

Abstract
Prior biological knowledge and phenotype information may help to identify disease genes from human whole-genome and whole-exome sequencing studies. We developed Phenolyzer (http://phenolyzer.usc.edu), a tool that uses prior information to implicate genes involved in diseases. Phenolyzer exhibits superior performance over competing methods for prioritizing Mendelian and complex disease genes, based on disease or phenotype terms entered as free text.
In a typical study using human whole-genome or whole-exome sequencing data, tens of thousands of single-nucleotide variants (SNVs), indels and structural variants (SVs) can be identified, but only a handful are relevant to the disease or phenotype of interest. Prioritizing candidate variants or genes from sequencing data therefore poses substantial challenges1. Several computational tools, including ANNOVAR2, snpEff3, VEP4, Jannovar5 and VAT6, address this problem mainly by employing a number of variant filtering steps, such as keeping nonsynonymous and splice variants and keeping variants with high conservation scores and low alternative allele frequencies7.
Prior biological knowledge and phenotype information may also help pinpoint genes that contribute to disease, and several prioritization tools utilize known genotype-phenotype relationship information. However, the input for many of these tools is limited to training gene lists (for example, ENDEAVOUR8), specific disease identifiers (for example, MedSim9 and Phevor10) or ontology identifiers (for example, Phen-Gen11). Such requirements may restrict usage and accessibility for average biologists. Several other tools can take keywords as input, including Genecards12, PosMed13 and SNPs3d14. Yet others use phenotype information to improve the prediction or prioritization of disease genes. For example, Phenomizer assesses an input query consisting of standard Human Phenotype Ontology (HPO) terms and generates a diagnosis with P values for each of the ~7,000 rare diseases in the HPO database15. PHIVE (ref. 16) improves the identification of disease genes by incorporating phenotype information from model organism studies in cross-species comparisons. Together, these approaches demonstrate the clear utility of using phenotype data to improve disease gene finding. However, no tool is able to utilize existing disease nomenclature systems to interpret the user input as free text, sometimes including multiple diseases or phenotype terms.
Here we introduce a computational tool called Phenolyzer to prioritize human disease genes based on disease or phenotype information provided by users as free text. Phenolyzer includes multiple components: (i) a tool to map user-supplied phenotypes to related diseases, (ii) a resource that integrates existing knowledge on known disease genes, (iii) an algorithm to predict previously unknown disease genes, (iv) a machine learning model that integrates multiple features to score and prioritize all candidate genes and (v) a network visualization tool to examine gene-gene and gene-disease relationships.
Phenolyzer works by an intuitive approach: it interprets user-supplied disease or phenotype terms as related disease names, which are then used to query precompiled databases to find and score relevant seed genes. The seed genes are then expanded to include related genes, on the basis of several types of gene-gene relationship logic such as exhibiting a protein-protein interaction, sharing a biological pathway or gene family, or transcriptionally regulating or being regulated by another gene. Finally, these different types of scores from seed gene ranking and gene-gene relationships are integrated to generate a ranked candidate gene list, together with detailed explanation to track the source of information used to compile the scores (Fig. 1). Phenolyzer is available as a web server at http://phenolyzer.usc.edu, and a command-line version can also be downloaded for batch processing (Supplementary Software). The user-friendly web server interface takes user input and generates results within minutes. The results page provides several tabs, including a summary tab with word cloud and links to output files, a gene-disease-term interaction network, a bar plot of the 500 most highly ranked genes with normalized scores, and detailed record-tracking information with raw scores and links to external databases (Supplementary Figs. 1–3).
Figure 1.
