

Abstract
IMPORTANCE
The identification of a patient with a rare form of severe dysbetalipoproteinemia allowed the study of the consequences of total absence of apolipoprotein E (apoE).
OBJECTIVES
To discover the molecular basis of this rare disorder and to determine the effects of complete absence of apoE on neurocognitive and visual function and on lipoprotein metabolism.
DESIGN, SETTING, AND PARTICIPANTS
Whole-exome sequencing was performed on the patient’s DNA. He underwent detailed neurological and visual function testing and lipoprotein analysis. Lipoprotein analysis was also performed in the Cardiovascular Research Institute, University of California, San Francisco, on blood samples from the proband’s mother, wife, 2 daughters, and normolipidemic control participants.
MAIN OUTCOME MEASURES
Whole-exome sequencing, lipoprotein analysis, and neurocognitive function.
RESULTS
The patient was homozygous for an ablative APOE frameshift mutation (c.291del, p.E97fs). No other mutations likely to contribute to the phenotype were discovered, with the possible exception of two, in ABCC2 (p.I670T) and LIPC (p.G137R). Despite complete absence of apoE, he had normal vision, exhibited normal cognitive, neurological, and retinal function, had normal findings on brain magnetic resonance imaging, and had normal cerebrospinal fluid levels of β-amyloid and tau proteins. He had no significant symptoms of cardiovascular disease except a suggestion of myocardial ischemia on treadmill testing and mild atherosclerosis noted on carotid ultrasonography. He had exceptionally high cholesterol content (760 mg/dL; to convert to millimoles per liter, multiply by 0.0259) and a high cholesterol to triglycerides ratio (1.52) in very low-density lipoproteins with elevated levels of small-diameter high-density lipoproteins, including high levels of prebeta-1 high-density lipoprotein. Intermediate-density lipoproteins, low-density lipoproteins, and very low-density lipoproteins contained elevated apoA-I and apoA-IV levels. The patient’s apoC-III and apoC-IV levels were decreased in very low-density lipoproteins. Electron microscopy revealed large lamellar particles having electron-opaque cores attached to electron-lucent zones in intermediate-density and low-density lipoproteins. Low-density lipoprotein particle diameters were distributed bimodally.
CONCLUSIONS AND RELEVANCE
Despite a profound effect on lipoprotein metabolism, detailed neurocognitive and retinal studies failed to demonstrate any defects. This suggests that functions of apoE in the brain and eye are not essential or that redundant mechanisms exist whereby its role can be fulfilled. Targeted knockdown of apoE in the central nervous system might be a therapeutic modality in neurodegenerative disorders.
Familial dysbetalipoproteinemia (type III hyperlipoproteinemia; OMIM 107741) is characterized by elevated levels of plasma cholesterol and triglycerides (TG) resulting from an accumulation of chylomicrons, very lowdensity lipoproteins (VLDL), and their remnants. Affected individuals may display palmar and/or tuberous xanthomas and develop premature atherosclerosis.1 This remnant removal disease is associated with APOE variants. Apolipoprotein E (apoE) plays a critical role in low-density lipoprotein (LDL) receptor (LDLR) and LDLR-related protein/heparin sulfate proteoglycan pathways for remnant clearance.1 The mature protein has 299 amino acids. It has LDLR-binding and lipid-binding domains at residues 130 through 150 and 200 through 280, respectively.1 Three common isoforms exist, apoE2, apoE3, and apoE4. Apolipoprotein E2 has low receptor-binding activity compared with apoE3. More than 90% of patients with dysbetalipoproteinemia2 are apoE2 homozygotes. The rest have rare deleterious APOE mutations.3 Most apoE2 homozygotes are unaffected, the presence of other genetic and/or environmental factors being necessary for development of dysbetalipoproteinemia. Cases of apoE deficiency have been reported,4-9 but none included neurocognitive or retinal function studies.
Next-generation DNA sequencing produces abundant, short, inexpensive reads (25-500 base pairs [bp]).10 Whole-exome sequencing was undertaken in an exceptionally severe case of dysbetalipoproteinemia to understand the molecular basis. Structural and compositional effects on lipoproteins were studied. Because apoE is believed to play a critical role in brain function11 and is expressed in the central nervous system and retinal pigment epithelium,12 extensive studies of retinal and neurocognitive function were performed.
Methods
Participants
A 40-year-old African American man was referred to the Lipid Clinic, University of California, San Francisco, with severe hyperlipidemia relatively unresponsive to statin and fibrate treatment. He had large tuberous xanthomas (Figure 1), especially on his elbows, knees, and ankles, Achilles tendon xanthomas, morbilliform xanthomas on his ears, alae nasae, and volar aspects of his hands and feet, and planar xanthomas on his gluteal region. Xanthomas were present on dorsal aspects of his hands over proximal and distal interphalangeal joints and metacarpophalangeal joints. Xanthomas first appeared at age 6 years. His body mass index (calculated as weight in kilograms divided by height in meters squared) was 30.4. He had been treated for modest hypertension, never smoked or used illicit drugs, and was not drinking alcohol. His diet, generally low in fat, contained little saturated and no trans fats and was low in rapidly absorbed carbohydrates. He had no signs or symptoms of neurological deficit, ophthalmological disease, or cardiovascular disease (CVD). He did not start talking until age 3 years. At school, he performed better in mathematics than reading, finishing 11th grade with a C+/B− average. His parents and 4 maternal half siblings have no evidence of CVD. His father, not available for study, is known to be well and to have a normal lipid panel. The patient has 3 healthy children aged 4, 5, and 7 years. They and his 57-year-old mother have no xanthomas. Written informed consent was obtained from participants for DNA isolation and plasma collection. Children were included with parental consent. The University of California, San Francisco Committee of Human Research Institutional Review Board approved these studies.
Figure 1. Xanthomas of a Patient With Homozygous Apolipoprotein E Deficiency.
