

Abstract
Maintenance of the correct level and organisation of nucleosomes is crucial for genome function. Here, we uncover a role for a conserved bromodomain AAA‐ATPase, Abo1, in the maintenance of nucleosome architecture in fission yeast. Cells lacking abo1 + experience both a reduction and mis‐positioning of nucleosomes at transcribed sequences in addition to increased intragenic transcription, phenotypes that are hallmarks of defective chromatin re‐establishment behind RNA polymerase II. Abo1 is recruited to gene sequences and associates with histone H3 and the histone chaperone FACT. Furthermore, the distribution of Abo1 on chromatin is disturbed by impaired FACT function. The role of Abo1 extends to some promoters and also to silent heterochromatin. Abo1 is recruited to pericentromeric heterochromatin independently of the HP1 ortholog, Swi6, where it enforces proper nucleosome occupancy. Consequently, loss of Abo1 alleviates silencing and causes elevated chromosome mis‐segregation. We suggest that Abo1 provides a histone chaperone function that maintains nucleosome architecture genome‐wide.
Keywords: Abo1, bromodomain AAA‐ATPases, chromatin, nucleosome mapping, Schizosaccharomyces pombe
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics
Introduction
Nuclear genomes are organised by assembly into chromatin. The fundamental repeating unit of chromatin is the nucleosome, which is composed of approximately 147 bp of DNA wrapped around an octamer of histone subunits. Nucleosomes represent an obstacle to DNA‐dependent processes such as transcription, and as a result, key steps in transcription involve regulated nucleosome removal and replacement 1. Transcription elongation by RNA polymerase II (RNAP II) may cause the displacement of H2A–H2B dimers from nucleosomes or even the eviction of entire histone octamers 1, 2, 3, 4. Therefore, RNAP II‐coupled nucleosome assembly is required to maintain the chromatin structure over gene bodies. A variety of factors have been shown to contribute to this process but prominent amongst them is FACT, an ATP‐independent histone chaperone complex that both facilitates nucleosome disassembly ahead of RNAP II and promotes reassembly behind it 5. Defects in transcription‐coupled chromatin assembly pathways result in a global decrease in nucleosome occupancy which leads to increased spurious intragenic transcription initiation 1.
Human ATAD2 (or ANCCA) is an evolutionarily conserved bromodomain containing ATPase that functions as a transcriptional co‐regulator via the control of chromatin 6, 7, 8, 9, 10. ATAD2 is predominantly expressed in the male germline but is systematically over‐expressed in tumours and is significantly associated with lung and breast cancers with poor prognoses 8, 11, 12, 13. A paralogous protein, ATAD2B, has been shown to be expressed during neuronal differentiation 14, but its molecular functions have not been determined. ATAD2 and ATAD2B belong to the AAA (ATPases associated with diverse cellular activities) ATPase family 15 and are distinct from the well‐characterised Snf2 class of ATP‐dependent chromatin remodelling enzymes. AAA‐ATPases typically form hexameric chaperones that utilise the energy from ATP hydrolysis to mediate the folding/unfolding of client proteins 15. The presence of a bromodomain in ATAD2 suggests that its targets include histones and this is supported by protein interaction studies 6, 16. As such, bromodomain AAA‐ATPases represent a novel group of ATP‐dependent histone chaperones.
