study of the LS12 and other unstable cell lines that examined mRNA and miR expression relative to the parental GM10115 cell line (unpublished data). The mRNA arrays identified approximately 460 genes for which expression was altered in the LS12 cells. Gene Ontology analysis for biological processes demonstrated that the annotation cluster with the highest level of enrichment was the ETC, followed by cellular redox homeostasis (Table 4). Although we did not necessarily observe significant changes in all ETC proteins, these data further support the hypothesis for ETC dysfunction and persistent oxidative stress in the genomic instability phenotype. When we integrated the mRNA and proteomics data, we identified two of the same mitochondrial genes, Cyt c and Lon protease. A search for miR regulators for these genes yielded hits for mmu-miR-207 and hsa-miR-33b, upregulated 0.38 and 0.54, respectively (log ratio LS12/GM). Both of these miR's are predicted to regulate Cyt c and display anticorrelated expression with the measured proteomics data. Further, when all of the differentially expressed mitochondrial genes on the mRNA array were merged with predicted targets for the 13 miR's we identified as altered in LS12 cells, 18 of 23 genes contained complementary sequences for altered miR's and many displayed anticorrelated expression (Table 5). These observations strongly suggest that epigenetic mechanisms play a role in perpetuating mitochondrial dysfunction and the genomic instability phenotype.
Table 4.
Pathways with enriched gene expression in LS12 cells relative to parental GM10115 cells.
| Annotation cluster | Enrichment score |
|---|---|
| Electron transport chain | 2.61 |
| Cell redox homeostasis | 2.35 |
| Intracellular protein transport | 1.89 |
| Protein metabolic process | 1.64 |
| Histone modification | 1.64 |
| Organelle organization | 1.27 |
| Mitosis | 1.22 |
| Cellular adhesion | 1.18 |
p<0.05.
Table 5.
Differentially regulated mitochondrial genes with complementary sequences for altered miR's in LS12 cells relative to parental GM10115 cells.
| Gene symbol | mRNA log ratio (LS12/GM) | MiR reporter | MiR log ratio (LS12/GM) |
|---|---|---|---|
| Acadl | −0.39 | Hsa-miR-33b | 0.54 |
| Acsl1 | −3.95 | Hsa-miR-134 | 0.22 |
| Hsa-miR-519d | −0.7 | ||
| Hsa-miR-517 | −0.5 | ||
| Hsa-miR-518e | −0.73 | ||
| Aifm1 | 0.41 | Hsa-miR-519d | −0.7 |
| Hsa-miR-572 | 0.95 | ||
| Hsa-miR-33b | 0.54 | ||
| Hsa-miR-96 | −1.33 | ||
| Atad3a | 0.22 | Hsa-miR-517 | −0.5 |
| Hsa-miR-518e | −0.73 | ||
| Hsa-miR-519d | −0.7 | ||
| Mmu-miR-483 | −1.1 | ||
| Atp5l | −0.24 | Mmu-miR-483 | −1.1 |
| Coq3 | 0.57 | Hsa-miR-483-5p | −0.95 |
| Mmu-miR-207 | 0.38 | ||
| Cox5a | 0.16 | Hsa-miR-134 | 0.22 |
| Cox6b1 | −0.25 | Mmu-miR-483 | −1.1 |
| Cycs | −0.39 | Mmu-miR-207 | 0.38 |
| Hsa-miR-33b | 0.54 | ||
| Hsa-miR-519d | −0.7 | ||
| Got2 | 0.46 | Mmu-miR-31 | 0.31 |
| Hsd17b10 | 0.21 | Hsa-miR-134 | 0.22 |
| Mrpl20 | −0.37 | Hsa-miR-518e | −0.73 |
| Ndufv2 | −0.46 | Hsa-miR-517 | −0.5 |
| Pitrm1 | 0.47 | Hsa-miR-518e | −0.73 |
| Ppp1cc | −0.19 | Hsa-miR-483-5p | −0.95 |
| Mmu-miR-483 | −1.1 | ||
| Sars2 | −0.60 | Hsa-miR-134 | 0.22 |
| Hsa-miR-483-5p | −0.95 | ||
| Spns1 | 0.32 | Hsa-miR-483-5p | −0.95 |
| Mmu-miR-207 | 0.38 | ||
| Uqcrfs1 | −0.32 | Hsa-miR-134 | 0.22 |
Conclusion
