

Article first Published online 18 December 2015
Key Words: enteric glial cells, motility, tipartite synapse, calcium signaling, gliotransmission, neuroglial communication, inflammatory bowel diseases, GI infection, human enteric glial cell, postoperative ileus, enteric nervous system, reactive hEGC phenotype, purinergic signaling
Abstract:
The word “glia” is derived from the Greek word “γλοια,” glue of the enteric nervous system, and for many years, enteric glial cells (EGCs) were believed to provide mainly structural support. However, EGCs as astrocytes in the central nervous system may serve a much more vital and active role in the enteric nervous system, and in homeostatic regulation of gastrointestinal functions. The emphasis of this review will be on emerging concepts supported by basic, translational, and/or clinical studies, implicating EGCs in neuron-to-glial (neuroglial) communication, motility, interactions with other cells in the gut microenvironment, infection, and inflammatory bowel diseases. The concept of the “reactive glial phenotype” is explored as it relates to inflammatory bowel diseases, bacterial and viral infections, postoperative ileus, functional gastrointestinal disorders, and motility disorders. The main theme of this review is that EGCs are emerging as a new frontier in neurogastroenterology and a potential therapeutic target. New technological innovations in neuroimaging techniques are facilitating progress in the field, and an update is provided on exciting new translational studies. Gaps in our knowledge are discussed for further research. Restoring normal EGC function may prove to be an efficient strategy to dampen inflammation. Probiotics, palmitoylethanolamide (peroxisome proliferator-activated receptor–α), interleukin-1 antagonists (anakinra), and interventions acting on nitric oxide, receptor for advanced glycation end products, S100B, or purinergic signaling pathways are relevant clinical targets on EGCs with therapeutic potential.
The first observation of enteric glial cells (EGCs) was made by Dogiel in 1899. Gabella1 provided a more detailed description of EGC-morphology and characteristics. EGCs resemble astrocytes in the central nervous system (CNS), and this has provided a conceptual framework to guide further research into EGCs. Glia is derived from the Greek word “γλοια,” glue of the enteric nervous system (ENS), and until recently, structural support for the ENS was believed to be their main function. However, this notion was incorrect, and we now know that they play a much more vital role in the ENS and in disease states of the gut. EGCs can be classified into 4 main types (I–IV) according to their shapes and locations in the gut wall,2,3 but the precise role of each type of EGCs in gastrointestinal (GI) physiology and pathophysiology is not well understood.3
EGCs are not excitable cells like neurons because they do not generate action potentials. However, they form vast communication networks through a complex repertoire of Ca2+ signals that enables EGCs to integrate information transmitted by neurons, glial cells, immune cells, or other cells in the gut microenvironment. EGCs communicate through Ca2+ signals, Ca2+ oscillations, or Ca2+ waves to modulate neural circuit activity and motility. Thus, their type of excitability is determined by their Ca2+ responses. In disease states, inflammation can convert the EGCs to a “reactive EGC phenotype.” The focus of this review is aimed at the physiology of EGCs and the role of the “reactive EGC phenotype” produced by inflammation in the pathogenesis of inflammatory bowel diseases (IBDs), and also bacterial and viral infections, postoperative ileus (POI), motility disorders, and functional gastrointestinal disorders (FGIDs) such as irritable bowel syndrome (IBS). The EGC is explored as a novel therapeutic target for GI diseases. Therefore, there is more emphasis in this review on translational and clinical studies and human EGC (hEGC). Studies in animal models serve to provide mechanistic insights into EGCs in GI physiology/pathophysiology, and in particular IBD, FGIDs, POI, GI infections, and motility disorders (i.e., slow transit/constipation). We focus our attention on the current knowledge about EGCs and emerging concepts about their functions in the normal gut or in disease states. Novel technologies and innovations are described and gaps in our knowledge identified, providing the basis for doing future basic, translational, and clinical research. The role of EGCs in motility disorders has been reviewed recently2,3 as has their role in protecting the intestinal epithelial barrier.4 The latter will therefore only be discussed briefly in the context of this review.
EGC FUNCTIONS AND MORPHOLOGY
EGCs are the major component of the ENS but, unlike neurons, their role in GI physiology and pathophysiology has only recently started to be unraveled. Nevertheless, we are starting to view EGCs as pivotal components of the ENS, also known as the “little brain in the gut” for its unique ability to autonomously regulate several GI functions, such as exocrine and endocrine secretions, motility, blood flow, and immune/inflammatory processes.5 The ENS with more than 100 million neurons comprises the intrinsic neural circuits of the GI tract that is organized into an extensive network of interconnected ganglia. The ENS orchestrates and coordinates neural, motor, secretomotor, and vasomotor functions.6–8 More than a dozen functionally distinct types of neurons exist in the ENS that are organized into 2 major nerve plexuses: the myenteric plexus (or Auerbach's plexus), that primarily provides motor innervations to the 2 muscle layers and secretomotor innervations to the mucosa; and the submucosal plexus (or Meissner's plexus), playing an important role in the control of secretion and coordination of motility and secretion.
EGCs are found in enteric ganglia, smooth muscle layers, and gut mucosa. In addition, a small number of extra-ganglionated enteric neurons and EGCs are present in the lamina propria of the mucosa, the mucosal plexus, and in the subserosal plexus in the connective-tissue layer.5,9 It has been estimated that EGC outnumber enteric neurons by 4:1 in the GI tract.2,10–12
Morphologically, EGCs are comparable with the astrocytes in the CNS. Like their CNS counterpart, they are small cells that envelop enteric neuronal cell bodies and axon bundles12 and extend their processes into the intestinal mucosa.13 Based on their localization within the layers of the gut, it has recently been proposed to morphologically characterize the EGCs into 4 types: a star-shaped, type I morphology within ganglia; a more elongated, type II, for interganglionic EGCs; type III for mucosal; and type IV for intramuscular EGCs.10
Functionally, EGCs have been traditionally considered as a mechanical support for enteric neurons, and they are able to release a wide range of factors responsible for the development, survival, and differentiation of peripheral neurons. Nowadays, besides their supportive role, it has been demonstrated that EGCs possess a more complex nature, playing a leading role in the maintenance of intestinal homeostasis.4
EGCs also resemble astrocytes in their expression of typical identification markers, which include intermediate filament glial fibrillary acidic protein (GFAP) and the S100B protein, other than the SRY box–containing gene 8/9/10 (Sox 8/9/10).12 GFAP expression in mature EGCs is modulated by cell differentiation, inflammation, and injury and, even if the functions of this protein are still obscure, its levels correlate with the functional state of EGCs.12 Sox 8/9/10, which can be expressed by multipotent ENS precursors, and/or by mature EGCs, can be used as EGC markers. In particular, Sox10, for its selective nuclear localization and expression in mature EGCs, is very well suited for quantification purposes.9
The diffusible S100B protein belongs to the S100 family that includes more than 20 EF-hand Ca2+-Zn2+–binding proteins and is specifically and physiologically expressed and released by EGCs in the gut.12 Both in gut and brain, S100B can be considered a “Janus-faced” protein, because in nanomolar concentrations, it regulates microenvironmental homeostasis, whereas in micromolar amounts it is correlated with a pathologic and inflammatory status.12 Especially in this latter case, S100B exerts its actions by binding to the receptor for advanced glycation end products (RAGE), with the downstream phosphorylation of the mitogen-activated protein kinase and the consequent activation of the nuclear factor-κB (NF-κB), which in turn leads to the transcription of different cytokines and inducible nitric oxide synthase (iNOS) protein.14 This is described further later.
It is worth noting that, although all populations of EGCs derive from a common pool of neural crest-derived progenitors, the unique microenvironments of the various layers of the gut designate the phenotype of these cells and also the different expression of their typical biomarkers. Indeed, multiple distinct types of glial cells expressing only S100B, only GFAP or both, and/or Sox 8/9/10 immunoreactivity in the gut, are encompassed by the term “enteric glia,” even if a functional distinction between these cells has never been demonstrated. In addition, 4 types of glial cells have been proposed according to their topography and distinct morphologies as noted earlier,3 and a better understanding of the functional phenotypes represented by these cells is clearly needed.
Adherent-Invasive Escherichia coli in IBD and Human EGCs
Adherent-invasive E. coli (AIEC) may play a role in the onset and propagation of IBD15 and their interactions with hEGC are likely a contributing factor. First of all, localized or generalized dysbiosis occurs in IBD and E. coli, and in particular, AIEC are found in ileal biopsies of 36.4% of patients with ileal-involvement of Crohn's disease (CD). In general, patients with IBD have higher concentrations of mucosa-associated bacteria than healthy controls.16 These pathogens invade and replicate extensively in intestinal epithelial cells (IECs) and macrophages; in the latter cell, they induce large amounts of tumor necrosis factor–α release. AIEC is restricted to CD biopsy (and not ulcerative colitis [UC] biopsy). Postsurgical exposure of terminal ileum to luminal contents is associated with an increase in inflammation, and diversion of the fecal stream is associated with improvement.17 Interaction between E. coli and host cells induces inflammatory responses by interacting with Toll-like receptors (TLRs).18 AIEC infection can result in high production of chemokines and cytokines, and they can exacerbate intestinal inflammation in animal models.19–21 Extracellular factors like flagellin interact with TLR5 linked to innate immunity in IBD. The presence of AIEC is implicated in the etiopathogenesis of ileal CD.
An important new discovery is that hEGCs can sense differences between pathogenic and probiotic bacteria in the gut. Pathogenic AIEC activate hEGC and induce cFos and major histocompatibility type II complex (MHC II). AIEC cause activation of the TLR/S100B-RAGE dependent iNOS-nitric oxide (NO) signaling pathway in hEGC. S100B release from hEGC is induced by pathogenic bacteria or by bacterial products (e.g., lipopolysaccharides).18 The mechanism is illustrated in Figure 1. EGCs can discriminate between pathogenic and probiotic (beneficial) bacteria (i.e., Lactobacillus paracasei F19) in their recognition and activation of TLR. It has been suggested that bacterial recognition may be tailored to the type of bacteria and localization of the bacteria (i.e., intracellular localization of AIEC versus extracellular for probiotic strain), and effects on EGCs are not restricted to soluble factors released by bacteria because differences still occur in heat-inactivated AIEC. Therefore, EGCs can sense the presence of pathogenic bacteria to mount an effective host-immune response through TLR. It has been suggested that targeting NO production in hEGCs may be a potential new therapeutic strategy to modulate abnormal NO responses in the gut.
FIGURE 1.
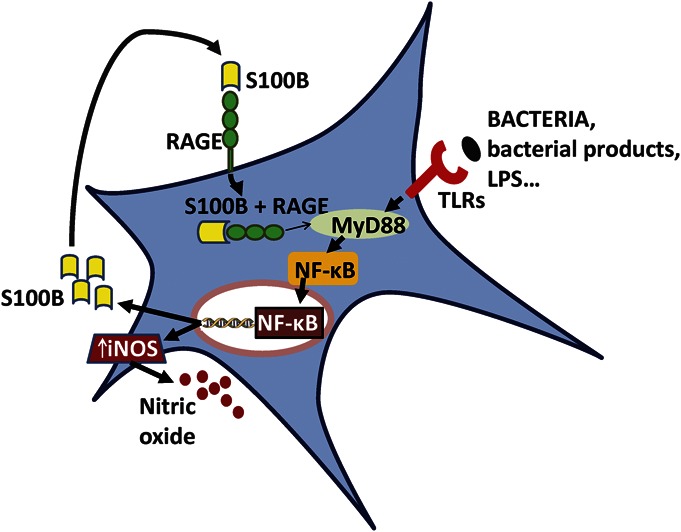
Human EGCs can sense differences between pathogenic and probiotic bacteria in the gut. Pathogenic AIEC activate hEGC and induce cFos and MHC II. AIECs cause activation of the TLR/s100B-RAGE dependent iNOS-NO signaling pathway in hEGC. S100B release from hEGC is induced by pathogenic bacteria or by bacterial products (e.g., lipopolysaccharides). EGCs can discriminate between pathogenic and probiotic (beneficial) bacteria (e.g., Lactobacillus paracasei F19) in their recognition and activation of TLR. AIEC induce S100B overexpression/release and NO release from hEGC. NO release depends on TLR/S100B signaling. Once released, S100B interacts with RAGE, which is expressed on the surface of hEGC. As proposed by various studies, the S100B/RAGE complex may be internalized and interact, directly or not, with MyD88, a downstream mediator of the TLR signaling pathway, thus inducing iNOS expression and NO release, through induction and translocation of the nuclear factor NF-κB in hEGCs. NF-κB also induces S100B synthesis and release that sustains NO signaling. A common pathway linking S100B, RAGE, and MyD88 is suggested to be involved. Overall, enteroglial-derived S100B protein integrates bacterial product(s) signaling through TLRs and the RAGE-iNOS-NO signaling pathways in the reactive hEGC phenotype (described in detail by Turco et al18). EGCs possess the molecular mechanisms and TLR signaling to be able to potentially trigger innate immune responses in the gut.
