

Abstract
We have examined the rapid reaction kinetics and spectroscopic properties of the molybdenum-containing, NAD+-dependent FdsABG formate dehydrogenase from Ralstonia eutropha. We confirm previous steady-state studies of the enzyme and extend its characterization to a rapid kinetic study of the reductive half-reaction (the reaction of formate with oxidized enzyme). We have also characterized the electron paramagnetic resonance signal of the molybdenum center in its MoV state and demonstrated the direct transfer of the substrate Cα hydrogen to the molybdenum center in the course of the reaction. Varying temperature, microwave power, and level of enzyme reduction, we are able to clearly identify the electron paramagnetic resonance signals for four of the iron/sulfur clusters of the enzyme and find suggestive evidence for two others; we observe a magnetic interaction between the molybdenum center and one of the iron/sulfur centers, permitting assignment of this signal to a specific iron/sulfur cluster in the enzyme. In light of recent advances in our understanding of the structure of the molybdenum center, we propose a reaction mechanism involving direct hydride transfer from formate to a molybdenum-sulfur group of the molybdenum center.
Keywords: electron paramagnetic resonance (EPR), enzyme catalysis, enzyme kinetics, enzyme mechanism, iron/sulfur protein, molybdenum
Introduction
The molybdenum-containing, NAD+-dependent formate dehydrogenases from bacteria such as Ralstonia eutropha catalyze the oxidation of formate to CO2, reducing NAD+ to NADH, and are members of the NADH dehydrogenase superfamily of enzymes. These cytosolic formate dehydrogenases are expressed under aerobic conditions and are distinct from the predominant bacterial formate dehydrogenases expressed under anaerobic conditions, which are typically membrane-associated and extremely O2-sensitive; they are also distinct from the cofactorless formate dehydrogenases from many eukaryotes, including humans, that catalyze the direct hydride transfer from formate to NAD+ (1). In R. eutropha, the trimeric FdsABG (soluble, NAD+-dependent, molybdenum-containing formate dehydrogenase, product of the fdsABG gene cluster) enzyme is encoded by the fdsGBACD operon and is predicted to contain seven iron/sulfur clusters, FMN, and a molybdenum center; each subunit bears significant sequence similarity (∼21% identity) and expected strong structural homology to a corresponding subunit of the matrix- or cytosol-exposed portion of NADH dehydrogenase, and the spatial layout of the several redox-active centers is particularly highly conserved. The 105-kDa FdsA is cognate to subunit Nqo3 in the crystallographically characterized Thermus thermophilus NADH dehydrogenase (2, 3) and is predicted to have four [4Fe-4S] clusters and one [2Fe-2S] cluster (4). The close structural homology predicted between FdsA and Nqo3 extends to the presence of a histidine ligand to one of the [4Fe-4S] clusters near the N terminus of the FdsA/Nqo3 subunit. The C terminus of FdsA contains a molybdenum center and has ∼60% sequence similarity to the structurally characterized molybdenum-containing formate dehydrogenase FdhF of Escherichia coli (part of the formate-hydrogen lyase complex), with Cys-378 occupying the homologous position to the molybdenum-coordinating Sec-140 in the latter enzyme (5). This domain is present in Nqo3 as well, although the molybdenum center itself and one (sometimes two) of the more C-terminal iron/sulfur clusters have been lost by the NADH dehydrogenases over the course of evolution (1, 3, 6). The 55-kDa FdsB subunit of the R. eutropha formate dehydrogenase has some 45% sequence identity to the FMN- and [4Fe-4S]-containing Nqo1 subunit of the T. thermophilus NADH dehydrogenase; the FMN is the site of NAD+ reduction. The 19-kDa FdsG subunit of the R. eutropha enzyme subunit has 34% sequence identity to the Nqo2 subunit of the T. thermophilus NADH dehydrogenase and has a [2Fe-2S] cluster that lies off the main electron transfer pathway connecting the FMN and molybdenum center.
The molybdenum center of FdsABG is the site of formate oxidation and is of the same general type seen in DMSO reductase (1, 7), with two equivalents of a pyranopterin cofactor coordinated (present as the guanosine dinucleotide in the case of FdsABG) to the metal via enedithiolate side chains. Cys-378 occupies a fifth ligand position in an expected trigonal prismatic coordination geometry of the oxidized enzyme, and on the basis of the requirement of the FdsC sulfide insertase for proper maturation of active FdsABG (8), the sixth ligand coordination position is most likely a terminal molybdenum-sulfur group. The makeup of the molybdenum coordination sphere of oxidized enzyme has recently been confirmed in an EXAFS study of the Rhodobacter capsulatus enzyme, specifically demonstrating a short molybdenum-sulfur bond at a distance of ∼2.17 Å in the oxidized enzyme (9).
Previous work with the R. eutropha FdsABG enzyme has demonstrated that the oxidized, as-isolated enzyme exhibits broad absorption throughout the visible region attributable to its FMN and iron/sulfur centers, which bleaches upon reduction by formate, NADH, or dithionite (10). The reduced enzyme exhibits multiple electron paramagnetic resonance (EPR)2 signals to the iron/sulfur clusters of the enzyme, although the g values for only three have been reported (11). In the present work, we examine the rapid reaction kinetics of the reaction of FdsABG with its reducing substrate formate and characterize additional iron/sulfur EPR signals of the enzyme. We have also characterized the EPR signal of the molybdenum center in its MoV state and demonstrate the direct transfer of the substrate Cα hydrogen to the molybdenum center, providing specific evidence for the enzyme operating via a hydride transfer mechanism. Finally, we observe a magnetic interaction between the molybdenum center and one of the iron/sulfur centers, permitting assignment of this signal to the [4Fe-4S] cluster nearest the molybdenum center (at a distance of 12.4 Å center to center, by analogy to the distance seen in the FdhF formate dehydrogenase).
Experimental Procedures
Organisms and Growth Conditions
R. eutropha strains H16 (ATCC 17699) and HF210 were grown as previously described (1), except that the final molybdenum concentration was increased to 0.15 mm. For precultures, up to four plates were washed with 0.5 liter of minimal media (supplemented with 0.1% (w/v) fructose and 0.2% (w/v) formate) and grown at 30 °C with shaking at 250 rpm in a 3-liter of Schott flask for ∼6 h. Large scale fermentation (12 liters) was performed at 30 °C in a 15-liter fermentor (New Brunswick BioFlo 415) with a stirring rate of 300–350 rpm and aeration at 6 liters/min (using media supplemented with formate 0.2% (w/v) only). pH was maintained at 6.9 by the automated addition of 44–50% (v/v) formic acid. Cells were harvested after the culture had reached an A436 of ∼6 for HF210 or ∼6.5 for H16. Cells grown to higher densities (>4 g/liters of medium) exhibited a significant decrease in FdsABG expression. Highest level of expression (60–80 units/g cells) was obtained from HF210 cells subjected to continuous growth, where cells from one growth cycle were used as precultures for subsequent cycles. Harvested cells were washed once at 4 °C with 75 mm potassium phosphate, 10 mm KNO3, pH 7.2, and stored at −80 °C until used.
Purification of FdsABG
All steps were performed at 0–4 °C with an Akta FPLC system (GE Healthcare) in a procedure modified from Friedebold et al. (10). Frozen cells, typically 48–52 g of wet weight, were thawed, suspended in 2–2.5 volumes of enzyme buffer, and disrupted by 1–2 passages through a French pressure cell at 20,000 p.s.i. Cell debris was removed by centrifugation for 45 min at 200,000 × g. The supernatant was made 29% saturated in ammonium sulfate and centrifuged for an additional 45 min at 200,000 × g. The 29% fraction was then brought to 46% ammonium sulfate saturation. After centrifugation, the pellet containing FdsABG was dissolved in 80 mm K-PO4, 20 mm KNO3, pH 8.0 (saturated with 15% ammonium sulfate), at 0.4 ml/g cells to maintain a relatively small loading volume to minimize peak spreading. Supernatant treated in this fashion was loaded onto a 2.6 × 16-cm butyl 4-Sepharose column (GE Healthcare), the column was washed with 0.5 column volume of 40 mm K-PO4, 10 mm KNO3, 0.7 m ammonium sulfate, pH 7.3 (HIC-A), and the enzyme was eluted with a linear gradient of 0.7–0 m ammonium sulfate over 4 column volumes. Active fractions (see assay conditions below) were pooled, and the protein was precipitated with 50% ammonium sulfate. After centrifugation the pellet was redissolved in 80 mm K-PO4, 20 mm KNO3, pH 8.0, and loaded onto a 2.6 × 20-cm G-50 desalting column equilibrated with 20 mm Tris-HCl, 10 mm KNO3, pH 7.4. Fractions from the desalting column were loaded onto a 2.6 × 14 cm Cibacron Blue-agarose 3GA, Type 3000-CL column (Sigma-Aldrich). Under these conditions, FdsABG did not bind to the gel matrix and eluted in the flowthrough. The resulting fractions were applied to a 2.6 × 14-cm Fractogel TMAE 650 (S) column (EMD-Millipore), washed with 2 column volumes of 40 mm K-PO4, 10 mm KNO3, pH 7.0, and eluted with a linear gradient of 0–0.6 m NaCl over 5 column volumes. Active fractions were pooled and divided into two halves, and the protein was precipitated with 50% ammonium sulfate. After centrifugation, one pellet was redissolved in 0.5 ml of HIC-A buffer, loaded onto a 5 ml butyl-Sepharose HP column (GE Healthcare), washed with 0.5 column volume HIC-A buffer, and eluted with a linear gradient of 0.5–0 m ammonium sulfate over 4 column volumes. The process was repeated with the second pellet. Both HIC steps were complicated by significant peak spreading; thus total column volumes were minimized to obtain optimal resolution. The active fractions were precipitated with 50% ammonium sulfate, redissolved, and loaded unto 1.6 × 60-cm Superdex 200 PG column (GE Healthcare) in buffer containing 300 mm KCl. Fractions containing only the dimer of the heterotrimer (with a molecular mass of ∼358 kDa) were precipitated with 50% ammonium sulfate, redissolved, and buffer-exchanged into storage buffer: 75 mm K-PO4, 10 mm KNO3, pH 7.5 via Amicon Ultra 4 (Millipore). Concentrated protein was flash frozen and stored in liquid N2.
