

Abstract
α- and β-neurexins are presynaptic cell-adhesion molecules implicated in autism and schizophrenia. We find that although β-neurexins are expressed at much lower levels than α-neurexins, conditional knockout of β-neurexins with continued expression of α-neurexins dramatically decreased neurotransmitter release at excitatory synapses in cultured cortical neurons. The β-neurexin knockout phenotype was attenuated by CB1-receptor inhibition which blocks presynaptic endocannabinoid signaling or by 2-arachidonoylglycerol synthesis inhibition which impairs postsynaptic endocannabinoid release. In synapses formed by CA1-region pyramidal neurons onto burst-firing subiculum neurons, presynaptic in vivo knockout of β-neurexins aggravated endocannabinoid-mediated inhibition of synaptic transmission and blocked LTP; presynaptic CB1-receptor antagonists or postsynaptic 2-arachidonoylglycerol synthesis inhibition again reversed this block. Moreover, conditional knockout of β-neurexins in CA1-region neurons impaired contextual fear memories. Thus, our data suggest that presynaptic β-neurexins control synaptic strength in excitatory synapses by regulating postsynaptic 2-arachidonoylglycerol synthesis, revealing an unexpected role for β-neurexins in the endocannabinoid-dependent regulation of neural circuits.
INTRODUCTION
Synaptic cell-adhesion molecules play critical roles in establishing and restructuring synaptic connections throughout life. Neurexins are evolutionarily conserved presynaptic cell-adhesion molecules that engage in trans-synaptic interactions with multifarious postsynaptic ligands, including neuroligins (NLs), cerebellins, and LRRTMs (Krueger et al., 2012; Sudhof, 2008). In mammals, neurexins are encoded by three genes, each of which contains independent promoters for longer α- and shorter β-neurexins (Rowen et al., 2002; Tabuchi and Sudhof, 2002; Ullrich et al., 1995; Ushkaryov et al., 1994; Ushkaryov et al., 1992). β-Neurexins are N-terminally truncated versions of α-neurexins that contain only a short (~40 residue) β-specific N-terminal sequence that then splices into the middle of the α-neurexin sequences (Ushkaryov et al., 1992). α- and shorter β-neurexin transcripts are extensively alternatively spliced at six canonical sites, resulting in over 1000 distinct neurexin mRNAs (Ullrich et al., 1995; Treutlein et al., 2014).
Although neurexins are well studied, little is known about their fundamental functions. Ligands that bind to either both α- and β-neurexins (e.g., neuroligins, LRRTMs, dystroglycan, and cerebellins; Ichtchenko et al., 1995, Ko et al., 2009; de Wit et al., 2009; Siddiqui et al., 2010, Uemura et al., 2010) or only to α-neurexins (e.g., neurexophilins; Petrenko et al., 1996) have been described, and constitutive knockouts (KOs) of α-neurexins were shown to severely impair neurotransmitter release (Missler et al., 2003). However, only limited understanding of α-neurexin functions is available, and little is known about β-neurexins. The lack of information on β-neurexin functions is particularly striking because nearly all biochemical studies on neurexins were performed with β-neurexins. Elucidating the synaptic actions of neurexins is a major technical challenge given their diversity and complexity. This challenge has taken on added importance given that hundreds of neurexin mutations were associated with several neuropsychiatric disorders (Südhof, 2008; Bang and Owczarek, 2013; Clarke and Eapen, 2014).
To specifically assess the function of β-neurexins, we generated mutant mice carrying conditional KO (cKO) alleles of all three β-neurexins. Despite low abundance of β-neurexin transcripts, we found that KO of β-neurexins in cultured neurons in vitro and in hippocampus in vivo impaired neurotransmitter release at excitatory synapses. Surprisingly, this decrease was due, at least in part, to enhanced tonic activation of presynaptic CB1-receptors (CB1Rs), caused by increased postsynaptic synthesis of the endocannabinoid 2-arachidonoylglycerol (2-AG). Moreover, synapses of hippocampal CA1 pyramidal cells onto pyramidal neurons in the subiculum – the major output pathway of the hippocampus – were differentially regulated by endocannabinoids, and deletion of β-neurexins selectively impaired the function of the more strongly endocannabinoid-regulated synapses in the subiculum. The importance of this circuit-specific synaptic alteration emerged from behavioral studies showing that the β-neurexin KO in the adult CA1 region produced an impairment of contextual fear memory. Thus, β-neurexins are produced as minor transcripts of neurexin genes that nevertheless are essential for the regulation of mammalian synaptic circuits due to modulation of endocannabinoid signaling via an unanticipated trans-synaptic mechanism.
RESULTS
Generation of β-neurexin-specific conditional triple KO mice
Using quantitative RT-PCR, we found that throughout the brain, all three β-neurexins were expressed at 10–100 fold lower levels than corresponding α-neurexins (Figs. 1A, S1). Despite their low abundance, however, β-neurexins are highly conserved, and could still perform essential functions. To test this hypothesis, we generated conditional and constitutive KO mice of all β-neurexin genes (Figs. 1B, S2A–S2C). In these mice, the 5′ exon that encodes the N-terminal β-neurexin-specific sequences was flanked by loxP sites, and epitope tags were inserted into the β-neurexin-specific sequences (EGFP for neurexin-1β and -3β; a hemagglutinin (HA) tag for neurexin-2β). We generated single, double, and triple conditional and constitutive β-neurexin KO mice (see Experimental Procedures for details) and focused our analyses on triple mutant mice targeting all β-neurexins to brace for potential redundancies among β-neurexins.
Figure 1. Conditional KO of β-neurexins impairs excitatory synaptic transmission.
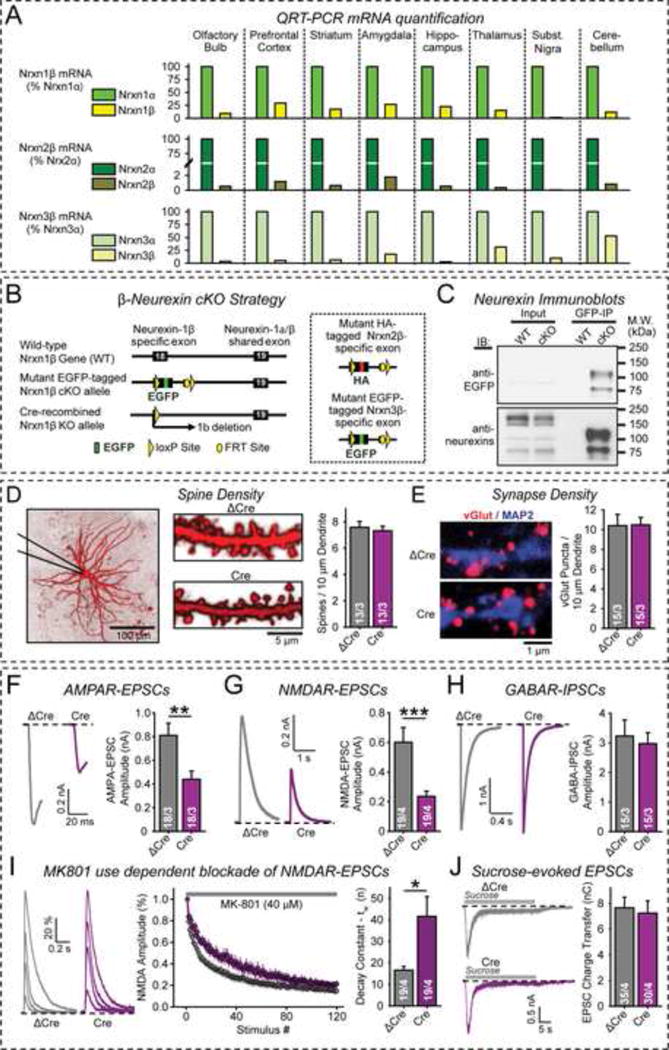
(A) β-Neurexins are expressed ~10–100 fold lower levels than α-neurexins. Data show relative α- and β-neurexin mRNA levels measured by quantitative RT-PCR (n=3 mice at P30).
(B) Conditional KO (cKO) strategy for neurexin-1β (left) and neurexin-2β and -3β (right dashed box). All cKOs involve floxing the 5′ β-neurexin-specific exon and adding an N-terminal epitope tag (EGFP for neurexin-1β and -3β; HA-tag for neurexin-2β).
(C) Immunoblots of tagged β-neurexins in cKO mice with antibodies to GFP (top) and to a conserved C-terminal neurexin epitope (bottom). Proteins in cortex homogenates (from 3-week old control and triple β-neurexin cKO mice; input) were immunoprecipitated with GFP antibodies (GFP-IP) to visualize the low-abundance β-neurexins in cKO mouse samples.
(D) β-Neurexin KO does not alter morphological parameters in cultured cortical neurons. Left, representative images of neurons filled with Alexa Fluor 594 via the patch pipette; right, summary graphs of spine density.
(E) β-Neurexin KO does not impair excitatory synapse density and size. Left, representative images; right, summary graphs of vGlut1-positive synapse density.
