

Abstract

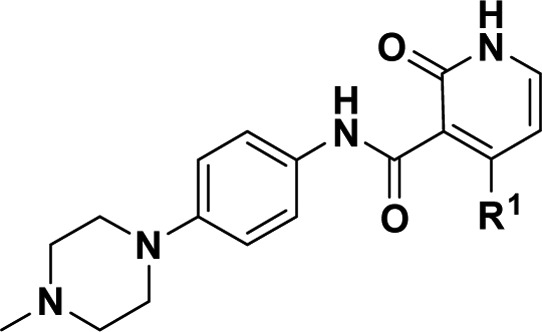
The rapid advancement of a series of noncovalent inhibitors of T790M mutants of EGFR is discussed. The optimization of pyridone 1, a nonselective high-throughput screening hit, to potent molecules with high levels of selectivity over wtEGFR and the broader kinome is described herein.
Keywords: Mutant EGFR, noncovalent, pyridone, T790M
Epidermal growth factor receptor (EGFR), a member of the ErbB family of tyrosine kinases, is involved in the activation of numerous crucial cell signaling pathways that play critical roles in cellular survival, proliferation, inhibition of apoptosis, and angiogenesis.1,2 EGFR overexpression has been found in 62% of nonsmall cell lung cancer (NSCLC) cases.3 While EGFR overexpression is considered a prognosticator of poor survival odds, NSCLCs harboring specific activating mutations in the tyrosine kinase domain of EGFR, including the point mutation L858R in exon 2 and deletions within exon 19 (e.g., residues 746–750), are known to confer hypersensitivity to tyrosine kinase inhibitors (TKIs) such as erlotinib and gefitinib.1 As inhibition of wild-type EGFR (wtEGFR) has been linked to dose-limiting toxicities, the increased inhibition of mutant EGFR relative to wtEGFR by TKIs allows for improved tolerability.4
While treatment of NSCLC harboring EGFR mutations with erlotinib or gefitinib offers initial strong clinical responses, these are followed by acquired resistance. Approximately 50–60% of such cases arise from a single secondary mutation within the kinase domain where the threonine gatekeeper residue, Thr790, is exchanged for methionine (T790M).1,5 This single point mutation, found along with an activating mutation, alters the binding pocket and renders erlotinib and gefitinib less effective.5−7 The compelling nature of the T790M EGFR mutants as therapeutic targets, along with an understanding of the relationship between wtEGFR inhibition and dose-limiting toxicities, led us to initiate efforts to identify inhibitors of the major resistance mutations of EGFR, namely, T790M/L858R (TMLR) and T790M/del(746–750) (TMdel), with selectivity over wtEGFR. In addition, a noncovalent inhibitor profile was selected to prevent the possible resistance mechanism known for covalent inhibitors, such as those against TMLR currently in the clinic.8−10 Herein we describe our efforts beginning with pyridone 1 to generate a potent and selective molecule for analysis of selectivity in cellular assays as well as develop SAR toward an in vivo tool compound.
A high throughput screen identified pyridone 1 as an inhibitor of TMLR with a Ki of 65 nM and LE11 of 0.3 (Table 1). Although relatively active and moderately ligand efficient, the initial lead had several liabilities requiring attention. Most importantly, compound 1 lacked selectivity over wtEGFR. In addition, improvement of the potency and selectivity over the kinome were essential as it showed greater than 70% percent inhibition for 67 kinases out of a panel of 220 kinases when tested at a 1 μM compound concentration (Supporting Information Table S1).
Table 1. Effects of C4 Modification on Potency and wtEGFR Selectivity.

Ki data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
In order to address liabilities and test key hypotheses, a synthetic route was developed allowing for rapid exploration at C3 and C4 (Scheme 1). For diversification at C4, pyridine 3 was first oxidized to carboxylic acid 4. Amide bond formation using various coupling conditions installed R1 at C3 and demethylation gave the pyridone. Final step diversification at C4 with R2 and R3 occurred via nucleophilic aromatic substitution (SNAr) to give 1–2, 5–22, and 38–39.
Scheme 1. Synthetic Routes Facilitating Diversification of Pyridone Scaffold.

