

Abstract
The heat shock proteins Hsp70 and Hsp90 require the help of tetratricopeptide repeat (TPR) domain-containing co-chaperones for many of their functions. Each monomer of Hsp70 or Hsp90 can only interact with a single TPR co-chaperone at a time and each member of the TPR co-chaperone family brings distinct functions into the complex. Thus, competition for TPR binding sites on Hsp70 and Hsp90 appears to shape chaperone activity. Recent structural and biophysical efforts have improved our understanding of chaperone-TPR contacts, focusing on the C-terminal EEVD motif that is present in both chaperones. To better understand these important protein-protein interactions on a wider scale, we measured the affinity of five TPR co-chaperones, CHIP, Hop, DnaJC7, FKBP51, and FKBP52, for the C-termini of four members of the chaperone family, Hsc70, Hsp72, Hsp90α, and Hsp90β, in vitro. These studies identified some surprising selectivity amongst the chaperone-TPR pairs, including the selective binding of FKBP51/52 to Hsp90α/β. These results also revealed that other TPR co-chaperones are only able to weakly discriminate between the chaperones or between their paralogs. We also explored whether mimicking phosphorylation of serine and threonine residues near the EEVD motif might impact affinity and found that pseudophosphorylation had selective effects on binding to CHIP but not other co-chaperones. Together, these findings suggest that both intrinsic affinity and post-translational modifications tune the interactions between Hsp70/90 and the TPR co-chaperones.
Introduction
The molecular chaperones heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) are essential regulators of cellular protein quality control, where they use ATP turnover to play broad roles in protein folding, trafficking, and degradation (1–6). In part, Hsp70 and Hsp90 are able to engage in so many different pathways because they collaborate with co-chaperones (7). Co-chaperones, including the tetratricopeptide repeat (TPR) domain-containing proteins, bind to chaperones and help determine whether “clients” will be folded, degraded, or sent to other fates. Because of this, there is great interest in studying the protein-protein interactions (PPIs) between chaperones and co-chaperones in order to better understand how these complexes form, how they are regulated, and how “decisions” are made. This knowledge is important because imbalances in protein quality control have been linked to a range of diseases, including cancer and neurodegeneration (8–13).
TPR domains are comprised of tandem 34 amino acid motifs, which form amphipathic antiparallel α-helix hairpins that stack on one another (14). Two of the most well studied TPR domain-containing proteins are Hop (Hsp70/Hsp90 organizing protein) and CHIP (carboxyl terminus of Hsp70 interacting protein) (15–18). Like all co-chaperones of this class, Hop and CHIP have no homology outside of the TPR domain. It is this diversity that allows the TPR co-chaperones to bring unique capabilities into chaperone complexes. For example, Hop uses multiple TPR domains to bind both Hsp70 and Hsp90 at the same time, coordinating these two chaperone systems and favoring client folding (19–24). In contrast, CHIP is an ubiquitin E3 ligase with a TPR domain and an effector U-box domain (25). This co-chaperone favors addition of polyubiquitin chains to Hsp70/90-bound clients, promoting their proteasomal degradation (26,27). Other important TPR proteins include FKBP51 and FKBP52, which work with Hsp70 and Hsp90 during the maturation and trafficking of steroid hormone receptors (28–30), and DnaJC7, which contains both a TPR domain and a J-domain that binds Hsp70s (31,32). Together, these observations suggest that the ultimate fate of chaperone-bound clients (e.g. whether they are folded, degraded, trafficked, or matured) may be guided, in part, by the “choice” of which TPR co-chaperone is bound.
The constitutive and heat-inducible paralogs of Hsp70 in the cytoplasm, termed Hsc70 (HSPA8) and Hsp72 (HSPA1A) respectively, contain a highly conserved EEVD motif at their respective C-termini. This four amino acid sequence binds to the concave face of the TPR domains, as revealed by mutagenesis and structural studies (33–35). In the bound form, key contacts are made between the chaperone’s carboxy terminus and conserved, cationic residues in the TPR domain. Like Hsp70s, both paralogs of Hsp90 in the cytosol (Hsp90α and Hsp90β) contain this same conserved, C-terminal EEVD motif. The striking thing about this observation is that, outside of this small motif, the two Hsp90s share no structural or sequence homology with the Hsp70s. Yet, the sequence of all four chaperones terminates with the same four amino acids, EEVD. In contrast, the other paralogs of Hsp70 and Hsp90, which are located in the endoplasmic reticulum, mitochondria and chloroplast, do not have EEVD motifs, suggesting that the TPR interactions may have evolved to specifically mediate quality control in the cytosol and nucleus.
More information about the interactions between TPR co-chaperones and cytoplasmic Hsp70s/Hsp90s, might help us understand a key step in protein quality control. Indeed, pioneering studies by multiple groups have examined the structures and affinities of important TPR-EEVD interactions, including Hop-Hsp70/90 (3,36,37), PP5-Hsp70/90 (38,39), CHIP-Hsp70/90 (40–43), and FKBP52-Hsp90 (44). This system has also been engineered to develop selective scaffolds for synthetic biology (45–47). However, a side-by-side comparison of the natural interactions has not yet been performed and is important because competition for TPR-domain proteins in the cytosol appears to be a major determinant of quality control. In this study, we used a fluorescence polarization (FP) platform to systematically compare the affinities of five different human TPR co-chaperones (Hop, CHIP, DnaJC7, FKBP51, and FKBP52) for the C-termini of the four cytosolic Hsp70/90s. We found that some co-chaperones, such as FKBP51 and FKBP52, have a strong preference for Hsp90s over Hsp70s. Other co-chaperones, including HOP, CHIP and DnaJC7, have a modest (~2-fold) preference for Hsp70s. Using chimeric peptides, we found that a single residue adjacent to the EEVD motif was important for some of this selectivity. Interestingly, none of the TPR co-chaperones could discriminate between the paralogs of Hsp70 or Hsp90, suggesting that they might work with both forms equally. Finally, we were inspired by recent reports (48) to explore the effects of mimicking C-terminal phosphorylation of Hsp70s and Hsp90s on their affinity for TPR co-chaperones. We found that binding to CHIP was strongly decreased, while other TPR co-chaperones were unaffected. Together, these studies provide a resource for understanding how interactions in this system are regulated.
Experimental Procedures
Plasmids
Human CHIP, Hop, FKBP51, and FKBP52 were expressed from a pET151 vector such that they contained an N-terminal His-tag and TEV cleavage site. Site-directed mutagenesis for Hop mutants (K8A, R77A, N223A, and R305A) was performed using the Phusion Site-Directed Mutagenesis Kit protocol (New England Biolabs, Ipswich, MA). Human DnaJC7, Hsp72 (HSPA1A), Hsp72ΔEEVD, and E. coli DnaJ were expressed from a pMCSG7 vector with an N-terminal His-tag and TEV cleavage site. Lastly, HIP was expressed from a pET28a vector with an N-terminal His-tag and Thrombin cleavage site.