(F–H) β-Neurexin KO in cultured cortical neurons impairs excitatory but not inhibitory synaptic transmission evoked by isolated action potentials (F, AMPAR-mediated EPSCs; G, NMDAR-mediated EPSCs; H, GABAR-mediated IPSCs).
(I) β-Neurexin KO decreases the presynaptic release probability, measured via the MK-801 induced progressive block of NMDAR-mediated synaptic responses. Left, representative EPSC traces for the 1st, 10th, 25th, and 125th stimulus; center, mean ESPC amplitudes; right, summary graphs of decay constants.
(J) β-Neurexin KO does not alter the readily-releasable vesicle pool as analyzed by stimulation with 0.5 M sucrose. Left, representative traces; right, total charge transfer.
Data in D–J are means ± SEM (numbers of neurons/independent cultures examined are shown in graphs). Statistical analyses were performed by Student’s t-test (*p<0.05, **p<0.01, ***p<0.001). For additional data, see Figs. S1–S4.
Conditional triple KO (cKO) mice were viable and fertile. The EGFP and HA tags did not alter expression of either α- or β-neurexin mRNAs (Figs. S2D, S2E). Probably because of their low expression levels, tagged β-neurexins could not be detected in total brain extracts, but were readily observed after immunoprecipitations of β-neurexins with EGFP antibodies (Fig. 1C).
Constitutive triple β-neurexin KO mice were also viable but were significantly smaller than wild-type mice and unable to reproduce (Figs. S2F). Even single neurexin-2β and -3β KO mice exhibited a significantly reduced body weight. Thus, β-neurexins – despite low abundance and in contrast to α-neurexins (Missler et al., 2003) – are important for animal health but not essential for animal survival.
Conditional β-neurexin KO impairs neurotransmitter release at excitatory synapses
We cultured cortical neurons from triple β-neurexin cKO mice and infected them with lentiviruses expressing active (Cre; to delete all β-neurexins) or inactive, truncated Cre-recombinase (ΔCre; as a control). β-neurexin KO neurons exhibited no change in dendritic arborization or synaptic morphology, suggesting normal neuronal development (Figs. 1D, 1E, S3, S4A–S4E). We evoked action potential-induced excitatory and inhibitory postsynaptic currents (EPSCs and IPSCs, respectively; Kaeser et al., 2011), and separately monitored pharmacologically isolated AMPA-receptor (AMPAR) and NMDA-receptor (NMDAR) mediated EPSCs and GABA-receptor (GABAR) mediated IPSCs. Strikingly, the β-neurexin KO decreased both AMPAR- and NMDAR-mediated EPSCs by ~50%, but had no effect on GABAR-mediated IPSCs (Figs. 1F–1H).
These results suggest that the β-neurexin KO caused a decrease in the probability of glutamate release at excitatory synapses. To directly test this hypothesis, we measured presynaptic release probability using the progressive use-dependent block of evoked NMDAR EPSCs by MK-801 (Hessler et al., 1993; Rosenmund et al., 1993). We observed in triple β-neurexin KO neurons a robust, ~2-fold decrease in the rate of synaptic NMDAR inactivation in the presence of MK-801, suggesting an ~2-fold decrease in release probability (Fig. 1I). This decrease was not due to a change in the readily-releasable pool of synaptic vesicles because the β-neurexin deletion had no effect on hypertonic sucrose evoked EPSCs (Fig. 1J).
β-Neurexin KO impairs action-potential induced Ca2+-influx into presynaptic terminals
The electrophysiological data suggest that β-neurexins are required for normal coupling at excitatory synapses of an action potential to Ca2+-triggered release, possibly because voltage-gated Ca2+-influx is impaired. To test this hypothesis, we constructed a chimeric protein (GCaMP5G-Syb2) containing an N-terminal GCaMP5G Ca2+-indicator fused to the synaptic vesicle protein synaptobrevin-2 (Fig. 2A). After lentiviral expression in neurons, GCaMP5G-Syb2 was efficiently targeted to presynaptic terminals (Fig. 2B). To restrict analyses of presynaptic Ca2+-transients to excitatory synapses, we sparsely transfected neurons with mCherry, and monitored action potential-elicited Ca2+-transients only in presynaptic boutons contacting postsynaptic dendritic spines (Fig. 2B). Electrical field stimulation (1–100 stimuli at 50 Hz) elicited robust Ca2+-induced fluorescence signals in these boutons that saturated after ~20 stimuli, consistent with accumulation of residual Ca2+ in presynaptic terminals during stimulus trains (Figs. 2B, 2C). Ca2+-signals were blocked by tetrodotoxin (TTX), confirming that they were induced by action potential-stimulated Ca2+-influx. For analyses, we normalized the Ca2+-induced fluorescence signals to the maximal fluorescence change induced by 100 stimuli (ΔFsat) which saturates the GCaMP5G-Syb2 Ca2+-sensor.
Figure 2. Conditional KO of β-neurexins impairs presynaptic Ca2+-influx.
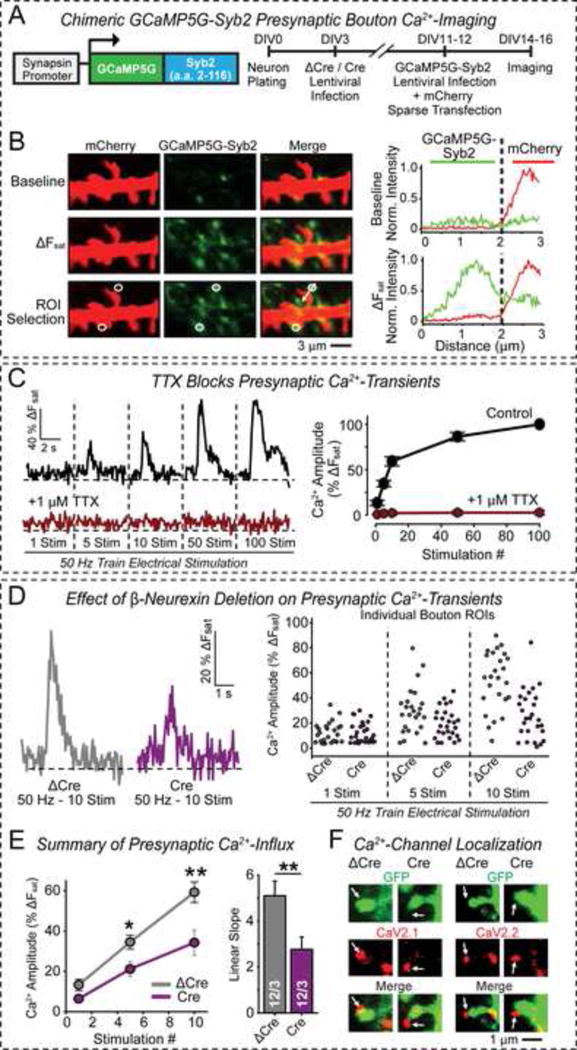
(A) Design of Ca2+-imaging experiments. Left, schematic of the GCaMP5G-Syb2 fusion protein that acts as a presynaptic Ca2+-probe; right, flow diagram of experiments.
(B) Representative Ca2+-imaging experiments. Left, sample images of presynaptic boutons containing GCaMP5G-Syb2 (green) that contact mCherry-containing dendritic spines (red; images were obtained before and after maximal stimulation to saturate Ca2+-transients [(ΔFsat]; white circles = regions of interest [ROIs] for quantitative analysis by line scans [arrows]). Right, summary graphs of line scans through ROIs at before (top) and after maximal stimulation (ΔFsat; bottom; green = presynaptic Ca2+-concentration; red = postsynaptic mCherry signal).
(C) Presynaptic Ca2+-transients saturate after 20 stimuli and are blocked by TTX. Left, representative Ca2+-transients; right, summary graphs (-TTX, n=12 neurons; +TTX, n=2 neurons).
(D) β-Neurexin KO impairs action potential-induced presynaptic Ca2+-influx in cultured cortical neurons (ΔCre = control; Cre = KO). Left, representative fluorescence traces of 10 stimuli applied at 50 Hz; right, scatter plots of individual bouton responses to 1, 5, and 10 stimuli.
(E) Summary plot of mean Ca2+-transients after 1, 5, and 10 stimuli. Left, n=3 independent experiments with 2–4 boutons per neuron; right, summary graph of the mean linear slopes fitted through the 1, 5, 10 stimuli plots.
(F) β-Neurexin KO does not alter levels or localization of presynaptic Ca2+-channels (representative images of cortical pyramidal neuron spines with sparse GFP expression that are stained for presynaptic P/Q-type (CaV2.1) or N-type (CaV2.2) Ca2+-channels). For quantitative analyses, see Figs. S4F–S4H.
Data are means ± SEM. Statistical analysis was performed by Student’s t-test (*p<0.05, **p<0.01).
