

Abstract
Controversy exists as to whether supplementation with the antioxidants vitamin E and vitamin C blocks adaptation to exercise. Exercise is a first-line means to treat obesity and its complications. While diet-induced obesity alters mitochondrial function and induces insulin resistance (IR), no data exist as to whether supplementation with vitamin E and vitamin C modify responses to exercise in pre-existing obesity. We tested the hypothesis that dietary supplementation with vitamin E (0.4 g α-tocopherol acetate/kg) and vitamin C (0.5 g/kg) blocks exercise-induced improvements on IR and mitochondrial content in obese rats maintained on a high-fat (45% fat energy (en)) diet. Diet-induced obese, sedentary rats had a 2-fold higher homeostasis model assessment of insulin resistance and larger insulin area under the curve following glucose tolerances test than rats fed a low-fat (10% fat en) diet. Exercising (12 weeks at 5 times per week in a motorized wheel) of obese rats normalized IR indices, an effect not modified by vitamin E and vitamin C. Vitamin E and vitamin C supplementation with exercise elevated mtDNA content in adipose and skeletal muscle to a greater extent (20%) than exercise alone in a depot-specific manner. On the other hand, vitamin C and vitamin E decreased exercise-induced increases in mitochondrial protein content for complex I (40%) and nicotinamide nucleotide transhydrogenase (35%) in a muscle-dependent manner. These data indicate that vitamin E and vitamin C supplementation in obese rodents does not modify exercise-induced improvements in insulin sensitivity but that changes in mitochondrial biogenesis and mitochondrial protein expression may be modified by antioxidant supplementation.
Keywords: insulin resistance, mitochondria, antioxidants, glucose tolerance, biogenesis, ascorbic acid, tocopherol
Introduction
There is controversy as to the effects of supplementation with vitamin E and vitamin C upon the physiologic responses to exercise with respect to adaptations in glucose homeostasis and mitochondrial function. It is well-documented that strenuous exercise induces the generation of reactive oxygen species (ROS) or reactive nitrogen species (RNS) and data suggest that ROS/RNS at the molecular level have a mechanistic role in promoting exercise adaptations, including elevated insulin sensitivity and mitochondrial biogenesis (Bailey et al. 2003; Ferreira and Reid 2008; Powers et al. 2011a, 2011b). Thus, the extent to which supplementation of the diet with vitamin E and vitamin C blocks these ROS-sensitive pathways and perhaps counteracts these pathways is a question with nutritional relevance.
Previous work examining the impact of vitamin E and vitamin C on exercise-induced adaptations has primarily focused on lean/healthy humans and rodents. Data by Ristow et al. and those by Gomez-Cabrera et al. indicate that antioxidant supplementation blocks exercise-induced increases in insulin sensitivity and mitochondrial biogenesis in healthy, lean humans who are untrained or trained (Gomez-Cabrera et al. 2008; Ristow et al. 2009). Similar findings were found in rats, where vitamin C supplementation blunted exercise-induced increases in endurance capacity, increases in ROS detoxification proteins (manganese superoxide dismutase and glutathione peroxidase 1), and increases in the mitochondrial biogenesis markers NRF-1 and mitochondrial transcription factor A (Gomez-Cabrera et al. 2008). The major difference between these studies was the negative impact of vitamin E and vitamin C supplementation on exercise-induced changes in endurance capacity in rodents, but a lack of an effect on a similar outcome measure (maximal oxygen uptake) in humans (Gomez-Cabrera et al. 2008). Paulsen and colleagues show a similar effect whereby vitamin E and vitamin C supplementation negatively impacts exercise-induced changes in skeletal muscle mitochondrial proteins, but had no effect on exercise endurance performance (Paulsen et al. 2014).
Contrary data are reported in studies in both humans and rodents in which vitamin E and vitamin C supplementation has no effect on exercise adaptations. Yfanti et al. reported that supplementation with vitamin E and vitamin C in healthy young adults did not alter exercise-induced increases in oxygen consumption, insulin sensitivity, and multiple molecular markers of muscle adaptation, including elevated citrate synthase and β-hydroxyacyl-CoA dehydrogenase, glucose transporter-4, and hexokinase II (Yfanti et al. 2010, 2011). Similar results occurred in lean rats, where acute and chronic exercise-induced changes in insulin sensitivity and mitochondrial protein content in skeletal muscle were not altered by vitamin E and vitamin C administration (Higashida et al. 2011). In conclusion, the current literature shows varying response to vitamin E and vitamin C supplementation upon exercise-induced effects in both humans and rodents. These effects could be due to differing doses, the quality of the supplementation, and subtle differences in outcome measures. It should also be emphasized that these studies were all conducted in healthy lean humans and rodents.
Currently, more than one-third of Americans are overweight or obese and >1:5 Americans are prediabetic or have type 2 diabetes (Centers for Disease Control and Prevention 2014). Clinical data demonstrate the heightened efficacy of exercise in resolving insulin resistance over pharmacological treatment with the mainstay drug, metformin (Knowler et al. 2002). The 2010 Dietary Guidelines for Americans and the 2008 Physical Activity Guidelines for Americans recommend moderate exercise to prevent overweight and obesity and to reduce the risk of cardiovascular disease and diabetes. Vitamin E and vitamin C supplements are used by >10% of the American population (Rock 2007). Therefore, it is novel and important to understand whether supplementation with vitamin E and vitamin C assists or inhibits the efficacy of exercise to reduce insulin resistance and other obesity-induced effects such as mitochondrial dysfunction in muscle and adipose.
In this work, we tested the hypothesis that supplementation with vitamin E and vitamin C would inhibit the exercise-induced elevation of insulin sensitivity and mitochondrial content in diet-induced obese rats. Furthermore, for the first time, we examined mitochondrial alterations in both muscle and adipose tissue, an important new step given that it is now known that exercise positively impacts adipose tissue mitochondrial in obesity (Laye et al. 2009a, 2009b; Smorlesi et al. 2012; Norheim et al. 2014). Our results indicate that maintenance of a high-fat diet during exercise, even with loss of adipose mass, blunted the predicted decreases in glucose area under the curve (AUC) but not insulin AUC and that supplementation with vitamin C and vitamin E had no effect upon the insulin response. On the other hand, antioxidant supplementation with exercise elevated levels of mtDNA in adipose and skeletal muscle to a greater extent than exercise alone. However, there was a decrease in mitochondrial protein content. These data indicate that mitochondrial biogenesis and mitochondrial protein expression may be separate events modified by antioxidant supplementation.
Materials and methods
All experiments were performed in accordance with the National Institutes of Health (NIH) guidelines for use of live animals and were approved by the Institutional Animal Care and Use Committee of the United States Department of Agriculture (USDA)/Agricultural Research Service, Grand Forks Human Nutrition Research Center. Fifty-six male, obese-prone Sprague–Dawley rats (Crl:OP(CD) strain code: 463) at 4 weeks of age were ordered from Charles River Laboratories International Inc. (Wilmington, Mass., USA). Animals were placed on a modified AIN-93G diet with 10% of the energy (%en) derived from fat for 2 weeks until the beginning of the study.
Diet intervention
A diagram of the study design is shown in Fig. 1. Beginning week 1, the animals were placed on 1 of 2 dietary treatments with 10%en fat (14 animals, control low-fat (LF) diet), or 45%en fat (56 animals, high-fat (HF) diet). Dietary components for LF and HF diets are shown in Table 1 and are essentially the same as previously published (Uthus and Picklo 2011; Picklo et al. 2013). LF rats received a diet composed of protein (20%en, from casein), fat (10%en), and carbohydrate (70%en). Carbohydrate consisted of cornstarch and sucrose. For group 1, the 10%en fat was supplied as lard with added linoleic acid (18:2n-6) and α-linolenic acid (18:3n-3) to meet nutritional requirements for polyunsaturated fatty acids. HF rats received a diet comprising protein (20%en, from casein), fat (45%en), and carbohydrate (35%en), with fat energy solely from lard. All diets were prepared in house. Fatty acid composition of the diets is provided in Table 2. All diets contained AIN93 mineral mix (Dyets Inc.; Bethlehem, Pa., USA) and AIN93-modified vitamin mix (Harlan/Teklad; Madison, Wis., USA). Previous data from our laboratory and others show that animals on the HF diet consumed between 15% to 20% less diet per day than their LF controls. For this reason, the HF diets contained 20% more vitamin mix and mineral mix than the LF diets. The AIN93-modified vitamin mix provided a final vitamin E content of 30 IU/kg diet and was provided as DL-α-tocopheryl acetate. While this level of vitamin E is lower than the AIN93G value (75 IU/kg diet) (Pitts and Bull 1977) the amount used is still within the values recommended for proper growth and immune response (Bendich et al. 1986; National Research Council 1995; Tovar et al. 2006).
Fig. 1.