Protein Expression and Purification
Hsp72 and Hsp72ΔEEVD proteins were expressed in Escherichia coli (E. coli) BL21 (DE3) cells. Liter cultures of terrific broth were grown at 37 °C until an OD600 of 0.6. Cultures were cooled to 25 °C and induced with isopropyl β-D-1-thiogalactopyranoside (IPTG; final concentration of 500 μM). Afterwards, cultures were grown overnight at 25 °C. For protein purification, cell pellets were re-suspended in His-binding buffer (50 mM TRIS, 10 mM Imidazole, 500 mM NaCl, pH 8) supplemented with protease inhibitors. Cells were lysed by sonication, pelleted by centrifugation, and the supernatant was applied to Ni-NTA His-Bind Resin (Novagen, Darmstadt, Germany). The resin was washed with His-binding buffer, followed by His-washing buffer (50 mM TRIS, 30 mM Imidazole, 300 mM NaCl, pH 8). The protein was then removed from the resin using His-elution buffer (50 mM TRIS, 300 mM Imidazole, 300 mM NaCl, pH 8). Before further purification by an ATP-agarose column (Sigma), MgCl2 and KCl was added to the eluted sample (final concentration: MgCl2 = 10 mM, KCl =10 mM). The sample was then applied to the ATP-agarose column, was first washed with buffer A (25 mM HEPES, 5 mM MgCl2, 10 mM KCl, pH 7.5) and then was washed with buffer B (25 mM HEPES, 5 mM MgCl2, 1M KCl, pH 7.5). The column was then washed a third time with buffer A and then eluted in buffer A containing 3 mM ATP. The pure protein was concentrated and exchanged into buffer A for storage. Note that the N-terminal His-tags were not removed.
Human CHIP, Hop, Hop mutants, FKBP51, and FKBP52 were expressed in E. coli BL21 (DE3) cells. Liter cultures of terrific broth were grown at 37 °C until an OD600 of 0.6. Cultures were cooled to 18 °C before induction with IPTG (final concentration of 500 μM) and then grown overnight. For protein purification, cell pellets were lysed and first purified using the batch Ni-NTA His-Bind resin protocol described above. The N-terminal His-tag was then removed using TEV protease. The sample was then further purified by size exclusion chromatography using a prep grade XK 16/100 Superdex 200 column (GE Healthcare Life Sciences) in a 50 mM HEPES, 10 mM NaCl, pH 7.4 buffer. Human HIP was purified using previously described methods (49).
The human TPR protein DnaJC7 and E.coli DnaJ were expressed in E. coli BL21 (DE3) cells. Liter cultures of terrific broth were grown at 37°C until an OD600 of 0.6 was reached. Cultures were then cooled to 18°C before induction with IPTG (500 μM) and then grown overnight. Cell pellets were re-suspended in DnaJC7/DnaJ His-binding buffer (50 mM TRIS, 10 mM Imidazole, 750 mM NaCl, pH 8) supplemented with protease inhibitors. Cells were lysed by sonication, subjected to centrifugation, and the supernatant was then applied to Ni-NTA His-Bind Resin (Novagen, Darmstadt, Germany). The resin was washed with the DnaJC7/DnaJ His-binding buffer, followed by an extensive wash with DnaJC7/DnaJ His-washing buffer 1 (50 mM TRIS, 30 mM Imidazole, 750 mM NaCl, 3% ethanol, pH 8). The resin was washed a third time with DnaJC7/DnaJ His-washing buffer 2 (50 mM TRIS, 30 mM Imidazole, 100 mM NaCl, 3% ethanol, pH 8). Finally, the protein was then removed from the resin with the His-elution buffer (50 mM TRIS, 300 mM Imidazole, 300 mM NaCl, pH 8). The purified protein was concentrated and exchanged into a 50 mM TRIS, 300 mM NaCl, pH 7.4 buffer for storage. Note that N-terminal His-tags were not removed.
Preparation of Apo Hsp70 protein
Hsp70 protein was made apo (e.g. nucleotide free) using extensive dialysis in 3 mL cassettes (catalog number = 66330, Life Technologies). First, the protein was dialyzed into 25 mM HEPES, 10 mM KCl, 5 mM EDTA, pH 7.5 at 4 °C for two days. Next, it was dialyzed into 25 mM HEPES, 10 mM KCl, pH 7.5 at 4 °C for another two days and then stored at −80 °C. Fresh buffers were made daily.
Fluorescence Polarization Assays
General Procedures
All Experiments were performed in 384-well, black, low volume, round-bottom plates (catalog number = 4511, Corning, NY). Polarization values in millipolarization units (mP) were measured at an excitation wavelength at 485 nm and an emission wavelength at 530 nm using a Molecular Devices Spectramax M5 plate reader (Sunnyvale, CA). For binding experiments, equilibrium-binding isotherms were constructed by plotting FP readings as a function of the protein concentration at a fixed concentration of a tracer. All experiments were performed at least twice in triplicate. Results are shown as the average and standard error of the mean (SEM) of all measurements. All experimental data were analyzed using GraphPad Prism 6 software.
TPR co-chaperones binding to Hsp70/90 C-terminal probes
Fluorescent C-terminal Hsp70 and Hsp90 peptides were custom ordered from GenScript (Piscataway, NJ) and designed to have an N-terminal 5-Carboxyfluorescein (5-Fam) via a 6-carbon spacer (aminohexanoic acid). These probes were stored as 5 mM DMSO stocks at −30 °C. Before use, the tracer solutions were diluted in the assay buffer (50 mM HEPES, 75 mM NaCl, 0.001% Triton X-100, pH 7.4 or 9.4) to a working concentration of 0.1 μM. Note that 5-Fam (pKa ~6.4) has pH-sensitive fluorescence, so no binding experiments were performed at low pH values. To each well was added 16 μL of a TPR co-chaperone (CHIP, Hop, DnaJC7, FKBP51, FKBP52, or HIP) from a 2-fold dilution series made using the assay buffer. Final concentrations of protein ranged from 0 to 125 μM. Next, 4 μL of a 0.1 μM 5-Fam-labeled C-terminal Hsp70/90 peptide was added to each well, to give a final concentration of 20 nM and a total assay volume of 20 μL. The plate was covered from light and allowed to incubate at room temperature for 30 minutes, which was determined to be at equilibrium.
FP competition experiment with C-terminal Hsp70/90 probes
We also determined the ability of full-length Hsp72 or Hsp72ΔEEVD to compete with the C-terminal Hsp72 probe (5-Fam-GSGPTIEEVD) for binding to a TPR protein (CHIP, Hop, or DnaJC7). First, 6 µL of a TPR co-chaperone was added to each well (final concentration: CHIP = 0.5 μM, Hop = 2.5 μM, or DnaJC7 = 2.5 μM). This amount equals the concentration of the TPR co-chaperone at which 50% of the FP probe (5-Fam-GSGPTIEEVD) is bound, based on binding experiments. Next, 10 µL of Hsp72 or Hsp72ΔEEVD from a 2-fold dilution made using the assay buffer (50 mM HEPES, 75 mM NaCl, 0.001% Triton X-100, pH 7.4) was added. Final concentrations of Hsp72/Hsp72ΔEEVD ranged from 0 to 40 μM. Finally, 4 μL of a 0.1 μM 5-Fam-GSGPTIEEVD was added to each well, to give final a concentration of 20 nM and a total assay volume of 20 μL. The plate was covered from light and allowed to incubate at room temperature for 30 minutes.
