

Abstract
Albomycin (ABM), also known as grisein, is a sulfur-containing metabolite produced by Streptomyces griseus ATCC 700974. Genes predicted to be involved in the biosynthesis of ABM and ABM-like molecules are found in the genomes of other actinomycetes. ABM has potent antibacterial activity, and as a result, many attempts have been made to develop ABM into a drug since the last century. Although the productivity of S. griseus can be increased with random mutagenesis methods, understanding of Streptomyces sulfur amino acid (SAA) metabolism, which supplies a precursor for ABM biosynthesis, could lead to improved and stable production. We previously characterized the gene cluster (abm) in the genome-sequenced S. griseus strain and proposed that the sulfur atom of ABM is derived from either cysteine (Cys) or homocysteine (Hcy). The gene product, AbmD, appears to be an important link between primary and secondary sulfur metabolic pathways. Here, we show that propargylglycine or iron supplementation in growth media increased ABM production by significantly changing the relative concentrations of intracellular Cys and Hcy. An SAA metabolic network of S. griseus was constructed. Pathways toward increasing Hcy were shown to positively impact ABM production. The abmD gene and five genes that increased the Hcy/Cys ratio were assembled downstream of hrdBp promoter sequences and integrated into the chromosome for overexpression. The ABM titer of one engineered strain, SCAK3, in a chemically defined medium was consistently improved to levels ∼400% of the wild type. Finally, we analyzed the production and growth of SCAK3 in shake flasks for further process development.
INTRODUCTION
The Streptomyces genus was established at the beginning of the golden age of antibiotic discovery (1). Streptomyces griseus, a representative organism of the genus (2), was shown during this time to produce streptomycin (3), which has been clinically used to treat bacterial infections and other human diseases. It is well known that Streptomyces organisms in general have a large biosynthetic potential, being capable of producing many bioactive secondary metabolites—the S. griseus strains producing albomycin (ABM) are no exception. ABM was named by former Soviet Union scientists and underwent clinical investigations well before the information was released to the English-speaking scientific world (4). The biological activity and chemical constitution of ABM were later reported to be nearly identical to those of grisein, which was isolated from a distinct subtype of S. griseus by U.S. scientists in the 1940s (5–7). Despite its remarkable properties, continued studies of ABM were not pursued, partly because of the poor yields (8) and unpublicized research on the microorganism (9, 10). At present, the wealth of Streptomyces genomic information that is publically available provides the possibility of using a systems biology approach to uncover new aspects of Streptomyces metabolism and potentially overcome the production bottleneck.
ABM and ABM-like secondary metabolites are structurally categorized as peptidyl nucleoside antibiotics (Fig. 1). The N terminus of the peptide is a ferrichrome siderophore moiety in ABM; thus, many human-pathogenic bacteria (e.g., pneumococci and enterics) actively take up ABM through an iron transport system as a mechanism to acquire iron from the environment (11). Once inside the pathogen, a serine-containing nucleosyl dipeptide, termed SB-217452 (12), at the C terminus of ABM is enzymatically released through hydrolysis. SB-217452 has been proposed to mimic seryl-AMP and therefore function as an antibiotic by inhibiting seryl-tRNA synthetase (SerRS), an essential enzyme involved in protein synthesis. The nucleoside component of SB-217452 consists of a highly unusual cyclic thioether group (—C—S—C—) that, along with several other features, including the C-3′ stereochemical configuration and additional base modifications, differentiates it from the canonical nucleosides. It was reported that the sulfur atom of ABM cannot be replaced with an oxygen; a chemically synthesized compound with the oxygen substitution completely lost biological activity (13). Studies aimed at defining ABM biosynthesis, such as the assembly of the empowered siderophore and the intriguing mechanism for the formation of this rare thiosugar-containing amino acid, including the identity of the precursor metabolite(s) in the ABM-producing S. griseus, could help alleviate the bottlenecks to bringing an ABM-inspired antibiotic to the clinic.
FIG 1.

Constructed sulfur amino acid metabolism and the linked metabolic network in Streptomyces griseus. Corresponding to the conventional name shown, the identified S. griseus ORF and its product are as follows: (1) sgr6646, homoserine O-acetyltransferase; (2) sgr6647, O-acetylhomoserine sulfhydrylase; (3) sgr2579, cystathionine-γ-synthase (CGS); (4) sgr3417, type II cystathionine-β-lyase (CBL); (5) sgr5847, B12-dependent methionine synthase; (6) sgr1212, B12-independent methionine synthase; (7) sgr6058, S-adenosylmethionine (SAM) synthetase; (8) sgr4513, S-adenosylhomocysteine (SAH) hydrolase; (9) sgr4452, cystathionine-β-synthase (CBS); and (10) sgr3660, cystathionine-γ-lyase (CGL). The abmD gene product is proposed to be a critical link between sulfur amino acid metabolism and ABM biosynthesis.
Bacterial sulfur amino acid (SAA) metabolism is highly adaptive and diversified (14). Biosynthesis of cysteine (Cys) and methionine (Met) via homocysteine (Hcy) are linked through bidirectional transsulfuration pathways. Each transsulfuration direction involves two sequential reactions that are catalyzed by different enzymes that often have relaxed specificities (Fig. 1). Interconversion between Hcy and Met occurs in the activated methyl cycle (AMC)—the so-called Met salvage pathway—which involves ATP and additional cofactors, such as cobalamin (vitamin B12) or 5-methyl-tetrahydrofolate. The intrinsic function and metabolic branching of AMC have been shown to lead to the production of some bacterial metabolites (e.g., quorum-sensing molecules, etc.) that can have a profound impact on the bacterial physiology and surrounding microbial community. SAA metabolism is also connected with aspartate metabolism via homoserine. However, most Streptomyces spp. and other actinobacteria do not encode a MetA enzyme, which acylates the hydroxyl of homoserine with succinyl coenzyme A (succinyl-CoA). Instead, a direct sulfhydrylation pathway comprising metX and metY is found in some actinomycetes (Fig. 1) (15, 16). Furthermore, Streptomyces coelicolor apparently lacks not only metA but also metXY (17); this aspect of Met synthesis in what is often considered the model Streptomyces organism remains unsolved. Most actinobacterial genomes also do not appear to have a canonical metC that codes for a cystathionine-β-lyase (14); it has been suggested that the malY gene product may perform this function (18). The genetically amenable ABM-producing species S. griseus is proposed as an excellent system for investigation into Streptomyces SAA metabolism and the interplay of primary and specialized metabolism, since the results may have a direct impact on industrial production of the ABM antibiotic.
We initiated research on ABM biosynthesis with one aim: to improve ABM production in a native producer, S. griseus ATCC 700974 (19–21). A draft genome sequence of S. griseus was obtained, and the ABM biosynthetic gene cluster (abm) was identified. Based on previous reports (22, 23), the purification and analytical determination of ABM were established, and a robust genetic system within S. griseus was developed to link the genetic information with ABM production. In collaboration with others, we also developed an in vitro enzyme assay for SerRS to show that SB-217452 cleaved from purified ABM is indeed a potent inhibitor of the enzyme activity. Interestingly, the abm gene cluster lacks an obvious pathway-specific regulatory gene, suggesting that ABM production might be controlled by the availability of a sulfur-containing primary metabolite. The protein encoded by abmD was identified as the best candidate to catalyze the first committed step in the biosynthesis of the nucleoside component of ABM using one of the known sulfur-containing primary metabolites. Here, we present experimental evidence to support this hypothesis, obtained by manipulating the newly identified genes of Streptomyces SAA metabolism, measuring the intracellular Hcy/Cys ratio, and performing fermentation optimization studies. The level of ABM production by S. griseus was increased 4-fold, and cell level analysis indicated that Hcy is directly involved in ABM biosynthesis.
MATERIALS AND METHODS
Bacterial strains, plasmids, reagents, and growth media.
