

Abstract
Sarcomeric myosins have the remarkable ability to form regular bipolar thick filaments that, together with actin thin filaments, constitute the fundamental contractile unit of skeletal and cardiac muscle. This has been established for over fifty years and yet a molecular model for the thick filament has not been attained. In part this is due to the lack of a detailed molecular model for the coiled-coil that constitutes the myosin rod. The ability to self-assemble resides in the C-terminal of the section of myosin known as light meromyosin (LMM) which exhibits strong salt dependent aggregation that has inhibited structural studies. Here we evaluate the feasibility of generating a complete model for the myosin rod by combining overlapping structures of five sections of coiled-coil covering 164 amino acid residues which constitute 20% of LMM. Each section contains ~7-9 heptads of myosin. The problem of aggregation was overcome by incorporating the globular folding domains, Gp7 and Xrcc4 which enhance crystallization. The effect of these domains on the stability and conformation of the myosin rod was examined through biophysical studies and overlapping structures. In addition, a computational approach was developed to combine the sections into a contiguous model. The structures were aligned, trimmed to form a contiguous model, and simulated for >700 ns to remove the discontinuities and achieve an equilibrated conformation that represents the native state. This experimental and computational strategy lays the foundation for building a model for the entire myosin rod.
Keywords: Coiled-coil, Protein structure, Molecular dynamics, X-ray crystallization, Cardiac muscle
Introduction
Cardiac and skeletal muscles contain sets of interdigitated thick and thin filaments. Muscle contraction occurs when the thick filaments, which are built primarily from myosin II, actively slide past the actin-containing thin filaments. Myosin plays both a structural and enzymatic role in this fundamental biological process. The globular N-terminal domains (heads) hydrolyze ATP and interact with actin to generate force, whereas the C-terminal region forms a long α-helix that dimerizes to form a coiled-coil. The coiled-coil is known as the myosin rod. The extended region of myosin can be split in two sections: Subfragment-2 (S2) and light meromyosin (LMM), where these are connected by a flexible section of the coiled-coil that functions as a hinge. In cardiac β-myosin LMM extends from approximately amino acid residues 1140 to 1935.1 LMM self-assembles to form the backbone of the thick filament at physiological ionic strength. At this time, there is considerable knowledge of the structure and function of the globular motor regions of myosin, but many aspects of the molecular organization of the myosin rods in the thick filament are still unresolved. This has important implications since a significant number of mutations that lead to skeletal and cardiac myopathies are located in the myosin rod.2-4
The reason that the structure of the myosin thick filament is not well understood is due to the lack of high resolution structural information for LMM and the complexity of the assembly. Although the myosin rod is predicted to consist of a coiled-coil, which is in principle a simple motif, there are distinct locations where the sequence deviates from a canonical structure.5 Indeed, there are four conserved locations that contain an additional “skip” residue that disrupts the coiled-coil heptad repeat. These skip residues have been proposed to introduce flexibility into the myosin rod to allow assembly into the thick filament.6,7 Surrounding these skip residues, the sequence exhibits a 28 amino acid repeat and contains an alternating pattern of positively and negatively charged amino acids, a feature that has been implicated in the staggered interaction between adjacent myosin molecules in the thick filament.6 In addition, the coiled-coil propensity in the proposed hinge region between S2 and LMM is considerably lower than the sequence signature for a stable coiled-coil. This region is adjacent to the first skip residue and has been proposed to provide an area of flexibility that allows the motor domains swing away from the myosin thick filament to interact with the actin thin filaments.6,8
Previous studies on tropomyosin and intermediate filaments have shown that the local pitch of a coiled-coil is profoundly influenced by the amino acid sequence, especially in those regions that deviate from a canonical heptad repeat.9,10 As such, it is not possible to accurately predict the structure of the myosin rod with the accuracy needed to build a realistic model for the rod and to understand the interactions between adjacent myosin molecules in the thick filament. Clearly there is a need for high resolution structural data.
The sheer size of the myosin rod at approximately 1600 Å in linear dimension, prohibits a direct crystallographic study. Thus, the only feasible approach is to divide the rod into smaller sections that might be more amenable to structural study. Indeed, this is the standard approach for investigating large intractable proteins, but it is more complicated for fragments of the myosin because the rod exhibits a strong salt-dependent aggregation. A direct result of this property is that fragments of myosin readily form paracrystals at low ionic strength.11-15 This is due in part to periodic clusters of positively and negatively charged residues that repeat every 28 amino acid residues for the entire length of the myosin rod noted above.5 In sarcomeric myosins the rod contains 38 of these repeats.6 The latter property has doubtlessly contributed to the difficulties of crystallizing short sections of the myosin rod that might otherwise appear amenable to structural study. Indeed, until recently there were no structures available for the myosin rod except for the N-terminal 126 amino acid residues of human cardiac β-myosin II S2, which exhibits for most of its length a canonical coiled-coil and has no tendency to aggregate at low ionic strength.16
Apart from the problems of self-assembly into paracrystals, a second issue is whether fragments of myosin contain all of the information necessary to fold correctly and yield a soluble protein. The folding of coiled-coils is generally highly cooperative where initiation of folding is often controlled by trigger sequences that have a higher coiled-coil propensity than the bulk of the protein. These sequence signatures usually lie towards the N-terminus of the coiled-coil.17-22 Removal of these motifs frequently prevents proper folding or loss of dimerization of the resultant fragment. Indeed, a recent paper by Wolny et al., shows that there appears to be a folding signal located in the myosin tail between 1301 and 1330 that leads to high helicity in even short peptides (7 heptads) that contain this sequence. Conversely, five other down-stream peptides that contained only seven heptads of the myosin rod showed limited α-helical content.23 This problem of stabilizing coiled coils that lack their native trigger sequence has been circumvented for some coiled-coils by incorporation of a short stable coiled-coil, such as that seen in the well-studied transcription factor GCN4.18,24,25 This strategy could be applied to sections of LMM but is not predicted to eliminate self-assembly into paracrystals at low ionic strength.
The problem of myosin self-assembly has now been solved by using globular fusion proteins in place of GCN4.26 This strategy was developed for investigating the overlap complex for tropomyosin and components of the yeast spindle pole body and has been recently applied to myosin.26-28 In those structural studies, the globular domains from human DNA ligase binding protein Xrcc4 and the bacteriophage φ29 scaffolding protein Gp7 were fused to the N-terminus of the targeted coiled-coil domains.29-31 The resultant fusion proteins were expressed with good solubility in Escherichia coli and crystallized readily. Following a similar strategy, the structure of the regions surrounding the four skip residues in human cardiac β-myosin II have been reported which now opens up the possibility of determining the structure of the entire myosin rod.26 The use of globular domains also solves the problem of the limited stability of short sections of the myosin rod.23
In principle, a model for the entire LMM (~800 amino acid residues) could be assembled from the structures of 15-20 overlapping fragments that each span 50-70 amino acid residues. The initial stages of this project are reported here and establishes proof of concept for the approach. This study reports a composite model for residues L1526-L1689 of human cardiac β-myosin generated from five overlapping structures, covering ~24% of LMM. This study also describes the degree of stabilization that can be obtained from a suite of folding domains and their effect on protein expression and crystallization. Molecular dynamic (MD) simulations of myosin coiled-coil with and without a folding domain provided a means to assess changes in dynamic properties brought about by the fusion. Additionally the fusion structures presented here have a large degree of sequence overlap that provides a basis for evaluating the effects of the fusion protein and crystallographic contacts on the targeted coiled-coil. Lastly, surface analysis shows that the composite model exhibits a substantial hydrophobic surface area which plays a role in the higher order assembly of bipolar thick filaments.
Materials and Methods
All cloning was performed as described previously.28 Briefly, QuikChange cloning was used to amplify and insert sections of the MYH7 gene into vectors containing the desired solubilization domain. DNA encoding human cardiac myosin rod was purchased as an image clone from Open Biosystems. The use of QuikChange cloning allowed all constructs to be made without introduction of cloning artifacts, which is critical for maintaining the correct coiled-coil registration between the folding domains and the target gene fragment. The sequences of all constructs were verified over their entire open reading frames.
Construct design
All fusion protein constructs were cloned into a modified pET24D (EMD) plasmid or pKLD37, a modified pET31b plasmid (EMD) by QuikChange cloning.32-35 A His-tag and an rTEV cleavage recognition site were introduced N-terminally to constructs that contained Gp7, Xrcc4, and GCN4.36 The Paircoil algorithm was used to predict regions of coiled-coil and their helical registration.37 A complete description of all constructs is given in Table 1.
Table 1.
