

Abstract
Background
The babA2 gene along with the cagA and vacA of Helicobacter pylori has been considered as a risk factor for the disease outcome in certain populations. This study was aimed to understand the role of babA2 of H. pylori with the background of cagA and vacA in disease manifestations in Indian sub population.
Methods
A total of 114 H. pylori strains isolated from duodenal ulcer (DU) (n = 53) and non-ulcer dyspepsia (NUD) patients (n = 61) were screened for the prevalence of these virulence markers by PCR. The comparative study of IL-8 production and apoptosis were done by co-culturing the AGS cell line with H. pylori strains with different genotypes. Adherence assay was performed with babA2 positive and negative strains. Two isogenic mutants of babA2 were constructed and the aforesaid comparative studies were carried out.
Results
PCR results indicated that 90.6 % (48/53), 82 % (50/61) and 73.6 % (39/53) strains from DU patients were positive for cagA, vacA, and babA2, respectively. Whereas the prevalence of these genes in NUD subjects were 70.5 % (43/61); 69.8 % (37/53), and 65.6 % (39/61), respectively. Although adherence to AGS cells was comparable among strains with babA2 positive and negative genotypes, but the triple positive strains could induce highest degree of IL-8 production and apoptosis, followed by the cagA−/vacA−/babA2+ strains and triple negative strains, respectively. The wild type strains showed significantly higher IL-8 induction as well as apoptosis in ex vivo than its isogenic mutant of babA2.
Conclusion
PCR study demonstrated that there was no significant association between the distribution of babA2 genotype or of triple positive strains and disease outcome in this sub population. The adherence assay showed that there was no significant difference in the extent of adherence to AGS cells among babA2 positive and negative strains. But the ex vivo study indicated that the triple positive or even the babA2 only positive strains are involved in increased virulence. The wild type strains also exhibited increased virulence compared to the babA2 mutant strains. This inconsistency demonstrated that bacterial genotype along with host genetic polymorphisms or other factors play important role in determining the clinical manifestation of H. pylori infections.
Keywords: Helicobacter pylori, Duodenal ulcer, BabA2, IL-8, Adhesion molecule, Apoptosis
Background
Helicobacter pylori is a Gram-negative, genetically diverse spiral bacteria that infects more than half of the population worldwide [1]. Infection with H. pylori is associated with duodenal ulcer (DU) or gastric ulcer, gastritis, and gastric adenocarcinoma [2]. About 65–70 % of the Indian population is infected with H. pylori [3, 4]. 15–20 % of overall infected population develop gastric or duodenal ulcer and less than 1 % develop gastric adenocarcinoma [5].
The genome of various H. pylori strains demonstrates significant genetic diversity. Genetic variation in specific virulence genes of H. pylori may participate in the pathogenic process of H. pylori infection in the stomach, thereby contributing to the variable risk of diverse clinical outcomes. So, in addition to the host immunological factors and environmental factors, another important reason for the diverse clinical outcomes is the differences in virulence factors among H. pylori strains [6].
Cytotoxin-associated gene (cagA) was the first reported gene that varies in H. pylori strains and is considered as a marker for the presence of the cag pathogenicity island (cag PAI), which includes a number of other genes associated with increased virulence [7–9]. About one-half to two-thirds of US and European strains carry the cag PAI and is associated with overt disease. In contrast, cag PAI is distributed among most of the Asian strains, irrespective of disease status. In India the asymptomatic individuals also carry the cag PAI [10]. It was found that most of the H. pylori strains in Indian subcontinent have cagA typeA and any particular type of cagA is not associated with disease outcome [11].
Some H. pylori strains can produce vacuoles in epithelial cells by the action of VacA protein which consists of a signal region (s1 or s2) and a middle region (m1 or m2). Strains harboring the s1m1 mosaic combination are said to be more cytotoxically potent than the strains with s1m2 genotype, while s2m2 strains do not secrete vacuolating cytotoxin [12]. In Western countries, H. pylori strains with s1m1 genotype usually also carry the cag pathogenicity island (cag PAI) and are more significantly associated with the disease than those strains which don’t have this cag PAI [7]. In Indian population, the strains with s1m1 genotype are predominant [13]. It concludes that the diseases are multi-factorial and thereby we need to identify other bacterial virulence factors which play important role in diseases manifestation.
It is generally known that bacterial adherence to the gastric epithelium is the first critical stage of colonization by H. pylori in the human stomach [14]. The blood group antigen binding adhesin (BabA) is one of the major outer membrane proteins of H. pylori that binds to the fucosylated Lewisb blood group antigens on the gastric epithelium and plays a key role in facilitating bacterial colonization to the stomach [15, 16]. It has been hypothesized that the adherent bacteria may be more successfully able to transfer their products to the host cells. Thus, a higher bacterial density coupled with more capable delivery of bacterial products to the host cells may provoke a stronger inflammatory response. It has been already shown that H. pylori is able to commence nonspecific immune responses [17]. H. pylori has various adhesins, including blood group antigen binding adhesin (BabA), which binds to the Lewis B (Leb) antigen [15, 18]. H. pylori adhesin-mediated colonization is multi-factorial and no single adhesin may be essential, although adherence of this bacteria to the gastric epithelium is a compulsory early step in colonization and renders H. pylori 100–1000 times more resistant to antibiotics than the non-adherent ones [19]. Blood group antigen binding adhesin is encoded by a polymorphic gene named babA2, while the babA1 allele is non-functional. Gerhard et al. [20] in 1999 first demonstrated a positive association between a babA2 carrying strain and duodenal ulcer (DU) and gastric cancer (GC). Subsequently, a series of studies of the association between babA2 gene and peptic ulcer diseases and GC have been performed, but with inconsistent or conflicting conclusions [21–24]. However, in Asian countries, most of the strains are babA2 positive irrespective of disease status [25, 26]. These data clearly indicates that the frequency of H. pyloribabA2 gene may vary geographically and their associations with disease outcome also vary accordingly.
Although Indian subcontinent constitutes about the 1/5th of the world population, but there was not much studies about the role of babA2 gene in disease outcome from this subcontinent. The contradictory conclusions from previous studies and the inadequate information from India provided the impetus to study (1) the clinical relevance of the babA2 gene by examining its association with H. pylori virulence-associated genes and with clinical outcome as well as (2) the comparative analysis of IL-8 production and apoptosis by co-culturing the AGS cell line with Indian H. pylori strains with variant genetic makeup.
Results
A total of 185 subjects underwent for endoscopy from which 114 H. pylori strains were isolated. The biopsies were divided into two groups according to their clinical symptoms: duodenal ulcer (DU) and non ulcer dyspepsia (NUD). Subjects with abdominal discomfort, acidity, loss of appetite but no frank ulceration were considered as non-ulcer dyspepsia (NUD) but those have endoscopically visible duodenal ulceration were considered as duodenal ulcer (DU) patients. The strains were isolated from 53 DU patients (male = 54 %, median age = 62 years, range 28–85 years) and 61 NUD patients (male = 52 %, median age = 67 years, range 27–87 years). The genomic DNA was isolated from these strains and was used for further downstream PCR-based analysis.