Binding of a florescent ATP analog to Hsp72/Hsp72ΔEEVD
The florescent ATP analog, Fam-ATP (N6-(6-Amino)hexyl-ATP-5Fam), was purchased from Jena Bioscience (catalog number = NU-805-5FM, Jena, Germany). To a plate, was added 16 μL of a 2-fold dilution series of protein (Hsp72 or Hsp72ΔEEVD). Dilution series were made using the assay buffer (100 mM TRIS, 20 mM KCl, 6 mM MgCl2, 0.01% Triton X-100, pH 7.4). Final concentrations of proteins ranged from 0 to 25 μM. Apo (nucleotide free) Hsp72/Hsp72ΔEEVD must be used in order to achieve substantial and reproducible binding to the Fam-ATP probe. Next, 6 μL of a 3.3 mM solution of ATP or ADP was added to each well, to give a final concentration 1 mM. Finally, 4 μL of a 0.1 μM Fam-ATP was added to each well, to give a final concentration of 20 nM and a total assay volume of 20 μL. The plate was allowed to incubate at room temperature covered from light for 30 minutes.
FP competition experiment with Fam-ATP
We also determined the ability of unlabeled ATP to compete with the Fam-ATP probe for binding to Hsp72/Hsp72ΔEEVD. First, apo Hsp72 or Hsp72ΔEEVD was added to each well to give a final concentration 0.5 µM. This amount equals the concentration of Hsp72/Hsp72ΔEEVD at which 50% of the FP probe (Fam-ATP) is bound base on binding experiments. Next, a 2-fold dilution of ATP made using the assay buffer (100 mM TRIS, 20 mM KCl, 6 mM MgCl2, 0.01% Triton X-100, pH 7.4) was added. Final concentrations of ATP ranged from 0 to 300 μM. Finally, Fam-ATP was added to each well, to give a final concentration of 20 nM and a total assay volume of 20 μL. The plate was covered from light and allowed to incubate at room temperature for 30 minutes to in order to reach equilibrium.
Binding of Fam-HLA substrate to Hsp72/Hsp72ΔEEVD
HLA substrate FP probe was custom ordered form the University of Michigan Proteomics & Peptide Synthesis Core. This probe was designed to have a 5-Fam N-terminal of the following sequence: RENLRIALRY. This probe the stored as 5 mM DMSO stocks at −30 °C. Before use, probes were diluted in the assay buffer (100 mM TRIS, 20 mM KCl, 6 mM MgCl2, 0.01% Triton X-100, pH 7.4) to a working concentration of 0.1 μM. To a plate, was added 16 μL of a 2-fold dilution series of protein (Hsp72, Hsp72ΔEEVD, or CHIP). Dilution series were made using the assay buffer. Final concentrations of the protein ranged from 0 to 25 μM. Apo (nucleotide free) Hsp72/Hsp72ΔEEVD must be used in order achieve substantial and reproducible binding to the Fam-HLA probe. Next, 4 μL of a 0.1 μM Fam-HLA was added to each well, to give a final concentration of 20 nM and a total assay volume of 20 μL. The plate was allowed to incubate at room temperature covered from light for 1 hour.
Luciferase refolding assay
Experiments were performed as described previously (50). Briefly, working stocks of denatured luciferase were prepared by mixing 10 μL of 200 μM native luciferase (Promega) with 30 μL of 8 M GnHCl for 1 hour at room temperature. Denatured luciferase stocks were stored at −80 °C until use. To white 96-well plates, was added denatured luciferase (final concentration of 100 nM), Hsp72 or Hsp72ΔEEVD (final concentration of 1 µM), and various concentrations of E. coli DnaJ or human DnaJC7 to give a final volume of 25 μL in refolding buffer (20 mM HEPES, 120 mM KAc, 1.2 mM MgAc, 15 mM DTT, 60 mM creatine phosphate, 35 U/mL creatine kinase, 5 ng/μL BSA, pH 7.4). The reaction was initiated by adding 10 μL of 2.5 mM ATP to give a final concentration of 1 mM. Plates were covered and incubated at 37 °C for 1 hour. Next, 25 μL of Steady-Glo reagent (Promega) was added to each well and luminescence values were measured immediately using a Molecular Devices Spectramax M5 plate reader (Sunnyvale, CA).
Results and Discussion
The binding of Hsp70 to TPR co-chaperones is largely mediated by Hsp70’s EEVD motif
Previous work, largely on CHIP and Hop, had shown that the EEVD motif provides the majority of the TPR interaction affinity, with less affinity (typically <20%) coming from secondary contacts (42,51–54). To ask whether this was also the case in other TPR-chaperone complexes, we generated a mutant Hsp72 construct that lacked an EEVD motif (Hsp72ΔEEVD). This mutant was normal in binding nucleotide (SI Fig 1A and 1B) and a client peptide derived from the MHC class I antigen HLA-B2702 (Fam-HLA) (SI Fig 1C), showing that the EEVD motif doesn’t directly contribute to these activities. We next tested whether this otherwise functional Hsp72ΔEEVD mutant could compete with the Hsp72 tracer (Fam-GSGPTIEEVD) for binding to TPR co-chaperones (SI Fig 2B). We found that Hsp72ΔEEVD was unable to compete for tracer binding, even at 40 µM, whereas wildtype (wt) Hsp72 could (SI Fig 2B). These results suggest that interactions outside of the canonical EEVD-TPR binding site are relatively weak, consistent with recent structural studies on Hsp70 and CHIP (42). Based on this result, we decided to focus strictly on the C-termini of Hsp70s and Hsp90s to further understand their interactions with TPR proteins.
Preferences of the TPR co-chaperones for binding to Hsp70 and Hsp90
In an effort to understand what factors influence binding to the molecular chaperones, we first determined the affinity of chaperone C-termini for full-length TPR proteins using a FP assay. In these studies, we focused on some of the best studied TPR co-chaperones: CHIP, Hop, DnaJC7, FKBP51 and FKBP52. In addition, we included HIP as a negative control because this co-chaperone binds Hsp70s in a region outside the EEVD motif (55). For our FP experiments, we measured the ability of TPR proteins to interact with fluorescently labeled peptides corresponding to the C-termini of Hsc70 (Fam-SSGPTIEEVD), Hsp72 (Fam-GSGPTIEEVD), Hsp90α (Fam-DDTSRMEEVD), and Hsp90β (Fam-EDASRMEEVD). Using this platform, we found that CHIP, Hop, and DnaJC7 bound to both Hsp70s and Hsp90s (Fig 1A). Of these complexes, CHIP had the tightest affinity, binding Hsc70 with a KD of 0.62 ± 0.06 μM and Hsp72 with a KD of 0.51 ± 0.03 µM. We also found that CHIP, Hop, and DnaJC7 bound Hsc70 and Hsp72 with ~2-fold tighter affinity than Hsp90α and Hsp90β (Fig 1 and SI Fig 3). Interestingly, we found that FKBP51 and FKBP52 did not interact with appreciable affinity (KD >75 µM) with Hsp70s. Rather, they specifically bound to Hsp90α and Hsp90β with KD values between 1 and 2 μM (Fig 1 and SI Fig 3). Another important observation was that no specificity was observed between paralogs (i.e. Hsc70 versus Hsp72), suggesting that TPR co-chaperones do not discriminate between them. The negative control, Hip, did not interact with any of the C-terminal tracers, as expected. Finally, a reversed Hsp90 peptide (Fam-DVEEM) had no affinity for any of the TPR co-chaperones (SI Figure 4), consistent with previous results (41).
Fig 1.

Binding of full-length TPR co-chaperones to the C-termini of cytosolic Hsp70s and Hsp90s. (A) Summary of affinity values, measured by FP. Experiments are the average of the results from at least two independent experiments performed in triplicate each. Error bars represent the standard error of the mean (SEM) of all measurements. Representative binding curves are shown for (B) DnaJC7 (C) FKBP52 and (D) the negative control Hip.