The bacterial strains and plasmids used are listed in Table 1. Primers for PCR amplification are in Table 2. Chemicals were purchased from standard commercial sources. General Streptomyces microbiological procedures were followed (24). The original ABM production broth (APB) consisted of the following (23): 20 g of starch, 5 g of l-ornithine HCl, 1.8 g of KH2PO4, 10.2 g of Na2HPO4, 2 g of (NH4)2SO4, 2 g of NaCl, 2 g of MgSO4·7H2O, 0.8 g of CaCl2·2H2O, 0.28 g of FeSO4·7H2O, and 0.02 g of ZnSO4·7H2O per liter of distilled water. To simplify ABM purification here, APB was modified to contain 20 g of glycerol as the sole carbon source, and ornithine and starch were omitted. Streptomyces spores and mycelia were routinely maintained on mannitol-soya flour (MS) agar plates. Streptomyces seed culture was in tryptic soy broth (TSB). Escherichia coli strains were cultured in Luria-Bertani (LB) broth or agar.
TABLE 1.
Bacterial strains and plasmids used or constructed in this study
| Strain or plasmid | Descriptiona | Source or referenceb |
|---|---|---|
| Strains | ||
| E. coli | ||
| JM109 | Strain used as a host for cloning and bioassay tester organism | ATCC |
| ET12567 | Strain harboring pUZ8002 plasmid; used for conjugating plasmid into Streptomyces | 24 |
| S. griseus | ||
| ATCC 700974 | Wild type | ATCC |
| SCAK1 | WT-derived strain containing pSET and hrdBp-metYXSO at attB | This study |
| SCAK2 | WT-derived strain containing pSET and hrdBp-abmD at attB | This study |
| SCAK3 | WT-derived strain containing pSET and hrdBp-metYXSO-hrdBp-abmD-hrdBp-malY-metB at attB | This study |
| SCAK4 | WT-derived strain containing pSET and hrdBp-ask-asd at attB | This study |
| SCAK5 | WT-derived strain containing pSETand hrdBp-metYXSO-hrdBp-ask-asd at attB | This study |
| S. albulus | ||
| NBRC 14147 | Genome used to amplify the genes for feedback-insensitive Ask and Asd | NBRC |
| Plasmids | ||
| pDrive | Used for subcloning (Kanr Ampr) | Qiagen |
| pSET(GUS) | Used for subcloning (Aprr) | 25 |
| pSE34-oriT | Multicopy plasmid in Streptomyces (Tsr) | 19 |
| pA10 | pSE34 derivative control | 19 |
| pCK1 | pSE34 derivative for ermE*p expression of metYXSO | This study |
| pCK2 | pSE34 derivative for ermE*p expression of malY-metB | This study |
| pCK3 | pSE34 derivative for ermE*p expression of mtcC-mtcB | This study |
| pCK4 | pSE34 derivative for ermE*p expression of sahH | This study |
| pCK5 | pSE34 derivative for ermE*p expression of metK-sahH | This study |
| pCK6 | pSE34 derivative for ermE*p expression of metE | This study |
| pCK7 | pSE34 derivative for ermE*p expression of metK-sahH-metE | This study |
Tsr, thiostrepton resistant; ermE*p, mutated ermE promoter.
ATCC, American Type Culture Collection; NBRC, Japanese NITE Biological Resource Center.
TABLE 2.
DNA primers used for PCR in this study
| Primer | Sequence (5′ to 3′) | Purpose |
|---|---|---|
| DirectS-F-XbaI | GAGGTCTAGAATGAGCCAGCCCCTCGACTCCGTCAC | Cloning metYXSO into pSE34 |
| DirectS-R-HindIII | AAACCAAGCTTTTCGGTCCCGTCCCGGGTGCCGCC | |
| CGL-F-XbaI | AAAAAATCTAGATGAGCACCATGGGCGACGGAACACG | Cloning mtcC into pSE34 |
| CGL-R-RBS-NheI | AAAAAAGCTAGCTCCTATCTACCGGACCGCCGCGTCCAGCGCCTG | |
| CBS-F-XbaI | AAAAAATCTAGAGTGCAATTCCACGATTCGATGATCAG | Cloning mtcB into pSE34 |
| CBS-R-HindIII | AAAAAAAAGCTTTCAGGCCTTGGCCGTGCCCGCGTCCT | |
| CBL-F-XbaI | AAAAAATCTAGAGTGGGCGCGCCCTACGACTTCGATAC | Cloning malY into pSE34 |
| CBL-R-RBS-NheI | AAAAAAGCTAGCTCCTATTCAATCGCCGGAGGGGCTTGTTCTCAG | |
| CGS-F-XbaI | AAAAAATCTAGAGTGGCCGGTAAGGGCCCTTGCGACG | Cloning metB into pSE34 |
| CGS-R-HindIII | AAAAAAAAGCTTTCAGCCCAGCGCCTGGGTGAGGTCG | |
| metK-F-XbaI | AGGTCTAGAGTGTCCCGCCGCCTTTTCACCTCGG | Cloning metK into pSE34 |
| metK-R-HindIII | CCTCCGAAGCTTTGTCCTGATGCTTGTACGGCCCTTACAGACC | |
| metE-F-XbaI | AAAAAATCTAGAGTGACAGCGAAGCCCGCAGCC | Cloning metE into pSE34 |
| metE-R-HindIII | AAAAAAAAGCTTCGGATCACGCCGCTTCGGTGGGC | |
| sahH-F-XbaI | CAGTCTAGAATGACGACGACGACCAACCGT | Cloning sahH into pSE34 |
| sahH-R-RBS-NheI | CTTCCGGCTAGCGGTCCTGGATCAGTAGCGGTAGTGGTC | |
| hrdBp-F-BamHI | AAAAAAGGATCCCATGCGTCACCTGCTCGTCGTCCATC | Cloning hrdBp-linked constructs into pSET |
| hrdBp-F-EcoRI | AAAAAAGAATTCCATGCGTCACCTGCTCGTCGTCCATC | Cloning hrdBp promoter into pSE34 |
| hrdBp-R-XbaI | AAAAATCTAGAACCTCTCGGAACGATGGAAACGGC | Cloning hrdBp promoter into pSE34 |
| DirectSinbet-F-BamHI | AAAAAAGGATCCGCCCGGTCGTCGCCGCTGGGC | mutating the BamHI within metYXSO |
| DirectSinbet-R-BglII | AAAAAAAGATCTCCCTCCCGTCGGCCACCGGAGGCCGGC | mutation and for PCR verification |
| DirectS-R-BglII | AAAAAAAGATCTTTCAGGTCACCGTCACCGGGTGCCGCAC | Cloning metYXO into pSET |
| abmD-F-XbaI | AAAAAATCTAGAATGACGGTCCTTCCCCT | Cloning abmD into pSE34 |
| abmD-R-HindIII | AAAAAAAAGCTTGTCTCCTCTCGGGTAC | Cloning abmD into pSE34 |
| abmD-R-NheI | AAAAAAGCTAGCGACGGGCGGTGGGC | Cloning abmD into pSET |
| ask-asd-F-XbaI | AAAAAATCTAGAGTGGGCCTTGTCGTGCAGAAGTACGGC | Cloning ask-asd into pSE34 |
| asK-D-Rm | CCGCGCGGACGGATGCGCGCCGCGCACCCGTTGACCTC | BamHI mutation primer |
| asK-D-Fm | GCGCGGCGCGCATCCGTCCGCGCGGCATCATCGCCAAC | BamHI mutation primer |
| ask-asd-R-RBS-HindIII | AAAAAAAAGCTTTCCTATCCGGAGAATCAGCCGCCCCCTAAGAGC | Cloning ask-asd from into pSE34 |
| hrdBp-F-HindIII | AAAAAAAAGCTTCCTCGTCGTCCATC | Cloning hrdBp-ask-asd from pSCAK2 to pSET |
| ask-asd-R-RBS-NheI | AAAAAAGCTAGCTCCTATCCGGAGAATCAGCCGCCCCCTAAGAGC |
Standard DNA manipulation and Streptomyces genetic techniques.