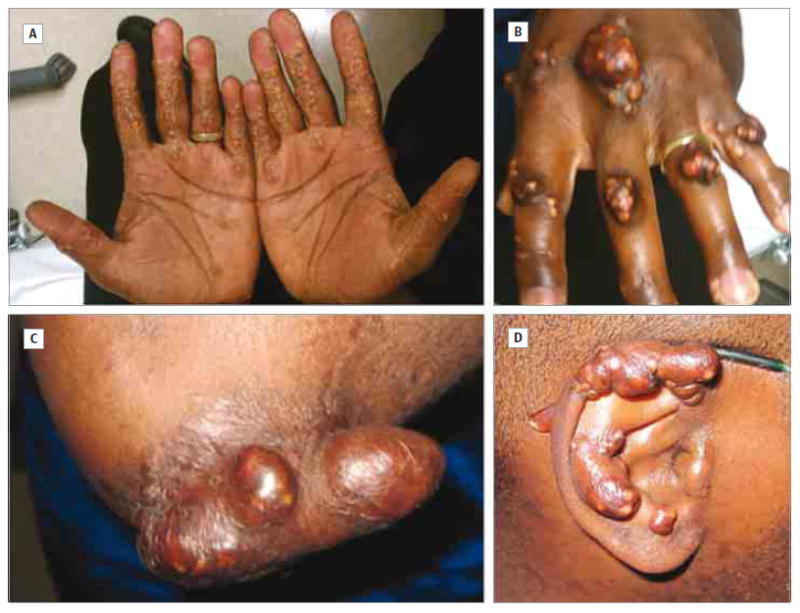
Palmar crease and morbilliform xanthomas (A) and tuberous xanthomas on the hands (B), elbow (C), and ear (D).
Clinical Testing
Ophthalmic examination included best-corrected visual acuity, automated perimetry with measurement of foveal thresholds, Goldmann kinetic perimetry, color vision examination, electro-oculography, and dark adaptation measurements. Color and autofluorescence fundus photographs and spectral-domain optical coherence tomographic images were obtained. Neurological examination included detailed physical examination by a neurologist, lumbar puncture, and brain magnetic resonance imaging. A full series of neuropsychological testing was undertaken. Cardiovascular examination included treadmill stress test (standard Bruce protocol), echocardiography, and bilateral duplex carotid ultrasonography.
Next-Generation Sequencing
We extracted DNA from blood using the Puregene kit (Qiagen). An Illumina TruSeq Exome Enrichment kit was used for library preparation and exome capture. The captured library was amplified with 10 polymerase chain reaction cycles and sequenced using an Illumina HiSeq 2000 sequencer. Paired 100-bp end reads (54million reads)were generated (mean coverage ×61) on targeted regions (89% coveredby ≥10 reads). Raw FASTQ data were aligned to the reference genome (hg19) by Burrows-Wheeler Aligner.13 Sequence Alignment/Map (SAM) files were converted to SAM Binary (BAM) files with SAMtools,14 sorted, marked for duplicates, and indexed with Picard (http://picard.sourceforge.net). Base quality score recalibration, local realignment, and duplicate removal were performed with Genome Analysis Toolkit.15,16 Single-nucleotide polymorphisms and indels were called by Genome Analysis Toolkit and quality filters were applied as recommended. The variant list was annotated and further filtered using summarize_annovar.pl from ANNOVAR.17 Rare and deleterious exonic variants were identified using the following filters: keep exonic or splicing variants, remove synonymous variants, remove variants not conserved and in segmental duplication regions, remove common variants (1000 Genomes Project; minor allele frequency >0.05), and keep damaging variants (Sorting Intolerant From Tolerant score <0.05). A list of 432 genes or loci (eTable 1 in the Supplement) related to lipid metabolism was compiled.
Lipoprotein Analysis
Blood was drawn from the patient, his mother, and a normolipidemic male participant after overnight fasting. Lipoprotein fractions were prepared from plasma by sequential ultracentrifugation.18 Cholesterol and TG contents were determined by automated chemical analysis. After negative staining with 2% potassium phosphotungstate, pH 6.5, using the drop method,19 lipoproteins were examined by electron microscopy at 80 kV in a JEOL 1230 electron microscope (JEOL USA, Inc) and photographed with a Gatan Ultrascan USC1000 camera (Gatan Inc), and particle diameters were measured using ImageJ64 (http://rsbweb.nih.gov/ij/index.html). Blood was also obtained from the patient’s wife and 2 daughters. Plasma from the patient and 8 control participants was analyzed using a Lipoprint high-density lipoproteins (HDL) sub-fractions kit (Quantimetrix). Nondenaturing 2-dimensional gradient gel electrophoresis followed by apoA-I immunoblotting was performed on plasma from the patient and 17 control participants.20
Lipoproteins were precipitated with ethanol and diethyl ether (3:1) and centrifuged. Pellets dissolved in10% sodium dodecyl sulfate were run on 4% to 20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis. Gels were stained with Coomassie Brilliant Blue R-250 or electrotransferred to nitrocellulose membranes. Following blocking, membranes were incubated with the appropriate antibody. Secondary antibodies were horseradish peroxidase linked. Blots were incubated with SuperSignal West Pico (Pierce). Chemiluminescence was detected using a ChemiDoc XRS+ imager (BioRad). Richard J. Havel, MD, University of California, San Francisco, provided apoB-100, apoE, and apoC-IV antibodies.21-23 The apoC-III goat antibody was ab27624 from Abcam. A trpE/apoA-IV fusion protein was generated to produce a goat antibody to human apoA-IV. The apoA-I antibody has been described.24
Cerebrospinal Fluid Analysis
Cerebrospinal fluid was obtained by lumbar puncture after an overnight fast. β-Amyloid 1-42 (Aβ42) and total tau protein levels were determined using the multiarray Aβ ultrasensitive kit and total tau kit, respectively (Meso Scale Discovery). Levels of tau phosphorylated at threonine 181 (P-tau181) were measured using the Innotest Phospho-Tau(181P) kit (Innogenetics). For Aβ42 and P-tau181, undiluted cerebrospinal fluidwas used. For total tau, it was diluted 4-fold. Normalization for plate-to-plate variation used reference samples provided by Kaj Blennow, MD, PhD, University of Gothenburg, Gothenburg, Sweden.
Results
Visual Examination
Visual function was normal by all tests. Foveal sensitivity and visual fields were normal bilaterally. Color vision was mildly abnormal, revealing a total color difference score of 193.5 OD and 168.3 OS with a color confusion index of 1.65 OD and 1.44 OS, consistent with mild tritanomaly or tritanopy affecting all 3 cone classes.25 The Arden ratio was normal, indicating normal retinal pigment epithelial depolarization in response to light. Recovery of dark-adapted visual sensitivity after exposure to bright light was normal, with normal cone thresholds, normal timing of the cone-rod break, and normal final rod-mediated thresholds. The retinal structure was normal, including retinal lamination and layer thickness throughout the central 20° of the macula in each eye. Fundus autofluorescence images were normal in the right eye but showed a single, small hyperautofluorescent spot corresponding to a white subretinal lesion on the color fundus photograph in the left eye. This finding could represent local evidence of abnormal lipid accumulation in or beneath the retinal pigment epithelium.