Orthologs of ATAD2 and ATAD2B have been identified in a range of eukaryotes including yeasts, worms and mammals. Like humans, Schizosaccharomyces pombe has two bromodomain AAA‐ATPases designated Abo1 and Abo2. In contrast, Saccharomyces cerevisiae has a single ortholog called Yta7, which was identified as a component of a boundary element that restricts the spread of silencing from the HMR locus 17. Yta7 is also required for transcriptional induction of histone genes, the GAL gene cluster and early meiotic genes 18, 19. Other evidence is suggestive of a global role in the control of nucleosome density. Yta7 exhibits both physical and genetic interactions with ATP‐independent histone chaperones and also core histones. The Yta7 bromodomain interacts with histone tails in an acetylation‐independent manner 16, and the N‐terminal domain also exhibits an affinity for histones in vitro, indicating the presence of a second chromatin binding region 16. Deletion of YTA7 results in histone over‐accumulation and increased nucleosome density within transcribed sequences 19, 20. Based on this, it has been proposed that Yta7 facilitates nucleosome disassembly. However, it is not known whether this chromatin disassembly function is conserved for other bromodomain AAA‐ATPases and the genome‐wide contribution of these proteins is not well understood. To address these issues, we have analysed the fission yeast, S. pombe, which is evolutionarily divergent from S. cerevisiae and has proved to be an excellent model for the analysis of chromatin function. Contrary to expectation, we find that S. pombe cells lacking Abo1, an ortholog of human ATAD2 and S. cerevisiae Yta7, experience a global reduction in nucleosome levels in addition to changes in nucleosome organisation. These defects in chromatin structure result in widespread transcriptional de‐repression, loss of heterochromatic silencing and increased chromosome mis‐segregation.
Results
Global role for Abo1 in the control of transcription
The S. pombe genome has two poorly characterised genes, abo1 + and abo2 +, that encode homologs of human ATAD2/ATAD2B and S. cerevisiae Yta7 (Fig 1A). Analysis of a genome‐wide haploid deletion collection has shown that neither abo1 + nor abo2 + are essential 21, and therefore, strains carrying null alleles in these genes were subjected to further characterisation. Loss of Abo1 resulted in an increase (~20%) in doubling time and also an elongated cell morphology that is characteristic of a cell cycle delay (Fig EV1A and B). The elongated morphology was retained in the absence of the ATR kinase, Rad3 (Fig EV1C), suggesting that the cell cycle delay is independent of the DNA replication and damage checkpoints. Cells lacking abo2 + did not display any obvious phenotypes; however, genetic crosses indicated that an abo1Δ abo2Δ double mutant strain is not viable (Fig EV1D).
Figure 1. Impact of Abo1 on transcriptome signatures.
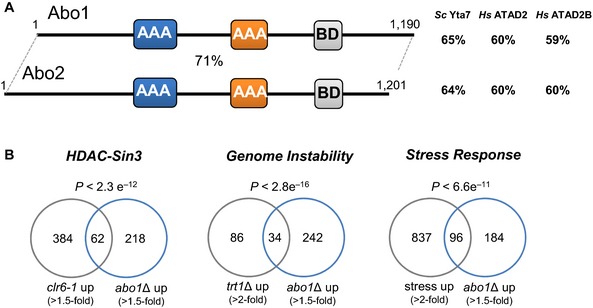
- Domain architecture of Schizosaccharomyces pombe bromodomain AAA‐ATPases. The D1 and D2 ATPase domains (AAA) are shaded blue and orange, respectively, and the bromodomain (BD) is grey. The percentage similarity to each other and the indicated proteins were determined by FASTA sequence comparison 73 using the scoring matrix BLOSUM50.
- Venn diagrams showing overlap between genes up‐regulated (≥ 1.5 fold) in abo1Δ mutants with genes up‐regulated under the indicated condition, along with the significance of the overlaps (based on hypergeometric distribution).
Figure EV1. Phenotypes associated with deletion of abo1 + .