We then used GCaMP5G-Syb2 to analyze Ca2+-transients in β-neurexin KO and control neurons. Ca2+-influx was induced by 1–10 action potential stimuli in the linear range of our Ca2+-sensor (Fig. 2C). We found that KO of β-neurexins significantly attenuated action potential-induced Ca2+-transients, with an overall ~2-fold decrease (Figs. 2D, 2E). KO of β-neurexins had no effect on presynaptic levels of voltage-gated N- or P/Q-type Ca2+-channels, suggesting a functional impairment (Figs. 2F, S4F–S4H).
Viewed together, these data show that the β-neurexin KO causes a ~2-fold decrease in three excitatory synapse parameters: EPSC amplitude, release probability, and action-potential induced Ca2+-influx. This suggests that β-neurexins, despite their low abundance compared to α-neurexins, are selectively essential for normal action-potential gated Ca2+-influx during neurotransmitter release at excitatory synapses.
β-Neurexin KO decreases spontaneous mini release at excitatory synapses: Selective rescue by neurexin-1β lacking an insert in SS#4
Neurotransmitter release occurs at synapses not only in response to action potentials, but also as spontaneous miniature EPSCs (mEPSCs) or miniature IPSCs (mIPSCs) that are also largely dependent on intracellular Ca2+ (Xu et al., 2009). The β-neurexin KO substantially depressed the mEPSC frequency (~2-fold), and slightly decreased the mEPSC amplitude and lowered the surface levels of GluA1 AMPARs (Figs. 3A, 3B, S4I, S4J). However, β-neurexin KO had no effect on mIPSC frequency and amplitude (Figs. 3C, 3D), consistent with the selective suppression of the presynaptic release probability by the β-neurexin KO in excitatory but not inhibitory synapses.
Figure 3. Neurexin-1β, but not neurexin-1α, and the CB1R antagonist AM251 rescue impaired spontaneous mini release in β-neurexin KO neurons.
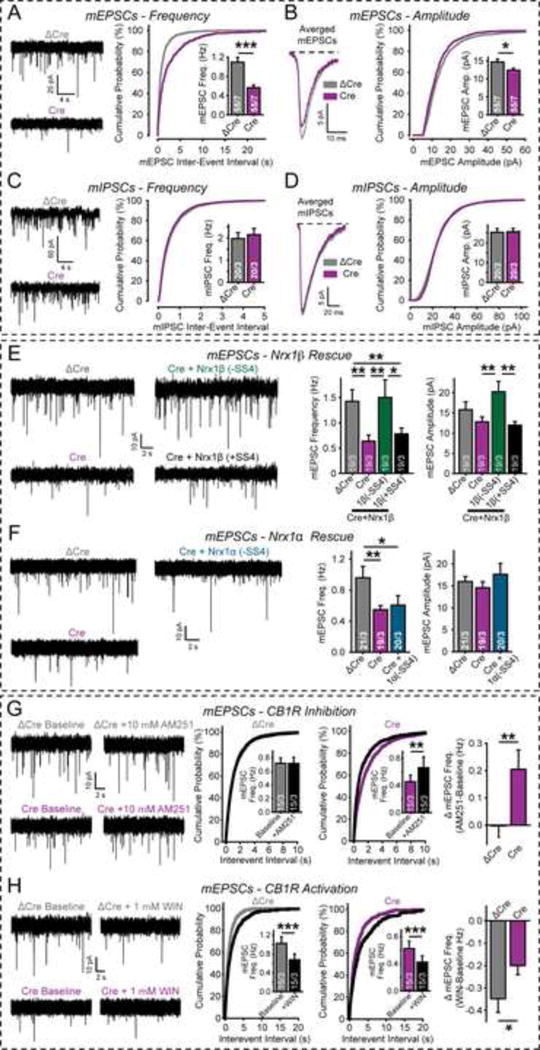
(A–D) β-Neurexin KO impairs mini release at excitatory but not inhibitory synapses (A & B, mESPCs; C & D, mIPSCs) in cortical neurons from triple β-neurexin cKO mice expressing inactive (ΔCre) or active Cre-recombinase (Cre). A & C, representative traces (left), cumulative distributions of inter-event intervals (right), and mean event frequencies (inset); B & D, average individual events (left), cumulative distributions of event amplitudes (right), and mean event amplitudes (inset).
(E) Neurexin-1β without an insert in SS#4 (−SS4), but not with an insert (+SS4), rescues the decreased mEPSC frequency in β-neurexin KO neurons.
(F) Neurexin-1α without an insert in SS#4 (−SS4) fails to rescue the decreased mEPSC frequency in β-neurexin KO neurons.
(G) Blocking CB1Rs with AM251 reverses the decrease in mEPSC frequency in β-neurexin KO neurons. Left, representative traces; center, cumulative distributions of event frequencies with insets showing mean frequencies; right, summary graphs of AM251-induced changes.
(H) Activating CB1Rs with WIN (WIN 55,212–2 mesylate) depresses mini release significantly more in control (ΔCre) than in β-neurexin KO neurons (Cre). Figure design is analogous to that of G.
Data are means ± SEM; numbers of neurons/independent cultures examined are shown in the graphs. Statistical analyses were performed using Student’s t-test (*p<0.05, **p<0.01, ***p<0.001). For additional data, see Figs. S4 and S5.
To validate the specificity of the β-neurexin KO effects on neurotransmitter release, we used ‘mini’ release as a measure of release probability, and tested the ability of neurexin-1β containing or lacking an insert in splice site #4 (SS4) and of neurexin-1α lacking an insert in SS4 to rescue the phenotype. Only neurexin-1β lacking an insert in SS4 rescued the decrease in excitatory synaptic transmission in β-neurexin KO neurons (Figs. 3E and 3F). Neurexin-1β lacking an insert in SS4 also enhanced mEPSC amplitude, probably because this neurexin splice variant stabilizes postsynaptic AMPARs (Aoto et al., 2013). The SS4-dependent rescue of the triple β-neurexin KO phenotype not only validates its overall specificity, but also suggests that β-neurexins control synaptic strength via a specific interaction with postsynaptic ligands that do not bind to α-neurexins. Alternative splicing at SS4 dramatically influences neurexin binding to postsynaptic ligands (Boucard et al., 2005; Chih et al., 2006; Ko et al., 2009; Siddiqui et al., 2010; Uemura et al., 2010; Matsuda et al., 2011). Indeed, we observed that at least some neuroligin-1 splice variants specifically bind only to β- but not to α-neurexins lacking an insert in SS4 (Fig. S5I).
β-Neurexin KO enhances basal endocannabinoid activity
How might deletion of β-neurexins influence presynaptic Ca2+-influx? A hint derives from the synaptic dysfunction caused by the neuroligin-3 KO, a postsynaptic cell-adhesion molecule that binds to presynaptic neurexins (Südhof, 2008). In CA1 pyramidal neurons, the neuroligin-3 KO decreases tonic endocannabinoid signaling, mediated by cannabinoid receptor type 1 (CB1R), at inhibitory synapses from CCK-positive basket neurons, thereby increasing GABA release (Foldy et al., 2013). This observation led us to ask whether the β-neurexin KO might cause the opposite change at excitatory synapses, i.e. an increase in basal endocannabinoid signaling, that could account for the decrease in Ca2+-influx and neurotransmitter release in β-neurexin KO synapses.
We tested the effect of the CB1R-antagonist AM251 on mEPSCs, again used as a measure of presynaptic release probability. AM251 had no effect on the mEPSC amplitude or frequency in control neurons (Figs. 3G, S5A–D). However, AM251 significantly enhanced the mEPSC frequency without changing the mEPSC amplitude in β-neurexin KO neurons (Fig. 3G, S5A–D), suggesting that KO of β-neurexins enhances basal endocannabinoid tone.
To further explore this hypothesis, we examined the effects of the CB1R-agonist WIN (WIN55,212-2 mesylate) on mEPSCs. While WIN produced a similar relative decrease in mEPSC frequency in triple β-neurexin KO and control neurons (Fig. S5G), we observed a significantly smaller absolute decrease in mEPSC frequency in KO neurons (Fig. 3H and S5H). This observation, consistent with the findings from the AM251 experiments, suggests that in β-neurexin KO synapses, CB1Rs are partially activated, and thus less additional inhibition is induced by WIN. Together, these data indicate that the β-neurexin KO caused an increase in the basal activity of presynaptic CB1Rs, which are known to inhibit presynaptic Ca2+-channels and to decrease neurotransmitter release (Twitchell et al., 1997: Kreitzer and Regehr, 2001; Brown et al., 2004; Szabo et al, 2014). Consistent with observations on neuroligin-3 (Földy et al., 2013), these data reveal a connection of the neurexin/neuroligin complex to endocannabinoid signaling. The direction of the effects, however, is diametrically opposite: whereas the β-neurexin KO suppresses the presynaptic release probability at excitatory synapses, the neuroligin-3 KO increases release at inhibitory synapses.