Fusion constructsa.
|
MyH7-1361-1406 MSYYHHHHHHDYDIPTSENLYFQGLSKANSEVAQWRTKYETDAIQRTEELEEAKKKLAQRLQEAEEAVEA |
|
GCN4-MyH7-1361-1406 MGSSHHHHHHHHDYDIPTSENLYFQGASMSVKELEDKVEELLSKNYHLENEVARLKKLLSKANSEVAQWRTK YETDAIQRTEELEEAKKKLAQRLQEAEEAVEAVNA |
|
Gp7-MyH7-1361-1406 MSYYHHHHHHDYDIPTSENLYFQGGSGPLKPEEHEDILNKLLDPELAQSERTEALQQLRVNYGSFVSEYNDL TKSLSKANSEVAQWRTKYETDAIQRTEELEEAKKKLAQRLQEAEEAVEAVNA |
|
Xrcc4-MyH7-1361-1406 MSYYHHHHHHDYDIPTSENLYFQGGSGERKISRIHLVSEPSITHFLQVSWEKTLESGFVITLTDGHSAWTGT VSESEISQEADDMAMEKGKYVGELRKALLSGAGPADVYTFNFSKESCYFFFEKNLKDVSFRLGSFNLEKVEN PAEVIRELICYCLDTTAENQAKNEHLQKENERLQRVLSKANSEVAQWRTKYETDAIQRTEELEEAKKKLAQR LQEAEEAVEAVNA |
|
Gp7-MyH7- MyH7-1526-1571 MSHHHHHHHHDYDIPTSENLYFQGGSGPLKPEEHEDILNKLLDPELAQSERTEALQQLRVNYGSFVSEYNDL TKSHEKLEKVRKQLEAEKMELQSALEEAEASLEHEEGKILRAQLEFNQIKAE |
|
Xrcc4-MyH7-1562-1622 MSYYHHHHHHDYDIPTSENLYFQGGSGERKISRIHLVSEPSITHFLQVSWEKTLESGFVITLTDGHSAWTGT VSESEISQEADDMAMEKGKYVGELRKALLSGAGPADVYTFNFSKESCYFFFEKNLKDVSFRLGSFNLEKVEN PAEVIRELICYCLDTTAENQAKNEHLQLEFNQIKAEIERKLAEKDEEMEQAKRNHLRVVDSLQTSLDAETRS RNEALRVKKKMEGDLb |
|
Xrcc4-MyH7-1590-1657 MSYYHHHHHHDYDIPTSENLYFQGGSGERKISRIHLVSEPSITHFLQVSWEKTLESGFVITLTDGHSAWTGT VSESEISQEADDMAMEKGKYVGELRKALLSGAGPADVYTFNFSKESCYFFFEKNLKDVSFRLGSFNLEKVEN PAEVIRELICYCLDTTAENQAKNEHHLRVVDSLQTSLDAETRSRNEALRVKKKMEGDLNEMEIQLSHANRMA AEAQKQVKSLQSLLKDTQIQL |
|
Xrcc4-MyH7-1631-1692 MSYYHHHHHHDYDIPTSENLYFQGGSGERKISRIHLVSEPSITHFLQVSWEKTLESGFVITLTDGHSAWTGT VSESEISQEADDMAMEKGKYVGELRKALLSGAGPADVYTFNFSKESCYFFFEKNLKDVSFRLGSFNLEKVEN PAEVIRELICYCLDTTAENQAKNEHANRMAAEAQKQVKSLQSLLKDTQIQLDDAVRANDDLKENIAIVERRN NLLQAELEELRAVVb |
The myosin segment is underlined.
The residues highlighted in bold were not observed in the crystal lattice though were present in the protein.
Protein expression and purification
Myosin fusion proteins were expressed in an E. coli BL21-CodonPlus (DE3)-RIL cell line (Stratagene). Cells were grown in lysogeny broth (LB) medium under an appropriate antibiotic selection at 37 °C with shaking to an A600 of ~1.0, then cooled on ice for 15 min; at this point, 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) was added and the cells were grown for an additional 16 h at 16 °C before harvesting by centrifugation. Cells were washed with 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.6, 50 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA) at a ratio of 1L of buffer per 6L of culture prior to flash freezing in liquid nitrogen and storage at −80 °C.
All protein purification steps were carried out at 4 °C. 10 g of cell were lysed in 100 mL of lysis buffer (50 mM HEPES pH 7.6, 50 mM NaCl, 20 mM imidazole, 0.5 mg/mL lysozyme) with 1 mM PMSF, 50 nM Leupeptin (Peptide International), 70 nM E-65 (Peptide International), 2 nM Aprotinin (ProSpec), and 2 μM AEBSF (Gold BioTechnology) by sonication. Lysate was clarified by centrifugation at 125,000 g for 30 min at 4 °C in a Ti-45 rotor. The supernatant was loaded onto a 5 mL Ni-NTA (nickel-nitrilotriacetic acid) column (QIAGEN) by gravity and washed with 25 column volumes of buffer A (25 mM HEPES pH 7.6, 300 mM NaCl, 20 mM imidazole). The column was then washed with an additional 5 column volumes buffer A with 40 mM imidazole. Protein was eluted in four column volumes of buffer A with 200 mM imidazole. 1 mM β-mercaptoethanol was added to all buffers for purification of constructs containing cysteine residues. The His-tag was removed by incubation with a 1:40 molar ratio of rTEV protease to myosin fusion protein at 4 °C in 25 mM HEPES pH 7.6, 100 mM NaCl, 0.1 mM EDTA, 0.5 mM tris(2-carboxyethyl)phosphine. The NaCl concentration was increased to 300 mM and the cleaved protein was then loaded onto a 2 mL Ni-NTA column equilibrated in buffer A without imidazole. Myosin constructs were eluted in four column volumes of buffer A, and rTEV protease was eluted with buffer A containing 200 mM imidazole. Fusion proteins were concentrated in an Amicon Ultra-15 30 KDa cutoff (Millipore) to between 10 and 25 mg/mL prior to overnight dialysis into storage buffer (10 mM HEPES pH 7.6, 100 mM NaCl). The protein was then flash-frozen in 30 μL droplets in liquid nitrogen and stored at −80 °C.
Crystallization
Crystals of Gp7-1526-1571 were grown at 4 °C by vapor diffusion from a 1:1 mixture of 15 mg/mL protein solution and a polyethylene glycol (PEG) solution consisting of 16% (w/v) PEG 8000, 400 mM malonate pH 7.2, and 100 mM triethanolamine pH 7.5. Rod-shaped crystals grew over the course of several days to final dimensions of 400 μm × 50 μm × 50 μm. All manipulations of crystals prior to freezing were carried out at 4 °C. Crystals were cryo-protected by first being transferred to synthetic mother liquor solution consisting of 18% (w/v) PEG 8000, 400 mM malonate pH 7.2, 100 mM triethanolamine pH 7.5, and 100 mM NaCl followed by stepwise transfer to a final solution of 18% (w/v) PEG 8000, 400 mM malonate pH 7.2, 100 mM triethanolamine pH 7.5, 100 mM NaCl, and 10% (w/v) ethylene glycol. Crystals were flash-frozen by plunging them into liquid nitrogen.
Crystals of reductively methylated38 Xrcc4-1562-1622 were grown at 4 °C by vapor diffusion from a 1:1 mixture of 13 mg/ mL protein solution and polyethylene glycol (PEG) solution consisting of 23% (w/v) PEG 4000, 500 mM NaCl, 100 mM triethanolamine pH 8.0. Shard-shaped crystals grew over the course of 7 days to an average dimension of 250 μm × 100 μm × 75 μm. All manipulations of the crystals were performed at 4 °C. Crystals were cryo-protected by first being transferred to synthetic mother liquor solution consisting of 23% (w/v) PEG 4000, 500 mM NaCl, 100 mM triethanolamine pH 8.0 followed by stepwise transfer to a final solution of 23% (w/v) PEG 4000, 500 mM NaCl, 100 mM triethanolamine pH 8.0, 12.5% (w/v) ethylene glycol, 250 mM CaCl2. Crystals were flash frozen by being rapidly plunged into liquid nitrogen.
Crystals of reductively methylated38 Xrcc4-1590-1657 were grown at 4 °C by vapor diffusion from a 1:1 mixture of 15 mg/ mL protein solution and methyl ether polyethylene glycol (MEPEG) solution consisting of 16% (w/v) MEPEG 2000, 250 mM KNO3, 100 mM MOPS pH 7.0. Hexagonal shaped crystals formed over the course of 7 days to an average dimension of 600 μm × 600 μm × 200 μm. Crystals were cryo-protected by transferring to an initial synthetic motherliqour solution consisting of 16% (w/v) MEPEG 2000, 200 mM KNO3, 100 mM MOPS pH 7.0 followed by a stepwise transfer into a final solution consisting of 20% (w/v) MEPEG 2000, 200 mM KNO3 and 12% (w/v) ethylene glycol. Crystals were flash frozen by being rapidly plunged into liquid nitrogen.
Crystals of Xrcc-1631-1692 were grown at 4 °C by vapor diffusion from a 1:1 mixture of 14 mg/ mL protein solution and MEPEG solution consisting of 14% (w/v) MEPEG 5000, 200 mM glycine, 100 mM bistrispropane pH 7.0. Crystals were cryoprotected by transferring to a cryoprotection solution of 20% (w/v) MEPEG 5000, 100 mM NaCl, 200 mM glycine, 100 mM bis-tris propane pH 7.0, followed by a stepwise transfer into a final solution consisting of 20% (w/v) MEPEG 5000, 200 mM glycine, 100 mM bis-tris propane pH 7.0, 12.5% (w/v) ethylene glycol, 200 mM CaCl2. Crystals were then flash frozen by plunging into liquid nitrogen.
Data collection and structure determination
X-ray diffraction data were collected at beam line SBC 19-ID (Advanced Photon Source). The datasets were integrated and scaled using HKL3000.39,40 X-ray data collection statistics are given in Table 2. The structures of Gp7-1526-1571, Xrcc4-1562-1622, Xrcc4-1590-1657, and Xrcc4-1631-1692 were solved by molecular replacement with Phaser41,42 using either residues 2 – 52 of Gp7 (1NO4) or residues 1 – 142 of Xrcc4 (1IK9) as search models.30,31 Following density modification by Parrot, initial models of Xrcc4-1562-1622, Xrcc4-1590-1657, and Xrcc4-1631-1692 were built in Buccaneer.43,44 Subsequent iterative cycles between manual model building in Coot followed by restrained refinement in Refmac 5.6 were used to generate the penultimate structural coordinates.45,46 Final refinements for Xrcc4-1590-1657 were performed by TLS and restrained refinement in Refmac 5.6. 42 Two chains were present in the asymmetric unit, and each chain was assigned as an individual TLS group for the entire chain. Final refinements on Xrcc4-1562-1622 and Xrcc4-1631-1692 structures were performed using Phenix Refine.47 For Xrcc4-1562-1622, four chains were present in the asymmetric unit, with chain A having 3 TLS groups, chain B having 2 TLS groups, chain C having 3 TLS groups, and chain D having 2 TLS groups. Xrcc4-1631-1692 utilized 2 TLS groups for each chain at the junction between the fusion protein and myosin, totaling 4 TLS groups.46 An initial model of Gp7-1526-1571 was constructed with Phenix AutoBuild and was refined with alternating rounds of manual model building in Coot followed by restrained refinement in Phenix.47 In the refinement two TLS groups per chain were defined by the junction between the fusion protein and myosin.48 The eight chains in the asymmetric unit were divided into a total of 16 TLS groups. Refinement statistics are presented in Table 2.