The Met residue of the Hsp90 C-terminus (MEEVD) influences binding preferences
Previous work (35) had suggested that the residue immediately N-terminal to the EEVD motif contributes to the differences between binding of Hsp70s and Hsp90s to TPR co-chaperones. The cytosolic Hsp70s end in either IEEVD or VEEVD, while Hsp90s terminate with MEEVD. To test whether this residue contributes to affinity, we generated a chimeric mutant in which the Ile residue of an Hsp70 tracer was replaced with a Met (Fam-GSGPTMEEVD). This chimera had a weakened interaction with CHIP, Hop, and DnaJC7 (Fig 2A and 2C), instead having an “Hsp90-like” affinity. Consistent with the model, this result suggested that the Met residue of Hsp90s might be important in the affinity differences between Hsp70s and Hsp90s. However, placing the Met in the context of Hsp70 C-terminus was not able to provide binding to FKBP51 and FKBP52 (KD > 75 μM) (Fig 2C and SI Fig 5), so other features must be responsible for the selectivity of FKBP51/52 for Hsp90s. To further explore the role of the Ile/Met residues, we generated the corresponding mutant Hsp90α tracer in which we switched the Met residue to an Ile (Fam-DDTSRIEEVD) and tested its binding. FKBP51 and FKBP52 no longer bound the mutant Hsp90α (KD >25 µM) (Fig 2C and SI Fig 5), reducing the affinity by at least 12-fold compared to wt Hsp90α. Co-crystal structures of FKBP52 bound to a MEEVD peptide show the Met of MEEVD forms a critical hydrogen bond with Lys-282 of FKBP52’s TPR domain, which is important in stabilizing the binding of this peptide (44). Similarly, the mutant had other binding preferences that mirrored those of Hsp70’s. For example, CHIP and DnaJC7 had increased affinity (≥2-fold) for the mutant in comparison to the wt (Fig 2C and SI Fig 5). However, the mutant did not bind Hop (KD >25 μM), so residues other than the Ile/Met must be critical. Taken together, these data illustrate that the Ile/Met position is a major contributor to differences between the binding affinities for Hsp70s and Hsp90s, but that other regions are important for specific pairs.
Fig 2.
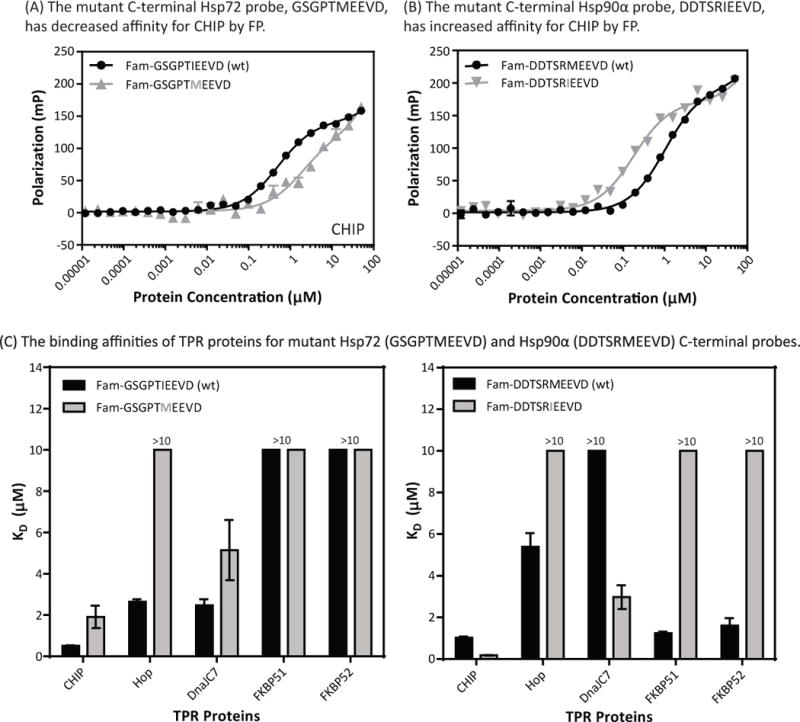
The Ile residue of Hsp70 (GSGPTIEEVD) and Met residue of Hsp90α (DDTSRMEEVD) strongly influence binding preferences. (A) CHIP preferentially binds the C-terminus of Hsp72 (GSGPTIEEVD) over the C-terminus of Hsp90α (DDTSRMEEVD). A mutant C-terminal Hsp72 probe (GSGPTMEEVD) has decreased affinity for CHIP. (B) A mutant C-terminal Hsp90α probe (DDTSRMEEVD) has increased affinity for CHIP. (C) The binding affinities of TPR proteins for mutant Hsp72 (GSGPTMEEVD) and Hsp90α (DDTSRMEEVD) C-terminal tracers. Affinities were measured by FP using full-length TPR proteins. Experiments are the average of the results from at least two independent experiments performed in triplicate each. Error bars represent SEM of all measurements.
Polar contacts dominate binding of TPR co-chaperones to Hsp70/Hsp90
The EEVD motif is strongly electronegative and the corresponding surface of the TPR domain tends to be electropositive (Fig 3A) (25,35,56). To explore the role of possible polar interactions in selectivity, we mutated the last Glu of the EEVD motif to either a neutral Ala (Fam-DDTSRMEAVD) or a cationic Lys (Fam-DDTSRMEKVD). Using FP, we found that all of the TPR proteins had slightly decreased affinity for the Ala mutant tracer (Fig 3C). FKBP51 was most sensitive to this change, binding the Ala mutant with a KD of 4.63 ± 0.38 μM, a ~4-fold decrease in affinity. The mutant Lys tracer (Fam-DDTSRMEKVD) had significantly decreased affinity for all TPR proteins (≥3-fold). Again, FKBP51 and FKBP52 were most sensitive to this change (KD > 25 μM). Next, we performed additional FP assays in which binding of wt Hsp70 and Hsp90 C-terminal tracers to TPR co-chaperones was measured at elevated pH. All of the TPR co-chaperones had reduced affinity at high pH (Fig 3D and SI Fig 6), supporting the idea that polar contacts are critical for the formation of EEVD-TPR domain complexes. However, the binding preferences did not dramatically switch, so pH seems unlikely to regulate TPR preferences.
Fig 3.

The binding of TPR co-chaperones to Hsp70/90 involves polar contacts in the EEVD motif. (A) The Hsp70/90 binding interface of TPR co-chaperones has a strong electropositive character. Surface representation of Hop’s TPR1 domain (PBD code = 1ELW) is shown as an example. Cationic residues (Lys and Arg) are highlighted in gray. Images were prepared using PyMOL. (B) Switching a glutamic acid in the EEVD motif to an alanine slightly decreased the affinity of TPR co-chaperones for Hsp70/90, while replacement with a lysine greatly decreased binding. (C) Affinities of TPR co-chaperones for Hsp90α mutant tracers DDTSRMEAVD and DDTSRMEKVD. (D) Binding of TPR co-chaperones to C-termini of Hsp70s and Hsp90s is pH dependent. Representative results are shown of CHIP binding GSGPTIEEVD. There was no change in the intrinsic fluorescence of the Fam fluorophore under these pH conditions (data not shown). All affinities were measured by FP using full-length TPR proteins. Experiments are the average of the results from at least two independent experiments performed in triplicate each. Error bars represent SEM of all measurements.
