

Abstract
Study Objective:
Comorbid sleep disorders occur in approximately one-third of people with epilepsy. Seizures and sleep disorders have an interdependent relationship where the occurrence of one can exacerbate the other. Orexin, a wake-promoting neuropeptide, is associated with sleep disorder symptoms. Here, we tested the hypothesis that orexin dysregulation plays a role in the comorbid sleep disorder symptoms in the Kcna1-null mouse model of temporal lobe epilepsy.
Methods:
Rest-activity was assessed using infrared beam actigraphy. Sleep architecture and seizures were assessed using continuous video-electroencephalography-electromyography recordings in Kcna1-null mice treated with vehicle or the dual orexin receptor antagonist, almorexant (100 mg/kg, intraperitoneally). Orexin levels in the lateral hypothalamus/perifornical region (LH/P) and hypothalamic pathology were assessed with immunohistochemistry and oxygen polarography.
Results:
Kcna1-null mice have increased latency to rapid eye movement (REM) sleep onset, sleep fragmentation, and number of wake epochs. The numbers of REM and non-REM (NREM) sleep epochs are significantly reduced in Kcna1-null mice. Severe seizures propagate to the wake-promoting LH/P where injury is apparent (indicated by astrogliosis, blood-brain barrier permeability, and impaired mitochondrial function). The number of orexin-positive neurons is increased in the LH/P compared to wild-type LH/P. Treatment with a dual orexin receptor antagonist significantly increases the number and duration of NREM sleep epochs and reduces the latency to REM sleep onset. Further, almorexant treatment reduces the incidence of severe seizures and overall seizure burden. Interestingly, we report a significant positive correlation between latency to REM onset and seizure burden in Kcna1-null mice.
Conclusion:
Dual orexin receptor antagonists may be an effective sleeping aid in epilepsy, and warrants further study on their somnogenic and ant-seizure effects in other epilepsy models.
Citation:
Roundtree HM, Simeone TA, Johnson C, Matthews SA, Samson KK, Simeone KA. Orexin receptor antagonism improves sleep and reduces seizures in Kcna1-null mice. SLEEP 2016;39(2):357–368.
Keywords: dual orexin receptor antagonist, epilepsy, Kcna1-null, orexin, sleep
Significance.
Failure to obtain optimal sleep may not only worsen seizures themselves, but may lower the susceptibility for comorbid cognitive, affective, gastrointestinal and/or immune problems, all of which have a significantly higher incidence in people with epilepsy. Our study supports the hypothesis that optimizing sleep may provide a preventative and restorative treatment option to improve seizure syndromes and/or attenuate other comorbidities. Our experimental findings suggest that comorbid sleep disorders in epilepsy may result from seizure–mediated changes in the neurobiology of brain regions which regulate sleep. As each type of epilepsy is unique, future studies will determine the generalizability of our findings in additional preclinical models of epilepsy.
INTRODUCTION
Epilepsy is a serious neurological disorder characterized by recurrent, unprovoked seizures that has a lifetime incidence of 1:26 people in the United States.1 Epilepsy is associated with increased risk for many comorbid conditions including sleep disorders.2,3 The resulting insufficient sleep is associated with worsening seizure frequency and/or severity, thus exacerbating the core syndrome.4,5 Insufficient sleep can also further reduce the quality of life of people with epilepsy by promoting other comorbid conditions, including cognitive impairments and/or psychological disorders, which are also dependent on sleep efficiency.4,6–13 Thus, there is a critical need to better understand and ameliorate the sleep disorder symptoms experienced by patients with epilepsy. Restoring sleep may improve the seizure profile and/or reduce other comorbid conditions.
Symptoms of sleep disorders in epilepsy frequently include increased latency to sleep onset, reduced sleep duration, and increased fragmentation.4,6,7 Similar symptoms in people without epilepsy and in animal models are associated with changes in orexin neurotransmission (hypocretin is also known as orexin; because of the use of an orexin receptor antagonist in this study, we chose to use the term orexin).14–17 Orexin neurons are located in the lateral hypothalamus and perifornical region (LH/P) and when activated, promote wakefulness, attention, and arousal.18 Orexin neurons funnel diverse afferent signals and send projections to the ascending reticular activating system to increase activation of wake-promoting norepinephrine, acetylcholine, serotonin, histamine, and dopamine neuronal populations.18–21
Orexin neurons are also directly and indirectly connected to the seizure-generating hippocampal network via monosynaptic and polysynaptic efferent and afferent projections.22–25 Considering that orexin activation promotes wakefulness, wakefulness is increased in people with epilepsy and sleep disorders, and orexin neurons are connected to the hippocampus, we hypothesized that dysregulated orexin neurotransmission plays a role in the sleep disorders associated with epilepsy.
To test this hypothesis, we used Kcna1-null mice, a model of multiple epilepsy syndromes,26 that we have previously demonstrated have highly variable diurnal rest activity patterns, specifically their peak activity and diurnal period.27,28 Here, we further determined that Kcna1-null mice express additional sleep disorder symptoms including increased latency to rest onset and rest fragmentation, and reduced nonrapid eye movement (NREM) and rapid eye movement (REM) sleep during periods of rest. Thus, Kcna1-null mice represent an appropriate model of epilepsy with comorbid sleep disorder symptoms to test the hypothesis that dysregulated orexin neurotransmission plays a role in the sleep disorders associated with epilepsy.
METHODS
Animals
C3HeB/FeJ Kcna1-null and wild-type littermates were bred and reared at Creighton University. Mice were entrained to a strict 12-h light/dark cycle with access to food and water ad libitum. Lights on occurred at Zeitgeber time (ZT) 00:00 h. Tails clips were collected on postnatal day (P) 12–15 and genotype was determined by Transnetyx, Inc (Cordova, TN, USA). On ∼P21 mice were weaned onto standard diet. Adult mice (P35–P50) were used for these experiments. All experiments conformed to National Institutes of Health guidelines in accordance with the United States Public Health Service's Policy on Humane Care and Use of Laboratory Animals and were approved by Creighton University's Institutional Animal Care and Use Committee.
Drugs and Reagents
All reagents were purchased through Sigma-Alderich (St. Louis, MO, USA) unless otherwise noted. Almorexant was provided by Acetlion Pharmaceuticals (San Francisco, CA, USA).
Actigraphy
Behavioral rest-activity cycles were assessed with integrated radio telemetry technology and switch-closure activity monitoring (Vital View data acquisition system, Mini Mitter Company, Inc; Bend, OR, USA). Adult mice (∼P40) were placed in an 8″ × 8″ × 16″ transparent Plexiglass cage and allowed to habituate for 3–4 h. Infrared beam breaks by activity were monitored in 6-min epochs and scored on a scale of 0–150 over a 4- to 7-d period, as we have previously reported.27,28
Electroencephalography and Electromyography Neurosurgeries
Neurosurgeries were conducted as we have previously described.29 Adult Kcna1-null mice (∼P35) were anesthetized with isoflurane (5% initiation and 3% maintenance). Electroencephalography (EEG) and electromyography (EMG) electrodes were implanted in all mice. For cortical EEG surgeries, two subdural, ipsilateral cortical electrodes were implanted at 1.2 mm anterior to bregma and 1 mm lateral to midline, and at 1.5 mm posterior to bregma and 1 mm lateral to midline. A reference electrode was implanted 1.5 mm posterior to bregma and 1 mm lateral to midline, contralateral from recording electrodes. For surgeries that implanted both cortical and depth electrodes, the anterior cortical electrode was implanted (as described previously) and a depth electrode (PFA-coated tungsten wire, A-M Systems, Carlsborg, WA, USA) was placed 1 mm posterior to bregma and 1 mm lateral to midline at a depth of 5 mm. EMG electrode wires were inserted into the nuchal muscles in all mice. Electrodes were soldered to the headmount (Pinnacle Technologies, Lawrence, KS, USA), and secured to the skull with dental cement. Mice were allowed to recover for 1 w. During recovery and throughout the experiment, mice were housed individually and were given access to food and water ad libitum.