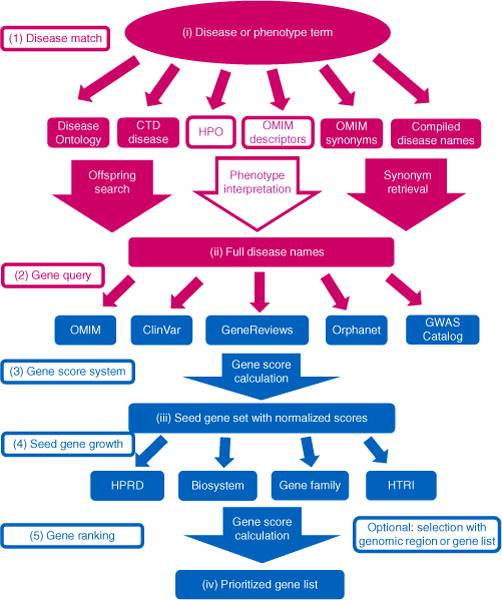
Workflow of Phenolyzer. (1) Disease match: each disease or phenotype query term is separately translated into sets of disease names by word match, offspring search, synonym retrieval and phenotype interpretation in disease name databases. (2) Gene query: each retrieved disease name is queried in the gene-disease databases based on an exact match, to get a list of genes. (3) Gene score system: a score based on the type and confidence of the gene-disease relationship is generated for each gene corresponding to each disease name. Then, for each input term, a weighted sum score is calculated for each reported gene by adding all the scores retrieved in previous step. The seed gene set is generated by collating all the genes of all input terms, and each gene score is normalized. (4) Seed gene growth: candidate disease genes are expanded beyond the seed gene set based on four types of gene-gene relationships; scores are calculated for all genes that connect with seed genes. (5) Gene ranking: all the information is integrated to generate a score for each gene, with the weights trained from a logistic regression model. The scores are renormalized to the final prioritized gene list. HPRD, Human Protein Reference Database; HTRI, Human Transcriptional Regulation Interaction Database.
We first tested Phenolyzer on a small dataset of 14 very well-established Mendelian diseases that are each known to be caused by mutations in only one or two genes17. Given the input phenotype, we examined how Phenolyzer ranks the disease causal gene against other genes in the genome, as “top 1,” “top 10,” “top 20” and “below top 20.” Note that if a disease has no corresponding record, it is categorized as “below top 20”; for the two diseases with two causal genes, the rank for two genes is averaged and rounded to the smaller integer. Because of the relatively small size of the dataset, we also manually tested several other tools (most of which do not offer batch processing), including Phenomizer, GeneCards, SNPs3d, PosMed and Phevor. A detailed comparison of functionality between Phenolyzer and other tools is given (Supplementary Table 1). Phenolyzer successfully prioritized all disease causal genes as “top 1” (Fig. 2a). SNPs3d failed only on one gene, which was ranked “top 3.” PosMed does not work well for scoring genes for monogenic diseases, probably because its algorithm relies purely on co-occurrence of gene-disease pairs from the literature. Phevor did not work well, likely because its algorithm does not weight different ontology scores. Phenomizer works well, considering that its input is the phenotype terms for each disease rather than the disease name itself (Supplementary Dataset 1).
Figure 2.
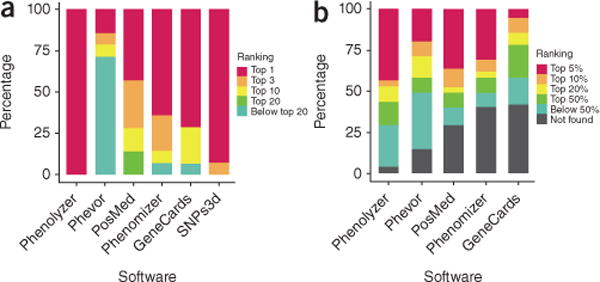
Comparison between Phenolyzer and other tools to find well-known monogenic disease genes and predict recently published novel disease genes. (a) The ranking distribution of genes for 14 monogenic diseases. (b) The ranking distribution of 55 recently published disease genes from four human genetics journals.
We next expanded our evaluation to 590 known inherited disease genes compiled in a newborn sequencing study18 (Supplementary Table 2). Because of the large size of the dataset, we were unable to compare to other tools on their web servers. We found that 81.2% of the genes were ranked as “top 1,” 90.7% of the genes were ranked as “top 10,” and overall 93.4% of the genes could be identified by Phenolyzer in its ranked list of candidate genes (Supplementary Dataset 2). Thus, our analysis clearly demonstrated that Phenolyzer is adept at finding genes that are known to be associated with Mendelian diseases.