Reagents and conditions: (a) 2-methyl-2-butene, NaClO2, NaPO4H2–H2O; (b) HBTU or oxalyl or thionyl chloride, NH2R1; (c) HCl; (d) NHR2R3, Δ; (e) R1C1NH2C2NH2, MeOH, Δ.
Diversification at C3 followed similar steps to those described above for compounds 23-26 and 28–36. From intermediate 4, demethylation followed by SNAr at C4 gave the pyridone late stage intermediate. The amide bond was then formed via the acid chloride. Benzimidazoles 27 and 37 were formed upon condensation of 3 with the required diamine prior to pyridone formation and coupling to HNR2R3.
An X-ray structure of compound 2 bound to TMLR (Figure 1B) informed our strategy to improve potency and selectivity over wtEGFR. The pyridone of compound 2 binds the protein hinge region through residues Met793 and Gln791. Two intramolecular hydrogen bonds, one between the amide NH and the pyridone carbonyl and a second between the amide carbonyl and the pyridone NHR, position the phenyl amide and the pyridone in a coplanar conformation, directing the phenyl-piperazine toward solvent. The C4 phenethyl substituent is then positioned toward a back pocket above the Met790 gatekeeper.
Figure 1.
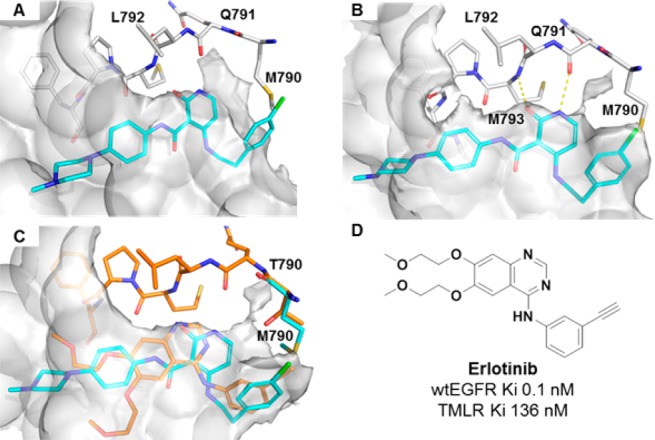
(A) Crystal structure of the apo-TMLR double mutant (PDB 5EDP). The pocket surface is shown in gray. Compound 2 is overlaid. (B) X-ray structure of compound 2 with the TMLR double mutant. Hinge residues Met793 and Gln791 are shown. The pocket surface is shown in gray. (C) Overlay of compound 2 (cyan) with the erlotinib crystal structure with wtEGFR (orange) showing the pocket created by Thr790 (gray surface) (PDB 1M17). (D) Structure of erlotinib with biochemical potency in wtEGFR and TMLR.
Comparison of the apo crystal structure of TMLR with that of 2 bound to TMLR shows an induced pocket above the Met gatekeeper upon binding 2 (Figure 1A,B). This region is similar to the crystal structure of erlotinib with wtEGFR (Figure 1C, orange with surface) where a large pocket above the gatekeeper is occupied by the phenyl alkyne.12 Overlay of 2 into the erlotinib wtEGFR crystal structure shows that the phenethyl group of 2 could occupy this space. This pocket appears important to activity for erlotinib, which is significantly more potent for wtEGFR than the double mutant (Figure 1D).13,14 By not inducing the “wtEGFR-like” pocket, and through effectively occupying the back pocket observed in the TMLR apo structure, we hypothesized selectivity would be gained over wtEGFR. Modeling suggested that by eliminating the ethylene linker between C4 and the phenyl ring (i.e., replacing the phenethylamine with an aniline) should effectively achieve this goal.
Indeed, truncating phenethyl (5) to the aniline (7) led to the first compound with selectivity over wtEGFR (Table 1), while the benzyl (6) was equipotent, yet not selective over wtEGFR. Compound 7’s loss in potency for wtEGFR while maintaining TMLR potency is presumably due to the shortened chain, leaving solvent-exposed hydrophobic space not filled by the ligand in wtEGFR. This space is occupied by the Met790 side chain in the T790M protein. Removal of the phenyl group altogether (8) decreased TMLR potency by less than 3-fold while maintaining selectivity over wtEGFR.