In this study we evaluated the mitochondrial subproteome to better define the mitochondrial dysfunction observed in the radiation-induced chromosomally unstable LS12 cells. We have validated the molecular and phenotypic observations of others for this genomically unstable cell line. Our data validate those of others suggesting that LS12 cells have a defect in the ETC function [6,7,15]. Our data demonstrate significantly decreased levels of mitochondrial Cyt c that probably contribute to electron leakage and explain the decrease in complex IV activity observed by Kim and colleagues [15]. These disruptions in electron flow through the ETC generate H2O2 oxidative stress. However, rather than succumb to oxidative stress-induced apoptosis, LS12 cells have upregulated gene expression and proteins that combat oxidative stress via multiple mechanisms and that protect against caspase-9-mediated apoptosis. We hypothesize that this generates a feedback loop that significantly contributes to perpetuation of the instability phenotype (Fig. 8). ETC dysfunction leads to increased levels of oxidative stress, triggering Cyt c release to initiate apoptosis, which further compromises mitochondrial function and perpetuates the pro-oxidant environment. Adaptation via expression of antiapoptotic and antioxidant proteins contributes to cellular survival. Compensation for suboptimal ETC function by subtle, consistent upregulation of the TCA cycle further contributes to this survival. Mitochondrial dysfunction manifests not only as altered protein levels and enzyme activities, but also as altered gene expression. Further, our data indicate that these changes may be mediated, at least in part, by epigenetic mechanisms. We suggest that mechanisms adapted by LS12 cells permit survival under suboptimal, pro-oxidant conditions and contribute to persistent genomic instability. Radiation-induced genomic instability is clearly a multifactorial phenomenon and each unstable cell line may have its own spectrum of alterations that allow for the unstable phenotype to persist. Analysis of the mitochondrial subproteome for other unstable cell lines will determine whether this is the case.
Fig. 8.
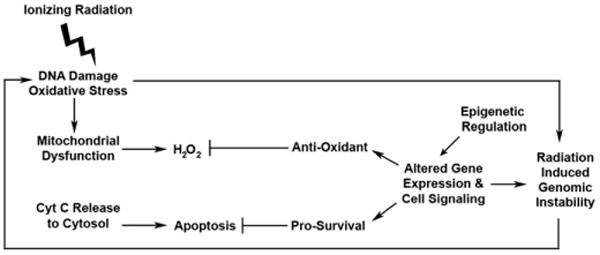
Proposed feedback loop that contributes to the instability phenotype of LS12 cells.
Supplementary Material
Acknowledgments
We are grateful to Dr. Umut Aypar for his scientific and intellectual input. This work was supported by Department of Energy Low Dose Program Glue Grant DE-FG02-07ER64339 (W.F.M./J.E.B.), NASA Grant NNX07AT42G (J.E.B.), NIH R01AG25323 (A.J.Y.), and NIH P30CA134274 (Greenebaum Cancer Center Support Grant) as well as by Battelle Memorial Institute, Pacific Northwest Division, under Contract DE-AC05-76RL0 1830 with the U.S. Department of Energy, Office of Biological and Environmental Research Low Dose Science Program. The U.S. Government retains and the publisher, by accepting the article for publication, acknowledges that the U.S. Government retains a nonexclusive, paid-up, irrevocable, worldwide license to publish or reproduce the published form of this article, or allow others to do so, for U.S. Government purposes.
Footnotes
Appendix A. Supplementary material: Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.freeradbiomed.2012.03.025.
Contributor Information
Austin J. Yang, Email: ayang@som.umaryland.edu.
Janet E. Baulch, Email: jbaulch@som.umaryland.edu.