Dual Orexin Receptor Antagonist Experimental Design
The design for the experiment using the dual orexin receptor drug almorexant is outlined in Figure 1. Due to the shortened lifespan of these mice, we use a timeline similar to those we have previously reported.27–29 Adult mice (∼P30) were placed in an 8” × 8” × 16” transparent Plexiglass cage and allowed to habituate for 5 d. On ∼P35, mice were implanted with EEG/EMG electrodes as described previously. Following a 5- to 7-d recovery, mice were injected with vehicle (intraperitoneally [i.p.], 25% dimethylsulfoxide in sterile saline) once daily for 3 consecutive days, followed by 3 consecutive days of once-daily 100 mg kg−1 almorexant (i.p., 25 mg/ mL in vehicle). Injections began at ZT 00:00. For each treatment, time-synchronized video-EEG-EMG recordings were started immediately after the injection on the second day and continued for 48 h. To allow the animals to habituate to and reduce the confounding effects of being tethered, handling, and injections, recordings from the third day of each treatment were analyzed 30 min after the injection. The analyses were limited to the first 5.5 h (∼ZT 00:30–06:00) due to the previously reported pharmacokinetics and dynamics of almorexant.30–32
Figure 1.

Schematic of the experimental design for the in vivo almorexant study. The ages for each experimental procedure are listed along the bottom of the figure. Animals were placed in recording cages on ∼P30 and allowed to habituate for 5 d before surgery. After 7 d of post-surgical recovery, mice were injected once daily with vehicle (for 3 d) then almorexant (for 3 d). Continuous video-EEG-EMG recordings were conducted for 48 h, starting on the second day of treatment.
Sleep Analyses
Using Sirenia Sleep Pro (v1.4.2, Pinnacle Technologies, Lawrence, KS, USA), recordings were divided into 10-sec epochs for sleep state scoring and analyzed using a semiautomated method. Recordings were initially scored into different sleep states based on EEG power in the delta band of the anterior cortical electrode, located over the motor cortices and the corpus callosum, and EMG power (10–50 Hz). Sleep states were defined as follows: wake = low delta power, high EMG; NREM = high delta power, low to medium EMG power; REM = low delta power, low EMG. Scoring based on EEG and EMG was then manually verified with video recording. The epochs during which an animal was seizing was excluded from the analyses. The percentage of epochs Kcna1-null mice spent seizing was minimal when compared to the total number of analyzed epochs (vehicle: 0.98% ± 0.44%; almorexant: 0.48% ± 0.08%).
Several endpoints were analyzed for wake, NREM sleep, and REM sleep: (1) the number of epochs in each state of vigilance (NREM sleep, REM sleep or wake), normalized to the total number of epochs analyzed within each animal; (2) the number of bouts; and (3) the number of transitions. Bouts were defined as three or more consecutive epochs uninterrupted by a bout of a different state. Transitions were defined as any change between states and categorized based on initial and subsequent state. (4) The length of bouts for NREM sleep and (5) the latency to REM sleep were also measured. The latency to REM sleep was defined as the amount of transpired time until the first REM sleep epoch following the start of the analysis.
Seizure Analyses
Seizures were identified based on ictal cortical EEG activity, high EMG activity, and previously defined seizure behaviors for this mouse strain.27–29 Seizure behavior was scored using a modified Racine scale, as we have previously described27–29: Stage 1-myoclonic jerk; Stage 2-head stereotypy; Stage 3-fore-limb/hindlimb clonus, tail extension, a single rearing event; Stage 4-continuous rearing and falling; Stage 5-severe tonic-clonic seizures. Stage 1 behavior is associated with an EEG spike wave discharge. Stages 2 through 5 are associated with a concordant and sustained increase in EMG activity and EEG amplitude and time-dependent frequencies. Seizure burden scores (SBS) were generated using the following equation: SBS = ∑(σ i θi), where σindicates the seizure severity score using the modified Racine scale, θ indicates the duration of each seizure, and i indicates each seizure.
Immunoglobulin G Immunohistochemistry
Mice were quickly anesthetized with isoflurane, transcardi-ally perfused with ice-cold 0.9% saline followed by 4% paraformaldehyde in 0.1 M PB (pH 7.4), decapitated and brains were post-fixed overnight. Brains were cryoprotected in 15% and 30% sucrose/phosphate buffered solutions before frozen in methyl-butane on dry ice. Sections containing the lateral hypothalamus (−1.30 thru −2.06 mm bregma) were examined. Sections affixed to slides were fixed in 4% paraformaldehyde in 0.1 M PB (pH 7.4), dehydrated and rehydrated in a series of ethanol baths, washed with 0.01 M PB saline/0.3% Triton X-100, pH 7.4 (PBST), blocked with 10% normal goat serum/ PBST, and incubated with AF-350 conjugated anti-mouse immunoglobulin G (IgG) (1:2000, Invitrogen, Grand Island, NY, USA) overnight at room temperature. Sections were scored on an intensity scale of punctate and diffuse IgG-positive staining (1–5, with 1 = low intensity, 5 = high intensity) by five blinded investigators. Sections with less punctate and more diffuse IgG are considered to have a leaky or more permeable blood-brain barrier (BBB) as previously described.33 Data were expressed as the mean punctate intensity.
Glial Fibrillary Acidic Protein and Orexin Immunohistochemistry
Mice were quickly anesthetized with isoflurane and decapitated, and brains were removed and frozen in methyl-butane on dry ice. Sections affixed to slides were fixed in 4% paraformalde-hyde in 0.1 M PB (pH 7.4), washed with PBST and blocked with 10% normal goat serum/PBST. Sections incubated with rabbit antiorexin (1:1000, Peninsula Laboratories, San Carlos, CA, USA) overnight at room temperature followed by AF-594 conjugated goat antirabbit IgG (1:600, Invitrogen) for 3 h at room temperature. In one cohort, sections were immediately cover-slipped with mounting media containing 4',6-diamidino-2-phenylindole (DAPI) and orexin was quantified using stereology (described in the next paragraph). In the second cohort, sections were incubated additionally in AF-488 conjugated mouse anti-GFAP (1:500, Invitrogen) overnight at room temperature and cover-slipped with DAPI mounting media. GFAP is used as a marker for astrocytes. The number of reactive astrocytes in the LH/P was quantified. Reactive astrocytes were operationally defined as astrocytes with morphological hypertrophy and extended processes, as previously described.34,35
Stereology
LH/P sections with orexin positive labeling were selected using unbiased systematic random sampling.36 The total number of immunopositive cells was estimated using the numerical density (NV) method: NV = (∑Q-/#Q-)/DAt; NV, numerical density; ∑Q-/#Q-, average number of cells per dissector; DAt, dissector area over the specified thickness, t.
Mitochondrial Isolation and Oxygen Polarography
Mice were anesthetized with isoflurane, and hypothalamic tissue was quickly microdissected on ice. Tissue was homogenized in isolation buffer (5×V/V in mM: 215 mannitol, 75 sucrose, 1 ethylene glycol tetraacetic acid (EGTA) 20 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and 0.1% bovine serum albumin (BSA), pH 7.2 adjusted with potassium hydroxide (KOH) and mitochondria were isolated using differential centrifugation as we have described.29 Isolated mitochondria were resuspended in isolation buffer without EGTA. Protein concentrations were determined with Bradford assay. Mitochondria (100 μg) were resuspended in potassium chloride (KCl) respiration buffer (in mM: 125 KCl, 20 HEPES, 2 MgCl2, 2.5monopotassium phosphate (KH2PO4, pH 7.2) and placed in a sealed, thermostatically controlled chamber at 37°C. Oxygen polarography was measured using a standard Clark-type electrode (Hanstech Instruments, Ltd., Norfolk, England) as we have described.29,37,38 The following sequential parameters were measured in duplicates per sample: Mitochondria were energized via adenosine triphosphate (ATP)-producing nicotinamide adenine dinucleotide hydride (NADH):coenzyme Q oxidoreductase (complex I)-driven state III respiration with 5 mM pyruvate and 2.5 mM malate (PM) and concurrent activation of adenosine triphosphate synthase by 150 μM adenosine diphosphate; state IV respiration was initiated with oligomycin-induced inhibition of ATP synthase (1 μM); maximal respiratory rate (state V) was measured in the presence of the chemical protonophore p-triflourometh-oxyphenylhydrazone (FCCP; 1 μM); and mitochondrial respiratory complex (MRC) I-respiratory dependency was verified in the presence of the MRCI inhibitor rotenone. Only samples with respiratory control ratios greater than 4 were analyzed. Unbiased assessment of the rate of oxygen consumption was determined using computer-generated line of best-fit algorithm and nmol of oxygen mg−1 min−1 was calculated.