We next explored the aniline SAR. Substitution with chlorine at C4 negatively impacted potency relative to C2 and C3 (11 versus 9 and 10). Alternate C2 substitution led to increases in both potency and selectivity for tolyl 12 and methoxy 13, with 13 possessing 58-fold selectivity over wtEGFR. Comparing the crystal structures of compound 13 soaked into either TMLR (cyan) or wtEGFR (orange, Supporting Information Figure S1A) reveals the phenyl group leaving the large back pocket of wtEGFR free of ligand. In TMLR, the smaller pocket allows for hydrophobic contacts between the phenyl ring and the Met790 side chain and directs the methyl group of the methoxy along the side of the back pocket where it may achieve van der Waals contacts (Supporting Information Figure S1B). When the amine linker was substituted with a methyl group (tertiary amine 14), measurable potency for both the mutant and wild type protein was lost, presumably due to loss in planarity and hydrogen bonding with the amide.
Replacement of the phenyl ring with 6-membered heteroaryls was then undertaken. 2-Pyridine 16 and 2,4-pyrimidine 17 were equipotent to phenyl 7. Addition of an amine to the C3 position of 17, designed to target polar interactions with the side chains of catalytic Lys745 and Glu762 (Supporting Information Figure S2), generated the first single digit nanomolar compound of the series, with >200× selectivity over wtEGFR (compound 19). However, removal of one of the pyrimidine nitrogens of 19 erased this gain in potency (pyridine 20) potentially due to the diaminopyridine being positively charged at physiological pH, unlike the pyrimidine, and clashing with Lys745.15
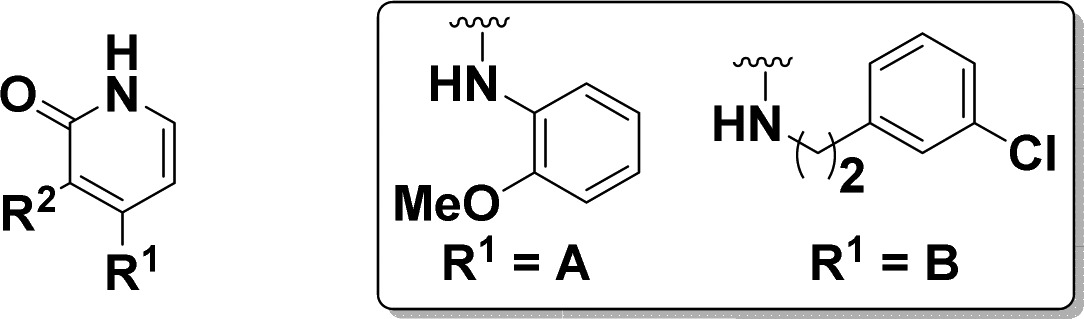
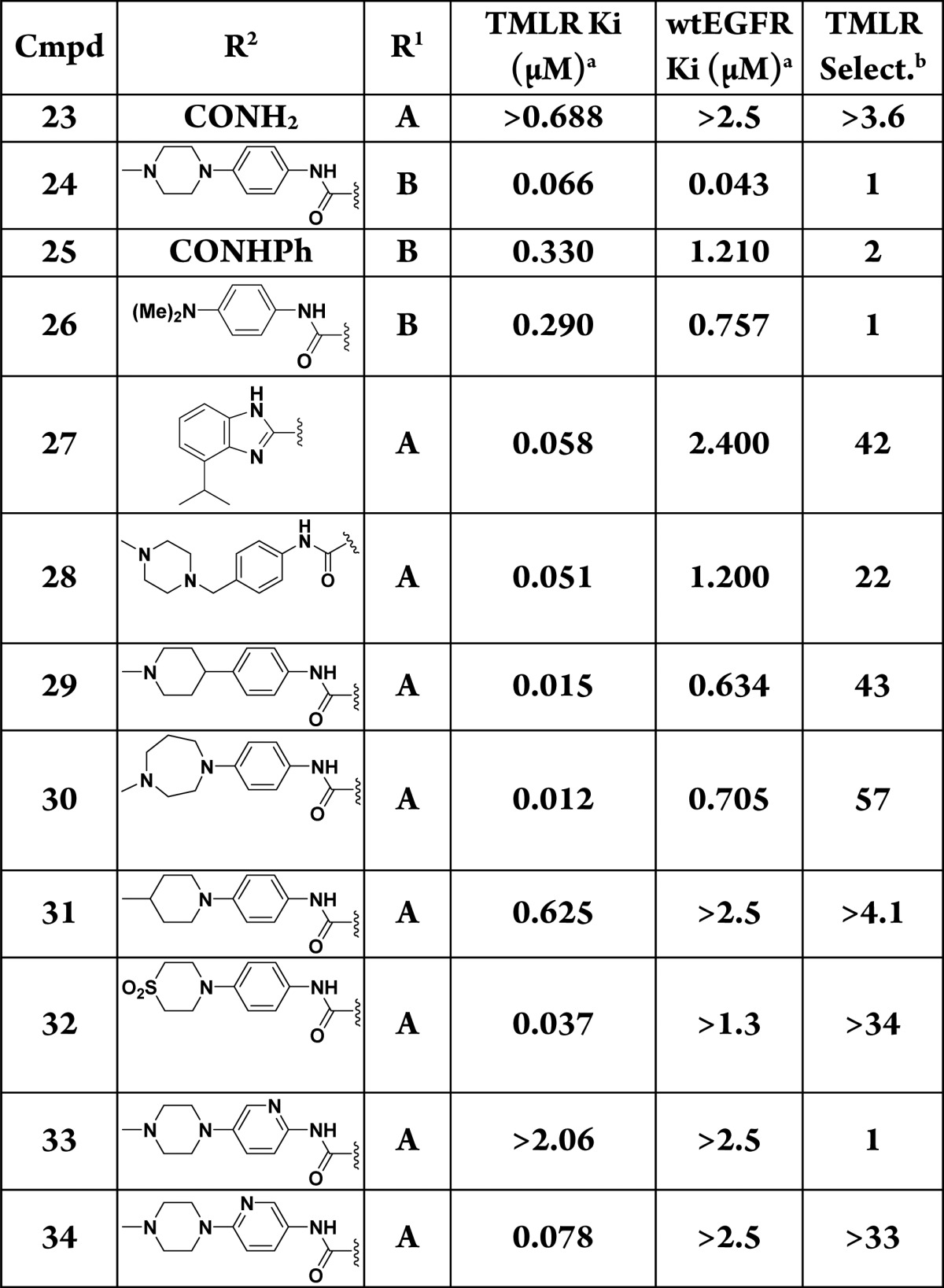