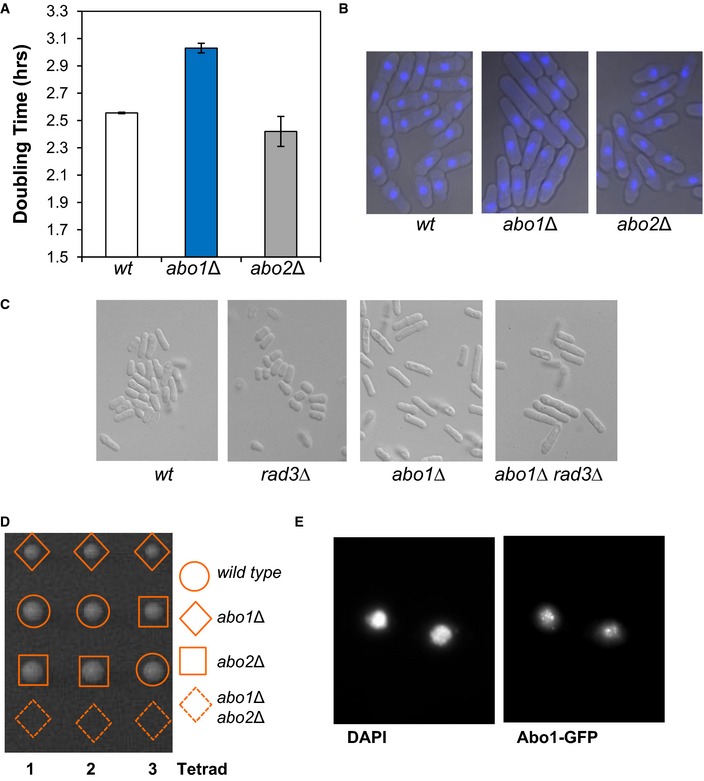
- Doubling time was estimated by measuring the OD595 of the indicated strains grown in YE5S medium at 30°C. Data are the mean of two independent biological repeats, and error bars denote the range of the data.
- abo1Δ cells have an elongated morphology. Microscopic analysis of DAPI‐stained cells. Data are representative of three biological repeats.
- The elongated morphology of abo1Δ cells is independent of the ATR checkpoint kinase, Rad3. Microscopic analysis of the indicated strain. Images are representative of duplicate experiments.
- An abo1Δ abo2Δ double mutant strain is not viable. Tetrad dissection of a genetic cross between abo1Δ and abo2Δ. Colonies arising from the spores of three asci are shown. Data are representative of three biological repeats.
- Subcellular localisation of Abo1. Cells expressing Abo1‐GFP were stained with DAPI and visualised using fluorescence microscopy. Data are representative of two biological repeats.
We next determined the global transcriptional contributions of Abo1 and Abo2 using microarray analyses. Only a small number of transcripts were reproducibly misregulated in the absence of abo2 +. This is consistent with the phenotypic analysis and data indicating that abo2 + is expressed at very low levels 22. In contrast, 280 transcripts were reproducibly up‐regulated upon abo1 + deletion. These transcripts included mRNAs and also a variety of non‐coding RNAs such as antisense transcripts and snoRNAs. Only 8 transcripts were found to be reproducibly down‐regulated in the abo1Δ background, suggesting that Abo1 predominantly functions to suppress transcription, at least in rapidly dividing cells. Consistent with this view, RNAs that were up‐regulated in the abo1Δ mutant significantly overlapped (P < 2.3 × 10−12) with targets of HDAC‐Sin3 co‐repressor complexes 23, 24 (Fig 1B). Further examination of gene expression patterns also revealed a significant overlap (P < 2.8 × 10−16) between genes that are up‐regulated in the abo1Δ mutant and those that are induced in response to genetic instability resulting from telomere crisis 25. It has been established that eukaryotic cells respond to diverse threats to genome integrity by inducing the expression of stress–response genes 26 and accordingly, the abo1Δ profile was enriched for core environmental stress–response (CESR) genes 27 (P < 6.6 × 10−11) (Fig 1B). Overall, the transcriptional signature of the abo1Δ mutant is indicative of a stress response resulting from impaired genome integrity.