A recent study indicates that inhibiting iNOS in EGCs restores electrogenic ion transport in mice with colitis.21a AIEC induce S100B overexpression/release and NO release from hEGC. NO release depends on TLR/S100B signaling. Once released, S100B interacts with RAGE, which is expressed on the surface of hEGC. As proposed by various studies, the S100B/RAGE complex may be internalized and interact, directly or not, with MyD88, a downstream mediator of the TLR signaling pathway, thus inducing iNOS expression and NO release, by induction of the nuclear factor NF-κB and nuclear translocation in hEGCs. NF-κB also induces S100B synthesis and release that sustains NO signaling. Overall, enteroglial-derived S100B protein integrates bacterial product(s) signaling through TLRs and the RAGE-iNOS-NO signaling pathways in the reactive hEGC phenotype (described in detail by Turco et al18). The signaling pathway is illustrated in Figure 1. In addition to its role in infection, AIEC (or LPS) activation of the TLR/MyD88–S100B/RAGE-iNOS-NO signaling pathway19 is an important signaling pathway for bacterial infection relevant to patients with CD, and it deserves further exploration to study hEGC function.
RAGE IN INFLAMMATION AND THERAPEUTICS
RAGE is usually upregulated in disease states, and it may play an important role in the development of the disease. RAGE senses a variety of signaling molecules such as advanced glycation end products, HMGB1, s100, calgranulins, C3a, phosphatidylserine, β-amyloid, and advanced oxidation protein products. Therapeutic strategies to block RAGE may be relevant for IBD and gut infections with AIEC involving a “reactive EGC phenotype,” since interference with the RAGE pathway can block inflammation, apoptosis, proliferation, and autophagy. Further studies are needed to unravel the complex molecular signaling pathways by which RAGE may alter hEGC functions in response to infection or inflammation and consequences to neuron-to-glial interactions and motility as described later. Signaling pathways that are regulated by RAGE have been reviewed by Xie et al.22
ROLE OF EGCs IN GI DISEASES
All reports about EGCs highlight their ability to regulate homeostasis. For this reason, EGC alterations could participate in the initiation/perpetuation of digestive diseases and their associated symptoms, especially those related to an inflammatory scenario.
Several early studies highlighted the importance of EGCs in regulating and protecting enteric neurons and the consequences of their dysfunction. In the first, Bush et al showed that the selective ablation of EGCs in transgenic mice led to significantly compromised epithelial and vascular integrity within the GI tract, resulting in a fulminating inflammation, hemorrhage, and necrosis13; similarly, in the second study, autoimmune depletion of enteric glia caused severe enterocolitis.23 Since then, EGC alterations have been shown to be associated to inflammatory and functional disorders in the gut. Notwithstanding, whether the observed EGC alterations are primary or secondary to inflammatory, immunologic or neurodegenerative processes are not clarified yet.
In patients with UC (compared with healthy controls), there is an increased GFAP and GDNF expression and an increased S100B immunoreactivity, associated with enhanced NO production through the specific stimulation of iNOS, through RAGE activation.24–26 These data support the hypothesis that EGCs are activated during UC, thus contributing to the spreading of the inflammation in the intestinal mucosa. Nevertheless, EGC activation may not be a specific event for UC because patients with infectious colitis showed increased GFAP and GDNF levels, and also infectious and inflammatory stimuli increased GFAP expression, and also S100B release.27,28
Differently from UC, in patients with CD, there is a reduced GFAP immunoreactivity than healthy control and a lower GFAP and GDNF expression than patients with UC. In addition, when comparing inflamed tissues from patients with UC and CD, although gliosis is evident in both ileal and colonic sites, it is much less pronounced in patients with CD than in patients with UC.23,26 Therefore, noninvolved intestinal mucosa in patients with CD is associated with a smaller EGC network that seems to respond poorly to inflammatory signals. Because EGC markers are differentially altered in CD and UC, with a decrease in EGC density in CD and a gliosis-like phenomenon in UC, it is tempting to speculate that these cells exert a dual and opposite role in the pathogenesis of CD and UC.29
The expression of the glial S100B protein is increased in the early inflammatory processes in response to the signal triggered by disruption of the intestinal barrier.25 Also in the duodenal mucosa of celiac disease patients, an increased S100B protein expression and release have been observed, and also in this case, S100B upregulation was accompanied by enhanced iNOS protein expression and consequent NO release, both representing crucial features in CD, suggesting that through S100B upregulation, EGCs directly participate in NO-dependent inflammation.24 In the same way, palmitoylethanolamide (PEA), by interacting with peroxisome proliferator-activated receptor–α expressed by glial cells, is able to counteract the increased expression of TLR4/S100B proteins, together with p38/p-ERK/pJNK-pathway signaling molecules, NF-κB expression, and NO release, in patients with UC.30
In the ENS, as in the brain, innate immunity may be regulated by TLR expressed on glial cells. Human EGCs possess the molecular mechanisms and TLR signaling to be able to potentially trigger innate immune responses in the gut. Human EGCs regulate host-bacterial crosstalk in the gut through TLRs as noted earlier. Viruses can also infect hEGC, and it would be of great interest and clinical relevance to unravel mechanisms of host-viral crosstalk.
Deleterious effects have also been ascribed to EGCs in in vitro and in vivo models of colorectal cancer, where tumor cells activated EGCs to acquire protumorigenic abilities, through prostaglandin E2 (PGE2)-dependent pathways.31 Overall, a dual protective and harmful role is possible for EGCs, as they are capable of releasing a wide range of protective (e.g., NT-3, GDNF, and GNSO) and/or destructive factors (e.g., interleukin [IL]-1β, IL-6, tumor necrosis factor-α, chemokines, and NO) involved in the regulation of gut homeostasis and in gut inflammation.12
Also for diverticular disease (DD), increasing evidence associates distinct abnormalities of the ENS and EGC with the disturbed motility patterns underlying DD. Patients with DD showed a decreased EGC and interstitial cells of Cajal density and S100B immunoreactivity, in myenteric ganglia.32 Although the overall glial cell density was reduced in DD, a study in 2010 showed that a subgroup of myenteric ganglia displayed bulbous protrusions almost exclusively composed of glial cells,33 but this reported enteric gliosis (an EGC activation phenomenon) has not yet been confirmed by other investigations. A loss of EGC, interstitial cells of Cajal, and neurons has also been observed in patients undergoing surgery for idiopathic severe slow transit constipation.34 The same group also reported a similar decrease in EGC number in the colons of patients with chagasic and idiopathic megacolon.35 Moreover, a study of EGC role in dilated and nondilated portions of colon from chagasic patients suggests that GFAP-positive EGCs prevent dilatation of the colon and protect the ENS against the inflammatory process and neuronal destruction.36
Glial dysfunction in both the ENS and CNS has been implicated in neurodegenerative diseases such as Parkinson's disease (PD), prion diseases, or metabolic diseases (e.g., associated with disruption of mitochondrial metabolism in mice) common to neurodegenerative disease.37–44 A common feature of these diseases is dysmotility. In the large bowel of patients with PD, there is elevation of proinflammatory cytokines and glial markers (e.g., GFAP, S100B, Sox10), suggesting an association between enteric inflammation and glial abnormalities.44 In patients with PD, aggregates of α-synuclein are found in both CNS and ENS in neurons or glial cells (e.g., called as Lewy bodies),39 and colonic biopsy studies suggest that α-synuclein may represent a biomarker of premotor PD.45 A study provided evidence for a reactive glial phenotype in colonic biopsy of patients with PD. They found elevated GFAP expression and a reduction in phosphorylation of GFAP, changes that have been associated to degenerative CNS diseases.46 A pathologic hallmark of PD, that is, also observed in an animal model of PD47 is constipation (and hyposmia) and this nonmotor symptom of PD (i.e., associated with a gut motor abnormality) can occur in the absence of classical CNS PD-motor symptoms and is therefore distinct from PD-motor abnormalities.48 Therefore, EGCs could potentially be involved in constipation in PD.
Loss of neurons and glial cells in the ENS is a characteristic of necrotizing enterocolitis in premature infants.49,50 Patients with CD have also been reported to have a smaller glial network, and plexitis has been implicated in CD recurrence after surgery.25,26,51
Overall, enteric glial networks react to gut inflammation by converting to a “reactive EGC phenotype.” Therefore various distinct events, such as IBD, glial-cell injury, infection with E. coli, Clostridium difficile (or viruses), can all cause alterations in proliferation of EGC and the expression of glial proteins (GFAP, S100B, and GDNF) and also alterations in glial function.26,52 In gut inflammatory states, EGCs are known to also increase the expression of neurotrophic factors (GDNF, BDNF, and NGF) that can have a protective influence.53,54
EMERGING ROLE OF EGC IN MOTILITY, COLONIC MOTOR MIGRATING COMPLEXES, NEURAL-GLIAL COMMUNICATION, IBS, POI, AND Ca2+ WAVES
Experimental evidence in animals and humans (in vitro studies) suggests that EGCs play a pivotal role in the modulation of neural-motor function, motility, and intestinal transit. We propose that a “reactive EGC phenotype” contributes to abnormal motility in GI diseases including IBD, IBS, POI, DD, and neurodegenerative diseases like PD, and also infection-induced inflammation of the GI tract. The general concept is illustrated in Figure 2 and discussed in detail throughout the article.
FIGURE 2.
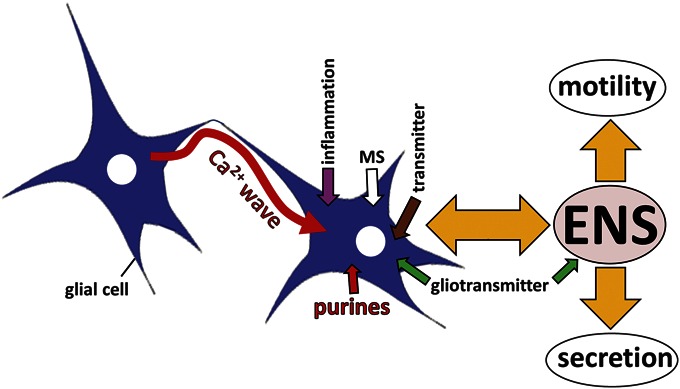
Working Hypothesis of Glial Modulation of Motility. EGCs are involved in bidirectional communication in the ENS. Neurons release neurotransmitters such as ACh, 5-HT, and ATP that activate glial cells by evoking a Ca2+ response. Glial cells do not have action potentials, but use Ca2+ signaling for cell-to-cell communication. EGCs communicate with each other by transient changes in intracellular free Ca2+ levels. Ca2+ waves can transmit information to distant sites within the ENS and convey long-distance communication. Information is decoded in the type of Ca2+ transient, kinetic profile, magnitude, duration, occurrence of Ca2+ oscillations, and distance, velocity, and propagation of Ca2+ waves. Glia release gliotransmitters such as ATP, glutamate, lipids to communicate with other glial cells and neurons (gliotransmission). EGC networks are connected by gap junctions and an elaborate set of channels, including P2X channels, TRP-channels, and hemichannels for cell-to-cell communication. EGCs represent sites of signal integration and processing. EGCs are highly sensitive to mechanical stimulation and are, therefore, very likely to respond to activation during peristaltic activity and movements of the GI tract. Bidirectional communication between neurons and glia is involved in the modulation of ENS neural activity and motor behavior of the gut (including motility and secretion). Inflammation can disrupt motility and coordination of motor-secretory events by influencing the normal behavior of EGCs.
Motility
Disruption of EGCs by a selective gliotoxin fluorocitrate inhibited neuromuscular transmission and transit in the intestinal tract.55 EGC disruption by genetic manipulation caused a delay in GI motility and an increase in intestinal barrier permeability associated with a disruption in cholinergic and nitrergic neurons of the myenteric plexus.56 Enteric gliopathy induced by a CNS gliotoxin (6-aminonicotinamide) was shown to be associated with symptoms of diarrhea. Abnormalities in secretomotor neural reflexes could cause such diarrhea, although the mechanisms remain unknown. Recent preliminary studies from our group using the gliotoxin fluorocitrate in vitro strongly suggest that human EGCs are involved in regulation of neuromuscular transmission and motility.57 An elegant study published in Gastroenterology by Brian Gulbransen's group58 used pharmacological agents or glia-specific disruption of the gene encoding connexin-43 (hGFAP::CreERT2+/−/C x 43f/f/mice) to show that Ca2+ waves in EGCs that are mediated by connexin-43 hemichannels regulate intestinal motility and gut transit. Furthermore, their study suggested that abnormalities in EGC Ca2+ responses and the Cx43 mechanism may be involved in age-related changes in motility.58
Colonic motor migrating complexes are associated with activation of EGC networks.59 In those rodent studies, it was shown that nerve activity may be necessary to drive EGC activity. However, it must be more complicated than that, for instance, gliofilaments are abundant in EGC surrounding neurons in ganglia. Gliofilaments are attached (anchored) to the surface of ganglia, and it has long been speculated that EGC play a major role in the structural rearrangement of ganglia exposed to intense mechanical stress, such as that occuring in myenteric ganglia during peristaltic activity associated with mechanical/contractile activity of the muscularis externa.1 Furthermore, we now know that hEGCs are very sensitive to mechanical stimulation (i.e., touch, pulsatile flow, and pressure),60 suggesting that the peristaltic activity during the colonic motor migrating complexes could also activate them to induce Ca2+ waves that can influence or modulate neural-motor activity; speculatively, this could potentially serve as a feedback loop from muscle to glia to fine-tune neural circuit activity.
