

Summary
Although compensatory islet hyperplasia in response to insulin resistance is a recognized feature in diabetes, the factor(s) that promote β-cell proliferation have been elusive. We previously reported that the liver is a source for such factors in the liver insulin receptor knockout (LIRKO) mouse, an insulin resistance model which manifests islet hyperplasia. Using proteomics we show that serpinB1, a protease inhibitor, which is abundant in the hepatocyte secretome and sera derived from LIRKO mice, is the liver-derived secretory protein that regulates β-cell proliferation in humans, mice and zebrafish. Small molecule compounds, that partially mimic serpinB1 effects of inhibiting elastase activity, enhanced proliferation of β-cells, and mice lacking serpinB1 exhibit attenuated β-cell compensation in response to insulin resistance. Finally, SerpinB1-treatment of islets modulated proteins in growth/survival pathways. Together, these data implicate serpinB1 as an endogenous protein that can potentially be harnessed to enhance functional β-cell mass in patients with diabetes.
Graphical abstract

Introduction
While the etiopathogenesis of type 1 and type 2 diabetes are different (Boitard, 2012; Muoio and Newgard, 2008), a paucity of functional β-cell mass is a central feature in both diseases (Butler et al., 2003; Henquin and Rahier, 2011). Currently there is considerable interest in developing safe approaches to replenish bioactive insulin in patients with diabetes by deriving insulin-producing cells from pluripotent cells (D'Amour et al., 2006; Kroon et al., 2008; Pagliuca et al., 2014; Rezania et al., 2014) or promoting proliferation of pre-existing β-cells (Dor et al., 2004; El Ouaamari et al., 2013; Yi et al., 2013). While the former approach continues to evolve, several groups have focused on identifying growth factors, hormones and/or signaling proteins to promote β-cell proliferation (cited in (El Ouaamari et al., 2013) and (Dirice et al., 2014)). Compared to rodents, adult human β-cells are contumacious to proliferation and have been suggested to turnover very slowly with the β-cell mass reaching a peak by early adulthood (Butler et al., 2003; Gregg et al., 2012; Kassem et al., 2000). Attempts to enhance human β-cell proliferation have also been hampered by poor knowledge of the signaling pathways that promote cell cycle progression (Bernal-Mizrachi et al., 2014; Kulkarni et al., 2012; Stewart et al., 2015). While two recent studies have reported the identification of a small molecule, harmine (Wang et al., 2015) and denosumab, a drug approved for the treatment of osteoporosis (Kondegowda et al., 2015) to increase human β-cell proliferation the identification of endogenous circulating factors that have the ability to replenish insulin-secreting cells is attractive for therapeutic purposes. We previously reported that compensatory β-cell growth in response to insulin resistance is mediated, in part, by liver-derived circulating factors in the liver-specific insulin receptor knockout (LIRKO) mouse, a model that exhibits significant hyperplasia of islets without compromising β-cell secretory responses to metabolic or hormonal stimuli (El Ouaamari et al., 2013). Here we report the identification of serpinB1 as a liver-derived secretory protein that promotes proliferation of human, mouse and zebrafish β-cells.
Results
Identification of serpinB1 as a hepatocyte-derived circulating protein in LIRKO mice
To identify the putative β-cell trophic factor in the LIRKO model, we performed mass spectrometry (MS)-based proteomics analyses of liver, liver explant-conditioned media (LECM), hepatocyte-conditioned media (HCM) and plasma from control or LIRKO animals (Figure 1A). Data analysis pointed to serpinB1 as the top significantly up-regulated protein in all samples with substantial increases in liver (~3.3-fold), LECM (~3.7-fold), HCM (~54-fold) and plasma (~3.3-fold) (Figure 1B; red bars indicate serpinB1). To validate the proteomics data, we examined liver expression and circulating levels of serpinB1 in the LIRKO mouse. RT-PCR and western blotting experiments using cross-reactive antibody to human SerpinB1 revealed that serpinB1 mRNA (LIRKO 2.4±0.6 vs. control 0.6±0.1, p<0.05, n=6) and protein levels (LIRKO 5.1±0.9 versus control 1.1±0.06, p<0.05, n=4–5) were elevated by 5-fold in 12-week-old LIRKO mice compared to age-matched controls (Figure 1C–E). Western blot analyses showed increased levels of serpinB1 in LIRKO-LECM (Figure 1F). SerpinA1 (also called α1-antitrypsin), which has partially overlapping biochemical activity, was not increased in LECM of LIRKO mice (Figure 1G). Importantly, we observed that serpinB1 is increased in LIRKO hepatocyte lysates where neutrophil markers such as proteinase-3 (PR-3) and neutrophil elastase (NE) were not detected, therefore excluding contaminating blood cells as a significant source of serpinB1 (Figure 1H). We used recombinant human SerpinB1 (rSerpinB1) to introduce a standard curve in western blotting experiments to provide a semi-quantitative measure of serpinB1 in serum samples (Figure 1I). Circulating serpinB1 was elevated in sera from 6 month-old LIRKO mice (78±7.9 versus control 24.2±4.2 ng equivalents/ml, p<0.01, n=10–12) (Figure 1J).
Fig. 1. Identification of serpinB1 in the LIRKO model.
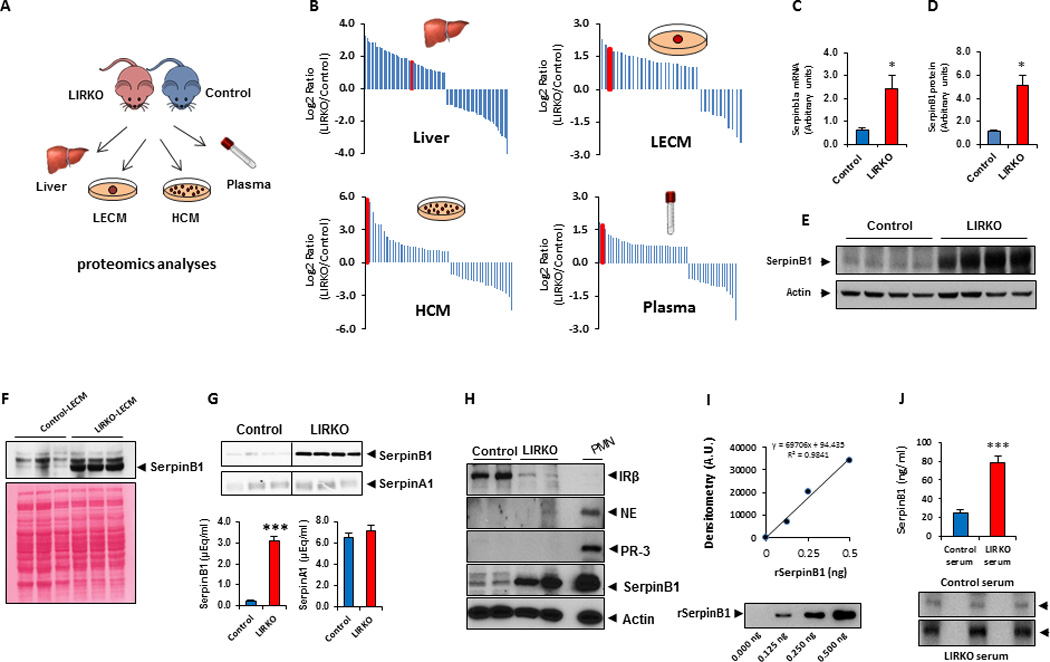
A. Experimental workflow for analysis of proteins from liver, liver explant conditioned media (LECM), hepatocyte-conditioned media (HCM), and plasma. B. Identification of serpinB1 by LC-MS/MS proteomics. Protein abundances were quantified based on spectral counts, and top differentially expressed proteins were plotted as log2 ratio of LIRKO vs control. Red bars correspond to serpinB1. C. Relative quantification of liver serpinb1a mRNA by quantitative RT-PCR (normalized to TBP). Data represent mean ± SEM. *p ≤ 0.05, (n=6 per group). D. Quantification of serpinB1 protein (in E) in 12 wk-old male control and LIRKO mice. E. Western blot of serpinB1 in liver. SerpinB1 protein was normalized to actin, and data represent mean ± SEM. *p ≤ 0.05, (n=4–5 per group). F. Western blot (top panel) of serpinB1 in LECM from 12 wk-old male control and LIRKO mice. Bottom panel shows Ponceau S staining of protein. G. Western blot of serpinB1 and serpinA1 (α1–antitrypsin) in LECM from control or LIRKO mice (10 wk old males). The bands (top panel) were quantified (bottom panel) relative to human SerpinB1 and human SerpinA1 run in parallel as standards. Data represent mean ± SEM, ***p ≤ 0.001 (n=3 per group). H. Western blot of insulin receptor, serpinB1, neutrophil elastase (NE) and proteinase-3 (PR-3) in hepatocytes from 12-wk-old male control or LIRKO mice. IR-β, insulin receptor beta subunit; NE, neutrophil elastase; PR-3, proteinase-3; PMN; polymorphonuclear leukocytes. I–J. Analysis of serpinB1 by western blot in serum derived from 12-wk-old male control or LIRKO mice. Quantification of serpinB1 bands in (J) is based on parallel standard curve of recombinant human SerpinB1 shown in (I). Data represent mean ± SEM. ***p ≤ 0.01, (n=10–12 per group)
Serpins are a highly conserved superfamily of ~45-kDa proteins, which are classified in 16 clades from A to P, and 36 members have been identified in humans (Silverman et al., 2001) and are known to regulate important proteolytic events. SerpinB1 is an evolutionarily conserved member of serpin clade B (Benarafa and Remold-O'Donnell, 2005) and inhibits the activity of several proteases including neutrophil elastase, cathepsin G and proteinase-3 (Cooley et al., 2001). While serpinB1 lacks the hydrophobic signal peptide commonly harbored by secretory proteins (Remold-O'Donnell, 1993), the protein is detectable in hepatic-conditioned media and serum, suggesting that its release is mediated by an unconventional pathway (Nickel, 2010). Since previous studies reported a caspase-1-dependent mechanism of unconventional secretion (Becker et al., 2009; Chakraborty et al., 2013; Keller et al., 2008), we used human primary keratinocytes to investigate whether SerpinB1 secretion requires intact caspase-1. Consistent with previous reports (Chakraborty et al., 2013; Feldmeyer et al., 2007), irradiation of human keratinocytes with Ultraviolet B light (UVB) activated the inflammasome and induced release of several pro-inflammatory cytokines including IL-1β and IL-18, concomitant with caspase-1 activation. SerpinB1 is released in culture media when keratinocytes were UVB-irradiated; when caspase-1 was downregulated by a siRNA approach the SerpinB1 secretion was abolished, as was secretion of IL-1β and IL-18 (Figure S1A). Similar observations were evident when cells were treated with the caspase-1 inhibitor YVAD or pan-caspase inhibitor VAD prior to UVB treatment (Figure S1B). We also detected SerpinB1 in supernatants from cultured HepG2 cells and observed that several inflammatory molecules stimulate its release upon short (5 h) or long (24 h) term treatment (Figure S1C). Consistent with increased levels of serpinB1 in LECM and serum from LIRKO mice, we found that caspase-1 mRNA and protein levels are increased in liver derived from LIRKO versus control groups (Figure S1D). Active caspase-1 (p20) was also highly abundant in LIRKO-LECM when compared to control conditions (Figure S1E).