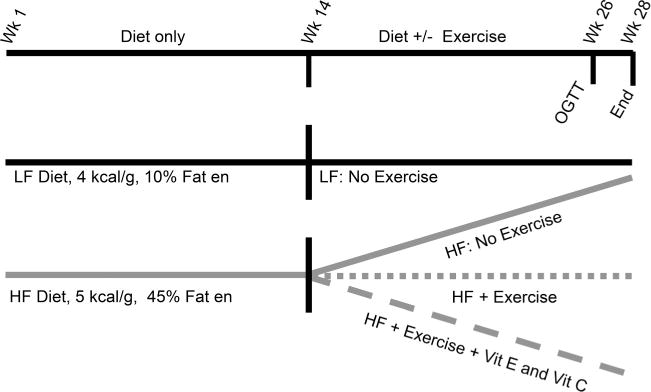
Diagram of study timeline. en, energy; HF, high fat; LF, low fat; OGTT, oral glucose tolerance test; Vit, vitamin.
Table 1.
Diet compositions.
| Diet | LF | HF | HF+CE |
|---|---|---|---|
| Energy, kcal/g diet | 4.2 | 5.0 | 5.0 |
| % Energy | |||
| Carbohydrate | 70 | 35 | 35 |
| Protein | 20 | 20 | 20 |
| Fat | 10 | 45 | 45 |
| Vitamin C, mg/kg | 0 | 0 | 500 |
| Vitamin E, IU/kg | 30 | 36 | 633 |
Note: Data are the mean of 3 independent samples. Values were determined by bomb calorimetry. SD ≤ 0.1 kcal/g. HF, high fat; HF+CE, high fat with added vitamin C and vitamin E; LF, low fat.
Table 2.
Percent energy of individual fatty acids.
| Major fatty acids | 10% fat energy | 45% fat energy |
|---|---|---|
| C4:0–C14:0 | 0.2 | 0.9 |
| C16:0 | 1.5 | 9.6 |
| C18:0 | 0 | 6.6 |
| C18:1n-9 | 2.4 | 15.8 |
| C18:2n-6 | 4.6 | 8.9 |
| C18:3n-3 | 0.4 | 0.5 |
| Total n-3 PUFA | 0.4 | 0.6 |
| Total n-6 PUFA | 4.9 | 9.1 |
| Total MUFA | 2.9 | 24.1 |
| Total saturated | 1.7 | 10.5 |
Note: Data are the mean of 3 samples assayed independently with SD ≤ 0.2% energy. Fatty acid content of the diets were determined by gas chromatography-based fatty acid methyl ester analysis. MUFA, monounsaturated fatty acids; PUFA, polyunsaturated fatty acids.
At week 13 of the study, evidence of hyperglycemia in the HF group was determined by measuring fasting blood glucose concentration in blood withdrawn from the tail. HF animals were evenly distributed by fasting blood sugar into 3 groups: HF sedentary, HF with exercise (HF+Ex), and HF with exercise with added vitamin C and vitamin E in the diet (HF+Ex+CE) with 14 animals in each group. The HF+Ex+CE diet contained 500 mg/kg of added L-ascorbic acid (Acros Organics, Geel, Belgium) and 400 mg/kg (597 IU/kg of D-α-tocopheryl acetate (Sigma–Aldrich, St. Louis, Mo., USA) as the source of vitamin E. The LF group remained on its same diet and was not exercised.
Exercise intervention
At 14, HF+Ex and HF+Ex+CE groups started a forced exercise training regimen using motor driven exercise wheels (Model 80805A; Lafayette Instruments Co, Lafayette, Ind., USA) for a total of 12 weeks. The regimen started at 4 m/min for a total of 200 m/day, 5 times/week, and progressed to 8.7 m/min for a total 1200 m/day, 5 times a week over a 7-week period with this same level of activity maintained for a 5-week period. Following completion of the exercise portion, animals did not exercise for 48 h prior to performance of an oral glucose tolerance test (OGTT).
After the OGTT, the animals were placed back into their respective diet and exercise groups for another 2 weeks prior to euthanasia. Animals were euthanized under overnighted fasting conditions by administration of a 1.37:1 mixture of ketamine (100 mg/mL):xylazine (100 mg/mL) at 1 mL/kg body weight, intra-peritoneal, and exsanguinated by descending vena cava blood draw. Tissues (muscle and adipose) and plasma were quickly frozen in liquid nitrogen and stored at −80 °C until use.
Food consumption was measured twice weekly. Body composition (lean vs fat mass) was measured weekly by a EchoMRI Whole Body Composition Analyzer (Echo Medical Systems, Houston, Texas, USA).
Glucose tolerance and insulin resistance
Glucose tolerance was determined by OGTT. After a 15-h fast, animals were administered glucose (1.4 g glucose/kg lean body mass). Blood was drawn from the tail vein prior to glucose and then at 7, 14, 21, 30, 60, and 120 min postglucose feeding. At each time point glucose was measured using a TRUEresult blood glucose meter and TRUEtest blood glucose test strips (NIPRO Diagnostics, Fort Lauderdale, Fla., USA). Plasma samples were taken for determination of insulin concentration and stored at −80 °C. Insulin was quantified in plasma obtained during the OGTT using the Rat Ultrasensitive Insulin ELISA (ALPCO Diagnostics, Salem, N.H., USA). Insulin sensitivity was determined using the homeostasis model assessment of insulin resistance (HOMA-IR).
Plasma antioxidant analyses
Tocopherol analysis was performed on EDTA plasma collected at the time of euthanasia and stored at −80 °C. Analyses were performed as described previously for this laboratory using a high-performance liquid chromatography method based upon that of Thurnham et al. (Holmstrom et al. 2012). Plasma ascorbate was quantified using a plate reader-based method developed by Vislisel et al. (2007).
Other plasma analyses
Plasma triglyceride and plasma cholesterol content were determined using a COBAS Integra Analyzer with the respective assay kits (Hoffman–LaRoche). Leptin concentration in plasma was determined by enzyme-linked immunosorbent assay using a kit purchased from (ALPCO Diagnostics). Nonesterified fatty acids (NEFA) were determined using the Serum/Plasma Fatty Acid Kit (cat no. SFA-5, Zen-Bio Inc., Research Triangle Park, N.C., USA).
mtDNA determination
mtDNA content in adipose tissue and skeletal muscle was analyzed by quantitative polymerase chain reaction (PCR) of the mitochondrially located Nd1 gene (a resulting protein that contributes to complex 1), which was normalized by quantitative PCR to nuclear DNA as assessed by β-actin gene content (Hancock et al. 2008; Sutherland et al. 2008; Benton et al. 2010). Tissues were digested in proteinase K, and DNA was extracted using the Qiacube automated nucleic acid purification system combined with the DNeasy Blood and Tissue kit (Qiagen, Valencia, Calif., USA) according to manufacturer’s instructions. Quantification and purity of the DNA were tested using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Wilmington, Del., USA). PCR reactions were performed on an ABI 7300 Real-time PCR System with SYBR Green PCR master mix (Applied Biosystems, Life Technologies, Carlsbad, Calif., USA) using default 2-step (95°–60°) amplification. Primer sequences are as follows: β-actin – forward, 5′-TTTACTTTGGGAGTGGCAGCCCTA-3′; reverse, 5′-TGGTGACAATGCCGTGTTCAATGG-3′ and mitochondrially encoded Nd1 gene of the NADH:quinone oxidoreductase (complex 1) – forward, 5′-AGCCGTTGCCCAAACCATCTCTTA-3′; reverse, 5′-TTTGTAGGGAGAAGGAGCCGCTTA-3′. Melting curves were performed with each run to ensure specific Melting curves amplification products.
Mitochondrial protein determination
Tissue (adipose and skeletal muscle) content of mitochondrial respiratory proteins was determined by dot blot analysis or Western blot analysis. Muscle samples were homogenized in the buffer consisting of 20 mmol/L Na2HPO4 (pH 7.0), 1 mmol/L diethyl-enetriaminepentaacetic acid and 50 μmol/L 2,6-di-tert-butyl-4-methy phenol (BHT), 0.5% Triton X-100, and protease inhibitor cocktail (Sigma–Aldrich). All samples were reduced with 0.1 mol/L dithiothreitol prior to use. Skeletal muscle samples (0.5 μg/0.1 mL) were applied to a polyvinylidene fluoride (PVDF) membrane using the Bio-Rad Dot Blot Apparatus. Following application of vacuum, each well was washed in Tris-buffered saline (TBS). For adipose samples, tissues were prepared using acetone extraction of adipose proteins to remove excessive lipid. Adipose tissue was sonicated in homogenizing buffer (1:2, w/v) and then centrifuged (5000g, 10 min, 4 °C). The infranatant was pipetted into a separate tube, sonicated, and centrifuged (10 000g, 10 min, 4 °C). Acetone was added to the supernatant (9:1, v/v), mixed by inversions and incubated on ice for 15 min. The samples were then centrifuged (2000g, 10 min, 4 °C), decanted, and protein dried under nitrogen. Adipose proteins were resolved by SDS-PAGE followed by transfer of the proteins to PVDF membranes. Protein loading was normalized through reversible staining of the membranes with MemCode reversible stain (ThermoFisher; Rockford, Ill., USA) and quantitation of the resulting signal.