Table 2.
Crystallographic data collection and refinement statistics
| Data Collection | Gp7-1526-1571 | Xrcc4-1562-1622 | Xrcc4-1590-1657 | Xrcc4-1631-1692 |
|---|---|---|---|---|
| Space group | P1 | P21 | C2 | C2 |
| Cell dimensions | ||||
| a, b, c (Å) | 56.1, 64.5, 70.5 | 83.1, 57.3, 112.1 | 223.6, 84.0, 39.4 | 200, 44.4, 74.7 |
| α,β,γ (°) | 70.5, 77.5, 74.2 | β = 99.9 | β=92.8 | β =108.2 |
| Wavelength (Å) | 0.9791 | 0.9791 | 0.97924 | 0.9791 |
| Resolution (Å)a | 50-2.1 (2.14-2.10) | 50-3.1 (3.15 −3.1) | 50-2.1 (2.14-2.10) | 50-2.3 (2.31-2.27) |
| Rmergea,b | 0.065 (0.43) | 0.070 (0.37) | 0.090 (0.37) | 0.068 (0.33) |
| <I>/<σI>a | 23.1 (2.67) | 27.3 (4.9) | 34.1 (2.7) | 40.2 (3.45) |
| Completeness (%)a | 97.6 (96.3) | 99.1 (99.4) | 99.4 (98.1) | 99.9 (99.6) |
| Redundancya | 3.8 (2.3) | 6.0 (5.9) | 4.8 (3.1) | 6.3(4.9) |
| Beamline | 19-ID | 19-ID | 19-ID | 19-ID |
|
| ||||
| Refinement | ||||
|
| ||||
| Resolution (Å)a | 50 – 2.1 (2.13-2.09) | 50 –3.1(3.18-3.10) | 111.7-2.3 (2.36-2.3) | 50-2.3 (2.36-2.30) |
| No. reflectionsc | 67072 (3282) | 18959 (948) | 30554 (1645) | 28089 (1404) |
| Rwork / Rfreed | 0.21/0.26 | 0.24/0.28 | 0.21/0.26 | 0.22/0.24 |
| No. atoms | ||||
| Protein | 6360 | 6341 | 3392 | 3191 |
| Water | 164 | 2 | 54 | 108 |
| Ligand | 0 | 0 | 0 | 0 |
| Average B-factors (Å2) | 43.0 | 82.0 | 59.1 | 63.4 |
| R.m.s. deviations | ||||
| Bond lengths (Å) | 0.008 | 0.004 | 0.012 | 0.004 |
| Bond angles (°) | 0.935 | 0.741 | 1.414 | 0.780 |
| Ramachandran (%) | ||||
| Most favored | 99.7 | 98.5 | 98.8 | 98.7 |
| Allowed | 0.3 | 1.5 | 1.2 | 1.3 |
| Disallowed | 0 | 0 | 0 | 0 |
| TLS Groups | 16 | 10 | 2 | 4 |
| PDB Accession Number | 5CJ1 | 5CJ4 | 5CHX | 5CJ0 |
Data in parentheses represent the highest resolution shell.
Data in parentheses represent the number of reflections used for the calculation of Rfree.
Rfactor = ∑ ∣ Fobs-Fcalc ∣ / ∑ ∣ Fobs ∣
Where Rwork refers to the Rfactor for the data used in the refinement and Rfree refers to the Rfactor for 5% of the data that were excluded from the refinement.
Reconstruction of myosin from structural fragments
The initial structure for the simulations was created in the MODELLER Homology Modeling Package49-53 starting from the aligned structures for Gp7-1526-1571, Xrcc4-1562-1622, Xrcc4-1590-1657, Xrcc4-1631-1692 and Xrcc4-1551-1609. After alignment, overlapping segments of the structures were trimmed and the orientation of the side chains were energy minimized and the conformation of the trimmed chain ends were optimized to obtain a complete segment consisting of 164 amino acids (MyH7-1526-1689) of the myosin rod surrounding the Skip 3 residue.
All molecular dynamics simulations were carried out using the AMBER v14 MD program.54-57 The Generalized Born (GB), an implicit solvent approach was chosen because it provides ~100 fold efficiency compared to the explicit solvent when graphical processing units are used,58-60 and long simulations are required to allow adequate structural relaxations of models constructed based on crystal structures. Specifically, the ff99SB force field improved with NMR observables (ff99SBnmr) was chosen61-66 along with the gb7 model.58 Production gb7 simulations were carried out for a minimum of 500 ns using a 1 fs time step (2 fs was used for the independent runs). Details of the simulations are summarized in Table 3. Langevin dynamics was followed with a collision frequency of 20 ps−1 at 300 K. The SHAKE algorithm was applied to bonds with hydrogen atoms with a tolerance of 10−5 Å.67 The non-bonded cutoff was set as 9999 Å, and the maximum distance between atom pairs (rgbmax) for Born radii calculations was kept at 12 Å. Salt was treated implicitly via Debye-Hückel theory and the concentration was set to the physiological concentration of 0.15 M. The collected trajectories were analyzed with AmberTools v15 and the Multiscale Modeling Tools for Structural Biology (MMTSB) package.57,68,69
Table 3.
Simulation summary
| Model | Trajectory Length (ns) |
|---|---|
| MyH7-1551-1602a | 1000 |
| Gp7-MyH7-1551-1602 | 1000 |
| Xrcc4-MyH7-1551-1603 | 1400 |
| MyH7-1526-1689 | 1100 |
| MyH7-1526-1689 | 520 |
| MyH7-1526-1689 | 540 |
Residues 1551-1602 or 1551-1603 were used in the simulation since these constituted the structurally ordered residues in the crystal structure for Xrcc4-1551-1609 (Accession number 4XA4).
To investigate the effect of the head groups, the isolated Skip 3 region, residues 1551-1603 from the MyH7-1551-1609 structure, was simulated along with fusions with Gp7 and Xrcc4 (Table 3).
In order to examine the convergence of the simulation of the contiguous model, three independent simulations were carried out for a minimum of 520 ns each. The analysis was performed over the combined trajectory of the three simulations where all averages were calculated for the entire pool of conformations. To determine if the simulations had converged the distribution of the clusters among the three individual trajectories was plotted after performing clustering using the k-clust algorithm in the MMTSB package.70 (Figure S1). It was observed that each cluster was sampled in every simulation. Additionally, the averaged DCOM and super helical pitch for the individual simulations (Figures S2 and S3) were in qualitative and quantitative agreement with the averages for the aggregate simulations.
CD and fluorescence spectroscopy
Circular dichroism spectra were recorded from 190 to 300 nm in an AVIV model 420 CD spectrophotometer. Protein samples were diluted to an A280 of 0.35 in buffer containing 50 mM sodium phosphate pH 7.0, 0.5 mM tris(2-carboxyethyl)phosphine. Thermal melting curves were recorded by monitoring the change in tryptophan emission intensity in a QuantaMaster Model C-60/2000 spectrofluorometer. Fluorescence emissions were recorded from 300 to 500 nm. The excitation wavelength was 290 nm. The temperature was increased by 4 degree increments starting at 4 °C. The sample was equilibrated at each temperature point for 10 minutes before measurement.
Results and Discussion
Fusion proteins do not significantly influence the backbone of the target coiled-coil
Four crystal structures of human cardiac β-myosin LMM are reported here that in combination with the previously determined structure for Skip 3 (Xrcc4-1551-1609 containing ~8 heptads of myosin)26 encompass residues 1526-1688 (Figure 1). In total this section extends over repeat 25 to the final heptad of repeat 30. These new fragments are defined as Gp7-1526-1571, Xrcc4-1562-1622, Xrcc4-1590-1657, and Xrcc4-1631-1692 that contain ~7, 9, 10, and 8 heptads of coiled-coil respectively. The four new structures overlap with Xrcc4-1551-1609 on the N- or C-terminal side for a total coverage of LMM of 164 residues (~23 heptads). These structures are N-terminal fusions with either Gp7 or Xrcc4 and were designed such that the overlapping regions of LMM coiled-coil could be used to assemble a larger composite structure. The junctions between the folding domain and myosin were designed to preserve the coiled-coil registry and minimize disruption of inter-chain salt bridges as has been previously described.27
Figure 1.

Structures of myosin fusion proteins and preliminary composite model. (A) A representation of myosin in which each 28 amino acid repeat of the C-terminal coiled-coil is an oval. S1, S2, and LMM are colored white, yellow and grey respectively. The numbering is shown for every fifth repeat and the positions of the skip residues are indicated. Repeats 25 through 30 are colored differently and the third skip residue, E1582, is shown in red. The fusion proteins are colored in grey while the myosin repeats are colored as in panel A for (B) Gp7-L1526-E1571, (C) Xrcc4-L1551-N1609, (D) Xrcc4-Q1562-L1622. (E) Xrcc4-H1590-L1657, (F) Xrcc4-A1632-R1689 and (G) A simple composite model for L1526-R1689 of human cardiac β-myosin. This was assembled from four of the five overlapping structures taken from Gp7-L1526-E1571, Xrcc4-L1551-N1609, Xrcc4-H1590-L1657, and Xrcc4-A1632-R1689. The residues incorporated from each structure are listed below. The coordinates for Xrcc4-L1551-N1609 were taken from the RCSB with accession 4XA4. Figures 1, 2, 3, 4, 5, 8, 9, and 10 were prepared in part with Pymol (http://www.pymol.org/).