Cardiovascular Examination
Baseline electrocardiographic findings were normal. On treadmill stress testing (standard Bruce protocol), 2- to3-mm upsloping ST depressions at peak stress that resolved to the baseline level during recovery were noted. This suggests, but is not diagnostic for, exercise-induced ischemia at high workload and high double product. Echocardiography revealed normal bi-ventricular size and systolic and diastolic function. There was no evidence of valve disease. Bilateral duplex carotid ultrasonography revealed mild intimal thickening involving the middle and distal common carotid arteries and extending to the bulb. Peak systolic and diastolic velocities were normal. Both internal carotid arteries demonstrated grade B (0%-15%) stenosis.
Neurological Examination, Cerebrospinal Fluid Analysis, and Brain Magnetic Resonance Imaging
Neurological examination findings were essentially normal. Findings on neuropsychological testing, including memory function and the Mini-Mental State Examination, were also within normal limits. Details of this testing are provided in the eAppendix in the Supplement. Brain magnetic resonance imaging obtained under research protocols showed normal brain volumes without any significant pattern of atrophy or white matter lesions. Full hippocampal volumes bilaterally and a robust-appearing corpus callosum were noted. Basilar artery architecture was normal.
Based on reliable thresholds,26,27 cerebrospinal fluid Aβ42 concentrations lower than 500 pg/mL predict the presence of brain amyloid. High concentrations of tau and P-tau181 (>450-600 and >60-85 pg/mL, respectively27,28) are typical in patients with Alzheimer disease and suggest ongoing neurodegeneration. Concentrations of Aβ42, total tau, and P-tau181 for the apoE-deficient patient were normal and did not indicate the presence of amyloid or neurodegeneration. Normalized concentrations were 829, 165, and 43.4 pg/mL, respectively. An independent run of the samples on a different lot of plates yielded values in the normal range27,28 for all biomarkers and were similar to values for cognitively normal elderly control participants on the same plate.
Laboratory Results
Fasting glucose level, complete blood cell count, and renal, liver, and thyroid function test results were normal. At his initial visit, the patient reported taking fenofibrate (145 mg/d), atorvastatin calcium (80 mg/d), and ω-3–acid ethyl esters (Lovaza; 3 g/d). His total cholesterol level was 426 mg/dL (to convert to millimoles per liter, multiply by 0.0259), his TG level was 298 mg/dL (to convert to millimoles per liter, multiply by 0.0113), and his HDL cholesterol (HDL-C) level was 65 mg/dL (to convert to millimoles per liter, multiply by 0.0259). His lipoprotein(a) level was 238nM (reference range <75nM). His levels of plasma phytosterols were normal. Histopathology of xanthomas revealed multiple nodules of histiocytes with foamy cytoplasm. Some were multinucleate with areas of fibrosis, granulomatous reaction, cholesterol cleft formation, and free lipid.
Analysis of Lipoprotein Fractions
For lipoprotein studies, no participants were taking hypolipidemic medication. The patient’s total cholesterol level was760 mg/dL, his TG level was 534 mg/dL, and his HDL-C level was 63 mg/dL. By immunoassay, his apoA-I level was 161 mg/dL and his apoB level was 84 mg/dL (to convert to grams per liter, multiply by 0.01). His apoB-48 level was greater than 20 mg/dL (upper limit of reference range, 1.4 mg/dL). His mother’s total cholesterol level was 193 mg/dL, her TG level was 112 mg/dL, her LDL cholesterol level was 129 mg/dL, and her HDL-C level was 42 mg/dL. His 4- and 5-year-old daughters, respectively, had total cholesterol levels of 177 and 158 mg/dL, TG levels of 102 and 132 mg/dL, LDL cholesterol levels of 75 and 54 mg/dL, and HDL-C levels of 81 and 78 mg/dL. Composition of lipoprotein fractions is shown in Table 1. For the patient, the bulk of the plasma cholesterol and TG was found in VLDL. The ratios of both total and unesterified cholesterol to TG were considerably higher in the patient’s VLDL than in the other samples. This was not observed in the other lipoproteins. In the patient’s and his mother’s LDL, both ratios were lower than in the control.
Table 1.
Lipid and Protein Composition of Ultracentrifugally Isolated Lipoprotein Fractions From the Proband, His Mother, and a Normolipidemic Male Control Participant
| Sample | Protein, mg/dL | TG, mg/dL | Cholesterol, mg/dL
|
Ratio
|
||
|---|---|---|---|---|---|---|
| Total | Unesterified (%) | Total Cholesterol to TG | Free Cholesterol to TG | |||
| VLDL | ||||||
|
| ||||||
| Proband | 151 | 408.9 | 621.1 | 181.1 (29.2) | 1.52 | 0.44 |
|
| ||||||
| Mother | 20 | 34.2 | 7.9 | 3.6 (45.1) | 0.23 | 0.11 |
|
| ||||||
| Control | 27 | 47.6 | 16.1 | 6.6 (40.7) | 0.34 | 0.14 |
|
| ||||||
| IDL | ||||||
|
| ||||||
| Proband | 20 | 31.1 | 54.8 | 15.3 (28.0) | 1.76 | 0.49 |
|
| ||||||
| Mother | 11 | 8.3 | 13.5 | 4.7 (34.8) | 1.63 | 0.57 |
|
| ||||||
| Control | 6 | 5.7 | 6.8 | 2.5 (37.5) | 1.19 | 0.44 |
|
| ||||||
| LDL | ||||||
|
| ||||||
| Proband | 27 | 12.2 | 31.6 | 11.5 (36.3) | 2.59 | 0.94 |
|
| ||||||
| Mother | 73 | 32.8 | 94.5 | 27.0 (28.6) | 2.88 | 0.82 |
|
| ||||||
| Control | 68 | 15.6 | 110.9 | 32.3 (29.2) | 7.11 | 2.07 |
|
| ||||||
| HDL | ||||||
|
| ||||||
| Proband | 117 | 19.5 | 59.0 | 15.8 (26.8) | 3.03 | 0.81 |
|
| ||||||
| Mother | 114 | 15.2 | 44.8 | 9.7 (21.6) | 2.95 | 0.63 |
|
| ||||||
| Control | 104 | 11.7 | 52.8 | 10.2 (19.2) | 4.51 | 0.87 |
Abbreviations: HDL, high-density lipoproteins; IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; TG, triglycerides; VLDL, very low-density lipoproteins.
