

Abstract
The room temperature radical decarboxylative allylation of N-protected α-amino acids and esters has been accomplished via a combination of palladium and photoredox catalysis to provide homoallylic amines. Mechanistic investigations revealed that the stability of the α-amino radical, which is formed by decarboxylation, dictates the predominant reaction pathway between competing mechanisms.
Homoallylic amines are robust building blocks used to construct a wide variety of natural products and other bioactive molecules.1 Classically, the addition of stoichiometric organometallic nucleophiles to an electrophilic aldimine has furnished these versatile molecules.1,2 More recently, catalytic asymmetric methods including metal-free variants have been described in the literature.1,3
Alternatively, the palladium-catalyzed decarboxylative coupling of α-imino esters has provided an umplong approach that couples 2-azaallyl anions with allyl electrophiles (Scheme 1).4 This method is advantageous because it uses abundant, inexpensive carboxylic acid derivatives to access reactive intermediates under neutral conditions via loss of CO2.5 One drawback is that the amine must be activated to stabilize the α-amino anion to facilitate decarboxylation which leads to regio- and chemoselectivity issues that limit the substrate scope (Scheme 1).6 We endeavored to extend this mode of reactivity towards synthetically useful N-protected amino acids that do not undergo anionic decarboxylation due to the formation of highly basic alkyl amino anions by utilizing alternate single electron pathways to facilitate decarboxylation.
Scheme 1.

Anionic versus radical decarboxylation pathways
The radical decarboxylation of carboxylic acids has historically been accomplished via electrochemical,7 photochemical,8 and reagent-based methods.9,10 Recently, the combination of transition metal and photoredox catalysis has also been used to overcome high-energy two electron processes in catalysis via single-electron-transfer (SET) events.11,12 For example, MacMillan12g,12h and Molander12i,12j have utilized photoredox events to facilitate the generation of alkyl radicals which undergo nickel-catalyzed cross-coupling. In addition, our lab has used palladium and photoredox catalysis to effect the decarboxylative allylation of aminoalkanoic acids and esters.11 A similar approach for the α-allylation of secondary amines and N-aryl tetrahydroisoquinolines has also been employed by Lu and Xiao.13 Herein we report that a dual catalytic approach allows the decarboxylative allylation of protected amino acids and peptides.
One obstacle encountered in our lab during development of the decarboxylative coupling of p-(aminophenyl)acetic acid esters was the products were formed in moderate yields due to the suspected formation of free radicals.11 DFT calculations on nickel-catalyzed radical cross-coupling by Molander and Kozlowski indicate that free benzylic radicals and the nickel-bound benzyl radical are nearly equienergetic14 and radical addition to the metal is reversible. Thus, we hypothesized that accessing higher energy radicals should disfavor free radical coupling by favoring formation of metal-bound radical intermediates which can undergo reductive elimination. Readily available amino acid derivatives bearing synthetically useful electron withdrawing nitrogen protecting groups, which generate less stable alkyl radicals upon decarboxylation, were chosen to test this hypothesis.
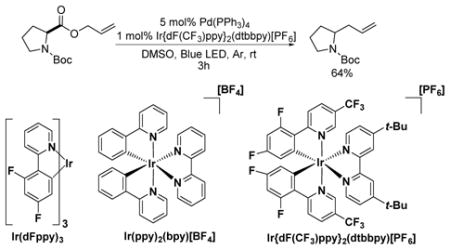 |
(1) |
Optimization studies were initiated by combining Boc-proline-allylester with Pd(PPh3)4 and various visible-light-mediated photoredox catalysts (eq. 1). Low conversion was observed by GC/MS when the strongly reducing Ir(dFppy)315 photocatalyst was employed. Substituting more oxidizing cationic heteroleptic iridium complexes led to an increase in conversion, but numerous byproducts were also detected by GC/MS analysis. A solvent screen revealed that the combination of the highly oxidizing photocatalyst Ir{dF(CF3)ppy}2(dtbbpy)[PF6] (Ered1/2[Ir*III/IrII] = + 1.21 V vs. SCE)16 with DMSO led to rapid conversion of the starting material without the previously observed byproduct formation or significant deactivation of the active catalytic species.17 Control reactions confirmed that no conversion of starting material to product occurred when the reaction was conducted without palladium, photocatalyst, or light.
We next evaluated the scope of α-amino allyl-esters that underwent decarboxylative allylation to furnish homoallyl amines (Scheme 2). A brief survey of nitrogen protecting groups revealed that tert-butyl and benzyl carbamates along with acetamides were compatible with the reaction conditions (2a–2c). A variety of phenylalanine derivatives bearing electron withdrawing (2d–2f), halogen (2g, 2h), and electron donating (2i), substituents on the aryl ring were allylated in good yields. The reaction was also performed on gram scale, but it required a longer reaction time when it was performed under more convenient concentrated conditions (2h). A phenylalanine allyl ester bearing a tertiary amine and an N-Boc phenylglycine allyl ester were also tolerated (2j, 2k).
Scheme 2.