Plasmids and DNA fragments were purified according to the provided protocol (Qiagen). Sequence analysis for cloning was performed with CLC Genomics Workbench 4.0. All restriction enzymes, DNA ligase, and other molecular biology reagents were from New England BioLabs (NEB) and used by following the provided instructions. PCR was performed with Q5 high-fidelity DNA polymerase from NEB. The annealing temperature was calculated based on the supplier's information (Integrated DNA Technologies). DNA was introduced into S. griseus ATCC 700974 by conjugation using E. coli ET12567/pUZ8002. Briefly, while E. coli in 10 ml of LB was grown to an optical density at 600 nm (OD600) of ∼0.4, 10 μl of S. griseus spores in 0.5 ml of 2× yeast extract-tryptone (YT) medium was subjected to heat shock for 10 min at 50°C and then allowed to cool to room temperature. After a washing with fresh LB, 0.5-ml quantities each of E. coli and Streptomyces were mixed, centrifuged at high speed, and resuspended in 50 μl of 2× YT. The resuspension was diluted with sterile water from 10−1 to 10−4; 100 μl of each dilution was plated onto an MS plate. After 16 to 20 h, the plates were overlaid with 25 μg/ml of nalidixic acid and 12 μg/ml of thiostrepton for replicating plasmids like pSE34 or with 50 μg/ml of apramycin for integration mutants. After an additional 5 days, individual colonies were picked for bioassay and PCR verification. Spores from at least three genetically confirmed transformants were pooled to create a strain for biochemical analysis.
Construction of pCK for plasmid-based gene overexpression.
The pCK plasmids were constructed from pSE34-oriT, which has a mutated ermE promoter (ermE*p) to drive the expression of genes cloned between XbaI and HindIII. Because XbaI and NheI cuts generate compatible cohesive ends and a religated sequence will no longer be recognized by the two enzymes, PCR fragments restricted with XbaI and NheI at the 5′ and 3′ ends, respectively, can be sequentially inserted into the XbaI site in the pSE34-derived vector. Genes of interest were amplified using S. griseus ATCC 700974 genomic DNA and a pair of primers, the names of which indicate the target, direction, and a built-in restriction site for cloning (Table 2). In some cases, the ribosomal binding site (RBS) of a target gene was included within the 3′ end of a fragment following the restriction site. Control plasmid pA10 is the pSE34-oriT vector containing abmK (19). The direct sulfhydrylation genes metYXSO were amplified as a single 4-kb DNA fragment and cloned into pSE34-oriT to obtain pCK1. To assemble the 2.4-kb malY-metB DNA fragment, PCR products were first assembled in pDrive (Qiagen) and then moved into pSE34-oriT to yield pCK2. To assemble the 2.6-kb mtcC-mtcB DNA fragment, PCR products were directly cloned into pSE34-oriT to yield pCK3. Cloning of the PCR-amplified 1.4-kb sahH and 2.3-kb metE DNA fragments into pSE34-oriT yielded the pCK4 and pCK6 plasmids. The 1.2-kb metK DNA fragment was first assembled with sahH as a two-gene operon in pDrive; then the two-gene fragment was cloned into pSE34-oriT to obtain pCK5. A metE fragment was also used to construct an ∼3.9-kb XbaI-HindIII sahH-metE DNA fragment, which was then cloned downstream of the XbaI-NheI metK DNA fragment to obtain a 5.1-kb XbaI-HindIII metK-sahH-metE DNA fragment in pDrive. The three-gene assembly was cloned into pSE34-oriT to obtain pCK7.
Construction of SCAK strains for integrative gene overexpression.
All pSCAK plasmids have a pSETGUS (25) vector backbone, and DNA fragments were cloned using either BamHI or XbaI or both sites. The hrdBp promoter was amplified from the genome in the form of an ∼0.3-kb EcoRI-XbaI DNA fragment to replace the ermE*p sequence in the pCK plasmids mentioned above. Since the ∼4.3-kb hrdBp-metYXSO DNA fragment of the pCK1 derivative has a BamHI located in the intergenic region of metY-metX, a 1.4-kb XbaI-BglII DNA fragment from hrdBp to the BamHI site was amplified using primers DirectS-F-XbaI and DirectSinbet-R-BglII and used to replace the same-size XbaI-BamHI fragment in the pCK1 derivative. The original BamHI was thus mutated due to the religatable sticky ends from BamHI and BglII restriction. Using this plasmid as a template and primers hrdBp-F-BamHI and DirectS-R-BglII, the 4.3-kb BamHI-BglII DNA fragment was amplified and cloned into pSET to obtain pSCAK1. Gene abmD was amplified as an ∼1-kb XbaI-HindIII DNA fragment with primers abmD-F-XbaI and abmD-R-HindIII and cloned into the hrdBp-containing pSE34 vector. Using this plasmid as the template, a 1.3-kb BamHI-NheI hrdBp-abmD DNA fragment was amplified with primers hrdBp-F-BamHI and abmD-R-NheI and cloned into pSET to obtain pSCAK2. The pDrive vector was used to first assemble a fragment with genes from multiple pathways. A 4.3-kb BamHI-BglII hrdBp-metYXO fragment and a 1.3-kb EcoRI-HindIII PhrdB-abmD fragment were amplified from pSCAK1 and pSCAK2, respectively, using primer pairs hrdBp-F-BamHI/DirectS-R-BglII and hrdBp-F-EcoRI/abmD-R-HindIII. A 2.7-kb HindIII-NheI hrdBp-malY-metB DNA fragment was amplified from a pCK2 derivative. These fragments were sequentially cloned into the pDrive vector to generate a six-gene construct. The ∼8.3-kb DNA insert was recovered as a BamHI-NheI fragment and cloned into pSET to obtain pSCAK3. The ask-asd operon has an internal BamHI site at position 416 of asd. Site-directed mutagenesis was performed to make a G416C mutation of the BamHI while switching to a synonymous codon. It involved overlapping of two PCR fragments, 1.68-kb and 0.753-kb DNA, amplified from the left and right sides of the mutation site, using primers ask-asd-F-XbaI/asK-D-Rm and asK-D-Fm/ask-asd-R-RBS-HindIII, respectively. The fragments were then mixed and used as a template for another round of PCR using the two inward primers. The amplified 2.4-kb XbaI-HindIII DNA fragment was coligated with the 0.3-kb EcoRI-XbaI hrdBp DNA fragment into pSE34. The resulting plasmid was used to amplify a 2.7-kb BamHI-NheI hrdBp-ask-asd DNA fragment using primers hrdBp-F-BamHI and ask-asd-R-RBS-NheI, which was then cloned into pSET to obtain pSCAK4. A 2.7-kb HindIII-NheI hrdBp-ask-asd DNA fragment was amplified from pSCAK2 using hrdBp-F-HindIII and ask-asd-R-RBS-NheI. This fragment and the 4.3-kb BamHI-BglII hrdBp-metYXOS DNA fragment from pSCAK4 were sequentially cloned into pDrive to generate a five-gene construct. The BamHI-NheI 7.1-kb DNA insert was recovered from the pDrive plasmid and cloned into pSET to obtain pSCAK5. The pSCAK plasmids were introduced into S. griseus ATCC 700974 by conjugation. Chromosomally integrated SCAK mutants, selected by apramycin resistance and verified by PCR with purified genomic DNA, were confirmed to be stable through several generations and minimally 7 months.
Growth of the wild type in PPGL-supplemented or iron-deficient medium and CDW.