SI conversion factors: To convert protein to grams per liter, multiply by 0.01; to convert TG to millimoles per liter, multiply by 0.0113; and to convert cholesterol to millimoles per liter, multiply by 0.0259.
The ratio of core lipid (TG plus cholesteryl esters) to protein was much higher for the patient’s VLDL compared with his mother and a control participant (5.6 vs 1.9 and 2.1, respectively). It was also considerably higher in the patient’s intermediate-density lipoprotein (IDL) (3.5 vs 1.6 and 1.7, respectively) but not in LDL or HDL. The patient’s IDL total lipid content was much less than in VLDL, although it was higher than in the other samples. The patient’s LDL cholesterol level was low compared with the other samples. His levels of VLDL, IDL, and LDL are characteristic of severe dysbetalipoproteinemia. There were no profound differences in HDL.
Coomassie-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis showed considerable amounts of apoB-48 in the patient’s VLDL, IDL, and LDL with trace amounts in the other samples (eFigure 1 in the Supplement). No apoB-48 was seen in the HDL fractions (data not shown). A significant amount of apoA-IV was seen in the patient’s VLDL, less in IDL, and a trace in LDL. Apolipoprotein A-I was seen in his VLDL, IDL, and LDL. There was no apoE in his lipoproteins. The level of apoE in his mother’s VLDL and IDL was about half that in the control. An immunoblot of the patient’s plasma showed an absence of apoE. His 2 daughters had about half the normal levels (eFigure 2 in the Supplement). Immunoblots confirmed the presence of high amounts of apoA-I and apoA-IV in the patient’s VLDL, IDL, and LDL (Figure 2). Particularly striking was the much higher amount of apoA-IV in VLDL and of apoA-I in LDL. There were decreased amounts of apoC-III and apoC-IV in VLDL compared with the control. The mother’s VLDL showed intermediate levels. Immunoblots showed no apoE in the patient’s fractions and reduced levels in his mother’s VLDL and IDL compared with the control (Figure 2). No truncated apoE species were detected.
Figure 2. Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis Immunoblots.
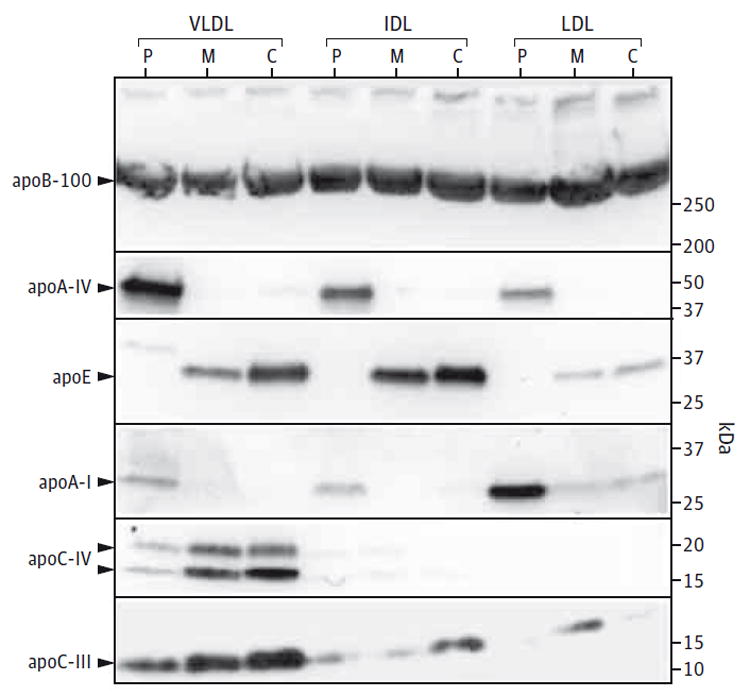
The abnormal composition of apolipoproteins (apo) in the lipoprotein fractions from the patient (P) is compared with the compositions of his mother (M) and a normolipidemic control participant (C). Equal amounts (20 μg) of total protein were run in each lane. Six antibodies were used against the following: apoB-100, apoA-IV, apoE, apoA-I, apoC-IV, and apoC-III. IDL indicates intermediate-density lipoprotein; LDL, low-density lipoprotein; and VLDL, very low-density lipoprotein.
HDL Analysis
The patient’s plasma showed an abnormal HDL size distribution (Figure 3) with fewer large particles (fractions 1, 2, and 3), totaling 45% of the HDL-C compared with a mean (SE) of 70.0% (2.0%) for control participants (n = 8). A higher percentage of small particles was seen in fractions 8, 9, and 10, representing 16% of total HDL-C compared with a mean (SE) of 3.4% (1.1%) for control participants. There was a higher percentage of intermediate-sized particles (fractions 4, 5, 6, and 7), comprising 38% of total HDL-C compared with a mean (SE) of 26.5% (1.5%) for control participants. These Lipoprint data were complemented by the 2-dimensional apoA-I immunoblot (eFigure 3 in the Supplement) showing elevated levels of prebeta-1 HDL. Quantitation by densitometry revealed a level of 22 mg/dL compared with a mean (SE) of 7 (0.6) mg/dL for control participants (n = 17).
Figure 3. Quantitative Scans of Lipoprint Polyacrylamide Gels.
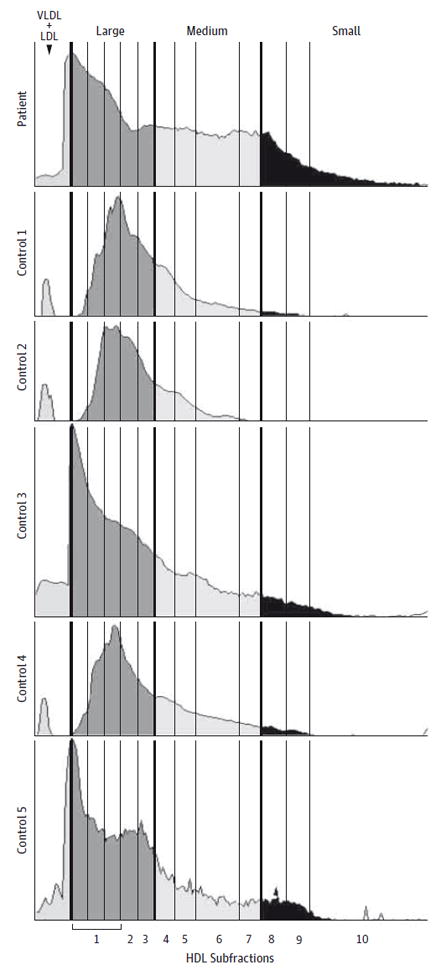
High-density lipoprotein (HDL) subfractions from the patient are compared with 5 representative control samples, measured as the scanned density of Sudan black–stained lipids. There is an abnormal HDL size distribution compared with control samples, with the patient’s HDL having a much higher proportion of small particles (fractions 8, 9, and 10). There is some increase in medium-sized particles (fractions 4, 5, 6, and 7). There is a correspondingly lower percentage in the largest particles (fractions 1, 2, and 3). LDL indicates low-density lipoprotein; VLDL, very low-density lipoprotein.