Activity assays were performed at 30 °C in 75 mm K-PO4, pH 7.7, with 2 mm NAD+ and 40 mm sodium formate; formation of NADH was monitored at 340 nm (ϵ = 6,220 m−1 cm−1). One unit of activity was defined as the amount of enzyme catalyzing the reduction of 1 μmol of NAD+ per min. Typically, 10 s were used to calculate the initial slope. Enzyme concentrations were determined using an extinction coefficient of E410 nm = 51,500 m−1 cm−1 estimated from the oxidized enzyme spectrum published by Friedebold et al. (10); activities were calculated with respect to one trimer with a molecular mass of 178,800 Da. Purified protein exhibited 70 units of activity per mg of protein, in good agreement with 80 units per mg obtained by Friedebold et al. (10).
UV-visible Absorbance Measurements
Activity measurements and absorbance spectra were performed using a Hewlett-Packard 8452A diode array spectrophotometer equipped with a thermostatted cell holder. Steady-state measurements collected at a range of pH values yielded a bell-shaped curve that was fitted to the equation for a double-ionization mechanism,
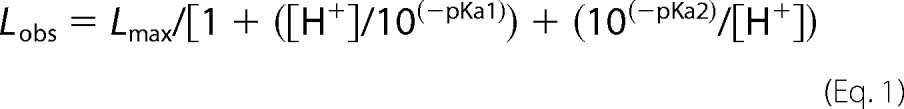 |
with the maximum theoretical value for L (Lmax), and the two pKa values (pKa1 and pKa2) obtained using PeakFit ver. 4 (Jandel Scientific). Reductive titrations were performed with 20 mm buffered sodium dithionite and 20 μm enzyme in 75 mm K-PO4, pH 7.7, under anaerobic conditions at RT. Prior to titrations the enzyme was exchanged into anaerobic 75 mm K-PO4, pH 7.7, to remove nitrate via Amicon Ultra 4 (Millipore).
Rapid Reaction Kinetics
The reaction of oxidized FdsABG with formate was followed using a SX-20 stopped flow spectrophotometer (Applied Photophysics, Inc.) equipped with photodiode array and photomultiplier tube detection and running ProData SX 2.2.5.6 acquisition software. The enzyme was exchanged into anaerobic 75 mm K-PO4, pH 7.7, to remove nitrate via Amicon Ultra 4 (Millipore) prior to use. As long as the enzyme was kept under anaerobic conditions, it could be maintained for 3–4 h without significant loss of activity. Friedebold et al. (10) showed that nitrate binding followed a mixed competitive-noncompetitive type inhibition. It is possible that nitrate binding at the molybdenum site blocks O2 from affecting the removal of the catalytically essential apical sulfur ligand (9) by some as yet unknown mechanism. Time courses for the reaction were monitored at 450 nm at 10 °C and fitted to a sum of three exponentials by nonlinear least squares regression analysis to the following equation,
where n refers to the number of kinetic phases observed. Analysis of time courses was performed using the software ProData Viewer 4.2.0 (Applied Photophysics, Inc). The observed rate constants, kobs, were plotted against substrate concentrations to obtain the limiting rate constant for reduction, kred, and the dissociation constant, Kd, using the following equation.
EPR Spectroscopy
MoV-containing EPR samples were prepared in 75 mm potassium phosphate, pH 7.5, in the presence of 10 mm KNO3 unless otherwise stated. MoV-containing spectra could be obtained upon reduction of the enzyme with either sodium dithionite or sodium formate, although the relative signal intensities of the MoV and several iron/sulfur signals depended to some degree on both the extent of reduction and the reductant used. Maximum accumulation of MoV EPR signal was obtained when the enzyme was ∼50% reduced (by addition of ∼5e equivalents), although additional reduction did not significantly affect MoV yield as long as the sample was frozen promptly. Nitrate had no effect on the shape of the EPR signals, although the yield of MoV EPR signal was typically reduced in the absence of nitrate, even when nitrate was removed under anaerobic conditions (likely because of a loss of activity during nitrate removal). No MoV EPR signal was observed when samples were prepared from protein that had been incubated aerobically in the absence of nitrate for at least 6 h at 4 °C (at which point virtually all of the enzymatic activity was lost). When necessary, buffer exchange was accomplished by concentrating protein samples, diluting with appropriate buffer, and reconcentrating via Amicon Ultra 4 (Millipore). Samples were made anaerobic at 4 °C (5–7 °C for deuterated samples) and transferred to argon-flushed, septum-sealed EPR tubes. Samples prepared in this way were subsequently frozen in an ethanol/dry ice bath and transferred to liquid nitrogen. Further details of sample preparation are included in figure captions. EPR spectra were recorded using a Brüker EMX spectrometer equipped with a Brüker ER 4119HS high sensitivity X-band cavity and gaussmeter, operating WinEPR version 4.33 acquisition software. Temperature was controlled using a Brüker variable temperature unit and liquid nitrogen or liquid helium cryostat. For purposes of comparison, all spectra were adjusted to the same microwave frequency by converting the magnetic field values for each individual spectrum to g values using the equation,
where v is in GHz, and B is in mT, and then back to mT using a microwave frequency of 9.475 GHz. Detailed instrument settings are included in figure captions. Simulations were performed using the EasySpin 4.5.5 software package (12). For MoV-containing spectra, Euler angles relating the g and A tensor frames were empirically obtained and fixed during final simulations. During simulations of MoV-containing spectra, g values for the iron/sulfur clusters were allowed to vary by no more than ± 0.001, whereas anisotropic line broadening (simulated as HStrain in the EasySpin software package) varied by no more than ± 10 MHz. Iron/sulfur linewidths used in the molybdenum-containing spectra were initially estimated from experimental spectra that contained the various iron/sulfur components at the temperatures where the molybdenum-containing spectra were obtained. These estimates were then used as starting points for simulation of the molybdenum-containing spectra. Neither the g nor the A tensors were generally affected by the way in which the linewidths were treated; only the relative contribution of each component to the composite spectra was sometimes affected. Because of the complexity of the composite spectra, no attempts were made to quantify the effects of temperature on the linewidths of the various signals. Every multicomponent simulation included a “weight” term, which could be used to estimate the relative contribution of each component to the composite spectrum.
Electron-electron interactions between MoV and Fe/S3 were simulated using an S = 1/2 spin system. The electron-electron spin-spin interaction module in EasySpin does not allow variable linewidths for the interacting sites, and at 20 K the linewidth of the g2 tensor of Fe/S3 is 1.5× greater than of the corresponding MoV. Initially, only the anisotropic splitting at g2 of the MoV (no interactions were apparent at g1 or g3) was simulated. After the initial simulation, the resulting value for the MoV could be fixed for the interacting Fe/S3 as well. Because of the broad nature of the iron/sulfur centers, the exclusion of electron-electron coupling for the Fe/S3 did not have a discernible effect on either the accuracy of the simulation or the resultant parameters.
Results
Enzyme Spectroscopy and Kinetics
FdsABG, isolated as described under “Experimental Procedures,” has the absorption spectrum shown in Fig. 1A, in good agreement with previous reports (4, 10, 11). The oxidized, as-isolated enzyme absorbs throughout the visible region, as expected given the large number of chromophores present, and has a broad shoulder at ∼450 nm that is attributable to the FMN cofactor. A reductive titration with sodium dithionite results in a systematic bleaching of this absorbance throughout the UV-visible region (Fig. 1B). A plot of the fractional absorbance change at 550 nm, where reduction of the iron/sulfur clusters is expected to predominate, versus that at 450 nm, where reduction of the FMN predominates, provides information regarding the relative order of reduction (because of the relative reduction potentials) of the FMN relative to the iron/sulfur clusters. As has been shown previously in related systems (13), a deflection from the diagonal (reflecting strict proportionality in the absorbance changes at the two wavelengths) up and to the left reflects earlier reduction of Fe/S clusters relative to FMN and a deflection down and to the right the reverse. As indicated in the inset to Fig. 1B, the plot shows a deflection up and to the left from the diagonal, indicating that the FMN becomes fully reduced later rather than earlier in the course of the titration relative to the iron/sulfur clusters. This suggests that the FMN has a relatively low reduction potential relative to the iron/sulfur clusters. Also consistent with previous reports (4, 10, 11), our enzyme preparation exhibits a broad pH optimum at ∼7.5; fits of this bell-shaped curve to Equation 1 yielded pKa values for the acidic and alkaline limbs of the plot of 5.6 and 9.3 (Fig. 2A).
FIGURE 1.
Reductive titration of FdsABG. A, oxidized (blue) and sodium dithionite-reduced (black) spectra. B, change in absorbance as a function of reduction. The inset plots the relative absorbance at 550 nm versus relative absorbance at 450 nm, with the diagonal (reflecting strict proportionality in the absorbance change at the two wavelengths) indicated. Details of titration are described under “Experimental Procedures.”
FIGURE 2.

A, pH dependence of kcat for FdsABG. The reactions were performed at 30 °C in a buffer containing 75 mm of malate, K2HPO4, Tris, and glycine, brought to pH 5–10. The fit to the data (solid line) yielded two pKa values of 5.6 and 9.3, respectively. B, Lineweaver-Burk plots for the reaction of FdsABG with formate at the following [NAD+]: 1.62 mm (closed circles), 0.54 mm (open circles), 0.18 mm (closed triangles), and 0.06 mm (open triangles). C, Lineweaver-Burk plots for the reaction of FdsABG with NAD+ at the following [formate]: 3.24 mm (closed circles), 1.08 mm (open circles), 0.36 mm (closed triangles), and 0.12 mm (open triangles). D, secondary plots of 1/y intercept taken from B versus 1/[NAD+]. E, secondary plots of 1/y intercept taken from C. versus 1/[formate]. The reactions in B and C were performed in 75 mm K-PO4, pH 7.7, at 30 °C. Linear regression analysis of plots D and E yielded a KmNAD+ = 130 μm and Kmformate = 310 μm, respectively, and an average kcat of 201 s−1.