Neural-Glial Communication
There is a very close functional relationship between the ENS and EGC. Thus far, we known that neuron-to-glial communication occurs in both rodent61 and human ENS,60 and purinergic signaling is implicated in such transmission. EGCs in human duodenal biopsy (or rat EGC) respond to several neurotransmitters including adenosine triphosphate (ATP), ACh, and 5-HT with a Ca2+ response.62 In animal models of gut inflammation (IBD), it has been shown that ATP release from enteric neurons through pannexin channels can activate EGCs that is suggested to be involved in neural-glial communication through Ca2+ signals.63 A better understanding of the bidirectional communication between neurons and glial cells is needed to fully appreciate the role of EGC in motility, GI motor disorders (or gut inflammatory diseases described later in this section).
Some evidence also supports functional anatomical connections between neurons and glial cells. Gabella1,64 first reported that specialized contacts between vesicle-containing varicose nerve endings and EGC are very common in enteric ganglia. These were presumed to be transmitter release sites for neuroglial communications. In fact, EGCs received cholinergic and serotonergic innervation, provided by enteric neurons.65 In the colon, individual serotonergic neurons of the myenteric plexus have been shown to innervate EGCs, submucosal neurons, interstitial cells of Cajal, and blood vessels, suggesting that they represent command neurons involved in the coordination of motility, secretion, and blood flow. In our recent preliminary studies, we could trigger a Ca2+ wave by local “puff” application of 5-HT to a single hEGC, indicating that 5-HT is involved in long-distance communication in the ENS gut reflexes and motility (unpublished, Linan-Rico & Christofi). Future studies on serotonergic modulation of neuron-to-glial communication can provide novel insights into ENS and motor regulation in the gut in normal and inflamed conditions.
Unlike the CNS, enteric communication between glia and neurons is poorly understood.66–70 As shown for the CNS, a single glial cell in the cerebellum, for example, contacts as many as 6000 synapses and therefore may influence the complex neural circuit behavior of that region of the brain. In the CNS, astrocytes modulate synaptic transmission, neuroplasticity,71–75 neuronal differentiation,75,76 and perhaps heterocellular signaling by Ca2+ dependent release of gliotransmitters.72,77,78 Among the many putative gliotransmitter substances that can be contained in glial synaptic vesicles include ATP, both inhibitory and excitatory amino acids, cytokines, neurotrophins, and tissue plasminogen activator.79–84 The role of these and other potential gliotransmitters remains elusive in the ENS of both animals and humans.
Glia have a “form of excitability based on sophisticated Ca2+ signaling mechanisms.” Glia form intracellular and intercellular Ca2+ waves for long-distance communication with synapses. Therefore, EGC like astrocytes may act as integrators of neural activity59 and to synchronize neural behavior. If that is so, glial cells should respond to synaptic activity (neuron-to-glial communication), transmit information to distant synapses (through Ca2+ waves), and glial cells should have a mechanism for communicating back to synapses (gliotransmission). These aspects remain poorly understood in the ENS of both animals and humans.
Overall, it is proposed that glial cells participate in synaptic signaling may regulate synaptic plasticity and network excitability, and inflammation in the ENS as they do in the brain.85 A complete understanding of the neuropathophysiology of IBD (or other GI diseases or disorders) requires a better understanding of the functional role of entering glial cells in health and diseases.
IBS
EGCs may be implicated in motor abnormalities associated with IBS. A study by Fujikawa et al86 in an IBS model of maternal separation revealed that EGCs were associated with stress-induced colonic hyper-contractility. The authors showed that a higher dose of the gliotoxin was required to inhibit both EFS-induced contractions and EGC activation in the stress model than in controls. In addition, GFAP-positive EGCs exhibited structural changes according to the stress intensity in the IBS model (i.e., maternal separation + acute stress). This study suggested that EGCs are involved in abnormal motility associated with FGIDs.
POI
Although EGCs do not have direct access to luminal environment their close proximity to enterocytes, projections into the most distant parts of the villi and their organization around the crypt bottom indicate that EGCs are involved in regulation of microbial, immunological, and environmental interactions. Recent work demonstrated that early postnatal EGC development depends on the bacterial colonization as germ-free housed mice or mice treated with broad-spectrum antibiotics lack EGC in the lamina propria.87 Therefore, it is likely that alterations in the luminal commensal and pathogenic flora, as observed during IBD, also affect EGC function. EGCs have been shown to fulfill antigen-presenting function after cytokine stimulation. Indeed, EGC area fully armored with well-known receptors of the innate immune system, including TLR 2, 3, 4, 7, and 9,18,30,88,89 and recent work demonstrated expression of the IL-1 receptor type I (IL1R1) and IL1R1-mediated release of IL-6 and MCP-1 from mouse EGC cultures in vitro.90
As indicated earlier, EGCs regulate motility in the intestinal tract, although the exact mechanisms still remain elusive. Indirect contribution of EGC in the maintenance of GI motility during inflammation has been shown by a T cell–mediated depletion of EGC in mice resulting in reduced gastric emptying, jejunal contractility, and overall GI transit simultaneously with a severe small bowel inflammation.56 The numbers of neurons expressing specific neurotransmitters or their producing enzymes, such as VIP, SP, or NOS and ChAT, respectively, were also affected by EGC ablation in the submucosal and myenteric plexus and therefore motility disturbances by EGC ablation likely originated from an altered neurochemical coding and function of the ENS including EGCs. Overall, it is known that inflammation activates EGC and converts them into a reactive glial cell phenotype, that is expected to cause alterations in motility, and this is also the pathogenic mechanism proposed for postoperative ileus (POI).
POI is a common symptom of abdominal surgery, associated with an increase in the risk of postoperative complications and morbidity. The annual health care costs due to extended hospitalization in patients with POI are into the billions. During abdominal surgery, intestinal manipulation leads to inflammation that disrupts motility. Furthermore, hEGCs are very sensitive to mechanical stimulation (Christofi, unpublished observations), and such intestinal manipulations are very likely to activate EGC. In a mouse model of POI, intestinal manipulation is used to mimic the clinical scenario to study the cellular mechanisms involved in the pathogenesis of POI. Surgical manipulation of the bowels causes inflammation of the muscularis externa and disrupts motility. In contrast to infection of EGC with AIEC or exposure to bacterial products (such as LPS), development of POI did not require TLRs but did require MyD88 indicating involvement of IL1R1. IL1R1−/−–knockout mice, or mice injected with IL-1 receptor antagonist (anakinra) or treated with antibodies to deplete IL-1-α and IL-1β before surgical manipulation, were protected against development of POI. IL1R1 and IL1-β stimulation caused an increase in the expression of IL-6 and chemokine monocyte chemotactic protein 1 in EGC.90 Despite the often extensive manipulation during abdominal surgery, and the postoperative abundance of extravasated immunocytes, the plexuses of the GI tract remain largely intact. POI in humans resolves with time (1–7 days postoperatively), and the patient is able to resume normal peristaltic activity associated with normal bowel movements, and therefore ENS dysfunction is temporary, and does not seem to be due to cellular loss of EGC and neurons. A drug that could interfere with the IL-1 signaling pathway is a potential novel therapy for treatment of POI. Further work involving EGC specific ablation of IL-1 signaling is needed to show whether pathogenesis of POI, particularly cytokine production, indeed requires IL-1 signaling in EGC.
Significance of Glial Ca2+ Waves in Gut Communication and Motility
Ca2+ signaling is very important in glial-to-glial communication, regulation of synaptic activity, neuromuscular activity, peristalsis, propulsive motility, and abnormal GI functions. In astrocytes, spatial properties of Ca2+ signaling can give rise to a brief Ca2+ spark in response to a gliotransmitter like ATP or to more complex activity including intracellular Ca2+ waves.91,92 In astrocytes, DAG/Ca2+ and protein kinase C translocations (and CaM) are important Ca2+-sensitive proteins involved in decoding Ca2+ signals.93 Astrocytes exhibit Ca2+ oscillations that are modulated by the frequency of the neuronal stimulation and they can persist for some time after the neuronal stimuli. Ca2+ oscillations involve synaptic release of transmitter to activate mGluR in astrocytes.94 Astrocytes and neurons communicate with each other based on their specific patterns of Ca2+ oscillations (e.g., calcium oscillations in glia occur in adjacent neurons). Glial Ca2+ oscillations may represent a major mechanism for controlling release of neuromodulatory transmitters. In addition to Ca2+ oscillations, astrocytes produce propagating Ca2+ waves for long-distance communication. Astrocyte Ca2+ signaling could influence the strength of synaptic transmission and the firing frequency of neurons.92,95–97 Ca2+ waves can be generated by receptor activation (ATP, glutamate, and endothelin-1) or intense neuronal firing in cultured organotypic slices.95,96 Astrocytes express P2Y1, P2Y2, P2Y4, and P2X7,98 and several purinergic receptors are likely to contribute to wave propagation in astrocytes.99
An emerging concept is the complex nature of hEGC activity. Our early preliminary findings from Ca2+ imaging studies in cultured hEGCs harvested from human intestinal surgical specimens indicate that hEGCs display Ca2+ oscillations, dynamic upward or downward changes in intracellular free-Ca2+ levels in response to different stimuli, Ca2+ waves involved in long-distance communication, respond to mechanical stimulation, purinergic mediators (i.e., ATP, uridine triphosphate), and neurotransmitters.57 Ca2+ oscillations and waves are also seen to occur spontaneously in intact human submucous plexus preparations with intact networks of ganglia (unpublished observations, Linan-Rico & Christofi, 2015). Glia in culture or intact networks of ganglia may share similar properties. An example is shown in Figure 3 to illustrate that hEGC in intact networks of ganglia or culture display baseline Ca2+ oscillations without stimulating them. Overall, our observations suggest that the hEGC is a suitable model to study glial behavior in normal or inflammed states.
FIGURE 3.
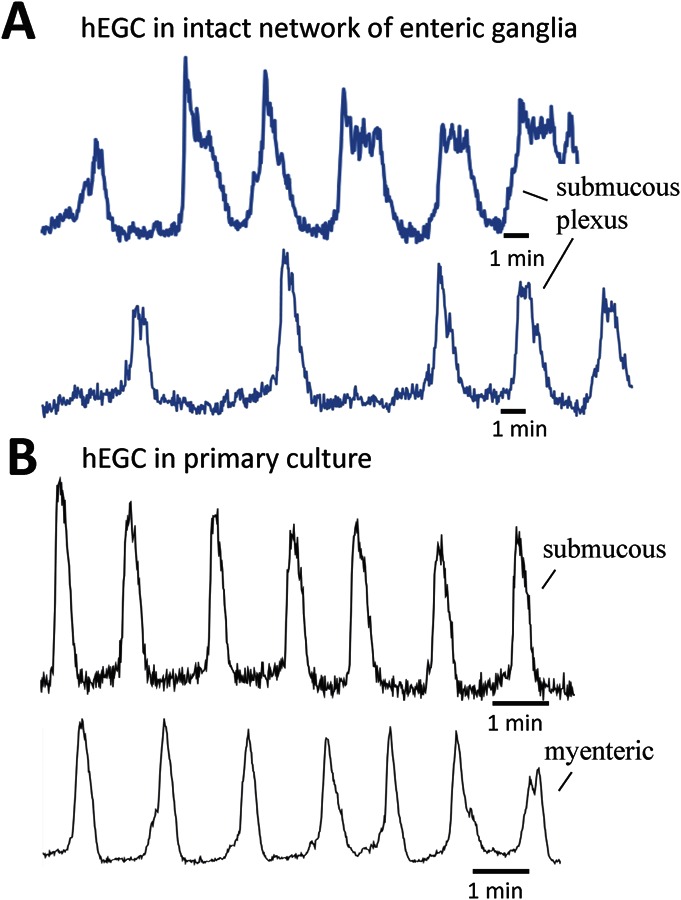
Human EGCs in culture share similar properties with naive hEGC in intact networks of enteric ganglia in microdissected preparations from GI surgical specimens. A, Ca2+ oscillations recorded in hEGC of the submucous plexus (hSMP); hSMP was microdissected from a surgical colon specimen. B, Ca2+ oscillations recorded in hEGC in primary culture, grown from ganglia isolated from a colon surgical specimen. Recording of fluo-4/Ca2+ oscillations in hEGC was made using an LSM Zeiss confocal/PMT imaging system (1 frame/s time-lapse imaging); hEGCs in situ were excited with an Ar-Kr laser at 488 nm, and fluorescence Ca2+ emissions were passed through a 505-nm dichroic and BP 510- to 550-nm filter/photomultiplier tube imaging system (pilot experiment performed by Andromeda Linan-Rico in F. L. Christofi's Purine Neuromodulation laboratory).
In the CNS, most studies show that astroglial Ca2+ signaling modulates neuronal activity through complex and poorly understood mechanisms.100,101 Overall, astroglial Ca2+ signaling increases neuronal activity by gliotransmitter release.102–105 Different gliotransmitters involved include glutamate,105 D-serine,103 or ATP that breaks down to adenosine and activates A1 or A2 receptors at presynaptic sites.102,104,106,107 It is speculated that astroglial regulation of synaptic efficacy may be regulated by calcium-dependent release of inhibitory gliotransmitters, including adenosine (A1 receptors), cannabinoids (CB1 receptors), or GABA (GABAA or GABAB receptors), acting to decrease presynaptic glutamate release.108 Other mechanisms may play a role, not related to gliotransmitter release,109–114 such as, for example, calcium-dependent potassium uptake through astroglial Na+/K+ ATPase to decrease extracellular K+ levels and thereby reduce excitatory synaptic transmission.114 These aspects have not been studied in the ENS.