For all blots, after blocking with 5% nonfat dry milk in TBS, membranes were incubated overnight at 4 °C with the appropriate primary antibodies diluted in 5% nonfat dry milk in Tris-buffered saline Tween-20 (TBST). Following washing of the blots in TBST, they were incubated for 2 h at 20 °C in the anti-mouse secondary antibody (Promega, Madison, Wis., USA) conjugated to horseradish peroxidase diluted in 5% nonfat dry milk in TBST (1:6000). Blots were developed and quantified as described above. Mouse anti-complex 1 subunit, predicted mass 39 kDa (Anti-NDUFA9, 1:2,000), mouse anti-complex II, predicted mass 70 kDa (SDHA; 1:10,000), and mouse anti-complex IV subunit, predicted mass 16 kDa (1:4000) were purchased from Abcam (Cambridge, Mass., USA). Mouse anti-nicotinamide nucleotide transhydrogenase, predicted mass 114 kDa (anti-NNT; 1:2000) was purchased from Abnova (Taiwan). Prior to development of the dot blot assays, it was verified by Western blot that the antibodies used recognized only a single protein of the expected mass (see Supplementary Figs. S1 and S21).
Diet analysis
Energy content of the diets was determined by bomb calorimetry. Samples were compressed into a pellet and then ignited in an oxygen-rich closed environment using a Parr Bomb Calorimeter (Parr Instrument Company, Moline, Ill., USA). Calibration of the bomb calorimeter was standardized using benzoic acid (Parr cat. no. 3413). Samples were analyzed in triplicate and a National Institute of Standards and Technology (NIST) standard, Typical Diet SRM 1548A (NIST Gaithersburg, Md., USA), was used for quality control. Fatty acid content of the diet was determined through fatty acid methyl ester/gas chromatography analysis (Uthus and Picklo 2011).
Statistics
Glucose AUCs and insulin AUC determination was performed using GraphPad Prism version 5.00 for Windows (GraphPad Software; San Diego, Calif., USA (available from www.graphpad.com)). Glucose and insulin curves derived from the OGTT were compared using a repeated-measures ANOVA with Tukey contrasts. Other data were analyzed using 1-way ANOVA or a 2-way repeated measures ANOVA as appropriate. Two-way repeated measures ANOVA was used to compare the effects on muscle group, dietary treatment, and their interactions. Statistical comparisons were made using GraphPad software or SAS (version 9.3; SAS Institute Inc., Cary, N.C., USA). Statistical significance was taken as p ≤ 0.05. All data are expressed as the mean ± SD unless otherwise stated.
Results
In this work, we examined the extent to which supplementation with vitamin E and vitamin C modulated exercise adaptations in animals made obese by an HF hypercaloric diet prior to the onset of exercise. Rats in the sedentary HF group developed elevated adiposity in the 13-week run-in prior to exercise (Table 3). Animals in the initial HF grouping had a significantly higher blood glucose content (73 ± 6 mg/dL; n = 42) than the control animals (63 ± 6 mg/dL; n = 14) prior to assignment into the exercise intervention groupings. Following the start of exercise, there was limited weight gain in the HF+Ex or HF+Ex+CE groups as compared with the control LF and HF sedentary groups. The percent adiposity of the HF+Ex and HF+Ex+CE groups remained constant in contrast with the LF and HF sedentary groups that increased the percentage of fat mass (Table 3). Following the onset of exercise, all groups increased lean body mass; however, by the end of the exercise period, the HF sedentary group had a minor but significantly higher lean body mass than the LF and HF+Ex groups. Detailed data regarding the time course of body composition during the study are provided in Supplementary Fig. S31.
Table 3.
Body composition before the exercise period (wk 12) and prior to the OGTT (wk 25).
| Group | Body mass, g
|
Lean body mass, g
|
Body fat mass, g
|
Body fat %
|
||||
|---|---|---|---|---|---|---|---|---|
| Wk 12 | Wk 25 | Wk 12 | Wk 25 | Wk 12 | Wk 25 | Wk 12 | Wk 25 | |
| LF, n = 14 | 482a (33) | 588a* (40) | 336a (24) | 377a* (32) | 85a (17) | 139a* (22) | 18a (3) | 22a* (3) |
| HF, n = 13 | 571b (40) | 729a* (62) | 358ab (19) | 401a* (29) | 146b (35) | 237b* (50) | 26b (5) | 32b* (5) |
| HF+Ex, n = 14 | 558b (38) | 592a* (29) | 358b (20) | 380a* (19) | 135b (26) | 135a (20) | 24b (3) | 23a (3) |
| HF+Ex+CE, n = 13 | 587b (39) | 611a* (24) | 367ab (25) | 387a* (27) | 153b (25) | 147a (32) | 26b (4) | 24a (5) |
Note: Values are means (SD). Data were compared using a repeated measures ANOVA with Tukey contrasts. Values with different letters are significantly different (p < 0.05) compared with another groups within the respective week for the given parameter. HF, high fat; HF+Ex, high fat with exercise; HF+Ex+CE, HF with exercise with added vitamin C and vitamin E in the diet; LF, low fat; OGTT, oral glucose tolerance test.
Significant difference (p < 0.05) between wk 12 and wk 25 for a given group and parameter.
Consistent with the higher levels of adiposity, HF sedentary rats had higher masses for liver, perirenal adipose, and epididymal adipose (Table 4). Elevations in hepatic mass and adipose mass were blunted by exercise although the perirenal adipose tissue of the HF+Ex+CE was significantly higher than that of the LF groups. Consistent with elevated adiposity, HF animals had higher plasma levels of leptin, cholesterol, and high-density lipoprotein than the LF group, with effects blunted in the exercising groups (Table 5). Plasma content of NEFA and triglycerides were not elevated in the HF groups versus the LF group; however, values for these endpoints were significantly reduced in the HF+Ex+CE group.
Table 4.
Final body and tissue masses.
| Animal group | LF | HF | HF+Ex | HF+Ex+CE |
|---|---|---|---|---|
| n | 14 | 13 | 14 | 13 |
| Final mass, g | ||||
| Mean | 589a | 764b | 598a | 617a |
| SD | 39 | 72 | 30 | 32 |
| Liver, g | ||||
| Mean | 14.1a | 17.9b | 14.3a | 15.5ab |
| SD | 2.5 | 2.9 | 2.2 | 2.2 |
| Perirenal adipose, g | ||||
| Mean | 22.3a | 57.2b | 29.7ac | 34.2c |
| SD | 4.6 | 14.1 | 7.3 | 8.8 |
| Epididymal adipose, g | ||||
| Mean | 14.8a | 20.7b | 14.2a | 15.1a |
| SD | 2.4 | 2.6 | 2.2 | 2.9 |
Note: Data were compared using a 1-way ANOVA with Tukey’s contrasts. Values with different superscripts are significantly different (p < 0.05) from values within the same endpoint group/row. HF, high fat; HF+Ex, high fat with exercise; HF+Ex+CE, HF with exercise with added vitamin C and vitamin E in the diet; LF, low fat.
Table 5.
Plasma levels of obesity-related and antioxidant endpoints.
| Animal group | LF | HF | HF+Ex | HF+Ex+CE |
|---|---|---|---|---|
| n | 14 | 13 | 14 | 13 |
| NEFA, μmol/L | ||||
| Mean | 452ab | 516a | 459ab | 338b |
| SD | 171 | 196 | 146 | 134 |
| Triglycerides, mg/dL | ||||
| Mean | 247ab | 381a | 209ab | 291b |
| SD | 85 | 145 | 101 | 176 |
| Cholesterol, mg/dL | ||||
| Mean | 91a | 171b | 99a | 137a |
| SD | 26 | 92 | 40 | 53 |
| HDL, mg/dL | ||||
| Mean | 64a | 100b | 67a | 80a |
| SD | 14 | 46 | 22 | 13 |
| Leptin, ng/mL | ||||
| Mean | 0.69a | 2.37b | 0.78a | 0.93a |
| SD | 0.36 | 1.01 | 0.27 | 0.46 |
| Ascorbate, μmol/L | ||||
| Mean | 43 | 52 | 53 | 53 |
| SD | 21 | 28 | 44 | 18 |
| α-Tocopherol:cholesterol | ||||
| Mean | 0.28a | 0.22a | 0.26a | 0.65b |
| SD | 0.04 | 0.02 | 0.04 | 0.09 |
Note: Plasma was taken at the end of study following anesthesia. α-Tocopherol is expressed normalized to plasma cholesterol content. Values with different letters are significantly different (p < 0.05) from values within the same analyte row. HF, high fat; HF+Ex, high fat with exercise; HF+Ex+CE, HF with exercise with added vitamin C and vitamin E in the diet; LF, low fat; NEFA, nonesterified fatty acids.