As noted earlier, isolated fragments of LMM exhibit a strong dependence salt dependence on solubility and tendency to form paracrystals which confound the ability to crystallize these proteins for high resolution structural studies. This problem was solved by including a globular domain excised from either Xrcc4 or Gp7. These proteins were selected from the protein data bank as small but structurally distinct domains that lead into well ordered dimeric coiled-coils. The intact Xrcc4 is involved in DNA repair and binds to both DNA ligase IV (RCSB accession number 1IK9)30 and DNA. Here, only the globular domain of the protein that consists of ~140 residues and is composed mostly of β-strands was used, but is referred to as Xrcc4 for simplicity in this context. Likewise, the bacteriophage φ29 scaffolding protein Gp7 consists of a small helical bundle domain that leads into an extended coiled-coil (RCSB accession number 1NO4)31 where only the first ~50 residues that contain the globular domain were incorporated in these fusions. These are denoted as Gp7-fusions for simplicity also.
Experimentally it was determined that the fusion proteins that exhibited the best characteristics for structural and biophysical studies contained ~8-10 heptads of LMM. Longer fusions did not crystallize well and demonstrated increasing salt-dependent solubility. On average, six constructs were prepared for every successful structural determination.
A major question in assembling structures from fragments derived from fusion proteins is whether the conformation of the resultant pieces is influenced by crystal packing forces or by the interface between the folding domain and the fragment of myosin. In the case of Xrcc4-1562-1622 there are two dimers in the asymmetric unit that have a Cα root mean square deviation of 0.63 Å2 over 89 α-carbon atoms of the target myosin coiled-coil. This indicates that crystallographic packing does not have a major influence on the resultant structure in this instance. In the same vein, the two fusions to Xrcc4 (Figure 1C and 1D) both overlap between residues 1562-1609 of myosin. This significant overlap is more than the minimum required for assembly into a larger model, but serves as a means to probe the influences of the fusion protein on the target section of coiled-coil. Each of the two Xrcc4-1562-1622 dimers aligns to Xrcc4-1551-1609 with a Cα root mean square deviation of 1.0 Å2 and 1.2 Å2 over 76 and 80 α-carbon atoms respectively (Figure 2). This strongly supports the hypothesis that the backbone conformations of the selected fragment of the coiled-coil are not notably perturbed by crystallographic contacts or the fusion to a folding domain.
Figure 2.

Stereo view of a structural alignment between Xrcc4-1551-1609 and Xrcc4-1562-1622. For Xrcc4-1551-1609 the Xrcc4 portion is colored in black and 1551-1609 of myosin is in blue. For Xrcc4-1562-1622 the Xrcc4 portion is colored in grey and 1562-1622 of myosin is in green. The structures are represented in cartoon and the skip residue is shown in red spheres. The primary point of divergence of the structures in the target coiled-coil is indicated with an asterisk.
To further test this hypothesis, MD simulations were performed on the isolated section of MyH7-1551-1602 and fused to either Gp7 or Xrcc4 (Table 3). Structures for the MyH7-1551-1602 segment were extracted from all three simulations to form a pool of conformations. Hierarchical clustering was performed over the ensemble of conformations for MyH7-1551-1603 using the k-clust algorithm in the MMTSB package.70 Using a Cα root mean square distance (RMSD) of 4.75 Å as the clustering threshold, four clusters were observed. Figure 3A displays distribution of clusters amongst the simulations along with Cα-RMSD with respect to the Xrcc4-1551-1609 crystal structure. The folding domains have a slight stabilizing effect on the conformational ensembles. This is manifested in the average Cα-RMSD. The individual MyH7-1551-1602 simulation has an average Cα-RMSD of 3.8 Å as opposed to 2.4 Å and 3.3 Å from Gp7- and Xrcc4-bound forms. Additionally, the individual MyH7-1551-1602 simulation samples a slightly larger range of conformations; this simulation contains two extra clusters that are not found in the simulation of the Gp7- and Xrcc4-bound constructs. However, those clusters are within 2.7 Å and 2.0 Å Cα-RMSD with respect to the crystal structure (possibly due to the fluctuations at the N-terminal) and have low populations. Cα root mean square fluctuations (Cα-RMSF) exhibit similar trends (Figure 3B). Thus, it can be concluded that the folding domain has a modest stabilizing effect (as supported by the biophysical measurements discussed below), and that the two most dominant conformational ensembles of the coiled-coils are not disturbed.
Figure 3.

Analysis of the molecular dynamics simulations for Skip 3 in the absence and presence of a fusion partner. (A) Clustering of the Skip 3 region (MyH7-1551-1602) conformations extracted from Skip 3 alone, Gp7-Skip 3 and Xrcc4-Skip 3 simulations, is presented in three individual panels along with the Cα-RMSD with respect to the Xrcc4-1551-1609 crystal structure. Hierarchical clustering was carried out with the k-clust protocol in MMTSB using a Cα-RMSD of 4.75 Å as the similarity measure. Different colors represent different clusters. (B) Root Mean Square Fluctuations (RMSF) based on the Cα-atoms (Black- isolated Skip 3 simulation, Red: Gp7-Skip 3, Turquoise: Xrcc4-Skip 3). (C) DCOM trend for Skip 3 (Black), Gp7-Skip 3 (red) and Xrcc4-Skip 3 (turquoise) simulations. (D) Representative members from each cluster are shown along with their overall population percentages and Cα-RMSD with respect to the Xrcc4-1551-1609 crystal structure.
The conformations sampled in all three simulations are well within comparable RMSD and RMSF values, however this does not necessarily quantify the changes in the degree of coiling. To explicitly quantify the degree of coiling, the distance between center of masses of the two α-helices (DCOM) was calculated as a moving average of seven consecutive α-carbon atoms (Figure 3C). All three simulations of the Skip 3 region show similar DCOM trends, except for a single heptad repeat at both the C- and N-termini. Hence, the MD simulations support the conjecture that the folding domains and C-terminal truncations do not alter the coiling patterns or the super-helical pitch except for the first and last heptad of the target section of coiled-coil.
Differences in the super-helical pitch at the fusion junction do not propagate into the target coiled-coil
Xrcc4-1562-1622 and Xrcc4-1551-1609 encompass a stretch of LMM that contains the third skip residue, E1582, located between repeat 26 and 27 that breaks the phase of the coiled-coil heptad repeat. The skip residue causes a significant local increase in the super-helical pitch encompassing 17 and 11 residues N- and C-terminal to the insertion. This distorted region of coiled-coil clearly starts at F1565, which places a bulky hydrophobic residue in a core d position (Figures 2 and 4). The two Xrcc4 constructs described herein both contain this residue, but in different positions relative to the N-terminal fusion interface. The junction between Xrcc4 and myosin in Xrcc4-1551-1609 is located two full heptads N-terminal to F1565. In contrast, the junction in Xrcc4-1562-1622 is only three residues N-terminal to F1565, or about one turn of an α-helix. As a consequence, the fusion junctions of these two constructs occur at different positions in the coiled-coil registry and include different Xrcc4 residues. The first myosin residue in Xrcc4-1551-1609 is a d position occupied by a leucine, while the first myosin residue in Xrcc4-1562-1622 is a glutamine in an e position (Figure 4).
Figure 4.

Stereo representation of the coiled coil centered on F1565 in the Xrcc4-1551-1609 and Xrcc4-1562-1622 fusion proteins. Xrcc4-1551-1609 is represented with blue cartoon helices and white stick side chains. Xrcc4-1562-1622 is represented with green cartoon helices and dark grey side chains. Only the side chains of residues along the interface are displayed. Residues from the Xrcc4 folding domain are not shown.
The major difference between these two structures occurs in the region surrounding F1565 near the fusion junction in Xrcc4-1562-1622, as indicated by an asterisk in Figures 2 and 4. For approximately two turns of the α-helix, just C-terminal to the folding domain interface, the coiled-coil is more tightly wound in Xrcc4-1562-1622 as compared to that region of myosin in Xrcc4-1551-1609. The coiled-coil of Xrcc4, measured from residues 118 to 153, has a super-helical pitch of about 160 Å, while the skip region of myosin’s super-helical pitch is measured at about 900 Å as calculated by the Crick coiled-coil parameterization (CCCP) server.71 It is unlikely that the structural difference is the result of crystallographic packing, given that conformational variability between crystal structures is typically observed in loops and surface residues, and not in packing residues such as those in the coiled-coil.72 Rather, this distortion appears to be the result of a mismatch of super-helical pitch between Xrcc4 and myosin. Except for the first five residues adjacent to the fusion protein the target myosin coiled-coils are highly similar. Fundamentally this implies that the structural influence of the folding domain does not propagate significantly beyond the first heptad of the target coiled-coil.
Xrcc4-1551-1609 is the highest resolution structure for that section of myosin coiled-coil and hence used in the construction of the composite model below (Figure 1C and F). The junction between the Xrcc4 folding domain and the target myosin coiled has a smooth transition, where the coiled-coil parameters are well matched. Both Xrcc4-1551-1609 and Xrcc4-1562-1622 exhibit stable coiled-coil C-termini with no significant differences over regions of overlap.