A full steady-state analysis, not previously reported, is shown in Fig. 2 (B–E). Lineweaver-Burk plots for the steady-state reaction of FdsABG at 30 °C (as used in previous studies) yield approximately parallel lines, as is frequently seen with enzymes in which the sites of reduction and oxidation are physically separated (14, 15). We obtain an apparent KmNAD+ of 130 μm, consistent with both the determination of Friedebold et al. (10) of 90 μm for the same enzyme, as well as the recent report of 173 μm for the enzyme from R. capsulatus (16). Our apparent Kmformate of 310 μm is in very good agreement with the 281 μm determined for the R. capsulatus enzyme (16) but nearly 10-fold smaller than the value of 3.3 mm determined by Friedebold et al. (10). The plots in Fig. 2 (B–E) yield an average kcat of 201 s−1, which corresponds to ∼67 units/mg of protein calculated using a molecular mass of 178,800 daltons for the heterotrimer, consistent with earlier reports of 80 units/mg (4, 10, 11).
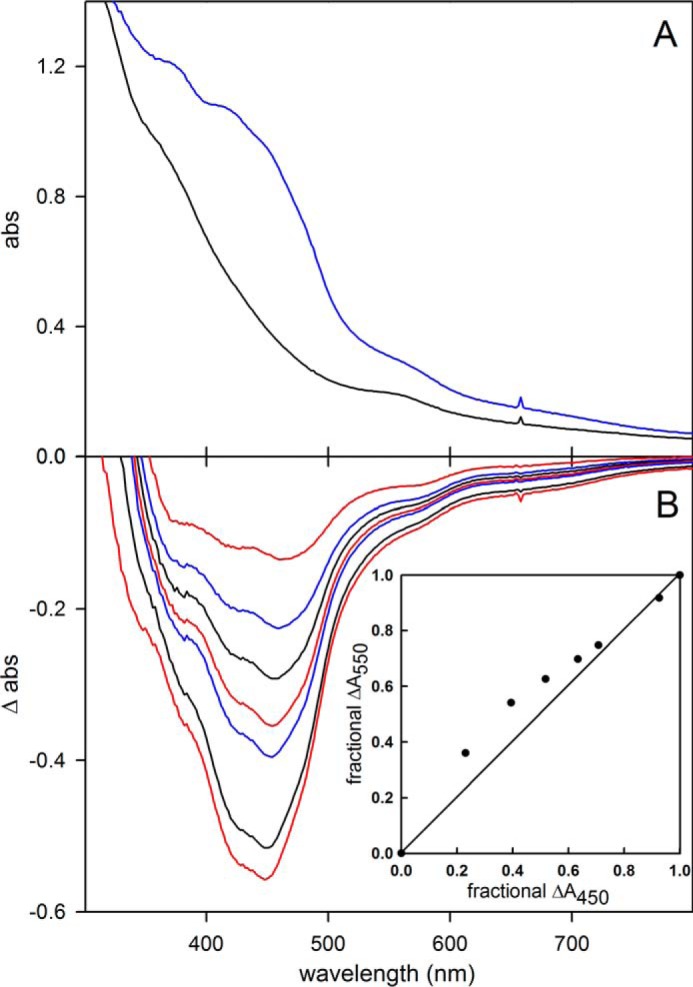
We next examined the rapid reaction kinetics of enzyme reduction by formate at pH 7.7, 10 °C, with the lower temperature being dictated by the high velocity of the reaction. As shown in Fig. 3, the overall reaction is triphasic, with a fast phase that accounts for ∼32% of the total observed absorbance change at 450 nm. Consistent with the reductive titration indicating a relatively low reduction potential for the FMN, the majority of the absorbance change at 450 nm, caused by reduction of FMN, occurs in the intermediate phase of the reaction (Fig. 3A). A plot of the observed rate constant for the fast phase of the reaction as a function of formate concentration is hyperbolic (Fig. 3C), yielding a limiting kfast at high [formate] of 140 s−1, and a Kdformate of 82 μm. Assuming a doubling of rate constant for every 10 °C increase in temperature, this corresponds to a limiting kfast of ∼600 s−1 at 30 °C, indicating that the reductive half of the catalytic sequence is only partially rate-limiting. The intermediate phase of the reaction is also [formate]-dependent, with a limiting kintermediate of 19 s−1 and Kdformate of 230 μm. We attribute this phase to the reaction of formate with partially reduced enzyme that accumulates in the course of the reaction, with the reaction rate attenuated because of the increasing likelihood that the molybdenum will be reduced (and therefore unreactive) on binding formate. The slowest phase of the reaction is [formate]-independent and accounts for only ∼15% of the total absorbance change. It is most likely due to the slow intermolecular transfer of reducing equivalents from the formate-reduced functional enzyme to a small population of nonfunctional enzyme.
FIGURE 3.
Pre-steady-state kinetics for the reduction of FdsABG with formate or [2H]formate. A, rapid scanning stopped flow traces for the reaction of 6 μm of FdsABG with 5 mm formate performed at 10 °C in 75 mm K-PO4, pH 7.7. B, a representative kinetic trace of the reaction of FdsABG with 0.8 mm sodium formate monitored at 450 nm. The trace is best represented by three phases with kobs = 124, 14, and 2.5 s−1, with ΔA = 0.011, 0.017, 0.006, respectively. C, plots of kfast (black circles) and kintermediate (red squares) versus formate concentrations or kfast (black triangles) versus [2H]formate concentrations. All reactions were performed at 10 °C in 75 mm K-PO4, pH 7.7, with 2.5 μm FdsABG. Hyperbolic fits (solid lines) yielded kfast = 140 s−1, Kdformate = 82 μm, kintermediate = 19 s−1, and Kdformate = 230 μm for reaction with formate and kfast = 66 s−1, and Kd[2H]formate = 193 μm for reaction with [2H]formate.
When deuterated formate is used in the rapid reaction kinetics, the same multiphasic kinetics is observed; kfast decreases to 66 s−1 and yields an isotope effect on kfast of 2.1. This is consistent with a primary isotope effect, but one in which the isotope-sensitive step of the reaction is only partially rate-limiting in the reductive-half-reaction.
MoV EPR and Demonstration of Direct Hydrogen Transfer from Substrate to the Molybdenum Center
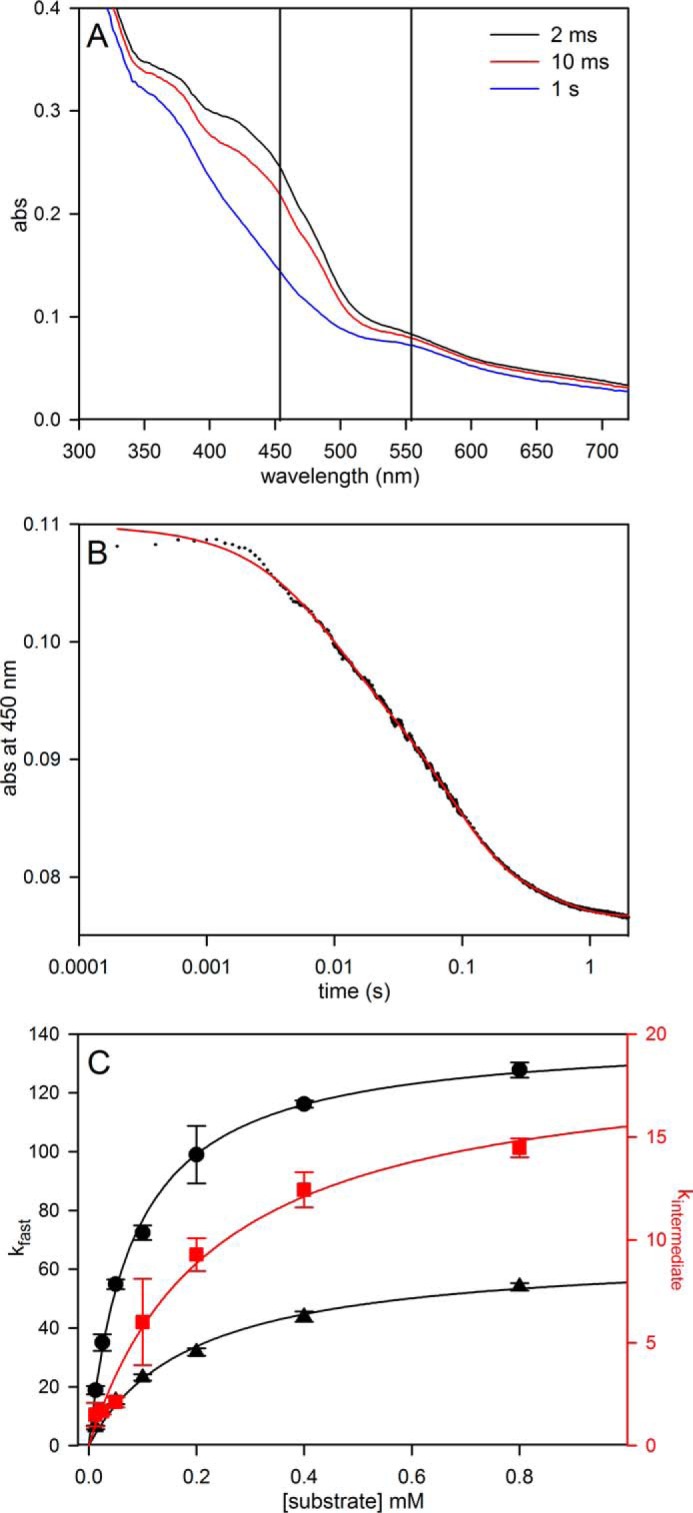
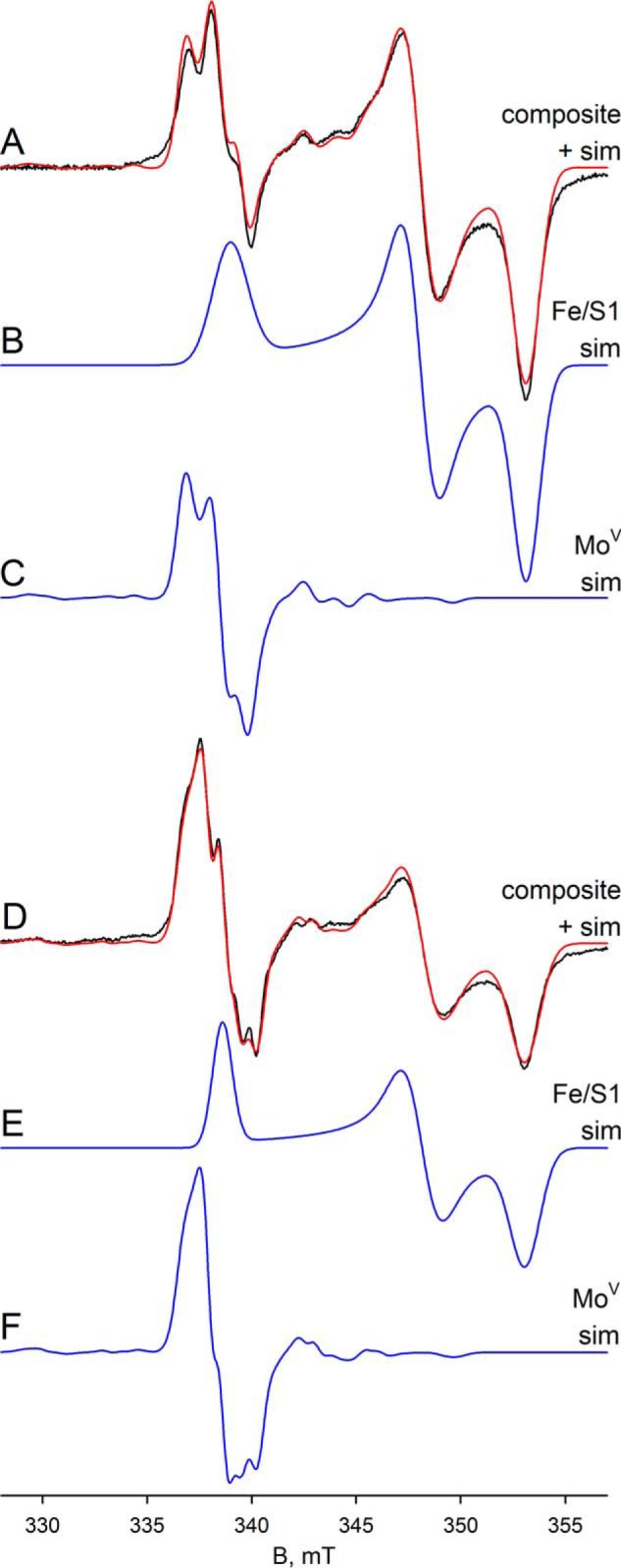
We next examined the EPR properties of the enzyme, beginning with the molybdenum center of the enzyme. Fig. 4A (black line) shows the EPR spectrum seen at 150 K of 100 μm FdsABG that has been partially reduced with sodium dithionite. The spectrum is composed of a narrow MoV signal centered at ∼338 mT and clearly showing hyperfine coupling caused by the 25% naturally occurring 95,97Mo (I = 5/2), as well as a much broader signal attributable to an iron/sulfur center. Simulations of the MoV component yielded g1,2,3 = 2.009, 2.001, 1.992 and 95,97Mo A1,2,3 = 138, 82, 45 MHz, with one strongly coupled proton having approximately isotropic 1H A1,2,3 = 18, 21, 18 MHz (Table 1); a simulation using these parameters is shown in Fig. 4C. A 5-fold expanded spectrum of the MoV EPR signal (Fig. 4C) shows in detail the molybdenum hyperfine splitting; the 1H hyperfine splitting is unresolved and apparent only in the broadening of the peaks. By comparison to other molybdenum-containing enzymes (e.g. DMSO reductase (17) and xanthine oxidase (18, 19)), the strength of the 1H coupling clearly indicates that the proton is part of the molybdenum center. Simulation of the iron/sulfur component yields g1,2,3 = 2.001, 1.946, 1.918 (Fig. 4B). A sum of the simulations for the individual MoV and iron/sulfur components is shown with the experimental spectrum shown in Fig. 4A (red spectrum). As can be seen, the fit to the experimental spectrum is excellent, and we conclude that just two signal-giving species are present: a single MoV species exhibiting strong coupling to a single proton and one iron/sulfur signal. The EPR signals attributable to other iron/sulfur centers of the enzyme are considered further below.