We next examined several datasets to evaluate the ability of Phenolyzer to prioritize candidate genes for complex diseases. For simplicity, we tested four complex diseases with input as “cancer,” “autism,” “rheumatoid arthritis” and “anemia,” including 517 genes in the Cancer Gene Census from COSMIC (Catalog of Somatic Mutations in Cancer)19, 22 genes strongly associated (false-discovery rate <0.05) with autism from exome sequencing20, 634 genes from RADB (Database of Rheumatoid Arthritis related Polymorphisms)21 and 121 genes at 75 loci associated with red blood cell phenotype and anemia22. All the reported genes were used as positive genes, and all other genes were used as a negative set (though we acknowledge that some may still be genuine disease genes). We used ROC (receiver operating characteristic) curves and AUC (area under the curve) values to evaluate each tool (Supplementary Fig. 4). Our results demonstrate that Phenolyzer compares favorably against all the other tools. For example, for autism-associated genes that were identified from an exome sequencing study, Phenolyzer achieved an AUC score above 0.85, but none of the other tools has AUC scores higher than 0.81. PosMed performs nearly as well as Phenolyzer, but it does not find as many true positive genes as Phenolyzer, which leads to lower AUC value. Phevor’s ontology propagation algorithm works well to retrieve a large list of genes and has the ability to discover novel genes. SNPs3d, Genecards and Phenomizer can generate only a limited number of candidate genes in the output and thus do not perform well, though tools such as Phenomizer were not designed to prioritize genes of complex disease. In addition, we also evaluated Phenolyzer with only the seed gene list (without the seed gene growth step), and found that its performance was greatly reduced, suggesting the importance of seed gene growth to find new genes not documented in Phenolyzer’s disease-gene knowledgebase (Supplementary Fig. 4 and Supplementary Dataset 3).
In addition to known disease genes, we further evaluated the performance of Phenolyzer to prioritize novel disease genes. For this analysis, we identified 55 disease genes published between June and August 2014 in four scientific journals (Nature Genetics, American Journal of Medical Genetics, American Journal of Human Genetics and Human Molecular Genetics). These discoveries represented novel findings at the time of our analysis, so it was less likely that Phenolyzer would be able to use the exact gene-disease records in any knowledgebase. We submitted the phenotypes or diseases described in each of the original publication to Phenolyzer, and examined whether the reported genes could be found in the ranked list of candidate genes. Compared to competing tools, Phenolyzer achieved the highest response rate, top 5% ratio and top 50% ratio. Phevor worked well to identify a large list of candidate genes but did not excel in ranking the causal genes at the top of the list. PosMed performed nearly as well as Phenolyzer as a result of its implementation of a powerful literature-mining engine, but it had a lower response rate. Phenomizer performed well on the genes for which it can generate predictions. GeneCards worked poorly on this task. SNPs3d was not included here since it cannot generate a large number of candidate genes (Fig. 2b, Supplementary Table 2 and Supplementary Dataset 4).
Compared to Phenomizer, Phenolyzer was able to analyze both concrete disease names as well as a list of phenotype terms. We performed a reanalysis of the 14 monogenic and four complex diseases using only a list of phenotype terms as the input and found that Phenolyzer had similar performance to Phenomizer for the known monogenic disease genes (Supplementary Fig. 5); for complex diseases, they also performed similarly, but Phenomizer was limited by its number of candidate genes in the results (Supplementary Fig. 4).
Although Phenolyzer has been developed as a phenotype analysis tool only, it is straightforward to link it with functional annotation software for next-generation sequencing data or for copy-number variation (CNV) data. To demonstrate this, we illustrated four user cases that combine Phenolyzer with exome sequencing data or CNV data to identify disease causal genes (Supplementary Note, Supplementary Fig. 6 and Supplementary Datasets 5–7). To facilitate users with sequencing data, we also implemented an automated Phenolyzer analysis pipeline in the wANNOVAR server23.
Compared to similar tools, Phenolyzer is better at prioritizing candidate genes for Mendelian diseases and complex diseases. In addition, the tool also works well for finding novel disease-gene associations, highlighting its ability to help formulate new biological hypotheses. For the ever-increasing number of human disease sequencing studies, Phenolyzer will help users leverage prior biological knowledge and phenotype information to expedite scientific discovery.