To explore the clinical significance of SerpinB1 in humans, we developed an ELISA to measure plasma levels of SerpinB1 and observed that its concentration in healthy individuals ranges between 10–20 ng/ml (Figure S2A). Furthermore, a multivariate analysis in a cohort of 49 individuals with risk factor(s) for type 2 diabetes revealed that the range in concentration was greater, generally from 4–56 ng/ml, however interestingly, one individual with morbid obesity without diabetes (BMI=59) exhibited extremely high levels (299 ng/ml) of circulating serpinB1. In a multivariate analysis, excluding this outlier, a positive correlation between circulating serpinB1 and insulin resistance (R2=0.15, p=0.026) was observed, using BMI and the composite insulin sensitivity index (CISI, Matsuda index) (Matsuda and DeFronzo, 1999) as covariates for measures of insulin sensitivity (Figure S2B). Furthermore, a search for missense variants of the corresponding gene in whole-exome sequencing data generated for 52 Joslin families with autosomal dominant diabetes showed that one of the families (for individual characteristics, see Table S1) carried a previously described variant (rs114597282, c.A269G, p.N90S) having a frequency of 1.7% among African-Americans and 0.01% among Europeans in the NHLBI Exome Sequencing Project (ESP) database. The variant segregated with diabetes in this family, with all four diabetic members being heterozygous for this substitution (transmission disequilibrium test p value=0.046) and only one non-penetrant individual being present in the youngest generation (Figure S2C). This variant is conserved among species (GERP score=5.44) and is predicted as “probably damaging” by Polyphen (score=0.98) and other prediction algorithms. Taken together, the significantly elevated serpinB1 in serum and hepatocyte secretome (HCM) in the LIRKO model, its presence in human sera, and its elevation in insulin resistant states in humans, as well as the segregation of a genetic variant of serpinb1 with human diabetes prompted us to focus on this protein as a potential β-cell growth factor.
SerpinB1 and its partial mimics promote proliferation of pancreatic β-cells in multiple species
To address whether serpinB1 promotes β-cell proliferation, we cultured mouse islets in the presence of recombinant human serpinB1 or ovalbumin and evaluated proliferation by Ki67 immunofluorescence staining. Ovalbumin, encoded by serpinb14, was chosen as control because it is a serpin closely related in structure to serpinB1 but lacks protease inhibitory activity (Benarafa and Remold-O'Donnell, 2005). Ovalbumin-treated mouse islets displayed low β-cell proliferation comparable to non-treated islets; rSerpinB1-treated islets exhibited a dose-dependent effect, and a 2-fold increase in the percentage of Ki67+ insulin+ cells was observed at the dose of 1µg/ml (Figure 2A and 2B). We next tested whether small molecule pharmacological agents that inhibit elastinolytic proteases and thus partially mimic serpinB1 activity, GW311616A (Macdonald et al., 2001) and sivelestat (Kawabata et al., 1991), would affect β-cell proliferation. Treatment of islets freshly isolated from male C57Bl/6 mice with GW311616A or sivelestat increased β-cell proliferation (Figure 2C and 2D). To further explore the role of serpinB1 in vivo, we administered 7–8 week old C57Bl/6 male mice with GW311616A, a partial mimic of serpinB1, by oral gavage (2 mg/kg/day for 2 weeks). Morphometric analyses showed that GW311616A treatment enhanced β-cell mass (Figure 2E and 2F) by increasing β-cell but not α-cell proliferation as assessed by BrdU incorporation (Figure 2G and 2H). The lack of proliferation in extra-pancreatic tissues including liver, skeletal muscle, visceral and subcutaneous adipose tissues, spleen and kidney (Figure S3A and S3B) suggests that GW311616A promotes selective β-cell proliferation. The proliferative action of SerpinB1 was also evident in human β-cells using islets obtained from 7 cadaveric organ donors (for donor characteristics, see Table S2). Quantification of Ki67+ insulin+ cells revealed that the number of proliferating β-cells increased in islets cultured in serpinB1-containing media (Figure 2I and 2J). The percent of proliferating human β-cells is in a similar range to those reported previously (El Ouaamari et al., 2013; Jiao et al., 2014; Rieck et al., 2012; Rutti et al., 2012; Walpita et al., 2012). Similar to its effect on mouse islets, sivelestat also increased proliferation of human β-cells (Figure 2K–M; for donor characteristics, see Table S3). To test whether sivelestat induces human β-cell proliferation in vivo, we transplanted human islets (obtained from the Integrated Islet Distribution Program, IIDP) under the kidney capsule of 10-week-old male non-obese diabetic-severe combined immunodeficiency-IL2rγnull (NSG) mice (Greiner et al., 2011). Ten days post-transplantation, osmotic pumps loaded with sivelestat (300 µg/kg/day) or vehicle were implanted into the mice and allowed to infuse for 14 days. Mice were provided BrdU in drinking water (80mg/ml) during the fourteen-day treatment period and received intraperitoneal injections of BrdU (100 mg/kg body weight) on days 8, 11 and 14 post-transplantation. Five hours after the last BrdU injection islet grafts and endogenous pancreases were harvested to assess β-cell proliferation (Figure 2N). As assessed by co-immunostaining with anti-insulin and anti-BrdU antibodies, human islet grafts retrieved from mice treated with sivelestat exhibited higher β-cell proliferation compared to vehicle-treated controls (Figure 2O and 2P). In parallel, islet β-cell proliferation was also increased in endogenous pancreases harvested from NSG mice infused with sivelestat (Figure 2Q and 2R). The in vivo effect of sivelestat on β-cell proliferation was also evident in C57/BL6J mice (Figure S4A and S4B).
Fig. 2. SerpinB1 and its partial mimics promote proliferation of mouse and human pancreatic β-cells.