Prevalence of babA2, cagA and vacA
The integrity and specificity of these isolated H. pylori DNAs were confirmed by PCR amplification of the urease gene, which yielded an expected 480 bp amplicon from all the DNAs (data not shown). The distribution of babA2 gene in the 114 Indian H. pylori strains were studied first using the PCR-based genotyping. The fragment of babA2 was amplified primarily by using forward primer nthu_babA2F [26] and reverse primer babA2R [20]. PCR assay with these primers yielded an amplicon of 832 bp (Fig. 1A) and 60.5 % (69/114) strains produced positive amplicon for babA2 gene. Another two sets of primers (namely babA2F and babA2R; babA2F and babA2R607) were used to amplify the babA2 gene from the strains that did not give positive amplicon with the primary set of primers [20, 27]. PCR with these two new combination of primers yielded amplicon of babA2 fragment (832 and 607 bp respectively) from 3 (72/114, Fig. 1B) and 5 (77/114, Fig. 1C) more clinical isolates, respectively. Finally, on the basis of the combined results with these three sets of primers, our analysis depicted that 67.5 % (77/114) Indian H. pylori strains were positive for babA2 gene (Table 1).
Fig. 1.
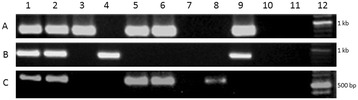
Genotyping of babA2 gene in Indian H. pylori isolates. The images shown are from a representative gel electrophoresis of a PCR amplification product of babA2 gene from Indian isolates and J99 control strain with A nthu_babA2F and babA2R primers [26], B babA2F and babA2R primers [20], and C babA2F and babA2R607 primers [27]. Where lane 1 positive control, lanes 2–10 clinical isolates, lane 11 negative control, and lane 12 DNA ladder
Table 1.
Prevalence of the babA2, cagA and vacA genes in different disease groups
| Gene | Total (n = 114) | DU (n = 53) | NUD (n = 61) |
|---|---|---|---|
| babA2 | 77 (67.5 %) | 37 (69.8 %) | 40 (65.6 %) |
| cagA | 98 (86 %) | 48 (90.6 %) | 50 (82 %) |
| vacA s1m1 | 82 (71.9 %) | 39 (73.6 %) | 43 (70.5 %) |
The cagA and vacA status of these 114 strains were determined using the previously described primers and protocols [5]. The cagA positive strains produced an amplicon of 350 bp (data not shown) and 86 % (98/114) strains were positive for cagA (Table 1). The strains that were negative for cagA, yielded a 550 bp amplicon for cag-empty site using the primers located in the flanking region of the cag PAI. vacA s and m alleles were also studied and it was shown that the most potentially toxic vacA s1m1 allele was predominantly present in the tested strains and it showed a frequency of 71.9 % (82/114).
The babA2 gene was determined among the two groups of patients [DU (53) and NUD (61)]. PCR results showed that 69.8 % (37/53) strains were babA2 positive in DU and 65.6 % (40/61) babA2 positive strains were present in NUD subjects (Table 1). The babA2 gene was almost equally distributed among strains from DU patients and NUDs in this region. So, the prevalence of babA2 in this region showed no significant correlation with disease outcome. Our study demonstrated that the babA2 genotype was predominant among H. pylori strains irrespective of disease status.
Among the 98 cagA positive strains used in this study, 41 of 48 DU patients and 40 of 50 NUD individuals produced a typical ~642-bp amplicons, designated as type A by Yamaoka et al. [28] (Table 2). Six strains from DU and nine strains from NUD yielded ~756 bp amplicon in PCR using the same set of primers and considered as having cagA type B/D (Table 2). Two strains (one each from DU and NUD) produced a ~810 bp amplicon corresponding to type C (Table 2). No correlation with disease outcome was found with any of the cagA types. Sequencing analysis of the representative strains showed that all of them carried Western-CagA-specific sequences (WSS) FPLKRHDKVDDLSKV. These strains contained 1–5 EPIYA motifs in their cagA gene sequence. Sequence analysis also revealed that more than 80 % of the cagA positive strains carried 3 EPIYA motifs (A-B-C type) at the 3′ end of the gene and they were almost equally distributed between DU and NUD. Similarly, vacAs1m1 allele was present in almost equal frequencies in DU patients and NUDs (Table 1).
Table 2.
Prevalence of cagA subtypes in different disease groups
| CagA types | DU (n = 48) | NUD (n = 50) |
|---|---|---|
| Type A | 41 (85.4 %) | 40 (80 %) |
| Type B/D | 6 (12.5 %) | 9 (18 %) |
| Type C | 1 (2 %) | 1 (2 %) |
The association study of these virulence genes in each strain reveals no statistically significant (P < 0.05) correlation between the distribution of the triple positive strains (combined presence of cagA, vacA s1m1 allele and babA2) and disease status. Additionally, 84 isolates were classified as type 1 strains (positive for both cagA and vacAs1m1) and 61 were found as triple positive strains among them. These type 1 strains also exhibited an almost uniform distribution among DUs (40/53, 75.47 %, Table 3) and NUDs (44/61, 72.13 %, Table 3). It was found that these 61 triple positive strains distributed almost equally in DU (29/53, 54.7 %, Table 3) patients and NUDs (32/61, 52.4 %, Table 3). Among the 16 cagA negative strains tested, 6 (37.5 %) were found as babA2 positive strains.
Table 3.
Prevalence of the type 1 and triple positive strains in different disease groups
| Strains | Total (n = 114) | DU (n = 53) | NUD (n = 61) |
|---|---|---|---|
| Type 1 strains | 84 (73.7 %) | 40 (75.5 %) | 44 (72.1 %) |
| Triple positive strains | 61 (53.5 %) | 29 (54.7 %) | 32 (52.4 %) |
Adherence of H. pylori to the gastric epithelial (AGS) cells is independent of babA2
Adherence of H. pylori to the AGS cells was performed as described in methods section and was found comparable among the strains with babA2 positive and babA2 negative genotypes (data not shown). It was also observed that there was significantly no difference in the ability of adherence between the wild type and isogenic babA2 mutants (data not shown).
Triple positive strains show greater IL-8 induction in AGS cells
Based on the presence or absence of these three virulence genes, the tested strains were divided into four groups: (1) triple positive strains (cagA+/vacA+/babA2+), (2) cagA+/vacA+/babA2− strains, (3) cagA−/vacA−/babA2+ strains, and (4) triple negative strains (cag−/vac−/babA2−). After co-cultured with H. pylori strains for 8 h the IL-8 secretion was significantly (P < 0.05) higher from those cells which were infected with the triple positive strains (827.3 ± 80.88 pg mL−1, n = 3) than the cells infected with cagA+/vacA+/babA2− strains (523.1 ± 43.84 pg mL−1, n = 3, Fig. 2a). Similarly, significantly (P < 0.05) higher amount of IL-8 induction was recorded when the cells were infected with cagA−/vacA−/babA2+ strains (462.7 ± 47.81 pg mL−1, n = 3) compared to triple negative strains (62.80 ± 2.30 pg mL−1, n = 3, Fig. 2b).