Statistics
Sleep data were analyzed using an analysis of variance (ANOVA) with Sidak post hoc test. Actigraphic, immunohistochemical, and mitochondrial data were analyzed using an unpaired Student t test. Statistical dependence was determined using Spearman non-parametric rank correlation coefficient. A value of P < 0.05 was considered statistically significant.
RESULTS
Kcna1-null Mice have Increased Activity during Periods of Rest
We have previously reported that Kcna1-null mice have altered diurnal periods and prolonged peak activity as determined by acti-graphic periodogram and cosinor analyses.27,28 Here, three endpoints common in sleep disorders including (1) latency to rest onset, (2) rest duration, and (2) fragmentation were analyzed during the rest period following actigraphy. We found that 100% of wild-type controls were able to achieve continuous rest (conservatively defined by uninterrupted rest for at least 30 min). In contrast, only 28% of epileptic mice were able to achieve continuous rest and of this cohort, the latency to rest onset was significantly increased (P < 0.05, Figure 2A). During the entire rest periods, Kcna1-null mice spent significantly less time resting (P < 0.05, Figure 2B) and were more active (P < 0.05, Figure 2C).
Figure 2.

Kcna1-null mice exhibit sleep disorder symptoms. Activity was scored during each 6-min epoch on a scale of 0–150 using actigraphy. Scores were summed during each rest and wake phase and the data were collapsed across days. (A) Latency to onset of 30 min of continuous rest was determined for each subject and normalized to the mean wild-type values within each experiment. The number of (B) inactive epochs and (C) active epochs during the rest phase are graphed. Data are mean ± standard error of the mean; n = 7–8; *P < 0.05.
Kcna1-null Mice have Reduced NREM Sleep and REM Sleep Epochs during Periods of Rest
The increased time spent active during the rest period suggested that Kcna1-null mice may either have longer durations of time spent awake or may transition to wake more often than wild-type controls. To distinguish between these possibilities, we examined the sleep architecture of Kcna1-null mice, and analyzed NREM sleep, REM sleep, and wakefulness using continuous time-synchronized video-EEG-EMG recordings. In this experiment, mice were treated with vehicle for 3 d followed by a treatment with an orexin receptor antagonist for 3 d. Analyses were conducted using two-factor ANOVA. The differences between genotypes (i.e. the vehicle data) will be discussed in this section. The differences between treatments will be discussed in the next section. To avoid plotting vehicle data twice, Figures 4 through 6 will be referenced in both sections.
Figure 4.

Representative hypnograms of Kcna1-null and wild-type mice. Kcna1-null and wild-type mice were treated with vehicle for 3 d then almorexant for 3 d (100 mg/kg, intraperitoneally). Video-EEG-EMG) data were scored in 10-sec epochs as wake, NREM sleep, or REM sleep for this first 5.5 h of the rest phase, 30 min following injection (Zeitgeber time [ZT] 00:30–06:00). (A) Sleep architecture is shown in representative hypnograms of a wild-type mouse and Kcna1-null mouse on vehicle, compared to (B) the same mice during almorexant treatment. Each hypnogram data point identifies the behavioral state of the mouse at each epoch over time (i.e. during each 10-sec epoch from ZT 00:30–06:00).
Figure 6.
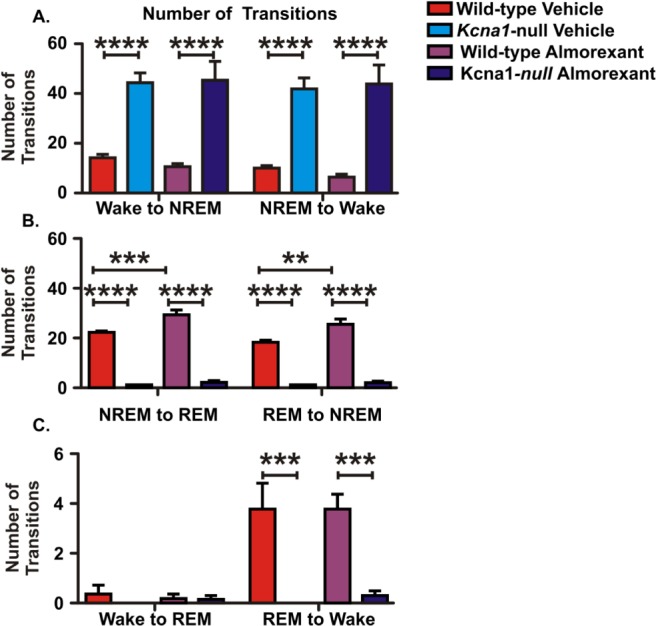
The transitions between behavioral states in Kcna1-null mice differ from wild-type controls. Transitions between behavioral states were grouped into six categories. Kcna1-null mice have significantly more transitions between wake and nonrapid eye movement (NREM) sleep, but have fewer transitions to or from rapid eye movement (REM) sleep. Almorexant treatment does not affect the number of transitions between any behavioral states in Kcna1-null mice. Almorexant treatment does increase the number of transitions between REM sleep and NREM sleep in wild-type controls. Data are mean ± standard error of the mean; n = 5–6; **P < 0.01, ***P < 0.001, ****P < 0.0001.
Raw EEG and EMG traces were divided into 10-sec epochs. As depicted in Figure 3, dominant frequencies and EMG activity were used to classify each epoch as NREM sleep (high EEG delta power with low to medium EMG power), REM sleep (low EEG delta power with low EMG activity) or awake (low EEG delta power with high EMG power). Hypnograms of vehicle-treated wild-type and Kcna1-null mice are depicted in Figure 4A. The amount of time mice spent in each behavioral state was determined by totaling the number of epochs and normalizing to the total number of epochs analyzed for that animal. During the rest period, Kcna1-null mice spent more time awake (F(1,9) = 42.2, P < 0.0005) and less time in both NREM sleep (F(1,9) = 20.3, P < 0.005) and REM sleep (F(1,8) = 229.3, P < 0.0001) when compared to wild-type controls (Figure 5A, left two bars in each graph). These data indicate that Kcna1-null mice have a deficiency in both NREM sleep and REM sleep.
Figure 3.

Representative traces of behavioral states. Sleep architecture was scored using video-EEGEMG data in Sirenia Sleep Pro using a semiautomated method. Each automated EEG-EMG score was manually verified with time-synchronized video data. Traces from time-matched subdural EEG electrodes (blue and orange traces) and EMG electrode (green) show typical characteristics of wake (low amplitude EEG, high amplitude EMG), nonrapid eye movement (NREM) sleep (high amplitude EEG, low-medium amplitude EMG), and rapid eye movement (REM) sleep (low amplitude EEG and EMG). EEG rhythms for each example epoch are shown in corresponding spectrograms.
Figure 5.

Almorexant increases nonrapid eye movement (NREM) sleep and reduces wake during the rest phase of Kcna1-null mice. The total percentage of epochs spent in each behavioral state was analyzed using two-way analysis of variance with Sidak post hoc test. (A) Kcna1-null mice spend significantly less time in NREM sleep and rapid eye movement (REM) sleep and more time awake compared to wild-type. Almorexant treatment restores NREM sleep of Kcna1-null mice to wild-type levels and significantly decreases time awake. (B) Bar graphs depicting the total number of bouts for each genotype and treatment. A bout is defined as three or more consecutive 10 s epochs of the same behavioral state. Kcna1-null mice have a greater number of wake bouts and fewer REM sleep bouts than wild-type mice. Data are mean ± standard error of the mean; n = 5–6; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n.s., not significant.
We began to assess how fragmented the rest period was by comparing (1) the number of bouts of and (2) the number of transitions among the three vigilant states. One bout was defined as three or more consecutive epochs of the same state. Kcna1-null mice had significantly more wake bouts (F(1,9) = 10.64, P < 0.01) and fewer REM sleep bouts (F(1,9) = 200, P < 0.0001) when compared to wild-type mice (Figure 5B, left two bars in each graph).
The transitions between behavioral states (NREM sleep-wake, NREM sleep-REM sleep, wake-REM sleep, and vice versa) were sorted into six categories based on the initial and subsequent behavioral states (Figure 6). Kcna1-null mice had significantly more transitions from NREM sleep to wake (F(1,9) = 61.3, P < 0.0001) when compared to wild-type mice (Figure 6A). Kcna1-null mice also had significantly fewer transitions between REM sleep and NREM sleep (REM versus NREM: F(1,9) = 260.5, P < 0.0001 and NREM versus REM: F(1,9) = 421.5, P < 0.0001, Figure 6B), and between REM sleep and wake (F(1,9) = 24.0, P < 0.001, Figures 6C). This is likely reflecting the REM sleep deficiency apparent during the period of analysis. These data suggest that Kcna1-null mice may have more fragmented rest when compared to wild-type mice.