Electron Microscopy
Electron microscopy revealed that particles in the patient’s VLDL were normal in appearance (Figure 4B) and particle diameter (eFigure 4 and eTable 2 in the Supplement). His IDL, displaying normal diameters, contained some large lamellar particles having electron-opaque cores attached to electronlucent zones often surrounded by membranes (Figure 4D). These lamellar structures were also seen in LDL (Figure 4F). A notable difference in LDL was a bimodal size distribution, although the overall mean and median diameters were similar to those in control participants. No HDL abnormalities were observed.
Figure 4. Electron Photomicrographs of Fasting Lipoprotein Fractions Visualized by Negative Staining.
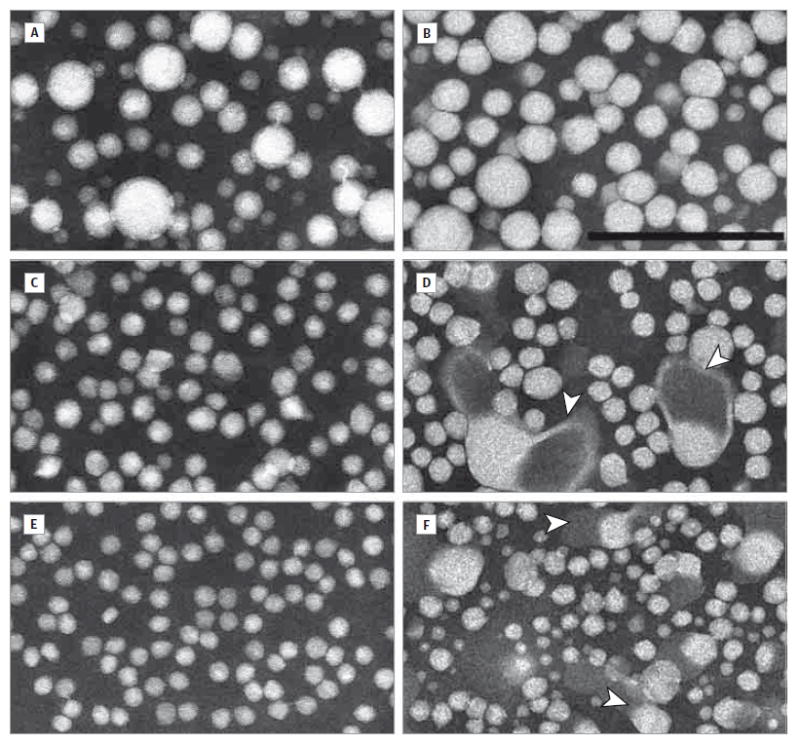
Very low-density lipoprotein fractions from a control participant (A) and the patient (B) are similar and contain spherical particles of 15 to 70 nm in diameter. The intermediate-density lipoprotein fraction from a control participant shows mainly round particles of 15 to 35 nm in diameter (C); intermediate-density lipoprotein from the patient appears similar but also has lamellar particles with electron-opaque cores attached to electron-lucent zones (arrowheads) (D). The low-density lipoprotein fraction from a control participant displays smaller, largely spherical particles of 15 to 25 nm in diameter (E); in contrast, low-density lipoprotein from the patient has a much more heterogeneous size distribution from 10 to 40 nm in diameter, again with the presence of lamellar particles (arrowheads) (F). Scale bar = 200 nm.
Exome Sequencing Results
To identify mutations causing the severe dysbetalipoproteinemia phenotype, we performed whole-exome sequencing and identified 645 rare and deleterious variants. Further filtering against lipid metabolism pathway genes yielded 9 variants (Table 2). A homozygous 1-bp frameshift deletion was identified in APOE exon 4 (c.291delG, p.E97fs) at chr19: 45411844 (GenBankNT_011109.16), predicting a change of residues 97 and 98 followed by a stop codon. Sanger sequencing confirmed the APOE mutation. The resulting truncated protein retains only one-third of the wild-type apoE and lacks the LDLR-binding and lipoprotein-binding domains. The patient’s mother and 2 daughters are heterozygotes. Genotypes of rs429358 and rs7412 indicated that the mutation is on an ε3 allele background. The homozygous variant in ABCC2 and heterozygous variant in LIPC (hepatic lipase), although very rare, may affect phenotype severity.
Table 2.
Rare, Deleterious Variants Identified From the Proband Using Exome Sequencing
| Gene | Variant Type | DNA | Amino Acid Change | Genotype | dbSNP No. or Chromosome Positiona | SIFT Score | Allele Frequencyb |
|---|---|---|---|---|---|---|---|
| ABCC2 | nsSNV | c.T2009C | p.I670T | Homozygous | rs17222632 | 0 | 0.0092 |
| APOE | fs del | c.291delG | p.E97fs | Homozygous | chr19: 45411844 | NA | NA |
| BUD13 | nsSNV | c.C1223T | p.P408L | Heterozygous | rs61730763 | 0.03 | 0.04 |
| FUT2 | fs del | c.811delC | p.P271fs | Heterozygous | rs1799761 | NA | NA |
| LIPC | nsSNV | c.G409A | p.G137R | Heterozygous | rs199787635 | 0 | 0 |
| LRP2 | nsSNV | c.T1817C | p.V606A | Heterozygous | rs116332504 | 0 | 0.03 |
| MCEE | nsSNV | c.G428A | p.R143H | Heterozygous | rs115175255 | 0 | 0.05 |
| PCCA | nsSNV | c.G925A | p.V309M | Heterozygous | chr13: 100925538 | 0 | NA |
| PLB1 | nsSNV | c.A2402T | p.N801I | Heterozygous | rs115240682 | 0.01 | 0.04 |
Abbreviations: fs del, frameshift deletion; NA, not applicable; nsSNV, nonsynonymous single-nucleotide variant; SIFT, Sorting Intolerant From Tolerant.