Plasma α-tocopherol levels and vitamin C were measured in fasting blood taken at the end of the study (Table 5). The plasma level of vitamin C was not elevated in the exercising, supplemented group versus the nonsupplemented groups. Plasma α-tocopherol levels were elevated in the HF+E+CE group compared with the other groups.
Prior to exercise, the mass of diet consumed by the HF-fed animals was similar to the LF animals (Table 6). Given the higher caloric content of the HF diet (5.0 kcal/g) versus the LF diet (4.2 kcal/g), the HF groups consumed a significantly higher amount of kilocalories per day prior to the onset of exercise. By the end of the exercise period, the exercising animals ate less diet than LF and the HF; however, when corrected for the total amount of kilocalories per day that was eaten, the exercising rats and the LF rats consumed a similar amount of energy. When caloric intake was normalized to body mass before the onset of exercise and after the exercise period, all groups had a similar intake of calories per day. All groups had a decrease in energy consumed per day per gram body weight in the period when some animals exercised and other did not. These data indicate that exercise was not a factor in the decrease in total energy intake in this period.
Table 6.
Food intake and caloric intake before the exercise period (wk 12) and prior to the oral glucose tolerance test (wk 25).
| Group | Food intake, g/d
|
Caloric intake, kcal/d
|
Caloric intake/body mass, kcal/(d·g)−1
|
|||
|---|---|---|---|---|---|---|
| Wk 12 | Wk 25 | Wk 12 | Wk 25 | Wk 12 | Wk 25 | |
| LF | 18.8a (1.2) | 18.2a (2.4) | 79.2a (5.2) | 76.5a (10.1) | 0.16 (0.01) | 0.13* (0.02) |
| HF | 18.5b (1.5) | 20.7a (2.5) | 92.5b (7.3) | 103.7b* (12.4) | 0.16 (0.01) | 0.14* (0.01) |
| HF+Ex | 18.4b (1.7) | 16.6b (1.4) | 92.1b (8.6) | 82.9a (7.1) | 0.17 (0.01) | 0.14* (0.02) |
| HF+Ex+CE | 19.4b (2.5) | 15.2b* (4.6) | 97.2b (12.4) | 82.0a* (6.5) | 0.17 (0.01) | 0.13* (0.01) |
Note: Data are means (SD); n = 13 or 14 for each group. Values with different superscript letters are significantly different (p< 0.05) compared with other groups within the respective week for the given parameter. HF, high fat; HF+Ex, high fat with exercising; HF+Ex+CE, high fat with exercise with vitamin C and vitamin E; LF, low fat. Note that the LF and HF groups did not exercise.
Values were significantly different (p < 0.05) at wk 12 vs wk 25 for the given parameter.
Following exercise, we examined glucose intolerance and insulin resistance through use of an OGTT with determination of blood glucose levels and plasma insulin levels (Fig. 2). Baseline fasting glucose levels were not significantly different between the treatment groups, whereas fasting insulin levels were 2.5-fold greater in the HF group versus the LF group. Fasting hyperinsulinemia was ameliorated in both exercising groups. As expected, based upon fasting levels of glucose and insulin, animals on the HF diet had a higher HOMA-IR than animals in the other groups. Analysis of glucose versus time from the OGTT showed an elevated glucose AUC in the HF versus LF animals, an effect that was not reversed in either exercising groups. This response is supported by the elevated levels of glucose at the latter time point of the AUC in the HF, HF+Ex, and HF+Ex+CE groups versus the LF group. On the other hand, insulin levels were significantly higher in the sedentary HF group versus the exercising and LF groups at all time-points during the OGTT. The greater insulin response during an OGTT is indicative of worsening insulin resistance.
Fig. 2.
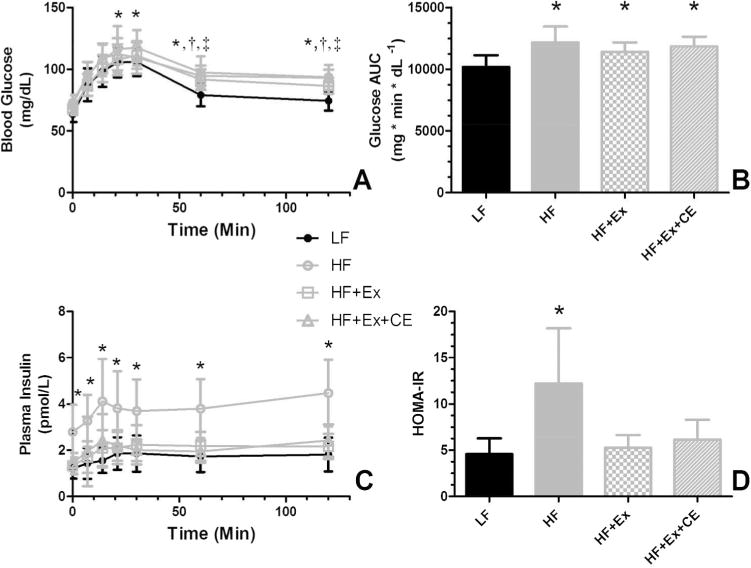
Supplementation with vitamin C and vitamin E does not block restoration of insulin sensitivity. Following 12 weeks of exercise or sedentary activity on the respective diets, rat underwent an OGTT. (A) Blood glucose levels over time. Note that the glucose over time curves for all HF-fed groups were significantly different than the LF control group. (B) Glucose AUC determinations derived from the glucose responses. Note that exercise while consuming the HF diet did not reduce the glucose AUC. (C) Plasma insulin levels during the OGTT. The insulin versus time curves for the HF sedentary groups was significantly different from those of the other groups. The LF and the exercising groups (HF+Ex and HF+Ex+CE) did not differ. (D) HOMA-IR scores derived from time zero data of the glucose and insulin curves. For (A) and (C), curves were compared using a repeated measures ANOVA with Tukey contrasts. For (B) and (D), data were compared using a 1-way ANOVA with Tukey contrasts and bars with different letters are significantly different (p < 0.05). Data in all graphs are the mean ± SD (n = 13 or 14). *, HF different from LF; †, HF+Ex different from LF; and ‡, HF+Ex+CE different from LF points. AUC, area under the curve; HF, high fat; HF+Ex, high fat with exercise; HF+Ex+CE, high fat with exercise with vitamin E and vitamin C; HOMA-IR, homeostasis model assessment of insulin resistance; LF, low fat; OGTT, oral glucose tolerance test.
We next examined the extent to which exercise or exercise plus vitamin E and C altered mitochondrial protein content and mitochondrial copy number in skeletal muscle and adipose depots. Analysis of skeletal muscle mtDNA and mitochondrial proteins were performed on gastrocnemius, soleus, and tricep (Fig. 3). Our data demonstrate an increase in mtDNA content as a result of exercise plus supplementation with vitamin C and vitamin E in gastrocnemius and tricep compared with exercise alone or HF-diet alone treatments. Analysis of the data indicates that maintenance of the animals on an HF diet resulted in a decrease of mtDNA, an effect reversed in the group receiving exercise and antioxidants (Fig. 3A).
Fig. 3.
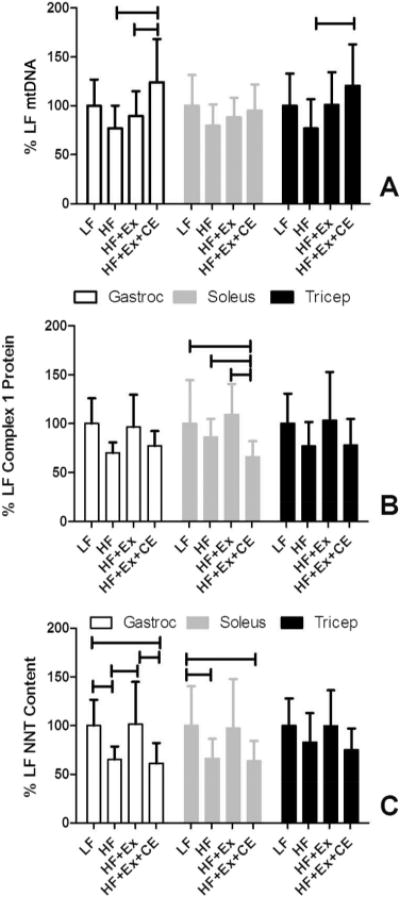
Supplementation with vitamin C and vitamin E modifies mitochondrial changes in response to exercise in skeletal muscle. mtDNA content (A) and mitochondrial protein content (B, C) of multiple muscles were determined following the end of the exercise period for sedentary (LF and HF) and exercising rats (HF+Ex and HF+Ex+CE). The high-fat diet in sedentary rats significantly suppressed mtDNA and mitochondrial protein content. Antioxidant supplementation blocked the ability of exercise to increase mitochondrial protein content. Two-way repeated-measures ANOVA was used to compare the effects on muscle group, dietary treatment, and their interactions. Horizontal bars denote the experimental groups that are significantly different (p < 0.05) within a specific muscle type. HF, high fat; HF+Ex, high fat with exercise; HF+Ex+CE, high fat with exercise with vitamin E and vitamin C; LF, low fat. Data are the mean ± SD (n = 13 or 14).