In the brain, gliotransmission or heterocellular (electrotonic) coupling occurs between glia and neurons through connexin hemichannels.115 Dye-coupling experiments in enteric ganglia provided evidence for glial-to-glial but not glial-to-neuron communication. Structural connections exist between neurons and glia through gap junctions and glia can be shown to be dye coupled.64 Dye coupling occurs in hEGCs, and injection of lucifer yellow into a single glial cell through a patch-pipette spreads to other glial cells (Christofi & Ochoa-Cortes, unpublished observations). It is very likely that signals from a single hEGC can transmit information to much longer distances within and between the ganglia because activation of a single hEGC in a pure culture can be shown to transmit information to another hEGC located ≥1000 μm away recorded by patch-clamp.60 Spread of Ca2+ waves from a single EGC in an intact human enteric ganglion has not been studied yet. Glia modulate the synaptic transmission of CNS (NTS) neurons through a purinergic/ATP signaling mechanism,116,117 are implicated in synaptic plasticity and memory,117 and have a functional role in neuronal excitability,118–122 heterosynaptic depression,119 coupling of neuronal activity to the cerebral microcirculation,120 neuropathophysiology such as in epilepsy,121–123 neuronal inhibitory activity,124,125 and cerebral vascular tone.126,127 ATP release from glia inhibits synaptic activity by adenosine production in the CNS.127 How gliotransmission modulates the ENS is not well understood,10,62 and much less is known than in the CNS. The mediators involved in gliotransmission in ENS remain unknown. Gliotransmission studies in both astrocytes and EGCs have analyzed glial Ca2+ responses by applying endogenous ligands that directly activate neuronal receptors or those that do not discriminate between neurons and glial cells, making it difficult to evaluate the contribution of glia and gliotransmission to neuronal responses. More innovative approaches are needed to address this question.
Specifically, the role of gliotransmission (glial-to-neuron communication) has not been adequately explored in situ in animals and not at all in human gut ENS preparations. Astrocyte Ca2+ signaling and gliotransmission occur in both rodent115 and human brain tissues.128 Glutamate release from astrocytes activates N-methyl-d-aspartate–R on neurons; reciprocal signaling between glia and neurons is suggested to occur in human brain tissue, and it involves a variety of transmitters as noted earlier. These experiments should be feasible in mixed human glial-neural cultures or intact microdissected mouse ganglia from S100B-GFP tagged glial cells using laser confocal Ca2+ imaging techniques129 in dual recordings from a GFP-glial cell and an unlabeled neuron to study communication. In the CNS, changes in membrane potential in astrocytes (glia) can modulate neuronal excitation through heterocellular-coupling,115 and structural connections between neurons and glia also exist in the ENS,64 but it remains unknown whether such connections are functional in the gut.
GAP JUNCTIONS AND CONNEXINS IN hEGCs AND INFLAMMATION
Mammals express 20 or more connexins and 3 pannexins. These proteins represent the main gap junction-forming proteins. Physiologic or pathophysiologic stimulation can cause opening of hemichannels. Inflammation has an impact on the functional expression of hemichannels in glial cells and neurons.130 IL-1β and tumor necrosis factor–α opens connexin (Cx43) channels in astrocytes that can release large molecules such as ATP, glutamate (others) in astrocytes, which can kill neurons in coculture through activation of pannexin hemichannels (P x 1). Overall, studies in astrocytes implicate 2 kinds of gap junction hemichannels in inflammatory responses and cell death. The role of these hemichannels and ATP release in gliotransmission, glial-to-glial communication, and cell death is not yet well characterized in animals (mice), and it is not known in human ENS.
Inflammation leads to alterations in gap junction communication in astrocytes131 with enhanced responses occurring in acute states and attenuated responses in the chronic state. In astrocytes, inflammation regulates expression of connexin-43. The connexin-43 expression and function are both sensitive to inflammation. These channels are involved in purine release in astrocytes. The role of connexin-43 channels or purinergic signaling in the human ENS is unkown although some of our preliminary studies are beginning to unravel these mechanisms.57,60 Gap junction channels allow intercellular exchange of nucleotides, ions, and small molecules between adjacent cells. Two connexons interact in the extracellular space to form the complete intercellular channel; each connexon is composed of 6 proteins, connexins. More than a dozen distinct connexin genes exist belonging to 2 distinct lineages, class I (β group) with Cx26, Cx30, Cx31, Cx31.1, and Cx32, or class II (α group) with Cx33, Cx37, Cx40, Cx43, and Cx46. They differ in their unitary conductance and channel gating properties.132–134 Cx43 is a phosphoprotein expressed in astrocytes but not neurons in the mature brain.135 Hemichannel protein levels change in response to disruption of tissue architecture. For instance, there is increased expression of Cx43 in early stage atherosclerosis.136 A decrease in expression of connexins correlates with tumor progression and metastasis. Expression of connexins and pannexins is likely to be altered in response to intestinal inflammation, resulting from infection or IBD and this aspect deserves further consideration. Cx43 is expressed in EGCs58 and its disruption in mouse EGCs blocks Ca2+ waves and slows intestinal transit but their role in the inflamed gut is unknown.
Several cell lines expressing Cx43m, Cx32, or Cx26 released 5- to 20-fold more ATP than mock-transfected Cx-deficient controls.99 Therefore, Cx expression potentiates ATP release to enlarge calcium waves. Ongoing studies in our laboratory are exploring ATP and purinergic signaling in Ca2+ responses, oscillations, and calcium waves in hEGC propagated through Cx channels in culture and intact enteric neural plexus preparations in normal and inflamed states.60
PUTATIVE MOLECULAR MECHANISMS OF ABNORMAL GLIAL FUNCTION IN INFLAMMED STATES
The molecular mechanisms regulating cytokine-activation of EGC are largely unknown. Particularly, purinergic signaling in EGC is well known to regulate essential mechanisms of cell proliferation, differentiation, death, and neuron-to-EGC communication.63,137 Furthermore, ATP is known to be a proinflammatory stimulus. Although purine release in POI (from the muscularis externa) has never been investigated, it is likely that dying immunocytes (in late phase POI), other dying cells, inflammatory cells, ischemia to the bowel, or extensive physical manipulation during the abdominal surgery, may release large amounts of purines, subsequently leading to sustained or excessive modulation of neuron-to-glial cell crosstalk or cytokine-induced immune responses. As enteric glia are also fully equipped with adrenergic and cholinergic receptors, modulating effects of the extrinsic, parasympathetic, and sympathetic innervation may also be involved, but so far have not been investigated.
Extracellular ATP concentrations remain low (nM-submicromolar) under physiological situations to act on glia to generate Ca2+ waves. However, under pathophysiological conditions or tissue injury, massive release of ATP into the extracellular environment could shift levels to high micromolar/submillimolar range.138,139 It has been proposed that ATP acts as a danger-associated molecular pattern,140 that promotes inflammasome activation141,142 in astrocytes143 or other cells144,145 leading to IL1β secretion that stimulates release/production of other proinflammatory cytokines146,147 and promotes the induction of a reactive astrocyte phenotype.148 NLR family members (NLRP1, 2, 3, 6, 12, NOD2), the HIN-200 family member AIM2, the ATP-release pannexin 1 channel, and ATP gated P2X7 receptor also induce inflammosome activation and IL-1β secretion.149,150 The NLRP2 inflammasome is a multiprotein complex consisting of NLRP2, the adapter protein apoptosis speck-like protein containing a caspase recruitment domain (ASC) and caspase-1. NLRP2 also interacts with pannexin 1 channel and P2X7 receptor. Stimulation of human astrocytes with ATP activates NLRP2 inflammasome leading to processing of caspase-1 and IL-1β. ATP-induced activation of inflammasome is inhibited by pannexin 1 inhibition (with probenecid) and by a P2X7 antagonist; siRNA of NLRP2 significantly decreases NLRP2 levels and caspase-1 processing in response to ATP. The role of ATP in inflammasome activation in EGC is not known.
Our ongoing studies indicated that hEGCs respond to inflammation by increasing the expression of cytokines, chemokines, and various components of the purinergic signaling pathways including P1, P2X receptors, P2Y receptors, and enzymes involved in the metabolism of endogenous purines (unpublished observations, Christofi & Linan-Rico). Differential upregulation of gene expressions that are induced by inflammation are expected to disrupt glial communication, purinergic signaling, and Ca2+ waves (unpublished observations, Linan-Rico, Ochoa-Cortes, Arsenescu & Christofi). These represent potential new therapeutic targets for GI motor disorders, associated with abnormal functions of EGC and neurons—they are relevant to GI infections associated with mucosal inflammation, IBD, POI, and FGIDs (e.g., slow transit constipation of chronic intestinal pseudo-obstruction).
GLIAL PROGENITORS, NEUROGENESIS, AND MOTILITY DISORDERS
Neurons and glial cells of the ENS, like CNS astrocytes, are derived from stem cells in the neural crest, a transient structure present during embryonic development.151 Neural crest progenitors that colonize the gut during embryogenesis give rise to enteric neurons and glia that are organized into intrinsic neural networks of the ENS. Genetic lineage tracing studies performed by Kabouridis et al87 indicate that renewal of mucosal EGCs is from incoming glial cells originating in the plexuses of the gut wall. They also showed that indigenous gut microbiota regulate the homeostasis of EGCs in gut mucosa. Glia are suggested as a potential way to regenerate the ENS. Therefore, EGCs can function as neuronal precursors, giving rise to neurons in vitro in the adult mouse; and it is also possible to induce neurogenesis in vivo in the adult mouse ENS from glial progenitors.152,153 An intriguing possibility in future research is to use glia to replace enteric neurons as an attempt to restore normal motility in diseases involving aganglionosis of the bowel such as Hirschsprung's disease or Chagas disease, both associated with intestinal obstruction. The reader is referred to a commentary on glial progenitors.154
EGC INTERACTIONS IN THE GUT MICROENVIRONMENT
Among their numerous functions, EGCs not only surround and protect neurons but also mediate interactions between neurons and other cell types resident in gut wall. Thanks to their numerous processes, EGCs contact immune effector cells, enteroendocrine cells, epithelial cells, and blood vessels, forming a network specialized in integrating bidirectional signaling from neurons to other cells.
As mentioned above, EGCs may be considered key components of the immune response in the gut against invading microorganisms. Indeed, EGCs release and respond to a wide range of cytokines and chemokines, express MHC I and II and TLRs, the costimulatory proteins CD80 and CD86, interact with lymphocytes, and are in close contact with mast cells and other inflammatory effectors.36,155
Recently, various microbial and environmental interactions with EGCs have been documented. Human EGCs are activated by exogenous stimuli, such as LPS,14 and in vitro and ex vivo models of Shigella flexneri invasion of IECs suggest a protective role of EGCs and enteroglial-derived factors against invading pathogens, probably thus contributing to the regulation of the host–bacteria interaction.156 A more mechanistic insight into EGC role in host–bacteria crosstalk came from the demonstration that EGC express TLR in response to AIEC contact, and NO production18 depends on both TLR and S100B\RAGE signaling. As abnormal NO production is a main feature in IBD and considering that various pathogenic and commensal bacteria have been implicated in the pathogenesis of gut inflammatory diseases (i.e., IBD),157 exploring the role of EGC–bacteria interaction in the context of IBD and/or inflammatory GI disorders may open up new and interesting scenarios. In line with this, Kabouridis et al,87 in a recent article, found that mucosal EGCs were profoundly reduced in number in the germ-free mice and that restoring normal gut microbiota rescued this difference. They also found that gut microbiota is required for sustaining generation of new EGCs in adult gut, once again highlighting the close relationship between EGC, bacteria, and the microbiome.
In the CNS, reactive astrogliosis is a general response in astrocytes to disease and brain injury associated with trauma, major surgery (postsurgical neuroinflammation), infection (and neuroinflammation), ischemia, and neurodegeneration.158–160 Reactive astroglia are emerging as pivotal regulators of inflammatory responses—they can produce a variety of proinflammatory molecules including cytokines, chemokines, growth factors, NO, and Prostaglandin E (PGE) similar in many ways to EGCs. A striking distinction is that the reactive astroglial phenotype strongly depends on the type of injury or inflammation.159 For example, genomic analysis of reactive astrogliosis indicated that astroglia induced by LPS exhibited transcriptosome changes and a molecular phenotype toward a proinflammatory and cytotoxic profile. In contrast, ischemia caused by cerebral artery occlusion shifted the astrocyte transcriptosome toward a neuroprotective profile. Inflammatory mediators (LPS, IFNγ, and TGF-β1) could alter the astrocyte transcriptosome and calcium signaling elicited by multiple G protein-coupled receptors including purinergic (P2Y14, P2Y1, A1), adenosinergic (ADRA2A), CXCR4 (CXCL12), and endothelin-1 receptors (EDNRA/B). The downstream functional consequences of such changes in GPCRs in astrocytes are not clear, but 1 study indicated that reactive astrogliosis in response to viral infection could significantly alter neuronal function.161 These aspects have also not been explored for GI diseases such as IBD, bacterial or viral infections, POI, FGIDs, or gut ischemia. A better understanding of the reactive EGC phenotype offers the potential for selective pharmacological manipulation and therapeutic intervention in GI diseases and disorders.