A detailed user manual is available online at http://phenolyzer.usc.edu/download/Phenolyzer_manual.pdf and will be regularly updated.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
Compilation of gene-disease databases
Phenolyzer incorporates a list of gene-disease databases, pre-compiled from several data sources, including OMIM24, Orphanet25, ClinVar26, Gene Reviews27 and GWAS Catalog28. This list may expand in the future. It also integrates four different gene-gene relationship databases: HPRD29 contains the human protein interaction data; Biosystem30 contains records from KEGG, BioCyc, Reactome, Pathway Interaction Database, WikiPathways and Gene Ontology; HGNC Gene Family31 contains the gene family information; and HTRI32 contains the human transcription factor interaction information. Gene symbols are standardized to Entrez Gene identifiers. If a gene symbol in any gene-disease or gene relationship database has no corresponding symbols or synonyms in the Entrez Gene database, it is removed from the results.
Disease and phenotype term interpretation
Currently, Phenolyzer uses CTD Medic vocabulary33, Disease Ontology34, precompiled OMIM disease synonyms24, Human Phenotype Ontology database35 and OMIM descriptors to expand and interpret a given term into a full set of specific disease names (Supplementary Fig. 7).
A disease or phenotype term set, Term = {Termk} with one or multiple terms, is used as input to describe diseases or phenotypes for a given patient. Each Termk is interpreted into a set of specific disease and phenotype names, Disease = {Diseasei}. For example, {‘Muscular dystrophy’, ‘Duchenne’} is an input term set. The separation into two parts increases the chance of matching a larger list (and hence more candidate genes), since the fine-grained term ‘Duchenne Muscular Dystrophy’ may not be the most accurate diagnosis for the patient (there are dozens of types of muscular dystrophy), and it reduces the chance of false-negative results due to an excessively fine-grained query term that is not in the Phenolyzer data sources. On the other hand, the final score of the true candidate gene will not be affected substantially. As another example, the interpreted disease names of ‘autism’ is illustrated (Supplementary Dataset 8). Basic regular expression techniques are used here to process each term into a format that all the continuous non-word characters are transformed into a single white space.
Word matching is conducted between each term and the names or synonyms in the several disease databases mentioned above and the disease names from all the pre-compiled gene-disease databases. Each term will be treated as a single phrase without tokenization, to examine whether it is contained within the disease or phenotype records in the databases. Even though no tokenization is conducted here, the large number of available synonyms in disease vocabularies results in a high chance of a final match in the disease-gene databases.
For matched disease names, all their synonyms and descendent disease names or synonyms are returned with a reliability score of 1.00. For matched phenotype names, disease reliabilities are calculated differently for HPO and OMIM descriptors. For HPO, reliability is dependent on the phenotype-disease mapping descriptions given by the HPO annotation file retrieved from its website: “rare” to 0.05, “occasional” to 0.075, “frequent” to 0.33, “typical” 0.5, “variable” to 0.5, “common” to 0.75, “hallmark” to 0.90, “obligate” to 1.00. If such description is not available, it is treated as “frequent.” For OMIM descriptors, the conditional probability P(Disease|Descriptor) is used as the reliability, which is the probability of the disease given the matched descriptors, based on the OMIM disease description file. Finally, if a disease name has several reliability scores from several sources, only the highest reliability score will be saved, thus avoiding duplicated calculation for the same disease.
Seed gene set generation
Each disease name Diseasei in the extended full disease name set is queried in the pre-compiled gene-disease databases. A case-insensitive match will be conducted. Each time a gene is found to be directly associated with a disease, a score is calculated. We calculate a weighted score for each gene given individual term,
The Score(Gene, Diseasei) is the sum of corresponding scores for all the five sources in the pre-compiled gene-disease database, with individual scores calculated by Gene Disease Score System (see methods below). Count(Disease) is the overall record number of the diseases occurring in gene-disease databases corresponding to the term. Reliability(Diseasei) is the reliability of the retrieved full disease name.