Regions of stable coiled-coil interface should be selected for C-terminal truncations
The most N-terminal segment described in this study, Gp7-1526-1571, contains myosin residues spanning repeat 25 and half of repeat 26 and also includes F1565. Unlike in the previous two Xrcc4 constructs, F1565 is located six residues from the C-terminus of the coiled-coil. Interestingly, the last two heptads of this fusion protein splay apart forming an antiparallel four-helix bundle between crystallographically related dimers (Figure 5). Given the redundancy of structural information for this section of the coiled-coil it is safe to assume that this arrangement is an artifact of crystal packing and was excluded from consideration in the composite model. This tetramerization probably arises because of a weak dimerization interface near the C-terminus of the coiled-coil that is readily counterbalanced by crystallographic packing forces (Figure 5). Interestingly, the coiled-coil prediction for this region is less than 100% in the region leading up to F156573 which suggests that a minimum of one heptad of canonical coiled-coil should be included at the C-terminus to avoid formation of antiparallel four helix bundles. The observation that truncated fragments of coiled-coils can come apart at their ends raises the question to what extent the folding domains stabilize these segments of coiled-coil. This was addressed experimentally by determining the folding and unfolding characteristics of a short section of myosin fused to three different folding domains. The same question was also examined earlier in the MD studies.
Figure 5.

Stereo diagram showing the antiparallel four helix bundle formed by Gp7-1526-1571. One dimer is colored in green while the symmetry related dimer is colored in white. The clustering of F1565 in all four chains is critical to the formation of the antiparallel helix bundle. The C-termini of the polypeptide chains are indicated.
Fusion proteins modulated the level of expression and increased the total α-helical content of the chimera
Fusions were constructed between the short coiled-coil of GCN4, Gp7, or Xrcc4 and a region of human cardiac myosin, that encompasses the second skip residue (MyH7 residues 1361 to 1406). The fusions were compared to a construct that did not include any folding domain. A mutant form of the GCN4 leucine zipper that was designed to have greater coiled-coil stability (GCN4-pMSE hereafter abbreviated to GCN4) was used as a comparison to two other folding domains.74 The particular region of myosin was selected because it contains a tryptophan residue enabling fluorescence-based measurements. All MyH7 fusions were purified by Ni-NTA chromatography under identical conditions and the amount of soluble protein was compared using relative band intensity on a Coomassie-stained SDS-PAGE gel. All of the constructs yielded a substantial quantity of soluble protein, where the Xrcc4 fusion yielded the greatest mass of soluble material (Figure 6). Interestingly, although the native coiled-coil protein was soluble without a folding domain, the expression levels do not convey the dramatic differences in biophysical behavior exhibited by these constructs. While MyH7-1361-1406 appeared folded, as indicated by a single sharp peak in gel filtration (data not shown), it had low α-helical content as measured by circular dichroism. The mean residue molar ellipticity was -12733 deg.cm2.dmol−1 at 222 nm which was much lower than a value of ~33,000 expected for a protein that is predicted to be entirely α-helical.75 This is consistent with the observation by Wolny et al., that many isolated short sections of the myosin rod exhibit low α-helical content.23 The addition of the folding domain greatly increases the mean residue molar ellipticity, as listed in Table 4, indicating that the stability conferred by the folding domain propagates into the target coiled-coil.
Figure 6.
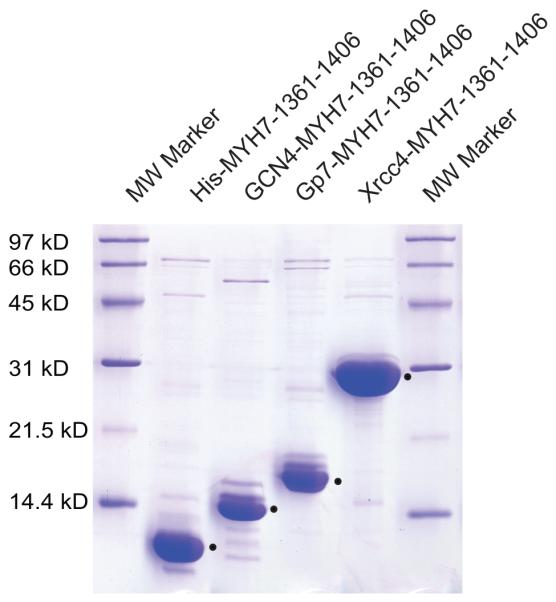
15% SDS-PAGE gel demonstrating soluble expression of MyH7 constructs. (A) All lanes contain 1.25 μL of a Ni-NTA purified MyH7 construct (equivalent of 0.625 μg of cells). Bands corresponding to MyH7 are marked with a dot to the right of the band. All MyH7 constructs show soluble overexpression, including fusion-less MyH7, and only minimal changes in soluble expression are noted with different fusions. Xrcc4-MyH7 shows the highest soluble expression level.
Table 4.
Circular Dichroism Theta Values for myosin fusion proteins
| Construct | Θ (deg cm2 dmol−1) at 222 nm |
|---|---|
| MyH7-1361-1406 | −12,733 |
| Gp7-MyH7-1361-1406 | −23,247 |
| GCN4-MyH7-1361-1406 | −27,766 |
| Xrcc4-MyH7-1361-1406 | −14,873 |
Fusion proteins enhance thermal stability
All fusions greatly enhanced the thermal stability of the target coiled-coil as shown by tryptophan autofluorescence (Figure 7). The control construct lacking an N-terminal fusion had an emission maximum of 355 nm indicating the tryptophan residue is solvent exposed and did not undergo a cooperative unfolding transition in temperature scanning measurements. The emission maximum of the fusion proteins were blue shifted to 342 nm (Gp7) and 340 (GCN4) due to increased shielding from solvent. GCN4 conferred the greatest enhancement of thermal stability. The analysis of Xrcc4 constructs, based on autofluorescence, is complicated by multiple tryptophan moieties and was therefore not included. The CD (Figure 7) and autofluorescence data taken together demonstrate that the fusions increase the total α-helical content and thermal stability of the target coiled-coil suggesting that these constructs are more suitable for biophysical analysis. This was reflected in the crystallization properties of these proteins.
Figure 7.

(A) Circular dichroism spectra of MYH7-1361-1406 (black) and Gp7 (Red), GCN4 (blue), Xrcc4 (black dashed line) fusions to MYH7-1361-1406. (B) Temperature scanning autofluorescence emission maximum for each construct is plotted versus temperature. Gp7 (Red ∎) and GCN4 (Blue ●) fusions to MYH7-1361-1406 show temperature dependent transition. MYH7-1361-1406 (Black ●) without fusion is shown in the inset and does not display a cooperative temperature depend change in tryptophan fluorescence. A line connecting the measured data has been added to each trace as a guide.
The fusion protein facilitates crystallization
Constructs were evaluated for crystallization in a 144 condition sparse matrix screen at 4°C and 25°C that samples a range of polyethylene glycol, hexylene glycol, and salt, based conditions. The screen was prepared in-house. No crystals have been observed for MyH7-1361-1406 alone, while the Xrcc4, GCN4, and Gp7 fusions readily crystallized, but even here among the fusions there were significant differences. GCN4 yielded visually stunning crystals, but these exhibited highly anisotropic diffraction. Conversely, the inclusion of a structurally more complex motif such as Gp7 yielded more ordered crystals that were readily amenable to structural analysis. In general, Gp7 fusions crystallize under a wider range of conditions than Xrcc4 fusions. The increased thermal stability of the fusion relative to the target coiled-coil coupled with a tendency for crystallization provide strong support for the use of these folding domains in the context of myosin.
Assembly of structural fragments into a contiguous model
There is considerable structural overlap between the five structures shown in Figure 1. However, a model built by simple alignment represents a crude approximation considering that overlapping regions exhibit slightly different conformations (Cα root mean square deviation of < 1 Å). Although previous studies that focus on modeling canonical coiled-coils have been reported, modeling the myosin rod based on those studies is unrealistic due to the presence the skip residues that lead to deviations from an ideal coiled-coil76. Our studies of the skip residues have shown that the deviation from an ideal coiled-coils surrounding a skip residue extends over ~four heptads where the distortions is dependent on the surrounding amino acid sequence.26 Due to the irregularities in the structure a model based on a typical canonical coiled-coil is incapable of defining the structure of the myosin rod76. Consequently a more systematic approach described here was developed to assemble a contiguous structure from the individual structures with the homology modeling program MODELLER.52
The initial model from this procedure was simulated through molecular dynamics to remove steric overlaps and allow adequate structural relaxations through three separate simulations. The trajectory length for each simulation is listed in Table 3. MD simulations of the composite model yielded a conformational ensemble rather than a single conformation and thus provide information on the flexibility of the molecule. This was derived from the sum of three independent trajectories (summing up to 2.2 μs).
Hierarchical clustering was performed on the ensemble of structures generated from the simulation to distinguish major conformers (Figure 8). The RMSD cut off for clustering was selected after trying several values varying from 6 Å to 10 Å. Within that range, 8 Å was found to yield the most robust clustering where this best reflected the diversity of the conformations sampled by the simulations. Six clusters were formed when the ensembles were sorted with this Cα–RMSD threshold (Figure 8G). The representative structure of a given cluster was taken as that which had the lowest overall Cα–RMSD to the other members of the cluster. A comparison of the six representative structures, one from each cluster, revealed that the major difference between the ensembles is the degree of supercoiling. At one extreme the Cα–RMSD increases to 10.7 Å between the 4th cluster (shown yellow) relative to the initial model, where the structure is less tightly wound and more flexuous than the initial model. The slight bend towards C-terminal is expected since the structure becomes more flexuous due to length. This particular cluster was only sampled in 5.5% of the overall population.
Figure 8.

Molecular dynamics analysis of the MyH7-1526-1689 segment of the myosin rod. (A) Cα-RMSD with respect to the initial model that was assembled directly from the crystal structures. Color coding represent different clusters obtained through hierarchical clustering using the k-clust protocol in MMTSB. The Cα-RMSD cut off for clustering was set to 8 Å which gives the most robust clusters. (B) Root Mean Square Fluctuations (RMSF) based on the Cα-atoms. (C) Estimated super-helical pitch (Å) trend over each heptad-repeat (Skip 3 residue: E1582) based on a single heptad (black) and two heptad repeats (red). The values shown are calculated through averaging over conformations from all 3 trajectories. (D) The length of each heptad repeat calculated as a moving average to reveal local fluctuations more closely (E) Coiled-coil propensities are calculated with a 28 residue sliding window using COILS (red) and MARCOIL (black) servers using only the sequence information (F) DCOM for the composite model calculated through averaging over conformations from all 3 trajectories. (G) Representative members from each cluster are shown along with their overall population percentages and Cα-RMSD with respect to the initial model for MyH7-1526-1689.