We further examined the extent to which exercise and supplementation with vitamin C and vitamin E modified mitochondrial protein content in these muscles through analysis of complex I and nicotinamide nucleotide transhydrogenase (NNT). We chose complex I as its activity is rate limiting for the electron transport chain. NNT is an inner mitochondrial membrane protein that catalyzes the reversible hydride transfer between NADH and NADPH and whose deletion in C57BL/6 mice is associated with insulin resistance (Freeman et al. 2006; Anderson et al. 2009; Parker et al. 2009; Nicholson et al. 2010; Wong et al. 2010). Contrary to the findings with mtDNA, there was no significant decrease in the complex I protein subunit measured in the HF diet-induced obese animals versus LF diet-fed animals (Fig. 3B). On the other hand, in soleus muscle, we observed a decrease in the levels of the target complex I subunit protein in exercising animals receiving vitamin C and vitamin E supplementation versus the LF controls, obese sedentary animals, and the exercising animals with antioxidant supplementation.
A similar, but more pronounced effect was observed with NNT (Fig. 3C). In this case, obesity itself reduced levels of NNT in gastrocnemius and soleus muscle. In gastrocnemius, exercise returned NNT levels of obese rats back to that of the LF animals, an effect blocked by supplementation with vitamin E and vitamin C. In the soleus muscle of supplemented animals, NNT content was significantly lower than that of the control (LF group).
Previous data indicate that an obese state decreases mitochondrial copy number in white adipose tissue and that conversely, exercise may increase the number of mitochondria leading to “browning” of adipose tissue (Smorlesi et al. 2012; Palou et al. 2013; Norheim et al. 2014). Exercise significantly enhanced mtDNA content in epididymal adipose over nonexercising HF animals but not in peri-renal adipose (Fig. 4A). For analysis of mitochondrial protein in adipose tissue, the Western blot signals for the complex I subunit and NNT were too faint to quantify reliably. Thus, analysis of mitochondrial proteins was performed by Western blot using antibodies against complex II and complex IV subunits. Content of mitochondrial proteins in perirenal adipose and epididymal adipose, complex II and complex IV, were not altered between the groups (Fig. 4B, 4C). Representative Western blots are provided in Supplementary Fig. S41.
Fig. 4.
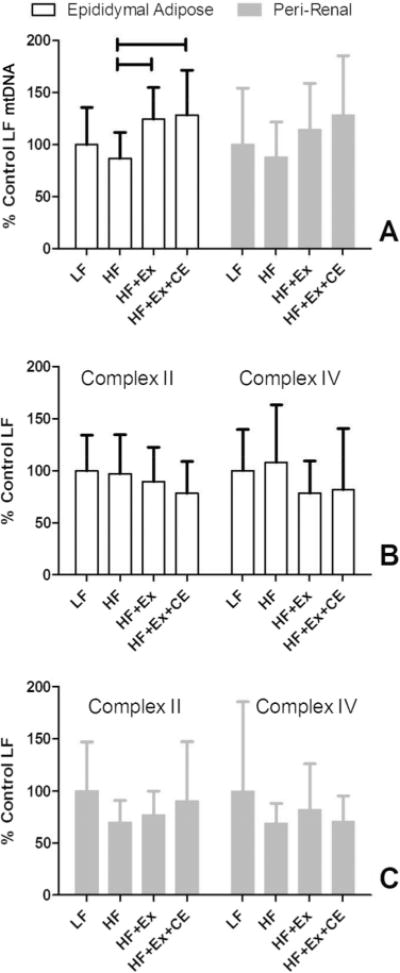
Supplementation with vitamin C and vitamin E modifies mtDNA changes in visceral adipose. mtDNA content (A) and mitochondrial protein content (B, C) of visceral, epididymal adipose depots were determined following the end of the exercise period for sedentary (LF and HF) and exercising rats (HF+Ex and HF+Ex+CE). Horizontal bars denote the specific groups that are significantly different (p < 0.05) within a specific muscle type. HF, high fat; HF+Ex, high fat with exercise; HF+Ex+CE, high fat with exercise with vitamin E and vitamin C; LF, low fat. Data are the mean ± SD (n = 13 or 14). Data were compared using a 1-way ANOVA with Tukey contrasts.
Discussion
Controversy exists as to whether supplementation with the antioxidants vitamin C and vitamin E blunt exercise-induced changes in insulin sensitivity and mitochondrial content. Studies addressing this topic so far have been conducted in lean healthy adults or in lean rodents. However, exercise is commonly recommended for 60% of the population who are overweight and obese as a method to prevent and treat insulin resistance, a condition that is estimated to affect >70 million Americans (Centers for Disease Control and Prevention 2014). Thus is it important to know the extent to which dietary antioxidant supplementation modifies exercise-induced adaptations in the context of obesity. In this work, we tested the hypothesis that supplementation with vitamin E and vitamin C blunts exercise-induced increases in insulin sensitivity and mitochondrial changes in obese rats. Our data indicate that vitamin C and vitamin E supplementation does not block improvements in insulin sensitivity but does blunt exercise-induced increases in mitochondrial protein content in skeletal muscle, particularly the NNT protein.
We performed OGTTs to analyze glucose intolerance and our experiment was sufficiently powered to detect to detect a 12% change in the glucose AUC. We found that HF-diet induced obesity did increase glucose AUCs compared with our LF group. However, fasting glucose was maintained in the HF-diet obese group because of pancreatic β-cell compensation or the production of more insulin to offset diminished peripheral insulin sensitivity (Kahn 2001). The maintained glucose tolerance because of increased postprandial insulin levels matches what occurs in the development of obesity in human subjects, as obese individuals often maintain glucose control for long periods of time because of β-cell compensation (Kahn 2001; Rizza 2010). However, the magnitude of the insulin response during an OGTT can be used as a surrogate of peripheral of insulin sensitivity in rodents prior to β-cell dysfunction found in frank diabetes (Noland et al. 2007; Naples et al. 2010).
Insulin AUC responses during the OGTT were used as a surrogate of insulin sensitivity, and the data support previous findings that supplementation with vitamin E and vitamin C does not impair the ability of exercise to improve insulin sensitivity (Higashida et al. 2011; Yfanti et al. 2011). Both insulin AUC during the OGTT and fasting insulin levels were identical between the LF, HF+Ex, and HF+Ex+CE groups. These data are in contrast with those reported by Ristow et al. (2009) but do agree with those data reported by Yfanti et al. (2011). An important difference in our study compared with those performed previously is that we induced both obesity and insulin resistance with an HF-diet and then treated with exercise whereas in contrast, existing clinical studies had tested the influence of vitamin E and vitamin C on exercise-induced improvements in insulin sensitivity in normal weight, otherwise healthy individuals (Ristow et al. 2009; Yfanti et al. 2011). Although mechanism(s) are unknown, there are undoubtedly differences in the mechanism(s) by which exercise treats or impacts an insulin-resistant condition in muscle and adipose versus compared with its effects on an already insulin-sensitive system. For example, insulin resistance is associated with impaired insulin signaling, lower mitochondrial content or function, and increased adiposity and inflammation, which are factors that are likely not present in nonobese individuals (Koves et al. 2008; Makki et al. 2013; Esser et al. 2014; Guo 2014; Martin and McGee 2014; Rutkowsky et al. 2014).
In contrast with insulin sensitivity, our data indicate that supplementation with vitamin E and vitamin C modified mitochondrial protein content. While exercise for 12 weeks (following 16 sedentary weeks on an HF diet) returned skeletal muscle levels of the complex I subunit and NNT to the LF levels in gastrocnemius and soleus, antioxidant supplementation blocked this effect such that the animals in the HF+Ex+CE groups had mitochondrial protein levels that were the same as the HF sedentary group that had been on the HF diet for over 6 months. Our data disagree with results obtained by Yfanti et al. and by Higashida et al. (Higashida et al. 2011; Yfanti et al. 2011) in which supplementation with vitamin C and vitamin E had no effect upon training-induced increases in mitochondrial respiratory endpoints. However, our experimental context, existing obesity, again differs from those studies and others in which healthy lean humans or untrained lean rats were studied (Ristow et al. 2009; Yfanti et al. 2010, 2011; Higashida et al. 2011; Paulsen et al. 2014). While we recognize that our studies did not include analysis of exercise performance endpoints, our data do agree with those demonstrating a blunting of exercise-induced mitochondrial changes with antioxidant supplementation (Gomez-Cabrera et al. 2008; Ristow et al. 2009; Paulsen et al. 2014).