One advantage of using the above formula is to support queries with multiple terms. For example, if ‘Alzheimer’ and ‘brain’ are entered as different terms, the genes corresponding to the more specific word ‘Alzheimer’ will have higher scores, due to its smaller number of corresponding records. Thus by adding each term’s scores, the most specific term will dominate. Additionally, the intersecting genes will always have higher scores. The final weighted sum score of a seed gene considering all reported gene-disease relationship is calculated by:
It is then normalized to be between 0 and 1 by dividing by the maximal score,
Seed gene set growth and model training
The seed gene set is grown based on four different types of gene relationship databases—HPRD, NCBI’s Biosystem, HGNC Gene Family and HTRI databases. For each seed gene Genei, each gene Genej gets a score for each type of relationship with the seed gene. For each type of relationship,
is the normalized seed gene score generated in the previous step. The logic here is that, if a seed gene has a higher score, then its related genes will also have higher scores. Scorerelation(Genei, Genej) comes from the Gene Prediction Score System (see methods below).
Each gene is processed to generate a score vector, x = [X1, X2, X3, X4, X5, X0], among which , X2 = SHPRD (Genei), X3 = SBiosystem(Genei) X4 = SHGNC(Genei), X5 = SHTRI(Genei), and X0 = 1. The initial weight vector is set w = [1.0, 0.1, 0.05, 0.05, 0.05, −0.5]. A positive gene has yn = 1 and a negative gene has yn = −1. Negative genes are selected randomly in Entrez Gene databases excluding true positive genes, as 10 times of the number of true positive genes. Logistic regression is used to model the score vector into yn:
A gradient descent algorithm is used, with 10,000 steps and learning rate η = 1 (Supplementary Fig. 8), to minimize the cost function,
For each step,
The training dataset comes from four different sources and includes four different diseases: type 1 diabetes (MIM 222100) with 439 Entrez genes from the Type 1 Diabetes Database36, type 2 diabetes (MIM 125853) with 522 Entrez genes from the Type 2 Diabetes Database37, Crohn’s disease (MIM 266600) with 207 Entrez genes from the literature38, and coronary artery disease with 604 Entrez genes from the Coronary Artery Disease Gene Database39. These represent some of the diseases most well studied by genome-wide and candidate gene association studies.
After the learning, the optimized weight vector is identified and the score with trained weights is calculated by Slogistic(Genei, Term) = wTxn − X0, and is then normalized to be between 0 and 1.
Gene disease score system
For each gene-disease pair in the gene-disease databases, the score is multiplied by two parameters: α represents what kind of study is used to infer the gene-disease relationship, and β represents the extent to which such relationship is confirmed. These parameters are defined by ad hoc measures specific for each database as described below.
For OMIM, α is 0.25, 0.5, 0.75 and 1 for Disorder Code 1, 2, 4 and 3, respectively, yet β is 0.25, 0.5, 0.75 and 1 for Status Code I, L, P, and C, respectively. Disorder Code is defined as below: 1 for “the disorder is placed on the map based on its association with a gene, but the underlying defect is not known,” 2 for “the disorder has been placed on the map by linkage; no mutation has been found,” 3 for “the molecular basis for the disorder is known; a mutation has been found in the gene,” and 4 for “a contiguous gene deletion or duplication syndrome, multiple genes are deleted or duplicated causing the phenotype.” Status Code is defined as below: I for “inconsistent— results of different laboratories disagree,” L for “limbo—evidence not as strong as that provisional, but included for heuristic reasons,” P for “provisional—based on evidence from one laboratory or one family,” C for “confirmed—observed in at least two laboratories or in several families.” For ClinVar, α is set at 0.25, and β is the reference count divided by the maximum number of reference counts. For Orphanet, α is 0.25 for “role in the phenotype of” or “candidate gene tested in,” 0.50 for “part of a fusion gene in,” “modifying germline mutation in” or “modifying somatic mutation in,” 0.75 for “major susceptibility in” and 1.00 for “disease-causing germline mutations in” or “disease-causing somatic mutations in,” whereas β is the reference count divided by the maximum number of reference count. For GWAS, α is set at 0.25, and β is 1 − P value. For GeneReviews, α and β are both set as 1. The score for a gene-disease pair from a specific data source is then calculated as αβ.