a,b Decarboxylative allylation of α-amino esters [a] Reactions performed on a 0.25 mmol scale at 0.13 M. [b] Isolated yields. [c] Gram scale, 0.37 M. [d] 95% pure. [e] Average yield of two runs.
Cyclic amino acid allyl esters derived from proline, 4-hydroxy-, thio-, and homoproline (2l–2o) were also successfully allylated. Other acyclic tertiary homoallyl amines were constructed from their amino allyl ester precursors including alanine (2p), methionine (2q), and aspartic acid (2r). A protected glycine allyl ester provided a secondary homoallylic amine, albeit in reduced yield (2s). Nitrogen- (2u, 2v) and sulfur-containing (2t) heterocycles also tolerated the reaction conditions, although lower yields were observed when pyridine (2v) or an unprotected indole (2u) was present. Lastly, a β-methallyl ester provided the desired homoallylic amine (2w), although a slightly higher reaction temperature (45 °C) was required.
One of the hallmarks of anionic decarboxylative couplings is that they are typically site-specific, leading to coupling only at the position that bears the carboxylate.5a With the goal of demonstrating that the typical thermodynamic selectivity of α-amino radical formation can be overridden by the structure of the starting material used in decarboxylative coupling, the allyl ester of N-Boc tetrahydro-3-isoquinoline carboxylic acid was prepared. Tetrahydroisoquinolines are widely used in photoredox couplings due to the ease of access of the benzylically stabilized α-amino radical; an excellent example involving the allylation of tetrahydroquinoline by Xiao is shown in Scheme 3.13 When our tetrahydroquinoline carboxylic ester was subjected to the standard reaction conditions, site-specific decarboxylative coupling to form a C–C bond occurred without isomerization to the thermodynamically more stable radical occurred (Scheme 3, >95% selectivity by crude 1H NMR spectroscopy).
Scheme 3.

Site-specific allylation
We also recognized that, in many cases, it would be beneficial to generate homoallylic amines directly from carboxylic acids via an intermolecular process. It was expected that treatment of the free acid with Pd(PPh3)4 and allylmethyl carbonate would provide the same ionic intermediates (a carboxylate anion and Pd-π-allyl cation) that were generated from the amino acid allyl esters. Indeed, the Boc-protected amino acids phenylalanine and proline provided the corresponding products 2a and 2l in higher yields than those generated from their allylic ester counterparts (Scheme 4). Next, several Boc-protected dipeptides, which contained both amide and carbamate moieties that could potentially undergo oxidation by the excited photocatalyst, were examined. The allylated peptides were isolated in moderate to good yields although extended reaction times were required.
Scheme 4.

Decarboxylative allylation of α-amino acidsa,b,c [a] Reactions performed on a 0.25 mmol scale at 0.13 M. [b] Isolated Yields. [c] Yields in parentheses refer to entries in Scheme 2.
Our initial hypothesis was that higher yields of radical decarboxylative allylation may be achieved by favoring metal-mediated C–C bond formation while avoiding free radical coupling. On the basis of observations by MacMiillan,12b,12g Molander,12i,12j and us,11 two different potential mechanistic pathways were proposed (Scheme 5). In either case, the cycle begins with oxidative addition of the allyl ester to Pd(0) which provides the cationic Pd-π-allyl species and the carboxylate counterion. Since more oxidizing photocatalysts are more effective for the transformation, we propose that the amino carboxylate (for example, Boc-Pro-OCs, E1/2 red = +0.95 V vs SCE)18 is then oxidized by the photoexcited iridium complex (Ered1/2[Ir*III/IrII] = + 1.21 V vs. SCE)16 which triggers radical decarboxylation to form an α-amino radical. If the radical is suitably stabilized then electron transfer from the reduced iridium catalyst to the the Pd(II) complex (A) can result in homolysis to provide an allyl radical (path a, Scheme 5).19,20 The ensuing free radical coupling can produce the allylated amine as well as diamine and hexadiene homocoupling products. Alternatively, if the α-amino radical is less stable, it may prefer to coordinate to palladium generating a Pd(III) intermediate (B)12b,12g,12i,12j that behaves as a persistent radical.21 Electron transfer from the reduced iridium complex prior to or after reductive elimination would form the observed product and regenerate Pd(0) (path b, Scheme 5).12b,12g,12i,12j A similar possibility was proposed by Molander and Kozlowski on the basis of DFT calculations of the pathways for nickel-catalyzed photoredox coupling of borates and aryl halides.14
Scheme 5.