Blocking Orexin Receptors Improves the NREM Sleep in Kcna1-null Mice
To determine whether orexin activation contributed to sleep disturbances, following vehicle treatment, Kcna1-null mice were treated with the dual orexin receptor antagonist (DORA) almorexant (100 mg/kg, i.p.).31,32 Based on the pharmacokinetics and pharmacodynamics of almorexant in rodents,30 the analyses were limited to the first 5.5 h, 30 min following injection. Representative EEG-EMG hypnograms of wild-type and Kcna1-null mice treated with vehicle then almorexant are depicted in Figures 4A and 4B, respectively.
Blocking orexin receptors during periods of rest significantly reduced the percentage wake epochs (F(1,9) = 15.2, P < 0.01) and increased the percentage of NREM sleep epochs in Kcna1-null mice (F(1,9) = 17.1, P < 0.001) to levels that were similar to those of wild-type controls (Figure 5A). The deficit in REM sleep was not rescued by 3 d of almorexant treatment in Kcna1-null mice. Almorexant did not have a significant effect on the percentage of wake, NREM sleep, or REM sleep epochs in wild-type mice during the rest phase. Unexpectedly, these data suggest the dose of almorexant increased the amount of NREM sleep of Kcna1-null mice; however, it did not have the same effect in wild-type controls.
Next, we determined whether almorexant influenced the number of bouts of or transitions among states of vigilance. There was no change in the number of NREM sleep or wake bouts of Kcna1 -null or wild-type mice (Figure 5B). Further, almorexant treatment had no effect on the number of transitions among vigilant states of Kcna1-null mice. In contrast, almorexant-treated wild-type mice had significantly more REM sleep bouts (F(1,9) = 12.9, P < 0.01) and more transitions between NREM and REM (NREM to REM: F(1,9) = 21.8, P < 0.001; REM to NREM: F(1,9) = 16.6, P < 0.01) (Figure 6B).
The increased number of NREM sleep epochs and the lack of change in NREM bout incidence suggest that the duration of each bout may be increased. Figure 7A depicts the distribution of the NREM sleep bout length. Goodness-of-fit-analysis indicates that treatment with almorexant increased the probability of longer NREM sleep bout durations in Kcna1-null mice (P < 0.0001). Almorexant did not change NREM sleep duration in wild-type controls. These data indicate that almorexant may improve the ability of Kcna1-null mice to remain in NREM sleep.
Figure 7.
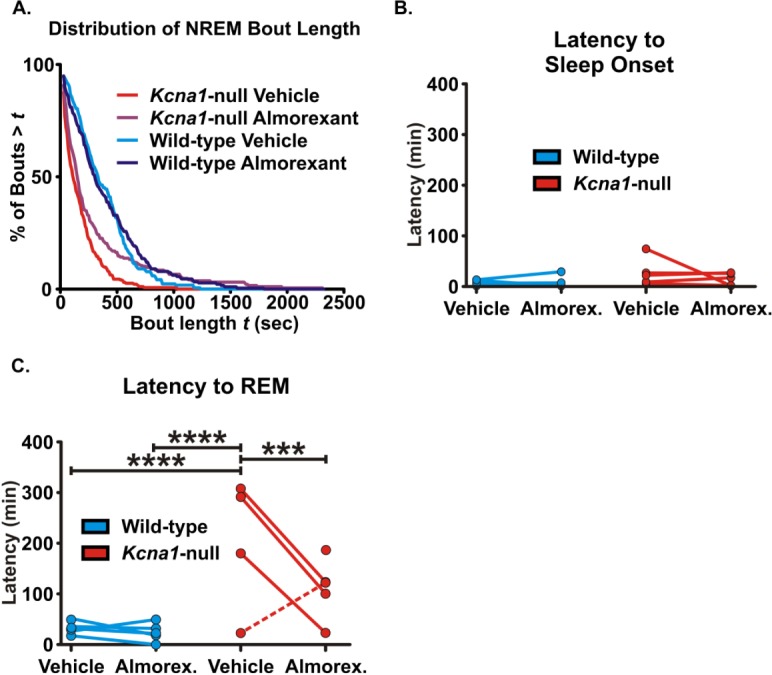
Almorexant increases the probability of longer nonrapid eye movement (NREM) sleep bout durations and reduces the latency to rapid eye movement (REM) sleep onset in Kcna1-null mice. (A) Probability curves of NREM sleep bout durations were plotted for each genotype and treatment. Kcna1-null mice are less likely to have longer duration of continuous NREM sleep. Almorexant increases the probability of longer NREM sleep bout durations in Kcna1-null mice. (B) The latency to sleep onset was determined as the time before the first occurrence of NREM sleep or REM sleep in each animal. The within-subject comparisons are plotted. (C) The latency to REM sleep was determined as the time before the first occurrence of REM sleep in each animal and within-subject comparisons are plotted. The latency to REM sleep in Kcna1-null mice was higher in all but one animal (statistical outlier; dashed line) compared to wild-type controls. In all Kcna1-null mice with increased latency to REM sleep, almorexant reduced the time to REM sleep onset. REM sleep was not detected in one subject during vehicle, however was apparent during almorexant treatment (data point with no associated line). Data are mean ± standard error of the mean; n = 5–6; ***P < 0.001, ****P < 0.0001.
In contrast to actigraphy data, there was no significant difference in latency to sleep onset (Figure 7B), defined as the time that transpired between the start of analysis and the first epoch of NREM sleep or REM sleep epoch (in all but one case, NREM sleep occurred first). This is likely due to the conservative definition of 30 min of continuous rest used in the actigraphy study. However, Kcna1-null mice do have an increased latency to REM sleep (Figure 7C, P < 0.0001 compared to wild-type on vehicle), defined as the time that transpired between the start of analysis and the first epoch of REM sleep only. (Note: because there was no statistical difference in the latency to sleep onset, the latency to REM sleep onset was similar whether defined with respect to the start of analysis or the start of sleep onset. Thus, we used the start of the analysis to maintain focus on REM sleep alone). Although it had no effect on latency to REM sleep in wild-type mice, almorexant appeared to reduce the latency to REM sleep onset when compared to vehicle-treated Kcna1-null mice (Figure 7C, also apparent in the hypnograms in Figure 4). Using the extreme studentized deviate (ESD) method, analyses of the percent reduction in latency to REM sleep onset following almorexant treatment identified one subject (with an increase in latency to REM onset during almorexant treatment) as a statistical outlier (P < 0.05, identified by a dashed line in Figure 7B). Quantification of the remaining four Kcna1-null mice indicated that the latency to REM sleep onset was significantly reduced during almorexant treatment (F(1,7) = 161.2, P < 0.0001). REM sleep rhythms were absent in one animal during vehicle treatment and then became apparent during almorexant treatment (Figure 7B, the data point above almorexant with no line). This interesting observation will be discussed more in the section entitled “Correlations between seizure and sleep profiles.” These data suggest that almorexant may reduce the latency to REM sleep onset, specifically in the Kcna1-null mice with prolonged onset when compared to wild-type controls.
Seizure-Like Activity and Pathology is Apparent in the Wake-Promoting LH/P of Kcna1-null Mice
The improvement in NREM sleep following almorexant suggested that activation of wake-promoting orexin neurocircuitry may be increased in Kcna1-null mice. Thus, we analyzed seizure propagation, markers of pathology, and orexin expression in the LH/P.
First, we determined whether seizures propagate to the orexin-rich LH/P. Subdural cortical electrodes and an LH/P depth electrode were implanted in Kcna1-null mice. Seizure activity, which originates from the hippocampus and propagates to cortex of Kcna1-null mice,35 was detected in the wake-promoting LH/P. Figure 8 depicts representative cortical and depth electrode traces of a seizure that electro-clinically manifested from a stage 1 to a stage 4 event. In all seizures analyzed, epileptiform activity was apparent in both the cortical as well as the depth electrode traces during severe seizures (stages 3–5), whereas the less severe seizures (stages 1–2) did not consistently propagate to the LH/P.