We chose levels of vitamin C and vitamin E that were comparable to those used in previous supplementation trials in human and rodents. In our study, daily intake of vitamin C was calculated to be approximately 16 mg/(kg body mass·day)−1 and vitamin E at 21 IU/(kg·day)−1. Studies in rats by Vinayagamoorthi and colleagues demonstrated an increase in insulin sensitivity in obese rats that had estimated intake of 50 mg/(kg·day)−1 and 26 IU/(kg·day)−1 for vitamin C and vitamin E, respectively, in addition to 50 mg/(kg·day)−1 of α-lipoic acid. (Vinayagamoorthi et al. 2008). Experiments performed in rats by Higashida and colleagues used vitamin C at 135 mg/(kg·day)−1 and vitamin E at 36 IU/(kg·day)−1 and demonstrated no effect of supplementation on mitochondrial and insulinemic endpoints related to exercise (Higashida et al. 2011). Studies in rats by Gomez-Cabrera and colleagues showing an exercise-inhibiting effect of vitamin C used vitamin C at 500 mg/(kg·day)−1 (Gomez-Cabrera et al. 2008). In relation to human studies, those performed by Yfanti and colleagues and by Ristow and colleagues use vitamin C at approximately 6.3 mg/(kg·day)−1 and vitamin E at 5 IU/(kg·day)−1 (Ristow et al. 2009; Yfanti et al. 2010, 2011). In a clinical trial examining the exercise-related effects of vitamin C supplementation, Gomez-Cabrera and colleagues used vitamin C at 13 mg/(kg·day)−1. We point out that supplementation levels used in our studies and those described above are several-fold higher that the recommended daily allowance of vitamin C (90 mg/day) and vitamin E (22.4 IU/day), about 1.3 mg/(kg·day)−1 and 0.32 IU/(kg·day)−1, for a 70-kg individual (Institute of Medicine 2000).
The plasma level of ascorbate was not elevated in the rats receiving the diet containing 500 mg/kg of sodium ascorbate. One potential explanation is that the overnight fasting period prior to collection of blood following sacrifice resulted in the clearance of the additional sodium ascorbate from the plasma through excretion or distribution into tissue pools (Rumsey and Levine 1998; Corti et al. 2010). Data from human studies indicate that a bolus dose of sodium ascorbate is cleared from the plasma within 4 h (Graumlich et al. 1997; Rumelin et al. 2005).
Varying data exist as to the effects of obesity in humans and rodents on an HF diet upon mitochondrial outcomes. Our data demonstrate decreases in the content of complex I and NNT in skeletal muscle as a result of HF, diet-induced obesity and are in agreement with data derived in human studies (Menshikova et al. 2005; Toledo et al. 2007, 2008; Ritov et al. 2010; Toledo and Goodpaster 2013) and rodents (Sparks et al. 2005; Bonnard et al. 2008). Some studies in rodents demonstrate increases in mitochondrial respiratory chain proteins (Turner et al. 2007; Hancock et al. 2008) while other have shown changes (either increases and decreases) in the tissue content of mitochondria only in selective proteins and in muscle-dependent manners in obese animals (Holmstrom et al. 2012; Stephenson et al. 2012). This variability may be attributed to differences in the percent energy from fat, the different fatty acid sources, and the length of time over which the diets were fed. Alterations in circulating fatty acids are may influence mitochondrial adaptations secondary to activation of nuclear transcriptional factors, such as the peroxisome proliferator activated receptors (Garcia-Roves et al. 2007; Hancock et al. 2008). However, nonesterified fatty acids were not different between the LF and HF groups in our study.
In obese humans, exercise does not increase mtDNA content in skeletal muscle although mitochondrial protein content increases, results of which are reflected in our study (Menshikova et al. 2005, 2007). That supplementation with vitamin C and vitamin E along with exercise significantly increased mtDNA content in the gastrocnemius muscle over the HF and HF+Ex groups is intriguing. Our data demonstrate that mtDNA content is not positively associated with mitochondrial protein content in that supplementation with vitamin E and vitamin C reduced content of mitochondrial proteins while not diminishing mtDNA. We are cautious in our interpretation of the data from skeletal muscle given the rather small increase observed and variability of the data. However, our work is consistent with literature showing discrepancies between mtDNA copy number from mitochondrial protein content (Menshikova et al. 2007; Laye et al. 2009b; Fernandez-Vizarra et al. 2011; Larsen et al. 2012; Pohjoismaki et al. 2012).
Our data demonstrate that NNT expression in skeletal muscle is dynamic, being modulated by consumption of an HF diet, exercise, and antioxidants. The literature indicates that the mitochondrial protein NNT is related to the development of insulin resistance (Freeman et al. 2006; Aston-Mourney et al. 2007; Anderson et al. 2009; Parker et al. 2009; Nicholson et al. 2010; Wong et al. 2010). These data are based upon initially upon work showing that age-related insulin resistance in C57Bl/6J mice was located to the NNT locus. Data by Heiker et al. show that the mRNA for NNT is elevated in the visceral, but not subcutaneous adipose tissue of obese individuals (Heiker et al. 2013). To our knowledge, these are the first data showing that NNT content in skeletal muscle is modulated by obesity. The extent to which NNT expression is regulated by pathways modulated by ROS or RNS such as the Nrf2/Antioxidant Response Element pathway has not been investigated. However, a recent paper demonstrated that ablation of NNT reduces mitochondrial detoxification of hydrogen peroxide, inhibition of the mitochondrial thioredoxin and peroxiredoxin, reduction of cellular free glutathione, and decreases in cellular respiration (Lopert and Patel 2014). Our data are limited in that we cannot assess whether the levels of NNT per mitochondrion are altered or whether these changes are a reflection of changes in mitochondrial content per tissue; however, our data suggest that measurement of NNT may be a more robust indicator of mitochondrial content than respiratory complexes.
We are cognizant that the caloric intake of the exercising animals decreased and may have contributed to the small but significant decrease in percentage of adiposity, effects observed in other studies (Oscai and Holloszy 1969; Applegate et al. 1982, 1984; Noland et al. 2007; Naples et al. 2010). Furthermore, the changes in insulin sensitivity may be the result of a reduction in adipose tissue in addition to physical activity. Our data show that during the time during the 12-week period in which animals were sedentary or exercising, a decrease in the energy intake when normalized to body mass was observed in all groups. These data indicate that changes in adiposity and insulin sensitivity are not merely the result of exercise-induced inhibition of food consumption. Studies by Toledo et al. indicate that dietary restriction plus exercise stimulate mitochondrial function and insulin sensitivity in obese individuals, whereas dietary restriction alone, while improving insulin sensitivity, does not improve mitochondrial function (Toledo et al. 2008). As decrements in food intake resulting from exercise is noted in male rats, but not female rats, subsequent studies in female rats are warranted to understand the response of exercise without decrements in food intake (Applegate et al. 1982).
In conclusion, our data demonstrate that supplementation with vitamin E and vitamin C does not prevent normalization of insulin sensitivity in response to exercise in preexisting obesity. However, supplementation does prevent exercise-induced elevations in mitochondrial proteins. Subsequent data are needed to determine molecular mechanisms and chemical signaling pathways that are modified by antioxidant supplementation.
Supplementary Material
Acknowledgments
The authors thank Joseph Idso, Kim Michelsen, and Brian Gregoire for their excellent technical assistance. Funding was provided through USDA-ARS Project 5450-51000-048-00D. The U.S. Department of Agriculture, Agricultural Research Service, Plains Area, is an equal opportunity/affirmative action employer and all agency services are available without discrimination. Salary support for J.P.T. was funded by NIH RO1DK088940. Mention of trade names or commercial products in this article is solely for providing specific information and does not imply recommendation or endorsement by the USDA.
Footnotes
Supplementary data are available with the article through the journal Web site at http://nrcresearchpress.com/doi/suppl/10.1139/apnm-2014-0302.
Contributor Information
Matthew J. Picklo, USDA-ARS Grand Forks Human Nutrition Research Center, 2420 2nd Avenue North, Grand Forks, ND 58201, USA
John P. Thyfault, Department of Nutrition and Exercise Physiology and Medicine, Division of Gastroenterology and Hepatology, University of Missouri, Columbia, MO 65212, USA.