Divergent pathways for photoredox decarboxylative coupling
The allyl esters of N,N-dibenzyl phenyalanine (4), N-Boc phenylglycine (5), and N-Boc phenylalanine (6) were prepared to attempt to distinguish if radical stability influenced whether C–C bond formation was occurring within the Pd coordination sphere (path b) or by an undesired free radical (path a) or radical chain process. Initial evaluation of BDEs revealed that the α-amino radical species derived from 6 is ~5–6 kcal/mol higher in energy than those derived from 4 and 5.22 Moreover, the single electron oxidation potential of the radical derived from 6 is expected to be endergonic whereas oxidations of the radicals formed from 4 and 5 are exergonic.19 In short, substrates 4 and 5 generate relatively stable radicals while substrate 6 generates a less stable radical.
To probe whether C–C bond formation occurs within the coordination sphere of the metal, 4–6 were subjected to slightly modified reaction conditions using a chiral non-racemic palladium complex (Scheme 6). Substrate 6, which produces a more unstable radical post decarboxylation, provided enantioenriched product which provides evidence that the C–C bond is forming within the coordination sphere of the chiral palladium complex. In contrast, the reactions of 4 and 5, which generate more stabilized radical intermediates, led to racemic or nearly racemic products.
Scheme 6.

Stereochemical test for palladium involvement in C-C formation.
The results of reaction with a chiral palladium catalyst suggest that substrate 6 reacts through a Pd-bound intermediate while substrates 4 and 5 react primarily through free radical coupling. One way to potentially test for free radical coupling is to measure products of free-radical homocoupling. Toward this end, 1H NMR spectroscopy was used to monitor the formation of 1,5-hexadiene via allyl-allyl coupling in reactions of 4–6. That free radical coupling is the primary mechanism for the reaction of 4 is supported by the observation of nearly statistical amounts homodimerization was observed in much lower quantity when 6, which produces a less stable α-amino radical, was utilized (product:hexadiene = 1.0 : 0.08). Similar NMR spectroscopic studies were used to examine the effect of palladium of homodimerization products, including 1,5-hexadiene (product:hexadiene = 1.0 : 0.50).23 In contrast, free radical concentration on the homodimerization of allyl radicals (Scheme 7). The amount of hexadiene observed did not appreciably change when the concentration of Pd was varied in the reaction of 5, which indicates a Pd-independent C–C bond formation (path a). Conversely, when 6, which forms a less stable radical, was subjected to similar inquiry, the quantity of hexadiene was affected by the concentration of palladium. This observation suggests pathways a and b are competitive, but pathway b is favored at higher palladium concentrations. Ultimately, the simplest interpretation of the observations is that, under our standard reaction conditions, less stable α-amino radicals react primarily via metal-mediated C–C bond formation, while more stabilized benzylic and α-tert-amino radicals form C–C bonds via radical coupling.
Scheme 7.

Does Pd concentration affect homodimerization? [a] At a constant substrate concentration of 0.0125M. [b] Reaction did not reach complete conversion
In conclusion, we have developed a direct method for the decarboxylative allylation of α-amino acid derivatives using dual palladium and photoredox catalysis to circumvent traditional limitations in decarboxylative couplings. The coupling is site-specific for the position that bears CO2, allowing the kinetic formation of contra-thermodynamic radical species. Detailed studies of product and byproduct formation implicate competing mechanisms for the coupling, with more stable radicals preferring to react via radical coupling and less stabilized radicals preferring to react via metal-mediated coupling.
Supplementary Material
Figure 1.