The simulated composite model for the MyH7-1526-1689 section of the myosin rod was also compared to the simulation of MyH7-1551-1602 extracted from the crystal structure of Xrcc4-1551-1609. This analysis probed how the increased length of the composite model alters the structures relative to simulations of smaller sections. It is important to emphasize that the conformational space visited in each simulation does not change between the simulations of the smaller section and the composite model. Clusters obtained from the composite model simulation remain within 2.1 Å Cα–RMSD of the crystal structure of Xrcc4-1551-1609. The composite model is expected to be flexuous due to its length. MyH7-1526-1689 exhibits higher Cα-RMSF than the MyH7-1551-1603 segment (Figure 8), which is typically in the range of 3-4 Å for the composite model and ~2 Å for Xrcc4-1551-1602 simulation. The highest degree of RMSF is observed between residues 1582-1640, following the skip residue. This prediction from the simulations is consistent with the observed structure for Xrcc4-1590-1657 which shows that the α-helices separate between residues 1608-1615 consistent with a lower predicted coiled-coil propensity.
Since the clustering results pointed to a difference in the degree of coiling among the representative structures, the super-helical pitch of the myosin rod was calculated over the full simulation. Super helical pitch was calculated based on a sliding window of 7 (black) or 14 (red) amino acids. As expected, there is an increased super-helical pitch around the skip residue, extending 20 amino acids upstream from the skip location where the computed super-helical pitch averages over 1300 Å as compared to the value of 170 Å for other regions (Figure 8C) for both approximations (single heptad vs 2-heptads). Both MyH7-1551-1602 and MyH7-1526-1689 follow the same qualitative trend (data not shown). These data taken together strongly support the feasibility of assembling a complete model for LMM and illustrate the difficulties associated with building a model based on a canonical coiled-coil.
The average length of each heptad repeat, or local pitch, was calculated over the trajectory (Figure 8D), to further emphasize the local disruption due to the skip residue. While the local pitch averages to 10 Å for most of the coiled-coil, it rises up to 12.5 Å around the skip residue. DCOM (See Methods for details) also shows increased distance between the centers of masses of the two helices around the skip residue and provides further evidence that the thick filament is less tightly wound around the skip residue.
Prediction software, such as that provided by servers such as COILS77 and MARCOIL,78 are commonly used to provide an estimate of how well a sequence matches a canonical coiled-coil. While these are outstanding at predicting potential coiled-coils they do not provide direct evidence of the structure adopted by a polypeptide chain in those regions where the sequence deviates from a canonical heptad pattern. There is also considerable discrepancy between predictions for those segments that do not exhibit a standard pattern. As shown in Figure 8E, the predictions from COILS and MARCOIL are not only inconsistent with each other but do not predict the observed structural features seen in our structures and simulations. MARCOIL predicts that the entire region should be coiled-coil with over 99% propensity whereas COILS predicts low coiled-coil propensity around residues 1558 and 1590-1613 region. This comparison demonstrates that the thick filament cannot be modeled using sequence information alone or built on a canonical coiled-coil; the presence of the skip residues disrupts the expected pattern such that structural information is required to accurately model these regions.
Finally, the interactions within the coiled-coil in the simulated composite model were examined. The electrostatic interactions were analyzed by calculating the average distances between the center of mass of the nitrogen atoms of arginine (NH1, NH2) and lysine (NZ) and the center of mass of oxygen atoms of glutamate (OE1, OE2) and aspartate (OD1, OD2). The distances are averaged over an ensemble of conformations sampled by three independent simulations rather than a static model. Residues with atoms that lie within 4.5 Å distance were selected as interacting pairs. Only a few electrostatic interactions were predicted to be occur between the two helices; those pairs are E1536-K1537, K1579-D1580 and R1604-E1608 (Figure 9A and Table S1). It is important to note that the charged interactions predicted do not follow the well known e-g’ interactions observed in typical parallel coiled-coils, where e type residue on one chain interacts with the preceding g’ type residue from the other chain.79 None of the pairs identified in this study match the e-g’ category. There are however a few ionic interactions between residues in the same chain, notably between the 1602D-1606R and 1615K-1619E pairs, that might contribute to the stability of the assembly (Figure 9B). No repulsive interactions were observed in this 164 amino acid segment of the myosin rod in the simulations.
Figure 9.

Analysis of the charged interactions and exposed hydrophobic surface of the composite model for MyH7-1526-1689. (A) Charged interactions between two helices evaluated by the average distances between center of mass of nitrogen atoms (NH1, NH2) of Arginine, nitrogen atom (NZ) of Lysine and center of mass of oxygen atoms from Glutamate (OE1, OE2) and Aspartate (OD1, OD2). The pairs of residues within 4.5 Å distance are shown (Table S1). The Skip 3 residue is shown in orange. Positively charged amino acids are displayed in blue whereas negatively charged amino acids are displayed in red. (B) Similarly, charged interactions within each chain are shown in sphere representation. (C) Solvent accessible surface area (SASA) (Å2) of the hydrophobic amino acid residues in the composite model (Table S2). (D) Side and top view of MyH7-1526-1689 are displayed with the predicted hydrophobic interactions. The hydrophobic interactions within the rod were evaluated from the average minimum distances between the hydrophobic side-chains. The pairs of residues that are closer than 5 Å are shown (Table S3). To distinguish residues from different helices, the predicted residues are colored in yellow and green for different helices. (E) Hydrophobic residues that have a SASA over 90 Å2 are shown on the composite model in sphere representation.
The stability of the coiled-coil is expected to be driven by the hydrophobic effect. The hydrophobic interactions between two helices of the coiled-coil were evaluated from the average distances between the center of masses of each hydrophobic side-chain (Figure 9C and Table S2 for a full list of interactions identified). The distances were averaged over the ensemble of conformations from three independent trajectories. All hydrophobic pairs whose center of mass of the side chains fall within 5 Å were selected and shown in orange Figure 9D, where these interactions are strictly in the interface between the two α-helices. Almost all interactions can be classified as contacts between a–d’ or a–g’ residues.
Unlike most typical soluble globular protein structures, the myosin coiled-coil exhibits a large number of hydrophobic side chains on the surface. There are numerous solvent exposed hydrophobic residues as indicated by the solvent accessible surface area (SASA, Å2) for each hydrophobic side chain (Figure 9A) including, L1526, M1538, L1559, L1563, L1591, L1612, I1627, M1635, L1649, I1655, I1673, and L1680. These all have SASA values over 90 Å2 (Figure 9E and Table S3). The composite model contains 132 hydrophobic residues, of which 24 are almost completely solvent exposed. This solvent accessibility is by far higher than it is expected for hydrophobic residues in α-helices; usually hydrophobic amino acids are only 1% to 5% accessible, except for Trp and Tyr which are 10% accessible.80 We hypothesize that these residues are important for assembly of the thick filament beyond the simple ionic interactions81 and will be important in future efforts to construction higher ordered structures. Interestingly, in most instances the hydrophobic character of these positions in the myosin rod is highly conserved across sarcomeric myosins.
Distribution of cardiomyopathy mutations
More than eighteen mutations that lead to cardio or skeletal myopathies have been identified in the segment of myosin modeled here.3,82 The location of these residues is shown in Figure 10, which reveals a broad distribution of sites. Of these, in vivo and in vitro measurements have only been reported for the effect of the E1555K substitution.23 These show reduced incorporation of the mutant protein into the sarcomere and decreased helicity in the coiled-coil which, together with studies of mutations outside the segment investigated here, suggest that mutations that reduce the helix stability lead to lower incorporation into the sarcomere. As shown on the model, E1555 is a surface residue that does not appear to be involved in any inter or intramolecular ionic interactions in the wild-type structure. Thus, the loss of stability cannot be attributed to the loss of a stabilizing interaction, but rather must be due to gain of a destabilizing contact within the coiled-coil. The reduced incorporation into the thick filament could be due to either defects in interactions between myosin rods or by reduced stability of the protein itself. Resolution of this question can now be approached through simulation of the mutant structure and through structural determination of segments that carry mutations.
Figure 10.

Disposition of mutations that lead to cardio or skeletal myopathies in MyH71526-1689. The mutations themselves are widely distributed along the length of the coiled-coil and occur at all heptad positions.
It is noteworthy that there are a considerable number of mutations to proline that lead to skeletal myopathies.82,83 In vitro studies of two mutations outside the region studied here (R1500P and L1706P) suggest that prolines can be introduced into the myosin rod and yet still lead to protein that is incorporated into thick filaments even though the assembly appears is less stable83. It is expected that introduction of a proline into a helical backbone will lead to loss of hydrogen bonding opportunities. However, proline residues are occasionally found in α-helices where they lead to a pronounced bend in their path. For example, there is a proline found in α-helix 2 of thioredoxin from E. coli (PDB accession code 2TRX) that leads to a prominent kink in the helix that alters the hydrogen bonding pattern surrounding the proline. This results in an increased rise per residue and loss of an additional hydrogen bond beyond that caused by the proline itself.84 There are at least eight examples of proline residues in dimeric or trimeric coiled-coils85 where these also lead to over-winding of the alpha helices.85 Thus, it is difficult to predict whether proline mutations in myosin lead to alterations in the stability of the coiled-coil itself or to changes in the manner the myosin rods interact with each other in the thick filament. Here again, the computational and structural approach outlined here is well suited to investigating the biophysical consequences of these mutations.