The chromosome position of the variant is given if the reference single-nucleotide polymorphism (rs) identification number is unavailable.
Allele frequency of the variant based on the 1000 Genomes Project May 2011 release.29
Discussion
We identified a novel 1-bp homozygous APOE frameshift deletion leading to apoE deficiency in a patient with severe dysbetalipoproteinemia. Exome sequencing proved valuable as other functionally damaging variants were identified in genes involved in lipid metabolism, providing clues to additional factors possibly contributing to the phenotype.
A neurological examination showed no evidence of neurodegenerative disease. Neuropsychological testing was essentially unremarkable, without any progressive concerns for cognitive decline or behavioral change. One notable exception pertains to neurodevelopment that reveals some learning disability, possibly dyslexia, given the history of delayed speech. It is unknown what effects his dyslipidemia may have on overall cognitive performance. The possible learning disability would be expected to have the greatest impact on the interpretation of results of cognitive testing. In general, patients with histories of learning disabilities such as dyslexia often have other untested strengths including excellent social and interpersonal skills, which this patient clearly possesses. Overall, he displayed intact global performance as evidenced by his Mini-Mental State Examination score. However, on subdomain tests, he had difficulties in multiple domains including memory, language, visuospatial abilities, and executive functions. Despite this, his functional status was intact and considered to be clinically normal. Further supporting this, brain imaging showed no pattern of atrophy or burden of white matter disease. The cerebrospinal fluid content of Aβ42 and tau proteins was normal.
An 11.3-kDa truncated apoE protein, predicted by the APOE mutation (p.E97fs), was absent in plasma and lipoproteins. The apoE ligand domain for the LDLR would be missing as would the C-terminal domain, which binds lipid and promotes tetramer formation. Apolipoprotein E, synthesized in liver and intestine, is essential for chylomicron and VLDL processing. As the TG cores are hydrolyzed, additional apoE is acquired. Resulting remnants pass the liver sieve30 and enter the space of Disse, gaining additional apoE before uptake via the LDLR, LDLR-related protein, and heparin sulfate proteoglycan.1 Apolipoprotein E is also involved in converting some VLDL remnants, via LIPC (lipase, hepatic), to LDL. The level of VLDL cholesterol seen here was considerably higher than seen in typical dysbetalipoproteinemia associated with ε2/ε2 homozygosity. The mean (SD) ratio of cholesterol to TG in VLDL is 0.53 (0.18) (n = 45) (C. R. Pullinger, PhD, and J. P. Kane, MD, PhD, unpublished data, 1989-2014). Therefore, the ratio of 1.52 in our patient is exceptionally high.
The increased amounts of apoA-IV in VLDL and IDL we observed were seen in other reports of apoE deficiency4,5,9 and in apoE-deficient mice.31 We additionally found apoA-IV in LDL. Increased amounts of apoA-I were observed in LDL from an apoE-deficient patient9 and in VLDL from ApoE knockout mice.31 The association of apoE deficiency with decreased apoC-III and apoC-IV in VLDL is novel. Abnormal lamellar lipoprotein particles similar to those we observed were seen in electron micrographs of IDL and LDL from apoE and lipc doubly deficient mice.32 These particles with electron-opaque cores and electron-lucent zones were thought to arise in the absence of lipc due to high amounts of β-VLDL. Apolipoprotein E–deficient mice exhibited less pronounced LDL lamellar particles.32 The absence of lipc possibly exacerbates the process but is not a prerequisite. Notably, our patient is heterozygous for a potentially significant mutation in LIPC.
We observed a large increase in small HDL species in our patient’s plasma, particularly prebeta-1 HDL, analogous to observations with ApoE knockout mice.33 Prebeta-1 HDL levels positively associate with increased risk of new CVD events,34 with ischemic heart disease,35 and with both coronary artery disease and myocardial infarction.36
We discovered 8 other apparently deleterious DNA variants apart from the APOE mutation. The homozygous variant inABCC2, a drug transporter, is not unique. Mutations inABCC2, also known as the multidrug resistance–associated protein 2 gene, associate with Dubin-Johnson syndrome. The mutation could possibly contribute to the partially lamellar character of IDL and LDL because cholestasis is associated with formation of lamellar lipoprotein-X. This variant, absent among Caucasian individuals, is present in populations of African origin. The rare heterozygous p.G137R variant in LIPC may contribute to the severity of the dysbetalipoproteinemia, similar to the increased phenotype severity in ApoE/Lipc knockout mice.32 The significance of the LRP2 (lipoprotein receptor–related protein 2) variant is unclear. It is found with African descent at a minor allele frequency of 3.4%. By genome-wide association studies, LRP2 is related to kidney function. Mutations associate with Donnai-Barrow facio-oculo-acoustico-renal syndrome but not with lipoprotein disorders. The MCEE (methylmalonyl–coenzyme A epimerase) variant occurs with African descent (minor allele frequency of 6.8%). Homozygous defects are linked to methylmalonic aciduria. How the PCCA (propionyl–coenzyme A carboxylase α subunit) mutation could contribute to the phenotype is unknown. Mutations in PCCA are associated with propionic acidemia. ThePLB1 (phospholipase B1) single-nucleotide polymorphism occurs with African descent (minor allele frequency of 3.4%). No diseases have been linked to this locus. Circulating vitamin E and B12 levels associate with a locus near BUD13 and FUT2, respectively.
The alterations in lipoproteins in the absence of apoE are manifold. Accumulation of VLDL and remnants reflects absence of the ligand for hepatic uptake. The observed morphologic changes appear to reflect abnormal accumulation of polar constituents in surface monolayers of VLDL, chylomicrons, and their daughter particles, particularly LDL. Enrichment of apoA-IV and apoA-I in these particles, possibly replacing apoE, is notable. Deficiency of apoC-III and apoC-IV could promote instability of surface monolayers. The striking increase of prebeta-1 HDL, regarded as the quantum particle among HDL species, suggests that apoE may play heretofore unappreciated roles in the generation, interconversion, or removal of HDL species.
Given the severity of his dyslipidemia, extensive xanthomas, and high potential for premature CVD, strategies aimed at reducing the generation of chylomicrons and VLDL should be aggressively pursued. Low dietary fat intake and use of fibrates and/or niacin coupled with statin therapy usually control hyperlipidemia in dysbetalipoproteinemia but have been relatively ineffective in the management of this patient. Inhibitors of proprotein convertase subtilisin/kexin type 9 and microsomal triglyceride transfer protein, apoB antisense oligonucleotides, and enhancers of adenosine monophosphate kinase may be of additional benefit. Bone marrow transplantation corrects dyslipidemia in apoE deficient mice37 and might be considered in the face of severe CVD.