Substrates and radical intermediates
Acknowledgments
Support for this research was provided by the National Science Foundation (CHE-1465172). Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grant # S10OD016360, NIH Shared Instrumentation Grant # S10RR024664, and NSF Major Research Instrumentation Grant # 0320648.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- 1.Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2011;111:7774–7854. doi: 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]
- 2.For allyl addition to imines: Yus M, González-Gómez JC, Foubelo F. Chem Rev. 2013;113:5595–5698. doi: 10.1021/cr400008h.Puentes CO, Kouznetsov V. J Heterocycl Chem. 2002;39:595–614.Ding H, Friestad GK. Synthesis. 2005;17:2815–2829.Ramadhar TR, Batey RA. Synthesis. 2011;9:1321–1346.
- 3.(a) Vieira EM, Snapper ML, Hoveyda AH. J Am Chem Soc. 2011;113:3332–3335. doi: 10.1021/ja200311n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Silverio DL, Torker S, Pilyugina T, Vieira EM, Snapper ML, Haeffner F, Hoveyda AH. Nature. 2013;494:216–221. doi: 10.1038/nature11844. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gandhi S, List B. Angew Chem Int Ed. 2013;52:2573–2576. doi: 10.1002/anie.201209776. and references therein. [DOI] [PubMed] [Google Scholar]
- 4.(a) Burger EC, Tunge JA. J Am Chem Soc. 2006;128:10002–10003. doi: 10.1021/ja063115x. [DOI] [PubMed] [Google Scholar]; (b) Yeagley AA, Chruma JJ. Org Lett. 2007;9:2879–2882. doi: 10.1021/ol071080f. [DOI] [PubMed] [Google Scholar]
- 5.For recent reviews, see: Weaver JD, Recio A, III, Grenning AJ, Tunge JA. Chem Rev. 2011;111:1846–1913. doi: 10.1021/cr1002744.Rodriguez N, Goossen LJ. Chem Soc Rev. 2011;40:5030–5048. doi: 10.1039/c1cs15093f.Dzik WI, Lange PP, Goosen LJ. Chem Sci. 2012;3:2671–2678.
- 6.Bordwell FG. Acc Chem Res. 1988;21:456–463. [Google Scholar]
- 7.(a) Anderson JM, Kochi JK. J Am Chem Soc. 1970;92:2450–2460. [Google Scholar]; (b) Trahanovsky WS, Cramer J, Brixius DW. J Am Chem Soc. 1974;96:1077–1081. [Google Scholar]
- 8.(a) Joschek HI, Grossweiner LI. J Am Chem Soc. 1966;88:3261–3268. doi: 10.1021/ja00966a017. [DOI] [PubMed] [Google Scholar]; (b) Davidson RS, Steiner PR. J Chem Soc C. 1971:1682–1689. [Google Scholar]; (c) Davidson RS, Steiner PR. J Chem Soc, Perkin Trans. 1972;2:1357–1362. [Google Scholar]; (d) Meiggs TO, Grossweiner LI, Miller SI. J Am Chem Soc. 1972;94:7981–7986. [Google Scholar]; (e) Battacharyya SN, Das PK. J Chem Soc, Faraday Trans 2. 1984;80:1107–1116. [Google Scholar]; (f) D’Alessandro N, Albini A, Mariano PS. J Org Chem. 1993;58:937–942. [Google Scholar]; (g) Habibi MH, Farhadi S. J Chem Res (S) 1998:776–777. [Google Scholar]; (h) Su Z, Mariano PS, Falvey DE, Yoon UC, Oh SW. J Am Chem Soc. 1998;120:10676–10686. [Google Scholar]; (i) Xu M, Wan P. Chem Commun. 2000:2147–2148. [Google Scholar]; (j) Gould IR, Lenhard JR, Farid S. J Phys Chem A. 2004;108:10949–10956. [Google Scholar]; (k) Poupko R, Rosenthal I, Elad D. Photochem Photobiol. 1973;17:395–402. [Google Scholar]
- 9.(a) Barton HRD, Hervé Y, Potier P, Thierry J. J Chem Soc Chem Commun. 1984:1298–1299. [Google Scholar]; (b) Barton HRD, Hervé Y, Potier P. J Thierry Tetrahedron. 1987:4297–4308. [Google Scholar]; (c) Vidal-Cros A, Bory S, Gaudry M, Marquet A. Tetrahedron Lett. 1989:1799–1802. [Google Scholar]; (d) Aveline BM, Kochevar IE, Redmond RW. J Am Chem Soc. 1995;117:9699–9708. [Google Scholar]; (e) Boto A, Hernández R, Suárez E. J Org Chem. 2000;65:4930–4937. doi: 10.1021/jo000356t. [DOI] [PubMed] [Google Scholar]
- 10.Miyake Y, Nakajima K, Nishibayashi Y. Chem Commun. 2013;49:7854–7856. doi: 10.1039/c3cc44438d. [DOI] [PubMed] [Google Scholar]