Conditional β-Neurexin KO increases postsynaptic 2-AG synthesis
To begin to explore how the β-neurexin KO increases the basal endocannabinoid ‘tone’ at excitatory synapses, we examined CB1R levels in β-neurexin KO neurons by immunoblotting and immunocytochemistry, but detected no changes, suggesting that presynaptic β-neurexins may influence endocannabinoid synthesis which is postsynaptic (Figs. 4A–4D; Murataeva et al. 2014, Castillo et al. 2012, Di Marzo et al. 2004). To test which of the brain’s two major endocannabinoids – 2-AG and anandamide – is active at the synapses that are affected by the β-neurexin KO, we measured the effects of exogenous 2-AG and anandamide on mEPSCs in control and β-neurexin KO neurons.
Figure 4. Blocking postsynaptic 2-AG synthesis rescues presynaptic β-neurexin KO phenotype in cultured neurons.
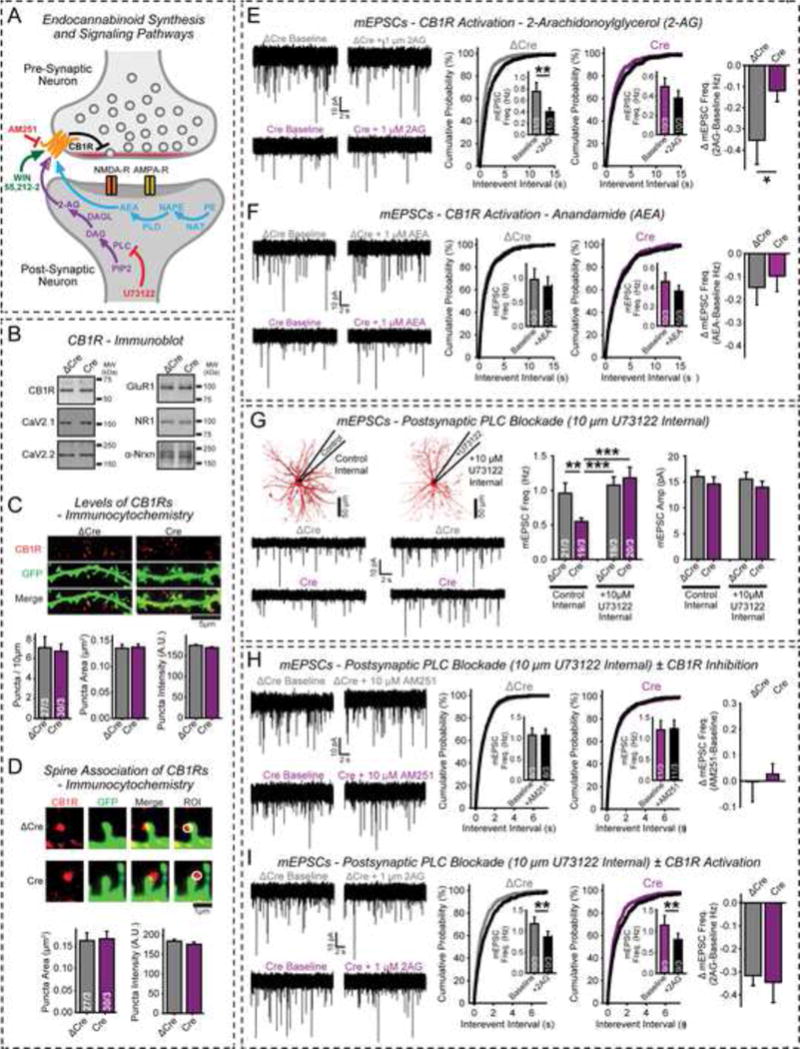
(A) Diagram of endocannabinoid signaling pathways. Two distinct endocannabinoids (2-arachidonoylglycerol [2-AG] and anandamide [AEA, N-arachidonoylethanolamine]) are synthesized by different postsynaptic enzymes; both act on presynaptic CB1Rs (abbreviations: PLC, phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate; DAG, diacylglycerol; DAGL, diacylglycerol lipase; NAT, N-acyltransferase; PE, phosphatidylethanolamine; NAPE, N-arachidonoyl phosphatidylethanolamine; PLD, phospholipase D). Modes of action of the 2-AG synthesis inhibitor U73122 and the CB1R agonist WIN and antagonist AM251 are indicated.
(B–D) CB1R levels are similar in cortical neurons from triple β-neurexin cKO mice expressing inactive (ΔCre) or active Cre-recombinase (Cre). B, representative immunoblots with antibodies to CB1R, CaV2.1 and CaV2.2 Ca2+-channels, GluR1, NR1 (an NMDAR subunit), and α-neurexins; C & D, immunocytochemistry quantifications of CB1R levels (C) and CB1R localization (D); note that neurons were sparsely transfected with EGFP for visualization of neuronal morphology.
(E & F) 2-AG causes a larger depression of mini release in control than in β-neurexin KO neurons (E), whereas anandamide is ineffective likely because it is only a partial agonist (F). Left, representative traces; center, cumulative distributions of mEPSC inter-event intervals (insets = mean frequencies); right, summary graphs of anandamide- or 2-AG-induced changes.
(G) Selective postsynaptic block of 2-AG synthesis by U73122 in the patch pipette rescues decreased mini release in β-neurexin KO neurons. Left, experimental set-up and representative mEPSC traces; right, bar diagrams of mEPSC frequencies and amplitudes.
(H & I) Selective postsynaptic block of 2-AG synthesis by U73122 prevents CB1R activation in β-neurexin KO neurons as measured by blocking CB1Rs with bath applied AM251 (H) or activating CB1Rs with bath-applied 2-AG (I). Left, representative traces; center, cumulative distributions of mEPSC inter-event intervals (insets = mean frequencies); right, summary graphs of AM251- or 2-AG-induced changes in mEPSC frequencies.
Data are means ± SEM; numbers of neurons/independent cultures examined are shown in graphs. Statistical analyses were with Student’s t-test (*p<0.05, **p<0.01, ***p<0.001).
Bath-applied anandamide had only a modest effect on mEPSCs in cultured cortical neurons, with no significant difference between control and β-neurexin KO neurons. In contrast, 2-AG robustly suppressed mEPSC frequency in control neurons but not in β-neurexin KO neurons (Figs. 4E, 4F). This observation, consistent with the findings from the AM251 and WIN experiments (Fig. 3G, 3H), suggests that the β-neurexin KO partially activates CB1Rs by increasing basal levels of 2-AG, which prevents the additional inhibition by exogenous 2-AG. Anandamide is likely relatively inactive because it is a partial agonist, and may not primarily act via CB1Rs (Freund et al., 2003).
2-AG is synthesized via a postsynaptic phospholipase C-dependent pathway (Fig. 4A). To test whether postsynaptic 2-AG synthesis may be up-regulated upon loss of presynaptic β-neurexins, we blocked phospholipase C-dependent 2-AG synthesis specifically in postsynaptic neurons by introducing the phospholipase C inhibitor U73122 via the patch pipette into postsynaptic neurons. U73122 had only minimal effects in control neurons, but caused full rescue of the mEPSC frequency in β-neurexin KO neurons (Fig. 4G).
Together, these data suggest that increased postsynaptic 2-AG synthesis produces the presynaptic β-neurexin KO phenotype. To consolidate this conclusion, we assayed the state of CB1Rs as a function of postsynaptic 2-AG synthesis inhibition. We bath-applied the CB1R-antagonist AM251 (Fig. 4H) or the CB1R-agonist 2-AG (Fig. 4I) while recording from postsynaptic neurons containing the 2-AG synthesis inhibitor U73122. U73122 eliminated the enhanced basal activity of CB1Rs in β-neurexin KO neurons (Figs. 3G, 4H) and restored the sensitivity of CB1Rs to exogenous 2-AG (Figs. 4F, 4I). Thus, postsynaptic inhibition of 2-AG synthesis in β-neurexin KO neurons restores the signaling set-point of presynaptic CB1Rs to that observed in control neurons, confirming that β-neurexins activate presynaptic CB1Rs via an up-regulation of postsynaptic 2-AG synthesis.
Conditional β-neurexin KO in vivo suppresses release at hippocampal output synapses
Modulation of endocannabinoid signaling by β-neurexins would have significant circuit implications, and could be relevant not only for understanding abnormalities of circuit dynamics in neuropsychiatric disorders, but also for possible future therapeutic options. Thus, we tested whether β-neurexins also control endocannabinoid signaling in vivo.
We stereotactically injected AAVs encoding Cre-EGFP or ΔCre-EGFP into the hippocampal CA1 region of triple β-neurexin cKO mice at P21, and analyzed acute subiculum slices at P35–P40 (Fig. 5A). We monitored synaptic transmission at synapses formed by CA1-region axons onto pyramidal neurons of the subiculum, which is the major output pathway of the hippocampus. In this approach, only presynaptic neurons are genetically manipulated (Aoto et al., 2013). Pyramidal subiculum neurons comprise two broad classes, ‘regular-firing’ and ‘burst-firing’ neurons that can be readily distinguished by the pattern of action potentials induced by current injections (Fig. 5B; Graves et al., 2012; Staff et al., 2000; van Welie et al., 2006). All analyses were performed separately in these two classes of neurons. Upon whole-cell break-in, we identified the neuron type in current-clamp mode, and then recorded EPSC’s induced by stimulating CA1-region axons in voltage-clamp mode (Fig. 5B).