Fig. 2.
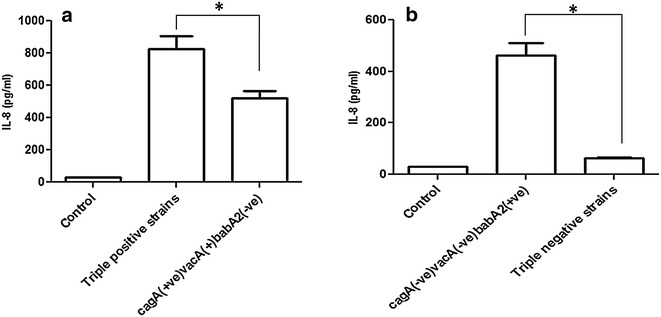
Triple positive strains and even the babA2 positive only strains enhance IL-8 production in AGS cells. In vitro IL-8 production from AGS cells co-cultured with randomly selected a triple positive and cagA +/vacA +/babA2 − strains, and b cagA −/vacA −/babA2 + and triple negative H. pylori strains (MOI is 100) for 8 h. IL-8 from culture supernatant was measured using ELISA as described in “Methods”. Data are expressed as mean ± standard error of mean (SEM) of 3 experiments in duplicates. *P < 0.05 as compared between groups
babA2 mutant strains show decreased level of IL-8 induction in AGS cells
To further establish our data we carried out the IL-8 assay in cell culture as described previously by infecting the cells with I-121, 135(1) wild type strains and their babA2 mutants separately. The results indicated that the significantly (P < 0.05) higher amount of IL-8 induction was recorded from the cells infected with the wild type strains (729.18 ± 22.39 and 433.95 ± 12.41 pg mL−1 respectively, Fig. 3) rather than its babA2 mutants (465.06 ± 16.19 and 202.77 ± 12.33 pg mL−1 respectively, Fig. 3).
Fig. 3.
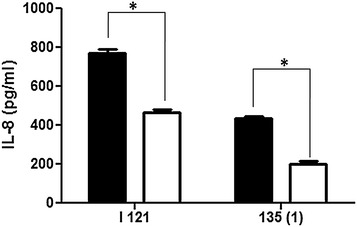
Wild type H. pylori strains show higher IL-8 production than their isogenic mutant of babA2 in AGS cells. IL-8 from culture supernatant was measured using ELISA as described in “Methods”. Data are expressed as mean ± standard error of mean (SEM) of 3 experiments in duplicates. Wild type is denoted with black filled bar (◼) and mutants denoted with empty bar (◻). *P < 0.05 as compared between groups
Triple positive strains trigger more apoptosis in AGS cell line
The cell cycle analysis with propidium iodide reveals distribution of cells in three major phases of the cell cycle (G1 vs. S vs. G2/M, Fig. 4a) and makes it possible to detect unhealthy cells with fractional DNA content. The cells in the sub-G0 phase represent apoptotic cells. After co-culturing the AGS cells with H. pylori for 24 h significantly (P < 0.05) higher amount of apoptotic cell death was found when the cells infected with triple positive strains (64.40 ± 2.485 %, n = 3, Fig. 4aii, b) in comparison to the cells infected with cagA+/vacA+/babA2− strains (47.17 ± 2.916 %, n = 3, Fig. 4aiii, b). Similarly, cagA−/vacA−/babA2+ strains (36.17 ± 1.955 %, n = 3, Fig. 4av, c) showed significantly (P < 0.05) higher apoptotic cell death than the triple negative strains (19.33 ± 2.038 %, n = 3, Fig. 4avi, c) after 24 h infection.
Fig. 4.
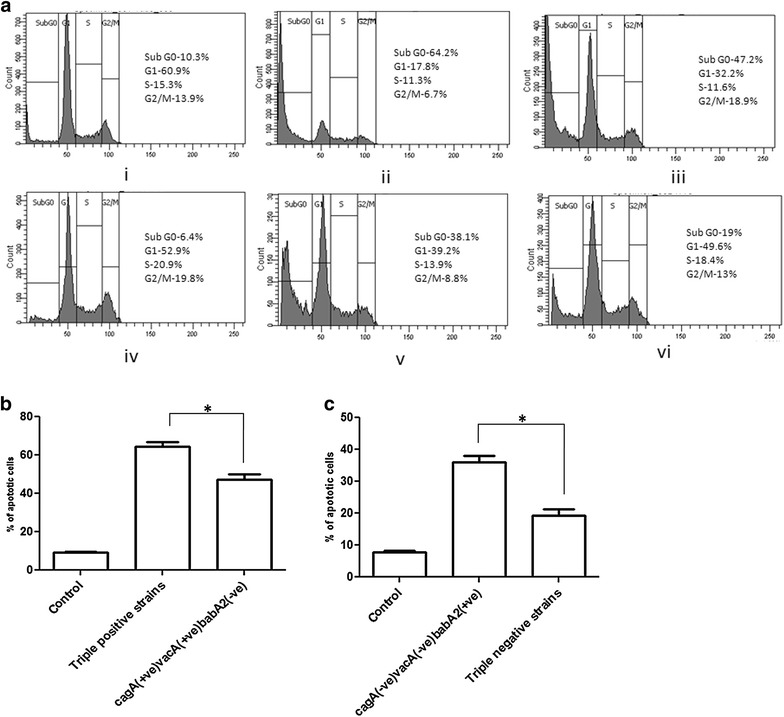
a–c Triple positive strains and even the babA2 positive only strains cause more apoptosis in AGS cells. a Apoptosis of AGS cells (a i, iv worked as controls) co-cultured with different genotypic variant i.e. a ii triple positive; a iii cagA +/vacA +/babA2 −; a v cagA −/vacA −/babA2 +; and a vi triple negative strains of H. pylori strains for 24 h (MOI is 100), stained with propidium iodide and analysed by flow cytometry. These figures are representative profile of at least three experiments. b, c Graphical representation of % apoptotic cells (Sub G0 phase) infected with same group of strains were expressed as mean ± SEM. *P < 0.05 as compared between groups
Mutation of babA2 gene causes reduced apoptosis in ex vivo
For reconfirming the apoptotic potential of the babA2 gene we carried out the cell cycle analysis experiment in AGS cells by infecting the cells with two wild type strains [I-121 and 135 (1) respectively] and their babA2 deletion mutants as described previously. After 24 h of infection it was found that the wild type strains were capable of significantly (P < 0.05) more apoptotic cell death (61.8 ± 1.88, 53.56 ± 1.946 % respectively, Fig. 5) than their isogenic mutants of babA2 (36.43 ± 1.486, 24.76 ± 1.328 %, respectively, Fig. 5).
Fig. 5.