Figure 8.

Severe seizures in Kcna1-null mice propagate to the LH/P. Representative lateral hypothalamus/perifornical region (LH/P) and cortical electroencephalographic (EEG) traces of one seizure that manifests from a less severe stage 1 to a severe stage 5 seizure. Arrows indicate an EEG event with a behavioral correlate. Seizure severity scores are in parentheses. A single arrow indicates the event was detected in the subdural channel. Double arrows indicate the event was detected in both subdural and LH/P electrodes. Stage 5 severe tonic-clonic period detected in both electrodes is indicated with brackets. Cort, subdural cortical electrode trace; LH, lateral hypothalamic and perifornical region depth electrode trace.
The hyperexcitability and hypersynchrony associated with seizures can induce pathology that significantly changes the neurobiological and metabolic landscape, which are notably apparent in the hippocampus of Kcna1-null mice.26,29,35,39 Pathological markers were used to assess injury in the LH/P of Kcna1-null mice. Blood-brain barrier (BBB) permeability and astrocyte morphology were examined with immunofluorescent histochemistry, and mitochondrial function was assessed with oxygen polarography.35 There was a significant reduction of punctate immunostaining and a concordant increase in IgG extravasation, indicative of an increase in BBB permeability in the LH/P of Kcna1-null mice (P < 0.05, Figure 9A). There was also an increase in the incidence of reactive astrocytes in this region of Kcna1-null mice when compared to wild-type controls (78.6 ± 7.9% compared to 34.8 ± 10.2%, P < 0.01, Figure 9B). We have previously reported that mitochondrial respiratory rates are reduced in the hippocampus and cortex of these mice.29 Here we found a similar reduction in ATP-producing state III, maximal state V, and state II respiratory rates of mitochondria isolated from the hypothalamus of Kcna1-null (P < 0.05, Figure 9C). Collectively, these data suggest that the more severe seizures propagate to the LH/P and that injury is apparent in the hypothalamus of Kcna1-null mice.
Figure 9.
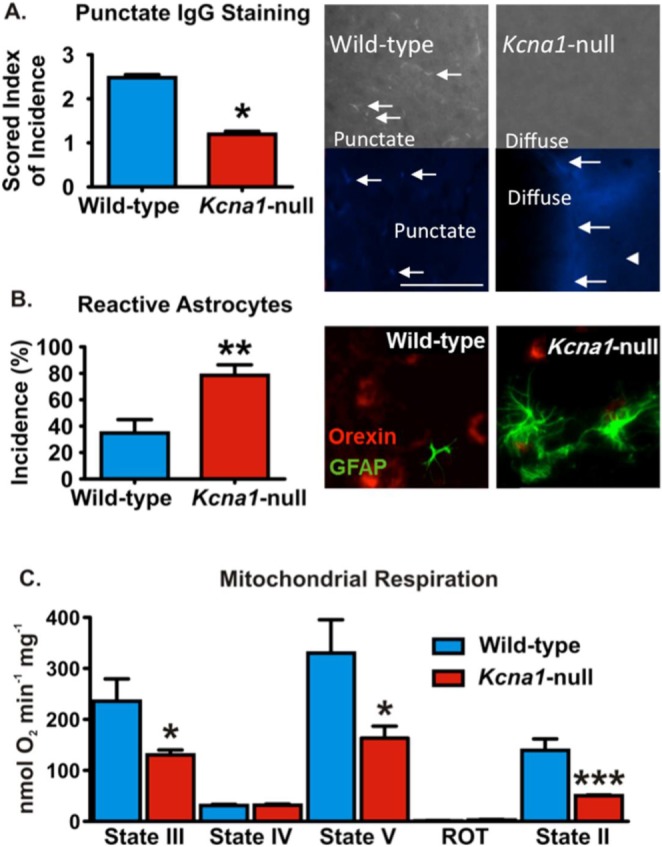
Pathology is apparent in the lateral hypothalamus/perifornical region (LH/P) of Kcna1-null mice. (A) Photomicrographs depict immunoglobulin G (IgG) labeling (white/blue) in LH/P parenchyma. In wild-type animals, punctate staining (arrows) is more pronounced, compared to diffuse labeling in Kcna1-null mice. Bar graph of punctate mouse IgG staining as scored on an arbitrary 1–5 scale by five blinded investigators (n = 4 animals/group, 3–4 LH/P sections/animal). (B) Reactive astrocytes are GFAP-positive (green) and exhibit extended processes and hypertrophy (see the Kcna1-null darkfield photomicrograph) when compared to typical astrocytes as depicted in the wild-type section. Coronal sections containing LH/P had a significantly higher incidence of reactive astrocytes compared to wild-type LH/P (n = 4 animals/group, 3–4 LH/P sections/animal). Sections are double-labeled for orexin (red). (C) Hypothalamic mitochondria isolated from Kcna1-null mice have reduced state III, state V, and state II respiratory rates when compared to wild-type controls (n = 3–4). Data are mean ± standard error of the mean. *P < 0.05, **P < 0.01, ***P < 0.001.
Finally, we assessed orexin protein levels in the LH/P to support or refute the hypothesis that orexin activity may be increased in Kcna1-null mice. The number of neurons expressing orexin throughout the anterior-posterior axis of the LH/P was significantly increased in Kcna1-null mice when compared to wild-type controls (P < 0.05, Figure 10). This increase existed in neurons both in the medial (P < 0.05) and lateral (P < 0.05) aspects of the posterior LH/P between −1.0 mm and −1.2 mm bregma (Figure 10, right inset). Collectively, these data indicate long-term changes in the neurobiological environment of the LH/P that supports the notion of increased orexinergic tone of Kcna1-null mice.
Figure 10.
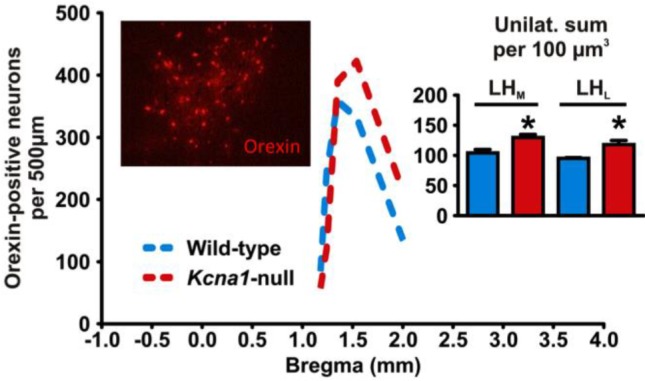
The number of orexin-positive neurons is increased in Kcna1-null LH/P. The distribution of orexin-positive neurons along the anterior-posterior axis for coronal brain sections of wild-type and Kcna1-null mice is shown. Kcna1-null mice show significantly more total orexin-positive cells. Left inset: Darkfield photomicrograph depicting orexin-positive (red) immunofluorescence in the lateral hypothalamus/ perifornical region (LH/P). Right inset: The increase of orexin-positive neurons occurred in both the medial and lateral aspects of the LH/P (n = 4–5 animals, 3–4 sections/animal). Data are mean ± standard error of the mean, *P < 0.05.
Dual Orexin Receptor Antagonism Decreases Seizures in Kcna1-null Mice
The association between sleep deprivation and the worsening of seizure frequency and/or severity suggests that improving the sleep profile may be associated with an improved seizure profile. We examined the same time period used for the sleep analyses to analyze seizure frequency, duration, and severity using the modified Racine scale, as we have described previously in the Methods section.27–29
Data were stratified into two groupings based on seizure severity (less severe stage 1 seizures and moderate/severe stage 2–5 seizures). We observed a 75% ± 6% reduction in the incidence of stage 2–5 seizures during almorexant treatment in four of five mice (P < 0.01; ESD analysis identified the mouse with the lowest seizure profile during vehicle as an outlier, dashed line in Figure 11A). The incidence of stage 1 seizures did not change during almorexant treatment (Figure 11B). Seizure durations range from less than 30 sec to several minutes. To take into account the severity and duration of each seizure, a seizure burden score was calculated for each animal (see Methods section). Seizure burden was reduced by 75% ± 9% during almorexant treatment in the same cohort (four of five mice, P < 0.01, Figure 11C). Plotting the initial seizure burden scores against the percent reduction during almorexant treatment revealed a significant correlation (Spearman rho = 1.00, P < 0.05, Figure 11D, fit with a logarithmic growth curve). These data indicate a potentially greater anti-seizure efficacy of almorexant with more severe seizures.