Figure 5. Presynaptic KO of β-neurexins in CA1 pyramidal neurons decreases synaptic strength at burst-firing neuron synapses in the subiculum.
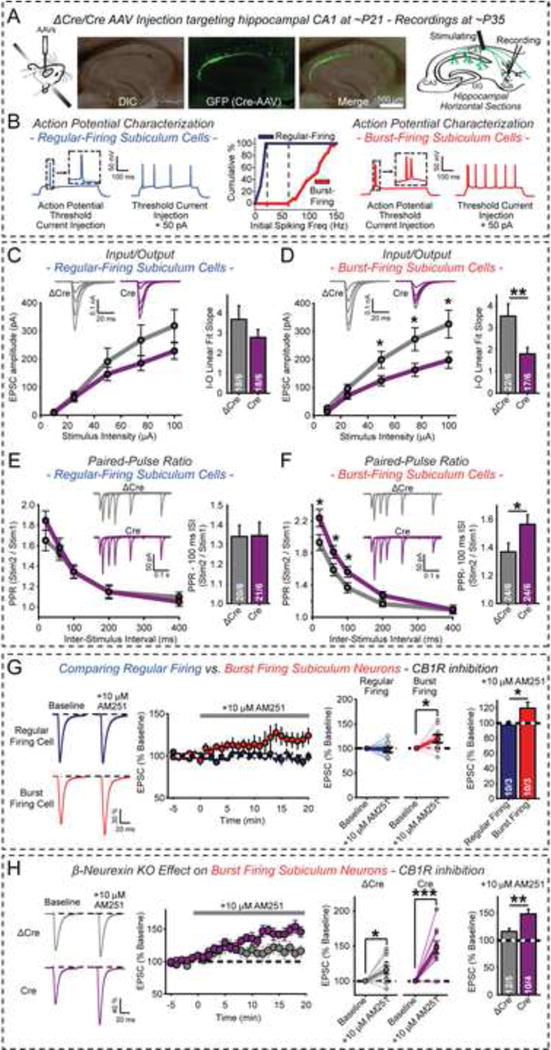
(A) Experimental design. Left, diagram of stereotactic injections into the CA1 region; center, representative images of slices from stereotactically injected mice at P35 to visualize AAV infections (slices with <90% EGFP expression in the CA1 region were rejected); right, electrophysiological recording configuration in acute subiculum slices (Aoto et al., 2013).
(B) Identification of regular- and burst-firing pyramidal subiculum neurons in current-clamp mode. Left and right, representative traces; center, summary graph of the initial spiking frequency.
(C & D) Input/output (I/O) relations of AMPAR-mediated EPSCs elicited by stimulation of CA1-derived axons and recorded in regular- (C) or burst-firing subiculum neurons (D). Left, summary plots with representative traces on top; right, summary graphs of fitted linear input/output slopes.
(E & F) Paired-pulse ratio (PPR) measurements of AMPAR EPSCs in regular- (E) and burst-firing subiculum neurons (F). Left, summary plots of PPRs vs. inter-stimulus intervals with representative traces on top; right, summary graphs of PPRs at 100 ms inter-stimulus intervals.
(G) Only burst-firing but not regular-firing subiculum neurons exhibit tonic endocannabinoid signaling in wild-type mice. EPSCs were elicited at 0.1 Hz before and after bath-application of the CB1R antagonist AM251. Left, representative EPSC traces; center, plots of relative EPSC amplitudes and AM251-induced EPSC amplitude changes in individual neurons; right, summary graphs of AM251-induced EPSC amplitude changes.
(H) Presynaptic β-neurexin KO increases tonic endocannabinoid signaling in burst-firing subiculum neurons. Experiments were performed as in G, except that burst-firing neurons were analyzed in slices from β-neurexin cKO mice with presynaptic expression of ΔCre- or Cre-EGFP in the CA1 region (see A).
Data are means ± SEM; numbers of neurons/mice examined are shown in the summary graphs. Statistical analysis was by paired Student’s t-test for single cell plots, and unpaired Student’s t-test for comparisons in other summary graphs (*p<0.05, **p<0.01, ***p<0.001).
We assessed excitatory synaptic strength by measuring the input-output relationship (Figs. 5C, 5D). In regular-firing neurons, the β-neurexin KO produced a trend towards decreased synaptic strength (Fig. 5C). In burst-firing neurons, however, the β-neurexin KO caused a ~2-fold decrease in synaptic strength, similar to cultured cortical neurons (Fig. 5D). Moreover, we measured paired-pulse ratios (PPRs) that inversely correlate with the presynaptic release probability (Kaeser and Regehr, 2014). Again, the presynaptic β-neurexin KO caused no change in regular-firing neurons (Fig. 5E), but increased the PPR in burst-firing neurons, consistent with a decrease in release probability (Fig. 5F). Thus, the presynaptic β-neurexin KO selectively decreases neurotransmitter release in burst-firing neuron synapses.
β-Neurexins control endocannabinoid regulation of subiculum synapses
Does the presynaptic β-neurexin KO impair neurotransmitter release in burst-firing subiculum neuron synapses by an endocannabinoid-dependent mechanism similar to cultured cortical neurons? To address this question, we first needed to learn whether tonic endocannabinoid signaling normally regulates release at subiculum synapses. Bath application of the CB1R antagonist AM251 had no effect on EPSC amplitude in regular-firing neurons, but caused a significant enhancement of EPSCs in burst-firing neurons (Fig. 5G). Bath application of the CB1R agonist WIN, conversely, induced a modest depression of EPSCs in regular-firing neurons but a significantly stronger depression of EPSCs in burst-firing neurons (Fig. S6A). Thus, endocannabinoids tonically modulate excitatory synapses formed onto burst-firing neurons, and perform a smaller role at synapses formed onto regular-firing neurons.
We next explored whether the β-neurexin KO decreases neurotransmitter release in burst-firing neuron synapses by increasing basal endocannabinoid tone. To this end, we bath-applied AM251 to subiculum slices without or with presynaptic KO of β-neurexins. Presynaptic β-neurexin KO caused a significant increase in the AM251-dependent enhancement of EPSCs (Fig. 5H), suggesting that similar to cultured neurons in vitro (Fig. 3G), the β-neurexin KO increased endocannabinoid-dependent inhibition of release in burst-firing neuron synapses in vivo (Fig. 5H). The β-neurexin KO did not change the relative magnitude of the EPSC depression by WIN (Figs. S5G, S6B), likely because the enhanced basal CB1R activity is not saturated by loss of β-neurexins. Together, these findings suggest that the β-neurexin KO decreases excitatory synaptic strength in burst-firing subiculum neurons at least in part by enhancing tonic activation of CB1Rs in presynaptic terminals of CA1 pyramidal neurons.
β-Neurexins control long-term plasticity in burst-firing neuron synapses
In regular-firing neurons of the subiculum, LTP is induced by postsynaptic activation of NMDARs, whereas in burst-firing neurons LTP is induced by presynaptic activation of PKA (Wozny et al., 2008; Behr et al., 2009). We asked whether LTP may also be differentially affected by the presynaptic β-neurexin KO in regular- and burst-firing neurons similar to endocannabinoid signaling. We observed no effect of the β-neurexin KO on LTP in regular-firing neurons (Fig. 6A). Strikingly, however, the β-neurexin KO blocked LTP in burst-firing neurons (Fig. 6B).
Figure 6. Presynaptic KO of β-neurexins selectively impairs LTP at burst-firing subiculum neurons by enhancing basal endocannabinoid activity.
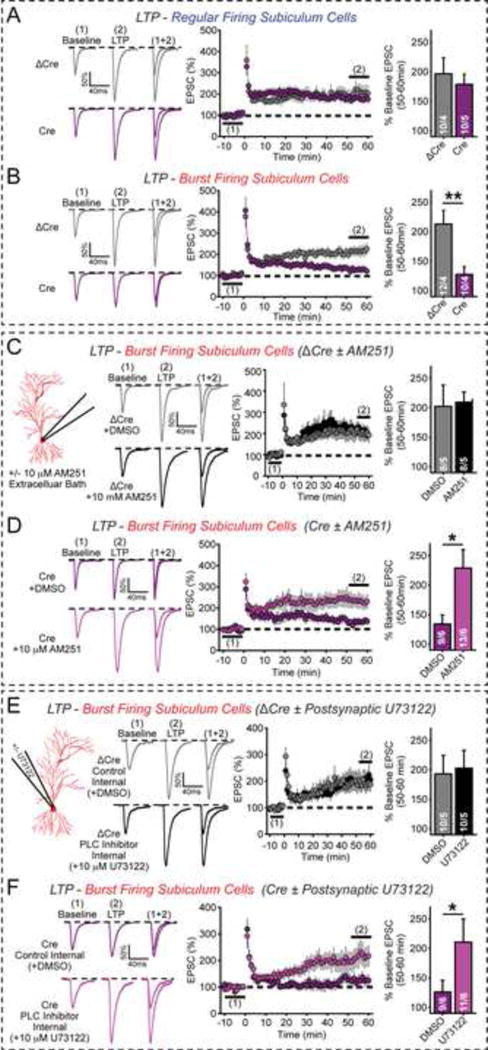
(A) KO of β-neurexins does not change LTP of CA1 EPSCs onto regular-firing subiculum neurons. LTP was induced by 4 × 100 Hz/1 s stimulation with 10 s intervals in current-clamp mode at resting potential in acute slices from CA1-region specific β-neurexin KO mice obtained as described in Fig. 5A. Left, representative traces; center, average EPSC amplitudes (1 min bins); right, summary graphs of mean LTP magnitude 50–60 min after induction.