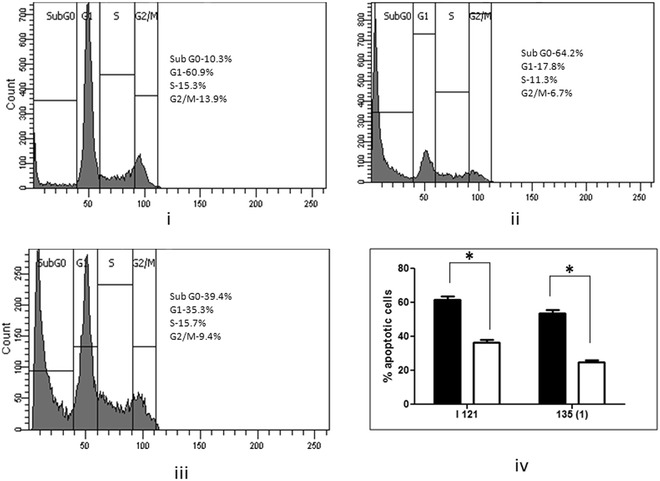
Wild type H. pylori strains cause more apoptotic cell death than their isogenic mutant of babA2 in AGS cells. Apoptosis of AGS cells (i worked as controls) co-cultured with different genotypic variant i.e. ii wild type I-121 strain; and iii isogenic babA2 mutant of I-121 strain for 24 h (MOI is 100), stained with propidium iodide and analysed by flow cytometry. i–iii is only representative profile of at least three experiments carried out with I-121 strain and its babA2 mutant. iv Graphical representation of percentage of apoptotic cells (sub G0 phase) infected with two wild type and their babA2 mutant strains were expressed as mean ± SEM. Wild type is denoted with black filled bar (◼) and mutants denoted with empty bar (◻). *P < 0.05 as compared between groups
Discussion
Various studies have indicated that the incidence and/or severity of gastroduodenal pathologies related to H. pylori may vary according to geographical regions [27]. There is also evidence for the existence of different strains of H. pylori with different degrees of virulence indicating the variation in the distribution of the different virulence attributes of H. pylori in different populations [29–31]. The cagA gene was found at a frequency of 86 % among the 114 tested strains of this region and this virulence marker was found at almost equal frequencies in strains from DU patients (90.6 %) and NUD patients (82 %), indicating that the prevalence of the cagA gene cannot be considered as a key virulence marker for determination of the clinical status of the host, as has been reported in other Indian studies [13, 32]. In our study most of the strains have type A cagA without any association in disease outcome. All the strains used in sequence analysis consist of Western-CagA-specific sequence and it is in agreement with our previous report [11]. We also found that highly virulent s1m1 allele of vacA gene was present among 71.9 % of Indian H. pylori strains and presence of this allele did not show any correlation with the disease outcome (DU = 73.6 %, NUD = 70.5 %) of the host. This data is in accord with the previous report having 70 % presence of s1m1 allele in Indian H. pylori isolates [5].
Here we found that the Indian H. pylori strains exhibited 67.5 % prevalence of babA2 gene and the babA2 gene was uniformly distributed (DU = 69.8 %, NUD = 65.6 %) irrespective of disease status. These results indicate that, unlike in the Western countries [33], presence of babA2 is not associated with clinical outcome in India. In our study among 84 double positive strains, 61 were found as triple positive. These 61 triple positive strains also showed an almost equal distribution in DU (29/53, 54.7 %) patients and NUDs (32/61, 52.4 %), which is inconsistent with the report from few Western countries [34].
More recent analyses of babA2 as a virulence marker have produced conflicting data on the usefulness of babA2 expression in predicting clinical outcome, which is most likely dependent on the geographic origin of the H. pylori strains. In Portuguese and Thai populations, babA2 is not a biomarker for peptic ulcer disease or gastric cancer [35, 36]. However, for strains isolated from Germany, Turkey, or northern Portugal, babA2 expression is associated with the severity of gastric disease [18, 37, 38]. This inconsistency may be due to the specific geographic variation of the circulating bacterial lineages. Another explanation for this lack of association may be due to the allelic variation of the babA2 gene which was studied by Pride et al. [39].
An inflammatory response is one of the main pathophysiological events in H. pylori mediated infection. It has been shown that epithelial cells secrete IL-8 as a result of H. pylori infection [40–42]. IL-8 acts as a potent chemo-attractant for neutrophils and is thought to play a crucial role in H. pylori induced tissue damage [43]. Several studies have shown that induction of IL-8 secretion from host cells is dependent on the presence of CagA, VacA, OMPs (OipA) and LPS [43–46]. In order to see the effect of babA2 gene on the IL-8 induction, triple positive (cagA−/vacA−/babA2+) strains and triple negative strains were cultured with AGS cells. The triple positive strains induced the highest amount of IL-8 secretion from AGS cells, and the double negative strains but with babA2 gene caused significantly higher amount of IL-8 secretion than the triple negative ones (Fig. 2, P < 0.05). Therefore, it may be emphasized that the triple positive strains and also the presence of babA2 somehow enhances the ability of H. pylori to induce IL-8 in the gastric mucosa.
IL-8 secretion and inflammation due to H. pylori infection lead to epithelial cell damage or apoptosis. Apoptosis is a genetically programmed form of cell death which is mainly characterized by some distinct morphological and molecular features. Microbial pathogens or their products can directly activate the apoptotic pathway which plays a role in pathogenesis [47]. It has been reported that cagA, and vacA have an effect on H. pylori mediated apoptosis [48, 49]. In order to elucidate the role of babA2 in apoptosis, the triple positive, cagA+/vacA+/babA2−, cagA−/vacA−/babA2+ and triple negative strains were used independently to infect the AGS cells. It was observed that the triple positive strains caused more epithelial cell death than the other strains, whereas the cagA−/vacA−/babA2+ strains caused significantly higher level of apoptosis than the triple negative strains (P < 0.05).
As the triple positive strains exhibit highest level of IL-8 induction as well as apoptosis that is why we made isogenic babA2 mutants of these strains [I-121 and 135(1) respectively] to reconfirm the role of babA2 in H. pylori virulence. The IL-8 induction assay and cell cycle analysis by flow cytometry were performed with these wild type and mutant strains as described in methods section. In agreement, it was found that the wild type strains were significantly (P < 0.05) more capable of IL-8 induction as well as mediating the apoptotic cell death in AGS cells (Figs. 3, 5). These data more strongly establish the virulence potential of the strains with babA2 positive genotype.
It has been already reported that ex vivo IL-8 production requires the adherence of viable H. pylori to the AGS cells [50], and as the induction of apoptosis and IL-8 secretion are often linked [51], so it can be said that the apoptosis in AGS cells also require the adherence of H. pylori. In this study, it was found that the adherence of babA2 positive and babA2 negative strains were comparable in AGS cells. In agreement, it was also found that the extent of adherence to the AGS cells were comparable between the wild type strains and isogenic babA2 mutants. This report suggests that although adherence is crucial, the babA2 gene products may regulate the IL-8 secretion and apoptosis by an adherence independent manner. H. pylori has at least five different adhesin factors which use different receptors for adhering on gastric epithelial cells [43], suggesting that presence or absence of only one of these genes may not affect its adherence to gastric cells. Further studies will be required to explain what other factors may impact the extent of adherence.