- 11.Lang SB, O’Nele KM, Tunge JA. J Am Chem Soc. 2014;136:13606–13609. doi: 10.1021/ja508317j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For representative examples of dual catalytic systems employing photoredox catalysts see: Osawa M, Nagai H, Akita M. Dalton Trans. 2007:827–829. doi: 10.1039/b618007h.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J Am Chem Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w.Ye Y, Sanford MS. J Am Chem Soc. 2012;134:9034–9037. doi: 10.1021/ja301553c.Rueping M, Koenigs RM, Poscharny K, Fabry DC, Leonori D, Vila C. Chem —Eur J. 2012;18:5170–5174. doi: 10.1002/chem.201200050.Sahoo B, Hopkinson MN, Glorius F. J Am Chem Soc. 2013;135:5505–5508. doi: 10.1021/ja400311h.Shu X, Zhang M, He Y, Frei H, Toste FD. J Am Chem Soc. 2014;136:5844–5847. doi: 10.1021/ja500716j.Zuo Z, Ahneman D, Chu L, Terrett J, Doyle AG, MacMillan DWC. Science. 2014;345:437–440. doi: 10.1126/science.1255525.Noble A, McCarver SJ, MacMillan DWC. J Am Chem Soc. 2014;137:624–627. doi: 10.1021/ja511913h.Tellis JC, Primer DN, Molander GA. Science. 2014;345:433–436. doi: 10.1126/science.1253647.Primer DN, Karakaya I, Tellis JC, Molander GA. J Am Chem Soc. 2015;137:2195–2198. doi: 10.1021/ja512946e.
- 13.Xuan J, Zeng TT, Feng ZJ, Deng QH, Chen JR, Lu LQ, Xiao WJ, Alper H. Angew Chem Int Ed. 2015;54:1625–1628. doi: 10.1002/anie.201409999. [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez O, Tellis JC, Primer DN, Molander GA, Kozlowski MC. J Am Chem Soc. 2015;137:4896–4899. doi: 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tamayo AB, Alleyne BD, Djurovich PI, Lamansky S, Tsyba I, Ho NN, Bau R, Thompson ME. J Am Chem Soc. 2003;125:7377–7387. doi: 10.1021/ja034537z. [DOI] [PubMed] [Google Scholar]
- 16.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr, Malliaras GG, Bernhard S. Chem Mater. 2005;17:5712–5719. [Google Scholar]
- 17.Devery JJ, III, Douglas JJ, Nguyen JD, Cole KP, Flowers RA, II, Stephenson CR. Chem Sci. 2015;6:537–541. doi: 10.1039/c4sc03064h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuo Z, MacMillan DWC. J Am Chem Soc. 2014;136:5257–5260. doi: 10.1021/ja501621q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The single electron reduction of cationic Pd(II) allyl species has been measured (~ −1.4 V versus SCE) Cantat T, Génin E, Giroud C, Meyer G, Jutand A. J Organomet Chem. 2003;687:365–376.Carturan G, Biasiolo M, Daniele S, Mazzocchin GA, Ugo P. Inorg Chim Acta. 1986;119:19–24. We do not expect Pd(II) species to be reduced by α-amino radicals given that α-amino radicals of trimethylamine (−0.61 V versus SCE) and N-methylacetamide (0.60 V versuse SCE) are weaker reductants than the reduced photocatalyst (−1.37 V versus SCE)15.Fu Y, Liu L, Yu HZ, Wang YM, Guo QX. J Am Chem Soc. 2005;127:7227–7234. doi: 10.1021/ja0421856.
- 20.Kunkely H, Vogler A. Inorg Chim Acta. 2003;344:262–264. [Google Scholar]
- 21.For persistent radicals in transition metal-catalyzed reactions see Jahn E, Jahn U. Angew Chem Int Ed. 2014;53:13326–13328. doi: 10.1002/anie.201408748.Jahn E. Top Curr Chem. 2012;320:121–190. 323–452. doi: 10.1007/128_2011_261.For reviews on the persisten radical effect see: Studer A. Chem Soc Rev. 2004;33:267–273. doi: 10.1039/b307652k.Fischer H. Chem Rev. 2001;101:3581–3610. doi: 10.1021/cr990124y.
- 22.The benzylic stabilization was estimated by comparing the BDE of trimethylamine (91 kcal/mol) to N,N-dimethylbenzylamine (84.9 kcal/mol): Luo, Y-R. Handbook of Bond Dissociation Energies in Organic Compound; CRC Press: Boca Raton, United States, 2003. The destabilization attributed to a carbamate was estimated by comparing the BDE of triethylamine (91kcal/mol): Wayner DDM, Clark KB, Rauk A, Yu D, Armstrong DA. J Am Chem Soc. 1997;119:8925–8932.to the BDE of methyl propylcarbamate (96 kcal/mol): Berry RJ, Wilson AL, Schwartz M. J Mol Struct (Theochem) 2000;496:121–129.
- 23.The diamine homocoupling product was also observed (product:diamine:hexadiene = 1.0:0.43:0.50) (see supporting information).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.