As we turned our attention to optimization of C3, the 2-methoxyaniline of compound 13 was retained due to its combination of potency and selectivity over wtEGFR. In order to understand the minimal motifs necessary for inhibition, a number of abridged amides were generated (Table 2). Truncation to the primary amide gave significant loss in activity (23) when compared to 13. Similar reductions in potency were found with the unsubstituted phenyl ring analogue (25) and the dimethylaniline (26), which lost 4 to 5 times that when compared to fully elaborated 24. Efforts to enforce conformational restriction of the amide led to benzimidazole 27, which maintained selectivity over wtEGFR with only a modest decrease in potency relative to compound 13. The isopropyl group on the phenyl ring was predicted to occupy the lipophilic space within the ribose pocket.9
Table 2. Effects of C3 Modification on TMLR Potency.
Ki data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
Focusing on the piperazine, we discovered several changes were tolerated (Table 2). Extension of the linker between the aryl ring and the piperazine (28), replacement of the linking nitrogen with carbon (29), and increase in the ring size (30) were all tolerated by TMLR. Interestingly, replacement of the terminal nitrogen with carbon (31) was not tolerated but replacement with a sulfone (32) was, with less than 2× loss in potency relative to 13, suggesting that polarity was beneficial in this region but a positive charge was not required.
Heteroaromatic replacements at C3 were also evaluated to reduce lipophilicity of compound 13 (LogD7.4 = 3.5) (Table 2). While pyridine 34 suffered a modest reduction in activity relative to compound 13, it did improve overall lipophilicity (LogD7.4 = 2.6) and kinetic solubility (65 μM vs 4 μM for compound 13). Pyridine 33 was not active, presumably due to a clash of the nitrogen lone-pair electrons with either the backbone carbonyl of Met793 (Figure 1) or the carbonyl of the amide depending on its conformation.