FIGURE 4.
EPR of molybdenum center of FdsABG collected at 150 K. A, MoV-Fe/S EPR spectrum (black) and simulation (red) of FdsABG collected with modulation amplitude = 2 Gauss and microwave power = 4 mW. The sample was prepared under anaerobic conditions by reduction of 100 μm of FdsABG in 75 mm K-PO4, pH 7.5, in the presence of 10 mm KNO3, with 5 mm buffered sodium dithionite. The MoV component represents ∼33% of total spin density. B, simulation of the Fe/S1 contribution to the spectrum in A. C, simulation of the MoV contribution to the spectrum in A. The hyperfine splitting caused by the 25% naturally occurring 95,97Mo (I = 5/2) is shown in the 5-fold expanded spectrum above. D, MoV-Fe/S EPR spectrum (black) and simulation (red) of deuterated FdsABG collected with modulation amplitude = 2 Gauss and microwave power = 4 mW. The sample was prepared under anaerobic conditions by reduction of 100 μm of FdsABG in 75 mm K-PO4, pD 7.1, in the presence of 10 mm KNO3, with 0.4 mm sodium formate. The MoV component represents ∼38% of total spin density. E, simulation of the Fe/S1 contribution to the spectrum in D. F, simulation of the MoV contribution to the spectrum in D. The hyperfine splitting caused by the 25% naturally occurring 95,97Mo (I = 5/2) is shown in the 3-fold expanded spectrum above. Simulation parameters are summarized in Table 1. sim, simulation.
TABLE 1.
EPR simulation parameters for MoV and reduced [2Fe-2S] and [4Fe-4S] centers of FdsABG
| Center |
g tensors |
Molybdenum coupling constantsa |
1H coupling constantsa |
e-ea | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| g1 | g2 | g3 | gave | A1 | A2 | A3 | A1 | A2 | A3 | ||
| MoVH, 150 Kb,c | 2.009 | 2.001 | 1.992 | 2.000 | 138d | 82 | 45 | 18 | 21 | 18 | |
| MoVD, 150 Kb,c | 2.009 | 2.001 | 1.992 | 2.000 | 139 | 84 | 49 | ||||
| MoVD, 60 Kb,c | 2.009 | 2.001 | 1.991 | 2.000 | 139 | 85 | 44 | ||||
| MoVD, 20 Kb,c | 2.009 | 2.001 | 1.992 | 2.001 | 138 | 84 | 46 | 19 | |||
| Fe/S1, 100 Kc | 2.001 | 1.946 | 1.918 | 1.955 | |||||||
| Fe/S2, 60 Kc | 2.017 | 1.947 | 1.933 | 1.966 | |||||||
| Fe/S3, 20 Kc | 2.044 | 1.937 | 1.898 | 1.959 | 19d | ||||||
| Fe/S4, 20 Ke | 2.095c | 1.888 | 1.862 | 1.948 | |||||||
| Fe/S4′, 20 Ke | 2.095c | 1.885 | 1.882 | 1.954 | |||||||
a In MHz; in the absence of multifrequency data, coupling constants are approximate.
b Euler angles (α = 10.8°, β = 0.2°, γ = 152.5°) were fixed during simulations; in the absence of multifrequency data, angles are approximate.
c g tensor uncertainty, ± 0.001.
d Fixed during simulations.
e g tensor uncertainty not determined (see text).
Upon preparation of the sample in D2O rather than H2O, the proton coupling to the MoV signal disappears (because of the very much weaker nuclear magnetic moment of 2H relative to 1H), clearly demonstrating the solvent exchangeability of the coupled proton and illustrating the intrinsic anisotropy of the MoV g tensor (Figs. 4D and 5). As in the proteated sample, the experimental spectrum again includes the EPR signal of a single iron/sulfur cluster. A 3-fold expanded spectrum in Fig. 4F shows in detail the molybdenum hyperfine splitting, narrowed by the absence of proton hyperfine splitting. Upon partial reduction of enzyme with deuterated formate in H2O (pH 8.5), the MoV signal seen at short times lacks the strong proton coupling (Fig. 6A) evident in the dithionite-reduced sample, although the g values for the signal are unchanged; over the course of 1 min, however, the proton coupling grows in (Fig. 6D), yielding an EPR signal indistinguishable from that seen in the dithionite-reduced enzyme. These results clearly demonstrate that the Cα hydrogen of formate is transferred to the molybdenum center in the course of the reaction and that the site is solvent-exchangeable. As discussed further below, we take this to reflect direct hydride transfer in the course of the reaction. There is strong mechanistic precedence for molybdenum-sulfur groups being good hydride acceptors in molybdenum-containing enzymes (20–22) and for molybdenum-SH protons being both strongly coupled and solvent-exchangeable (23–26). The mechanistic implications of these results are considered further under “Discussion.”
FIGURE 5.
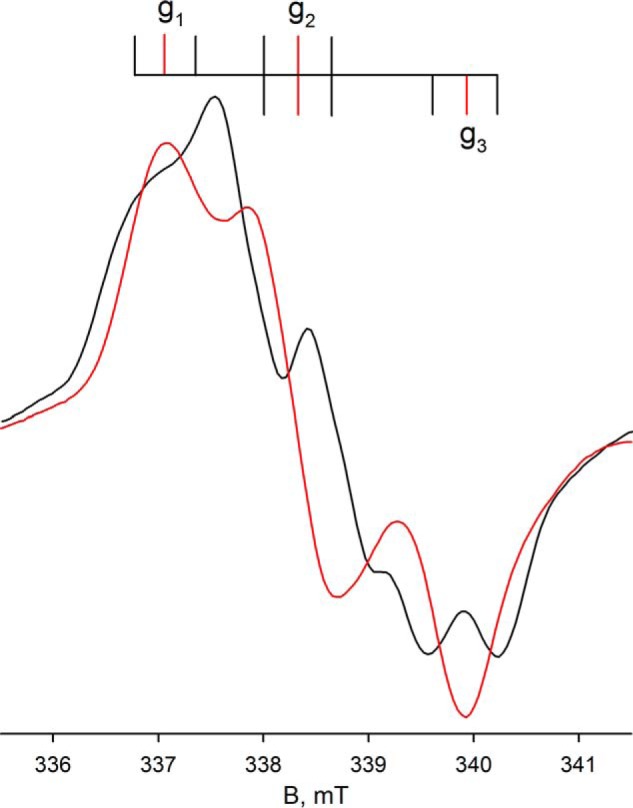
Expanded view of the EPR of molybdenum center of FdsABG from Fig. 4. Superposition of the composite MoV-Fe/S EPR spectra from Fig. 4A (black) with Fig. 4D (red). The line diagram above indicates the location of the principal g tensors (red vertical lines) and the approximate location of the 1H hyperfine splitting (I = 1/2) in the spectrum from Fig. 4A (black vertical lines).