In addition to bacteria, viruses can target EGCs. Selgrad et al162 found an active lytic JC virus infection in myenteric glial cells derived from patients with chronic idiopathic intestinal pseudo-obstruction, a rare syndrome characterized by severely impaired GI motility. Moreover, as summarized by Galligan,163 HIV disrupts nervous system function by infecting EGCs. Infected glia release the HIV transactivated factor, Tat, which could alter action potentials in enteric neurons by increasing Na+ channel expression. Tat can also interact synergistically with morphine, worsening GI dysfunction in HIV-infected narcotic users and in HIV-infected patients using opioid drugs to treat diarrhea. Bacteria, bacterial products and viruses, may induce activation of EGCs (e.g., produce a reactive glial phenotype) and disrupt ENS function also in patients with necrotizing enterocolitis, a pathology characterized by increased gut permeability and epithelial leakiness.164
Since it has been discovered that the targeted ablation of enteric glia in the small intestine caused a fulminant and fatal inflammation of the bowel with the consequent breakdown of the intestinal barrier, EGC–IEC interactions have been largely documented and discussed, going beyond the aims of this review. Briefly summarizing, from in vitro models, EGCs can decrease mucosal permeability and regulate ZO-1 and occludin expression, in part by the release of S-nitrosoglutathione.165 This serves to protect/maintain mucosal integrity. The release of transforming growth factor β-1 inhibits IEC proliferation,166 whereas, by the synthesis and release of the prostaglandin 15dPGJ2, EGCs are involved in the control of IEC proliferation/differentiation, through activation of PPARγ.167 At a genetic level, impact of EGCs on IEC transcriptome results in regulation of genes involved in cell-to-cell and cell-to-matrix adhesion and also cell differentiation/proliferation and cell motility.168
In a article, using serial-block face scanning electron microscope along with confocal microscopy, a close connection between EGCs and enteroendocrine cells has been revealed and it has been proposed that EGCs, by the release of neurotrophic factors (GDNF and/or S100B), modulate the proliferation and hormone content in enteroendocrine cells.169 This hypothesis needs to be confirmed but, because hormone secretion from enteroendocrine cells is altered by high-fat diets and also pancreatic beta cells express GDNF receptors,170 it may be speculated that EGCs could also be involved in the regulation of obesity.
Recent studies have shown that electrical stimulation of the vagus nerve can protect the intestinal barrier and alleviate inflammatory intestinal injury in animals after burn injury, through activation of EGCs.171 Moreover, electroacupuncture, through EGC activation, attenuates hemorrhage-induced intestinal inflammatory insults and protects the intestinal barrier integrity.172
Another environmental interaction with EGC is that induced by the antiprotozoal drug pentamidine, which is able to reduce inflammation, is DSS-induced colitis in mice likely by targeting EGC because drug treatment reduced both GFAP expression in EGC and reduced S100B-induced inflammation in the mucosa of treated mice.173
Taken together, these data seem to suggest a central role for EGCs in integrating signals coming from different cells involved in autonomic, neurologic, motility, secretion, endocrine, absorption, and barrier/immune functions to sustain gut homeostasis. It is conceivable, although not unraveled yet, that alterations in the integrated glial cells network of connection may be a contributing factor in the development of intestinal pathologies involving motility (FGIDs, slow transit constipation, POI, and PD) and inflammation (IBD, POI, bacterial or viral infections). Whether no specific EGC alterations have been linked to any pathology, it is probably because glial changes are so devious and backhanded and glial interactions with other cells are so promiscuous that they are difficult to pinpoint. It is also very difficult to prove because multiple cells are implicated in the pathogenesis of chronic diseases such as IBD or FGIDs, and a single cell like the EGCs cannot explain the clinical symptoms.
EGCs–ENS interactions may regulate motility, secretion, nutrient uptake, and blood flow in the intestinal tract. In disease states, emerging evidence suggests an important role in promoting inflammation—they produce inflammatory cytokines, substance P and neurokinin A to act on immune cells.174 Glial proliferation occurs in IBD in both animal models and humans.28 As noted earlier, EGC activation causes amplification of intestinal inflammation, in part through production of glial S100B.30 A study by Esposito et al30 provided novel evidence that PEA is an effective anti-inflammatory drug able to block inflammation in animals and humans by selective targeting of the S100B/TLR4 dependent PPAR activation pathway on EGCs, to cause inhibition of NFκB-dependent inflammation. A comprehensive analysis was performed in mouse models of DSS-induced colitis, colonic biopsies derived from patients with UC, and primary cultures of mouse and hEGCs. PEA represents a potential therapeutic target for UC. The anti-inflammatory effects of PEA depend on its ability to activate peroxisome proliferator-activated receptors (PPARs), representing ligand-activated transcription factors.175 The anti-inflammatory effects of PEA are blocked after silencing of PPAR-α expression in EGCs, in PPAR-α–knockout mice, indicating that effects of PEA are mainly dependent on PPAR-α. The reader is also referred to a commentary176 on the gut article by Esposito et al.30
EGCs—Reporter Mouse Models To Study GI Diseases
Intimate association with a variety of different enteric cell types makes the EGC a most interesting cell among all cells of the gut wall along the entire GI tract. The role of EGCs in physiological regulation of GI function and their role in GI disorders have been demonstrated in studies performed in several rodent models. With these models, some more or less EGC-specific molecules or their gene expression–driving promoters have been used to analyze EGC function. Among these markers, the most commonly used molecules are intermediate filament GFAP, the Ca2+-binding protein S100B and transcription factor Sox10. Studies have used GFAP to drive expression of artificial genes to cause EGC death.13 Other studies used an autoimmune model of CD8 T cell–driven EGC elimination driven by GFAP expression of a specific T-cell antigen.23,56 In both models, EGC ablation resulted in lethal gut pathology. However, collateral damage was also observed because of nonexclusive expression of GFAP, particularly in central astrocytes, the so far accepted counterpart of EGC. Although significance of the data received from GFAP-mediated genetic ablation is impaired because of the absence of a highly specific EGC reporter activity, the data indicate that EGCs play a crucial role in GI function.
Several transgenic mice have been developed using reporter genes, allowing for detection and in vivo and ex vivo functional analysis of EGCs. These mice express GFP either under control of the GFAP, S100B, or Sox-10 promoter. Most of these mouse strains were initially created to study astrocytes in the CNS. Recent elegant studies used a novel multicolor Cre reporter mouse for lineage tracing of Sox-10 expressing EGC, demonstrating that mucosal EGCs are continuously renewed from incoming glial cells originating in the ENS (neural plexuses) of the gut wall.87 Unexpectedly, extensive heterogeneity and plasticity of EGC has been shown by this lineage tracing studies.10 Of note, not all EGCs express all previously mentioned markers. They are rather seen as a heterogeneous cell population, particularly differing in their neurochemical coding and function between interganglionic and intraganglionic EGC. Although intraganglionic EGCs coexpress most of the prototypical EGC markers GFAP, S100β, and Sox10, some EGC found outside the myenteric ganglia do not label for these markers.
Although this heterogeneity makes EGC even more interesting, so far no reliable methods or markers, with the exception of Cre-driven lineage tracing experiments, exist allowing separation of different EGC subpopulations. In vitro experiments with primary EGCs are challenging because of exhausting and costly isolation procedures to obtain a pure yield of living EGCs to use in functional studies. An EGC line has been developed by Rühl et al177 and some homologies to primary EGC, particularly GFAP, S100, and vimentin expression were described. However, as for many transformed cell lines, an abnormal high proliferation rate and insufficient differentiation, and many missing homologies to primary EGCs for mouse rat and human, make these cells a nonideal cell culture model. With improved isolation procedures, enriched primary EGC cultures can be obtained from incomplete muscularis externa digestions allowing mechanical separation of individual ganglia.178 Outgrowth of EGCs in isolated ganglia generated EGC cultures of up to 80% purity. These cells were shown to express GFAP, S100B, and Sox10 and respond to ATP178 and IL-1β.90 However, these cells can be contaminated with faster growing smooth muscle cells and myofibroblasts, and special attention to specific culture conditions is required to eliminate erroneous signals. We are using immunoisolation techniques to provide a more pure hEGC population18 suitable for patch-clamp, Ca2+ imaging, Ca2+ waves, and molecular signaling studies.18,30,60
Despite progress to date with various rodent models, further in-depth EGC characterization of subpopulations, improved isolation techniques and molecular and functional fingerprinting of this heterogeneous cell population are required. The common strategy of knowledge translation from central astrocytes to EGC research should be carefully performed and interpreted because differences do exist between these cell types, determined by their individual niches and unique microenvironments. This is obvious as illustrated in as simple findings as failure to detect robust astrocyte markers such as glutamate receptor 1 (Glast-1) (S. Wehner, unpublished observations) or aldehyde dehydrogenase family member L1 (Aldh1L1)179 in most of the mouse EGC populations.
As mentioned before, technical limitations in EGC research are based on the absence of a precise set of markers, particularly cell surface markers, able to identify and isolate different subsets of EGC. In comparison to the broad spectrum of established immunocytes markers, allowing precise description of their activation status and function under naive and pathological conditions, those markers are mostly missing for EGC. Recent improvement of cellular isolation techniques from small animal and hEGC under naive and pathological conditions will fill this gap. A first experimental approach is a comparative gene expression array and flow cytometry. In line with insufficient presence of specific EGC markers, data about posttranslational and epigenetic alterations in EGC in inflammatory diseases but also GI cancer development are missing. Finally, it remains an open question if a dysregulated EGC function is the hen or the egg in individual GI pathologies or just a consequence of other mechanisms affecting the ENS.
FUTURE DIRECTIONS, TECHNICAL INNOVATIONS, AND NOVEL MODELS FOR STUDIES WITH HUMAN EGCs
We are applying real-time and laser confocal Ca2+ imaging (using a camera or PMT/resonant scanner detector) to monitor Ca2+ responses in hEGC to study Ca2+ oscillations, cell-to-cell communication, connexin hemichannel activity, and Ca2+ waves in isolated hEGC from human surgical specimens, mixed glial/neural cultures, isolated human enteric ganglia, or intact microdissected human myenteric or submucous plexus preparations. Human EGCs isolated from intestinal surgical specimens grown in culture (hEGC) share very similar functional properties to native hEGC in the intact ENS (e.g., microdissected human submucous plexus) of surgical specimens (as noted earlier, Fig. 3). Therefore, the hEGC culture model is emerging as a suitable model to investigate the physiology and pathophysiology of hEGCs in the human intestinal tract. Ca2+ responses and Ca2+ waves are required for normal intestinal transit/motility, and by disruption of glial-to-glial communication in hGFAP::CreERT2+/−/Cx43f/f/mice (by disruption of the gene encoding connexin-43 hemichannels), they were able to show that disruption of Ca2+ waves disrupts intestinal motility and gut transit. Cx43 channels are also involved in cell-to-cell communication in hEGCs (unpublished observations, Ochoa Cortes, Linan-Rico, Turco, Cuomo & Christofi). Therefore, motility is regulated by Ca2+ waves in EGCs. This provides a suitable target for translational studies in hEGC with clinical relevance to motility, IBD, and gut inflammation-induced by bacterial or viral infections. Thus, in-depth mechanistic studies related to Ca2+ excitability, in response to infection or inflammation in EGCs, ischemia, injury, excessive mechanical stimulation (i.e., stretch to mimic luminal distension), represent a key-piece of the puzzle in understand GI diseases and motility disorders.
Dual patch-clamp recordings in combination with fluorescent Ca2+ reporter techniques are under development for use in mixed hEGC/neuronal cultures or isolated ganglia are probing the questions related to neural-glial communication. Optogenetic calcium reporter mice for glia (GCAMP3/GFAP:Cre) or mixed neural/glial cultures (GCAMP3/Wnt1:Cre) are suitable for studies on glial calcium waves for analysis without any interference from calcium signals in neurons.10,64 GCAMP3/Wnt1:Cre mice can be used to study neuron-to-glial communication in normal or inflamed colon. An advantage of the optogenetic reporter mouse model is that there is no need any longer to load inflamed tissues with calcium dye to study calcium signaling. Loading is variable and depends on the health of the tissue/cells taking up the dye, and may provide some erroneous results.
Future studies are necessary to evaluate complex molecular signaling mechanisms that operate in EGCs and to reveal species differences between animals and humans (in normal or inflamed states). Our studies on glia transcriptosome analysis in hEGCs and riboTag/Rp122HA mouse lines could provide a plethora of information on gene expression and alterations of genes in response to infection, inflammation, or other diseases. Silencing RNAs are a suitable tool to study the role of specific receptor/genes in the function of hEGC,175,176 and future studies on physiologic and pathophysiologic mechanisms, including purinergic signaling mechanisms. Glial cells-specific–knockout mouse models are key to deciphering the molecular mechanisms involved glial regulation of motility and intestinal transit.58
CONCLUDING REMARKS
Emerging evidence suggests that EGC are critical regulators of gut homeostatic functions by modulating mucosal permeability, neural activity (of both intrinsic and extrinsic innervations), immune functions, motility, secretion (endocrine/electrolyte), absorption, and vascular tone. In disease states (i.e., infection, IBD, IBS/FGIDs, POI, ischemic bowel, slow transit constipation, DD, neurodegenerative diseases such as PD affecting the gut causing constipation), a reactive EGC phenotype can potentially have profound effects on gut behavior and this is not well understood. In diseases, EGCs promote inflammation and can therefore disrupt and contribute to the pathology and clinical symptoms by various mechanisms. Our emerging concept of the reactive EGC phenotype is illustrated in Figure 4. Notwithstanding the seminal contributions of a number of investigative groups around the world (i.e., Gabella, Hanani, Savidge, Sharkey, Ruhl, Gulbransen, Vanden Berghe, Smith, Wehner, Turco, Cirello, Cuomo, and Esposito), our understanding of neural-glial communication, gliotransmission, Ca2+ waves in normal and inflamed states is still at its infancy. And we know even less about the reactive EGC phenotype in specific diseases or disorders. A huge gap exists in knowledge of hEGCs, and the technology is now available to study them in situ or primary culture. SiRNA studies in hEGC can complement studies in mouse EGCs from genomic models to confirm whether mouse data can be translated to humans. Overall, hEGCs offer a unique translational model to study their function in health and disease. Purinergic signaling is emerging as a pivotal mechanism worth investigation with potential novel therapeutic targets in the horizon for diverse pathologies such as IBD, GI infection, POI, FGIDs, motility disorders (i.e., slow transit constipation), and neurodegenerative diseases (i.e., constipation in patients with PD). A recent review focused on the therapeutic potential of purines in IBD and FGIDs.180 The EGC is another potential target for intervention.