With C3 and C4 SAR in hand, we sought to understand if these changes improved selectivity over the broader kinome by examining the kinase selectivity against an abridged panel of those screened for compound 1. Kinases were selected based on strong inhibition seen with compound 1, diversity, or kinases known to show potential safety liabilities (Supporting Information, Table S2). While compounds 13 and 19 showed improved wtEGFR selectivity relative to 1, they maintained similar kinome selectivity and were potent inhibitors of JAK2 at a 1 μM concentration. In a full dose–response assay, 13 inhibited JAK2 with a 9 nM Ki (Table 3) and 10-fold selectivity relative to JAK1 (Ki 172 nM). That compounds with modest to high selectivity over wtEGFR would lack JAK selectivity is unsurprising as both TMLR and the JAK family contain a Met gatekeeper. Key amino acid differences between EGFR and numerous other kinases, including JAK family members, appeared to be more accessible from C3. This led us to hypothesize that further changes in this region would have greater impact on the kinome, our second selectivity goal.
Table 3. Selectivity over JAK Family Members.
| Cmpd | TMLR Ki (μM)a | wtEGFR Select.b | JAK1 Select.c | JAK2 Select.d |
|---|---|---|---|---|
| 13 | 0.019 | 57 | 9 | 0.5 |
| 19 | 0.004 | 213 | 3 | 2 |
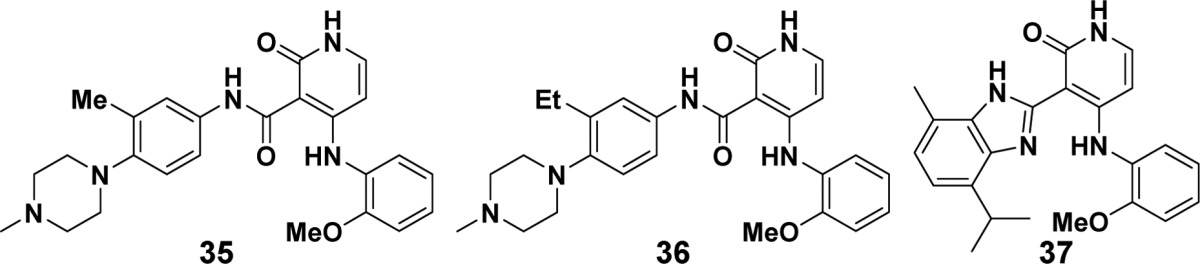
| 35 | 0.026 | 16 | 42 | 0.9 |
| 36 | 0.031 | > 80 | 104 | 4 |
| 27 | 0.058 | 42 | 2 | 0.2 |
| 37 | 0.088 | 27 | 6 | 0.3 |
Ki data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
Ratio of JAK1 Kiapp over EGFR(TMLR) Kiapp.
Ratio of JAK2 Kiapp over EGFR(TMLR) Kiapp.
In an effort to improve kinase selectivity, we focused on substitution of the phenyl ring to target key residue Leu792 in TMLR as has successfully been utilized for other kinase inhibitors seeking selectivity over JAK.16 This residue is a Tyr or Phe in the JAK family (Figure S3). By directing a methyl (35) or ethyl (36) substituent in this direction, TMLR potency and wtEGFR selectivity were comparable to 13, but markedly increased selectivity against JAK1 was realized, with a modest improvement in JAK2 selectivity for compound 36 (Table 3). Figure S3 shows the phenyl ring of 13 at the edge of the surface of an aligned JAK2 crystal structure. Substitution at C3 of the phenyl ring would clash with the surface of the JAK2 binding pocket. If the substitution occupied the alternate side of the phenyl ring, the piperazine ring would need to shift, and this in turn would clash with the surface. Either would result in a loss in affinity for JAK as is reflected in 36 and 35 to a lesser extent. The disconnect in selectivity between JAK1 and JAK2 may be due to plasticity of the hinge region for these two proteins due to their sequence variability.14
Benzimidazole 27 itself was not selective for TMLR over JAK family members but provided an alternate vector toward L792. Methyl substitution at benzimidazole C7 modestly improved selectivity over JAK1 to 6× (37), and while JAK2 selectivity was not improved, broader selectivity in an abbreviated kinase panel was realized.