References
- 1.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation. II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res. 2003;159:581–596. doi: 10.1667/0033-7587(2003)159[0581:nadeoe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 2.Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation. I.Radiation-induced genomic instability and bystander effects in vitro. Radiat Res. 2003;159:567–580. doi: 10.1667/0033-7587(2003)159[0567:nadeoe]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 3.Lorimore SA, et al. Chromosomal instability in the descendants of unirradiated surviving cells after α-particle irradiation. Proc Natl Acad Sci USA. 1998;95:5730–5733. doi: 10.1073/pnas.95.10.5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aypar U, Morgan WF, Baulch JE. Radiation-induced genomic instability: are epigenetic mechanisms the missing link? Int J Radiat Biol. 2011;87:179–191. doi: 10.3109/09553002.2010.522686. [DOI] [PubMed] [Google Scholar]
- 5.Streffer C. Strong association between cancer and genomic instability. Radiat Environ Biophys. 2010;49:125–131. doi: 10.1007/s00411-009-0258-4. [DOI] [PubMed] [Google Scholar]
- 6.Dayal D, et al. Hydrogen peroxide mediates the radiation-induced mutator phenotype in mammalian cells. Biochem J. 2008;413:185–191. doi: 10.1042/BJ20071643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dayal D, et al. Mitochondrial complex II dysfunction can contribute significantly to genomic instability after exposure to ionizing radiation. Radiat Res. 2009;172:737–745. doi: 10.1667/RR1617.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Limoli CL, et al. Persistent oxidative stress in chromosomally unstable cells. Cancer Res. 2003;63:3107–3111. [PubMed] [Google Scholar]
- 9.Limoli CL, et al. Apoptosis, reproductive failure, and oxidative stress in Chinese hamster ovary cells with compromised genomic integrity. Cancer Res. 1998;58:3712–3718. [PubMed] [Google Scholar]
- 10.Limoli CL, et al. Attenuation of radiation-induced genomic instability by free radical scavengers and cellular proliferation. Free Radic Biol Med. 2001;31:10–19. doi: 10.1016/s0891-5849(01)00542-1. [DOI] [PubMed] [Google Scholar]
- 11.Limoli CL, et al. Chromosomal instability and its relationship to other end points of genomic instability. Cancer Res. 1997;57:5557–5563. [PubMed] [Google Scholar]
- 12.Limoli CL, et al. Genomic instability induced by high and low LET ionizing radiation. Adv Space Res. 2000;25:2107–2117. doi: 10.1016/s0273-1177(99)01062-5. [DOI] [PubMed] [Google Scholar]
- 13.Limoli CL, et al. Chromosomal instability induced by heavy ion irradiation. Int J Radiat Biol. 2000;76:1599–1606. doi: 10.1080/09553000050201082. [DOI] [PubMed] [Google Scholar]
- 14.Spitz DR, et al. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–322. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- 15.Kim GJ, Fiskum GM, Morgan WF. A role for mitochondrial dysfunction in perpetuating radiation-induced genomic instability. Cancer Res. 2006;66:10377–10383. doi: 10.1158/0008-5472.CAN-05-3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JH, et al. Profiling mitochondrial proteins in radiation-induced genome-unstable cell lines with persistent oxidative stress by mass spectrometry. Radiat Res. 2008;169:700–706. doi: 10.1667/RR1186.1. [DOI] [PubMed] [Google Scholar]
- 17.Marder BA, Morgan WF. Delayed chromosomal instability induced by DNA damage. Mol Cell Biol. 1993;13:6667–6677. doi: 10.1128/mcb.13.11.6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attardi G, Ching E. Biogenesis of mitochondrial proteins in HeLa cells. Methods Enzymol. 1979;56:66–79. doi: 10.1016/0076-6879(79)56010-8. [DOI] [PubMed] [Google Scholar]
- 19.Duan X, et al. A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. J Proteome Res. 2009;8:2838–2850. doi: 10.1021/pr900001t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang G, Neubert TA. Automated comparative proteomics based on multiplex tandem mass spectrometry and stable isotope labeling. Mol Cell Proteomics. 2006;5:401–411. doi: 10.1074/mcp.T500021-MCP200. [DOI] [PubMed] [Google Scholar]
- 21.Dennis G, Jr, et al. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:R60. [PubMed] [Google Scholar]
- 22.Shah AR, et al. Enabling high-throughput data management for systems biology: the bioinformatics resource manager. Bioinformatics. 2007;23:906–909. doi: 10.1093/bioinformatics/btm031. [DOI] [PubMed] [Google Scholar]
- 23.Weber G, Wildgruber R. Free-flow electrophoresis system for proteomics applications. Methods Mol Biol. 2008;384:703–716. doi: 10.1007/978-1-59745-376-9_28. [DOI] [PubMed] [Google Scholar]