S. griseus ATCC 700974 spores were grown in tryptic soy broth (Bacto) for 2 days. Two milliliters of the seed culture was used to inoculate 50 ml of APB in a 300-ml flask with extradeep baffles. The flasks were shaken at 250 rpm and 28°C for 4 days and sampled at the desired time point. Propargylglycine (PPGL; Sigma) was added to a final concentration of 9 mM at the time of inoculation. Iron-deficient medium was prepared by excluding ferrous sulfate from the production medium. The cell mass of the bacterial culture was measured by a modified method (26). A bacterial culture (50 ml) was centrifuged to sediment the cells. The cell pellets were resuspended in 3 ml of water, transferred onto a preweighed glass dish, and then dried in a chamber at 80°C overnight. The weight difference was reported as the cell dry weight (CDW).
Bioassay for determination of ABM production and titer.
Agar diffusion bioassay is a standard method for detecting microbial antagonism. Since it was previously shown that ABM from S. griseus ATCC 700974 in the defined medium APB is the sole agent responsible for the specific bioactivity (19, 23), a bioassay was used to detect the production during fermentation. An E. coli JM109 culture grown overnight was diluted to an OD600 of ∼0.1. A 100-μl quantity of the diluted culture was pipetted onto a Mueller-Hinton (MH) agar plate and spread evenly over the surface of the agar with a sterile cotton swab. Wells were made in MH agar using the back end of a 20- to 200-μl tip, to which Streptomyces culture (20 μl) was added. The plate was incubated at 37°C, and a zone of inhibition which formed in the E. coli lawn was read at 18 h. A bioassay plate was loaded with serially diluted cultures or solutions for comparing and estimating ABM concentrations (7, 23). A standard curve was generated with freshly purified ABM, showing that the assay limit is about 2 ng in 20 μl of solution, which is 100 times more sensitive than high-performance liquid chromatography (HPLC) quantitation. A linear regression between the logarithm of the ABM concentration and the diameter of the zone was identical to that reported previously (23) (see Fig. S1 in the supplemental material).
HPLC-fluorescence detection of intracellular thiol amino acids.
The major intracellular thiols were extracted by a modification of a previously reported method (27). Cells from a 1-ml culture were pelleted by centrifugation and then quickly washed twice with 1 ml of 2.63% NaCl solution. HPLC-grade methanol prechilled at −20°C (1 ml of 60% solution) was added to the tube. The pellet was resuspended, frozen in liquid nitrogen, and stored at −80°C until extraction. A 20-μl quantity of 2 mM monobromobimane (Life Technologies), dissolved in acetonitrile (ACN), was added to the suspension after thawing of the cells. The mixture was vortexed for 1 min and then immediately frozen in liquid nitrogen for 5 min. This sequence of steps was repeated three times. The tube was centrifuged at 5,000 × g for 10 min at 4°C, and the supernatant was transferred to a new tube and mixed with an equivalent amount of chloroform. After centrifugation for 10 min at 13,000 × g, the aqueous phase was recovered and 10 μl was analyzed by HPLC. As a negative control, 20 μl of 2 mM N-ethylmaleimide (NEM) in ethanol was added to the tube during cell lysis. The data were collected with cultures grown for 4 days. HPLC was performed using a Shimadzu Prominence system that included a photodiode array detector (SPD-M20A) and a fluorescence detector (RF-20A). The HPLC system was equipped with an Alltech ALLSPHERE ODS-2 5μ 250- by 4.6-mm column, and the flow rate was maintained at 1.5 ml/min. The gradient program from solvent A (H2O) to solvent B (ACN) was 0 to 40% B in 10 min, 40% to 80% solvent B in 5 min, 80% to 100% solvent B in 2 min, 100% to 0% solvent A in 2 min, and 0% solvent A for 4 min.
ABM isolation and HPLC analysis of ABM production.
The procedures were modified from previous methods (19, 23). Cleared culture (50 ml) was mixed with 5 to 10 g of XAD-4 resin (Sigma). The resin was first washed with 100 ml of water and then washed with 100 ml of 15% methanol. ABM was eluted with 100 ml of 50% methanol, and the methanol was removed with a rotary evaporator. The extract was lyophilized to dryness and reconstituted in 1 ml of water. The extract (100 μl) was used for HPLC analysis in two steps. The first HPLC separation used a Phenomenex Sphereclone C18 ODS-2 10μ column (250 by 4.6 mm) with a flow rate of 1.5 ml/min. The gradient program from solvent A (H2O in 0.01% formic acid) to solvent B (ACN in 0.01% formic acid) was 0 to 40% solvent B in 15 min, 40% to 95% solvent B in 2 min, 95% solvent B for 3 min, 95% to 0% solvent B in 2 min, and 0% solvent B for 3 min. A Shimadzu FRC-10A fraction collector was used to collect 0.5-ml fractions, of which 20 μl was used in each bioassay. The active fractions (∼9.3 min) were combined, lyophilized, and reconstituted in 100 μl of water. ABM was further purified using HPLC with a Phenomenex Kinetex XB-C-18 2.6μ column (75 by 4.6 mm). The solvents were the same as described above, and the flow rate was maintained at 1 ml/min. The gradient was from 0 to 40% solvent B in 15 min, 40% to 95% solvent B in 2 min, 95% solvent B for 3 min, 95% to 0% solvent B in 3 min, and 0% solvent B for 7 min. ABM was eluted at ∼6.5 min and verified by high-resolution mass spectrometry, UV-visible (UV-VIS) absorption spectrum, and the corresponding bioactivity. A standard curve of HPLC peak area versus the amount of pure ABM analyzed was prepared (see Fig. S2 in the supplemental material).
RESULTS AND DISCUSSION
Intracellular flux of free-thiol amino acids in relation to ABM production.
Gene abmD was predicted to encode a pyridoxal phosphate (PLP)-dependent enzyme that is similar to ACCD (1-aminocyclopropane-1-carboxylic acid) deaminase (E value, 2e−55) (19) through sequence alignment or cystathionine-β-synthase (CBS; E value, 4e−67) through structural predictions. It also has weak homology to d-cysteine desulfhydrases. AbmD produced and purified from E. coli showed the expected absorbance maximum at ∼410 nm at pH 8.0 (see Fig. S3 in the supplemental material), which is indicative of a PLP-containing enzyme. AbmD is the only ABM biosynthetic enzyme that displayed absorption, suggesting that AbmD catalyzes a reaction that can be a key link between primary and secondary metabolism by utilizing one of the two free-thiol amino acids to perform the first committed step of the pathway. While functional investigation of AbmD is ongoing, we tested whether ABM biosynthesis would be controlled by the abmD gene product in a productive manner (28).
We examined the intracellular flux of Hcy and Cys in relation to ABM production (Fig. 2). An HPLC-fluorescence method was used to monitor the relative concentrations of Hcy and Cys since the two metabolites were the only major free-thiol amino acids detected in the cells under various fermentation conditions. It was previously reported that iron (Fe2+ or Fe3+) supplementation in the growth medium (up to 1 mM) significantly increased ABM production, and this increase was positively correlated (23). We therefore asked whether iron would also affect intracellular levels of Hcy and Cys. When wild-type (WT) S. griseus was grown in glycerol-based ABM production broth without iron supplementation (APB−iron), the residual iron in other medium components was sufficient to support growth to ∼70% of the cell dry weight (CDW) of cultures from normal APB (2.75 g versus 3.92 ± 0.08 g/liter), but very little ABM production was observed after a 4-day incubation (Fig. 2A). Intracellular Hcy was barely detected in the iron-deficient cells (Fig. 2B, HPLC trace iv), while the amount of Hcy was significantly greater in iron-supplemented cells (Fig. 2B, HPLC trace v). Hcy in the APB sample was three times that in the APB-iron sample when it was normalized to the Cys detected with the same method. We next examined the effect of supplementing the culture with propargylglycine (PPGL), which is known to inhibit SAA metabolism in vivo. PPGL functions as a mechanism-based inhibitor of the transsulfuration reactions, targeting both CGS and CGL proteins (Fig. 1) (29, 30), but it is less effective on direct sulfhydrylation (31). PPGL was previously shown to inhibit the synthesis of a cysteine-containing antibiotic, thienamycin, produced by Streptomyces cattleya NRRL 8057 (30). The WT S. griseus strain was grown in APB in the presence of PPGL (APB+PPGL). While the cells grew to a CDW (3.20 ± 0.17 g/liter) that was ∼81% of normal growth, the PPGL-treated culture produced 30 to 50% more ABM on day 4, as determined by both bioassay and HPLC quantification of the purified ABM (Fig. 2C; see also Fig. S4 in the supplemental material). Thus, unlike thienamycin production, albomycin production does not seem to depend upon the transsulfuration pathways. Furthermore, in the profile of SAA, both Cys and Hcy peaks decreased but the relative amount of Hcy increased to 19.7% (Fig. 2B, HPLC traces v and vi). Taking into consideration the three independent growth conditions, the results suggest that an increase in ABM production is correlated with an increase in the relative intracellular concentration of Hcy.