With preliminary SAR established around biochemical potency, wtEGFR selectivity, and broader kinome selectivity, a subset of pyridone inhibitors were progressed into cellular assays (Table 4). Compounds 13 and 29 had gains in biochemical potency, ligand efficiency, and wtEGFR selectivity relative to initial hit 1. While this did not translate into improved cellular activity, compounds 13 and 29 both showed low micromolar to submicromolar activity. Compound 29 also demonstrated 23-fold cellular selectivity over wtEGFR when comparing pEGFR inhibition in the TMLR cell line H1975 vs the wtEGFR-driven cell line H292 meeting the goal of a selective tool compound with improved kinase selectivity for evaluation in cellular assays. In addition, 29 showed desirable physicochemical properties, improved solubility, and moderate in vitro stability highlighting the promise of this series for further development.
Table 4. Physicochemical Properties and Cellular Potency for Key Analogues.
| Cmpd | 1 | 13 | 29 |
|---|---|---|---|
| LogD, TPSA, LEa | –, 100, 0.3 | 3.5, 89, 0.34 | 2.7, 86, 0.34 |
| Kinetic sol. (μM) | 11 | 4 | 61 |
| TMLR Ki (μM)b | 0.065 | 0.019 | 0.015 |
| wtEGFR Select.c | <1× | 58× | 29× |
| pEGFR H1975 EC50 (μM)b | 0.412 | 1.09 | 0.433 |
| pEGFR Select. vs H292d | 5× | >23× | |
| H/RLMe | 11/31 | 14/41 | 11/29 |
| H/R CLhepf | 13/35 | 12,24 | 8/25 |
LE calculated using TMLR Ki.
Ki and EC50 data are an average of at least two independent experiments.
Ratio of wtEGFR Kiapp over EGFR(TMLR) Kiapp.
Ratio of pEGFR EC50 in H292 cells over pEGFR EC50 in H1975 cells.
H/RLM defined as predicted hepatic clearance in liver microsomes for human (H) and rat (R). Reported in units of mL/min/kg.
H/R CLhep defined as predicted hepatic clearance in hepatocytes for human (H) and rat (R). Reported in units of mL/min/kg.
In conclusion, the compelling nature of the T790M EGFR mutants as therapeutic targets for oncology led us to initiate efforts to identify inhibitors of the major resistance mutations of EGFR, namely, T790M/L858R (TMLR) and T790M/del(746–750) (TMdel), with selectivity over wtEGFR. Beginning with high throughput screening hit 1, which lacked selectivity over wtEGFR, we identified a novel series of noncovalent pyridone-based T790M EGFR inhibitors. Replacement of the phenethyl motif with an aniline substructure provided a significant improvement in selectivity over wtEGFR and a desirable starting point for further optimization. Optimization of C3 provided a framework for addressing broad kinase selectivity. Several of the optimized inhibitors showed substantial improvement in biochemical potency and ligand efficiency selectivity across the kinome. Gratifyingly, this selectivity was recapitulated in a cellular format. Furthermore, improvements in physicochemical properties and kinetic solubility with maintained moderate in vitro metabolic stability exemplify the promise of the scaffold. Further optimization of this scaffold to achieve improved cellular potency will be the focus of future reports.
Glossary
ABBREVIATIONS
- wtEGFR
wild-type EGFR
- TMLR
T790M/L858R mutation
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.5b00428.