Abo1 maintains nucleosome organisation and occupancy at RNAP II transcribed sequences
Analysis of the subcellular localisation of Abo1 revealed that it was broadly distributed throughout the nucleus (Fig EV1E), consistent with a global role in the regulation of chromatin. Therefore, the impact of Abo1 on genome‐wide nucleosome organisation was assessed using a chromatin sequencing methodology in which both the position and size of micrococcal nuclease (MNase)‐resistant chromatin species are determined 28. Two bioreps were processed, within which MNase digestion levels for chromatin in abo1Δ cells were closely matched to isogenic wild‐type cells (Fig EV2). However, between bioreps, we employed two different levels of MNase digestion (Biorep1 low MNase digestion; Biorep2 high MNase digestion) to control for chromatin regions with enhanced MNase sensitivity 29. Data sets were stratified according to paired‐read end‐to‐end distance into ranges where read pairs of 150 bp (± 20%) derive primarily from mono‐nucleosomes 28. Frequency distributions of the read mid‐points were then mapped to the S. pombe genome, and peaks in these distributions were taken to imply the presence of positioned nucleosomes in the cell population (Fig EV2). Bulk positioned nucleosome distributions from wild‐type cells in this study closely matched those determined in a previous study 30, confirming the validity of our technological approach (Fig EV2C). However, the distributions of positioned nucleosomes in abo1Δ mutant cells were perturbed relative to wild‐type in both biorep comparisons, suggesting a significant defect in genomic chromatin organisation (Fig EV2C and D).
Figure EV2. Paired‐end mode chromatin‐seq of abo1Δ.
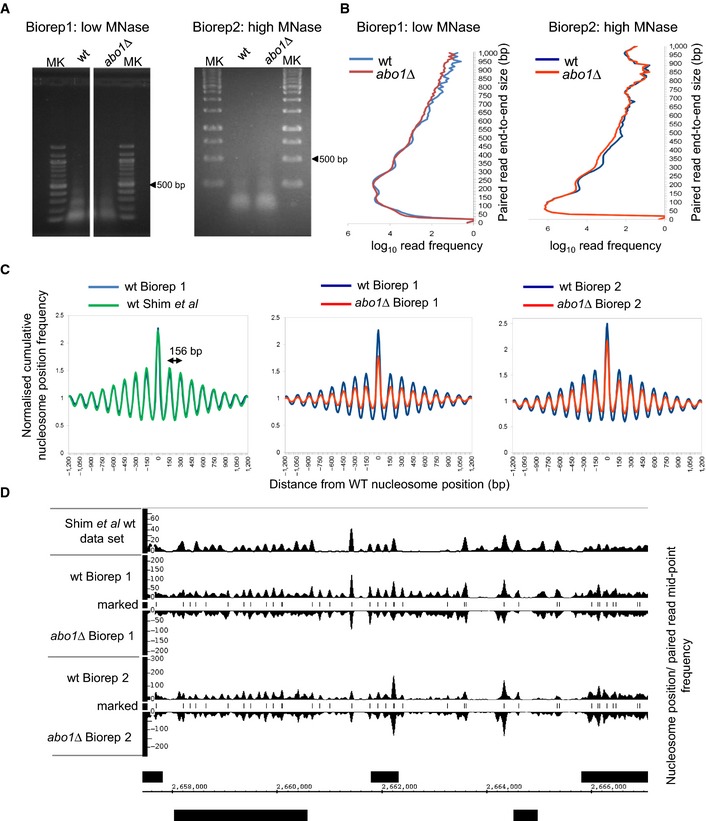
- Ethidium‐stained agarose gel separations of the two DNA pools (Biorep1 and Biorep2) extracted from MNase‐digested S. pombe chromatin and used for chromatin sequencing in this study.
- Frequency distributions of paired‐read end‐to‐end size values after chromatin‐seq of DNAs shown in (A). Note that increased MNase digestion used for Biorep2 samples shifts end‐to‐end size values downwards as expected.
- Nucleosome positions in wild‐type (Biorep1) cells were defined as the locations of 150 ± 30 bp (nucleosome) size class particle frequency peak summits (frequency value > 25). This simple heuristic procedure identifies 60,658 putative positioned nucleosomes in the S. pombe genome. The nucleosome size class particle frequency distributions centred on and surrounding (± 1,200 bp) these positions were then smoothed using an Epanechnikov kernel density estimate (to match that of a previously published data set (Gene Expression Omnibus GSE40451 30), summed and normalised to the average frequency value occurring in the ± 1,200 bp window, for each of the data sets. These cumulative distributions reveal the average nucleosome organisation surrounding positioned nucleosomes in the genome of each cell type. Three pair‐wise comparisons are shown. The nucleosome distribution from wild‐type Biorep1 overlaps with that in a previously published wild‐type data set (Gene Expression Omnibus GSE40451 30), confirming that our nucleosome mapping method yields similar results to those obtained using other technology. The abo1∆ mutant nucleosome distributions observed in Bioreps 1 and 2 both show a lower peak height and higher trough depth than the corresponding wild‐type. The wavelength of the peak pattern is shown and is equal to the known S. pombe nucleosome repeat length.