TPR1 and TPR2A of Hop selectively interact with the C-termini of Hsp70 and Hsp90 in the full-length protein
Hop is unique among the TPR co-chaperones studied here in that it contains three TPR domains that are termed: TPR1, TPR2A, and TPR2B (Fig 4A). Previous co-crystallographic and in vitro binding studies have shown that TPR1, when studied as an isolated protein, prefers to bind the C-terminus of Hsp70, whereas the isolated TPR2A domain binds tighter to Hsp90’s C-terminus (35). In the co-crystal structures, the N-terminal portions of the peptides seemed to dictate selectivity by occupying different hydrophobic patches within their respective TPR domains. We wanted to test whether this discrimination was preserved in full-length Hop because it seemed possible that the binding properties could be significantly altered in the context of the multi-domain protein, instead of isolated domains. Accordingly, we introduced single point mutations into full-length Hop that disrupt the critical “carboxylate clamps” required for EEVD binding. Two of the point mutations (K8A and R77A) were in the TPR1 domain, while the other mutations (N223A and R305A) were in Hop’s TPR2A domain (Fig 4A and 4B). Using our FP assay, we tested the ability of these mutant proteins to interact with Hsp70 and Hsp90 tracers. HopK8A and HopR77A did not interact with appreciable affinity (KD >25 μM) to Hsp70, but had normal affinity for Hsp90s (Kd values ~ 6 to 8 μM) (Fig 4C and SI Fig 7). Conversely, TPR2B mutants, HopN223A and HopR305A, selectively interacted with Hsp70 (KD of 2–4 μM) but not Hsp90s. Taken together, this work supports the conclusions made from studying individual domains of Hop.
Fig 4.

Hop’s TPR1 and TPR2A domains selectively interact with the C-termini of Hsp70 and Hsp90. (A) Schematic of the domain architecture of Hop. Gray lines indicate point mutations made in Hop’s TPR1 and TPR2A domains. (B) Structures of Hop’s TPR1 domain (PBD = 1ELW) and TPR2A domain (PBD = 1ELR). Residues that were mutated in these domains are highlighted in Gray. Hsp70/90 C-terminal peptides are shown in black. Structures were prepared using PyMOL. (C) Table summarizing binding affinities of Hop point mutants (K8A, R77A, N223A, and R305A) for chaperone tracers. Affinities were measured by FP using full-length Hop. The results are the average of triplicates and the error bars are SEM. Representative data are shown from two independent replicates. (D) Hop R77A binds specifically to Hsp90s. (E) Hop R305A binds specifically to Hsp70s.
The TPR domains and J domain of DnaJC7 are modular
DnaJC7 is unique among the TPR co-chaperones studied here in that it contains both TPR domains and a J-domain. Thus, it has two ways of interacting with Hsp70s and we were interested in understanding whether the TPR interaction could impact the J-domain activity. A common assay for studying J protein activity is the refolding of denatured firefly luciferase (50), which strictly requires addition of both an Hsp70 and a J protein. We found that a prototypical J protein, DnaJ, could stimulate refolding of denatured luciferase by both Hsp72 and Hsp72ΔEEVD (Fig 5A), as expected. Surprisingly, we found that DnaJC7, despite its TPR domains, also had equal activity for both Hsp72 and Hsp72ΔEEVD (Fig 5B). Compared to the positive control, DnaJC7 had relatively mild activity in this assay, but the results clearly showed that the TPR domain interaction does not impact the availability of the J domain. The C-terminal region of Hsp72 has a long disordered region between the parts of the chaperone that are involved in contacting the TPR- and J-domains, which might allow the two interactions to act independently.
Fig 5.
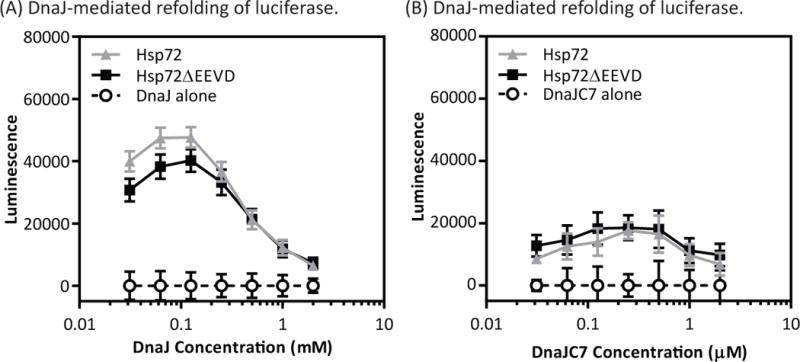
The TPR and J domain regions of DnaJC7 are modular. (A) Addition of DnaJ, a prototypical J protein, stimulated refolding of firefly luciferase by either Hsp72 or Hsp72ΔEEVD. (B) Likewise, DnaJC7 worked with both Hsp72 and Hsp72ΔEEVD to refold denatured luciferase. The results are the average of triplicates and the error bars are SEM. Representative data are shown from two independent replicates.
Phosphorylation of Hsp70/Hsp90 C-termini dramatically affects binding to CHIP, but not other TPR co-chaperones
Recently, Muller and co-workers have used an in vitro phosphorylation assays, coupled with mass spectrometry, to detect phosphorylation sites on the C-termini of both Hsp70 and Hsp90. They also showed that phosphorylation of serine and threonine residues in the C-termini of Hsp70 and Hsp90 weakens binding to CHIP (48). Thus, although it remaisn to be seen whether this phosphorylation event occurs in cells, we wondered if it might be a more general regulatory mechanism for TPR binding. To test this idea, we generated FP tracers that mimicked phosphorylation (Fig 6A) and measured their binding to TPR proteins. Consistent with previous data (48), the affinity of CHIP for the mutant Hsp70 Fam-SSGPEIEEVD and Hsp90 Fam-DDTERMEEVD tracers was reduced by more than 8-fold (Fig 6A). However, Hop had mildly enhanced binding (~2-fold) to pseudophosphorylated Hsp70 and Hsp90 C-termini (Fig 6B) and the binding of DnaJC7, FKBP51, and FKBP52 was unaffected (Fig 6B). Thus, mimicking phosphorylation of the C-termini of Hsp70 and Hsp90 seemed to tune the affinity for select TPR co-chaperones, but not others.
Fig 6.

Mimicking phosphorylation of Hsp70/90 selectively weakens binding to CHIP. (A) CHIP has decreased affinity for Hsp70/90 C-termini that contain phosphomimetic residues. (B) Binding affinities of other TPR co-chaperones for mutant Hsp70/90 C-termini. All affinities measured by FP using full-length TPR proteins. Experiments are the average of the results from at least two independent experiments performed in triplicate each. Error bars represent SEM of all measurements.
Conclusions
The molecular chaperones Hsp70 and Hsp90 work with TPR co-chaperones to mediate protein triage and quality control. In this study, we characterized how TPR co-chaperones, including CHIP, Hop, DnaJC7, FKBP51, and FKBP52, bind the C-termini of four cytosolic human Hsp70s and Hsp90s in vitro. Some TPR co-chaperones showed a preference for binding to the chaperones. For example, CHIP, Hop, and DnaJC7 had a 2-fold overall preference for Hsp70s over Hsp90s. Using point mutants, we learned that the Ile/Met residue adjacent to the EEVD motif was one feature that gives rise to these differences. Moreover, we found that mimicking phosphorylation of Ser/Thr residues in the C-termini reduced affinity for CHIP and modestly enhanced affinity for Hop, but that this modification had little effect on the interactions with other TPR proteins. This result was surprising, given the dramatic increase in size and charge at these sites and their proximity to the EEVD-TPR contact. Finally, no specificity was observed when comparing chaperone paralogs (i.e. Hsc70 versus Hsp72) in any of these platforms. This was also surprising because the expression of the paralogs is regulated by quite different mechanisms and a few reports have started to identify pathways that rely on one and not the other (57–59). However, from the TPR’s point-of-view, they appear to be degenerate. Together, these studies expand our understanding of chaperone-TPR interactions. It is important to emphasize that some of these conclusions have been suggested by previous studies (vide infra). The comprehensive approach taken here was designed to provide the full spectrum of interaction affinities and reveal broader patterns. Some of the surprising results from this approach include the findings that FKBP51 and FKBP52 do not bind Hsp70s and that pseudophosphorylation has no effect on binding to DnaJC7, FKBP51, and FKBP52. Thus, TPR interactions are perhaps tuned by unexpected mechanisms.