Additional figures, tables, and examples syntheses (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Remon J.; Morán T.; Majem M.; Reguart N.; Dalmau E.; Márquez-Medina D.; Lianes P. Acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in EGFR-mutant non-small cell lung cancer: A new era begins. Cancer Treat. Rev. 2014, 40, 93–101. 10.1016/j.ctrv.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Sordella R.; Bell D. W.; Haber D. A.; Settleman J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Sharma S. V.; Bell D. W.; Settleman J.; Haber D. A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. 10.1038/nrc2088. [DOI] [PubMed] [Google Scholar]
- Lacouture M. E. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat. Rev. Cancer 2006, 6, 803–812. 10.1038/nrc1970. [DOI] [PubMed] [Google Scholar]
- Yun C.-H.; Mengwasser K. E.; Toms A. V.; Woo M. S.; Greulich H.; Wong K.-K.; Meyerson M.; Eck M. J. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 2070–2075. 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutto L.; Gervasio F. L. Effects of oncogenic mutations on the conformational free-energy landscape of EGFR kinase. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 10616–10621. 10.1073/pnas.1221953110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowder M. A.; Doerner A. E.; Schepartz A. Structural Differences between Wild-Type and Double Mutant EGFR Modulated by Third-Generation Kinase Inhibitors. J. Am. Chem. Soc. 2015, 137, 6456–6459. 10.1021/jacs.5b02326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercan D.; Choi H. G.; Yun C.-H.; Capelletti M.; Xie T.; Eck M. J.; Gray N. S.; Jänne P. A. EGFR mutations and resistance to Irreversible pyrimidine based EGFR Inhibitors. Clin. Cancer Res. 2015, 21, 3913–3923. 10.1158/1078-0432.CCR-14-2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanan E. J.; Eigenbrot C.; Bryan M. C.; Burdick D. J.; Chan B. K.; Chen Y.; Dotson J.; Heald R. A.; Jackson P. S.; La H.; Lainchbury M. D.; Malek S.; Purkey H. E.; Schaefer G.; Schmidt S.; Seward E. M.; Sideris S.; Tam C.; Wang S.; Yeap S. K.; Yen I.; Yin J.; Yu C.; Zilberleyb I.; Heffron T. P. Discovery of Selective and Noncovalent Diaminopyrimidine-Based Inhibitors of Epidermal Growth Factor Receptor Containing the T790M Resistance Mutation. J. Med. Chem. 2014, 57, 10176–10191. 10.1021/jm501578n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederst M. J.; Hu H.; Mulvey H. E.; Lockerman E. L.; Garcia A. R.; Piotrowska Z.; Sequist L. V.; Engelman J. A. The allelic context of the C797S mutation acquired upon treatment with third generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 17, 3924–3933. 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins A. L.; Keseru G. M.; Leeson P. D.; Rees D. C.; Reynolds C. H. The role of ligand efficiency metrics in drug discovery. Nat. Rev. Drug Discovery 2014, 13, 105–121. 10.1038/nrd4163. [DOI] [PubMed] [Google Scholar]
- Stamos J.; Sliwkowski M. X.; Eigenbrot C. Structure of the Epidermal Growth Factor Receptor Kinase Domain Alone and in Complex with a 4-Anilinoquinazoline Inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- Walter A. O.; Sjin R. T. T.; Haringsma H. J.; Ohashi K.; Sun J.; Lee K.; Dubrovskiy A.; Labenski M.; Zhu Z.; Wang Z.; Sheets M.; Martin T. S.; Karp R.; van Kalken D.; Chaturvedi P.; Niu D.; Nacht M.; Petter R. C.; Westlin W.; Lin K.; Jaw-Tsai S.; Raponi M.; Dyke T. V.; Etter J.; Weaver Z.; Pao W.; Singh J.; Simmons A. D.; Harding T. C.; Allen A. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discovery 2013, 3, 1404–1415. 10.1158/2159-8290.CD-13-0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams N. K.; Bamert R. S.; Patel O.; Wang C.; Walden P. M.; Wilks A. F.; Fantino E.; Rossjohn J.; Lucet I. S. Dissecting Specificity in the Janus Kinases: The Structures of JAK-Specific Inhibitors Complexed to the JAK1 and JAK2 Protein Tyrosine Kinase Domains. J. Mol. Biol. 2009, 387, 219–232. 10.1016/j.jmb.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Huang C. Y.; Miller P. S. Triplex formation by an oligodeoxyribonucleotide containing N4-(6-aminopyridinyl)-2′-deoxycytidine. J. Am. Chem. Soc. 1993, 115, 10456–10457. 10.1021/ja00075a110. [DOI] [Google Scholar]
- Chen H.; Chan B. K.; Drummond J.; Estrada A. A.; Gunzner-Toste J.; Liu X.; Liu Y.; Moffat J.; Shore D.; Sweeney Z. K.; Tran T.; Wang S.; Zhao G.; Zhu H.; Burdick D. J. Discovery of Selective LRRK2 Inhibitors Guided by Computational Analysis and Molecular Modeling. J. Med. Chem. 2012, 55, 5536–5545. 10.1021/jm300452p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.