FIGURE 4.
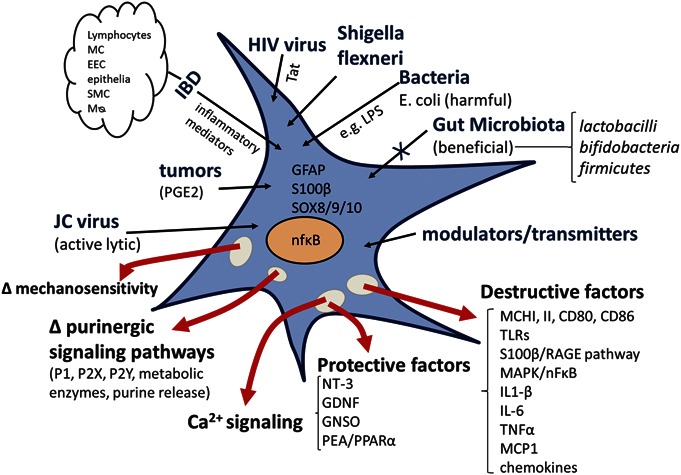
Working model of activation of EGC and induction of a “reactive EGC phenotype,” in ENS: a variety of neurotransmitters and potential gliotransmitters can activate EGC including 5-HT, ACh, GABA, glutamate, lipids, ATP, uridine triphosphate, ADP, and adenosine. Intestinal infection with bacteria or viruses, inflammatory mediators, IBD, or POI can induce a reactive glial cell phenotype. HIV virus or JC virus can infect hEGC and cause phenotypic changes, associated with changes in neural behavior. Bacterial infection with AIEC or Shigella flexneri can activate EGC, but they can discriminate between these pathogenic bacteria and probiotic bacteria (Lactobacillus paracasei). Inflammatory mediators released in the gut in response to infection, IBD, or POI can activate EGC. The “reactive glial phenotype” can release protective factors (neurotrophin-3, GDNF, GNSO, and PEA/PPAR-α) or destructive factors (proinflammatory mediators such as IL1β, IL-6, tumor necrosis factor-α, MCP-1, activation of the mitogen-activated protein kinase/NF-κB proinflammatory transcription pathway, upregulation of MHC class II molecules, and activation of TLRs). Such insults in various pathologic situations can alter EGC mechanosensitivity, Ca2+ signaling, and purinergic signaling. Abnormal glial function is expected to alter neuron-to-glial communication, neural circuit behavior in the ENS, neuromuscular transmission, and GI motility. Mø, macrophage; EEC, enteroendocrine cells; SMC, smooth muscle cells; TLR, Toll-like receptors.
Considering the almost consolidated role of EGC in intestinal homeostasis, disruption of EGC network, with the subsequent abnormal release of glial mediators (S100B, NO, cytokines, and many others not yet known), may account for the progression, if not the onset, of intestinal inflammation, also in IBD. Restoring proper EGC function may prove to be an efficient strategy to dampen inflammation and in this sense, the use of probiotics and PEA may be very useful to counteract S100B and other cytokines release from EGC, and also the use of S100B siRNA.
Future studies on EGCs should also focus on unraveling the mechanisms underlying the crosstalk among EGC, IEC, and intestinal neurons, and also the comprehension of EGC immune interactions. These studies may reveal the possibility to manipulate EGC functions to counteract intestinal diseases. Finally, an emerging concept is that, through the “gut-brain axis,” is possible to modulate central symptoms by acting in the periphery and vice versa. Establishing the role of EGC in this bidirectional communication may be of extreme interest, from both a clinical and research standpoint.
Footnotes
This work was supported by NINDS Diabetes and Kidney Diseases Grant DK093499 and NCRR S10RR11434 shared instrumentation Grant to F. L. Christofi; strategic initiative funds from the department of Anesthesiology and Neuroscience Signature Program at the Wexner Medical Center to F. L. Christofi toward developing a Neuromodulation Program. F. Turco was a visiting scholar in Dr. F. L. Christof's Purine Neuromodulation Lab from Dr. R. Cuomo's group at the University of Naples, Italy. This work was also supported by a Grant to F. Turco and R. Cuomo from the Italian Ministry of University and Research, COFIN project 2009HLNNRL. S. Wehner is a member of the DFG Excellence Cluster ImmunoSensation at the University of Bonn, Germany. A. Linan-Rico and F. Ochoa-Cortes are postdoctoral scientists in Dr. F. L. Christofi's laboratory who contributed to novel concepts on hEGCs described in the review based on their ongoing research. Dr. E. Whitaker is a physician scientist in Dr. Christofi's lab and is supported by an NIH Loan Repayment Program grant and a Davis Bremmer Foundation pre-K Award through the NIH CTSA. Many thanks to Peter Vacarrella who made figures and illustrations, and Ms Iveta Grants for excellent administrative support to prepare and submit the manuscript.
The authors have no conflict of interest to disclose
REFERENCES
- 1.Gabella G. Fine structure of the myenteric plexus in the guinea-pig ileum. J Anat. 1972;111:69–97. [PMC free article] [PubMed] [Google Scholar]
- 2.Gulbransen BD, Sharkey KA. Novel functional roles for enteric glia in the gastrointestinal tract. Nat Rev Gastroenterol Hepatol. 2012;9:625–632. [DOI] [PubMed] [Google Scholar]
- 3.Sharkey KA. Emerging roles for enteric glia in gastrointestinal disorders. J Clin Invest. 2015;125:918–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu YB, Li YQ. Enteric glial cells and their role in the intestinal epithelial barrier. World J Gastroenterol. 2014;20:11273–11280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goyal RK, Hirano I. The enteric nervous system. N Engl J Med. 1996;34:1106–1115. [DOI] [PubMed] [Google Scholar]
- 6.Bozarov A, Wang YZ, Yu JG, et al. Activation of adenosine low-affinity A3 receptors inhibits the enteric short interplexus neural circuit triggered by histamine. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1147–G1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christofi FL. Purinergic receptors and gastrointestinal secretomotor function. Purinergic Signal. 2008;4:213–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Furness JB. The enteric nervous system: normal functions and enteric neuropathies. Neurogastroenterol Motil. 2008;20:32–38. [DOI] [PubMed] [Google Scholar]
- 9.Hoff S, Zeller F, Von Weyhern CW, et al. Quantitative assessment of glial cells in the human and guinea pig enteric nervous system with an anti-Sox8/9/10 antibody. J Comp Neurol. 2008;509:356–371. [DOI] [PubMed] [Google Scholar]
- 10.Boesmans W, Lasrado R, Vanden Berghe P, et al. Heterogeneity and phenotypic plasticity of glial cells in the mammalian enteric nervous system. Glia. 2015;63:229–241. [DOI] [PubMed] [Google Scholar]
- 11.Gershon MD, Rothman TP. Enteric glia. Glia. 1991;4:195–204. [DOI] [PubMed] [Google Scholar]
- 12.Rühl A. Glial cells in the gut. Neurogastroenterol Motil. 2005;17:777–790. [DOI] [PubMed] [Google Scholar]
- 13.Bush TG, Savidge TC, Freeman TC, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. [DOI] [PubMed] [Google Scholar]
- 14.Cirillo C, Sarnelli G, Turco F, et al. Proinflammatory stimuli activates human-derived enteroglial cells and induces autocrine nitric oxide production. Neurogastroenterol Motil. 2011;23:e372–e382. [DOI] [PubMed] [Google Scholar]
- 15.Rolhion N, Darfeuille-Michaud A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:1277–1283. [DOI] [PubMed] [Google Scholar]
- 16.Swidsinski A, Ladhoff A, Pernthaler A, et al. Mucosal flora in inflammatory bowel disease. Gastroenterology. 2002;122:44–54. [DOI] [PubMed] [Google Scholar]
- 17.Rutgeerts P, Goboes K, Peeters M, et al. Effect of faecal stream diversion on recurrence of Crohn's disease in the neoterminal ileum. Lancet. 1991;338:771–774. [DOI] [PubMed] [Google Scholar]
- 18.Turco F, Sarnelli G, Cirillo C, et al. Enteroglial-derived S100B protein integrates bacteria-induced Toll-like receptor signalling in human enteric glial cells. Gut. 2014;63:105–115. [DOI] [PubMed] [Google Scholar]
- 19.Glasser AL, Boudeau J, Barnich N, et al. Adherent invasive Escherichia coli strains from patients with Crohn's disease survive and replicate within macrophages without inducing host cell death. Infect Immun. 2001;69:5529–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chassaing B, Darfeuille-Michaud A. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology. 2011;140:1720–1728. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen HT, Dalmasso G, Müller S, et al. Crohn's disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology. 2014;146:508–519. [DOI] [PubMed] [Google Scholar]
- 21a.MacEachern SJ, Patel BA, Keenan CM, et al. Inhibiting inducible nitric oxide synthase in enteric glia restores electrogenic ion transport in mice with colitis. Gastroenterology. 2015;149:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie J, Méndez JD, Méndez-Valenzuela V, et al. Cellular signalling of the receptor for advanced glycation end products (RAGE). Cell Signal. 2013;25:2185–2197. [DOI] [PubMed] [Google Scholar]
- 23.Cornet A, Savidge TC, Cabarrocas J, et al. Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn's disease? Proc Natl Acad Sci U S A. 2001;98:13306–13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cirillo C, Sarnelli G, Esposito G, et al. Increased mucosal nitric oxide production in ulcerative colitis is mediated in part by the enteroglial-derived S100B protein. Neurogastroenterol Motil. 2009;21:1209–e112. [DOI] [PubMed] [Google Scholar]
- 25.Cirillo C, Sarnelli G, Mango A, et al. Enteric glia stimulates inflammation-induced responses in human intestine and interacts with immune cells via S100B protein. Gastroenterology. 2009;136:A271–A272. [Google Scholar]
- 26.Von Boyen GB, Schulte N, Pflüger C, et al. Distribution of enteric glia and GDNF during gut inflammation. BMC Gastroenterol. 2011;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.von Boyen GB, Steinkamp M, Reinshagen M, et al. Proinflammatory cytokines increase glial fibrillary acidic protein expression in enteric glia. Gut. 2004;53:222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cirillo C, Sarnelli G, Esposito G, et al. S100B protein in the gut: the evidence for enteroglial-sustained intestinal inflammation. World J Gastroenterol. 2011;17:1261–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neunlist M, Rolli-Derkinderen M, Latorre R, et al. Enteric glial cells: recent developments and future directions. Gastroenterology. 2014;147:1230–1237. [DOI] [PubMed] [Google Scholar]
- 30.Esposito G, Capoccia E, Turco F, et al. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut. 2014;63:1300–1312. [DOI] [PubMed] [Google Scholar]
- 31.Valès S, Biraud M, Marionneau-Lambot S, et al. Enteric glial cells activate colon cancer stem cells to promote tumorigenesis. FASEB J. 2015;1:1000.2. [Google Scholar]
- 32.Bassotti G, Battaglia E, Bellone G, et al. Interstitial cells of Cajal, enteric nerves, and glial cells in colonic diverticular disease. J Clin Pathol. 2005;58:973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wedel T, Büsing V, Heinrichs G, et al. Diverticular disease is associated with an enteric neuropathy as revealed by morphometric analysis. Neurogastroenterol Motil. 2010;22:407–414, e93–94. [DOI] [PubMed] [Google Scholar]
- 34.Bassotti G, Villanacci V, Cathomas G, et al. Enteric neuropathology of the terminal ileum in patients with intractable slow-transit constipation. Hum Pathol. 2006;37:1252–1258. [DOI] [PubMed] [Google Scholar]
- 35.Iantorno G, Bassotti G, Kogan Z, et al. The enteric nervous system in chagasic and idiopathic megacolon. Am J Surg Pathol. 2007;31:460–468. [DOI] [PubMed] [Google Scholar]
- 36.Da Silveira AB, Freitas MA, de Oliveira EC, et al. Glial fibrillary acidic protein and S-100 colocalization in the enteroglial cells in dilated and nondilated portions of colon from chagasic patients. Hum Pathol. 2009;40:244–251. [DOI] [PubMed] [Google Scholar]
- 37.Erlich RB, Kahn SA, Lima FR, et al. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia. 2007;55:1690–1698. [DOI] [PubMed] [Google Scholar]
- 38.Lima FR, Arantes CP, Muras AG, et al. Cellular prion protein expression in astrocytes modulates neuronal survival and differentiation. J Neurochem. 2007;103:2164–2176. [DOI] [PubMed] [Google Scholar]
- 39.Braga CA, Follmer C, Palhano FL, et al. The anti-Parkinsonian drug selegiline delays the nucleation phase of α-synuclein aggregation leading to the formation of nontoxic species. J Mol Biol. 2011;405:254–273. [DOI] [PubMed] [Google Scholar]
- 40.Kujala P, Raymond CR, Romeijn M, et al. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog. 2011;7:e1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seelig DM, Mason GL, Telling GC, et al. Chronic wasting disease prion trafficking via the autonomic nervous system. Am J Pathol. 2011;179:1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cronier S, Carimalo J, Schaeffer B, et al. Endogenous prion protein conversion is required for prion-induced neuritic alterations and neuronal death. FASEB J. 2012;26:3854–3861. [DOI] [PubMed] [Google Scholar]
- 43.Fonseca AC, Romão L, Amaral RF, et al. Microglial stress inducible protein 1 promotes proliferation and migration in human glioblastoma cells. Neuroscience. 2012;200:130–141. [DOI] [PubMed] [Google Scholar]
- 44.Devos D, Lebouvier T, Lardeux B, et al. Colonic inflammation in Parkinson's disease. Neurobiol Dis. 2013;50:42–48. [DOI] [PubMed] [Google Scholar]