FIG 2.

Relationship of ABM production and intracellular free-thiol amino acids of WT S. griseus. (A) Bioassay with cells grown in the absence (APB-iron) or presence (APB) of 1 mM iron in a glycerol-based ABM production medium (APB). The size of the inhibition zone is proportional to the amount of ABM in the culture assayed (see Materials and Methods). (B) HPLC-fluorescence detection of monobromobimane-derived intracellular amino acids. Cysteine (●) and homocysteine (*) are the only two free-thiol amino acids detected under these conditions. The chromatograms show (i) and (ii) standards, (iii) N-ethylmaleimide (NEM)-treated intracellular metabolic extracts (the negative control), (iv) cells in APB without iron, (v) cells in normal APB with 1 mM iron, and (vi) cells in APB supplemented with 9 mM propargylglycine (APB+PPGL). Percentage ratios of the peak areas of the two thiols are shown for traces iv, v, and vi. The standard error of three replicates for each was ≤0.2. (C) ABM production of cells grown in APB or APB+PPGL. ABM was quantified by isolation and HPLC analysis. The statistical significance was derived from nine experimental replicates; the standard error is presented by an error bar. **, P < 0.01 (t test).
Key SAA metabolic genes in S. griseus and functional assignment.
We previously reported that the genome of the ABM producer is very similar to that of the well-studied S. griseus NBRC 13350 (19). A preliminary genome-scale comparison is presented in Fig. S5 in the supplemental material. Since there are few reports concerning SAA metabolic genes in Streptomyces (32), we used the RAST genome annotation server (33) to identify candidates in the whole genome of NBRC 13350 (34). Ten major SAA metabolic genes or open reading frames (ORFs) were identified (Fig. 1), suggesting that S. griseus is Cys and Met prototrophic. Their homologs were subsequently identified in the S. griseus ATCC 700974 genome. Nucleotide identity between the genes of NBRC 13350 and ATCC 700974 (see Table S1 in the supplemental material) is 93.7% on average, with the highest at 97% (sahH) and the lowest at 89% (malY). The two closely related S. griseus strains can be used to perform genomic research of Streptomyces, such as on housekeeping genes and the conserved functions (35), as well as small noncoding RNAs (36).
Several subclasses of PLP-dependent enzymes are involved in the formation of thiol amino acids (14, 37), and the functions of the assigned ORFs were interrogated. The amplified ORFs of each pathway were assembled into a cassette for expression under the control of the ermE*p promoter in Streptomyces multicopy plasmid pSE34-oriT (19). It should be noted that direct sulfhydrylation ORFs sgr6647 and sgr6646 (metY and -X) appear in both genomes and are located in a genetic operon with a third ORF, sgr6645 (metSO), encoding a putative sulfite oxidase, immediately downstream of sgr6646. The three ORFs were amplified and cloned as one DNA fragment for expression. The different plasmids were separately introduced into S. griseus, and the fermentation cultures of the positive transformants were analyzed for ABM production and Hcy/Cys levels. Overexpression of the direct sulfhydrylation ORFs in pCK1 and transsulfuration ORFs sgr3417 and sgr2579 (malY and metB) in pCK2 caused an increase of Hcy relative to Cys, while the reverse transsulfuration ORFs sgr3660 and sgr4452 (mtcC and mtcB) in pCK3 caused a reduction of intracellular Hcy, although the effect was less pronounced (Fig. 3A and B). These results are consistent with expectations for SAA metabolism. Overexpression of metYXSO increased overall intracellular thiol amino acid concentrations, and this increase was significantly greater than for recombinant strains expressing malY-metB (Fig. 3A). The recombinant strains also produced ABM at different levels (Fig. 3C). The additional metYXSO in pCK1 enabled the highest ABM production compared to the control plasmid without the genes, while the addition of mtcC-mtcB (pCK3) led to comparable levels of ABM production relative to the control. No difference in the CDWs of these strains was observed. The results agreed in general with the functional assignment of the SAA metabolic genes. Moreover, the results revealed that a higher level of Hcy caused by the gene expression is not toxic to S. griseus and, importantly, that increased Hcy improves ABM production. The overflow of Hcy could be redirected to the ABM pathway by overexpressing the gate-keeping enzyme AbmD. A comparable engineering strategy leading to improved production of microbial metabolites has been reported for other systems, such as cephamycin C-producing Streptomyces (28).
FIG 3.

Effect of plasmid-based overexpression of the identified sulfur amino acid metabolic genes in WT S. griseus. (A) HPLC-fluorescence chromatograms of intracellular Cys and Hcy detected in the transformed strains. (B) Relative percentages of intracellular Cys and Hcy. For each strain that is a mixture of 3 to 5 individual colonies, the percentage is an average of three independent measures. (C) ABM production level. The introduced plasmids are described in Table 1. Each production level is the average of at least three experimental replicates; the standard error is represented by an error bar. Statistical significance was determined by comparing the level of a specific strain to the control. **, P < 0.01; NS, not significant.
Overexpression of Hcy synthesis genes and abmD for improved and stable ABM production.
Hcy concentration can be increased by inserting genetic constructs directly into the chromosome. The S. griseus draft genome was searched for two genetic elements for chromosomal incorporation of exogenous DNA. The first element was a phage ΦC31 attB site that allows the integration of pSET vector-carried DNA into a host chromosome (38). A 51-bp sequence in a hypothetical protein gene (39), highly similar to the attB sequences found in other Streptomyces genomes, was identified (Fig. 4A). The second genetic element is a constitutive promoter, such as hrdBp, often used in Streptomyces genetic engineering (26). HrdB is a principal sigma factor that has been studied in S. griseus (40). The genes encoding HrdB (sgr1701) in the two S. griseus strains are 96% identical. The promoter hrdBp was defined as an ∼350-bp DNA upstream sequence. We initially constructed a chromosomally integrated mutant, SCAK1, that has the direct sulfhydrylation genes metYXSO under the control of the hrdBp promoter sequence (Fig. 4B; see also Fig. S6 in the supplemental material). As expected, the intracellular concentration of Hcy of SCAK1 increased to a level almost equal to that of Cys (see Fig. S7 in the supplemental material). While ABM production by SCAK1 increased (Fig. 4C and D), the CDW of SCAK1 at day 4 decreased to 2.22 ± 0.27 g/liter, compared to the CDW of the WT, 3.92 ± 0.08 g/liter. We also made an abmD-integrated mutant, SCAK2 (Fig. 4B), that has abmD under the control of the hrdBp promoter, and the exconjugants (attB::pSCAK2) were obtained at a much lower frequency than for SCAK1 generation and the double-crossover mutant previously prepared (19). After several trials, we managed to isolate one such mutant, SCAK2 (see Fig. S6 in the supplemental material), from the 10−2 to 10−3 dilution of a conjugation mixture. ABM production by SCAK2 was nearly double that of the wild-type strain (Fig. 4C and D).