In this report, although it was observed that Indian H. pylori strains harbored babA2 gene independent of disease status and also the triple positive strains showed no association with disease status, but on the other hand, the triple positive strains and even only the babA2 positive strains showed more virulence as they induced more IL-8 secretion and as well as caused more apoptosis in AGS cell line than the triple negative strains. In accordance, the wild type strains also exhibited more capability to induce IL-8 secretion as well as apoptosis than their isogenic babA2 mutants in the cell culture study. Apparently, it seems that these two statements are contradictory. What could be the probable reasons? H. pylori is one of the genetically diverse bacterial species with regard to genotyping [52–54]. Previous reports also suggested that H. pylori genotype varies geographically [2, 54]. The polymorphisms of some cytokine genes have been found to be associated with H. pylori mediated clinical consequences, probably because they modulate the amount of cytokine production in response to H. pylori infection [55]. It was already been established that dietary salt intake can play an important role in enhancing the likelihood of severe clinical consequences of H. pylori mediated infection [56]. Environmental iron level also brings about the changes in the composition of the H. pylori outer membrane which ultimately influences the pattern of clinical outcomes [57]. These above mentioned information clearly establish the role of host factors and environmental factors in the disease outcomes of H. pylori mediated infection. Importantly, H. pylori toxin gene expression is inconsistent with the general phenomenon observed for other pathogenic bacteria, that is, that expression of the important toxin genes are strongly associated with diseases [58]. The expressions of toxin genes by H.pylori are not solely dependent on the disease status. So, this result can not lead to the underestimation of the role of babA2 gene as a toxin gene. The genetic constitution of host may play a critical role in the successful colonization and ultimately in the disease consequences of H. pylori infection. We can say that the virulent effect of babA2 gene of H. pylori may come into action only after interplay with certain host factors. Our results also showed that babA2 gene in combination with the presence of cagA and vacA s1m1 allele become much more toxic as the triple positive strains showed highest degree of IL-8 induction as well as apoptotic cell death in cell culture study, although the PCR-based genotyping results showed a uniform distribution of triple positive strains independent of disease status. These discrepancies may be due to the fact that at the time of H. pylori isolation, the NUD people harboring the triple positive strains may be at the risk of developing an ulcer disease in future which cannot be come into our count.
Conclusion
In conclusion, although the presence of babA2 gene is not associated with disease outcome in India, but the babA2 positive strains showed more virulence than their negative counterparts in ex vivo study. Study also indicated that adherence of H. pylori to the gastric epithelial (AGS) cells is not solely dependent on babA2. It is evident from this study that bacterial genetic constitution cannot be considered as only responsible factor for disease consequences. Certain host factors along with bacterial genetic traits may play a crucial role in this aspect. Further studies are required to determine the function(s) of babA2 and its relationship with disease outcomes.
Methods
Collection of biopsy samples
Biopsy specimens were obtained as described previously [58] from a total of 185 adult subjects with upper gastrointestinal irregularities underwent endoscopy at the hospital of the Institute of Post Graduate Medical Education and Research, Kolkata, and St John’s Medical College Hospital, Bangalore, India, during 2009–2011. A detailed case study of each individual was done prior to endoscopy. The objective of the study was explained to each individual and informed consent was obtained from each of them under protocols approved by the ethical committees of respective institutes based on the Helsinki Declaration. These biopsy samples were transported to the laboratory under ice-cold condition in Brucella broth (0.6 ml, Difco Laboratories) with glycerol (15 %) and stored in −70 °C until culture.
Helicobacter pylori culture
The Brucella broth containing the biopsy samples were vortexed in laboratory for 2 min and 200 µl of the mixture was streaked onto brain heart infusion agar (BHIA) plate containing horse serum (7 %, Invitrogen, Grand Island, NY, USA), IsoVitalex (0.4 %, Becton–Dickinson, San Jose, CA, USA), trimethoprim (5 µg mL−1), vancomycin (6 µg mL−1), polymyxin B (10 µg mL−1) and nalidixic acid (8 µg mL−1) (all from Sigma Chemicals, MO, USA). These plates were incubated at 37 °C in microaerophilic condition (85 % N2, 10 % CO2, and 5 % O2) for 3–6 days [10] in a double gas incubator (Heraeus Instruments, Hanau, Germany) and were identified on the basis of their typical morphology and urease, oxidase and catalase test result.
DNA extraction and genotyping of cagA, vacA and babA2
Genomic DNA was extracted by using CTAB method with phenol/chloroform and isopropanol precipitation as described elsewhere [59]. Purified DNAs were stored at −20 ℃ until use. All the PCR reactions were carried out in a 25 µl reaction volume containing genomic DNA (50 ng), forward and reverse primers (25 pmol), each deoxynucleoside triphosphate (0.25 mM each, Roche, Berlin, Germany), Taq DNA polymerase (1 U, Genei, Bangalore, India), and Mgcl2 (1.5 mM) in a standard PCR buffer (Genei, Bangalore, India) for 35 cycles, generally under the following reaction condition: 95 ℃ for 1 min, 55 ℃ for 1 min, and 72 ℃ for a time chosen based on the size of the expected amplified fragment (1 min kb−1) in a Master Cycler apparatus (Eppendorf, Hamburg, Germany). The primers used in this study are listed in Table 4.
Table 4.
Primers used in this study for PCR amplification
| Primer | Sequence (5′–3′) | Amplicon (bp) | Reference |
|---|---|---|---|
| nthu_babA2F babA2R babA2F babA2R607 UreBF UreBR CagA5cF CagA3cR vacAsF vacAsR vacAmF vacAmR |
AATCGAAAAAGGAGAAAACATGAAAAA TGTTAGTGATTTCGGTGTAGGACA AATCCAAAAAGGAGAAAAAGTATGAAA GTTTTCTTTGAGCGCGGGTAAGC CGTCCGGCAATAGCTGCCATAGT GTAGGTCCTGCTACTGAAGCCTTA GTTGATAACGCTGTCGCTTCA GGGTTGTATGATATTTTCCATAA ATGGAAATACAACAAACACAC CTGCTTGAATGCGCCAAAC CAATCTGTCCAATCAAGCGAG GCGTCAAAATAATTCCAAGG |
832 607 480 350 s1-259/s2-286 m1-567/m2-642 |
[26] [20] [20] [61] [5] [5] [62] [62] [63] [63] [63] [63] |
Generation of babA2 mutant H. pylori
A 832 bp fragment of babA2 was cloned into TOPO TA cloning vector (Invitrogen, USA) to generate the plasmid pcr2.1: babA2. The isolated plasmid (pCR2.1: babA2) DNA was digested with restriction enzyme BsmI (New England Biolabs, USA) and ligated with chloramphenicol cassette amplified from DR2 plasmid by phusion enzyme (Thermo-scientific) to produce plasmid pcr2.1: babA2: cmp. This construct was then used to electroporate babA2 positive H. pylori cells. Transformed single colonies were sub cultured and screened for the interruption of babA2 gene by PCR with babA2F/babA2R set of primers. Transformed colonies (positive colonies) gave about 1.6 kb amplicon rather than 832 bp.