- 24.Drews O, et al. Mammalian proteasome subpopulations with distinct molecular compositions and proteolytic activities. Mol Cell Proteomics. 2007;6:2021–2031. doi: 10.1074/mcp.M700187-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Eubel H, et al. Free-flow electrophoresis for purification of plant mitochondria by surface charge. Plant J. 2007;52:583–594. doi: 10.1111/j.1365-313X.2007.03253.x. [DOI] [PubMed] [Google Scholar]
- 26.Zischka H, et al. Improved proteome analysis of Saccharomyces cerevisiae mitochondria by free-flow electrophoresis. Proteomics. 2003;3:906–916. doi: 10.1002/pmic.200300376. [DOI] [PubMed] [Google Scholar]
- 27.Ericson I. Determination of the isoelectric point of rat liver mitochondria by cross-partition. Biochim Biophys Acta. 1974;356:100–107. doi: 10.1016/0005-2736(74)90297-1. [DOI] [PubMed] [Google Scholar]
- 28.Nagahara N, Katayama A. Post-translational regulation of mercaptopyruvate sulfur transferase via a low redox potential cysteine–sulfenate in the maintenance of redox homeostasis. J Biol Chem. 2005;280:34569–34576. doi: 10.1074/jbc.M505643200. [DOI] [PubMed] [Google Scholar]
- 29.Bota DA, Davies KJ. Lon protease preferentially degrades oxidized mitochondrial aconitase by an ATP-stimulated mechanism. Nat Cell Biol. 2002;4:674–680. doi: 10.1038/ncb836. [DOI] [PubMed] [Google Scholar]
- 30.Montesano Gesualdi N, et al. Tumor necrosis factor-associated protein 1 (TRAP-1) protects cells from oxidative stress and apoptosis. Stress. 2007;10:342–350. doi: 10.1080/10253890701314863. [DOI] [PubMed] [Google Scholar]
- 31.Nagar S, Smith LE, Morgan WF. Variation in apoptosis profiles in radiation-induced genomically unstable cell lines. Radiat Res. 2005;163:324–331. doi: 10.1667/rr3287. [DOI] [PubMed] [Google Scholar]
- 32.Laiakis EC, Baulch JE, Morgan WF. Interleukin 8 exhibits a pro-mitogenic and pro-survival role in radiation induced genomically unstable cells. Mutat Res. 2008;640:74–81. doi: 10.1016/j.mrfmmm.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Lazarou M, et al. Inhibition of Bak activation by VDAC2 is dependent on the Bak transmembrane anchor. J Biol Chem. 2010;285:36876–36883. doi: 10.1074/jbc.M110.159301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ripple MO, Abajian M, Springett R. Cytochrome c is rapidly reduced in the cytosol after mitochondrial outer membrane permeabilization. Apoptosis. 2010;15:563–573. doi: 10.1007/s10495-010-0455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Won J, et al. Cleavage of 14-3-3 protein by caspase-3 facilitates Bad interaction with Bcl-x(L) during apoptosis. J Biol Chem. 2003;278:19347–19351. doi: 10.1074/jbc.M213098200. [DOI] [PubMed] [Google Scholar]
- 36.Beere HM, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 37.Zuo S, et al. 14-3-3 ε dynamically interacts with key components of mitogen-activated protein kinase signal module for selective modulation of the TNF-α-induced time course-dependent NF-κB activity. J Proteome Res. 2010;9:3465–3478. doi: 10.1021/pr9011377. [DOI] [PubMed] [Google Scholar]
- 38.Zha J, et al. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L) Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- 39.Schindler CK, Heverin M, Henshall DC. Isoform- and subcellular fraction-specific differences in hippocampal 14-3-3 levels following experimentally evoked seizures and in human temporal lobe epilepsy. J Neurochem. 2006;99:561–569. doi: 10.1111/j.1471-4159.2006.04153.x. [DOI] [PubMed] [Google Scholar]
- 40.Oyedotun KS, Lemire BD. The quaternary structure of the Saccharomyces cerevisiae succinate dehydrogenase: homology modeling, cofactor docking, and molecular dynamics simulation studies. J Biol Chem. 2004;279:9424–9431. doi: 10.1074/jbc.M311876200. [DOI] [PubMed] [Google Scholar]
- 41.Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143–1151. doi: 10.1042/bst0311143. [DOI] [PubMed] [Google Scholar]
- 42.Head RA, et al. Clinical and genetic spectrum of pyruvate dehydrogenase deficiency: dihydrolipoamide acetyltransferase (E2) deficiency. Ann Neurol. 2005;58:234–241. doi: 10.1002/ana.20550. [DOI] [PubMed] [Google Scholar]
- 43.Houten SM, Wanders RJ. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J Inherit Metab Dis. 2010;33:469–477. doi: 10.1007/s10545-010-9061-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Snyder AR, Morgan WF. Differential induction and activation of NF-κB transcription complexes in radiation-induced chromosomally unstable cell lines. Environ Mol Mutagen. 2005;45:177–187. doi: 10.1002/em.20092. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.