Apolipoprotein E is expressed in the central and peripheral nervous systems.38 Produced and secreted mainly by astrocytes,39 it is believed to be important for normal brain function. Apolipoprotein E was thought not to cross the blood-brain barrier, with all found in the central nervous system synthesized locally. However, recent evidence indicates that apoE4 expression compromises the blood-brain barrier (as does apoE deficiency).40 Studies of brain histology, neurodegenerative markers, cholinergic activity, and neuronal function in knockout mice have been equivocal.39 Failure of detailed neurocognitive and retinal studies to demonstrate defects in our patient suggests either that the functions of apoE in the brain and eye are not critical or that they can be fulfilled by a surrogate protein. Surprisingly, with respect to central nervous system function, it appears that having no apoE is better than having the apoE4 protein. Thus, projected therapies aimed at reducing apoE4 in the brain could be of benefit in neurodegenerative disorders such as Alzheimer disease.
Conclusions
The molecular basis for the severe dyslipidemia in our patient was found to be homozygosity for an ablative APOE frame-shift mutation (c.291del, p.E97fs). The absence of the apoE protein did not affect neurocognitive or retinal function.
Supplementary Material
Acknowledgments
Funding/Support: This work was supported in part by UCSF Dermatology Training Grant T32 AR007175 (Dr Mak), Memory and Aging Center grant P01AG019724, and the UCSF Academic Senate Award (Dr Pullinger) from the University of California, San Francisco, the Hellman Family Award (Dr Pullinger), the Campini Foundation, the Joseph Drown Foundation (Dr Malloy), and gifts from Peter Read, Donald Yellon, and the Mildred V. Strouss Charitable Trust.
Role of the Sponsor: The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Author Contributions: Dr Malloy had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Drs Mak and Pullinger contributed equally to this study.
Study concept and design: Mak, Pullinger, Tang, Deo, Schaefer, Asztalos, Miller, Kane, Kwok, Malloy.
Acquisition, analysis, or interpretation of data: Mak, Pullinger, Wong, Schwarz, Gugliucci, Movsesyan, Ishida, Chu, Poon, Kim, Stock, Schaefer, Asztalos, Castellano, Wyss-Coray, Duncan, Kane, Malloy.
Drafting of the manuscript: Mak, Pullinger, Deo, Movsesyan, Poon, Stock, Asztalos, Duncan, Kane, Malloy.
Critical revision of the manuscript for important intellectual content: Pullinger, Tang, Wong, Schwarz Gugliucci, Ishida, Chu, Kim, Stock, Schaefer, Castellano, Wyss-Coray, Duncan, Miller, Kane, Kwok, Malloy.
Statistical analysis: Mak.
Obtained funding: Kwok, Malloy.
Administrative, technical, or material support: Pullinger, Tang, Wong, Schwarz, Gugliucci, Movsesyan, Chu, Poon, Stock, Schaefer, Castellano, Kane, Malloy.
Study supervision: Mak, Pullinger, Deo, Ishida, Stock, Wyss-Coray, Miller, Kane, Kwok, Malloy.
Additional Contributions: Russell Caccavello,MS, Touro University California, Vallejo, provided expert technical assistance, Bart P. Duell, MD, Oregon Health and Science University, Portland, provided the phytosterol measurements, and Robin Ketelle, RN, MS, Memory and Aging Center, University of California, San Francisco, provided able assistance in coordinating this study. Richard J. Havel,MD, University of California, San Francisco, provided apoB-100, apoE, and apoC-IV antibodies. Normalization for plate-to-plate variation used reference samples provided by Kaj Blennow, MD, PhD, University of Gothenburg, Gothenburg, Sweden. No compensation was received for these contributions.
Supplemental content at jamaneurology.com
Conflict of Interest Disclosures: None reported.
References
- 1.Mahley RW, Rall SC. Type III hyperlipoproteinemia (dysbetalipoproteinemia): the role of apolipoprotein E in normal and abnormal lipoproteinmetabolism. In: Valle D, Beaudet AL, Vogelstein B, et al., editors. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw-Hill; 2007. [December 11, 2007]. [DOI] [Google Scholar]
- 2.Utermann G. Apolipoprotein E polymorphism in health and disease. Am Heart J. 1987;113(2, pt 2):433–440. doi: 10.1016/0002-8703(87)90610-7. [DOI] [PubMed] [Google Scholar]
- 3.de Knijff P, van den Maagdenberg AM, Frants RR, Havekes LM. Genetic heterogeneity of apolipoprotein E and its influence on plasma lipid and lipoprotein levels. Hum Mutat. 1994;4(3):178–194. doi: 10.1002/humu.1380040303. [DOI] [PubMed] [Google Scholar]
- 4.Ghiselli G, Schaefer EJ, Gascon P, Breser HB., Jr Type III hyperlipoproteinemia associated with apolipoprotein E deficiency. Science. 1981;214(4526):1239–1241. doi: 10.1126/science.6795720. [DOI] [PubMed] [Google Scholar]
- 5.Mabuchi H, Itoh H, Takeda M, et al. A young type III hyperlipoproteinemic patient associated with apolipoprotein E deficiency. Metabolism. 1989;38(2):115–119. doi: 10.1016/0026-0495(89)90249-7. [DOI] [PubMed] [Google Scholar]
- 6.Kurosaka D, Teramoto T, Matsushima T, et al. Apolipoprotein E deficiencywith a depressedmRNA of normal size. Atherosclerosis. 1991;88(1):15–20. doi: 10.1016/0021-9150(91)90252-x. [DOI] [PubMed] [Google Scholar]
- 7.Feussner G, Funke H, Weng W, Assmann G, Lackner KJ, Ziegler R. Severe type III hyperlipoproteinemia associated with unusual apolipoprotein E1 phenotype and epsilon 1/’null’genotype. Eur J Clin Invest. 1992;22(9):599–608. doi: 10.1111/j.1365-2362.1992.tb01511.x. [DOI] [PubMed] [Google Scholar]
- 8.Lohse P, Brewer HB, III, Meng MS, Skarlatos SI, LaRosa JC, Brewer HB., Jr Familial apolipoprotein E deficiency and type III hyperlipoproteinemia due to a premature stop codon in the apolipoprotein E gene. J Lipid Res. 1992;33(11):1583–1590. [PubMed] [Google Scholar]
- 9.Feussner G, Dobmeyer J, Grone HJ, Lohmer S, Wohlfeil S. A 10-bp deletion in the apolipoprotein epsilon gene causing apolipoprotein E deficiency and severe type III hyperlipoproteinemia. Am J Hum Genet. 1996;58(2):281–291. [PMC free article] [PubMed] [Google Scholar]