What are the implications of these results for understanding chaperone-mediated quality control? Before this work, one formal possibility was that different TPR co-chaperones might display a clear hierarchy of affinity constants. This scenario would have suggested a model in which certain TPR co-chaperones could effectively out-compete others to drive quality control “decisions”. However, with a few exceptions (e.g. FKBP51/52 binding exclusively to Hsp90s and selectivity within Hop TPR domains), there were no dramatic differences between the observed affinity constants (see Figure 1). So, what other factors might contribute to selectivity in this system? One possibility is that secondary contacts (e.g. those outside the EEVD motif) might help tune the interactions. However, there appears to be comparatively little energy in these interactions, so their contributions might be expected to be relatively small. Another possibility is that the expression levels of the individual TPR domain co-chaperones, rather than their intrinsic affinity values, may dominate which complexes are most likely to form. For example, Hop expression is known to be induced in response to certain stress conditions, such as infection (60), which could reshape the dynamics of which TPR interactions are favored. However, this model seems unsatisfying by itself. Rather, an addition to this model is suggested by the observations that mimicking phosphorylation dramatically weakens the affinity of the CHIP-Hsp70/Hsp90 complexes, while enhancing the corresponding Hop complexes. Specifically, it seems plausible that post-translational modifications (PTMs) might help guide which TPR co-chaperone is bound by the specific chaperone. In the case of phosphorylation, contact with CHIP is apparently disfavored, while interactions with other TPR co-chaperones are spared or even enhanced (in the case of Hop). This mechanism is appealing because it would allow quality control “decisions” to be shaped by signaling pathways, providing a way for cells and organisms to adjust their proteomes in response to cues or changing conditions. Even this model seems rather incomplete, so we also favor the idea that other features might ultimately be found to contribute to the choice of which TPR co-chaperone is bound. These features might include the structure of the client, whether it directly interacts with co-chaperones, and the subcellular co-localization of all the components. Future work will need to explore how these factors guide the selection of TPR-chaperone pairs. These results suggest that, except for the special cases of FKBPs and individual Hop domains, features other than affinity of the EEVD-TPR contacts might play dominant roles.
There has been great interest in targeting these protein-protein interactions to treat diseases (61–65). Many of these strategies are focused on inhibiting EEVD-TPR contacts, such as that between Hop and Hsp70/90, which are important in cancer. However, our results suggest that such approaches may have unintended consequences. For example, androgen receptor (AR) is dependent on Hop-Hsp70 for its maturation (66), but it also requires CHIP-Hsp70 for its degradation (67,68). Thus, it isn’t clear what effect an EEVD-TPR inhibitor might ultimately have on levels of that client. One might conceivably achieve greater selectivity by developing inhibitors of the secondary contacts between chaperones and TPR co-chaperones, which presumably occur at sites that are less degenerate than the EEVD-TPR contact. However, like others (42), we found that secondary contacts (e.g. those outside the EEVD) contribute relatively little binding free energy. Thus, it may be difficult to identify compounds that compete with the interactions by binding at these secondary contacts. Despite this challenge, some progress has been made with derivatives of the natural product sansalvamide A, which inhibit some Hsp90-TPR interactions, but not others (64,65). Although the mechanisms are not yet clear, these molecules are thought to act at allosteric sites on Hsp90, avoiding the problem of weak affinity in the secondary contacts. It is becoming more widely appreciated that allosteric inhibitors are effective against otherwise “undruggable” protein-protein interactions (69). Our results support the continued focus on allosteric sites, rather than TPR-EEVD inhibitors, in the pursuit of reagents for fine-tuning protein quality control.
Supplementary Material
Acknowledgments
V.A.A. was supported by a pre-doctoral fellowship from the NIH (F31 AG043266-02). J.E.G. acknowledges the financial support of NIH NS059690 and GM109896. We would like to thank Y. Osawa (University of Michigan) for his generous gift of the Hip expression vector. We would also like to thank I. Taylor (UCSF) for helpful conversations.
Abbreviations
- 5-Fam
5-Carboxyfluorescein
- FKBP51
51-kDa FK506-binding protein
- FKBP52
52-kDa FK506-binding protein
- CHIP
Carboxyl terminus of Hsc70 interacting protein
- FP
Fluorescence polarization
- Hsp70
Heat shock protein 70
- Hsp90
Heat shock protein 90
- Hop
Hsp70/90 organizing protein
- Hip
Hsp70-interacting protein
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- NBD
Nucleotide binding domain
- PTMs
Post-translational modifications
- TPR
Tetratricopeptide repeat
References
- 1.Kundrat L, Regan L. Balance between folding and degradation for Hsp90-dependent client proteins: a key role for CHIP. Biochemistry. 2010;49:7428–7438. doi: 10.1021/bi100386w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chiang HL, Terlecky SR, Plant CP, Dice JF. A Role for a 70-Kilodaton Heat-Shock Protein in Lysosomal Degradation of Intracellular Proteins. Science. 1989;246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 3.Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, Ciechanover A. Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J Biol Chem. 1997;272:9002–9010. doi: 10.1074/jbc.272.14.9002. [DOI] [PubMed] [Google Scholar]
- 4.Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–451. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 5.Hartl FU, Hayer-Hartl M. Protein folding – Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 6.Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone-mediated protein folding in the cytosol. Nat Rev Mol Cell Bio. 2004;5:781–791. doi: 10.1038/nrm1492. [DOI] [PubMed] [Google Scholar]
- 7.Hohfeld J, Cyr DM, Patterson C. From the cradle to the grave: molecular chaperones that may choose between folding and degradation. Embo Rep. 2001;2:885–890. doi: 10.1093/embo-reports/kve206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gong M, Hubner N. Molecular genetics of human hypertension. Clin Sci. 2006;110:315–326. doi: 10.1042/CS20050208. [DOI] [PubMed] [Google Scholar]
- 9.Cummings CJ, Sun YL, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- 10.Warrick JM, Chan HYE, Gray-Board GL, Chai YH, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- 11.Yaglom JA, Gabai VL, Sherman MY. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res. 2007;67:2373–2381. doi: 10.1158/0008-5472.CAN-06-3796. [DOI] [PubMed] [Google Scholar]