- 45.Shannon KM, Keshavarzian A, Dodiya HB, et al. Is alpha-synuclein in the colon a biomarker for premotor Parkinson's disease? Evidence from 3 cases. Mov Disord. 2012;27:716–719. [DOI] [PubMed] [Google Scholar]
- 46.Clairembault T, Kamphuis W, Leclair-Visonneau L, et al. Enteric GFAP expression and phosphorylation in Parkinson's disease. J Neurochem. 2014;130:805–815. [DOI] [PubMed] [Google Scholar]
- 47.Blandini F, Balestra B, Levandis G, et al. Functional and neurochemical changes of the gastrointestinal tract in a rodent model of Parkinson's disease. Neurosci Lett. 2009;467:203–207. [DOI] [PubMed] [Google Scholar]
- 48.Cersosimo MG, Benarroch EE. Pathological correlates of gastrointestinal dysfunction in Parkinson's disease. Neurobiol Dis. 2012;46:559–564. [DOI] [PubMed] [Google Scholar]
- 49.Sigge W, Wedel T, Kühnel W, et al. Morphologic alterations of the enteric nervous system and deficiency of non-adrenergic non-cholinergic inhibitory innervation in neonatal necrotizing enterocolitis. Eur J Pediatr Surg. 1998;8:87–94. [DOI] [PubMed] [Google Scholar]
- 50.Wedel T, Krammer HJ, Kühnel W, et al. Alterations of the enteric nervous system in neonatal necrotizing enterocolitis revealed by whole-mount immunohistochemistry. Pediatr Pathol Lab Med. 1998;18:57–70. [PubMed] [Google Scholar]
- 51.Sokol H, Polin V, Lavergne-Slove A, et al. Plexitis as a predictive factor of early postoperative clinical recurrence in Crohn's disease. Gut. 2009;58:1218–1225. [DOI] [PubMed] [Google Scholar]
- 52.Coelho-Aguiar Jde M, Bon-Frauches AC, Gomes AL, et al. The enteric glia: identity and functions. Glia. 2015;63:921–935. [DOI] [PubMed] [Google Scholar]
- 53.Steinkamp M, Gundel H, Schulte N, et al. GDNF protects enteric glia from apoptosis: evidence for an autocrine loop. BMC Gastroenterol. 2012;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinkamp M, Schulte N, Spaniol U, et al. Brain derived neurotrophic factor inhibits apoptosis in enteric glia during gut inflammation. Med Sci Monit. 2012;18:BR117–BR122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nasser Y, Fernandez E, Keenan CM, et al. Role of enteric glia in intestinal physiology: effects of the gliotoxin fluorocitrate on motor and secretory function. Am J Physiol Gastrointest Liver Physiol. 2006;291:G912–G927. [DOI] [PubMed] [Google Scholar]
- 56.Aubé AC, Cabarrocas J, Bauer J, et al. Changes in enteric neurone phenotype and intestinal functions in a transgenic mouse model of enteric glia disruption. Gut. 2006;55:630–637.16236773 [Google Scholar]
- 57.Linan Rico A, Grants I, Needleman BJ, et al. Gliomodulation of neuronal and motor behavior in the human GI tract. Gastroenterology. 2015;148:S-18. [Google Scholar]
- 58.McClain JL, Grubišić V, Fried D, et al. Ca2+ responses in enteric glia are mediated by connexin-43 hemichannels and modulate colonic transit in mice. Gastroenterology. 2014;146:497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Broadhead MJ, Bayguinov PO, Okamoto T, et al. Ca2+ transients in myenteric glial cells during the colonic migrating motor complex in the isolated murine large intestine. J Physiol. 2012;590:335–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Linan Rico A, Turco F, Soghomonyan S, et al. Modulation of Ca2+ waves in human enteric glial cells. Gastroenterology. 2015;148:S-79. [Google Scholar]
- 61.Gulbransen BD, Sharkey KA. Purinergic neuron-to-glia signaling in the enteric nervous system. Gastroenterology. 2009;136:1349–1358. [DOI] [PubMed] [Google Scholar]
- 62.Boesmans W, Martens MA, Weltens N, et al. Imaging neuron-glia interactions in the enteric nervous system. Front Cell Neurosci. 2013;7:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gulbransen BD, Bashashati M, Hirota SA, et al. Activation of neuronal P2X7 receptor-pannexin-1 mediates death of enteric neurons during colitis. Nat Med. 2012;18:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gabella G. Ultrastructure of the nerve plexuses of the mammalian intestine: the enteric glial cells Neuroscience. 1981:6:425–436. [DOI] [PubMed] [Google Scholar]
- 65.Okamoto T, Barton MJ, Hennig GW, et al. Extensive projections of myenteric serotonergic neurons suggest they comprose the central processing unit in th colon. Neurogastroenterolo Motil. 2014;26:556–570. [DOI] [PubMed] [Google Scholar]
- 66.Garcia-Abreu J, Silva LC, Tovar FF, et al. Compartmental distribution of sulfated glycosaminoglycans in lateral and medial midbrain astroglial cultures. Glia. 1996;17:339–344. [DOI] [PubMed] [Google Scholar]
- 67.Fróes MM, Correia AH, Garcia-Abreu J, et al. Gap-junctional coupling between neurons and astrocytes in primary central nervous system cultures. Proc Natl Acad Sci U S A. 1999;96:7541–7546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Faria J, Romão L, Martins S, et al. Interactive properties of human glioblastoma cells with brain neurons in culture and neuronal modulation of glial laminin organization. Differentiation. 2006;74:562–572. [DOI] [PubMed] [Google Scholar]
- 69.Izmiryan A, Cheraud Y, Khanamiryan L, et al. Different expression of synemin isoforms in glia and neurons during nervous system development. Glia. 2006;54:204–213. [DOI] [PubMed] [Google Scholar]
- 70.Romão LF, Mendes FA, Feitosa NM, et al. Connective tissue growth factor (CTGF/CCN2) is negatively regulated during neuron-glioblastoma interaction. PLoS One. 2013;8:e55605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Araque A, Parpura V, Sanzgiri RP, et al. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci. 1999;22:208–215. [DOI] [PubMed] [Google Scholar]
- 72.Romão LF, Sousa Vde O, Neto VM, et al. Glutamate activates GFAP gene promoter from cultured astrocytes through TGF-beta1 pathways. J Neurochem. 2008;106:746–756. [DOI] [PubMed] [Google Scholar]
- 73.Bezzi P, Volterra A. A neuron-glia signalling network in the active brain. Curr Opin Neurobiol. 2001;11:387–394. [DOI] [PubMed] [Google Scholar]
- 74.Diniz LP, Almeida JC, Tortelli V, et al. Astrocyte-induced synaptogenesis is mediated by transforming growth factor β signaling through modulation of D-serine levels in cerebral cortex neurons. J Biol Chem. 2012;287:41432–41445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gomes FC, Garcia-Abreu J, Galou M, et al. Neurons induce GFAP gene promoter of cultured astrocytes from transgenic mice. Glia. 1999;26:97–108. [DOI] [PubMed] [Google Scholar]
- 76.Gomes FC, Maia CG, de Menezes JR, et al. Cerebellar astrocytes treated by thyroid hormone modulate neuronal proliferation. Glia. 1999;25:247–255. [PubMed] [Google Scholar]
- 77.Malarkey EB, Ni Y, Parpura V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia. 2008;56:821–835. [DOI] [PubMed] [Google Scholar]
- 78.Araque A, Martín ED, Perea G, et al. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. Erratum in: J Neurosci. 2005;25:3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bergami M, Santi S, Formaggio E, et al. Uptake and recycling of pro-BDNF for transmitter-induced secretion by cortical astrocytes. J Cell Biol. 2008;183:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blum AE, Joseph SM, Przybylski RJ, et al. Rho-family GTPases modulate Ca(2+)-dependent ATP release from astrocytes. Am J Physiol Cell Physiol. 2008;295:C231–C241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fujita T, Tozaki-Saitoh H, Inoue K. P2Y1 receptor signaling enhances neuroprotection by astrocytes against oxidative stress via IL-6 release in hippocampal cultures. Glia. 2009;57:244–257. [DOI] [PubMed] [Google Scholar]
- 83.Zhuang Z, Yang B, Theus MH, et al. EphrinBs regulate D-serine synthesis and release in astrocytes. J Neurosci. 2010;30:16015–16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kang N, Peng H, Yu Y, et al. Astrocytes release D-serine by a large vesicle. Neuroscience. 2013;240:243–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Vijayaraghavan S. Glial-neuronal interactions—implications for plasticity and drug addiction. AAPS J. 2009;11:123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fujikawa Y, Tominaga K, Tanaka F, et al. Enteric glial cells are associated with stress-induced colonic hyper-contraction in maternally separated rats. Neurogastroenterol Motil. 2015;27:1010–1023. [DOI] [PubMed] [Google Scholar]
- 87.Kabouridis PS, Lasrado R, McCallum S, et al. Microbiota controls the homeostasis of glial cells in the gut lamina propria. Neuron. 2015;85:289–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brun P, Giron MC, Qesari M, et al. Toll-like receptor 2 regulates intestinal inflammation by controlling integrity of the enteric nervous system. Gastroenterology. 2013;145:1323–1333. [DOI] [PubMed] [Google Scholar]
- 89.Barajon I, Serrao G, Arnaboldi F, et al. Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J Histochem Cytochem. 2009;57:1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stoffels B, Hupa KJ, Snoek SA, et al. Postoperative ileus involves interleukin-1 receptor signaling in enteric glia. Gastroenterology. 2014;146:176–187. [DOI] [PubMed] [Google Scholar]
- 91.Li YX, Schaffner AE, Walton MK, et al. Astrocytes regulate developmental changes in the chloride ion gradient of embryonic rat ventral spinal cord neurons in culture. J Physiol. 1998;509:847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cotrina ML, Nedergaard M. Intracellular calcium control mechanisms in glia. In: Kettenman H, Ransom BR, eds. Neuroglia. 2nd ed Oxford, NY: Oxford University Press; 2004:230–242. [Google Scholar]
- 93.Codazzi F, Teruel MN, Meyer T. Control of astrocyte Ca(2+) oscillations and waves by oscillating translocation and activation of protein kinase. Curr Biol. 2001;11:1089–1097. [DOI] [PubMed] [Google Scholar]
- 94.Pasti L, Volterra A, Pozzan T, et al. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harris-White ME, Zanotti SA, Frautschy SA, et al. Spiral intercellular calcium waves in hippocampal slice cultures. J Neurophysiol. 1998;79:1045–1052. [DOI] [PubMed] [Google Scholar]
- 96.Arcuino G, Lin JH, Takano T, et al. Intercellular calcium signaling mediated by point-source burst release of ATP. Proc Natl Acad Sci U S A. 2002;99:9840–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cornell-Bell AH, Finkbeiner SM, Cooper MS, et al. Glutamate induces calcium waves in cultured astrocytes: long-range glial signaling. Science. 1990;247:470–473. [DOI] [PubMed] [Google Scholar]
- 98.John GR, Scemes E, Suadicani SO, et al. IL-1beta differentially regulates calcium wave propagation between primary human fetal astrocytes via pathways involving P2 receptors and gap junction channels. Proc Natl Acad Sci U S A. 1999;96:11613–11618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cotrina ML, Lin JH, Alves-Rodrigues A, et al. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci U S A. 1998;95:15735–15740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Volterra A, Liaudet N, Savtchouk I. Astrocyte Ca²⁺ signalling: an unexpected complexity. Nat Rev Neurosci. 2014;15:327–335. [DOI] [PubMed] [Google Scholar]
- 101.Sibille J, Zapata J, Teillon J, et al. Astroglial calcium signaling displays short-term plasticity and adjusts synaptic efficacy. Front Cell Neurosci. 2015;9:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Serrano A, Haddjeri N, Lacaille JC, et al. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Henneberger C, Papouin T, Oliet SH, et al. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Panatier A, Vallée J, Haber M, et al. Astrocytes are endogenous regulators of basal transmission at central synapses. Cell. 2011;146:785–798. [DOI] [PubMed] [Google Scholar]
- 105.Navarrete M, Perea G, Fernandez de Sevilla D, et al. Astrocytes mediate in vivo cholinergic-induced synaptic plasticity. PLoS Biol. 2012;10:e1001259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Benedetti B, Matyash V, Kettenmann H. Astrocytes control GABAergic inhibition of neurons in the mouse barrel cortex. J Physiol. 2011;589:1159–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andersson M, Hanse E. Astrocytes impose postburst depression of release probability at hippocampal glutamate synapses. J Neurosci. 2010;30:5776–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Navarrete M, Araque A. Endocannabinoids mediate neuron-astrocyte communication. Neuron. 2008;57:883–893. [DOI] [PubMed] [Google Scholar]
- 109.Iino M, Goto K, Kakegawa W, et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. [DOI] [PubMed] [Google Scholar]
- 110.Saab AS, Neumeyer A, Jahn HM, et al. Bergmann glial AMPA receptors are required for fine motor coordination. Science. 2012;337:749–753. [DOI] [PubMed] [Google Scholar]