FIGURE 6.
A, MoV-Fe/S EPR spectrum (black) and simulation (red) of FdsABG collected at 150 K with modulation amplitude = 3 Gauss and microwave power = 4 mW. The sample was prepared under anaerobic conditions by reduction of 50 μm of FdsABG in 200 mm Tris-HCl, pH 8.5, in the presence of 10 mm KNO3, with 0.25 mm sodium formate on ice and mixing for 5 s before freezing. The MoV component represents ∼10% of total spin density. B, simulation of the Fe/S1 contribution to the spectrum in A. C, simulation of the MoV contribution to the spectrum in A. D, MoV-Fe/S EPR spectrum (black) and simulation (red) of FdsABG collected at 150 K with modulation amplitude = 2 Gauss and microwave power = 4 mW. The sample in A above was thawed in a room temperature water bath and incubated for an additional 55 s before refreezing. The MoV component represents ∼19% of total spin density. E, simulation of the Fe/S1 contribution to the spectrum in D. F, simulation of the MoV contribution to the spectrum in D. sim, simulation.
Additional Fe/S EPR Signals
We have next examined additional EPR signals attributable to the iron/sulfur centers of FdsABG, which have been only incompletely analyzed previously (2). The spectra shown in the following figures have been generated from samples prepared under various conditions described in detail in the figure legends and under “Experimental Procedures.” In addition to the iron/sulfur signal seen at 150 K shown in Figs. 4 and 6, additional signals are observed as the temperature is progressively lowered to 20 K. Fig. 7 illustrates the various experimental spectra observed in the temperature range, 20–100 K. Fig. 7A shows the spectrum seen at 100 K, which again can be described by a single rhombic species with g1,2,3 = 2.001, 1.946, 1.918 and which we designate as Fe/S1 (this is the same iron/sulfur cluster that contributes to the molybdenum-containing spectra in Figs. 4 and 6). This species is generated by brief air reoxidation of the sample exhibiting the MoV signal in Fig. 6D. Fe/S1 can also be generated by reducing nonfunctional enzyme with less than 1 e− equivalent and is certainly the iron/sulfur cluster with the highest reduction potential. On the basis of its high reduction potential, we tentatively assign the cluster giving rise to the Fe/S1 signal as that coordinated by His-112 near the N terminus of the FdsA subunit, because substitution of His for Cys in iron/sulfur clusters is known to increase their reduction potentials. Nonfunctional enzyme that is unable to oxidize formate is prepared by incubation of the enzyme in nitrate-free buffer for at least 6 h at 4 °C. Upon exhaustive reduction of the nonfunctional enzyme with sodium dithionite, a second, nearly axial Fe/S signal is observed below 120 K with g1,2,3 = 2.017, 1.947, 1.933, which we designate Fe/S2 (Fig. 7B). At 60 K the integrated intensities of the two signals are identical within experimental error. The Fe/S1 and Fe/S2 signals are also observed in a sample prepared by reducing nonfunctional enzyme with 10 mm NADH and freezing promptly (data not shown). As the temperature is lowered to 20 K, two additional signals become apparent. The first, which we designate Fe/S3, can be simulated with g1,2,3 = 2.044, 1.937, 1.898 (Fig. 7C), although precise determination of g2 is difficult because it overlaps with features of other signals. The second additional signal apparent in the 20 K spectrum is very broad and can be observed up to 60 K. This signal can be better resolved at microwave powers greater than 20 mW, where Fe/S1 can be almost completely power-saturated, and Fe/S2 and Fe/S3 can be partially so. This broad signal, designated Fe/S4, is most clearly evident with 10-Gauss modulation amplitude and 100-mW microwave power (Fig. 7D). Simulation of the experimental spectrum yields g1,2,3 = 2.095, 1.888, 1.862 for Fe/S4. Upon closer inspection of the shape of the Fe/S4 signal (Fig. 7E), the asymmetry and the broadness of the g1 and g2 tensors suggest an additional very broad signal, and it is noteworthy that at nonsaturating power (Fig. 7C), the simulation yields a slightly different set of g values with g1,2,3 = 2.095, 1.885, 1.882, which we designate Fe/S4′ in Table 1. This lends additional evidence for the presence of a second very broad signal in the envelope in Fig. 7 (C–E). Although the total integrated intensity of the Fe/S4-Fe/S4′ contribution to the experimental spectrum is over 23% in Fig. 7C and 35% in Fig. 7D, the very broad nature of this signal makes precise determination of the g tensor difficult, particularly with regard to g2, which overlaps with features of other signals.
FIGURE 7.
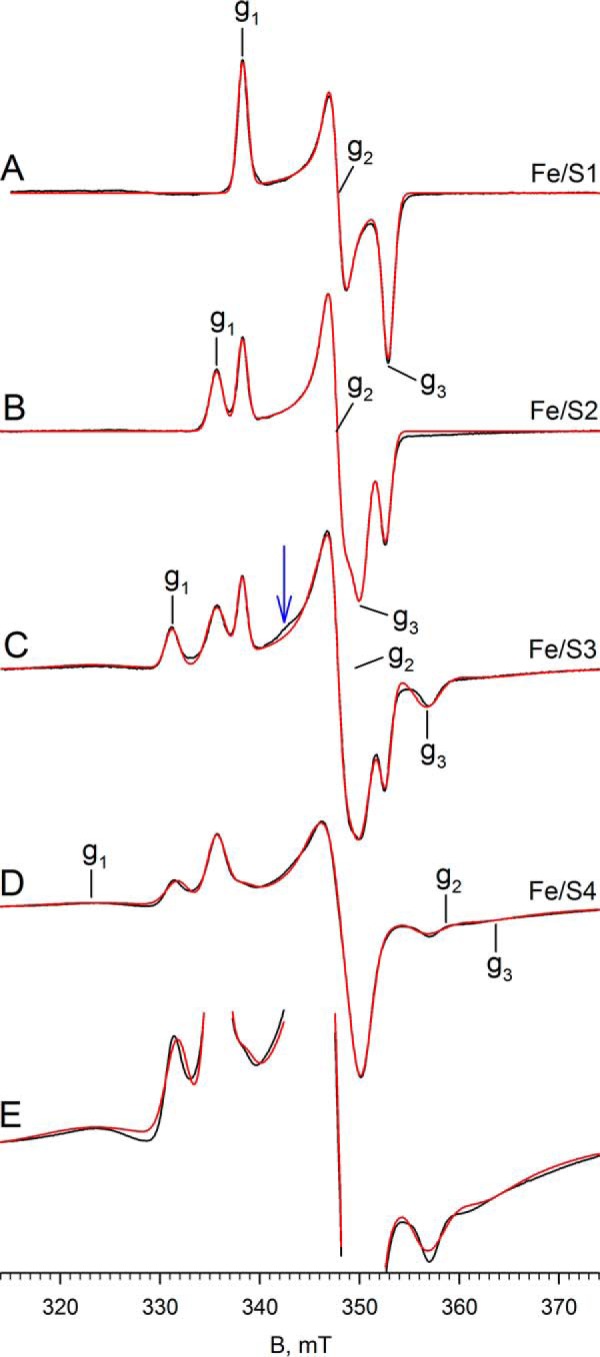
EPR of Fe/S centers of FdsABG. A, Fe/S1 EPR spectrum (black) and simulation (red) of FdsABG collected with modulation amplitude = 4 Gauss and microwave power = 4 mW at 100 K. The sample was prepared by thawing out the sample from Fig. 6D under aerobic conditions in a room temperature water bath and incubating for an additional 1 min before refreezing. Locations of g1, g2, and g3 are indicated. B, iron/sulfur EPR spectrum (black) and simulation (red) of FdsABG collected at 60 K with modulation amplitude = 4 Gauss and microwave power = 4 mW. The sample was prepared by incubation of 100 μm of FdsABG in 75 mm K-PO4, pH 7.7, in the absence of 10 mm KNO3 for >6 h. (aerobically at 4 °C), followed by reduction under anaerobic conditions with 5 mm buffered sodium dithionite and further incubation for 1 h before freezing. Locations of g1, g2, and g3 corresponding to the Fe/S2 component of the spectrum are indicated. C, Fe/S EPR spectrum (black) and simulation (red) of FdsABG sample from B collected at 20 K at with modulation amplitude = 3 Gauss and microwave power = 0.2 mW. Locations of g1, g2, and g3 corresponding to the Fe/S3 component of the spectrum are indicated. D, iron/sulfur EPR spectrum (black) and simulation (red) of FdsABG sample from B collected at 20 K at with modulation amplitude = 10 Gauss and microwave power = 100 mW. Locations of g1, g2, and g3 corresponding to the Fe/S4 component of the spectrum are indicated. E, spectrum from D enlarged to show the broad peaks corresponding to Fe/S4. Simulation parameters are summarized in Table 1.
Comparing the region centered at ∼342 mT in Fig. 7C with Fig. 7D, the simulations are unable to reproduce a minor feature of the experimental spectrum (see blue arrow in Fig. 7C), which appears to be power saturated in Fig. 7D. Attempts to manipulate both the position and linewidth parameters of the g2 tensor of either Fe/S3 or Fe/S4 to account for this small feature were unsuccessful, and it is possible that this feature may represent g2 of a sixth cluster with its g1 overlapping with one or another of the better described signals. Simulation of the composite spectrum in Fig. 7C yields integrated intensities for Fe/S1 through Fe/S4 in a ratio of ∼1:1.6:1.3:1.1, respectively. Simulations of the four well resolved iron/sulfur clusters are shown in Fig. 8.
FIGURE 8.