IL-8 assay
All the bacterial strains were cultured in 7 % serum containing BHIA plates for 24 h at 37 °C under microaerophilic conditions. In order to obtain ex vivo IL-8 secretion from gastric epithelial cells, AGS (human gastric adenocarcinoma cell line) cells were plated (2.5 × 105 cells mL−1) into 24 well plates and cultured for 24 h. H. pylori [multiplicity of infection (MOI) of 100] were added to cultured cells. After 8 h of infection, IL-8 levels in the supernatant were assayed in duplicate three times using a commercially available specific ELISA kit (Genetix, New Delhi, India) following the manufacturer’s protocols.
Cell cycle analysis
AGS cells (1 × 106 cells mL−1 in each well) were infected with 1 day old H. pylori culture. After 24 h of infection cells were fixed in 70 % chilled ethanol and were kept at 4 ℃ for further analysis. Prior to analysis cells were washed in 2 % fetal bovine serum (FBS) containing PBS (pH 7.4) and the cell pellets were stained with propidium iodide (50 µg mL−1) containing DNase-free RNase (0.1 mg mL−1). Cells were then acquired on flow cytometer and the data was analyzed in FACS Diva software (Becton–Dickinson, USA).
Adherence assay
AGS cells were cultured to about 70–80 % confluency in complete RPMI-1640 medium. AGS cells were washed three times with sterile PBS, and RPMI with 10 % FBS was added. 24 h old H. pylori culture was washed with sterile PBS and suspended in incomplete RPMI. Adherence assay was performed by adding the bacterial cells to the AGS monolayer (MOI 100) and incubated for 2 h [60]. After 2 h of infection unadhered bacterial cells were removed and washed three times with sterile PBS. The adhered bacteria were collected in PBS by scrapping the cell-line along with the infected bacteria. The CFU was determined by plating various serial dilutions of these bacterial suspensions on BHI agar plates. All experiments were repeated at least thrice for each strain.
Statistical analysis
Each experiment was performed at least thrice in duplicates and results expressed as mean ± standard error of the mean (SEM). Statistical analysis was done by T test and ANOVA (wherever applicable), using Graph Pad Prism software (version 5, Graph Pad Software Inc, La Jolla, CA, USA), P values <0.05 were considered to be significant.
Authors’ contributions
AKM and PG designed the experiment. PG, AS, MG, R, JA and RD performed experiment. PG, AS, R and AKM wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
PG and AS acknowledges Indian Council of Medical Research (ICMR) for providing a Senior Research Fellowship and ICMR centenary postdoctoral fellowship respectively. This work was supported in part by the Council of Scientific and Industrial Research (CSIR) [Ref. No. 37(1640)/14/EMR-II]; Department of Biotechnology, Government of India (No.: BT/240/NE/TBP/2011) and ICMR. Funders have no role in study design, data collection, analysis, interpretation and publication.
Compliance with ethical standards
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Prachetash Ghosh, Email: prache.bio@gmail.com.
Avijit Sarkar, Email: vijit2me@gmail.com.
Mou Ganguly, Email: moumukhopadhyay6@gmail.com.
Raghwan, Email: raghwankmr@gmail.com.
Jawed Alam, Email: jawedalam81@yahoo.com.
Ronita De, Email: dronita@gmail.com.
Asish K. Mukhopadhyay, Phone: +91 9830468362, Email: asish_mukhopadhyay@yahoo.com
References
- 1.Brown LM. Helicobacter pylori: epidemiology and routes of transmission. Epidemiol Rev. 2002;22:283–297. doi: 10.1093/oxfordjournals.epirev.a018040. [DOI] [PubMed] [Google Scholar]
- 2.Covacci A, Telford JL, Giudice GD, Parsonnet J, Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284:1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 3.Graham DY, Adam E, Reddy GT, Agarwal JP, Agarwal R, Evans DJ, Malaty HM, Evans DG. Seroepidemiology of Helicobacter pylori infection in India. Comparison of developing and developed countries. Dig Dis Sci. 1991;36:1084–1088. doi: 10.1007/BF01297451. [DOI] [PubMed] [Google Scholar]
- 4.Singh V, Trikha B, Nain CK, Singh K, Vaiphei K. Epidemiology of Helicobacter pylori and peptic ulcer in India. J Gastroenterol Hepatol. 2002;17:659–665. doi: 10.1046/j.1440-1746.2002.02746.x. [DOI] [PubMed] [Google Scholar]
- 5.Alam J, Maiti S, Ghosh P, De R, Chowdhury A, Das S, Macaden R, Devarbhavi H, Ramamurthy T, Mukhopadhyay AK. Significant association of the dupA gene of Helicobacter pylori with duodenal ulcer development in a South-east Indian population. J Med Microbiol. 2012;61:1295–1302. doi: 10.1099/jmm.0.038398-0. [DOI] [PubMed] [Google Scholar]
- 6.Suerbaum S, Michetti P. Helicobacter pylori infection. N Eng J Med. 2002;347:1157–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 7.Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. Cag pathogenicity island of Helicobacter pylori, encodes type I-specific and diseases associated virulence factors. Proc Natl Acad Sci. 1996;93:14648–14653. doi: 10.1073/pnas.93.25.14648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broutet N, Marais A, Lamouliatte H, de Mascarel A, Samoyeau R, Salamon R, Megraud F. cagA status and eradication treatment outcome of anti-Helicobacter pylori triple therapies in patients with nonulcer dyspepsia. J Clin Microbiol. 2001;39:1319–1322. doi: 10.1128/JCM.39.4.1319-1322.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rahman M, Mukhopadhyay AK, Nahar S, et al. DNA-level characterization of Helicobacter pylori strains from patients with overt disease and with benign infections in Bangladesh. J Clin Microbiol. 2003;41:2008–2014. doi: 10.1128/JCM.41.5.2008-2014.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De R, Kundu P, Swarnakar S, Ramamurthy T, Chowdhury A, Nair GB, Mukhopadhyay AK. Antimicrobial activity of curcumin against Helicobacter pylori isolates from India and during infections in mice. Antimicrob Agents Chemother. 2009;53:1592–1597. doi: 10.1128/AAC.01242-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chattopadhyay S, Patra R, Chatterjee R, De R, Alam J, Ramamurthy T, Chowdhury A, Nair GB, Berg DE, Mukhopadhyay AK. Distinct repeat motifs at the C-terminal region of CagA of Helicobacter pylori strains isolated from diseased patients and asymptomatic individuals in West Bengal, India. Gut Pathog. 2012;4:4. doi: 10.1186/1757-4749-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montecucco C, Papini E, de Bernard M, Telford JL, Rappuoli R. Helicobacter pylori vacuolating cytotoxin and associated pathogenic factors. In: Alouf JE, Freer JH, editors. The comparative source book of bacterial protein toxins. San Diego: Academic Press; 1999. pp. 264–283. [Google Scholar]