FIG 4.

Chromosomal integration of genetic constructs for increasing sulfur-carbon flow toward Hcy and the improved ABM production. (A) Comparison of attB sequences identified in the Streptomyces genomes. (B) Diagram of the major chromosomally integrated constructs. (C) HPLC chromatograms of purified albomycins of S. griseus ATCC 900794 WT and the mutants. Albomycin δ2 is the major product. Other albomycin congener (ferrichrome) or variants (δ1) were eluted from the HPLC column before or after the δ2. (D) ABM production of the analyzed chromosomal mutants. Each production level is the average of at least three experimental replicates; the standard error is represented by an error bar. **, P < 0.01.
To demonstrate that ABM production is jointly controlled by Hcy flux and abmD expression, the transsulfuration genes (malY-metB) under the control of hrdBp were included with abmD and the direct sulhydrylation genes (metXYSO) to generate the integrated mutant strain SCAK3 (Fig. 4B). The transformed colonies were readily prepared and verified by PCR (see Fig. S6 in the supplemental material). Thirty colonies were screened using the antibacterial bioassay, and ∼27 colonies gave a larger zone of inhibition than that of the WT strain. These colonies all produced similarly high levels of ABM. Several colonies were pooled to create strain SCAK3 for the subsequent metabolic analyses. The Hcy level in the SCAK3 cells was steadily low, and the CDW of SCAK3 was reproducibly 4.25 ± 0.24 g/liter, comparable to or better than that of the WT, suggesting that the growth problem of the two early mutants can be overcome by a combined action of the precursor supply pathways and overexpression of rate-limiting abmD. ABM production by SCAK3 was ∼400% that for the WT strain and ∼200% that for the SCAK1 or SCAK2 strain (Fig. 4C), indicating that the co-overexpression of abmD has efficiently diverted the augmented Hcy flux into ABM biosynthesis. Furthermore, no antibiotics were needed to maintain the ABM overproduction phenotype, although the SCAK3 strain was repeatedly passed through medium without selection. Thus, a stable, high-level ABM producer was generated. Although it is known that some bacteria cannot tolerate an elevation of intracellular Hcy (41), this strategy has the potential to be used to discover ABM-like molecules potentially produced by other actinomycetes, such as Streptomyces C (19).
Exploring metabolic switches and sulfur source for increasing ABM production.
Biosynthesis of methionine in bacteria is linked to central carbon metabolism and whole-cell physiology via additional metabolic pathways: one is the aspartate-to-homoserine path of C4 metabolism that starts with aspartokinase (Ask) and aspartate semialdehyde dehydrogenase (Asd), and another is the activated methyl cycle (AMC) of C1 metabolism (Fig. 1). The C4 carbon flow is controlled by a feedback inhibition mechanism of Ask and Asd. Streptomyces albulus NBRC 14147 encodes an inhibition-resistant Ask in the ask-asd operon, which is a major reason that it produces an aspartate-derived antibiotic product at a gram/liter level (42). AMC has two metabolic controlling elements as well. First, S-adenosyl-l-methionine (SAM)-dependent methyltransferases are subject to S-adenosyl-l-homocysteine (SAH) product inhibition. Without sufficient SAH hydrolase to degrade the coproduct generated by SAM-dependent enzymes, including AbmI (19), a cell's primary metabolism is impeded. Second, SAM is shared by many biological pathways, and the supply is limited. Thus, an exogenous supply of SAM or overexpression of SAM synthetase (MetK) has been reported to increase the production of antibiotics in Streptomyces (43). We attempted to explore these master metabolic switches for increasing ABM production by genetic manipulation. Single- or multiple-gene constructs were cloned into pSE34-oriT under the control of ermE*p for expression in WT S. griseus. As shown in Fig. 5, sahH overexpression with pCK4 improved ABM production, but the effect was less than with metK-sahH coexpression with pCK5. Overexpression of metE with pCK6 did not cause a change of ABM production, while the coexpression of the three genes with pCK7 only slightly increased ABM production levels (Fig. 5). The overexpression of metE was unable to compete with natural abmD but reduced the positive effect of metK-sahH coexpression. CDWs of strains carrying pCK4 and pCK5 were ∼15% more than for the control strain, indicating the important role of metK and sahH in cell fitness rather than having a specific effect on ABM biosynthesis. We finally made two more chromosomally integrated mutants, SCAK4 (hrdBp-ask-asd) and SCAK5 (hrdBp-metYXSO-hrdBp-ask-asd). Both of them showed a modest but reproducible increase of ABM production (Fig. 5). We have yet to confirm whether Ask/Asd enzyme activities are elevated in SCAK4 and SCAK5. However, in glycerol-based medium, the levels of intracellular Cys and Hcy were nearly identical, suggesting that the increased Hcy flux caused by the ask-asd played a direct role in the increase in ABM production. Future engineering efforts involving C1 and C4 metabolism for optimizing ABM production will likely need to consider the major carbon source of fermentation.
FIG 5.
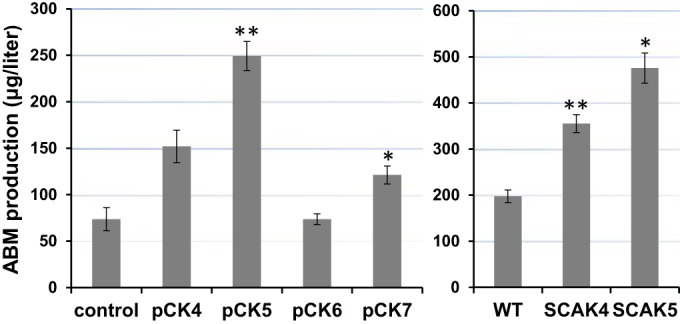
ABM production of the constructed strains. pCK series are strains containing replicating plasmids. SCAKs are strains with chromosomally integrated mutations. The genes cloned in each construct for overexperssion are as follows: for pCK4, sahH; for pCK5, metK-sahH; for pCK6, metE; for pCK7, metK-sahH-metE; for SCAK4, ask-asd; and for SCAK5, metY-X-SO-ask-asd. Each production level is the average of at least three experimental replicates; the standard error is represented by an error bar. **, P < 0.01; *, P < 0.05.
In addition to engineering the metabolic switches related to SAA metabolism, we also thought that a more reduced sulfur source in the medium might increase ABM production. Sodium sulfide cannot be used because it reacts immediately with iron to form a precipitate; thus, thiosulfate supplementation was examined. SCAK3 grew normally in thiosulfate-based medium, but ABM production was not increased. It remains unknown whether S. griseus has a thiosulfate reductase activity. Overall, the results are consistent with the higher-order metabolic regulatory circuits such as AMC, Ask/Asd, and sulfur assimilation in S. griseus having an indirect role in ABM production by altering Hcy flux.
SCAK3's growth and ABM production level in an optimized medium.