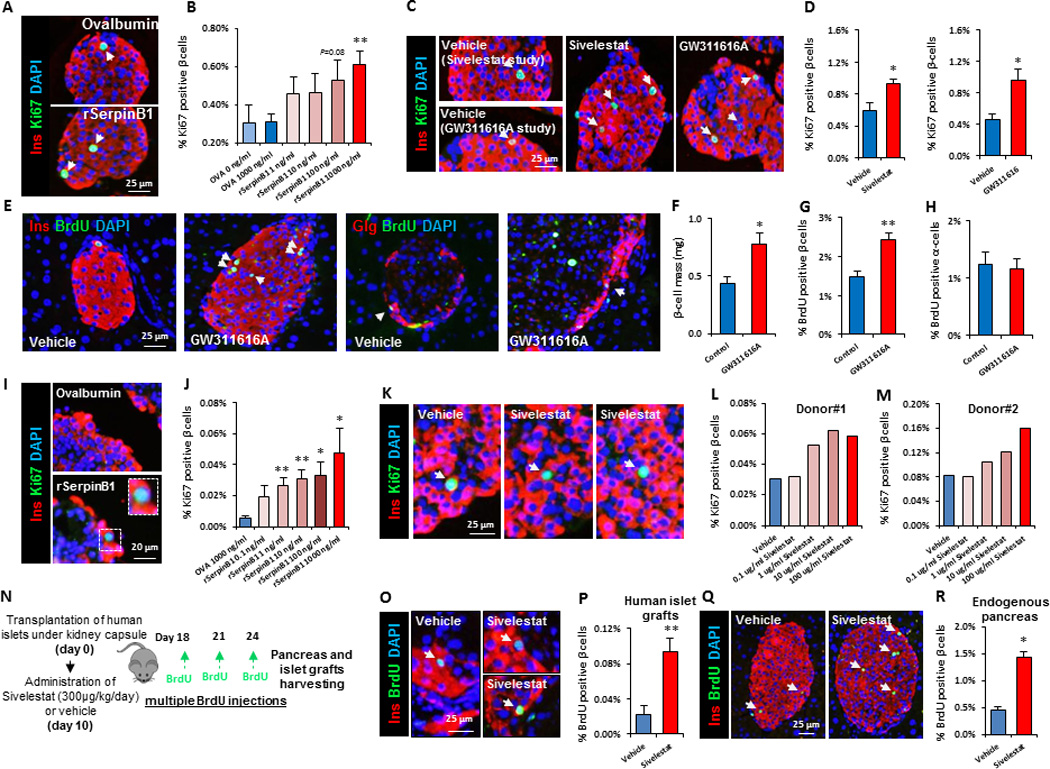
A. Representative images of mouse islets treated with ovalbumin or SerpinB1 and co-immunostained for Ki67, insulin and DAPI. B. Quantification of Ki67+ insulin+ cells (in A). Data represent mean ± SEM. **p ≤ 0.01, (n=6–12 per group). C. Representative images and quantitation of insulin+ Ki67+ cells of islets isolated from wild-type male mice and cultured for 48 hr in presence of 100 µg/ml of either sivelestat or GW311616A. D. Quantification of insulin+Ki67+ cells of sivelestat-treated islets (in C). Data represent mean ± SEM. *p ≤ 0.05, (n=3 per group for GW311616A studies and n=6 per group for sivelestat studies). Five to six-week old wild-type male mice were treated with GW311616A for 2 wks. Islet β-cell and α-cell proliferation was assessed by immunostaining. E. Pancreatic sections co-immunostained for BrdU and insulin and DAPI (two left panels) or co-immunostained for glucagon and BrdU and DAPI (two right panels). F. Quantification of beta cell mass (in E). Data represent mean ± SEM. *p ≤ 0.05, (n=4–5 per group). G. Quantification of insulin+ BrdU+ cells (in E). Data represent mean ± SEM. **p ≤ 0.01, (n=4–5 per group). H. Quantification of glucagon+ BrdU+ cells (in E). Data represent mean ± SEM. **p ≤ 0.01, (n=4–5 per group). I. Representative images of human islets treated with ovalbumin or SerpinB1 and co-immunostained for Ki67, insulin and DAPI. J. Quantification of Ki67+ insulin+ cells (in I). Data represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, (n=7 per group). For details of the human donors please see Table S2. K. Representative images of human islets treated with vehicle or sivelestat and co-immunostained for Ki67, insulin and DAPI. L and M. Quantification of Ki67+ insulin+ cells (in K). For details of human donors please see Table S3. N. Experimental workflow for transplantation studies to explore the effects of sivelestat on human β-cell proliferation in vivo. O. Representative images of human islet grafts retrieved from mice treated with sivelestat or vehicle and co-immunostained for BrdU, insulin and DAPI. P. Quantification of BrdU+ insulin+ cells (in O). Q. Representative images of endogenous pancreases harvested from mice treated with sivelestat or vehicle and co-immunostained for BrdU, insulin and DAPI. R. Quantification of BrdU+ insulin+ cells (in Q). For details of human donors please see Table S3. Data represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, (n=5–6 per group for retrieved human islet grafts and n=3 for endogenous pancreas). Arrows indicate proliferating cells.
Next, to determine whether the potentiation of β-cell proliferation by serpinb1 is conserved across species we examined serpinb1-overexpressing zebrafish larvae (Figure 3A). Whereas the human clade B serpin loci encode 13 proteins (serpinB1-13) with distinct functions, the corresponding locus in zebrafish is substantially simpler: it includes a distinct serpinb1 orthologous gene with a strikingly conserved reactive center loop, suggesting conserved function (Benarafa and Remold-O'Donnell, 2005). The overexpressing larvae were generated by cloning serpinb1 downstream of a ubiquitous promoter (beta-actin), i.e., generating widespread mosaic overexpression of serpinb1 (see details in supplemental experimental procedures). The same cloning procedure was performed for the controls H2A-mCherry and serpina7 (another member of the zebrafish Serpin family). We started by determining serpinb1’s effect on β-cell regeneration using different transgenic zebrafish larvae expressing nitroreductase (NTR)—an enzyme that converts metronidazole to a cytotoxic product—under the control of the insulin promoter; incubating these larvae in metronidazole results in the specific ablation of their β cells (Andersson et al., 2012). Each construct was injected, together with mRNA encoding transposase, into 1-cell-stage Tg(ins:CFP-NTR); Tg(ins:Kaede) embryos, giving rise to zebrafish larvae in which the β cells are visualized by the green-fluorescent protein Kaede. From 3–4 days post fertilization (dpf), we used metronidazole to ablate the β cells of mosaically overexpressing larvae and control larvae, and at 6 dpf we examined whether overexpression of any of the proteins had increased β-cell regeneration. Overexpression of serpinb1 strikingly increased regeneration of the β-cell mass by 50%, whereas none of the controls had a significant effect (Figure 3B–F). To determine serpinb1’s effect on β-cell proliferation, we examined the incorporation of EdU as an indicator of DNA replication. We exposed Tg(ins:flag-NTR);Tg(ins:H2B-GFP) larvae to metronidazole from 3–4 dpf to ablate the β cells and then incubated control and bactin:serpinb1-overexpressing larvae with EdU from 4–6 dpf (Figure 3G-H). Overexpression of serpinb1 significantly increased the total number of β cells, as well as doubling the number of β cells incorporating EdU, when compared to control larvae (Figure 3I and 3J). We next assessed the effect of serpinb1 on β-cell formation during development, rather than regeneration, of the pancreas. To examine the total number of β cells, as well as their proliferation, we exposed Tg(ins:H2B-GFP) control and bactin:serpinb1-overexpressing larvae to EdU from 4–6 dpf (Figure 3K and 3L). Serpinb1 did not significantly increase the total number of β cells, but it significantly increased the number of β cells that incorporated EdU (Figure 3M and 3N). Together, these data provide evidence for serpinB1 as a phylogenetically conserved protein that stimulates β-cell proliferation in multiple species including zebrafish, mouse and man.
Fig. 3. Overexpression of Serpinb1 in zebrafish enhances β-cell regeneration and proliferation.