(B) Same as (A), except that burst-firing subiculum neurons were analyzed.
(C & D) Same as (B), except that the effect of the CB1R antagonist AM251 on LTP was examined in slices from mice injected with inactive Cre-recombinase (C) or with active Cre-recombinase (D).
(E & F) Same as (C & D), except that the effect of the phospholipase C inhibitor U73122 introduced into the postsynaptic neuron via the patch pipette was examined.
Data shown are means ± SEM; numbers of neurons/mice examined are shown in the graphs. Statistical analysis was performed by Student’s t-test (* p<0.05, **p<0.01).
Presynaptic LTP in burst-firing neurons depends on adenylate cyclase/PKA signaling (Wozny et al., 2008). Interestingly, CB1Rs are coupled to Gαi which inhibits adenylate cyclase (Castillo et al., 2012; Kano et al., 2009). Thus, we hypothesized that enhanced basal CB1R activity caused by the β-neurexin KO may contribute to, or even cause, the LTP impairment. To test this hypothesis, we examined the effects of AM251 on LTP in burst-firing subiculum neurons. AM251 had no effect on LTP in control neurons (Fig. 6C), but rescued the blocked LTP in β-neurexin KO neurons (Fig. 6D). To examine whether the mechanism of CB1R activation by the β-neurexin KO in LTP mirrors that of CB1R activation in release, we asked whether postsynaptic inhibition of 2-AG synthesis also rescued the LTP impairment in β-neurexin KO synapses. Indeed, selective introduction of the 2-AG synthesis inhibitor U73122 into postsynaptic patched neurons (Fig. 4A) had no effect on LTP in control slices but fully rescued the block of LTP in β-neurexin KO slices (Figs. 6E, 6F). These results suggest that following the genetic ablation of β-neurexins, tonic postsynaptic 2-AG synthesis is enhanced and activates presynaptic CB1Rs, which impair this form of LTP.
Deletion of β-neurexins in CA1 region impairs contextual fear memory
To examine whether the function of β-neurexins in hippocampal CA1 neurons is important for learning and memory, we tested the behavioral effects of deleting β-neurexins from the hippocampal CA1 region (Figs. 7A and 7B). We observed no changes in the open field behavior of mice, as measured quantitatively on a force actometer (Fig. 7C, S7A). We then performed simultaneous cued and contextual fear conditioning. The CA1-region specific β-neurexin KO did not impair fear-learning acquisition (Fig. S7B), but strongly reduced freezing behavior when the mouse was placed in the context of the tone-footshock pairings (Fig. 7D). This phenotype was specific for contextual memory since the β-neurexin KO had no effect on freezing in response to an altered context or to the auditory cue, indicating that contextual fear memory – which is known to be dependent on hippocampal function (Fanselow and Dong, 2010) – is selectively impaired by the β-neurexin KO (Fig. 7D).
Figure 7. Conditional KO of β-neurexins in the hippocampal CA1 region impairs contextual memory: Model for β-neurexin action.
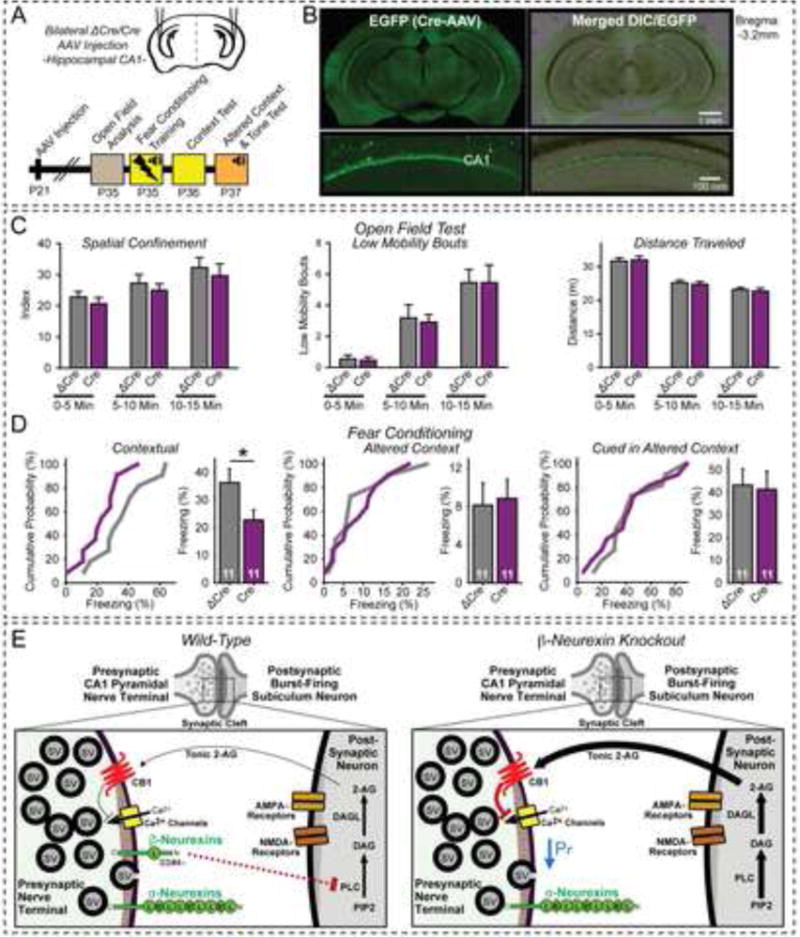
(A) Design of behavioral experiments (after Xu et al., 2012).
(B) Representative coronal images illustrating expression of Cre-EGFP in the CA1 region of the hippocampus after stereotactic injection (top) and zoomed CA1 image (bottom).
(C & D) Analysis of ΔCre- or Cre-injected mice in open field (C) and fear-conditioning tests (D). Open field behavior (analyzed in three 5 min segments) was quantified as spatial confinement (C, left), low mobility bouts (C, center), and total distance traveled (C, right). Fear-conditioning training exposed mice to three 30 s tones ending with 2 s electrical footshocks separated by 1 min intervals (D; left graphs, cumulative distributions; right summary graphs, mean fear-conditioning memory as measured by freezing). Data are means ± SEM; numbers of mice examined are shown in the graphs. Statistical analysis was performed by Student’s t-test (* p<0.05).
(E) Model for β-neurexin action. In wild-type excitatory synapses (left), presynaptic β-neurexins regulate endocannabinoid signaling by controlling postsynaptic 2-AG synthesis, possibly via trans-synaptic interaction with postsynaptic neuroligin isoforms that exclusively bind to β- but not α-neurexins lacking an insert in SS#4. In excitatory β-neurexin KO synapses (right), 2-AG synthesis is disinhibited, CB1Rs are activated, and synaptic strength is decreased; moreover, in burst-firing subiculum neurons LTP is blocked which may be responsible for the impairment in contextual memory.
DISCUSSION
We analyzed triple conditional KO mice that target all β-neurexin genes in two preparations: cultured cortical neurons after conditional KO of β-neurexins in vitro, and acute subiculum slices after conditional presynaptic KO of β-neurexins in vivo. Our data demonstrate that β-neurexins regulate the strength and long-term plasticity of a subset of excitatory synapses, that the β-neurexin KO impairs presynaptic Ca2+-influx triggered by an action potential in these synapses, and that presynaptic β-neurexins control tonic postsynaptic endocannabinoid signaling mediated by 2-AG (Fig. 7D)., Our in vivo results further suggest that β-neurexins regulate neural circuits by modulating the strength and plasticity of a subset of excitatory synapses via endocannabinoids, and that this regulation is behaviorally important. Given the complex nature of the many overlapping neural circuits that ultimately guide behavior, it is perhaps not surprising that the endocannabinoid-dependent modulation of synaptic circuits is controlled by trans-synaptic cell-adhesion molecules, and that this control is essential for the information processing capacity of the brain. However, it is unexpected that β-neurexins as relatively minor neurexin gene transcripts perform a pervasive regulatory role in synapses, a role that adds to previously defined other functions of neurexins (Missler et al., 2003; Aoto et al., 2013).