References
- Anderson AA, Helmering J, Juan T, Li CM, McCormick J, Graham M, et al. Pancreatic islet expression profiling in diabetes-prone C57BLKS/J mice reveals transcriptional differences contributed by DBA loci, including Plagl1 and Nnt. Pathogenetics. 2009;2:1. doi: 10.1186/1755-8417-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Applegate EA, Upton DE, Stern JS. Food intake, body composition and blood lipids following treadmill exercise in male and female rats. Physiol Behav. 1982;28:917–920. doi: 10.1016/0031-9384(82)90214-1. [DOI] [PubMed] [Google Scholar]
- Applegate EA, Upton DE, Stern JS. Exercise and detraining: effect on food intake, adiposity and lipogenesis in Osborne-Mendel rats made obese by a high fat diet. J Nutr. 1984;114:447–459. doi: 10.1093/jn/114.2.447. [DOI] [PubMed] [Google Scholar]
- Aston-Mourney K, Wong N, Kebede M, Zraika S, Balmer L, McMahon JM, et al. Increased nicotinamide nucleotide transhydrogenase levels predispose to insulin hypersecretion in a mouse strain susceptible to diabetes. Diabetologia. 2007;50:2476–2485. doi: 10.1007/s00125-007-0814-x. [DOI] [PubMed] [Google Scholar]
- Bailey DM, Davies B, Young IS, Jackson MJ, Davison GW, Isaacson R, Richardson RS. EPR spectroscopic detection of free radical outflow from an isolated muscle bed in exercising humans. J Appl Physiol. 2003;94:1714–1718. doi: 10.1152/japplphysiol.01024.2002. [DOI] [PubMed] [Google Scholar]
- Bendich A, Gabriel E, Machlin LJ. Dietary vitamin E requirement for optimum immune responses in the rat. J Nutr. 1986;116:675–681. doi: 10.1093/jn/116.4.675. [DOI] [PubMed] [Google Scholar]
- Benton CR, Holloway GP, Han XX, Yoshida Y, Snook LA, Lally J, et al. Increased levels of peroxisome proliferator-activated receptor gamma, coactivator 1 alpha (PGC-1alpha) improve lipid utilisation, insulin signalling and glucose transport in skeletal muscle of lean and insulin-resistant obese Zucker rats. Diabetologia. 2010;53:2008–2019. doi: 10.1007/s00125-010-1773-1. [DOI] [PubMed] [Google Scholar]
- Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, et al. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. doi: 10.1172/JCI32601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. 2014. [Google Scholar]
- Corti A, Casini AF, Pompella A. Cellular pathways for transport and efflux of ascorbate and dehydroascorbate. Arch Biochem Biophys. 2010;500:107–115. doi: 10.1016/j.abb.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Esser N, Legrand-Poels S, Piette J, Scheen AJ, Paquot N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res Clin Pract. 2014;105(2):141–150. doi: 10.1016/j.diabres.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Enriquez JA, Perez-Martos A, Montoya J, Fernandez-Silva P. Tissue-specific differences in mitochondrial activity and biogenesis. Mitochondrion. 2011;11:207–213. doi: 10.1016/j.mito.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Ferreira LF, Reid MB. Muscle-derived ROS and thiol regulation in muscle fatigue. J Appl Physiol (1985) 2008;104:853–860. doi: 10.1152/japplphysiol.00953.2007. [DOI] [PubMed] [Google Scholar]
- Freeman H, Shimomura K, Cox RD, Ashcroft FM. Nicotinamide nucleotide transhydrogenase: a link between insulin secretion, glucose metabolism and oxidative stress. Biochem Soc Trans. 2006;34:806–810. doi: 10.1042/BST0340806. [DOI] [PubMed] [Google Scholar]
- Garcia-Roves P, Huss JM, Han DH, Hancock CR, Iglesias-Gutierrez E, Chen M, Holloszy JO. Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc Natl Acad Sci USA. 2007;104:10709–10713. doi: 10.1073/pnas.0704024104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, et al. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr. 2008;87:142–149. doi: 10.1093/ajcn/87.1.142. [DOI] [PubMed] [Google Scholar]
- Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LR, Jr, Wang Y, Levine M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14:1133–1139. doi: 10.1023/A:1012186203165. [DOI] [PubMed] [Google Scholar]
- Guo S. Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J Endocrinol. 2014;220:T1–T23. doi: 10.1530/JOE-13-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock CR, Han DH, Chen M, Terada S, Yasuda T, Wright DC, Holloszy JO. High-fat diets cause insulin resistance despite an increase in muscle mitochondria. Proc Natl Acad Sci USA. 2008;105:7815–7820. doi: 10.1073/pnas.0802057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiker JT, Kern M, Kosacka J, Flehmig G, Stumvoll M, Shang E, et al. Nicotinamide nucleotide transhydrogenase mRNA expression is related to human obesity. Obesity (Silver Spring) 2013;21:529–534. doi: 10.1002/oby.20095. [DOI] [PubMed] [Google Scholar]
- Higashida K, Kim SH, Higuchi M, Holloszy JO, Han DH. Normal adaptations to exercise despite protection against oxidative stress. Am J Physiol Endocrinol Metab. 2011;301:E779–E784. doi: 10.1152/ajpendo.00655.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom MH, Iglesias-Gutierrez E, Zierath JR, Garcia-Roves PM. Tissue-specific control of mitochondrial respiration in obesity-related insulin resistance and diabetes. Am J Physiol Endocrinol Metab. 2012;302:E731–E739. doi: 10.1152/ajpendo.00159.2011. [DOI] [PubMed] [Google Scholar]
- Institute of Medicine. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. National Academy Press; Washington DC, USA: 2000. [PubMed] [Google Scholar]
- Kahn SE. Clinical review 135: The importance of beta-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. doi: 10.1210/jcem.86.9.7713. [DOI] [PubMed] [Google Scholar]
- Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, Nathan DM. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346:393–403. doi: 10.1056/NEJMoa012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7:45–56. doi: 10.1016/j.cmet.2007.10.013. [DOI] [PubMed] [Google Scholar]
- Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. 2012;590:3349–3360. doi: 10.1113/jphysiol.2012.230185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laye MJ, Rector RS, Borengasser SJ, Naples SP, Uptergrove GM, Ibdah JA, et al. Cessation of daily wheel running differentially alters fat oxidation capacity in liver, muscle, and adipose tissue. J Appl Physiol. 2009a;106:161–168. doi: 10.1152/japplphysiol.91186.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laye MJ, Rector RS, Warner SO, Naples SP, Perretta AL, Uptergrove GM, et al. Changes in visceral adipose tissue mitochondrial content with type 2 diabetes and daily voluntary wheel running in OLETF rats. J Physiol. 2009b;587:3729–3739. doi: 10.1113/jphysiol.2009.172601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopert P, Patel M. Nicotinamide nucleotide transhydrogenase (Nnt) links the substrate requirement in brain mitochondria for hydrogen peroxide removal to the thioredoxin/peroxiredoxin (Trx/Prx) system. J Biol Chem. 2014;289:15611–15620. doi: 10.1074/jbc.M113.533653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makki K, Froguel P, Wolowczuk I. Adipose tissue in obesity-related inflammation and insulin resistance: cells, cytokines, and chemokines. ISRN Inflamm. 2013;2013:139239. doi: 10.1155/2013/139239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SD, McGee SL. The role of mitochondria in the aetiology of insulin resistance and type 2 diabetes. Biochim Biophys Acta. 2014;1840:1303–1312. doi: 10.1016/j.bbagen.2013.09.019. [DOI] [PubMed] [Google Scholar]
- Menshikova EV, Ritov VB, Toledo FG, Ferrell RE, Goodpaster BH, Kelley DE. Effects of weight loss and physical activity on skeletal muscle mitochondrial function in obesity. Am J Physiol Endocrinol Metab. 2005;288:E818–E825. doi: 10.1152/ajpendo.00322.2004. [DOI] [PubMed] [Google Scholar]
- Menshikova EV, Ritov VB, Ferrell RE, Azuma K, Goodpaster BH, Kelley DE. Characteristics of skeletal muscle mitochondrial biogenesis induced by moderate-intensity exercise and weight loss in obesity. J Appl Physiol. 2007;103:21–27. doi: 10.1152/japplphysiol.01228.2006. [DOI] [PubMed] [Google Scholar]
- Naples SP, Borengasser SJ, Rector RS, Uptergrove GM, Morris EM, Mikus CR, et al. Skeletal muscle mitochondrial and metabolic responses to a high-fat diet in female rats bred for high and low aerobic capacity. Appl Physiol Nutr Metab. 2010;35:151–162. doi: 10.1139/H09-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Research Council. Nutrient Requirements of Laboratory Animals. National Academy Press; Washington, DC, USA: 1995. [Google Scholar]