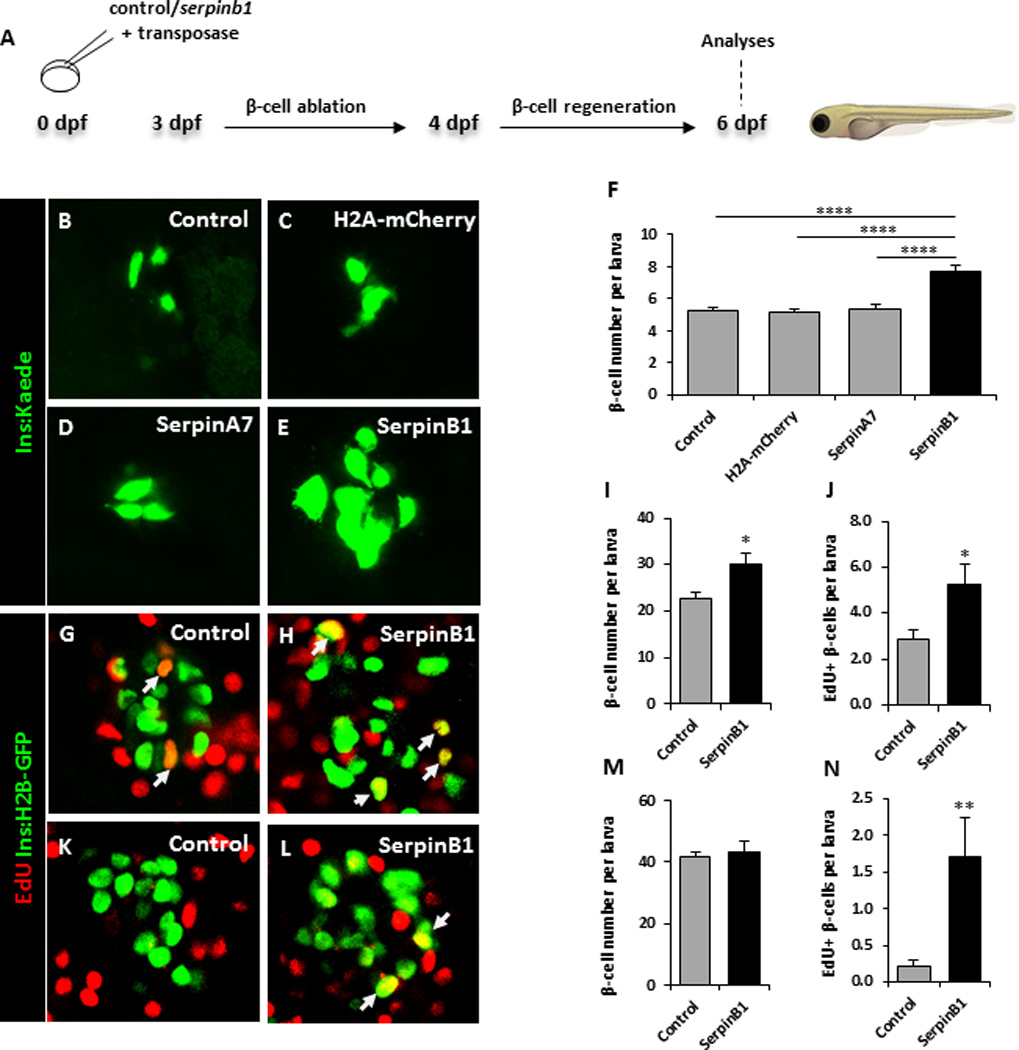
A. Schematic of experimental plan. B–E. Representative images at 6 dpf of Tg(ins:kaede);Tg(ins:CFP-NTR) transgenic larvae that had not been injected (control), or injected at the 1-cell stage with transposase mRNA + bactin:H2A-mCherry, bactin:serpina7, or bactin:serpinb1; subjected to β-cell ablation by metronidazole during 3–4 dpf, and subsequently allowed to regenerate for 2 days. F. Quantification of β-cell regeneration at 6 dpf in control (n=87), bactin:H2A-mCherry-overexpressing (n=61), bactin:serpina7-overexpressing (n=46), bactin:serpinb1-overexpressing (n=36) Tg(ins:kaede);Tg(ins:CFP-NTR) larvae. G–J. Control (n=27) and bactin:serpinb1-overexpressing (n=18) Tg(ins:H2B-GFP);Tg(ins:Flag-NTR) transgenics were treated with metronidazole from 3–4 dpf to ablate the β-cells, and subsequently incubated with EdU during regeneration from 4–6 dpf. G–H. Representative confocal images at 6 dpf of control and bactin:serpinb1-overexpressing larvae showing β-cells in green and the β-cells that had incorporated EdU in yellow (green and red overlap; arrowheads). I. Quantification of the total number of β-cells at 6 dpf. J. Quantification of β-cells that incorporated EdU during β-cell regeneration from 4–6 dpf. K–N. To determine whether Serpinb1 affects β-cell proliferation during regular development, we treated control (n=25) and bactin:serpinb1-overexpressing Tg(ins:H2B-GFP) (n=21) transgenic larvae with EdU from 4–6 dpf. K–L. Representative confocal images at 6 dpf of control and bactin:serpinb1-overexpressing larvae showing β-cells in green and the β-cells that had incorporated EdU in yellow (green and red overlap; arrowhead). M. Quantification of the total number of β-cells at 6 dpf. N. Quantification of β-cells that incorporated EdU from 4–6 dpf. Data shown are the mean ± SEM; **** P<0.0001, **P<0.01, * P<0.05. Arrows indicate proliferating cells.
SerpinB1 deficiency leads to maladaptive β-cell proliferation in insulin resistant states
To assess the in vivo relevance of serpinB1 in the adaptive β-cell response to insulin resistant states, we challenged control or serpinb1a-deficient (serpinB1KO) mice with stimuli that caused acute or chronic insulin resistance. To evaluate the response to acute insulin resistance we adopted two approaches: first, we treated sixteen-week-old control male mice with the insulin receptor antagonist S961 (10 nmoles/week) for two weeks (Figure S5A) and observed progressive hyperglycemia in the mice (Figure S5B) as previously described (Yi et al., 2013). SerpinB1KO mice treated with S961 peptide showed elevated random fed blood glucose that was higher when compared to S961-treated controls. No differences were observed in blood glucose levels between phosphate buffered saline (PBS)-treated control and serpinB1KO mice (Figure S5B). Quantitation of proliferation by co-immunostaining BrdU+ insulin+ cells revealed a ~10-fold increase in S961-infused mice when compared to respective PBS-treated controls (Figure S5C and S5D). Importantly, while PBS-infused serpinB1KO mice showed a low level of proliferating β-cells similar to PBS-infused controls, S961-treated serpinB1KO mice showed a detectable but attenuated response; the number of BrdU+ insulin+ cells were ~40% fewer compared with S961-treated control mice (Figure S5C and S5D). The reduction in adaptive β-cell proliferation was supported by an attenuated increase in the number of β-cells that co-stained positive for phospho-histone H3 (pHH3), an additional marker of cell proliferation (Figure S5E). In a second model we fed sixteen-week old control and serpinB1KO mice with 60% kcal HFD for 10 weeks and analyzed β-cell proliferation by BrdU incorporation and immunofluorescence staining. A ~50% reduction in the number of BrdU+ insulin+ cells in serpinB1KO-HFD mice compared to age-matched control-HFD mice (Figure S5F and S5G) suggested impaired compensatory β-cell proliferation; this was confirmed by staining for two additional proliferation markers including pHH3 (Figure S5H and S5I) and Ki67 (Figure S5J and S5K). However, we did not observe significant alterations in β-cell mass in serpinB1KO as compared to control mice in either the S961 or the short-term HFD models suggesting additional factors likely contribute to increasing the β-cell mass in these short-term insulin resistance models. In a third model we explored whether serpinb1 is critical for long term β-cell response by subjecting 8-week old control and serpinB1KO mice to low or high fat diets (LFD or HFD) for 30 weeks which led to chronic insulin resistance as shown by hyperinsulinemia in both groups (Control, LFD: 1.5±0.2 versus HFD: 9.6±1.5 ng/mL; p<0.05; serpinB1KO, LFD: 1.8±0.2 versus HFD: 3.7±0.7 ng/mL; p<0.05, n=4–5). As expected, control mice challenged with HFD, compared to the corresponding LFD cohort, showed enhanced β-cell proliferation and mass. In contrast, mice lacking serpinb1 challenged with a similar HFD showed significantly lower increases in β-cell proliferation and mass (Figure 4A–E). Taken together, these data suggest that the effects of serpinb1 for β-cell compensatory hyperplasia are more apparent in chronic insulin resistance.
Fig. 4. SerpinB1 deficiency leads to maladaptive β-cell proliferation in insulin resistant states.
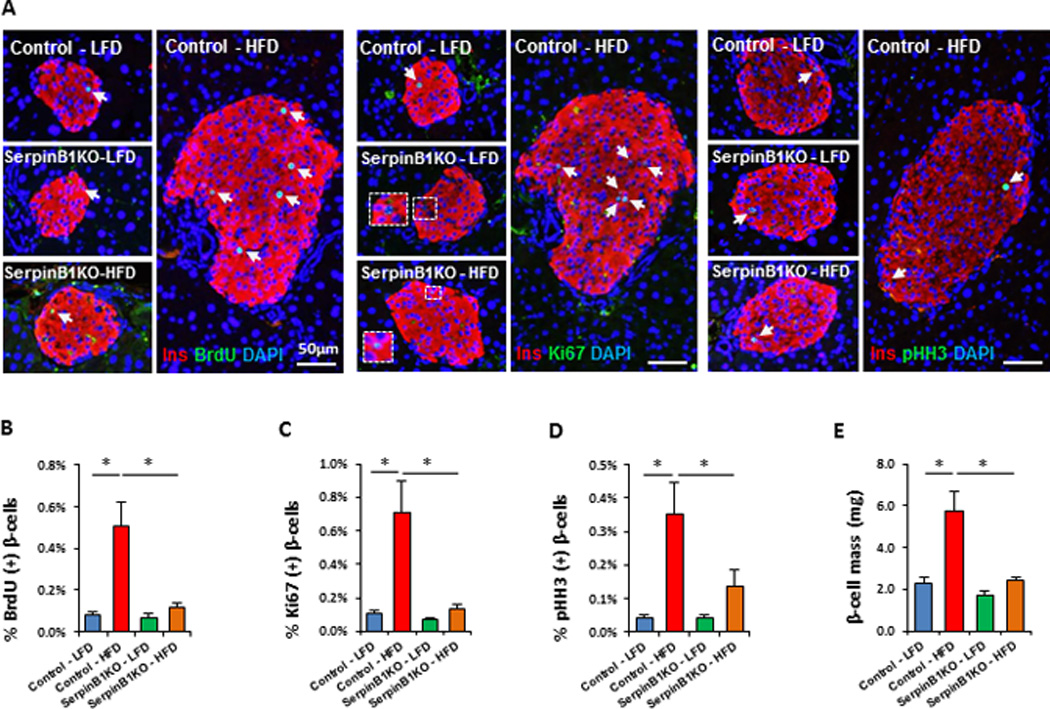
Eight week old control or serpinb1a−/− (serpinB1KO) male mice were challenged with low fat diet (LFD) or HFD for 30 weeks. Five hours before sacrificing, mice were injected with BrdU (100 mg/kg body weight). A. Representative images of pancreases co-immunostained for BrdU and insulin and DAPI (left panel). Representative images of pancreases co-immunostained for Ki67 and insulin and DAPI (middle panel). Representative images of pancreases co-immunostained for pHH3 and insulin and DAPI (right panel). B. Quantification of BrdU+ insulin+ cells (in A). C. Quantification of Ki67+ insulin+ cells (in A). D. Quantification of pHH3+ insulin+ cells (in A). E. Measurement of β-cell mass. Data represent mean ± SEM. *p ≤ 0.05, (n=4–6 per group). Immunostaining for BrdU and Ki67 markers, shown in (A), were performed on consecutive sections. Arrows indicate proliferating cells.