In cultured cortical neurons in vitro, KO of β-neurexins decreased the release probability at excitatory synapses ~2-fold by causing a ~2-fold decrease in presynaptic Ca2+-transients (Figs. 1F–1J, 2D, 2E, 3A–3D). The magnitude of this effect was surprising considering the low expression of β-neurexins (Fig. 1A) and the continued presence of the more abundant α-neurexins. The underlying mechanism consisted, at least in part, of an increase in tonic 2-AG endocannabinoid signaling, as evidenced by the reversal of the release phenotype both by bath-application of the CB1R-antagonist AM251 (Fig. 3G) and by postsynaptic inhibition of 2-AG synthesis (Fig. 4G). Thus, β-neurexins appear to be selectively essential for regulating excitatory synaptic strength via a control of tonic endocannabinoid signaling.
In synapses formed by CA1-region pyramidal neurons onto pyramidal neurons in the subiculum in vivo, the β-neurexin KO also impaired excitatory synaptic transmission. We separately examined synapses of regular- and burst-firing neurons, the two types of subiculum pyramidal neurons (Staff et al., 2000; Van Welie et al., 2006). The presynaptic β-neurexin KO produced a decrease of excitatory synaptic strength in postsynaptic burst-firing but not regular-firing neurons; this loss was reversed, at least in part, by the CB1R antagonist AM251 (Fig. 5). Moreover, only PKA-dependent presynaptic LTP in burst-firing but not NMDAR-dependent postsynaptic LTP in regular-firing neurons was impaired by the β-neurexin KO (Figs. 6A, 6B). Presynaptic LTP in presynaptic β-neurexin KO slices was restored by inhibiting CB1Rs (Figs. 6C, 6D) and, most importantly, by postsynaptic inhibition of 2-AG synthesis in burst-firing neurons (Figs. 6E, 6F). The regulatory function of presynaptic β-neurexins in release is likely physiologically important since the β-neurexin KO in the CA1 region severely impaired contextual fear conditioning (Fig. 7).
To the best of our knowledge, our findings represent the first description of trans-synaptic control of endocannabinoid signaling by neurexins, complementing previous observations of a role of postsynaptic neuroligin-3 in regulating the endocannabinoid tone (Földy et al., 2013). Tonic endocannabinoid signaling at excitatory synapses has not previously been documented, but anatomical evidence supports the presence of DGL-α (which synthesizes the endocannabinoid 2-AG; Fig. 4A) and of CB1Rs at excitatory spine synapses in the hippocampus (Katona et al, 2006; Kawamura et al., 2006). Moreover, several studies showed that the CB1R-agonist WIN inhibits extracellular stimulation-evoked glutamate release onto CA1 pyramidal neurons (Hajos et al., 2001, Ohno-Shosaku et al., 2002, Kawamura et al., 2006, Takahashi and Castillo, 2006). Excitatory synapses onto CA1 pyramidal neurons exhibit evidence of phasic but not tonic endocannabinoid signaling (Ohno-Shosaku et al., 2002; Chiu and Castillo, 2008; Földy et al., 2013), suggesting that the regulation of tonic 2-AG signaling by β-neurexins is synapse-specific and further emphasizing how the identity of trans-synaptic neurexin complexes can dictate function. No previous evidence has linked endocannabinoid signaling to modulation of presynaptic LTP of excitatory synapses, although CB1R activation has been implicated in LTD (Peterfi et al., 2012; Han et al., 2012).
Mechanistically, the large effects of the β-neurexin KO in the presence of the more abundant α-neurexins (Figs. 1A, S1) suggest that β-neurexins perform unique non-redundant functions. The presynaptic action of β-neurexins (Figs. 5, 6), the SS4-dependence of the rescue (Fig. 3E), and the rescue of the presynaptic β-neurexin KO phenotype by blocking postsynaptic synthesis of 2-AG (Figs. 4, 6) demonstrate that the functions of β-neurexins involve trans-synaptic ligand interactions. It is intriguing that some splice variants of neuroligin-1 only bind to β- but not to α-neurexins (Fig. S5I), and that deletion of neuroligin-3 abolishes tonic endocannabinoid signaling at inhibitory synapses of CCK basket cells (Foldy et al., 2013). Although the β-neurexin KO led to the opposite effect at excitatory synapses on subiculum burst-firing neurons, namely an enhancement of basal endocannabinoid signaling, it is possible that they are due to the same principal process: trans-synaptic regulation of endocannabinoid signaling that involves interactions of specific neurexin splice-variants with particular neuroligin isoforms. Alternatively, it is possible that as yet unknown β-neurexin specific ligands mediate their functions, or that α-and β-neurexins are localized to distinct synaptic sites in a neuron.
The β-neurexin KO phenotype resembles that of neurexin-3 SS4 knockin mice in that both genetic manipulations reveal a requirement for neurexins lacking an insert in SS4 (Fig. 3E and Aoto et al., 2013). However, the phenotypes of these mutations are very different. While the β-neurexin KO caused a decrease in presynaptic release probability and a loss of presynaptic LTP without changes in postsynaptic parameters, the opposite was observed in SS4 knockin mice expressing neurexin-3 with constitutively spliced-in SS4 (Aoto et al., 2013). This difference in phenotypes is likely due to the fact that the SS4 knockin affects ALL α- and β-transcripts of ONE particular neurexin gene, whereas the β-neurexin triple KO affects ALL β- but not α-transcripts of ALL neurexin genes (which primarily express α-neurexins). Thus, the only overlap between the two genetic manipulations involves relatively small amounts of neurexin-3β mRNAs. Any phenotypic overlap of these manipulations likely would have been occluded by their more dramatic general phenotypes, although the small decrease in mEPSC amplitude and AMPAR surface levels in β-neurexin KO synapses (Figs. 3A, S4F) may be due to the mechanism described by Aoto et al. for the neurexin-3 SS4 knockin (2013).
Independent of the molecular mechanism underlying the selective functional role of β-neurexins in regulating excitatory synaptic strength, this role likely has significant implications for neural circuit dynamics (Figs. 7A–7D). Information processing by neural circuits involves continuous modulation of synaptic strength at specific sites, such that the input/output relations of a circuit depend on how action potentials are transformed into synaptic signals that eventually cause firing – or inhibition of firing – of the circuit output neurons. Endocannabinoids have emerged as major regulators of circuit dynamics (Katona and Freund, 2008; Castillo et al., 2012; Melis et al., 2014). Thus, the control of endocannabinoid signaling by trans-synaptically acting β-neurexins which in turn are regulated by alternative splicing likely impacts circuit dynamics in many brain regions. Understanding such dynamics will be essential for understanding behavior in general, and the conditional β-neurexin KO mice provide a useful tool for region-specific modulation of circuit dynamics in order to probe its behavioral relevance.
EXPERIMENTAL PROCEDURES
In all experiments, the researcher was blinded to the genetic manipulation. All plasmids are available upon request, and the mice described here were deposited in Jackson Labs for distribution. Brief experimental procedures are listed here. For details, please see Extended Experimental Procedures.
Mouse generation and husbandry
Neurexin-β-floxed (NBF) mice were generated by homologous recombination targeting the 5′ unique exon for each of the three β-neurexin genes that is not shared with its α-neurexin counterparts. All procedures conformed to NIH Guidelines for the Care and Use of Laboratory Animals and were approved by the Stanford University Administrative Panel on Laboratory Animal Care.
mRNA measurements
mRNA measurements were performed using quantitative RT-PCR on RNA isolated from ~P30 mouse brain tissues using the RNAqueous-Micro RNA isolation kit (Invitrogen). Reactions for α- and β-neurexins and GAPDH (internal control) were run with primers and probes as described in the Extended Experimental Procedures.
Neuron cultures
Cortical neurons were cultured from newborn NBF mice, infected on DIV3-4 with lentiviruses, transfected using the calcium phosphate method when indicated, and analyzed at DIV 14–16.
Virus Preparations
Nuclear localized EGFP-Cre and EGFP-ΔCre fusion proteins deliverable by lentiviruses were from previously described vectors (Kaeser et al., 2011). All neurexin-1β rescue constructs were previously described mouse cDNAs expressed from separate lentiviruses (Aoto et al., 2013). For in vivo infections, we employed an AAV-DJ strain that is highly efficient in vivo as described (Xu et al. 2012).
Stereotactic Injections
Stereotactic injections of AAVs were performed as described (Xu et al. 2012). Efficiency and localization of AAV expression were confirmed by fluorescence of nuclear EGFP encoded by the expressed inactive and active EGFP-Cre-recombinase fusion proteins.
Ca2+-Imaging
A chimeric GCaMP5G-Syb2 was made and used in order to target GCaMP5G calcium sensor to presynaptic terminals as described in Extended Experimental Procedures. Neuronal morphology was visualized by sparse Ca2+-phosphate transfection with mCherry expression construct, and imaging was performed on a Zeiss LSM 510 confocal microscope.
Electrophysiology
For details of electrophysiological recordings from cultured neurons and acute slices, see the Extended Experimental Procedures.
Fear Conditioning
Fear conditioning and open field behavioral analysis was essentially performed as previously described (Xu et al., 2012).