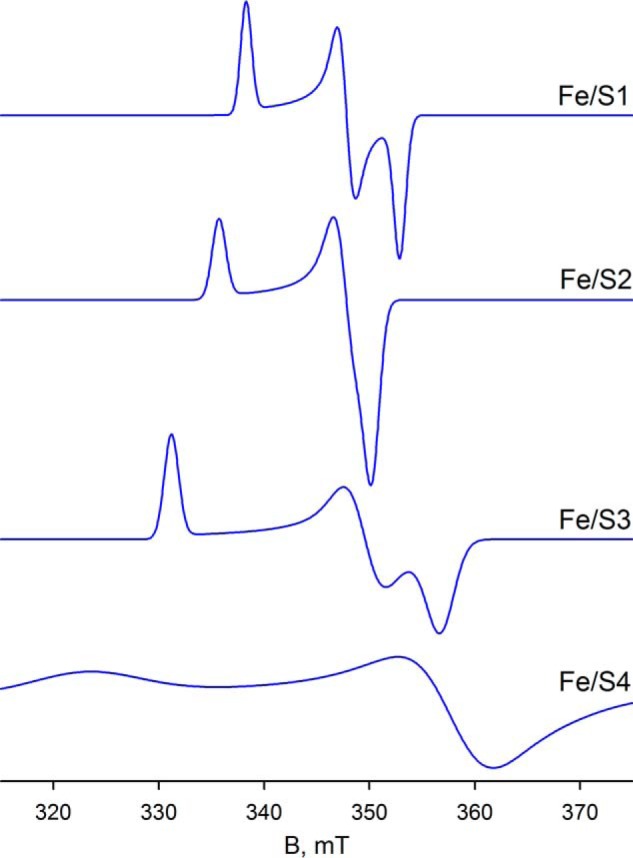
Simulations of EPR spectra for Fe/S1–Fe/S4 from parameters summarized in Table 1. The iron/sulfur clusters are classified by the increasing value of their g1 tensor.
Molybdenum-Iron/Sulfur Interaction
Finally, we analyzed the dithionite-reduced FdsABG in deuterated buffer at 20 and 60 K. At 60 K, the experimental spectrum contains contributions from both the narrow MoV signal and also three iron/sulfur signals: Fe/S1, Fe/S2, and Fe/S4′ in a ratio of ∼1:0.8:0.2, respectively (Fig. 9). The simulation in Fig. 9C shows the MoV, whereas a comparison of the sum of the MoV and iron/sulfur components with the experimental spectrum is shown in Fig. 9A (red versus black, respectively). At 20 K, the experimental spectrum differs from the data collected at 60 K by the appearance of an additional signal (Fig. 9D, black). Simulation of the iron/sulfur component yields four signals in a ratio of ∼1:0.9:0.9:0.7, assigned to Fe/S1, Fe/S2, Fe/S3, and Fe/S4′, respectively (Fig. 9E). Simulation of the MoV signal in the spectrum obtained at 20 K indicates that the signal is split with a hyperfine coupling constant of ∼19 MHz (Fig. 9, compare C with F), which we attribute to anisotropic coupling to an iron/sulfur cluster. Because the experimental spectra at 60 and 20 K differ only in the presence of Fe/S3 in the latter, we conclude that Fe/S3 is the magnetically coupled partner to the MoV center. The coupling on Fe/S3 caused by the MoV center is not observed because of the broad linewidths associated with Fe/S3. We do note, however, that the extreme anisotropy of the coupling is extremely unusual, although it appears not to represent a rapid passage artifact as the coupling persisted unchanged as the EPR modulation frequency was varied (data not shown). In addition to the simulation of the isolated MoV signals, Fig. 9D shows a comparison of the sum of the MoV and iron/sulfur components with the experimental spectrum (red versus black, respectively). As can be seen, the fit to the experimental spectrum is excellent, and we conclude that five signal-giving species are present in the experimental spectrum: a single MoV species, magnetically coupled to an adjacent iron sulfur, Fe/S3, and the EPR spectra of Fe/S3 and three other iron/sulfur signals.
FIGURE 9.
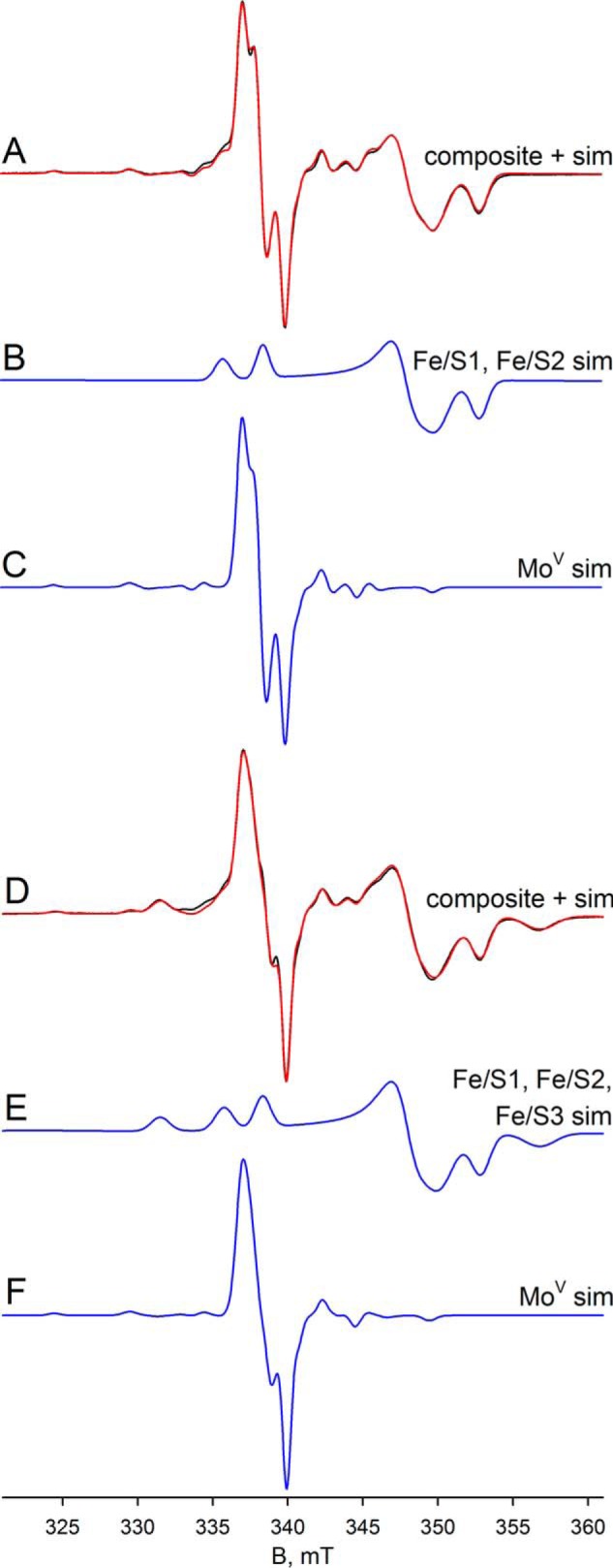
EPR of deuterated molybdenum center of FdsABG collected at 20–60 K. A, MoV-Fe/S EPR spectrum (black) and simulation (red) of FdsABG sample as described in Fig. 4D collected with modulation amplitude = 2 Gauss and microwave power = 0.2 mW at 60 K. The MoV component represents ∼27% of total spin density. B, simulation of the Fe/S1 and Fe/S2 contributions to the spectrum in A. C, simulation of the MoV contribution to the spectrum in A. D, MoV-Fe/S EPR spectrum (black) and simulation (red) of FdsABG sample from A. above collected with modulation amplitude = 2 Gauss and microwave power = 0.02 mW at 20 K. The MoV component represents ∼16% of total spin density. E, simulation of the Fe/S1–Fe/S3 contributions to the spectrum in D. F, simulation of the MoV contribution to the spectrum in D. The simulation parameters are summarized in Table 1. sim, simulation.
Discussion
Kinetics of the R. eutropha FdsABG Formate Dehydrogenase
The steady-state kinetics reported here demonstrate that our enzyme preparation has the characteristics previously reported and in particular has approximately the same high specific activity. Our rapid kinetic study of the reaction of enzyme with formate indicates that the reductive half-reaction is multiphasic. The fast and intermediate phases both exhibit hyperbolic dependence on [formate], with a limiting kfast of 140 s−1 and Kdformate of 82 μm for the fast phase and limiting kintermediate of 19 s−1 and Kdformate of 230 μm for the intermediate phase. Given the large number of redox-active centers and the fact that at least five equivalents of formate must react with enzyme to fully reduce it, we interpret the fast phase of the reaction as reflecting the intrinsic rate constant for reaction of formate with enzyme possessing an oxidized molybdenum center, with the intermediate phase reflecting the reaction of formate with enzyme that has already reacted with several equivalents of substrate and has a significant amount of molybdenum that is at least partially reduced and therefore unreactive. It is likely that electron transfer among the several redox-active centers of FdsABG is rapid relative to the rate of formate oxidation at the molybdenum center, so that in these partially reduced enzyme forms, the distribution of electrons will be dictated by the relative reduction potentials of the centers. On the basis of the absorbance change associated with the fast and intermediate phases of the reaction with formate, FMN reduction is associated with the latter, suggesting that the FMN has a relatively low reduction potential and accumulates reducing equivalents only later in the reaction. Although it is surprising that this might be the case given that the FMN is the site of reaction with NAD+, it is consistent with the results of the equilibrium reductive titration reported here, which indicates that the FMN is reduced only later in the course of the titration.
With regard to the fast phase of the reaction, which again is taken to reflect the intrinsic rate of reaction of formate with the fully oxidized molybdenum center, we note that the limiting rate constant of 140 s−1 at 10 °C corresponds to ∼600 s−1 at 30 °C, the temperature at which, as in past work, the steady-state work was done (assuming a doubling of rate constant for every 10 °C increase in temperature). This being the case, a comparison with the observed kcat of 201 s−1 indicates that the reductive half-reaction of the catalytic sequence is only partially rate-limiting. Similarly, the modest kinetic isotope of 2.1 seen when using [2H]formate as substrate indicates that the isotope-sensitive step is only partially rate-limiting in the reductive half-reaction.