SerpinB1 activates proteins in the growth factor signaling pathway
To demonstrate whether protease inhibitory activity is critical for enhancement of β-cell proliferation by SerpinB1, we tested different commercially available SerpinB1 recombinant proteins that bear a tag sequence at the N- or C-terminus. As reported previously, insect cell derived SerpinB1, which is identical to the native protein (Cooley et al., 1998), forms a covalent complex (approximately 66 kD) with each of its target proteases (Cooley et al., 2001); this is shown for human neutrophil elastase (NE) and porcine pancreatic elastase (PE) (Figure 5A). Secondly, peptidase assays demonstrated that insect cell-derived untagged SerpinB1 from two independent preparations dose-dependently decreased activity of these proteases (shown for NE); however, the commercial proteins that are tagged at the N-terminus (GeneCopoeia or OriGene) or C-terminus (OriGene) only minimally inhibited peptidase activity of NE (Figure 5B). GeneCopoeia N-tagged SerpinB1 and OriGene C-tagged serpinB1 formed small amounts of complex, which was maximal with <0.1 or 0.3 molar equivalents NE, respectively, consistent with low inhibition in the peptidase assay; the OriGene C-tagged serpinB1 was also partially degraded (Figure 5C). For OriGene N-tagged serpinB1, no complex was detected on incubation with 0.3 molar equivalents of NE, and the recombinant serpin was completely degraded by NE; proteolytic degradation of the serpin by NE was confirmed by inactivating NE with DFP (diisopropyl fluorophosphate) (Amrein and Stossel, 1980) before use (Figure 5C). Insect cell-derived untagged serpinB1 was nearly quantitatively converted to complex or was further converted to the post-complex species, and importantly, no active 26 kDa NE band remained (Figure 5C). GeneCopoeia serpinB1, which lacks the ability to form a complex with neutrophil elastase and is unable to reduce peptidase activity, did not stimulate β-cell proliferation as opposed to untagged serpinB1 (Figure 5D). These findings suggest that the ability to inhibit protease is a requirement that is necessary for the β-cell proliferation-enhancing action of SerpinB1.
Fig. 5. Protease inhibitory activity is involved in SerpinB1 enhancement of β-cell proliferation.
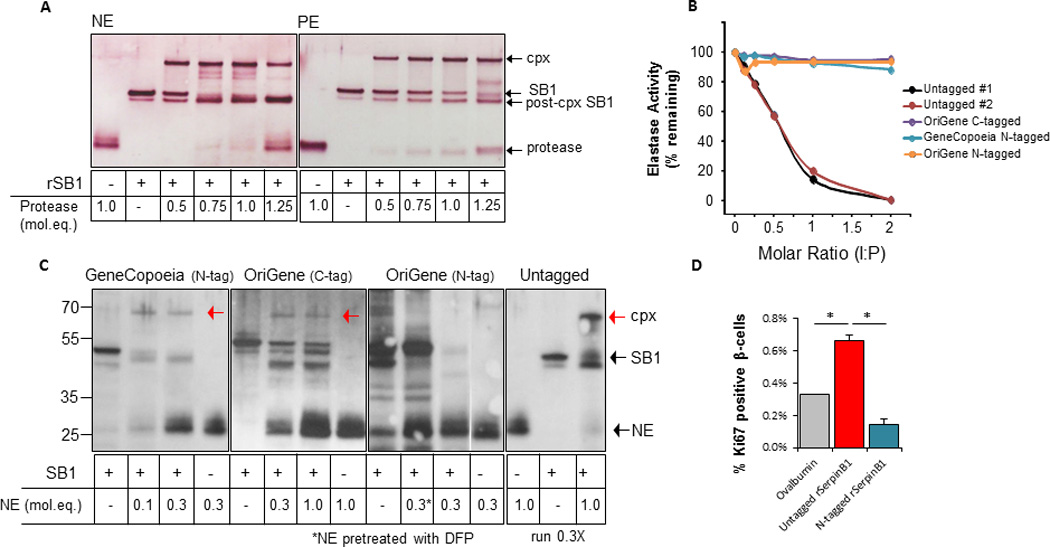
A. Activity of recombinant human SerpinB1 demonstrated by covalent complex formation with protease as previously described (Cooley et al., 2001). SerpinB1 (160ng), generated in insect cells (see Methods), was incubated with the indicated molar equivalents (mol.eq.) of human neutrophil elastase (NE) or porcine pancreatic elastase (PE) in 20 µl for 5 min at 37°C. Shown are reduced SDS gels gold-stained for total protein. Arrows indicate active SerpinB1 (42 kD), NE or PE (26 kD), complex (cpx, 66 kD) and post-cpx (inactive) SerpinB1 (38 kD). B. Activities of commercial recombinant SerpinB1 preparations and two preparations of insect cell-derived untagged SerpinB1 examined by peptidase inhibition. NE (500 ng) was combined with the indicated molar equivalents of SerpinB1 preparations in 150 µl, and the mixtures were incubated at 37C for 3 min. The substrate Ala-Ala-Pro-Val-p-nitroanilide was added and the change of OD405 nm was measured over 5 min. The abscissa shows the molar inhibitor:proteinase (I:P) ratio during the 3 min reaction. C. Activities of preparations of recombinant human serpinB1 examined by complex formation with NE. Equal amounts of SerpinB1 preparation (160ng based on suppliers' information) was incubated with the indicated molar equivalents of NE for 5 min at 37°C. Shown are gold-stained reduced SDS gels. The three commercial products were examined on separate gels; insect cell-derived untagged SerpinB1 was examined on the same gel as the GeneCopoeia preparation, but only 1/3 of the reaction was run to avoid overloading. The NE control lane is shown twice (lanes 4 and 13) in lane 10, NE was inactivated with DFP (diisopropyl fluorophosphate) prior to incubation with the serpin. Red arrows in lanes 3, 7 and 15 indicate the covalent SerpinB1-NE complex. D. Isolated islets of naive wild-type mice were stimulated with ovalbumin, insect cell derived untagged SerpinB1 or N-tagged SerpinB1 from GeneCopoeia (1µg/ml). Islets were embedded in agarose and immunostained for insulin and Ki67 and the nuclei were stained with DAPI. Quantification of Ki67+ insulin+ cells. Data represent mean ± SEM, *p < 0.05, (n= 3 per group).