Supplementary Material
Acknowledgments
We would like to thank Dr. Wei Xu and Dr. Theodoros Tsetsenis for advice on fear conditioning experiments, and Dr. Ken Mackie (Indiana University) for generous contribution of CB1R antibodies used in this study. This paper was supported by grants from the NIH (R37 MH052804 to T.C.S.; K99 MH103531 to J.A.; K99 DA034029 to C.F.; P50 MH086403 to R.C.M., T.C.S., and L.C.) and from Autism Speaks (7953 to G.R.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION
Supplemental Information includes detailed experimental procedures and seven supplemental figures.
AUTHOR CONTRIBUTIONS
G.R.A. designed, performed, and analyzed most experiments; J.A., C.F., A.X.Y., D.W., and S-J.L. contributed to specific experiments; K.T. generated the conditional KO mice; J.C. contributed Ca2+-imaging tools; G.R.A., L.C., R.C.M. and T.C.S. designed experiments and analyzed data; and T.C.S. wrote the paper with input from all authors.
References
- Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Südhof TC. Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell. 2013;154:75–88. doi: 10.1016/j.cell.2013.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang ML, Owczarek S. A matter of balance: role of neurexin and neuroligin at the synapse. Neurochemical research. 2013;38:1174–1189. doi: 10.1007/s11064-013-1029-9. [DOI] [PubMed] [Google Scholar]
- Behr J, Wozny C, Fidzinski P, Schmitz D. Synaptic plasticity in the subiculum. Prog Neurobiol. 2009;89:334–342. doi: 10.1016/j.pneurobio.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Boucard AA, Chubykin AA, Comoletti D, Taylor P, Südhof TC. A splice code for trans-synaptic cell adhesion mediated by binding of neuroligin 1 to alpha- and beta-neurexins. Neuron. 2005;48:229–236. doi: 10.1016/j.neuron.2005.08.026. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci. 2004;24:5623–5631. doi: 10.1523/JNEUROSCI.0918-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke RA, Eapen V. Balance within the Neurexin Trans-Synaptic Connexus Stabilizes Behavioral Control. Frontiers in human neuroscience. 2014;8:52. doi: 10.3389/fnhum.2014.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, Sylwestrak E, O’Sullivan ML, Otto S, Tiglio K, Savas JN, Yates JR, 3rd, Comoletti D, Taylor P, Ghosh A. LRRTM2 interacts with Neurexin1 and regulates excitatory synapse formation. Neuron. 2009;64:799–806. doi: 10.1016/j.neuron.2009.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Bifulco M, De Petrocellis L. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3:771–784. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Dong HW. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7–19. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foldy C, Malenka RC, Südhof TC. Autism-associated neuroligin-3 mutations commonly disrupt tonic endocannabinoid signaling. Neuron. 2013;78:498–509. doi: 10.1016/j.neuron.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D. Role of endogenous cannabinoids in synaptic signaling. Physiol Rev. 2003;83:1017–1066. doi: 10.1152/physrev.00004.2003. [DOI] [PubMed] [Google Scholar]
- Glass M, Northup JK. Agonist selective regulation of G proteins by cannabinoid CB1 and CB2receptors. Mol Pharmacol. 1999;56:1362–1369. doi: 10.1124/mol.56.6.1362. [DOI] [PubMed] [Google Scholar]
- Graves AR, Moore SJ, Bloss EB, Mensh BD, Kath WL, Spruston N. Hippocampal pyramidal neurons comprise two distinct cell types that are countermodulated by metabotropic receptors. Neuron. 2012;76:776–789. doi: 10.1016/j.neuron.2012.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Deng L, Wang Y, Dulubova I, Liu X, Rizo J, Südhof TC. RIM proteins tether Ca2+ channels to presynaptic active zones via a direct PDZ-domain interaction. Cell. 2011;144:282–295. doi: 10.1016/j.cell.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG. Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annual review of physiology. 2014;76:333–363. doi: 10.1146/annurev-physiol-021113-170338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nature medicine. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Ko J, Fuccillo MV, Malenka RC, Südhof TC. LRRTM2 functions as a neurexin ligand in promoting excitatory synapse formation. Neuron. 2009;64:791–798. doi: 10.1016/j.neuron.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Krueger DD, Tuffy LP, Papadopoulos T, Brose N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr Opin Neurobiol. 2012;22:412–422. doi: 10.1016/j.conb.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Matsuda K, Yuzaki M. Cbln family proteins promote synapse formation by regulating distinct neurexin signaling pathways in various brain regions. Eur J Neurosci. 2011;33:1447–1461. doi: 10.1111/j.1460-9568.2011.07638.x. [DOI] [PubMed] [Google Scholar]
- Melis M, Greco B, Tonini R. Interplay between synaptic endocannabinoid signaling and metaplasticity in neuronal circuit function and dysfunction. Eur J Neurosci. 2014;39:1189–1201. doi: 10.1111/ejn.12501. [DOI] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC. α-Neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature. 2003;423:939–948. doi: 10.1038/nature01755. [DOI] [PubMed] [Google Scholar]
- Murataeva N, Straiker A, Mackie K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol. 2014;171:1379–1391. doi: 10.1111/bph.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrenko AG, Ullrich B, Missler M, Krasnoperov V, Rosahl TW, Südhof TC. Structure and evolution of neurexophilin. J Neurosci. 1996;16:4360–4369. doi: 10.1523/JNEUROSCI.16-14-04360.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Rowen L, Young J, Birditt B, Kaur A, Madan A, Philipps DL, Qin S, Minx P, Wilson RK, Hood L, et al. Analysis of the human neurexin genes: alternative splicing and the generation of protein diversity. Genomics. 2002;79:587–597. doi: 10.1006/geno.2002.6734. [DOI] [PubMed] [Google Scholar]
- Siddiqui TJ, Pancaroglu R, Kang Y, Rooyakkers A, Craig AM. LRRTMs and neuroligins bind neurexins with a differential code to cooperate in glutamate synapse development. J Neurosci. 2010;30:7495–7506. doi: 10.1523/JNEUROSCI.0470-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staff NP, Jung HY, Thiagarajan T, Yao M, Spruston N. Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol. 2000;84:2398–2408. doi: 10.1152/jn.2000.84.5.2398. [DOI] [PubMed] [Google Scholar]
- Südhof TC. Neuroligins and neurexins link synaptic function to cognitive disease. Nature. 2008;455:903–911. doi: 10.1038/nature07456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo GG, Lenkey N, Holderith N, Andrasi T, Nusser Z, Hajos N. Presynaptic calcium channel inhibition underlies CB(1) cannabinoid receptor-mediated suppression of GABA release. J Neurosci. 2014;34:7958–7963. doi: 10.1523/JNEUROSCI.0247-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabuchi K, Südhof TC. Structure and evolution of neurexin genes: insight into the mechanism of alternative splicing. Genomics. 2002;79:849–859. doi: 10.1006/geno.2002.6780. [DOI] [PubMed] [Google Scholar]
- Treutlein B, Gokce O, Quake SR, Südhof TC. Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc Natl Acad Sci U S A. 2014;111:E1291–1299. doi: 10.1073/pnas.1403244111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- Uemura T, Lee SJ, Yasumura M, Takeuchi T, Yoshida T, Ra M, Taguchi R, Sakimura K, Mishina M. Trans-synaptic interaction of GluRdelta2 and Neurexin through Cbln1 mediates synapse formation in the cerebellum. Cell. 2010;141:1068–1079. doi: 10.1016/j.cell.2010.04.035. [DOI] [PubMed] [Google Scholar]
- Ullrich B, Ushkaryov YA, Südhof TC. Cartography of neurexins: more than 1000 isoforms generated by alternative splicing and expressed in distinct subsets of neurons. Neuron. 1995;14:497–507. doi: 10.1016/0896-6273(95)90306-2. [DOI] [PubMed] [Google Scholar]
- Ushkaryov YA, Hata Y, Ichtchenko K, Moomaw C, Afendis S, Slaughter CA, Südhof TC. Conserved domain structure of beta-neurexins. Unusual cleaved signal sequences in receptor-like neuronal cell-surface proteins. J Biol Chem. 1994;269:11987–11992. [PubMed] [Google Scholar]
- Ushkaryov YA, Petrenko AG, Geppert M, Südhof TC. Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- van Welie I, Remme MW, van Hooft JA, Wadman WJ. Different levels of Ih determine distinct temporal integration in bursting and regular-spiking neurons in rat subiculum. J Physiol. 2006;576:203–214. doi: 10.1113/jphysiol.2006.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozny C, Maier N, Fidzinski P, Breustedt J, Behr J, Schmitz D. Differential cAMP signaling at hippocampal output synapses. J Neurosci. 2008;28:14358–14362. doi: 10.1523/JNEUROSCI.4973-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Morishita W, Buckmaster PS, Pang ZP, Malenka RC, Südhof TC. Distinct neuronal coding schemes in memory revealed by selective erasure of fast synchronous synaptic transmission. Neuron. 2012;73:990–1001. doi: 10.1016/j.neuron.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.