Gene prediction score system
The parameters for Gene Prediction Score System are defined by ad hoc measures specific to each database. For HGNC gene family, the score is set at 1. For Biosystem, each entry has already been assigned an individual score by NCBI, which is normalized from 0 to 1 by dividing by the maximum score. For all the Biosystems containing the pair of genes of interest, the maximum normalized score is returned as the final score for this pair. For HPRD, the score is first set to 0; if the two genes have the interaction information as “in vivo,” the score is increased by 1, if “in vitro” increased by 0.5, if “yeast two hybrid” increased by 0.25. Then the final score is normalized to be from 0 to 1 by dividing by 1.75. For HTRI, the score is calculated by normalizing the PubMed reference count by the maximum count, to range from 0 to 1, and the transcription factors (the source of the regulating relation) are penalized by multiplication by 0.25.
Tool comparison
For short disease names, the disease name is directly used as input. For long disease names, the best effort is used to obtain optimal result. For example, if no matching is found using the full name, we will divide the long disease name into shorter terms. The details of the exact input and the ranks are included in for monogenic diseases (Supplementary Dataset 1), for novel disease gene prediction (Supplementary Dataset 4). The output for complex disease comparison is also included (Supplementary Dataset 3).
For Phevor, a variant file with all the genes mutated to the same extent was generously provided by M.V. Singleton (one of the developers of Phevor) specifically for testing Phevor. Thus we can first enter the disease terms in Disease Ontology field and phenotype terms in Human Phenotype Ontology field and generate the gene profile, then run Phevor with the supplied variant file and obtain a final gene list with scores. The gene ranking depends only on the disease or phenotype terms but not the mutations, so we can use the results for comparison and benchmarking. Since Phevor takes at most 5 terms for one type of input, in rare cases when there are more than 5 available terms, only the first 5 are selected. We observed that in Phevor’s result, a large number of genes may have the same rank. For example, in the output for ‘liver cirrhosis’, there are 7,715 genes with the same rank. To deal with this issue, we use the smallest rank on all these genes. To ensure a fair comparison with other tools, we keep using the smallest rank when encountering the same situation for other tools.
For Phenomizer, the disease or phenotype term is used to search for features and diseases. Then all the related features are added to the patient’s feature, and the clinical diagnosis is generated. For an extreme example of “intellectual disability,” there are 185 diseases that can be retrieved, and all the feature annotations of these diseases are manually added. These same features are also used as input for Phenolyzer to demonstrate its performance for 14 monogenic diseases and complex diseases.
We made best efforts to ensure that for each disease or phenotype, a gene list is retrieved for each tool. If a term cannot generate any record, we will try a more general term. The only exception is “longevity” for Phenomizer and Phevor, where “lifespan” and “life span” are also tried yet no result can be generated. All the comparison shown for Phenolyzer is conducted without the add-on databases (which are additional databases outside of the core Phenolyzer databases), and with the trained weights from logistic regression except when stated otherwise.
Supplementary Material
Acknowledgments
This work was supported by US National Institutes of Health grant R01-HG006465 to K.W. We thank members of the Wang laboratory for testing the Phenolyzer website.
Footnotes
Code availability. The latest version of the Phenolyzer command line tool is available at https://github.com/WangGenomicsLab/phenolyzer. A recent version is also included with the paper (Supplementary Software).
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
AUTHOR CONTRIBUTIONS
H.Y. compiled the data, performed the computational experiments, developed software tools and drafted the manuscript. P.N.R. advised on phenotype data analysis and interpretation. K.W. designed the study, supervised its execution and revised the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare competing financial interests: details are available in the online version of the paper.