Conclusions
This study establishes a methodology for determining the complete structure of the myosin rod. It shows that the use of fusion proteins allows restricted segments of the myosin rod to be expressed and purified which allows structural and biophysical characterization. It also demonstrates that the fusion domains themselves introduce only a small perturbation in the structure that is readily eliminated by structural determination of overlapping fragments. The study also shows that the fragments can be assembled into composite models through molecular dynamics simulations. The five fragments studied here contained a total of 292 residues of myosin which yielded a contiguous model of 164 residue fragment. This suggests that a complete model for LMM can be obtained by implementing a strategy that includes duplicate structures for every residue in the coiled-coil. The results thus far show a surprising number of exposed hydrophobic side chains which suggests they may play a role in the assembly of myosin rods into the thick filament. The methodology established here will facilitate construction of a model for the entire myosin rod, and lays the ground work for assembling the rods into a model for the thick filament. This study also creates a framework for understanding the biophysical consequences of mutations in myosin that lead to cardio and skeletal myopathies. These studies are in progress.
Supplementary Material
Acknowledgments
We also thank Dr. Darrell McCaslin and Dr. Ben Knowles for helpful discussions related to the biophysical characterization of coiled-coils. This work was supported by NIH grant R21 HL111237 to IR and QC. The computational work was also partially supported by NSF-CHE1300209 to QC. Use of the Structural Biology ID19 and BM19 beamlines, Argonne National Laboratory Advanced Photon Source was supported by the U.S. Department of Energy, Office of Energy Research, under Contract No. W-31-109-ENG-38.
Footnotes
Conflict of Interest Statement
The authors state that there are no conflicts of interest associated with this manuscript or study.
Accession codes.
Atomic coordinates have been deposited in the Protein Data Bank under accession codes 5CJ1, 5CJ4, 5CHX, and 5CJ0 corresponding to Gp7-1525-1571, Xrcc4-1562-1622, Xrcc4-1590-1657, and Xrcc4-1629-1692 respectively.
References
- 1.Lowey S, Slayter HS, Weeds AG, Baker H. Substructure of the myosin molecule. I. Subfragments of myosin by enzymic degradation. J Mol Biol. 1969;42(1):1–29. doi: 10.1016/0022-2836(69)90483-5. [DOI] [PubMed] [Google Scholar]
- 2.Oldfors A. Hereditary myosin myopathies. Neuromuscul Disord. 2007;17(5):355–367. doi: 10.1016/j.nmd.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 3.Buvoli M, Hamady M, Leinwand LA, Knight R. Bioinformatics assessment of beta-myosin mutations reveals myosin's high sensitivity to mutations. Trends Cardiovasc Med. 2008;18(4):141–149. doi: 10.1016/j.tcm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111(3):375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McLachlan AD, Karn J. Periodic features in the amino acid sequence of nematode myosin rod. J Mol Biol. 1983;164(4):605–626. doi: 10.1016/0022-2836(83)90053-0. [DOI] [PubMed] [Google Scholar]
- 6.McLachlan AD, Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982;299(5880):226–231. doi: 10.1038/299226a0. [DOI] [PubMed] [Google Scholar]
- 7.Offer G. Skip residues correlate with bends in the myosin tail. J Mol Biol. 1990;216(2):213–218. doi: 10.1016/S0022-2836(05)80309-2. [DOI] [PubMed] [Google Scholar]
- 8.Huxley HE. The mechanism of muscular contraction. Science. 1969;164(3886):1356–1365. doi: 10.1126/science.164.3886.1356. [DOI] [PubMed] [Google Scholar]
- 9.Brown JH. How sequence directs bending in tropomyosin and other two-stranded alpha-helical coiled coils. Protein Sci. 2010;19(7):1366–1375. doi: 10.1002/pro.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nicolet S, Herrmann H, Aebi U, Strelkov SV. Atomic structure of vimentin coil 2. J Struct Biol. 2010;170(2):369–376. doi: 10.1016/j.jsb.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 11.Bennett PM. The structure of spindle-shaped paracrystals of light meromyosin. J Mol Biol. 1981;146(2):201–221. doi: 10.1016/0022-2836(81)90432-0. [DOI] [PubMed] [Google Scholar]
- 12.Nyitray L, Mocz G, Szilagyi L, Balint M, Lu RC, Wong A, Gergely J. The proteolytic substructure of light meromyosin. Localization of a region responsible for the low ionic strength insolubility of myosin. J Biol Chem. 1983;258(21):13213–13220. [PubMed] [Google Scholar]
- 13.Cross RA, Vandekerckhove J. Solubility-determining domain of smooth muscle myosin rod. FEBS Lett. 1986;200(2):355–360. doi: 10.1016/0014-5793(86)81168-1. [DOI] [PubMed] [Google Scholar]
- 14.Atkinson SJ, Stewart M. Expression in Escherichia coli of fragments of the coiled-coil rod domain of rabbit myosin: influence of different regions of the molecule on aggregation and paracrystal formation. J Cell Sci. 1991;99(Pt 4):823–836. doi: 10.1242/jcs.99.4.823. [DOI] [PubMed] [Google Scholar]
- 15.Sohn RL, Vikstrom KL, Strauss M, Cohen C, Szent-Gyorgyi AG, Leinwand LA. A 29 residue region of the sarcomeric myosin rod is necessary for filament formation. J Mol Biol. 1997;266(2):317–330. doi: 10.1006/jmbi.1996.0790. [DOI] [PubMed] [Google Scholar]
- 16.Blankenfeldt W, Thoma NH, Wray JS, Gautel M, Schlichting I. Crystal structures of human cardiac beta-myosin II S2-Delta provide insight into the functional role of the S2 subfragment. Proc Natl Acad Sci U S A. 2006;103(47):17713–17717. doi: 10.1073/pnas.0606741103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dill KA, Fiebig KM, Chan HS. Cooperativity in protein-folding kinetics. Proc Natl Acad Sci U S A. 1993;90(5):1942–1946. doi: 10.1073/pnas.90.5.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kammerer RA, Schulthess T, Landwehr R, Lustig A, Engel J, Aebi U, Steinmetz MO. An autonomous folding unit mediates the assembly of two-stranded coiled coils. Proc Natl Acad Sci U S A. 1998;95(23):13419–13424. doi: 10.1073/pnas.95.23.13419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinmetz MO, Stock A, Schulthess T, Landwehr R, Lustig A, Faix J, Gerisch G, Aebi U, Kammerer RA. A distinct 14 residue site triggers coiled-coil formation in cortexillin I. Embo J. 1998;17(7):1883–1891. doi: 10.1093/emboj/17.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu KC, Bryan JT, Morasso MI, Jang SI, Lee JH, Yang JM, Marekov LN, Parry DA, Steinert PM. Coiled-coil trigger motifs in the 1B and 2B rod domain segments are required for the stability of keratin intermediate filaments. Mol Biol Cell. 2000;11(10):3539–3558. doi: 10.1091/mbc.11.10.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lupas AN, Gruber M. The structure of alpha-helical coiled coils. Advances in protein chemistry. 2005;70:37–78. doi: 10.1016/S0065-3233(05)70003-6. [DOI] [PubMed] [Google Scholar]
- 22.Ciani B, Bjelic S, Honnappa S, Jawhari H, Jaussi R, Payapilly A, Jowitt T, Steinmetz MO, Kammerer RA. Molecular basis of coiled-coil oligomerization-state specificity. Proc Natl Acad Sci U S A. 2010;107(46):19850–19855. doi: 10.1073/pnas.1008502107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolny M, Colegrave M, Colman L, White E, Knight PJ, Peckham M. Cardiomyopathy mutations in the tail of beta-cardiac myosin modify the coiled-coil structure and affect integration into thick filaments in muscle sarcomeres in adult cardiomyocytes. J Biol Chem. 2013;288(44):31952–31962. doi: 10.1074/jbc.M113.513291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Gao R, Lynn DG. Ratcheting up vir gene expression in Agrobacterium tumefaciens: coiled coils in histidine kinase signal transduction. Chembiochem : a European journal of chemical biology. 2002;3(4):311–317. doi: 10.1002/1439-7633(20020402)3:4<311::AID-CBIC311>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 25.Wolfe SA, Grant RA, Pabo CO. Structure of a designed dimeric zinc finger protein bound to DNA. Biochemistry. 2003;42(46):13401–13409. doi: 10.1021/bi034830b. [DOI] [PubMed] [Google Scholar]
- 26.Taylor KC, Buvoli M, Korkmaz EN, Buvoli A, Zheng Y, Heinze NT, Cui Q, Leinwand LA, Rayment I. Skip residues modulate the structural properties of the myosin rod and guide thick filament assembly. Proc Natl Acad Sci U S A. 2015 doi: 10.1073/pnas.1505813112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frye J, Klenchin VA, Rayment I. Structure of the tropomyosin overlap complex from chicken smooth muscle: insight into the diversity of N-terminal recognition. Biochemistry. 2010;49(23):4908–4920. doi: 10.1021/bi100349a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klenchin VA, Frye JJ, Jones MH, Winey M, Rayment I. Structure-function analysis of the C-terminal domain of CNM67, a core component of the Saccharomyces cerevisiae spindle pole body. The Journal of biological chemistry. 2011;286(20):18240–18250. doi: 10.1074/jbc.M111.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junop MS, Modesti M, Guarne A, Ghirlando R, Gellert M, Yang W. Crystal structure of the Xrcc4 DNA repair protein and implications for end joining. EMBO J. 2000;19(22):5962–5970. doi: 10.1093/emboj/19.22.5962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sibanda BL, Critchlow SE, Begun J, Pei XY, Jackson SP, Blundell TL, Pellegrini L. Crystal structure of an Xrcc4-DNA ligase IV complex. Nat Struct Biol. 2001;8(12):1015–1019. doi: 10.1038/nsb725. [DOI] [PubMed] [Google Scholar]
- 31.Morais MC, Kanamaru S, Badasso MO, Koti JS, Owen BA, McMurray CT, Anderson DL, Rossmann MG. Bacteriophage phi29 scaffolding protein gp7 before and after prohead assembly. Nat Struct Biol. 2003;10(7):572–576. doi: 10.1038/nsb939. [DOI] [PubMed] [Google Scholar]