To gain initial insights into the signaling pathways mediating β-cell proliferation in response to SerpinB1, we considered a phosphoproteomics approach. Protein phosphorylation has been long accepted as a major currency in signal transduction pathways and cell proliferation is known to be regulated by signaling modules that include the MAP kinase pathways. Further, measurement of phosphorylation dynamics represents a more direct way to identify potential pathways and regulatory targets compared to other techniques such as gene expression profiling. Briefly, isolated islets from C57Bl/6 male mice were cultured for 10, 30 or 120 min in the presence of 1µg/ml of ovalbumin or SerpinB1. Subsequently, islets were subjected to phosphopeptide enrichment and LC-MS/MS analysis (Mertins et al., 2014) (Figure 6A). As shown by the HeatMap (Figure 6B), a 10 min-treatment with SerpinB1 had a minimal effect on the islet phosphoproteome. However, islets incubated with SerpinB1 for 30 or 120 min exhibited an enhanced phosphorylation of ~250 proteins with at least two-fold change when compared to islets cultured with ovalbumin (for additional details, see Table S4). The modulation of several phosphoproteins identified at 30 min was sustained 2 hr after SerpinB1 treatment. Ingenuity Pathway Analysis (IPA) revealed that SerpinB1 activated key proteins in the growth factor (insulin/IGF-1) signaling cascade. In early events (within 10 min), SerpinB1 stimulated MAPK3 phosphorylation, a kinase previously implicated in the proliferation of β-cells (Hayes et al., 2013). Treatment for 30 or 120 min were characterized by activation of several proteins in the insulin/IGF-1 signaling cascade including IRS-2 (Kubota et al., 2004; Withers et al., 1998) and GSK3 (Liu et al., 2010). Finally, phosphoproteomics analyses also revealed increased phosphorylation of several proteins regulating cell survival and function including protein kinase cAMP-dependent regulatory subunits (PRKAR1A, PRKAR1B and PRKAR2B) (Hussain et al., 2006; Jhala et al., 2003), and phosphodiesterase 3B (PDE3B) (Harndahl et al., 2002). In independent experiments we confirmed, by western blots, the altered phosphorylation of MAPK, PRKAR2B and GSK3 subunits in response to treatment with SerpinB1 (Figure 6C–E; lower panels show quantification). Incubation of freshly isolated islets with serpinB1 did not significantly affect glucose-stimulated secretion compared to controls suggesting that effects of serpinB1 on islet β-cell proliferation do not adversely impact insulin secretion under the conditions tested (Figure 6F). Together, these data suggest that SerpinB1 enhances proliferation/survival by modulating proteins in the growth factor signaling pathway.
Fig. 6. SerpinB1 activates proteins in the growth factor signaling pathway.
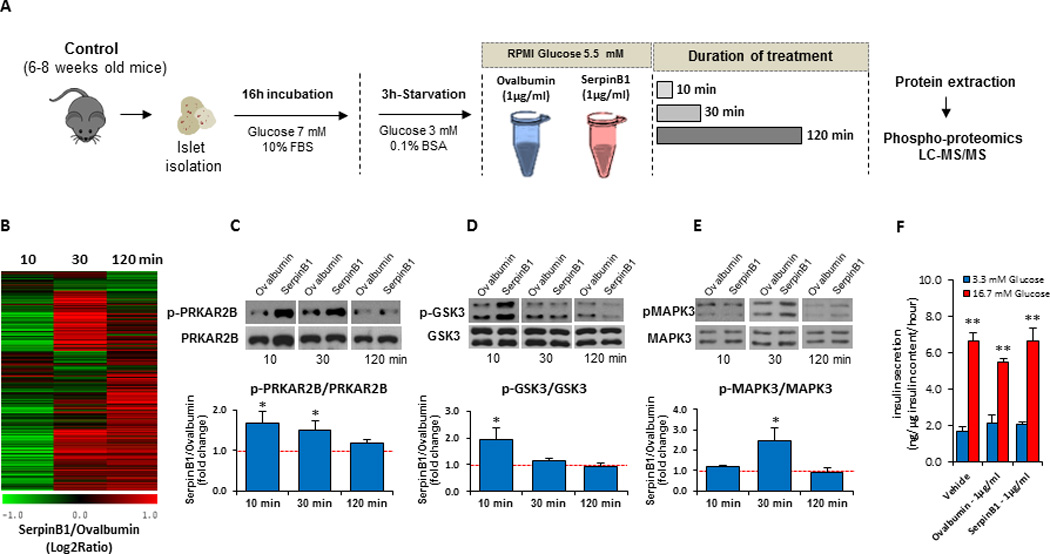
A. Schematic of experimental plan. Islets (100) isolated from male C57Bl/6 mice were treated for 10, 30 or 120 min (n=3) with (1 µg/ml) ovalbumin or rSerpinB1 (insect cell derived), and islet lysates were analyzed by LC-MS/MS phosphoproteomics. B. Heat map of the relative abundances of ~1,100 phosphopeptides in islets stimulated with SerpinB1 vs. ovalbumin. The relative abundances were displayed as Log2 Ratio (serpinB1/ovalbumin). C. Western blots (upper panel) and quantification (lower panel) of p-PRKAR2B/PRKAR2B in response to SerpinB1. Data represent mean ± SEM, *p < 0.05, (n= 5 per group) D. Western blots (upper panel) and quantification (lower panel) of p-GSK3/GSK3 in response to SerpinB1. Data represent mean ± SEM, *p < 0.05, (n= 5 per group). E. Western blots (upper panel) and quantification (lower panel) of p-MAPK3/MAPK3 in response to SerpinB1. Data represent mean ± SEM, *p < 0.05, (n= 5 per group). F. Glucose-stimulated insulin secretion (GSIS) in presence of vehicle, ovalbumin (1µg/ml) or rSerpinB1 (1µg/ml). Data represent mean ± SEM, **p < 0.01, (n=4 per group).
Discussion
Identification of molecules that have the ability to enhance proliferation of terminally differentiated cells is a desirable goal in regenerative medicine, particularly in diabetes where β-cell numbers are reduced. Here, we identified serpinB1 as an endogenous liver-derived secretory protein that stimulates human, mouse and zebrafish β-cell proliferation.
One interesting aspect of serpinB1 viewed as a secretory molecule is its lack of the classical hydrophobic signal peptide. Our data indicate that inflammation stimulates unconventional secretion of serpinB1 in a caspase-1-dependent manner. It is important to note, however, that the levels of several circulating cytokines in the LIRKO model are comparable to those observed in age-matched controls (El Ouaamari et al., 2013) and hence excludes systemic inflammation as a physiological factor triggering serpinB1 release in vivo. It is possible that the absence of insulin signaling in the liver interferes with caspase-1 activation and thus serpinB1 release. This notion is compatible with a previous report suggesting the suppressive role of insulin/IGF-1 in caspase-1 processing (Jung et al., 1996) and is consistent with increased levels of active caspase-1 in LIRKO-derived hepatocytes that are blind to insulin.
Since inhibition of proteases is SerpinB1’s reported biochemical function to date (Cooley et al., 2001), we postulated that the enhancing effect of SerpinB1 on β-cell proliferation involves the intermediacy of a protease. Indeed, recombinant SerpinB1 proteins lacking the ability to inhibit protease activity were unable to enhance β-cell proliferation in vitro. This observation suggests that SerpinB1 neutralizes a protease that would otherwise interfere with proliferation. In fact, the small molecule inhibitors of elastases, GW311616A and sivelestat, directly enhanced proliferation of mouse and human insulin-producing cells. The parallel findings for GW311616A, sivelestat and SerpinB1 make elastases strong candidates. While SerpinB1 action could be explained by its ability to modulate phosphorylation of key molecules (e.g. MAPK3, GSK3β/α, and PKA) of the insulin/IGF-1 growth/survival pathways, it is unclear how SerpinB1 precisely regulates these pathways. One possibility is that these pathways are activated through SerpinB1-mediated protease inhibition, particularly inhibition of elastase molecules known to be expressed in pancreatic β-cells (Kutlu et al., 2009). This idea is consistent with previous reports suggesting the role for neutrophil elastase in modulating proteins in the insulin/IGF-1 signaling pathway (Bristow et al., 2008; Houghton et al., 2010; Talukdar et al., 2012). Elucidation of interactions with other proteases such as proteinase-3 and cathepsin G in the β-cell and its potential role in regulating insulin sensitivity will further assist in deciphering the signaling pathways activated by SerpinB1. Alternative possibilities that require further investigation include interactions with protease activated receptors (PARs) which are expressed in islets (Shirakawa J, El Ouaamari A, Kulkarni RN, unpublished data).
Using zebrafish we determined that serpinB1’s ability to potentiate β-cell proliferation is conserved from fish to mammals. Moreover, in zebrafish we showed that serpinB1 can potentiate β-cell proliferation in vivo analogous to the in vivo effects we observed in mouse and human islets. By ablating the β-cells in zebrafish we also observed that serpinB1 can stimulate β-cell regeneration and warrants studies to examine its role during β-cell development.
In sum, the identification of SerpinB1 as a conserved endogenous secretory protein that promotes proliferation of β-cells across species constitutes an important step to achieve regeneration of functional β-cells. While it is likely that additional factors will be identified the next challenge will be to explore whether one or a combination of these factors can safely, specifically and reversibly enhance human β-cell mass with the long term goal of restoring normoglycemia in patients with diabetes.
Experimental procedures
Animals
All mice studied were 6–8-wk old males on the C57BL/6 background except where indicated otherwise. Mice were housed in pathogen-free facilities and maintained in the Animal Care Facilities at Joslin Diabetes Center, Boston, MA, Foster Biomedical Research Laboratory, Brandeis University, Waltham, MA or Boston Children’s Hospital. Studies conducted and protocols used were approved by the Institutional Animal Care and Use Committees of the Joslin Diabetes Center and/or Brandeis University and/or Boston Children’s Hospital and were in accordance with National Institute of Health guidelines. See the Supplemental Experimental Procedures for details of the animal genotypes. For short term studies, sixteen-week-old serpinB1KO and age-matched wild-type male mice were challenged with HFD (Research Diet, catalog# D12492) for 10 weeks. For long term studies, eight-week-old serpinB1KO and age-matched wild-type male mice were fed with low fat diet (Research Diet, catalog# D12450J) or HFD (Research Diet, catalog# D12492) for 30 weeks.