- 13.Chattopadhyay S, Datta S, Chowdhury A, Chowdhury S, Mukhopadhyay AK, Rajendran K, Bhattacharya SK, Berg DE, Nair GB. Virulence genes in Helicobacter pylori strains from West Bengal residents with overt H. pylori-associated disease and healthy volunteers. J Clin Microbiol. 2002;40:2622–2625. doi: 10.1128/JCM.40.7.2622-2625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheu BS, Yang HB, Yeh YC, Wu JJ. Helicobacter pylori colonization of the human gastric epithelium: a bug’s first step is a novel target for us. J Gastroenterol Hepatol. 2010;25:26–32. doi: 10.1111/j.1440-1746.2009.06141.x. [DOI] [PubMed] [Google Scholar]
- 15.Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, Berg DE, Covacci A, Engstrand L, Boren T. Helicobacter pylori adhesin binding fucosylated histo-blood group antigens revealed by retagging. Science. 1998;279:373–377. doi: 10.1126/science.279.5349.373. [DOI] [PubMed] [Google Scholar]
- 16.Yamaoka Y. Roles of Helicobacter pylori BabA in gastroduodenal pathogenesis. World J Gastroenterol. 2008;14:4265–4272. doi: 10.3748/wjg.14.4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rad R, Gerhard M, Lang R, Schöniger M, Rösch T, Schepp W, Becker I, Wagner H, Prinz C. The Helicobacter pylori blood group antigen-binding adhesin facilitates bacterial colonization and augments a nonspecific immune response. J Immunol. 2002;168:3033–3041. doi: 10.4049/jimmunol.168.6.3033. [DOI] [PubMed] [Google Scholar]
- 18.Boren T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 19.Megraud F, Trimoulet pascale, Lamouliatte H, Boyanova L. Bactericidal effect of amoxicillin on Helicobacter pylori in an in vitro model using epithelial cells. Antimicrob Agents Chemother. 1991;35:869–72. [DOI] [PMC free article] [PubMed]
- 20.Gerhard M, Lehn N, Neumayer N, Boren T, Rad R, Schepp W, Miehlke S, Classen M, Prinz C. Clinical relevance of the Helicobacter pylori gene for blood-group antigen-binding adhesion. Proc Natl Acad Sci. 1999;96:12778–12783. doi: 10.1073/pnas.96.22.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oliveira AG, Santos A, Guerra JB, et al. babA2-and cagA-positive Helicobacter pylori strains are associated with duodenal ulcer and gastric carcinoma in Brazil. J Clin Microbiol. 2003;41:3964–3966. doi: 10.1128/JCM.41.8.3964-3966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattar R, dos Santos AF, Eisig JN, Rodrigues TN, Silva FM, Lupinacci RM, Iriya K, Carrilho FJ. No correlation of babA2 with vacA and cagA genotypes of Helicobacter pylori and grading of gastritis from peptic ulcer disease patients in Brazil. Helicobacter. 2005;10:601–608. doi: 10.1111/j.1523-5378.2005.00360.x. [DOI] [PubMed] [Google Scholar]
- 23.Erzin Y, Koksal V, Altun S, Dobrucali A, Aslan M, Erdamar S, Dirican A, Kocazeybek B. Prevalence of Helicobacter pylorivacA, cagA, cagE, iceA, babA2 genotypes and correlation with clinical outcome in Turkish patients with dyspepsia. Helicobacter. 2006;11:574–580. doi: 10.1111/j.1523-5378.2006.00461.x. [DOI] [PubMed] [Google Scholar]
- 24.Mottaghi B, Safaralizadeh R, Bonyadi M, Latifi-Navid S, Somi MH (2014) Helicobacter pylori vacAi region polymorphism but not babA2 status associated to gastric cancer risk in northwestern Iran. Clin Exp Med (ahead of print). [DOI] [PubMed]
- 25.Kim SY, Woo CW, Lee YM, Son BR, Kim JW, Chae HB, Youn SJ, Park SM. Genotyping CagA, VacA subtype, IceA1, and BabA of Helicobacter pylori isolates from Korean patients, and their association with gastroduodenal diseases. J Korean Med Sci. 2001;16:579–584. doi: 10.3346/jkms.2001.16.5.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lai CH, Kuo CH, Chen YC, Chao FY, Poon SK, Chang CS, Wang WC. High prevalence of cagA and babA2 positive Helicobacter pylori clinical isolates in Taiwan. J Clin Microbiol. 2002;40:3860–3862. doi: 10.1128/JCM.40.10.3860-3862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Torres L, Melian K, Moreno A, Alonso J, Sabatier C, Hernandez M, Bermudez L, Rodriguez B. Prevalence of vacA, cagA and babA2 genes in Cuban Helicobacter pylori isolates. World J Gastroenterol. 2009;15:204–210. doi: 10.3748/wjg.15.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. Variants of the 3′ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. J Clin Microbiol. 1998;36:2258–2263. doi: 10.1128/jcm.36.8.2258-2263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blaser MJ, Perez-Perez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 1995;55:2111–2115. [PubMed] [Google Scholar]
- 30.Queiroz DM, Mendes EN, Carvalho AST, Rocha GA, Oliveira AMR, Soares TF, Santos A, Cabral MMDA, Nogueira AMMF. Factors associated with Helicobacter pylori infection by a cagA-positive strain in children. J Infect Dis. 2000;181:626–630. doi: 10.1086/315262. [DOI] [PubMed] [Google Scholar]
- 31.Yamaoka Y, Kato M, Asaka M. Geographic differences in gastric cancer incidence can be explained by differences between Helicobacter pylori strains. Intern Med. 2008;47:1077–1083. doi: 10.2169/internalmedicine.47.0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saxena A, Shukla S, Prasad KN, Ghoshal UC. Virulence attributes of Helicobacter pylori isolates & their association with gastroduodenal disease. Indian J Med Res. 2011;133:514–520. [PMC free article] [PubMed] [Google Scholar]
- 33.Chen MY, He CY, Meng X, Yuan Y. Association of Helicobacter pylori babA2 with peptic ulcer disease and gastric cancer. World J Gastroenterol. 2013;19:4242–4251. doi: 10.3748/wjg.v19.i26.4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Olfat FO, Zheng Q, Oleastro M, Voland P, Borén T, Karttunen R, Engstrand L, Rad R, Prinz C, Gerhard M. Correlation of the Helicobacter pylori adherence factor BabA with duodenal ulcer disease in four European countries. FEMS Immunol Med Microbiol. 2005;44:151–156. doi: 10.1016/j.femsim.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 35.Gatti LL, Modena JL, Payao SL, Smith Mde A, Fukuhara Y, Módena JL, de Oliveira RB, Brocchi M. Prevalence of Helicobacter pylori cagA, iceA and babA2 alleles in Brazilian patients with upper gastrointestinal diseases. Acta Trop. 2006;100:232–240. doi: 10.1016/j.actatropica.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 36.Chomvarin C, Namwat W, Chaicumpar K, Mairianq P, Sangchan A, Sripa B, Tor-Udam S, Vilaichone RK. Prevalence of Helicobacter pylori vacA, cagA, cagE, iceA and babA2 genotypes in Thai dyspeptic patients. Int J Infect Dis. 2008;12:30–36. doi: 10.1016/j.ijid.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 37.Erzin Y, Koksal V, Altun S, Dobrucali A, Aslan M, Erdamar S, Goksel S, Dirican A, Kocazeybek B. Role of host interleukin 1beta gene (IL-1B) and interleukin 1 receptor antagonist gene (IL-1RN) polymorphisms in clinical outcomes in Helicobacter pylori-positive Turkish patients with dyspepsia. J Gastroenterol. 2008;43:705–710. doi: 10.1007/s00535-008-2220-7. [DOI] [PubMed] [Google Scholar]