- 32.Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC. Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli. Plasmid. 2008;59(3):231–237. doi: 10.1016/j.plasmid.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen GJ, Qiu N, Karrer C, Caspers P, Page MG. Restriction site-free insertion of PCR products directionally into vectors. Biotechniques. 2000;28(3):498–500. 504–495. doi: 10.2144/00283st08. [DOI] [PubMed] [Google Scholar]
- 34.Miyazaki K, Takenouchi M. Creating random mutagenesis libraries using megaprimer PCR of whole plasmid. Biotechniques. 2002;33(5):1033–1034. 1036–1038. doi: 10.2144/02335st03. [DOI] [PubMed] [Google Scholar]
- 35.van den Ent F, Lowe J. RF cloning: a restriction-free method for inserting target genes into plasmids. J Biochem Biophys Methods. 2006;67(1):67–74. doi: 10.1016/j.jbbm.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 36.Blommel PG, Fox BG. A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif. 2007;55(1):53–68. doi: 10.1016/j.pep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22(3):356–358. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- 38.Rayment I. Reductive alkylation of lysine residues to alter crystallization properties of proteins. Macromolecular Crystallography, Pt A. Volume 276, Methods in Enzymology. 1997:171–179. [PubMed] [Google Scholar]
- 39.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 8):859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project N The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50(Pt 5):760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40(Pt 4):658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cowtan K. Fitting molecular fragments into electron density. Acta Crystallogr D. 2008;64:83–89. doi: 10.1107/S0907444907033938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cowtan K. Recent developments in classical density modification. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):470–478. doi: 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 46.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schomaker V, Trueblood KN. Correlation of internal torsional motion with overall molecular motion in crystals. Acta Crystallogr B. 1998;54:507–514. [Google Scholar]
- 49.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 50.Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- 51.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformatics. 2006 doi: 10.1002/0471250953.bi0506s15. Chapter 5:Unit 5 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using MODELLER. Current protocols in protein science / editorial board, John E Coligan [et al] 2007 doi: 10.1002/0471140864.ps0209s50. Chapter 2:Unit 2 9. [DOI] [PubMed] [Google Scholar]
- 53.Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods in molecular biology (Clifton, NJ. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]
- 54.Case D, Darden T, Cheatham TI, Simmerling C, Wang J, Duke R, Luo R, Merz K, Wang B, Pearlman D, Crowley M, Brozell S, Tsui V, Gohlke H, Mongan J, Hornak V, Cui G, Beroza P, Schafmeister C, Caldwell J, Ross W, Kollman P. Amber 8. University of California San Fransisco; California San Fransisco: 2004. [Google Scholar]
- 55.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. The Amber biomolecular simulation programs. J Comput Chem. 2005;26(16):1668–1688. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Amber, a Package of Computer-Programs for Applying Molecular Mechanics, Normal-Mode Analysis, Molecular-Dynamics and Free-Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comput Phys Commun. 1995;91(1-3):1–41. [Google Scholar]
- 57.Salomon-Ferrer R, Gotz AW, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. Journal of chemical theory and computation. 2013;9(9):3878–3888. doi: 10.1021/ct400314y. [DOI] [PubMed] [Google Scholar]
- 58.Mongan J, Simmerling C, McCammon JA, Case DA, Onufriev A. Generalized Born model with a simple, robust molecular volume correction. Journal of chemical theory and computation. 2007;3(1):156–169. doi: 10.1021/ct600085e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feig M, Brooks CL. Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol. 2004;14(2):217–224. doi: 10.1016/j.sbi.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 60.Gotz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. Journal of chemical theory and computation. 2012;8(5):1542–1555. doi: 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li DW, Bruschweiler R. Certification of Molecular Dynamics Trajectories with NMR Chemical Shifts. J Phys Chem Lett. 2010;1(1):246–248. [Google Scholar]
- 62.Li DW, Bruschweiler R. NMR-Based Protein Potentials. Angew Chem Int Edit. 2010;49(38):6778–6780. doi: 10.1002/anie.201001898. [DOI] [PubMed] [Google Scholar]
- 63.Showalter SA, Bruschweiler R. Validation of molecular dynamics simulations of biomolecules using NMR spin relaxation as benchmarks: Application to the AMBER99SB force field. Journal of chemical theory and computation. 2007;3(3):961–975. doi: 10.1021/ct7000045. [DOI] [PubMed] [Google Scholar]
- 64.Showalter SA, Bruschweiler R. Quantitative molecular ensemble interpretation of NMR dipolar couplings without restraints. J Am Chem Soc. 2007;129(14):4158. doi: 10.1021/ja070658d. + [DOI] [PubMed] [Google Scholar]
- 65.Beauchamp KA, Lin YS, Das R, Pande VS. Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. Journal of chemical theory and computation. 2012;8(4):1409–1414. doi: 10.1021/ct2007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins-Structure Function and Bioinformatics. 2006;65(3):712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of NAlkanes. J Comput Phys. 1977;23(3):327–341. [Google Scholar]
- 68.Case D, Darden T, Cheatham T, III, Simmerling C, Wang J, Duke R, Luo R, Walker R, Zhang W, Merz K. AMBER 12. 2. Vol. 1. University of California; San Francisco: 2012. p. 3. [Google Scholar]
- 69.Feig M, Karanicolas J, Brooks CL. MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graph Model. 2004;22(5):377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 70.Feig M, Karanicolas J, Brooks CL., 3rd MMTSB Tool Set: enhanced sampling and multiscale modeling methods for applications in structural biology. J Mol Graph Model. 2004;22(5):377–395. doi: 10.1016/j.jmgm.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 71.Grigoryan G, Degrado WF. Probing designability via a generalized model of helical bundle geometry. J Mol Biol. 2011;405(4):1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kondrashov DA, Zhang W, Aranda R, Stec B, Phillips GN. Sampling of the native conformational ensemble of myoglobin via structures in different crystalline environments. Proteins-Structure Function and Bioinformatics. 2008;70(2):353–362. doi: 10.1002/prot.21499. [DOI] [PubMed] [Google Scholar]
- 73.Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18(4):617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- 74.Lu M, Shu W, Ji H, Spek E, Wang L, Kallenbach NR. Helix capping in the GCN4 leucine zipper. J Mol Biol. 1999;288(4):743–752. doi: 10.1006/jmbi.1999.2707. [DOI] [PubMed] [Google Scholar]
- 75.Pelton JT, McLean LR. Spectroscopic methods for analysis of protein secondary structure. Anal Biochem. 2000;277(2):167–176. doi: 10.1006/abio.1999.4320. [DOI] [PubMed] [Google Scholar]
- 76.Offer G, Sessions R. Computer modelling of the alpha-helical coiled coil: packing of side-chains in the inner core. J Mol Biol. 1995;249(5):967–987. doi: 10.1006/jmbi.1995.0352. [DOI] [PubMed] [Google Scholar]
- 77.Lupas A, Van Dyke M, Stock J. Predicting coiled coils from protein sequences. Science. 1991;252(5010):1162–1164. doi: 10.1126/science.252.5009.1162. [DOI] [PubMed] [Google Scholar]
- 78.Delorenzi M, Speed T. An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics. 2002;18(4):617–625. doi: 10.1093/bioinformatics/18.4.617. [DOI] [PubMed] [Google Scholar]
- 79.Monera OD, Kay CM, Hodges RS. Electrostatic interactions control the parallel and antiparallel orientation of alpha-helical chains in two-stranded alpha-helical coiled-coils. Biochemistry. 1994;33(13):3862–3871. doi: 10.1021/bi00179a010. [DOI] [PubMed] [Google Scholar]
- 80.Lins L, Thomas A, Brasseur R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003;12(7):1406–1417. doi: 10.1110/ps.0304803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McLachlan AD, Karn J. Periodic charge distributions in the myosin rod amino acid sequence match cross-bridge spacings in muscle. Nature. 1982;299(5880):226–231. doi: 10.1038/299226a0. [DOI] [PubMed] [Google Scholar]
- 82.Lamont PJ, Wallefeld W, Hilton-Jones D, Udd B, Argov Z, Barboi AC, Bonneman C, Boycott KM, Bushby K, Connolly AM, Davies N, Beggs AH, Cox GF, Dastgir J, DeChene ET, Gooding R, Jungbluth H, Muelas N, Palmio J, Penttila S, Schmedding E, Suominen T, Straub V, Staples C, Van den Bergh PY, Vilchez JJ, Wagner KR, Wheeler PG, Wraige E, Laing NG. Novel mutations widen the phenotypic spectrum of slow skeletal/beta-cardiac myosin (MYH7) distal myopathy. Human mutation. 2014;35(7):868–879. doi: 10.1002/humu.22553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Buvoli M, Buvoli A, Leinwand LA. Effects of pathogenic proline mutations on myosin assembly. J Mol Biol. 2012;415(5):807–818. doi: 10.1016/j.jmb.2011.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katti SK, LeMaster DM, Eklund H. Crystal structure of thioredoxin from Escherichia coli at 1.68 A resolution. J Mol Biol. 1990;212(1):167–184. doi: 10.1016/0022-2836(90)90313-B. [DOI] [PubMed] [Google Scholar]
- 85.Deiss S, Hernandez Alvarez B, Bar K, Ewers CP, Coles M, Albrecht R, Hartmann MD. Your personalized protein structure: Andrei N. Lupas fused to GCN4 adaptors. J Struct Biol. 2014;186(3):380–385. doi: 10.1016/j.jsb.2014.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.