Figure 11.

Almorexant reduces severe seizures in Kcna1-null mice. Seizure incidence and severity during vehicle and almorexant treatment were scored and seizure burden was calculated. Within-scatterplots depict (A) the incidence of severe seizures (stages 2–5), (B) the incidence of the less severe stage 1 seizures, and (C) seizure burden during vehicle and almorexant treatments. (D) Percent reduction of seizure burden following treatment showed a positive Spearman correlation with initial seizure burden and was fit using a logarithmic growth curve. (E) Latency to rapid eye movememt (REM) sleep also showed a positive Spearman correlation with a linear fit with seizure burden both before and after treatment with almorexant. Dashed lines indicate an outlier, n = 5, **P < 0.01.
Correlation between Seizure and Sleep Profiles
A final interesting observation was in the extreme seizure and sleep profiles: (1) the animal with the highest seizure burden lacked REM sleep during vehicle treatment, and (2) the mouse with the least severe seizure profile had a latency to REM sleep onset that resembled wild-type latencies. Indeed, there was a positive linear correlation between REM sleep onset and seizure severity (r = 0.82, P < 0.05, Figure 11E).
DISCUSSION
A connection between epilepsy and sleep has long been recognized. Seizures and sleep disorders have an interdependent relationship where the occurrence of one can exacerbate the other. Research has primarily focused on the relationship of brain EEG rhythms (e.g., slow wave sleep, sharp waves, sleep spindles, ripples, etc.) and the development of seizures. Here, we used a novel strategy and focused on the sleep disorder comorbidity itself in an effort to increase understanding of the sleep and seizure dynamics and to identify a novel antiseizure and somnogenic treatment.
To our knowledge this study is the first to report a detailed characterization of the sleep architecture deficiencies of Kcna1-null mice, further supporting the use of this animal model to examine sleep disorder symptoms associated with epilepsy. Treatment with a DORA significantly increases NREM sleep and reduces the latency to REM onset in Kcna1-null mice. The more severe seizures propagate to the wake-promoting LH/P, where astrogliosis and BBB permeability are apparent and hypothalamic mitochondrial function is impaired. The number of orexin-positive neurons is increased in the LH/P. Blocking orexin receptors with almorexant reduces the severity of seizures and importantly, there is a significant correlation between latency to REM onset and seizure burden in Kcna1-null mice.
Seizure Propagation and Neurobiological Changes in the LH/P
Kcna1-null mice lack the potassium channel Kv1.1 alpha subunit and are a genetic model of temporal lobe epilepsy. This model offers diverse and severe seizure phenotypes involving multiple daily myoclonic jerks, head movement stereotypies, clonus, and tonic-clonic seizures.26–29,35,39,40 Seizures arise in the hippocampus and propagate to the cortex of Kcna1-null mice.35 Seizure severity and incidence increase with age and are associated with hippocampal sclerosis involving cell death, astrogliosis, mossy fiber sprouting, and mitochondrial impairment.29,35,40
To our knowledge, we are the first to provide evidence that severe seizures propagate to the LH/P. Consistent with the damage reported in the Kcna1-null hippocampus, we identified similar pathology including astrogliosis, BBB permeability in the LH/P, and impairment of hypothalamic mitochondrial function. Our findings support a limited number of previous studies, which have noted seizure-related injury in the hypothalamus and LH/P. Increased metabolic activity and injury have been reported in the hypothalamus following induction of status epilepticus in the pilocarpine epilepsy mode.22,41 Studies using kainic acid and lithium-pilocarpine to induce severe seizures have demonstrated neurodegeneration and cell death throughout the hypothalamus; injury specifically in the LH/P was noted in mice subjected to kainic acid-induced seizures.42 The limited available human data from MRI studies of mesial temporal lobe epilepsy indicate a strong concordance between changes in the hippocampus and degeneration of its major efferent tract, the fornix, and subsequent targets in the hypothalamus.43–45
In addition to the presence of pathology, we also found an increase in the number of orexin-positive neurons in the LH/P. Previous studies have reported mechanisms that promote orexin expression. Our data indicate one of two possibilities. First, there may be an increase in the amount of orexin protein expressed within neurons that previously expressed subdetectable levels. Many signals can influence the degree of orexin expression within a neuron. Prepro-orexin messenger RNA and plasma orexin protein levels are significantly increased under different metabolic conditions, such as 48 h of fasting or acute (6 h) hypoglycemia.46 Levels of orexin are also significantly higher in younger rats compared to older rats.47 A second possibility is that the number of orexin expressing cells may be increased. Temporal lobe seizures are associated with increased neurogenesis in the hippocampus.48 The number of orexin neurons can be increased by the stress hormone corticosterone.49 It is well documented that seizure activity promotes an acute spatial-temporal change in the metabolic landscape in the hippocampus and hypothalamus,22,23,42–45 including enhancing stress-regulating hormones and changing mitochondrial metabolism and function.29,37,50 Whether seizures increase expression in a low-expressing pool of neurons or increases the number of neurons is currently under investigation.
Regardless, the overall increase in orexin and other changes in the neurobiological landscape of the LH/P in Kcna1-null mice will influence neurotransmission. Similar changes within the Kcna1-null tri-synaptic hippocampal network promote excitability.26,29,39 Orexin neurons project to wake-promoting regions containing norepinephrine, acetylcholine, serotonin, histamine, and dopamine neurons. Previous studies report that experimental increase in orexin receptor activation induces wakefulness.18 Increased drive of this circuitry may participate in enhancing the propensity to wake in Kcna1-null mice during periods of rest.
Sleep and Seizures
Sleep disorder symptoms including increased latency to sleep onset, increased awakenings, and reduced sleep duration in humans and animal models without epilepsy are associated with a dysregulated orexinergic system,14–17 a system responsible for promoting wakefulness.18 If the Kcna1-null orexinergic system were overactive during rest periods, we would predict that sleep architecture changes may include increased time awake and decreased NREM sleep and/or REM sleep. Further, we would predict that administration of a DORA may restore some or all aspects of sleep architecture to resemble that of wild-type mice.
Indeed, Kcna1-null mice have increased wake during periods of rest. We previously reported that epileptic Kcna1-null mice exhibit highly variable diurnal rest-activity patterns, specifically their peak activity and diurnal period.27,28 Here, we further determined that Kcna1-null mice express additional sleep disorder symptoms during periods of rest, including increased activity (as measured by actigraphy) and latency to REM sleep onset and reduced NREM sleep and REM sleep (as determined using video-EEG-EMG recording). This phenotype resembles sleep disorder symptoms described in people with epilepsy, which include increased latency to sleep onset, increased entries into wakefulness, and decreased sleep efficiency and NREM sleep.4,6,7 These sleep disorder symptoms resemble those associated with a dysregulation of the orexinergic system.14,15 Therefore, we further tested the hypothesis that orexin plays a role in the sleep deficiency of Kcna1-null mice by administering a DORA, almorexant. We conducted experiments during the rest phase when Kcna1-null mice are more active compared to wild-type mice. Almorexant treatment reduced the time spent awake and reversed the NREM sleep deficiency by improving the ability to maintain NREM sleep (i.e. by increasing bout durations). While almorexant reduced the latency to REM onset in Kcna1-null mice, the acute treatment was unable to restore the number of REM sleep epochs.
Interestingly, these improvements to sleep following almorexant treatment occurred in the Kcna1-null mice only. Wild-type mice did not experience a change in the number of wake and NREM sleep epochs, or a change in latency to REM sleep onset. This is not surprising considering DORAs are typically tested in wild-type mice during the wake phase, a time when orexin neurons are more active and an inhibitory effect would be maximal.31,32 During normal sleep, orexinergic activity is suppressed, and hence the inhibitory effect of almorexant would be minimal during the rest phase. This may explain why acute almorexant treatment did not have a significant effect on these endpoints in wild-type mice.
In addition to improving sleep, almorexant treatment reduced the incidence of the more severe seizures (stage 2–5) and seizure burden by 75%. Whether almorexant exerted indirect or direct antiseizure effects has yet to be determined. Almorexant may have indirectly reduced seizures by improving sleep. It is well known that inadequate sleep/sleep deprivation worsens seizures in people with epilepsy.4,5,51 We specifically found that longer REM sleep latencies positively correlated with greater seizure burden. In one study, sleep deprivation associated with exacerbating seizures was also associated with an increase orexin concentration in cerebro- spinal fluid.51 Thus, almorexant may have indirectly reduced seizures by improving sleep.