It was reported that WT S. griseus produced ABM at 1 to 2 mg/liter during standard fermentation (23). In addition to iron and buffering agents, ornithine and specific carbon sources in the medium were shown to be important for high-level ABM production by selected clones. Nonetheless, high-level-producing strains could not be preserved for a long period. At the outset of our experiments that lasted >8 months, the strains were grown up in glycerol-APB medium without ornithine, and ABM was isolated and quantified with HPLC. The yield was 0.20 ± 0.013 mg/liter or 0.85 ± 0.046 mg/liter for WT or SCAK3, respectively, when fermented in flasks with rigorous shaking. Adding ornithine alone to the glycerol-APB medium did not improve ABM production in SCAK3. The previously optimized medium, which contains ornithine and 2% starch, supported a higher ABM production by SCAK3 with the shake flask method; however, the ABM purification process became significantly more complex under those conditions. We then used the bioassay to determine the ABM titer and estimated the production by SCAK3 in the optimized medium despite this problem (Fig. 6A). ABM production peaked at day 4, when the level for SCAK3 (∼11 mg/liter) was three times as much as for the WT (∼3.7 mg/liter) (Fig. 6B). The CDWs of both cultures reached the highest level at day 3 (Fig. 6C), when SCAK3's mass was about twice that of the WT in flasks. Starting at day 4, the CDW of SCAK3 decreased toward the end, while the CDW of the WT changed less even after ABM production ceased. This observation indicated that more SCAK3 cells might have lysed once it produced a large amount of ABM in the shake flask fermentation. Nonetheless, the SCAK3 strain produced more ABM than the WT strain in the optimized medium at a given time. Since ABM production by the WT could be increased up to 25 mg/liter in a fed-batch fermentor (23), we estimate that ABM production by SCAK3 will approach ∼100 mg/liter. The purification of highly water-soluble small-peptide ABM and ABM derivatives remains challenging but solvable. A more efficient ABM purification process will be needed to improve the yields for further preclinical studies or for generating ABM analogs by mutasynthesis or semisynthesis.
FIG 6.

(A) Bioassay plates for determining the ABM titer of SCAK3 and WT strains grown in starch- and ornithine-based APB medium. Cleared fermentation culture was diluted with water from 1:20 to 1:120, as indicated. (B) ABM production of SCAK3 and WT over time. Twenty milliliters of TSB seed culture grown for 2 days was equally distributed to 10 300-ml baffled flasks, each containing 50 ml of starch-ornithine-APB medium. The flasks were incubated at 28°C and 250 rpm. One flask was harvested every day for determining bioactivity and CDW. The experiment was performed in duplicates; the standard error of each data point is represented by an error bar. (C) Cell dry weight of the cultures.
In summary, ABM is an antibacterial agent that was chosen for pharmaceutical development long before the genomic era (6, 12, 13, 44). A rational metabolic engineering strategy for increasing ABM production was adopted in this study. We analyzed two free-thiol amino acids, Cys and Hcy, in the model producing strain, S. griseus ATCC 700974, and correlated the levels to ABM production. We showed that the intracellular concentration of Hcy relative to that of Cys could be dramatically increased by overexpression of genes involved in direct sulfhydrylation and transsulfuration pathways. Refactoring of the primary metabolic genes for constitutive expression and integration into the chromosome, along with providing a duplicate abmD gene for the proposed initial step of the biosynthesis of the nucleoside component of ABM, resulted in a genetically stable, high-level ABM producer. In a glycerol-based medium, the ABM production level of the resulting strain, SCAK3, quantified by isolation and HPLC, was three times more than that of the WT. Thus, although ABM biosynthesis in this strain is apparently not controlled by a pathway-specific regulatory gene, ABM production can be manipulated by genetic engineering of the Hcy branch point of SAA metabolism in Streptomyces. The present research has only focused on limited principles of metabolic engineering for increasing ABM production. Further improvement is expected upon a more thorough metabolomic analysis of S. griseus as well as whole-cell modeling of the production event. At the enzyme level, AbmD is proposed to catalyze S-C bond formation using two building blocks, a hydroxyl-activated serine and an Hcy (see Fig. S8 in the supplemental material). The reaction path should be interesting due to its deviation from the course of plant ACCD (45) or human CBS (46), offering yet another microbial example of the diversity of the PLP-dependent enzyme superfamily.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from U.S. National Institutes of Health (AI087849 to S.V.L.) and the National Basic Research Program of China (2011CBA00803 and 2012CB721101 to W.Z.).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02517-15.
REFERENCES
- 1.Waksman SA, Henrici AT. 1943. The nomenclature and classification of the actinomycetes. J Bacteriol 46:337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waksman SA, Reilly HC, Harris DA. 1948. Streptomyces griseus (Krainsky) Waksman and Henrici. J Bacteriol 56:259–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones D, Metzger HJ, Schatz A, Waksman SA. 1944. Control of Gram-negative bacteria in experimental animals by streptomycin. Science 100:103–105. doi: 10.1126/science.100.2588.103. [DOI] [PubMed] [Google Scholar]
- 4.Gause GF. 1955. Recent studies on albomycin, a new antibiotic. Br Med J 12:1177–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stapley EO, Ormond RE. 1957. Similarity of albomycin and grisein. Science 125:587–589. doi: 10.1126/science.125.3248.587. [DOI] [PubMed] [Google Scholar]
- 6.Kuehl FA, Bishop MN, Chaiet L, Folkers K. 1951. Isolation and some chemical properties of grisein. J Am Chem Soc 73:1770–1773. doi: 10.1021/ja01148a099. [DOI] [Google Scholar]
- 7.Reynolds DM, Waksman SA. 1948. Grisein, an antibiotic produced by certain strains of Streptomyces griseus. J Bacteriol 55:739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pramanik A, Stroeher UH, Krejci J, Standish AJ, Bohn E, Paton JC, Autenrieth IB, Braun V. 2007. Albomycin is an effective antibiotic, as exemplified with Yersinia enterocolitica and Streptococcus pneumoniae. Int J Med Microbiol 297:459–469. doi: 10.1016/j.ijmm.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 9.Waksman SA. 1957. Penalty of isolationism. Science 125:585–587. doi: 10.1126/science.125.3248.585. [DOI] [PubMed] [Google Scholar]
- 10.Anonymous. 1958. Albomycin and grisein. Br Med J 1:391. [PubMed] [Google Scholar]
- 11.Braun V, Pramanik A, Gwinner T, Koberle M, Bohn E. 2009. Sideromycins: tools and antibiotics. Biometals 22:3–13. doi: 10.1007/s10534-008-9199-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stefanska AL, Fulston M, Houge-Frydrych CSV, Jones JJ, Warr SR. 2000. A potent seryl tRNA synthetase inhibitor SB-217452 isolated from a Streptomyces species. J Antibiot 53:1346–1353. doi: 10.7164/antibiotics.53.1346. [DOI] [PubMed] [Google Scholar]
- 13.Paulsen H, Brieden M, Benz G. 1987. Synthese des Sauerstofanalogs der Desferriforn von δ1-Albomycine. Liebigs Ann Chem 1987:565–575. [Google Scholar]
- 14.Ferla MP, Patrick WM. 2014. Bacterial methionine biosynthesis. Microbiology 160:1571–1584. doi: 10.1099/mic.0.077826-0. [DOI] [PubMed] [Google Scholar]
- 15.Lee HS, Hwang BJ. 2003. Methionine biosynthesis and its regulation in Corynebacterium glutamicum: parallel pathways of transsulfuration and direct sulfhydrylation. Appl Microbiol Biotechnol 62:459–467. doi: 10.1007/s00253-003-1306-7. [DOI] [PubMed] [Google Scholar]