References
- 1.Lyon GJ, Wang K. Genome Med. 2012;4:58. doi: 10.1186/gm359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang K, Li M, Hakonarson H. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cingolani P, et al. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McLaren W, et al. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jäger M, et al. Hum Mutat. 2014;35:548–555. doi: 10.1002/humu.22531. [DOI] [PubMed] [Google Scholar]
- 6.Habegger L, et al. Bioinformatics. 2012;28:2267–2269. doi: 10.1093/bioinformatics/bts368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bamshad MJ, et al. Nat Rev Genet. 2011;12:745–755. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 8.Aerts S, et al. Nat Biotechnol. 2006;24:537–544. doi: 10.1038/nbt1203. [DOI] [PubMed] [Google Scholar]
- 9.Schlicker A, Lengauer T, Albrecht M. Bioinformatics. 2010;26:i561–i567. doi: 10.1093/bioinformatics/btq384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singleton MV, et al. Am J Hum Genet. 2014;94:599–610. doi: 10.1016/j.ajhg.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Javed A, Agrawal S, Ng PC. Nat Methods. 2014;11:935–937. doi: 10.1038/nmeth.3046. [DOI] [PubMed] [Google Scholar]
- 12.Safran M, et al. Database. 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makita Y, et al. Nucleic Acids Res. 2013;41:W109–W114. doi: 10.1093/nar/gkt474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yue P, Melamud E, Moult J. BMC Bioinformatics. 2006;7:166. doi: 10.1186/1471-2105-7-166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köhler S, et al. Am J Hum Genet. 2009;85:457–464. doi: 10.1016/j.ajhg.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson PN, et al. Genome Res. 2014;24:340–348. doi: 10.1101/gr.160325.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chial H. Nat Educ. 2008;1:192. [Google Scholar]
- 18.Saunders CJ, et al. Sci Translat Med. 2012;4:154ra135. doi: 10.1126/scitranslmed.3004041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Forbes SA, et al. Nucleic Acids Res. 2011;39:D945–D950. doi: 10.1093/nar/gkq929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Rubeis S, et al. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang R, et al. Database. 2014;2014:bau090. [Google Scholar]
- 22.van der Harst P, et al. Nature. 2012;492:369–375. [Google Scholar]
- 23.Chang X, Wang K. J Med Genet. 2012;49:433–436. doi: 10.1136/jmedgenet-2012-100918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amberger J, Bocchini C, Hamosh A. Hum Mutat. 2011;32:564–567. doi: 10.1002/humu.21466. [DOI] [PubMed] [Google Scholar]
- 25.Rath A, et al. Hum Mutat. 2012;33:803–808. doi: 10.1002/humu.22078. [DOI] [PubMed] [Google Scholar]
- 26.Landrum MJ, et al. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pagon RA, et al. GeneReviews. 1993 http://www.ncbi.nlm.nih.gov/books/NBK1116/?partid=1250.
- 28.Hindorff LA, Junkins HA, Mehta J, Manolio T. A Catalog of Published Genome-Wide Association Studies. National Human Genome Research Institute; 2011. https://www.genome.gov/26525384. [Google Scholar]
- 29.Peri S, et al. Nucleic Acids Res. 2004;32:D497–D501. doi: 10.1093/nar/gkh070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Geer LY, et al. Nucleic Acids Res. 2010;38:D492–D496. doi: 10.1093/nar/gkp858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seal RL, Gordon SM, Lush MJ, Wright MW, Bruford EA. Nucleic Acids Res. 2011;39:D514–D519. doi: 10.1093/nar/gkq892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bovolenta LA, Acencio ML, Lemke N. BMC Genomics. 2012;13:405. doi: 10.1186/1471-2164-13-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis AP, Wiegers TC, Rosenstein MC, Mattingly CJ. Database. 2012;2012:bar065. doi: 10.1093/database/bar065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schriml LM, et al. Nucleic Acids Res. 2012;40:D940–D946. doi: 10.1093/nar/gkr972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson PN, Mundlos S. Clin Genet. 2010;77:525–534. doi: 10.1111/j.1399-0004.2010.01436.x. [DOI] [PubMed] [Google Scholar]
- 36.Burren OS, et al. Nucleic Acids Res. 2011;39:D997–D1001. doi: 10.1093/nar/gkq912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lim JE, et al. BMC Med Inform Decis Mak. 2010;10:76. doi: 10.1186/1472-6947-10-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elding H, Lau W, Swallow DM, Maniatis N. Am J Hum Genet. 2013;92:107–113. doi: 10.1016/j.ajhg.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu H, et al. Nucleic Acids Res. 2011;39:D991–D996. doi: 10.1093/nar/gkq1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.