LECM and HCM preparation
The preparation of liver explant-conditioned media (LECM) and hepatocyte conditioned media (HCM) have been described previously (El Ouaamari et al., 2013). See Supplemental Experimental Procedures for additional information.
LC-MS/MS based proteomics
Proteomic analyses were performed as previously described (Zhou et al., 2010). See Supplemental Experimental Procedures for additional information.
Mouse Islet Studies
Islets were isolated from 6–8 week old male C57BL/6 mice using intra-ductal collagenase technique (El Ouaamari et al., 2013). Islets were handpicked and cultured overnight in RPMI 1640 media containing 7 mM glucose and 10% fetal bovine serum (FBS) and penicillin/streptomycin (1% v/v). After 3h-starvation in RPMI 1640 media containing 3 mM glucose and 0.1%BSA, islets were stimulated as indicated (with recombinant protein or small molecules) for 48h and then embedded in agarose and paraffin, sectioned and used for immunostaining studies as described below and in (El Ouaamari et al., 2013).
Human islet studies
Human islets were obtained from the Integrated Islet Distribution Program. All studies and protocols used were approved by the Joslin Diabetes Center’s Committee on Human Studies (CHS#5-05). Upon receipt, islets were cultured overnight in Miami Media #1A (Cellgro). The islets were then starved in Final Wash/Culture Media (Cellgro) for 3-hours before being stimulated with Miami Media #1A supplemented with sivelestat or GW311616A. Twenty four hrs later, islets were embedded in agarose and used for immunostaining studies (described below).
Immunostaining studies
Pancreases and in vitro stimulated islets were analyzed by immunostaining using anti-Ki67 (BD), anti-phospho-histone H3 (pHH3)(Millipore), anti-BrdU (Dako), anti-insulin (Abcam), or anti-glucagon (Sigma-Aldrich) antibodies. Quantification of replicating β- and α-cells and calculation of β-cell mass was performed as described previously (El Ouaamari et al., 2013).
Phosphoproteomics analysis
Phosphoproteomics analyses were performed as described in Supplemental Experimental Procedures. To validate the phosphoproteomics findings, frozen SerpinB1-treated and ovalbumin-treated islets were lysed in RIPA buffer (150mM NaCl, 10mM Tris, pH 7.2, 1% Triton X-100, 1% deoxycholate, 5mM EDTA) containing 200µM orthovanadate, protease and phosphatase inhibitors (Sigma-Aldrich) (Liew et al., 2014) and subjected to western blot analyses. pMAPK3, total MAPK3 and total GSK3 antibodies are from Cell Signaling. pPRKAR2B and total PRKAR2B are from Santa Cruz. pGSK3 antibody is from Millipore.
RT-PCR
Total RNA was extracted, reverse transcribed and quantitative PCR was performed as outlined in Supplemental Experimental Procedures.
Statistical analysis
All data are presented as mean ± SEM. Data were analyzed using unpaired, two-tailed Student’s ‘t’ test, ANOVA or multivariate analyses as appropriate and a “p” value < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgements
We thank C. Ronald Kahn MD for sharing the LIRKO mouse model, Ping Li MD for assistance with statistical analyses, Dr. Lauge Schäffer (Novo Nordisk) for providing the S961 compound, Drs. Wei-Min Chen and Michele Sale (University of Virginia) for useful discussions and Sandra Roger for technical assistance. This work was supported by NIH RO1 DK67536 and RO1 DK103215 (to R.N.K.); RO1 DK55523 (to A.D. and R.N.K.); NIH RO1 HL066548, R21 AI103407 and an RRRC award from Boston Children’s Hospital (to E.R.O), R01 DK 074795 and P41 GM103493 (to R.D.S.); UC4 DK104167 (to W.J.Q. and R.N.K.), Société Francophone du Diabète, Association Française des Diabétiques, American Diabetes Association and JDRF (3-APF-2014-182-A-N) (to A.E.) and a grant from the Juvenile Diabetes Research Foundation/Sanofi Aventis Strategic Alliance (17-2011-644) (R.N.K.), and P30 DK036836 (DRC). Zebrafish experiments were performed at the Karolinska Institute and supported by the Ragnar Söderbergs Foundation and the Swedish Research Council (O.A). Proteomics and phosphoproteomics experiments were performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by Department of Energy and located at Pacific Northwest National Laboratory, which is operated by Battelle Memorial Institute for the DOE under Contract DE-AC05-76RL0 1830.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Information
Supplemental information includes Supplementary Experimental Procedures, five figures, and three tables.
Author Contributions: A.E. and R.N.K. conceived of the idea, designed experiments, analyzed the data, and wrote/edited the manuscript. Individual experimental contributions are as follows: E.R.O. and W.J.Q. contributed equally to design of experiments providing reagents and writing/editing the manuscript. J.Z., M.A.G., R.D.S. and W.J.Q. were responsible for proteomics and phospho-proteomics experiments. E.D. contributed to islet isolation and transplantation studies. N.G., J.H., J.B., J.S, D.F.D., S.K., S.B., G.Q., C.W.L. provided technical assistance. H.D.B. conducted serpinB1 secretion studies in keratinocytes. R.M. assisted with mouse experiments. L.H, J.C., and Y.G. conducted mouse experiments, and ELISA and biochemical assays. A.B.G. provided human samples and contributed to analysis of human ELISA assays. A.D. and P.J. provided data on the serpinB1 variant. C.K. and O.A. conducted zebrafish studies. All authors read and approved the manuscript.
References
- Amrein PC, Stossel TP. Prevention of degradation of human polymorphonuclear leukocyte proteins by diisopropylfluorophosphate. Blood. 1980;56:442–447. [PubMed] [Google Scholar]
- Andersson O, Adams BA, Yoo D, Ellis GC, Gut P, Anderson RM, German MS, Stainier DY. Adenosine signaling promotes regeneration of pancreatic beta cells in vivo. Cell metabolism. 2012;15:885–894. doi: 10.1016/j.cmet.2012.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker CE, Creagh EM, O'Neill LA. Rab39a binds caspase-1 and is required for caspase-1-dependent interleukin-1beta secretion. The Journal of biological chemistry. 2009;284:34531–34537. doi: 10.1074/jbc.M109.046102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarafa C, Remold-O'Donnell E. The ovalbumin serpins revisited: perspective from the chicken genome of clade B serpin evolution in vertebrates. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:11367–11372. doi: 10.1073/pnas.0502934102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A. Human beta-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes. 2014;63:819–831. doi: 10.2337/db13-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitard C. Pancreatic islet autoimmunity. Presse Med. 2012;41:e636–e650. doi: 10.1016/j.lpm.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Bristow CL, Wolkowicz R, Trucy M, Franklin A, Di Meo F, Kozlowski MT, Winston R, Arnold RR. NF-kappaB signaling, elastase localization, and phagocytosis differ in HIV-1 permissive and nonpermissive U937 clones. J Immunol. 2008;180:492–499. doi: 10.4049/jimmunol.180.1.492. [DOI] [PubMed] [Google Scholar]
- Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102–110. doi: 10.2337/diabetes.52.1.102. [DOI] [PubMed] [Google Scholar]
- Chakraborty R, Bhatt KH, Sodhi A. Ultraviolet B induces high mobility group box 1 release from mouse peritoneal macrophages in vitro via caspase-1 mediated secretion pathway. Immunobiology. 2013;218:135–144. doi: 10.1016/j.imbio.2012.02.006. [DOI] [PubMed] [Google Scholar]
- Cooley J, Mathieu B, Remold-O'Donnell E, Mandle RJ. Production of recombinant human monocyte/neutrophil elastase inhibitor (rM/NEI) Protein expression and purification. 1998;14:38–44. doi: 10.1006/prep.1998.0951. [DOI] [PubMed] [Google Scholar]
- Cooley J, Takayama TK, Shapiro SD, Schechter NM, Remold-O'Donnell E. The serpin MNEI inhibits elastase-like and chymotrypsin-like serine proteases through efficient reactions at two active sites. Biochemistry. 2001;40:15762–15770. doi: 10.1021/bi0113925. [DOI] [PubMed] [Google Scholar]
- D'Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, Moorman MA, Kroon E, Carpenter MK, Baetge EE. Production of pancreatic hormone-expressing endocrine cells from human embryonic stem cells. Nature biotechnology. 2006;24:1392–1401. doi: 10.1038/nbt1259. [DOI] [PubMed] [Google Scholar]
- Dirice E, Kahraman S, Jiang W, El Ouaamari A, De Jesus DF, Teo AK, Hu J, Kawamori D, Gaglia JL, Mathis D, et al. Soluble factors secreted by T cells promote beta-cell proliferation. Diabetes. 2014;63:188–202. doi: 10.2337/db13-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- El Ouaamari A, Kawamori D, Dirice E, Liew CW, Shadrach JL, Hu J, Katsuta H, Hollister-Lock J, Qian WJ, Wagers AJ, et al. Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep. 2013;3:401–410. doi: 10.1016/j.celrep.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmeyer L, Keller M, Niklaus G, Hohl D, Werner S, Beer HD. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Current biology : CB. 2007;17:1140–1145. doi: 10.1016/j.cub.2007.05.074. [DOI] [PubMed] [Google Scholar]
- Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, Wright AJ, Atkinson MA, Rhodes CJ. Formation of a human beta-cell population within pancreatic islets is set early in life. The Journal of clinical endocrinology and metabolism. 2012;97:3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greiner DL, Brehm MA, Hosur V, Harlan DM, Powers AC, Shultz LD. Humanized mice for the study of type 1 and type 2 diabetes. Annals of the New York Academy of Sciences. 2011;1245:55–58. doi: 10.1111/j.1749-6632.2011.06318.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harndahl L, Jing XJ, Ivarsson R, Degerman E, Ahren B, Manganiello VC, Renstrom E, Holst LS. Important role of phosphodiesterase 3B for the stimulatory action of cAMP on pancreatic beta-cell exocytosis and release of insulin. The Journal of biological chemistry. 2002;277:37446–37455. doi: 10.1074/jbc.M205401200. [DOI] [PubMed] [Google Scholar]
- Hayes HL, Moss LG, Schisler JC, Haldeman JM, Zhang Z, Rosenberg PB, Newgard CB, Hohmeier HE. Pdx-1 activates islet alpha- and beta-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Molecular and cellular biology. 2013;33:4017–4029. doi: 10.1128/MCB.00469-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC, Rahier J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia. 2011;54:1720–1725. doi: 10.1007/s00125-011-2118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, Stolz DB, Land SR, Marconcini LA, Kliment CR, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nature medicine. 2010;16:219–223. doi: 10.1038/nm.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain MA, Porras DL, Rowe MH, West JR, Song WJ, Schreiber WE, Wondisford FE. Increased pancreatic beta-cell proliferation mediated by CREB binding protein gene activation. Molecular and cellular biology. 2006;26:7747–7759. doi: 10.1128/MCB.02353-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhala US, Canettieri G, Screaton RA, Kulkarni RN, Krajewski S, Reed J, Walker J, Lin X, White M, Montminy M. cAMP promotes pancreatic beta-cell survival via CREB-mediated induction of IRS2. Genes & development. 2003;17:1575–1580. doi: 10.1101/gad.1097103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y, Le Lay J, Yu M, Naji A, Kaestner KH. Elevated mouse hepatic betatrophin expression does not increase human beta-cell replication in the transplant setting. Diabetes. 2014;63:1283–1288. doi: 10.2337/db13-1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung Y, Miura M, Yuan J. Suppression of interleukin-1 beta-converting enzyme-mediated cell death by insulin-like growth factor. The Journal of biological chemistry. 1996;271:5112–5117. doi: 10.1074/jbc.271.9.5112. [DOI] [PubMed] [Google Scholar]
- Kassem SA, Ariel I, Thornton PS, Scheimberg I, Glaser B. Beta-cell proliferation and apoptosis in the developing normal human pancreas and in hyperinsulinism of infancy. Diabetes. 2000;49:1325–1333. doi: 10.2337/diabetes.49.8.1325. [DOI] [PubMed] [Google Scholar]
- Kawabata K, Suzuki M, Sugitani M, Imaki K, Toda M, Miyamoto T. ONO-5046, a novel inhibitor of human neutrophil elastase. Biochemical and biophysical research communications. 1991;177:814–820. doi: 10.1016/0006-291x(91)91862-7. [DOI] [PubMed] [Google Scholar]
- Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- Kondegowda NG, Fenutria R, Pollack IR, Orthofer M, Garcia-Ocana A, Penninger JM, Vasavada RC. Osteoprotegerin and Denosumab Stimulate Human Beta Cell Proliferation through Inhibition of the Receptor Activator of NF-kappaB Ligand Pathway. Cell metabolism. 2015;22:77–85. doi: 10.1016/j.cmet.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroon E, Martinson LA, Kadoya K, Bang AG, Kelly OG, Eliazer S, Young H, Richardson M, Smart NG, Cunningham J, et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nature biotechnology. 2008;26:443–452. doi: 10.1038/nbt1393. [DOI] [PubMed] [Google Scholar]
- Kubota N, Terauchi Y, Tobe K, Yano W, Suzuki R, Ueki K, Takamoto I, Satoh H, Maki T, Kubota T, et al. Insulin receptor substrate 2 plays a crucial role in beta cells and the hypothalamus. The Journal of clinical investigation. 2004;114:917–927. doi: 10.1172/JCI21484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni RN, Mizrachi EB, Ocana AG, Stewart AF. Human beta-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes. 2012;61:2205–2213. doi: 10.2337/db12-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlu B, Burdick D, Baxter D, Rasschaert J, Flamez D, Eizirik DL, Welsh N, Goodman N, Hood L. Detailed transcriptome atlas of the pancreatic beta cell. BMC medical genomics. 2009;2:3. doi: 10.1186/1755-8794-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CW, Assmann A, Templin AT, Raum JC, Lipson KL, Rajan S, Qiang G, Hu J, Kawamori D, Lindberg I, et al. Insulin regulates carboxypeptidase E by modulating translation initiation scaffolding protein eIF4G1 in pancreatic beta cells. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E2319–E2328. doi: 10.1073/pnas.1323066111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Tanabe K, Baronnier D, Patel S, Woodgett J, Cras-Meneur C, Permutt MA. Conditional ablation of Gsk-3beta in islet beta cells results in expanded mass and resistance to fat feeding-induced diabetes in mice. Diabetologia. 2010;53:2600–2610. doi: 10.1007/s00125-010-1882-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald SJ, Dowle MD, Harrison LA, Shah P, Johnson MR, Inglis GG, Clarke GD, Smith RA, Humphreys D, Molloy CR, et al. The discovery of a potent, intracellular, orally bioavailable, long duration inhibitor of human neutrophil elastase--GW311616A a development candidate. Bioorganic & medicinal chemistry letters. 2001;11:895–898. doi: 10.1016/s0960-894x(01)00078-6. [DOI] [PubMed] [Google Scholar]
- Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Molecular & cellular proteomics : MCP. 2014;13:1690–1704. doi: 10.1074/mcp.M113.036392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:193–205. doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- Nickel W. Pathways of unconventional protein secretion. Current opinion in biotechnology. 2010;21:621–626. doi: 10.1016/j.copbio.2010.06.004. [DOI] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remold-O'Donnell E. The ovalbumin family of serpin proteins. FEBS letters. 1993;315:105–108. doi: 10.1016/0014-5793(93)81143-n. [DOI] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O'Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nature biotechnology. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Rieck S, Zhang J, Li Z, Liu C, Naji A, Takane KK, Fiaschi-Taesch NM, Stewart AF, Kushner JA, Kaestner KH. Overexpression of hepatocyte nuclear factor-4alpha initiates cell cycle entry, but is not sufficient to promote beta-cell expansion in human islets. Mol Endocrinol. 2012;26:1590–1602. doi: 10.1210/me.2012-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human beta-cells. PloS one. 2012;7:e35801. doi: 10.1371/journal.pone.0035801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PG, Irving JA, Lomas DA, Luke CJ, Moyer RW, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. The Journal of biological chemistry. 2001;276:33293–33296. doi: 10.1074/jbc.R100016200. [DOI] [PubMed] [Google Scholar]
- Stewart AF, Hussain MA, Garcia-Ocana A, Vasavada RC, Bhushan A, Bernal-Mizrachi E, Kulkarni RN. Human beta-cell proliferation and intracellular signaling: part 3. Diabetes. 2015;64:1872–1885. doi: 10.2337/db14-1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talukdar S, Oh da Y, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nature medicine. 2012;18:1407–1412. doi: 10.1038/nm.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walpita D, Hasaka T, Spoonamore J, Vetere A, Takane KK, Fomina-Yadlin D, Fiaschi-Taesch N, Shamji A, Clemons PA, Stewart AF, et al. A human islet cell culture system for high-throughput screening. Journal of biomolecular screening. 2012;17:509–518. doi: 10.1177/1087057111430253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Alvarez-Perez JC, Felsenfeld DP, Liu H, Sivendran S, Bender A, Kumar A, Sanchez R, Scott DK, Garcia-Ocana A, et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nature medicine. 2015;21:383–388. doi: 10.1038/nm.3820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- Yi P, Park JS, Melton DA. Betatrophin: A Hormone that Controls Pancreatic beta Cell Proliferation. Cell. 2013 doi: 10.1016/j.cell.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhou JY, Schepmoes AA, Zhang X, Moore RJ, Monroe ME, Lee JH, Camp DG, Smith RD, Qian WJ. Improved LC-MS/MS spectral counting statistics by recovering low-scoring spectra matched to confidently identified peptide sequences. Journal of proteome research. 2010;9:5698–5704. doi: 10.1021/pr100508p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.