- 111.Lee HU, Yamazaki Y, Tanaka KF, et al. Increased astrocytic ATP release results in enhanced excitability of the hippocampus. Glia. 2013;61:210–224. [DOI] [PubMed] [Google Scholar]
- 112.Bernardinelli Y, Randall J, Janett E, et al. Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr Biol. 2014;24:1679–1688. [DOI] [PubMed] [Google Scholar]
- 113.Pannasch U, Freche D, Dallérac G, et al. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat Neurosci. 2014;17:549–558. [DOI] [PubMed] [Google Scholar]
- 114.Wang F, Smith NA, Xu Q, et al. Astrocytes modulate neural network activity by Ca²+-dependent uptake of extracellular K+. Sci Signal. 2012;5:ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alvarez-Maubecin V, Garcia-Hernandez F, Williams JT, et al. Functional coupling between neurons and glia. J Neurosci. 2000;20:4091–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Accorsi-Mendonça D, Zoccal DB, Bonagamba LG, et al. Glial cells modulate the synaptic transmission of NTS neurons sending projections to ventral medulla of Wistar rats. Physiol Rep. 2013;1:e00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.López-Hidalgo M, Salgado-Puga K, Alvarado-Martínez R, et al. Nicotine uses neuron-glia communication to enhance hippocampal synaptic transmission and long-term memory. PLoS One. 2012;7:e49998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. [DOI] [PubMed] [Google Scholar]
- 119.Andersson M, Blomstrand F, Hanse E. Astrocytes play a critical role in transient heterosynaptic depression in the rat hippocampal CA1 region. J Physiol. 2007;585:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Straub SV, Nelson MT. Astrocytic calcium signaling: the information currency coupling neuronal activity to the cerebral microcirculation. Trends Cardiovasc Med. 2007;17:183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Parpura V, Haydon PG. Physiological astrocytic calcium levels stimulate glutamate release to modulate adjacent neurons. Proc Natl Acad Sci U S A. 2000;97:8629–8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. [DOI] [PubMed] [Google Scholar]
- 124.Kang J, Jiang L, Goldman SA, et al. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. [DOI] [PubMed] [Google Scholar]
- 125.Liu QS, Xu Q, Kang J, et al. Astrocyte activation of presynaptic metabotropic glutamate receptors modulates hippocampal inhibitory synaptic transmission. Neuron Glia Biol. 2004;1:307–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zonta M, Angulo MC, Gobbo S, et al. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci. 2003;6:43–50. [DOI] [PubMed] [Google Scholar]
- 127.Newman EA. Glial cell inhibition of neurons by release of ATP. J Neurosci. 2003;23:1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Navarrete M, Perea G, Maglio L, et al. Astrocyte calcium signal and gliotransmission in human brain tissue. Cereb Cortex. 2013;23:1240–1246. [DOI] [PubMed] [Google Scholar]
- 129.Liñán-Rico A, Wunderlich JE, Enneking JT, et al. Neuropharmacology of purinergic receptors in human submucous plexus: Involvement of P2X1, P2X2, P2X3 channels, P2Y and A3 metabotropic receptors in neurotransmission. Neuropharmacology. 2015;95:83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bennett MV, Garré JM, Orellana JA, et al. Connexin and pannexin hemichannels in inflammatory responses of glia and neurons. Brain Res. 2012;1487:3–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Karpuk N, Burkovetskaya M, Fritz T, et al. Neuroinflammation leads to region-dependent alterations in astrocyte gap junction communication and hemichannel activity. J Neurosci. 2011;31:414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bennett MV, Barrio LC, Bargiello TA, et al. Gap junctions: new tools, new answers, new questions. Neuron. 1991;6:305–320. [DOI] [PubMed] [Google Scholar]
- 133.Beyer EC, Paul DL, Goodenough DA. Connexin family of gap junction proteins. J Membr Biol. 1990;116:187–194. [DOI] [PubMed] [Google Scholar]
- 134.Dermietzel R, Hwang TK, Spray DS. The gap junction family: structure, function and chemistry. Anat Embryol (Berl). 1990;182:517–528. [DOI] [PubMed] [Google Scholar]
- 135.Musil LS, Goodenough DA. Gap junctional intercellular communication and the regulation of connexin expression and function. Curr Opin Cell Biol. 1990;2:875–880. [DOI] [PubMed] [Google Scholar]
- 136.Blackburn JP, Peters NS, Yeh HI, et al. Upregulation of connexin43 gap junctions during early stages of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 1995;15:1219–1228. [DOI] [PubMed] [Google Scholar]
- 137.Lecca D, Ceruti S, Fumagalli M, et al. Purinergic trophic signalling in glial cells: functional effects and modulation of cell proliferation, differentiation, and death. Purinergic Signal. 2012;8:539–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Pearson T, Currie AJ, Etherington LA, et al. Plasticity of purine release during cerebral ischemia: clinical implications? J Cell Mol Med. 2003;7:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Di Virgilio F, Ceruti S, Bramanti P, et al. Purinergic signalling in inflammation of the central nervous system. Trends Neurosci. 2009;32:79–87. [DOI] [PubMed] [Google Scholar]
- 141.Chakraborty S, Kaushik DK, Gupta M, et al. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res. 2010;88:1615–1631. [DOI] [PubMed] [Google Scholar]
- 142.Li H, Ambade A, Re F. Cutting edge: necrosis activates the NLRP3 inflammasome. J Immunol. 2009;183:1528–1532. [DOI] [PubMed] [Google Scholar]
- 143.Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61:1113–1121. [DOI] [PubMed] [Google Scholar]
- 144.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. [DOI] [PubMed] [Google Scholar]
- 145.Hansen JD, Vojtech LN, Laing KJ. Sensing disease and danger: a survey of vertebrate PRRs and their origins. Dev Comp Immunol. 2011;35:886–897. [DOI] [PubMed] [Google Scholar]
- 146.Cahill CM, Rogers JT. Interleukin (IL) 1beta induction of IL-6 is mediated by a novel phosphatidylinositol 3-kinase-dependent AKT/IkappaB kinase alpha pathway targeting activator protein-1. J Biol Chem. 2008;283:25900–25912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ikejima T, Okusawa S, Ghezzi P, et al. Interleukin-1 induces tumor necrosis factor (TNF) in human peripheral blood mononuclear cells in vitro and a circulating TNF-like activity in rabbits. J Infect Dis. 1990;162:215–223. [DOI] [PubMed] [Google Scholar]
- 148.Dunn SL, Young EA, Hall MD, et al. Activation of astrocyte intracellular signaling pathways by interleukin-1 in rat primary striatal cultures. Glia. 2002;37:31–42. [DOI] [PubMed] [Google Scholar]
- 149.Tschopp J. Mitochondria: Sovereign of inflammation? Eur J Immunol. 2011;41:1196–1202. [DOI] [PubMed] [Google Scholar]
- 150.Gross O, Thomas CJ, Guarda G, et al. The inflammasome: an integrated view. Immunol Rev. 2011;243:136–151. [DOI] [PubMed] [Google Scholar]
- 151.Dupin E, Creuzet S, Le Douarin NM. The contribution of the neural crest to the vertebrate body. Adv Exp Med Biol. 2006;589:96–119. [DOI] [PubMed] [Google Scholar]
- 152.Joseph NM, He S, Quintana E, et al. Enteric glia are multipotent in culture but primarily form glia in the adult rodent gut. J Clin Invest. 2011;121:3398–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Laranjeira C, Sandgren K, Kessaris N, et al. Glial cells in the mouse enteric nervous system can undergo neurogenesis in response to injury. J Clin Invest. 2011;121:3412–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gershon MD. Behind an enteric neuron there may lie a glial cell. J Clin Invest. 2011;121:3386–3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Bassotti G, Villanacci V, Nascimbeni R, et al. Increase of colonic mast cells in obstructed defecation and their relationship with enteric glia. Dig Dis Sci. 2012;57:65–71. [DOI] [PubMed] [Google Scholar]
- 156.Flamant M, Aubert P, Rolli-Derkinderen M, et al. Enteric glia protect against Shigella flexneri invasion in intestinal epithelial cells: a role for S-nitrosoglutathione. Gut. 2011;60:473–484. [DOI] [PubMed] [Google Scholar]
- 157.Sartor RB, Mazmanian SK. Intestinal Microbes in inflammatory bowel diseases. Am J Gastroenterol. 2012;1:15–21. [Google Scholar]
- 158.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zamanian JL, Xu L, Foo LC, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–6410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hamby ME, Coppola G, Ao Y, et al. Inflammatory mediators alter the astrocyte transcriptome and calcium signaling elicited by multiple G-protein-coupled receptors. J Neurosci. 2012;32:14489–14510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ortinski PI, Dong J, Mungenast A, et al. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Selgrad M, De Giorgio R, Fini L, et al. JC virus infects the enteric glia of patients with chronic idiopathic intestinal pseudo-obstruction. Gut. 2009;58:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Galligan JJ. HIV, Opiates, and enteric neuron dysfunction. Neurogastroenterol Motil. 2015;27:449–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Fagbemi AO, Torrente F, Puleston J, et al. Enteric neural disruption in necrotizing enterocolitis occurs in association with myenteric glial cell CCL20 expression. J Pediatr Gastroenterol Nutr. 2013;57:788–793. [DOI] [PubMed] [Google Scholar]
- 165.Savidge TC, Newman P, Pothoulakis C, et al. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132:1344–1358. [DOI] [PubMed] [Google Scholar]
- 166.Neunlist M, Aubert P, Bonnaud S, et al. Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1-dependent pathway. Am J Physiol Gastrointest Liver Physiol. 2007;292:G231–G241. [DOI] [PubMed] [Google Scholar]
- 167.Bach-Ngohou K, Mahé MM, Aubert P, et al. Enteric glia modulate epithelial cell proliferation and differentiation through 15-deoxy-12,14-prostaglandin J2. J Physiol. 2010;588:2533–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Van Landeghem L, Mahé MM, Teusan R, et al. Regulation of intestinal epithelial cells transcriptome by enteric glial cells: impact on intestinal epithelial barrier functions. BMC Genomics. 2009;10:507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bohórquez DV, Samsa LA, Roholt A, et al. An enteroendocrine cell-enteric glia connection revealed by 3D electron microscopy. PLoS One. 2014;9:e89881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Mwangi S, Anitha M, Mallikarjun C, et al. Glial cell line-derived neurotrophic factor increases beta-cell mass and improves glucose tolerance. Gastroenterology. 2008;134:727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Costantini TW, Bansal V, Krzyzaniak M, et al. Vagal nerve stimulation protects against burn-induced intestinal injury through activation of enteric glia cells. Am J Physiol Gastrointest Liver Physiol. 2010;299:G1308–G1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hu S, Zhao ZK, Liu R, et al. Electroacupuncture activates enteric glial cells and protects the gut barrier in hemorrhaged rats. World J Gastroenterol. 2015;21:1468–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Esposito G, Capoccia E, Sarnelli G, et al. The antiprotozoal drug pentamidine ameliorates experimentally induced acute colitis in mice. J Neuroinflammation. 2012;9:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sharkey KA, Nasser Y, Ruhl A. Enteric glia. Gut. 2004;53:1390. [PMC free article] [PubMed] [Google Scholar]
- 175.Alhouayek M, Muccioli GG. The endocannabinoid system in inflammatory bowel diseases: from pathophysiology to therapeutic opportunity. Trends Mol Med. 2012;18:615–625. [DOI] [PubMed] [Google Scholar]
- 176.McKernan DP, Finn DP. An apPEAling new therapeutic for ulcerative colitis? Gut. 2014;63:1207–1208. [DOI] [PubMed] [Google Scholar]
- 177.Rühl A, Trotter J, Stremmel W. Isolation of enteric glia and establishment of transformed enteroglial cell lines from the myenteric plexus of adult rat. Neurogastroenterol Motil. 2001;13:95–106. [DOI] [PubMed] [Google Scholar]
- 178.Soret R, Coquenlorge S, Cossais F, et al. Characterization of human, mouse, and rat cultures of enteric glial cells and their effect on intestinal epithelial cells. Neurogastroenterol Motil. 2013;25:e755–e764. [DOI] [PubMed] [Google Scholar]
- 179.Boesmans W, Rocha NP, Reis HJ, et al. The astrocyte marker Aldh1L1 does not reliably label enteric glial cells. Neurosci Lett. 2014;566:102–105. [DOI] [PubMed] [Google Scholar]
- 180.Ochoa-Cortes F, Liñán-Rico A, Jacobson KA, et al. Potential for developing purinergic drugs for gastrointestinal diseases. Inflamm Bowel Dis. 2014;20:1259–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]