- Nicholson A, Reifsnyder PC, Malcolm RD, Lucas CA, MacGregor GR, Zhang W, Leiter EH. Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity (Silver Spring) 2010;18:1902–1905. doi: 10.1038/oby.2009.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noland RC, Thyfault JP, Henes ST, Whitfield BR, Woodlief TL, Evans JR, et al. Artificial selection for high-capacity endurance running is protective against high-fat diet-induced insulin resistance. Am J Physiol Endocrinol Metab. 2007;293:E31–E41. doi: 10.1152/ajpendo.00500.2006. [DOI] [PubMed] [Google Scholar]
- Norheim F, Langleite TM, Hjorth M, Holen T, Kielland A, Stadheim HK, et al. The effects of acute and chronic exercise on PGC-1alpha, irisin and browning of subcutaneous adipose tissue in humans. FEBS J. 2014;281:739–749. doi: 10.1111/febs.12619. [DOI] [PubMed] [Google Scholar]
- Oscai LB, Holloszy JO. Effects of weight changes produced by exercise, food restriction, or overeating on body composition. J Clin Invest. 1969;48:2124–2128. doi: 10.1172/JCI106179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palou A, Pico C, Bonet ML. Nutritional potential of metabolic remodelling of white adipose tissue. Curr Opin Clin Nutr Metab Care. 2013;16:650–656. doi: 10.1097/MCO.0b013e328365980f. [DOI] [PubMed] [Google Scholar]
- Parker N, Vidal-Puig AJ, Azzu V, Brand MD. Dysregulation of glucose homeostasis in nicotinamide nucleotide transhydrogenase knockout mice is independent of uncoupling protein 2. Biochim Biophys Acta. 2009;1787:1451–1457. doi: 10.1016/j.bbabio.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen G, Cumming KT, Holden G, Hallen J, Ronnestad BR, Sveen O, et al. Vitamin C and E supplementation hampers cellular adaptation to endurance training in humans: a double-blind, randomised, controlled trial. J Physiol. 2014;592:1887–1901. doi: 10.1113/jphysiol.2013.267419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picklo MJ, Sr, Idso JP, Jackson MI. S-Glutathionylation of hepatic and visceral adipose proteins decreases in obese rats. Obesity (Silver Spring) 2013;21:297–305. doi: 10.1002/oby.20002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitts GC, Bull LS. Exercise, dietary obesity, and growth in the rat. Am J Physiol. 1977;232:R38–R44. doi: 10.1152/ajpregu.1977.232.1.R38. [DOI] [PubMed] [Google Scholar]
- Pohjoismaki JL, Boettger T, Liu Z, Goffart S, Szibor M, Braun T. Oxidative stress during mitochondrial biogenesis compromises mtDNA integrity in growing hearts and induces a global DNA repair response. Nucleic Acids Res. 2012;40:6595–6607. doi: 10.1093/nar/gks301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers SK, Nelson WB, Hudson MB. Exercise-induced oxidative stress in humans: cause and consequences. Free Radic Biol Med. 2011a;51:942–950. doi: 10.1016/j.freeradbiomed.2010.12.009. [DOI] [PubMed] [Google Scholar]
- Powers SK, Talbert EE, Adhihetty PJ. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J Physiol. 2011b;589:2129–2138. doi: 10.1113/jphysiol.2010.201327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, et al. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc Natl Acad Sci USA. 2009;106:8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritov VB, Menshikova EV, Azuma K, Wood R, Toledo FG, Goodpaster BH, et al. Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am J Physiol Endocrinol Metab. 2010;298:E49–E58. doi: 10.1152/ajpendo.00317.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizza RA. Pathogenesis of fasting and postprandial hyperglycemia in type 2 diabetes: implications for therapy. Diabetes. 2010;59:2697–2707. doi: 10.2337/db10-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock CL. Multivitamin-multimineral supplements: who uses them? Am J Clin Nutr. 2007;85:277S–279S. doi: 10.1093/ajcn/85.1.277S. [DOI] [PubMed] [Google Scholar]
- Rumelin A, Humbert T, Luhker O, Drescher A, Fauth U. Metabolic clearance of the antioxidant ascorbic acid in surgical patients. J Surg Res. 2005;129:46–51. doi: 10.1016/j.jss.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Rumsey SC, Levine M. Absorption, transport, and disposition of ascorbic acid in humans. J Nutr Biochem. 1998;9:116–130. doi: 10.1016/S0955-2863(98)00002-3. [DOI] [Google Scholar]
- Rutkowsky JM, Knotts TA, Ono-Moore KD, McCoin CS, Huang S, Schneider D, et al. Acylcarnitines activate proinflammatory signaling pathways. Am J Physiol Endocrinol Metab. 2014;306:E1378–E1387. doi: 10.1152/ajpendo.00656.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smorlesi A, Frontini A, Giordano A, Cinti S. The adipose organ: white-brown adipocyte plasticity and metabolic inflammation. Obes Rev. 2012;13(Suppl. 2):83–96. doi: 10.1111/j.1467-789X.2012.01039.x. [DOI] [PubMed] [Google Scholar]
- Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54:1926–1933. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- Stephenson EJ, Camera DM, Jenkins TA, Kosari S, Lee JS, Hawley JA, Stepto NK. Skeletal muscle respiratory capacity is enhanced in rats consuming an obesogenic Western diet. Am J Physiol Endocrinol Metab. 2012;302:E1541–E1549. doi: 10.1152/ajpendo.00590.2011. [DOI] [PubMed] [Google Scholar]
- Sutherland LN, Capozzi LC, Turchinsky NJ, Bell RC, Wright DC. Time course of high-fat diet-induced reductions in adipose tissue mitochondrial proteins: potential mechanisms and the relationship to glucose intolerance. Am J Physiol Endocrinol Metab. 2008;295:E1076–E1083. doi: 10.1152/ajpendo.90408.2008. [DOI] [PubMed] [Google Scholar]
- Toledo FG, Goodpaster BH. The role of weight loss and exercise in correcting skeletal muscle mitochondrial abnormalities in obesity, diabetes and aging. Mol Cell Endocrinol. 2013;379:30–34. doi: 10.1016/j.mce.2013.06.018. [DOI] [PubMed] [Google Scholar]
- Toledo FG, Menshikova EV, Ritov VB, Azuma K, Radikova Z, DeLany J, Kelley DE. Effects of physical activity and weight loss on skeletal muscle mitochondria and relationship with glucose control in type 2 diabetes. Diabetes. 2007;56:2142–2147. doi: 10.2337/db07-0141. [DOI] [PubMed] [Google Scholar]
- Toledo FG, Menshikova EV, Azuma K, Radikova Z, Kelley CA, Ritov VB, Kelley DE. Mitochondrial capacity in skeletal muscle is not stimulated by weight loss despite increases in insulin action and decreases in intramyocellular lipid content. Diabetes. 2008;57:987–994. doi: 10.2337/db07-1429. [DOI] [PubMed] [Google Scholar]
- Tovar A, Ameho CK, Blumberg JB, Peterson JW, Smith D, Booth SL. Extrahepatic tissue concentrations of vitamin K are lower in rats fed a high vitamin E diet. Nutr Metab (Lond) 2006;3:29. doi: 10.1186/1743-7075-3-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Bruce CR, Beale SM, Hoehn KL, So T, Rolph MS, Cooney GJ. Excess lipid availability increases mitochondrial fatty acid oxidative capacity in muscle: evidence against a role for reduced fatty acid oxidation in lipid-induced insulin resistance in rodents. Diabetes. 2007;56:2085–2092. doi: 10.2337/db07-0093. [DOI] [PubMed] [Google Scholar]
- Uthus EO, Picklo MJ., Sr Obesity reduces methionine sulphoxide reductase activity in visceral adipose tissue. Free Radic Res. 2011;45:1052–1060. doi: 10.3109/10715762.2011.591793. [DOI] [PubMed] [Google Scholar]
- Vinayagamoorthi R, Bobby Z, Sridhar MG. Antioxidants preserve redox balance and inhibit c-Jun-N-terminal kinase pathway while improving insulin signaling in fat-fed rats: evidence for the role of oxidative stress on IRS-1 serine phosphorylation and insulin resistance. J Endocrinol. 2008;197:287–296. doi: 10.1677/JOE-08-0061. [DOI] [PubMed] [Google Scholar]
- Vislisel JM, Schafer FQ, Buettner GR. A simple and sensitive assay for ascorbate using a plate reader. Anal Biochem. 2007;365:31–39. doi: 10.1016/j.ab.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong N, Blair AR, Morahan G, Andrikopoulos S. The deletion variant of nicotinamide nucleotide transhydrogenase (Nnt) does not affect insulin secretion or glucose tolerance. Endocrinology. 2010;151:96–102. doi: 10.1210/en.2009-0887. [DOI] [PubMed] [Google Scholar]
- Yfanti C, Akerstrom T, Nielsen S, Nielsen AR, Mounier R, Mortensen OH, et al. Antioxidant supplementation does not alter endurance training adaptation. Med Sci Sports Exerc. 2010;42:1388–1395. doi: 10.1249/MSS.0b013e3181cd76be. [DOI] [PubMed] [Google Scholar]
- Yfanti C, Nielsen AR, Akerstrom T, Nielsen S, Rose AJ, Richter EA, et al. Effect of antioxidant supplementation on insulin sensitivity in response to endurance exercise training. Am J Physiol Endocrinol Metab. 2011;300:E761–E770. doi: 10.1152/ajpendo.00207.2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.