- 16.Nagasawa T, Kanzaki H, Yamada H. 1984. Cystathionine gamma-lyase of Streptomyces phaeochromogenes. The occurrence of cystathionine gamma-lyase in filamentous bacteria and its purification and characterization. J Biol Chem 259:10393–10403. [PubMed] [Google Scholar]
- 17.Kim SH, Lee B-R, Kim J-N, Kim B-G. 2012. NdgR, a common transcriptional activator for methionine and leucine biosynthesis in Streptomyces coelicolor. J Bacteriol 194:6837–6846. doi: 10.1128/JB.00695-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zdych E, Peist R, Reidl J, Boos W. 1995. MalY of Escherichia coli is an enzyme with the activity of a beta C-S lyase (cystathionase). J Bacteriol 177:5035–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zeng Y, Kulkarni A, Yang Z, Patil P, Zhou W, Chi X, Van Lanen S, Chen S. 2012. Biosynthesis of albomycin δ2 provides a template for assembling siderophore and aminoacyl-tRNA synthetase inhibitor conjugates. ACS Chem Biol 7:1565–1575. doi: 10.1021/cb300173x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeng Y, Roy H, Patil PB, Ibba M, Chen S. 2009. Characterization of two seryl-tRNA synthetases in albomycin-producing Streptomyces sp. strain ATCC 700974. Antimicrob Agents Chemother 53:4619–4627. doi: 10.1128/AAC.00782-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JY, Carlson BA, Xu X-M, Zeng Y, Chen S, Gladyshev VN, Lee BJ, Hatfield DL. 2011. Inhibition of selenocysteine tRNA[Ser]Sec aminoacylation provides evidence that aminoacylation is required for regulatory methylation of this tRNA. Biochem Biophys Res Commun 409:814–819. doi: 10.1016/j.bbrc.2011.05.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Benz G, Schröder T, Kurz J, Wünsche C, Karl W, Steffens G, Pfitzner J, Schmidt D. 1982. Constitution of the Deferri form of the albomycins δ1, δ2 and ε. Angew Chem Int Ed Engl 21:527–528. doi: 10.1002/anie.198205271. [DOI] [Google Scholar]
- 23.Fiedler HP, Walz F, Dohle A, Zahner H. 1985. Albomycin: studies on fermentation, isolation and quantitative determination. Appl Microbiol Biotechnol 21:341–347. [Google Scholar]
- 24.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. John Innes Centre, Norwich Research Park, Colney, Norwich, England. [Google Scholar]
- 25.Myronovskyi M, Welle E, Fedorenko V, Luzhetskyy A. 2011. β-Glucuronidase as a sensitive and versatile reporter in actinomycetes. Appl Environ Microbiol 77:5370–5383. doi: 10.1128/AEM.00434-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du D, Zhu Y, Wei J, Tian Y, Niu G, Tan H. 2013. Improvement of gougerotin and nikkomycin production by engineering their biosynthetic gene clusters. Appl Microbiol Biotechnol 97:6383–6396. doi: 10.1007/s00253-013-4836-7. [DOI] [PubMed] [Google Scholar]
- 27.Newton GL, Fahey RC, Cohen G, Aharonowitz Y. 1993. Low-molecular-weight thiols in streptomycetes and their potential role as antioxidants. J Bacteriol 175:2734–2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malmberg LH, Hu WS, Sherman DH. 1993. Precursor flux control through targeted chromosomal insertion of the lysine epsilon-aminotransferase (lat) gene in cephamycin C biosynthesis. J Bacteriol 175:6916–6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnston M, Jankowski D, Marcotte P, Tanaka H, Esaki N, Soda K, Walsh C. 1979. Suicide inactivation of bacterial cystathionine γ-synthase and methionine γ-lyase during processing of L-propargylglycine. Biochemistry 18:4690–4701. doi: 10.1021/bi00588a033. [DOI] [PubMed] [Google Scholar]
- 30.Williamson JM, Meyer R, Inamine E. 1985. Reverse transsulfuration and its relationship to thienamycin biosynthesis in Streptomyces cattleya. Antimicrob Agents Chemother 28:478–484. doi: 10.1128/AAC.28.4.478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piotrowska M, Paszewski A. 1986. Propargylglycine as a fungal inhibitor: effect on sulphur amino acid metabolism. J Gen Microbiol 132:2753–2760. [Google Scholar]
- 32.Chang Z, Vining LC. 2002. Biosynthesis of sulfur-containing amino acids in Streptomyces venezuelae ISP5230: roles for cystathionine γ-synthase and transsulfuration. Microbiology 148:2135–2147. doi: 10.1099/00221287-148-7-2135. [DOI] [PubMed] [Google Scholar]
- 33.Aziz R, Bartels D, Best A, DeJongh M, Disz T, Edwards R, Formsma K, Gerdes S, Glass E, Kubal M, Meyer F, Olsen G, Olson R, Osterman A, Overbeek R, McNeil L, Paarmann D, Paczian T, Parrello B, Pusch G, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. 2008. The RAST server: Rapid Annotations using Subsystems Technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ohnishi Y, Ishikawa J, Hara H, Suzuki H, Ikenoya M, Ikeda H, Yamashita A, Hattori M, Horinouchi S. 2008. Genome sequence of the streptomycin-producing microorganism Streptomyces griseus IFO 13350. J Bacteriol 190:4050–4060. doi: 10.1128/JB.00204-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rong X, Huang Y. 2010. Taxonomic evaluation of the Streptomyces griseus clade using multilocus sequence analysis and DNA-DNA hybridization, with proposal to combine 29 species and three subspecies as 11 genomic species. Int J Syst Evol Microbiol 60:696–703. doi: 10.1099/ijs.0.012419-0. [DOI] [PubMed] [Google Scholar]
- 36.Moody M, Young R, Jones S, Elliot M. 2013. Comparative analysis of non-coding RNAs in the antibiotic-producing Streptomyces bacteria. BMC Genomics 14:558. doi: 10.1186/1471-2164-14-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aitken SM, Kirsch JF. 2005. The enzymology of cystathionine biosynthesis: strategies for the control of substrate and reaction specificity. Arch Biochem Biophys 433:166–175. doi: 10.1016/j.abb.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 38.Baltz RH. 2012. Streptomyces temperate bacteriophage integration systems for stable genetic engineering of actinomycetes (and other organisms). J Ind Microbiol Biotechnol 39:661–672. doi: 10.1007/s10295-011-1069-6. [DOI] [PubMed] [Google Scholar]
- 39.Combes P, Till R, Bee S, Smith MCM. 2002. The Streptomyces genome contains multiple pseudo-attB sites for the φC31-encoded site-specific recombination system. J Bacteriol 184:5746–5752. doi: 10.1128/JB.184.20.5746-5752.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shinkawa H, Hatada Y, Okada M, Kinashi H, Nimi O. 1995. Nucleotide sequence of a principal sigma factor gene (hrdB) of Streptomyces griseus. J Biochem 118:494–499. [DOI] [PubMed] [Google Scholar]
- 41.Roe AJ, O'Byrne C, McLaggan D, Booth IR. 2002. Inhibition of Escherichia coli growth by acetic acid: a problem with methionine biosynthesis and homocysteine toxicity. Microbiology 148:2215–2222. doi: 10.1099/00221287-148-7-2215. [DOI] [PubMed] [Google Scholar]
- 42.Hamano Y, Nicchu I, Shimizu T, Onji Y, Hiraki J, Takagi H. 2007. ε-Poly-l-lysine producer, Streptomyces albulus, has feedback-inhibition resistant aspartokinase. Appl Microbiol Biotechnol 76:873–882. doi: 10.1007/s00253-007-1052-3. [DOI] [PubMed] [Google Scholar]
- 43.Zhao XQ, Gust B, Heide L. 2010. S-Adenosylmethionine (SAM) and antibiotic biosynthesis: effect of external addition of SAM and of overexpression of SAM biosynthesis genes on novobiocin production in Streptomyces. Arch Microbiol 192:289–297. doi: 10.1007/s00203-010-0548-x. [DOI] [PubMed] [Google Scholar]
- 44.Maehr H, Berger J. 1969. The production, isolation and characterization of a grisein-like sideromycin complex. Biotechnol Bioeng 11:1111–1123. doi: 10.1002/bit.260110608. [DOI] [PubMed] [Google Scholar]
- 45.Thibodeaux CJ, Liu H-W. 2011. Mechanistic studies of 1-aminocyclopropane-1-carboxylate deaminase: characterization of an unusual pyridoxal 5′-phosphate-dependent reaction. Biochemistry 50:1950–1962. doi: 10.1021/bi101927s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Miles EW, Kraus JP. 2004. Cystathionine β-synthase: structure, function, regulation, and location of homocystinuria-causing mutations. J Biol Chem 279:29871–29874. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.