A second possibility is that almorexant directly reduced seizures. Orexin neurons project to a multitude of neuronal populations including the hippocampus,21,52 raising the possibility that almorexant directly dampens seizure-generating mechanisms in the hippocampus. Excitatory orexin receptors OX1R and OX2R are coupled to Gs and Gq/Gs proteins, respectively, and are expressed in CA1–3 and the dentate gyrus.18,52,53 In vivo intracerebroventricular (i.c.v.) injection of orexin excites CA1 pyramidal cells and in vitro application induces longterm potentiation at the Schaffer collateral-CA1 synapse.52,54 Furthermore, i.c.v injection of high dose of orexins causes behavioral seizure activity in rats and bilateral hippocampal injection of selective OX1R or OX2R antagonists reduces acute pentylenetetrazole-induced seizures.55,56 Studies to distinguish the indirect and/or direct mechanism of the seizure-dampening effects of almorexant are currently underway.
CONCLUSION
Our findings suggest that pathology in the LH/P of Kcna1-null mice, possibly caused by seizure propagation from the hippo-campus, reflects long-term changes in the neural environment that may contribute to comorbid sleep disorders and the worsening of seizures in Kcna1-null mice. Inhibiting orexin receptors circumvents the pathology and reverses the NREM deficit and reduces seizure severity. DORAs are currently under evaluation for clinical use in sleep disorders. One example is suvorexant, which is approved by the US Food and Drug Administration for treating insomnia. Here, we provide evidence that DORAs may be an effective sleeping aid in epilepsy, and warrant further study on their effects in other epilepsy models, both to improve comorbid sleep disturbances as well as reduce seizure severity.
DISCLOSURE STATEMENT
This was not an industry supported study. Funding was provided by Nebraska LB692, Epilepsy Foundation of America and National Institutes of Health (NS072179). This project was also supported by Grant Number G20RR024001 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the national Center for Research Resources or the National Institutes of Health. Please note, Kristina A. Simeone has published under the names KA Dorenbos, KA Fenoglio, and KA Simeone. The authors have indicated no financial conflicts of interest.
ACKNOWLEDGMENTS
The authors wish to thank the Nebraska LB692, Epilepsy Foundation of America and National Institutes of Health (NS072179) for funding this research. This project was also supported by Grant Number G20RR024001 from the National Center for Research Resources.
REFERENCES
- 1.Hesdorffer DC, Logroscino G, Benn EK, Katri N, Cascino G, Hauser WA. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology. 2011;76:23–7. doi: 10.1212/WNL.0b013e318204a36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manni R, Terzaghi M. Comorbidity between epilepsy and sleep disorders. Epilepsy Res. 2010;90:171–7. doi: 10.1016/j.eplepsyres.2010.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Gilliam FG, Mendiratta A, Pack AM, Bazil CW. Epilepsy and common comorbidities: improving the outpatient epilepsy encounter. Epileptic Disord. 2005;7:27–33. [PubMed] [Google Scholar]
- 4.Kotagal P, Yardi N. The relationship between sleep and epilepsy. Semin Pediatr Neurol. 2008;15:42–9. doi: 10.1016/j.spen.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 5.Grigg-Damberger MM, Ralls F. Sleep disorders in adults with epilepsy: past, present, and future directions. Curr Opin Pulm Med. 2014;20:542–9. doi: 10.1097/MCP.0000000000000101. [DOI] [PubMed] [Google Scholar]
- 6.Malow BA. Sleep and epilepsy. Neurol Clin. 2005;23:1127–47. doi: 10.1016/j.ncl.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Malow BA, Bowes RJ, Lin X. Predictors of sleepiness in epilepsy patients. Sleep. 1997;20:1105–10. doi: 10.1093/sleep/20.12.1105. [DOI] [PubMed] [Google Scholar]
- 8.Rocamora R, Sanchez-Alvarez JC, Salas-Puig J. The relationship between sleep and epilepsy. Neurologist. 2008;14:S35–43. doi: 10.1097/01.nrl.0000340790.15295.59. [DOI] [PubMed] [Google Scholar]
- 9.de Weerd A, de Haas S, Otte A, et al. Subjective sleep disturbance in patients with partial epilepsy: a questionnaire-based study on prevalence and impact on quality of life. Epilepsia. 2004;45:1397–404. doi: 10.1111/j.0013-9580.2004.46703.x. [DOI] [PubMed] [Google Scholar]
- 10.Steinsbekk S, Berg-Nielsen TS, Wichstrom L. Sleep disorders in preschoolers: prevalence and comorbidity with psychiatric symptoms. J Dev Behav Pediatr. 2013;34:633–41. doi: 10.1097/01.DBP.0000437636.33306.49. [DOI] [PubMed] [Google Scholar]
- 11.Jacoby A, Snape D, Lane S, Baker GA. Self-reported anxiety and sleep problems in people with epilepsy and their association with quality of life. Epilepsy Behav. 2015;43:149–58. doi: 10.1016/j.yebeh.2014.09.071. [DOI] [PubMed] [Google Scholar]
- 12.Killgore WD. Effects of sleep deprivation on cognition. Prog Brain Res. 2010;185:105–29. doi: 10.1016/B978-0-444-53702-7.00007-5. [DOI] [PubMed] [Google Scholar]
- 13.Plihal W, Born J. Effects of early and late nocturnal sleep on declarative and procedural memory. J Cogn Neurosci. 1997;9:534–47. doi: 10.1162/jocn.1997.9.4.534. [DOI] [PubMed] [Google Scholar]
- 14.Prober DA, Rihel J, Onah AA, Sung RJ, Schier AF. Hypocretin/orexin overexpression induces an insomnia-like phenotype in zebrafish. J Neurosci. 2006;26:13400–10. doi: 10.1523/JNEUROSCI.4332-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brisbare-Roch C, Dingemanse J, Koberstein R, et al. Promotion of sleep by targeting the orexin system in rats, dogs and humans. Nat Med. 2007;13:150–5. doi: 10.1038/nm1544. [DOI] [PubMed] [Google Scholar]
- 16.Horvath TL, Gao XB. Input organization and plasticity of hypocretin neurons: possible clues to obesity's association with insomnia. Cell Metab. 2005;1:279–86. doi: 10.1016/j.cmet.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Vgontzas AN, Chrousos GP. Sleep, the hypothalamic-pituitary-adrenal axis, and cytokines: multiple interactions and disturbances in sleep disorders. Endocrinol Metab Clin North Am. 2002;31:15–36. doi: 10.1016/s0889-8529(01)00005-6. [DOI] [PubMed] [Google Scholar]
- 18.Sakurai T. The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci. 2007;8:171–81. doi: 10.1038/nrn2092. [DOI] [PubMed] [Google Scholar]
- 19.Szymusiak R, McGinty D. Hypothalamic regulation of sleep and arousal. Ann N Y Acad Sci. 2008;1129:275–86. doi: 10.1196/annals.1417.027. [DOI] [PubMed] [Google Scholar]
- 20.Sakurai T, Mieda M, Tsujino N. The orexin system: roles in sleep/wake regulation. Ann N Y Acad Sci. 2010;1200:149–61. doi: 10.1111/j.1749-6632.2010.05513.x. [DOI] [PubMed] [Google Scholar]
- 21.Peyron C, Tighe DK, van den Pol AN, et al. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, Liu Y, Huang Y, Chen L. Time-course of neuronal death in the mouse pilocarpine model of chronic epilepsy using Fluoro-Jade C staining. Brain Res. 2008;1241:157–67. doi: 10.1016/j.brainres.2008.07.097. [DOI] [PubMed] [Google Scholar]
- 23.Dube C, Boyet S, Marescaux C, Nehlig A. Relationship between neuronal loss and interictal glucose metabolism during the chronic phase of the lithium-pilocarpine model of epilepsy in the immature and adult rat. Exp Neurol. 2001;167:227–41. doi: 10.1006/exnr.2000.7561. [DOI] [PubMed] [Google Scholar]
- 24.Yoshida K, McCormack S, España RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. 2006;494:845–61. doi: 10.1002/cne.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kukkonen JP, Holmqvist T, Ammoun S, Akerman KE. Functions of the orexinergic/hypocretinergic system. Am J Physiol Cell Physiol. 2002;283:C1567–91. doi: 10.1152/ajpcell.00055.2002. [DOI] [PubMed] [Google Scholar]