MoV EPR and Mechanistic Implications
With regard to the EPR of the molybdenum center, we note that the very high gave of the MoV EPR signal seen here is consistent with an all-sulfur molybdenum coordination sphere, as concluded in a recent XAS analysis of the homologous FdsABG from R. capsulatus (9). The signal is also characterized by strong coupling to a solvent-exchangeable proton, with A1,2,3 = 18, 21, and 18 MHz. This coupling is in fact comparable with that exhibited by the molybdenum-SH proton of the so-called “rapid type 1” EPR signal seen with xanthine oxidase, another molybdenum-containing enzyme possessing a molybdenum-sulfur in the oxidized state. Like the strongly coupled proton in the “rapid type 1” signal seen with xanthine oxidase, that which is coupled in the MoV signal of FdsABG is substrate-derived and, ultimately, solvent-exchangeable. We conclude that the substrate-derived, solvent-exchangeable, and strongly coupled proton seen here with the FdsABG formate dehydrogenase is similarly due specifically to the proton of a molybdenum-SH group, formed by protonation of the molybdenum-sulfur group of oxidized enzyme upon reduction of the molybdenum. Although it is conceivable that this hydride transfer might be mediated by some intervening group in the active site, this site would have to be both redox-active and not solvent-exchangeable. An examination of the active site of the E. coli FdhF formate dehydrogenase (Fig. 10), indicates that no such group is readily identifiable. We specifically note that of the amino acid residues surrounding the molybdenum center, only two are not conserved between FdhF and FdsA: Sec-140 that coordinates the metal (and is a Cys in FdsA) and Val-338, which is a Leu in FdsA. His-141 is in the substrate binding pocket but is not redox-active; similarly, the pyranopterin cofactors of the molybdenum center are known not to be formally redox-active (1). The demonstration here that the strongly coupled proton seen in the cysteine-containing R. eutropha FdsABG is substrate-derived is also consistent with previous work with the selenocysteine-containing FdhF formate dehydrogenase from E. coli, which also exhibits a strongly coupled and substrate-derived proton (27) (we also confirm the strong pH dependence of the rate of solvent exchange seen with the FdsF protein, which slows significantly at higher pH; data not shown). These earlier workers, however, assigned the coupled proton to a conserved active site histidine of the FdhF enzyme (His-141, lying some 6.1 Å from the molybdenum) rather than to a ligand of the molybdenum coordination sphere (which at the time was believed to possess molybdenum-oxygen rather than molybdenum-sulfur in the oxidized state and to possess only the selenocysteine and pyranopterin ligands in a five-coordinate reduced state (5)). Protonation of the selenocysteine ligand to the molybdenum of the E. coli FdhF was thus the only option considered in this earlier work). We note that our present assignment of the strongly coupled proton to a MoV-SH group is in fact fully consistent with all results reported previously for the E. coli FdhF enzyme and provides the more plausible explanation as to why the observed proton coupling should be so strong.
FIGURE 10.
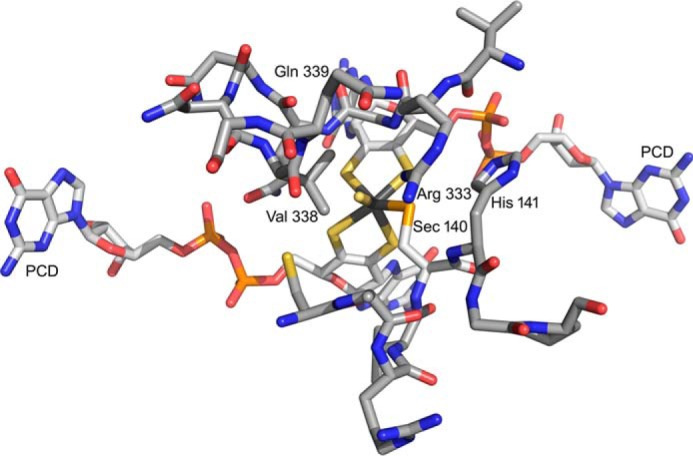
The active site of the FdhF formate dehydrogenase from E. coli (PDB code 1FDO). The molybdenum is at the center of the figure, coordinated by the two equivalents of pyranopterin cytidine dinucleotide (PCD), a terminal sulfido (represented as a coordinated water in the original PDB file), and Sec-140. The substrate binding site is delineated by two loops consisting of 137ARVUHGP143 and 328GVNPLRGQNNVQG340. Arg-333 has been proposed to interact with the negatively charged substrate. In these two stretches, only two residues differ in the FdsA subunit of the R. eutropha formate dehydrogenase: Sec-140, which is a Cys that coordinates the molybdenum in FdsA, and Val-338, which is a Leu in FdsA.
On the basis of the interpretation that the substrate proton is abstracted by an active site histidine, mechanisms for the E. coli FdhF (and by implication other molybdenum-containing formate dehydrogenases) have been proposed in which formate coordinates to the molybdenum via its oxyanion group and displaces the selenocysteine, followed by abstraction of the Cα proton by His-141 (possibly involving the now-dissociated SeCys) and transfer of an electron pair into the redox-active molybdenum dxy orbital (5). Given that the pKa of a molybdenum-coordinated formate is expected to be at least as high as that of formamide (pKa = 23.5) (28), proton abstraction by a histidine residue is extremely thermodynamically unfavorable. We propose instead that the reaction for both the cysteine- and selenocysteine-containing molybdenum formate dehydrogenases (and probably also the related tungsten-containing enzymes) proceeds by a simple hydride transfer from formate, with molybdenum-sulfur serving as the hydride acceptor, as shown in Fig. 11. In this mechanism, it must be remembered that in a formal valence count, all four electrons in the molybdenum-sulfur bond of oxidized enzyme belong to the sulfur, and the last arrow in the sequence shown represents the formal reduction of the six-coordinate molybdenum from MoVI to MoIV. Retention of the catalytically essential sulfur as a ligand to the molybdenum in the reduced species is necessary because the subsequent transfer of one electron out of the molybdenum center yields the MoV-SH species observed by EPR (its loss in any case would lead to inactivation of the enzyme). Similarly, it is likely (although not required by the mechanism) that the cysteine ligand (or selenocysteine in the case of FdhF) is retained. We note that XAS analysis of FdhF from both E. coli (29) and Desulfovibrio desulfuricans (30) indicates that the molybdenum center is six-coordinate in the reduced state with the selenocysteine of these enzymes specifically retained; the reduced FdsABG from R. capsulatus is also six-coordinate (9), with the cysteine retained. The catalytic sequence is completed from the EPR-active MoV state by a second electron transfer and loss of the substrate-derived proton to yield the initial MoVI species.
FIGURE 11.

A proposed hydride transfer mechanism for the formate dehydrogenases. Beginning with the ionized substrate, the second carbon-oxygen double bond forms displacing hydride, which attacks the molybdenum-sulfur group, bringing about the formal two-electron reduction of the metal with concomitant transfer of the Cα-H to the sulfur.
Supporting this mechanism, we note that: 1) formate is intrinsically a good hydride donor (as reflected in the fact that the eukaryotic formate dehydrogenases are cofactorless and catalyze direct hydride transfer from formate to NAD+ through a ternary E-formate-NAD+ complex); and 2) molybdenum-sulfur groups are facile hydride acceptors (virtually all members of the xanthine oxidase family of molybdenum enzymes possess a molybdenum-sulfur group that serves as a hydride acceptor in the course of the reaction (14–16)). Other elements of this mechanism are also chemically reasonable, namely that formate need not coordinate to the molybdenum in the course of the reaction and that the molybdenum itself is coordination-stable (apart from the conversion of a MoVI-S group to a MoIV-SH group in the course of the reaction). As in the case of xanthine oxidase and related enzymes (31, 32), the MoV state forms only at the completion of the reductive half-reaction, with transfer of a single electron to the proximal [4Fe-4S] cluster to yield the EPR-active MoV-SH species. It is to be emphasized that the proton egress pathway from the molybdenum center identified by Stadtman and co-workers (5, 27), beginning with His-141 (analogous to His-379 in the FdsABG enzyme), indeed likely plays an important catalytic role in facilitating deprotonation of the molybdenum center as it is reoxidized to the MoVI state upon subsequent transfer of the second electron out of the molybdenum center.
EPR of the Iron/Sulfur Centers of FdsABG
As summarized in Fig. 8 and Table 1, we have identified and characterized four EPR signals, designated Fe/S1-Fe/S4, that are attributable to the four iron/sulfur clusters of FdsABG; a fifth signal is tentatively identified but less well characterized (Fe/S4′). Of the four well characterized signals, two are almost axial and two are rhombic; of these, two exhibit a particularly high g anisotropy (Fe/S3 and Fe/S4). Fe/S1 and Fe/S2, the more modestly anisotropic signals, are observed at liquid nitrogen temperatures and exhibit rather narrow linewidths that suggest slow spin lattice relaxation rates. Although Fe/S1 is observed at temperatures from 200 K down to below 5 K, Fe/S3 is only seen below 120 K. The two more highly rhombic Fe/S3 and Fe/S4 signals are seen only at liquid helium temperatures. All of the iron/sulfur signals persist down to the limit of helium temperatures, below 5 K.
Based on the sequence (33) and structural (2) similarities of the (cofactorless) C-terminal region of Nqo3 of the T. thermophilus NADH dehydrogenase (2, 3) to the FdhF formate dehydrogenase (5), a model can be constructed for FdsABG that gives some indication of the disposition of its nine redox-active centers, as shown in Fig. 12 (1). Interestingly, in the structure of the T. thermophilus Nqo3 subunit, the C-terminal [4Fe-4S] cluster (that nearest the position of the erstwhile molybdenum-containing domain) is some 20 Å from the nearest iron/sulfur cluster in the subunit. The alignment of Nqo3 with FdsA, however, provides clear evidence for an intervening [4Fe-4S] cluster (to the N-terminal side of the isolated cluster in Nqo3) in FdsA (1). The position of this additional cluster in FdsABG in the context of the structure of Nqo3 of the T. thermophilus NADH dehydrogenase is readily defined by noting that the last [4Fe-4S] cluster before the gap in Nqo3 is found in a classic 2 × [4Fe-4S] bacterial ferredoxin-like structural motif that lacks a second [Fe-4S] cluster (because of loss of cysteine residues necessary to coordinate it). This circumstance allows the approximate position of the additional [4Fe-4S] cluster of FdsA to be identified in the context of the structure of the T. thermophilus NADH dehydrogenase (Fig. 12, circled) (1).
FIGURE 12.
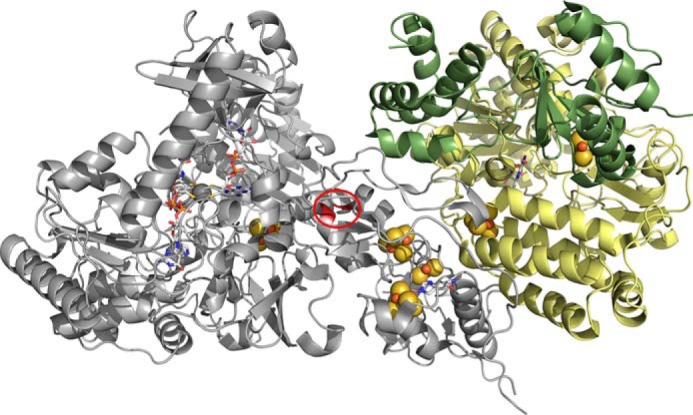
A structural model for the R. eutropha formate dehydrogenase. The molybdenum center is at left, and the FMN is at right, with the several iron/sulfur clusters intervening. The model is based on homologies to the T. thermophilus NADH dehydrogenase (PDB code 3IAM) and the E. coli formate dehydrogenase (PDB code 1AA6), with the position of the additional [4Fe-4S] cluster present in FdsA but absent in Nqo3 indicated by the red circle.