- 12.Neckers L, Mimnaugh E, Schulte TW. Hsp90 as an anti-cancer target. Drug Resist Update. 1999;2:165–172. doi: 10.1054/drup.1999.0082. [DOI] [PubMed] [Google Scholar]
- 13.Waza M, Adachi H, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. Modulation of Hsp90 function in neurodegenerative disorders: a molecular-targeted therapy against disease-causing protein. J Mol Med. 2006;84:635–646. doi: 10.1007/s00109-006-0066-0. [DOI] [PubMed] [Google Scholar]
- 14.D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Nicolet CM, Craig EA. Isolation and Characterization of Sti1, a Stress-Inducible Gene from Saccharomyces-Cerevisiae. Mol Cell Biol. 1989;9:3638–3646. doi: 10.1128/mcb.9.9.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith DF, Sullivan WP, Marion TN, Zaitsu K, Madden B, Mccormick DJ, Toft DO. Identification of a 60-Kilodalton Stress-Related Protein, P60, Which Interacts with Hsp90 and Hsp70. Mol Cell Biol. 1993;13:869–876. doi: 10.1128/mcb.13.2.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballinger CA, Connell P, Wu YX, Hu ZY, Thompson LJ, Yin LY, Patterson C. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–4545. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ballinger CA, Hu ZY, Connell P, Patterson C. Identification of CHIP, a novel striated muscle-restricted TPR-containing protein that negatively regulates chaperone functions. Faseb J. 1999;13:A442–A442. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez MP, Sullivan WP, Toft DO. The assembly and intermolecular properties of the hsp70-Hop-hsp90 molecular chaperone complex. J Biol Chem. 2002;277:38294–38304. doi: 10.1074/jbc.M206566200. [DOI] [PubMed] [Google Scholar]
- 20.Johnson BD, Schumacher RJ, Ross ED, Toft DO. Hop modulates hsp70/hsp90 interactions in protein folding. J Biol Chem. 1998;273:3679–3686. doi: 10.1074/jbc.273.6.3679. [DOI] [PubMed] [Google Scholar]
- 21.Chen SY, Smith DF. Hop as an adaptor in the heat shock protein 70 (Hsp70) and Hsp90 chaperone machinery. J Biol Chem. 1998;273:35194–35200. doi: 10.1074/jbc.273.52.35194. [DOI] [PubMed] [Google Scholar]
- 22.Morishima Y, Murphy PJM, Li DP, Sanchez ER, Pratt WB. Stepwise assembly of a glucocorticoid receptor center dot hsp90 heterocomplex resolves two sequential ATP-dependent events involving first hsp70 and then hsp90 in opening of the steroid binding pocket. J Biol Chem. 2000;275:18054–18060. doi: 10.1074/jbc.M000434200. [DOI] [PubMed] [Google Scholar]
- 23.Hutchison KA, Dittmar KD, Czar MJ, Pratt WB. Proof That Hsp70 Is Required for Assembly of the Glucocorticoid Receptor into a Heterocomplex with Hsp90. J Biol Chem. 1994;269:5043–5049. [PubMed] [Google Scholar]
- 24.Smith DF. Dynamics of Heat-Shock Protein 90-Progesterone Receptor-Binding and the Disactivation Loop Model for Steroid-Receptor Complexes. Mol Endocrinol. 1993;7:1418–1429. doi: 10.1210/mend.7.11.7906860. [DOI] [PubMed] [Google Scholar]
- 25.Zhang MH, Windheim M, Roe SM, Peggie M, Cohen P, Prodromou C, Pearl LH. Chaperoned ubiquitylation – Crystal structures of the CHIPU box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol Cell. 2005;20:525–538. doi: 10.1016/j.molcel.2005.09.023. [DOI] [PubMed] [Google Scholar]
- 26.Connell P, Ballinger CA, Jiang JH, Wu YX, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 27.Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–555. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies TH, Ning YM, Sanchez ER. A new first step in activation of steroid receptors – Hormone-induced switching of FKBP51 and FKBP52 immunophilins. J Biol Chem. 2002;277:4597–4600. doi: 10.1074/jbc.C100531200. [DOI] [PubMed] [Google Scholar]
- 29.Jaaskelainen T, Makkonen H, Palvimo JJ. Steroid up-regulation of FKBP51 and its role in hormone signaling. Curr Opin Pharmacol. 2011;11:326–331. doi: 10.1016/j.coph.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 30.Riggs DL, Cox MB, Tardif HL, Hessling M, Buchner J, Smith DF. Noncatalytic role of the FKBP52 peptidyl-prolyl isomerase domain in the regulation of steroid hormone signaling. Mol Cell Biol. 2007;27:8658–8669. doi: 10.1128/MCB.00985-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brychzy A, Rein T, Winklhofer KF, Hartl FU, Young JC, Obermann WMJ. Cofactor Tpr2 combines two TPR domains and a J domain to regulate the Hsp70/Hsp90 chaperone system. Embo J. 2003;22:3613–3623. doi: 10.1093/emboj/cdg362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moffatt NSC, Bruinsma E, Uhl C, Obermann WMJ, Toft D. Role of the cochaperone Tpr2 in Hsp90 chaperoning. Biochemistry-Us. 2008;47:8203–8213. doi: 10.1021/bi800770g. [DOI] [PubMed] [Google Scholar]
- 33.Russell LC, Whitt SR, Chen MS, Chinkers M. Identification of conserved residues required for the binding of a tetratricopeptide repeat domain to heat shock protein 90. J Biol Chem. 1999;274:20060–20063. doi: 10.1074/jbc.274.29.20060. [DOI] [PubMed] [Google Scholar]
- 34.Carrello A, Ingley E, Minchin RF, Tsai S, Ratajczak T. The common tetratricopeptide repeat acceptor site for steroid receptor-associated immunophilins and Hop is located in the dimerization domain of hsp90. J Biol Chem. 1999;274:2682–2689. doi: 10.1074/jbc.274.5.2682. [DOI] [PubMed] [Google Scholar]
- 35.Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU, Moarefi I. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- 36.Southworth DR, Agard DA. Client-Loading Conformation of the Hsp90 Molecular Chaperone Revealed in the Cryo-EM Structure of the Human Hsp90:Hop Complex. Mol Cell. 2011;42:771–781. doi: 10.1016/j.molcel.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014;157:1685–1697. doi: 10.1016/j.cell.2014.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cliff MJ, Harris R, Barford D, Ladbury JE, Williams MA. Conformational diversity in the TPR domain-mediated interaction of protein phosphatase 5 with Hsp90. Structure. 2006;14:415–426. doi: 10.1016/j.str.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 39.Connarn JN, Assimon VA, Reed RA, Tse E, Southworth DR, Zuiderweg ERP, Gestwicki JE, Sun DX. The Molecular Chaperone Hsp70 Activates Protein Phosphatase 5 (PP5) by Binding the Tetratricopeptide Repeat (TPR) Domain. J Biol Chem. 2014;289:2908–2917. doi: 10.1074/jbc.M113.519421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L, Liu YT, Hao R, Chen L, Chang ZJ, Wang HR, Wang ZX, Wu JW. Molecular Mechanism of the Negative Regulation of Smad1/5 Protein by Carboxyl Terminus of Hsc70-interacting Protein (CHIP) J Biol Chem. 2011;286 doi: 10.1074/jbc.M110.201814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smith MC, Scaglione KM, Assimon VA, Patury S, Thompson AD, Dickey CA, Southworth DR, Paulson HL, Gestwicki JE, Zuiderweg ERP. The E3 Ubiquitin Ligase CHIP and the Molecular Chaperone Hsc70 Form a Dynamic, Tethered Complex. Biochemistry-Us. 2013;52:5354–5364. doi: 10.1021/bi4009209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang HQ, Amick J, Chakravarti R, Santarriaga S, Schlanger S, McGlone C, Dare M, Nix JC, Scaglione KM, Stuehr DJ, Misra S, Page RC. A Bipartite Interaction between Hsp70 and CHIP Regulates Ubiquitination of Chaperoned Client Proteins. Structure. 