- 38.Azevedo M, Eriksson S, Mendes N, Serpa J, Resende LP, Ruvoen-Clouet N, Haas R, Boren T, Le Pendu J, David L. Infection by Helicobacter pylori expressing the BabA adhesin is influenced by the secretor phenotype. J Pathol. 2008;215:308–316. doi: 10.1002/path.2363. [DOI] [PubMed] [Google Scholar]
- 39.Pride DT, Meinersmann RJ, Blaser MJ. Allelic variation within Helicobacter pylori babA and babB. Infect Immun. 2001;69:1160–11671. doi: 10.1128/IAI.69.2.1160-1171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crabtree JE, Wyatt JI, Trejdosiewicz LK, Peichl P, Nichols PH, Ramsay N, Primrose JN, Lindley IJ. Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol. 1994;47:61–66. doi: 10.1136/jcp.47.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moss SF, Legon S, Davies J, Calam J. Cytokine gene expression in Helicobacter pylori associated antral gastritis. Gut. 1994;35:1567–1570. doi: 10.1136/gut.35.11.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull. 1998;54:139–150. doi: 10.1093/oxfordjournals.bmb.a011664. [DOI] [PubMed] [Google Scholar]
- 43.Yamaoka Y, Kwon DH, Graham DY. A M(r) 34,000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci. 2000;97:7533–7538. doi: 10.1073/pnas.130079797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gionchetti P, Vaira D, Campieri M, Holton J, Menegatti M, Belluzzi A, Bertinelli E, Ferretti M, Brignola C, Miglioli M. Enhanced mucosal interleukin-6 and -8 in Helicobacter pylori-positive dyspeptic patients. Am J Gastroenterol. 1994;89:883–887. [PubMed] [Google Scholar]
- 45.Algood HMS, Cover TL. Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin Microbiol Rev. 2006;19:597–613. doi: 10.1128/CMR.00006-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kusters JG, van Vliet AHM, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zychlinsky A, Sansonetti P. Perspectives series: host/pathogen interactions. Apoptosis in bacterial pathogenesis. J Clin Invest. 1997;100:493–495. doi: 10.1172/JCI119557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wagner S, Beil W, Westermann J, Logan RP, Bock CT, Trautwein C, Bleck JS, Manns MP. Regulation of gastric epithelial cell growth by Helicobacter pylori: offdence for a major role of apoptosis. Gastroenterology. 1997;113:1836–1847. doi: 10.1016/S0016-5085(97)70003-9. [DOI] [PubMed] [Google Scholar]
- 49.Rudi J, Kuck D, Strand S, von Herbay A, Mariani SM, Krammer PH, Galle PR, Stremmel W. Involvement of the CD95 (APO-1/Fas) receptor and ligand system in Helicobacter pylori-induced gastric epithelial apoptosis. J Clin Invest. 1998;102:1506–1514. doi: 10.1172/JCI2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma SA, Tummuru MK, Miller GG, Blaser MJ. Interleukin-8 response of gastric epithelial cell lines to Helicobacter pylori stimulation in vitro. Infect Immun. 1995;63:1681–1687. doi: 10.1128/iai.63.5.1681-1687.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan X, Crowe SE, Behar S, Gunasena H, Ye G, Haeberle H, Van Houten N, Gourley WK, Ernst PB, Reyes VE. The effect of class II major histocompatibility complex expression on adherence of Helicobacter pylori and induction of apoptosis in gastric epithelial cells: a mechanism for T helper cell type 1-mediated damage. J Exp Med. 1998;187:1659–1669. doi: 10.1084/jem.187.10.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akopyanz N, Bukanov NO, Westblom TU, Kresovich S, Berg DE. DNA diversity among clinical isolates of Helicobacter pylori detected by PCR-based RAPD fingerprinting. Nucleic Acid Res. 1992;20:5137–5142. doi: 10.1093/nar/20.19.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Achtman M, Azuma T, Berg DE, Ito Y, Morelli G, Pan ZJ, Suerbaum S, Thompson SA, van der Ende A, van Doorn LJ. Recombination and clonal groupings within Helicobacter pylori from different geographical regions. Mol Microbiol. 1999;32:459–470. doi: 10.1046/j.1365-2958.1999.01382.x. [DOI] [PubMed] [Google Scholar]
- 54.Kersulyte D, Mukhopadhyay AK, Velapatiño B, et al. Differences in genotypes of Helicobacter pylori from different human populations. J Bacteriol. 2000;182:3210–3218. doi: 10.1128/JB.182.11.3210-3218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Basso D, Plebani M. H. pylori infection: bacterial virulence factors and cytokine gene polymorphisms as determinants of infection outcome. Crit Rev Clin Lab Sci. 2004;41:313–337. doi: 10.1080/10408360490472804. [DOI] [PubMed] [Google Scholar]
- 56.Gancz H, Jones KR, Merrell DS. Sodium chloride affects Helicobacter pylori growth and gene expression. J Bacteriol. 2008;190:4100–4105. doi: 10.1128/JB.01728-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Keenan JI, Allardyce RA. Iron influences the expression of Helicobacter pylori outer membrane vesicle-associated virulence factors. Eur J Gastroenterol Hepatol. 2000;12:1267–1273. doi: 10.1097/00042737-200012120-00002. [DOI] [PubMed] [Google Scholar]
- 58.Mukhopadhyay AK, Kersulyte D, Jeong JY, et al. Distinctiveness of genotypes of Helicobacter pylori in Clcutta, India. J Bacteriol. 2000;182:3219–3227. doi: 10.1128/JB.182.11.3219-3227.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K. Current Protocols in Molecular Biology. New York: Greene publishing and Wiley-Interscience; 1993. [Google Scholar]
- 60.Raghwan Chowdhury R. Host cell contact induces fur-dependent expression of virulence factors CagA and VacA in Helicobacter pylori. Helicobacter. 2013;19:17–25. doi: 10.1111/hel.12087. [DOI] [PubMed] [Google Scholar]
- 61.Zambon CF, Navaglia F, Basso D, Rugge M, Plebani M. Helicobacter pyloribabA2, cagA, and s1 vacA genes work synergistically in causing intestinal metaplasia. J Clin Pathol. 2003;56:287–291. doi: 10.1136/jcp.56.4.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chattopadhyay S, Patra R, Ramamurthy T, Chowdhury A, Santra A, Dhali GK, Bhattacharya SK, Berg DE, Nair GB, Mukhopadhyay AK. Multiplex PCR assay for rapid detection and genotyping of Helicobacter pylori directly from biopsy specimens. J Clin Microbiol. 2004;42:2821–2824. doi: 10.1128/JCM.42.6.2821-2824.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Atherton JC, Cao P, Peek RM, Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori. Association of specific vacA types with cytotoxin production and peptic ulceration. J Biol Chem. 1995;270:17771–17777. doi: 10.1074/jbc.270.30.17771. [DOI] [PubMed] [Google Scholar]