The above model provides a frame of reference for assigning the EPR signals seen with FdsABG to specific clusters, as correlated with its amino acid sequence. First, the iron/sulfur cluster that is magnetically coupled to the molybdenum center is undoubtedly that lying closest to it. We therefore assign the Fe/S3 signal with g1,2,3 = 2.044, 1.937, 1.898 EPR to the most C-terminal [4Fe-4S] cluster of FdsA, coordinated by Cys-249, Cys-252, Cys-256, and Cys-285. This cluster is homologous to the sole iron/sulfur cluster seen in the E. coli FdhF enzyme, which lies to the N-terminal side of the molybdenum-binding portion of the enzyme (and whose EPR signal has g1,2,3 = 2.045, 1.957, 1.840 (27)). Treating the electron-electron interaction as strictly dipolar in nature, the experimentally observed coupling of 19 MHz would correspond to a 14 Å center to center distance (12) that is generally consistent with the 12.4 Å distance seen in the E. coli FdhF formate dehydrogenase (PDB code 1AA6). The next most straightforward assignment is Fe/S1, which with g1,2,3 = 2.001, 1.946, 1.918 has both a relatively low gave and a high reduction potential. We thus tentatively assign this signal to the [4Fe-4S] cluster of FdsA that is coordinated by His-112, Cys-116, Cys-119, and Cys-125, noting that the histidine for sulfur substitution is expected to have the dual effect of decreasing gave and increasing the reduction potential of the cluster. Assignment of the remaining EPR signals is much more problematic and must await future site-directed mutagenesis studies knocking out one or another center to allow definitive assignments.
Author Contributions
D. N. performed all EPR and rapid reaction kinetic work and assisted in preparing the manuscript. J. D. and M. E. assisted with the steady-state kinetics work. R. H. conceived the project, directed the research and was principally responsible for preparation of the manuscript.
This work was supported by U.S. Department of Energy Grant DE-FG02-13ER16411 (to R. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- EPR
- electron paramagnetic resonance
- PDB
- Protein Data Bank.
References
- 1.Hille R., Hall J., and Basu P. (2014) The mononuclear molybdenum enzymes. Chem. Rev. 114, 3963–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sazanov L. A., and Hinchliffe P. (2006) Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436 [DOI] [PubMed] [Google Scholar]
- 3.Efremov R. G., Baradaran R., and Sazanov L. A. (2010) The architecture of respiratory complex I. Nature 465, 441–445 [DOI] [PubMed] [Google Scholar]
- 4.Oh J. I., and Bowien B. (1998) Structural analysis of the fds operon encoding the NAD+-linked formate dehydrogenase of Ralstonia eutropha. J. Biol. Chem. 273, 26349–26360 [DOI] [PubMed] [Google Scholar]
- 5.Boyington J. C., Gladyshev V. N., Khangulov S. V., Stadtman T. C., and Sun P. D. (1997) Crystal structure of formate dehydrogenase H: catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. Science 275, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 6.Rothery R. A., Workun G. J., and Weiner J. H. (2008) The prokaryotic complex iron-sulfur molybdoenzyme family. Biochim. Biophys. Acta 1778, 1897–1929 [DOI] [PubMed] [Google Scholar]
- 7.Hille R. (1996) The mononuclear molybdenum enzymes. Chem. Rev 96, 2757–2816 [DOI] [PubMed] [Google Scholar]
- 8.Thomé R., Gust A., Toci R., Mendel R., Bittner F., Magalon A., and Walburger A. (2012) A sulfurtransferase is essential for activity of formate dehydrogenases in Escherichia coli. J. Biol. Chem. 287, 4671–4678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schrapers P., Hartmann T., Kositzki R., Dau H., Reschke S., Schulzke C., Leimkühler S., and Haumann M. (2015) Sulfido and cysteine ligation changes at the molybdenum cofactor during substrate conversion by formate dehydrogenase (FDH) from Rhodobacter capsulatus. Inorg. Chem. 54, 3260–3271 [DOI] [PubMed] [Google Scholar]
- 10.Friedebold J., and Bowien B. (1993) Physiological and biochemical characterization of the soluble formate dehydrogenase, a molybdoenzyme from Alcaligenes eutrophus. J. Bacteriol. 175, 4719–4728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedebold J., Mayer F., Bill E., Trautwein A. X., and Bowien B. (1995) Structural and immunological studies on the soluble formate dehydrogenase from Alcaligenes eutrophus. Biol. Chem. Hoppe-Seyler 376, 561–568 [DOI] [PubMed] [Google Scholar]
- 12.Stoll S., and Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 [DOI] [PubMed] [Google Scholar]
- 13.Olson J. S., Ballou D. P., Palmer G., and Massey V. (1974) Mechanism of action of xanthine oxidase. J. Biol. Chem. 249, 4363–4382 [PubMed] [Google Scholar]
- 14.Edmondson D., Ballou D., Van Heuvelen A., Palmer G., and Massey V. (1973) Kinetic studies on substrate reduction of xanthine oxidase. J. Biol. Chem. 248, 6135–6144 [PubMed] [Google Scholar]
- 15.Brody M. S., and Hille R. (1999) The kinetic behavior of chicken liver sulfite oxidase. Biochemistry 38, 6668–6677 [DOI] [PubMed] [Google Scholar]
- 16.Hartmann T., and Leimkühler S. (2013) The oxygen-tolerant and NAD+-dependent formate dehydrogenase from Rhodobacter capsulatus is able to catalyze the reduction of CO2 to formate. FEBS J. 280, 6083–6096 [DOI] [PubMed] [Google Scholar]
- 17.Bennett B., Benson N., McEwan A. G., and Bray R. C. (1994) Multiple states of the molybdenum center of dimethylsulfoxide reductase from Rhodobacter capsulatus revealed by EPR. Eur. J. Biochem. 225, 321–331 [DOI] [PubMed] [Google Scholar]
- 18.Bray R. C., Knowles P. F., Pick F. M., and Vänngård T. (1968) Electron spin resonance evidence for interaction of protons with Mo(V) in reduced froms of xanthine oxidase. Biochem. J. 107, 601–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bray R. C., and Vänngård T. (1969) “Rapidly appearing” molybdenum electron paramagnetic resonance signals from xanthine oxidase. Biochem. J. 114, 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hille R. (1994) The reaction mechanism of oxomolybdenum enzymes. Biochim. Biophys. Acta 1184, 143–169 [DOI] [PubMed] [Google Scholar]
- 21.Huber R., Hof P., Duarte R. O., Moura J. J., Moura I., Liu M.-Y., LeGall J., Hille R., Archer M., and Romão M. J. (1996) A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proc. Natl. Acad. Sci. U.S.A. 93, 8846–8851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xia M., Dempski R., and Hille R. (1999) The reductive half-reaction of xanthine oxidase: reaction with aldehyde substrates and identification of the catalytically labile oxygen. J. Biol. Chem. 274, 3323–3330 [DOI] [PubMed] [Google Scholar]
- 23.Gutteridge S., Tanner S. J., and Bray R. C. (1978) The molybdenum center of xanthine oxidase: evidence for proton transfer from substrates to the center and for the existence of an anion binding site. Biochem. J. 175, 869–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gutteridge S., Tanner S. J., and Bray R. C. (1978) Comparison of the molybdenum centers of native and desulfo xanthine oxidase: nature of the cyanide-labile sulfur atom and nature of the proton-accepting group. Biochem. J. 175, 887–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Malthouse J. P., Gutteridge S., and Bray R. C. (1980) Rapid type-2 molybdenum(V) electron paramagnetic resonance signals from xanthine oxidase and the structure of the active center of the enzyme. Biochem. J. 185, 767–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gutteridge S., Bray R. C. (1980) O-17 splitting of the very rapid molybdneum(V) electron paramagnetic resonance signal from xanthine oxidase: rate of exchange with water of the coupled oxygen. Biochem. J. 189, 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khangulov S. V., Gladyshev V. N., Dismukes G. C., and Stadtman T. C. (1998) Selenium-containing formate dehydrogenase H from Escherichia coli: a molybdopterin enzyme that catalyzes formate oxidation without oxygen transfer. Biochemistry 37, 3518–3528 [DOI] [PubMed] [Google Scholar]
- 28.Bordwell F. G., Bartmess J. E., and Hautala J. A. (1978) Alkyl effects on equilibrium acidities of carbon acids in protic and dipolar aprotic media and the gas phase. J. Org. Chem. 43, 3113–3116 [Google Scholar]
- 29.George G. N., Colangelo C. M., Dong J., Scott R. A., Khangulov S. V., Gladyshev V. N., and Stadtman T. C. (1998) X-ray absorption spectroscopy of the molybdenum site of Escherichia coli formate dehydrogenase. J. Am. Chem. Soc. 120, 1267–1273 [Google Scholar]
- 30.George G. N., Costa C., Moura. J. J. G., and Moura I. (1999) Observation of ligand-based redox chemistry at the active site of a molybdenum enzyme. J. Am. Chem. Soc. 121, 2625–2626 [Google Scholar]
- 31.McWhirter R. B., and Hille R. (1991) The reductive half-reaction of xanthine oxidase: identification of spectral intermediates in the hydroxylation of 2-hydroxy-6-methylpurine. J. Biol. Chem. 266, 23724–23731 [PubMed] [Google Scholar]
- 32.Hille R., Kim J. H., and Hemann C. (1993) The reductive half-reaction of xanthine oxidase: mechanistic role of the rapid type-1 MoV electronparamagnetic resonance signal. Biochemistry 32, 3973–39780 [DOI] [PubMed] [Google Scholar]
- 33.Finel M. (1998) Organization and evolution of structural elements within complex I. Biochim. Biophys. Acta 1364, 112–121 [DOI] [PubMed] [Google Scholar]