- 10.Metzker ML. Sequencing technologies: the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 11.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103(15):5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ishida BY, Bailey KR, Duncan KG, et al. Regulated expression of apolipoprotein E by human retinal pigment epithelial cells. J Lipid Res. 2004;45(2):263–271. doi: 10.1194/jlr.M300306-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li H, Handsaker B, Wysoker A, et al. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Havel RJ, Eder HA, Bragdon JH. The distribution and chemical composition of ultracentrifugally separated lipoproteins in human serum. J Clin Invest. 1955;34(9):1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamilton RL, Jr, Goerke J, Guo LSS, Williams MC, Havel RJ. Unilamellar liposomes made with the French pressure cell: a simple preparative and semiquantitative technique. J Lipid Res. 1980;21(8):981–992. [PubMed] [Google Scholar]
- 20.Asztalos BF, Sloop CH, Wong L, Roheim PS. Two-dimensional electrophoresis of plasma lipoproteins: recognition of new apo A-I-containing subpopulations. Biochim Biophys Acta. 1993;1169(3):291–300. doi: 10.1016/0005-2760(93)90253-6. [DOI] [PubMed] [Google Scholar]
- 21.Campos E, Nakajima K, Tanaka A, Havel RJ. Properties of an apolipoprotein E-enriched fraction of triglyceride-rich lipoproteins isolated from human blood plasmawith a monoclonal antibody to apolipoprotein B-100. J Lipid Res. 1992;33(3):369–380. [PubMed] [Google Scholar]
- 22.Kotite L, Zhang L-H, Yu Z, Burlingame AL, Havel RJ. Human apoC-IV: isolation, characterization, and immunochemical quantification in plasma and plasma lipoproteins. J Lipid Res. 2003;44(7):1387–1394. doi: 10.1194/jlr.M300087-JLR200. [DOI] [PubMed] [Google Scholar]
- 23.Leary ET, Wang T, Baker DJ, et al. Evaluation of an immunoseparation method for quantitative measurement of remnant-like particle-cholesterol in serum and plasma. Clin Chem. 1998;44(12):2490–2498. [PubMed] [Google Scholar]
- 24.Kunitake ST, O’Connor P, Naya-Vigne J. Heterogeneity of high-density lipoproteins and apolipoprotein A-I as related to quantification of apolipoprotein A-I. Methods Enzymol. 1996;263:260–267. doi: 10.1016/s0076-6879(96)63018-3. [DOI] [PubMed] [Google Scholar]
- 25.Bowman KJ. Amethod for quantitative scoring of the Farnsworth Panel D-15. Acta Ophthalmol (Copenh) 1982;60(6):907–916. doi: 10.1111/j.1755-3768.1982.tb00621.x. [DOI] [PubMed] [Google Scholar]
- 26.Fagan AM, Head D, Shah AR, et al. Decreased cerebrospinal fluid Abeta(42) correlates with brain atrophy in cognitively normal elderly. Ann Neurol. 2009;65(2):176–183. doi: 10.1002/ana.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Humpel C. Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol. 2011;29(1):26–32. doi: 10.1016/j.tibtech.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302(4):385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 29.Clarke L, Zheng-Bradley X, Smith R, et al. 1000 Genomes Project Consortium. The 1000 Genomes Project: data management and community access. Nat Methods. 2012;9(5):459–462. doi: 10.1038/nmeth.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fraser R, Cogger VC, Dobbs B, et al. The liver sieve and atherosclerosis. Pathology. 2012;44(3):181–186. doi: 10.1097/PAT.0b013e328351bcc8. [DOI] [PubMed] [Google Scholar]
- 31.Plump AS, Smith JD, Hayek T, et al. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71(2):343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 32.Bergeron N, Kotite L, Verges M, et al. Lamellar lipoproteins uniquely contribute to hyperlipidemia in mice doubly deficient in apolipoprotein E and hepatic lipase. Proc Natl Acad Sci U S A. 1998;95(26):15647–15652. doi: 10.1073/pnas.95.26.15647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mezdour H, Jones R, Dengremont C, Castro G, Maeda N. Hepatic lipase deficiency increases plasma cholesterol but reduces susceptibility to atherosclerosis in apolipoprotein E-deficient mice. J Biol Chem. 1997;272(21):13570–13575. doi: 10.1074/jbc.272.21.13570. [DOI] [PubMed] [Google Scholar]
- 34.Asztalos BF, Collins D, Cupples LA, et al. Value of high-density lipoprotein (HDL) subpopulations in predicting recurrent cardiovascular events in the Veterans Affairs HDL Intervention Trial. Arterioscler Thromb Vasc Biol. 2005;25(10):2185–2191. doi: 10.1161/01.ATV.0000183727.90611.4f. [DOI] [PubMed] [Google Scholar]
- 35.Sethi AA, Sampson M, Warnick R, et al. High pre-beta1 HDL concentrations and low lecithin: cholesterol acyltransferase activities are strong positive risk markers for ischemic heart disease and independent of HDL-cholesterol. Clin Chem. 2010;56(7):1128–1137. doi: 10.1373/clinchem.2009.139931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guey LT, Pullinger CR, Ishida BY, et al. Relation of increased prebeta-1 high-density lipoprotein levels to risk of coronary heart disease. Am J Cardiol. 2011;108(3):360–366. doi: 10.1016/j.amjcard.2011.03.054. [DOI] [PubMed] [Google Scholar]
- 37.Van Eck M, Herijgers N, Yates J, et al. Bone marrow transplantation in apolipoprotein E-deficient mice. Effect of ApoE gene dosage on serum lipid concentrations, (beta)VLDL catabolism, and atherosclerosis. Arterioscler Thromb Vasc Biol. 1997;17(11):3117–3126. doi: 10.1161/01.atv.17.11.3117. [DOI] [PubMed] [Google Scholar]
- 38.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM. Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest. 1985;76(4):1501–1513. doi: 10.1172/JCI112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hauser PS, Narayanaswami V, Ryan RO. Apolipoprotein E: from lipid transport to neurobiology. Prog Lipid Res. 2011;50(1):62–74. doi: 10.1016/j.plipres.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.