2015;23:472–482. doi: 10.1016/j.str.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kundrat L, Regan L. Identification of Residues on Hsp70 and Hsp90 Ubiquitinated by the Cochaperone CHIP. J Mol Biol. 2010;395:587–594. doi: 10.1016/j.jmb.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu BL, Li PY, Liu YW, Lou ZY, Ding Y, Shu CL, Ye S, Bartlam M, Shen BF, Rao ZH. 3D structure of human FK506-binding protein 52: Implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. P Natl Acad Sci USA. 2004;101:8348–8353. doi: 10.1073/pnas.0305969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackrel ME, Valverde R, Regan L. Redesign of a protein-peptide interaction: characterization and applications. Protein Sci. 2009;18:762–774. doi: 10.1002/pro.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cortajarena AL, Yi F, Regan L. Designed TPR modules as novel anticancer agents. ACS Chem Biol. 2008;3:161–166. doi: 10.1021/cb700260z. [DOI] [PubMed] [Google Scholar]
- 47.Cortajarena AL, Liu TY, Hochstrasser M, Regan L. Designed proteins to modulate cellular networks. ACS Chem Biol. 2010;5:545–552. doi: 10.1021/cb9002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Muller P, Ruckova E, Halada P, Coates PJ, Hrstka R, Lane DP, Vojtesek B. C-terminal phosphorylation of Hsp70 and Hsp90 regulates alternate binding to co-chaperones CHIP and HOP to determine cellular protein folding/degradation balances. Oncogene. 2013;32:3101–3110. doi: 10.1038/onc.2012.314. [DOI] [PubMed] [Google Scholar]
- 49.Morishima Y, Lau M, Peng HM, Miyata Y, Gestwicki JE, Pratt WB, Osawa Y. Heme-dependent activation of neuronal nitric oxide synthase by cytosol is due to an Hsp70-dependent, thioredoxin-mediated thiol-disulfide interchange in the heme/substrate binding cleft. Biochemistry. 2011;50:7146–7156. doi: 10.1021/bi200751t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wisen S, Gestwicki JE. Identification of small molecules that modify the protein folding activity of heat shock protein 70. Analytical Biochemistry. 2008;374:371–377. doi: 10.1016/j.ab.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 51.Carrigan PE, Nelson GM, Roberts PJ, Stoffer JN, Riggs DL, Smith DF. Multiple domains of the co-chaperone Hop are important for Hsp70 binding. J Biol Chem. 2004;279:16185–16193. doi: 10.1074/jbc.M314130200. [DOI] [PubMed] [Google Scholar]
- 52.Brinker A, Scheufler C, von der Mulbe F, Fleckenstein B, Herrmann C, Jung G, Moarefi I, Hartl FU. Ligand discrimination by TPR domains – Relevance and selectivity of EEVD-recognition in Hsp70 center dot Hop center dot Hsp90 complexes. J Biol Chem. 2002;277:19265–19275. doi: 10.1074/jbc.M109002200. [DOI] [PubMed] [Google Scholar]
- 53.Cheung-Flynn J, Roberts PJ, Riggs DL, Smith DF. C-terminal sequences outside the tetratricopeptide repeat domain of FKBP51 and FKBP52 cause differential binding to hsp90. J Biol Chem. 2003;278:17388–17394. doi: 10.1074/jbc.M300955200. [DOI] [PubMed] [Google Scholar]
- 54.Chen SY, Sullivan WP, Toft DO, Smith DF. Differential interactions of p23 and the TPR-containing proteins Hop, Cyp40, FKBP52 and FKBP51 with Hsp90 mutants. Cell Stress Chaperon. 1998;3:118–129. doi: 10.1379/1466-1268(1998)003<0118:diopat>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, Hartl FU, Bracher A. Structure and function of Hip, an attenuator of the Hsp70 chaperone cycle. Nat Struct Mol Biol. 2013;20:929–+. doi: 10.1038/nsmb.2608. [DOI] [PubMed] [Google Scholar]
- 56.Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. P Natl Acad Sci USA. 2003;100:868–873. doi: 10.1073/pnas.0231020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patel PD, Yan PR, Seidler PM, Patel HJ, Sun WL, Yang CH, Que NS, Taldone T, Finotti P, Stephani RA, Gewirth DT, Chiosis G. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol. 2013;9:677–+. doi: 10.1038/nchembio.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fontaine SN, Rauch JN, Nordhues BA, Assimon VA, Stothert AR, Jinwal UK, Sabbagh JJ, Chang L, Stevens SM, Zuiderweg ERP, Gestwicki JE, Dickey CA. Isoform-selective Genetic Inhibition of Constitutive Cytosolic Hsp70 Activity Promotes Client Tau Degradation Using an Altered Co-chaperone Complement. J Biol Chem. 2015;290:13115–13127. doi: 10.1074/jbc.M115.637595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jinwal UK, Akoury E, Abisambra JF, O’Leary JC, Thompson AD, Blair LJ, Jin Y, Bacon J, Nordhues BA, Cockman M, Zhang J, Li PF, Zhang B, Borysov S, Uversky VN, Biernat J, Mandelkow E, Gestwicki JE, Zweckstetter M, Dickey CA. Imbalance of Hsp70 family variants fosters tau accumulation. Faseb J. 2013;27:1450–1459. doi: 10.1096/fj.12-220889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heine H, Delude RL, Monks BG, Espevik T, Golenbock DT. Bacterial lipopolysaccharide induces expression of the stress response genes hop and H411. J Biol Chem. 1999;274:21049–21055. doi: 10.1074/jbc.274.30.21049. [DOI] [PubMed] [Google Scholar]
- 61.Yi F, Regan L. A Novel Class of Small Molecule Inhibitors of Hsp90. Acs Chem Biol. 2008;3:645–654. doi: 10.1021/cb800162x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang T, Hamza A, Cao XH, Wang B, Yu SW, Zhan CG, Sun DX. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008;7:162–170. doi: 10.1158/1535-7163.MCT-07-0484. [DOI] [PubMed] [Google Scholar]
- 63.Pimienta G, Herbert KM, Regan L. A Compound That Inhibits the HOP-Hsp90 Complex Formation and Has Unique Killing Effects in Breast Cancer Cell Lines. Mol Pharmaceut. 2011;8:2252–2261. doi: 10.1021/mp200346y. [DOI] [PubMed] [Google Scholar]
- 64.Vasko RC, Rodriguez RA, Cunningham CN, Ardi VC, Agard DA, McAlpine SR. Mechanistic Studies of Sansalvamide A-Amide: An Allosteric Modulator of Hsp90. Acs Medicinal Chemistry Letters. 2010;1:4–8. doi: 10.1021/ml900003t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ardi VC, Alexander LD, Johnson VA, McAlpine SR. Macrocycles That Inhibit the Binding between Heat Shock Protein 90 and TPR-Containing Proteins. Acs Chem Biol. 2011;6:1357–1366. doi: 10.1021/cb200203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: A diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 67.Sarkar S, Brautigan DL, Parsons SJ, Larner JM. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene. 2014;33:26–33. doi: 10.1038/onc.2012.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Adachi H, Waza M, Tokui K, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci. 2007;27:5115–5126. doi: 10.1523/JNEUROSCI.1242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thompson AD, Dugan A, Gestwicki JE, Mapp AK. Fine-tuning multiprotein complexes using small molecules. ACS chemical biology. 2012;7:1311–1320. doi: 10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.