- 26.Simeone TA, Simeone KA, Samson KK, Kim do Y, Rho JM. Loss of the Kv1.1 potassium channel promotes pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices. Neurobiol Dis. 2013;54:68–81. doi: 10.1016/j.nbd.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fenoglio-Simeone K, Mazarati A, Sefidvash-Hockley S, et al. Anticonvulsant effects of the selective melatonin receptor agonist ramelteon. Epilepsy Behav. 2009;16:52–7. doi: 10.1016/j.yebeh.2009.07.022. [DOI] [PubMed] [Google Scholar]
- 28.Fenoglio-Simeone KA, Wilke JC, Milligan HL, Allen CN, Rho JM, Maganti RK. Ketogenic diet treatment abolishes seizure periodicity and improves diurnal rhythmicity in epileptic Kcna1-null mice. Epilepsia. 2009;50:2027–34. doi: 10.1111/j.1528-1167.2009.02163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Simeone KA, Matthews SA, Samson KK, Simeone TA. Targeting deficiencies in mitochondrial respiratory complex I and functional uncoupling exerts anti-seizure effects in a genetic model of temporal lobe epilepsy and in a model of acute temporal lobe seizures. Exp Neurol. 2014;251:84–90. doi: 10.1016/j.expneurol.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morairty SR, Revel FG, Malherbe P, et al. Dual hypocretin receptor antagonism is more effective for sleep promotion than antagonism of either receptor alone. PLoS One. 2012;7:e39131. doi: 10.1371/journal.pone.0039131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morairty SR, Wilk AJ, Lincoln WU, Neylan TC, Kilduff TS. The hypocretin/orexin antagonist almorexant promotes sleep without impairment of performance in rats. Front Neurosci. 2014;8 doi: 10.3389/fnins.2014.00003. Article 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Black SW, Morairty SR, Fisher SP, Chen TM, Warrier DR, Kilduff TS. Almorexant promotes sleep and exacerbates cataplexy in a murine model of narcolepsy. Sleep. 2013;36:325–36. doi: 10.5665/sleep.2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcon J, Gagliardi B, Balosso S, et al. Age-dependent vascular changes induced by status epilepticus in rat forebrain: implications for epileptogenesis. Neurobiol Dis. 2009;34:121–32. doi: 10.1016/j.nbd.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 34.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–47. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wenzel HJ, Vacher H, Clark E, et al. Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia. 2007;48:2023–46. doi: 10.1111/j.1528-1167.2007.01189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.West MJ. University of Aarhus, Denmark: Cold Spring Harbor Laboratory Press; 2012. Basic stereology for biologists and neuroscientists. [Google Scholar]
- 37.Sullivan PG, Dube C, Dorenbos K, Steward O, Baram TZ. Mitochondrial uncoupling protein-2 protects the immature brain from excitotoxic neuronal death. Ann Neurol. 2003;53:711–7. doi: 10.1002/ana.10543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sullivan PG, Rippy NA, Dorenbos K, Concepcion RC, Agarwal AK, Rho JM. The ketogenic diet increases mitochondrial uncoupling protein levels and activity. Ann Neurol. 2004;55:576–80. doi: 10.1002/ana.20062. [DOI] [PubMed] [Google Scholar]
- 39.Simeone TA, Samson KK, Matthews SA, Simeone KA. In vivo ketogenic diet treatment attenuates pathologic sharp waves and high frequency oscillations in in vitro hippocampal slices from epileptic Kv1.1α knockout mice. Epilepsia. 2014;55.5:e44–9. doi: 10.1111/epi.12603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim do Y, Simeone KA, Simeone TA, et al. Ketone bodies mediate anti-seizure effects through mPT. Ann Neurol. 2015;78:77–87. doi: 10.1002/ana.24424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fernandes MJ, Dube C, Boyet S, Marescaux C, Nehlig A. Correlation between hypermetabolism and neuronal damage during status epilepticus induced by lithium and pilocarpine in immature and adult rats. J Cereb Blood Flow Metab. 1999;19:195–209. doi: 10.1097/00004647-199902000-00011. [DOI] [PubMed] [Google Scholar]
- 42.Covolan L, Mello LE. Temporal profile of neuronal injury following pilocarpine or kainic acid-induced status epilepticus. Epilepsy Res. 2000;39:133–52. doi: 10.1016/s0920-1211(99)00119-9. [DOI] [PubMed] [Google Scholar]
- 43.Ng S, Lau T, Hui F, et al. MRI of the fornix and mammillary body in temporal lobe epilepsy. Neuroradiology. 1997;39:551–5. doi: 10.1007/s002340050465. [DOI] [PubMed] [Google Scholar]
- 44.Karakas HM. The role of the mammillary body in the propagation of the ictal activity. Neurobiology (Bp) 2001;9:81–9. doi: 10.1556/neurob.9.2001.2.2. [DOI] [PubMed] [Google Scholar]
- 45.Ozturk A, Yousem DM, Mahmood A, El Sayed S. Prevalence of asymmetry of mammillary body and fornix size on MR imaging. AJNR Am J Neuroradiol. 2008;29:384–7. doi: 10.3174/ajnr.A0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai XJ, Widdowson PS, Harrold J, et al. Hypothalamic orexin expression: modulation by blood glucose and feeding. Diabetes. 1999;48:2132–7. doi: 10.2337/diabetes.48.11.2132. [DOI] [PubMed] [Google Scholar]
- 47.Desarnaud F, Murillo-Rodriguez E, Lin L, et al. The diurnal rhythm of hypocretin in young and old F344 rats. Sleep. 2004;27:851–6. doi: 10.1093/sleep/27.5.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–38. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jalewa J, Wong-Lin K, McGinnity TM, Prasad G, Hölscher C. Increased number of orexin/hypocretin neurons with high and prolonged external stress-induced depression. Behav Brain Res. 2014;272:196–204. doi: 10.1016/j.bbr.2014.05.030. [DOI] [PubMed] [Google Scholar]
- 50.Patel M, Li QY, Chang LY, Crapo J, Liang LP. Activation of NADPH oxidase and extracellular superoxide production in seizure-induced hippocampal damage. J Neurochem. 2005;92:123–31. doi: 10.1111/j.1471-4159.2004.02838.x. [DOI] [PubMed] [Google Scholar]
- 51.Ni LY, Zhu MJ, Song Y, Liu XM, Tang JY. Pentylenetetrazol-induced seizures are exacerbated by sleep deprivation through orexin receptor-mediated hippocampal cell proliferation. Neurol Sci. 2014;35:245–2. doi: 10.1007/s10072-013-1495-5. [DOI] [PubMed] [Google Scholar]
- 52.Selbach O, Doreulee N, Bohla C, et al. Orexins/hypocretins cause sharp wave-and θ-related synaptic plasticity in the hippocampus via glutamatergic, gabaergic, noradrenergic, and cholinergic signaling. Neuroscience. 2004;127:519–28. doi: 10.1016/j.neuroscience.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 53.Marcus JN, Aschkenasi CJ, Lee CE, et al. Differential expression of orexin receptors 1 and 2 in the rat brain. J Comp Neurol. 2001;435:6–25. doi: 10.1002/cne.1190. [DOI] [PubMed] [Google Scholar]
- 54.Riahi E, Arezoomandan R, Fatahi Z, Haghparast A. The electrical activity of hippocampal pyramidal neuron is subjected to descending control by the brain orexin/hypocretin system. Neurobiol Learn Mem. 2015;119:93–101. doi: 10.1016/j.nlm.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 55.Ida T, Nakahara K, Katayama T, Murakami N, Nakazato M. Effect of lateral cerebroventricular injection of the appetite-stimulating neuropeptide, orexin and neuropeptide Y, on the various behavioral activities of rats. Brain Res. 1999;821:526–9. doi: 10.1016/s0006-8993(99)01131-2. [DOI] [PubMed] [Google Scholar]
- 56.Goudarzi E, Salmani ME, Lashkarbolouki T, Goudarzi I. Hippocampal orexin receptors inactivation reduces PTZ induced seizures of male rats. Pharmacol Biochem Behav. 2015;130:77–83. doi: 10.1016/j.pbb.2015.01.006. [DOI] [PubMed] [Google Scholar]