- Genome browser plot of wild‐type nucleosome occupancy data set (Gene Expression Omnibus GSE40451 30) plotted in relation to the 150 ± 30 bp paired‐read mid‐point frequency data obtained in this study (smoothed using an Epanechnikov kernel density estimate). Peak positions match between the two wild‐type data sets, confirming that the 150 ± 30 bp class of paired sequence reads accurately represents nucleosomal species from chromatin. Nucleosome positions in the wild‐type Biorep1 data set defined by our heuristic peak marking procedure are shown as “marked nucleosomes”.
Given the change in transcript profile observed in the abo1Δ mutant, we focused on the nucleosome architecture surrounding RNA pol II transcription start sites (TSS). Chromatin at the 5ʹ end of eukaryotic genes is typically organised with a nucleosome‐depleted region (NDR) located immediately upstream of the TSS, which is flanked by an ordered nucleosomal array extending into the coding sequence 31, 32. Comparison of average nucleosome positions surrounding TSSs from wild‐type and abo1Δ revealed that the NDR and upstream gene regulatory chromatin structure were retained in cells lacking Abo1, as was the average spacing of the reading frame nucleosomal array (Fig 2A). However, the peak amplitude under both MNase digestion conditions was significantly reduced in the mutant background in coding region nucleosomes. Furthermore, there is also a reduction in the depth of the troughs between peaks, suggesting that a re‐distribution of nucleosomes is occurring in the abo1Δ mutant cells. We note that under high MNase digestion conditions, the frequency of nucleosome‐sized particles detected in both wild‐type and abo1Δ mutant samples dropped in the region directly surrounding the TSS relative to the values observed under low MNase conditions. This result is consistent with the observation that eukaryotic chromatin in this region is labile to nuclease digestion 29.
Figure 2. Deletion of abo1 + results in the perturbation of nucleosomal organisation at coding sequences.
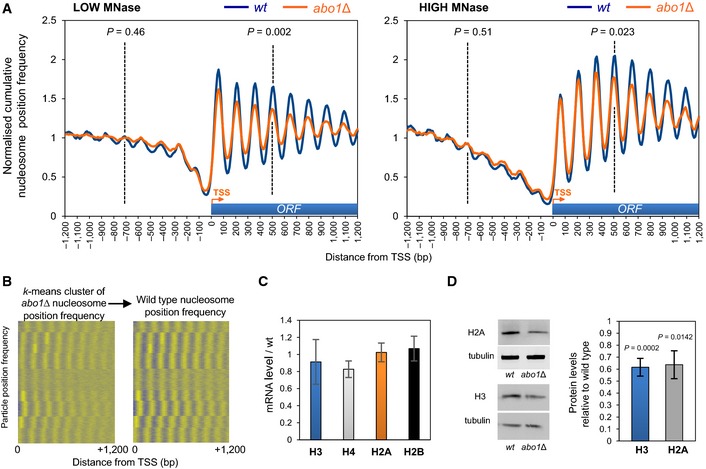
- Normalised cumulative nucleosome (150 ± 30 bp size class) position frequency profiles for 4,013 Schizosaccharomyces pombe genes aligned at the transcription start site (TSS) plotted from low MNase (biorep1) and high MNase (biorep2) data sets. P‐values (calculated using a two‐tailed unpaired t‐test) for the difference in means between random n = 100 subsets of the frequency values at the points indicated by the dotted lines (“−4” nucleosome and “+4” nucleosome) are shown.
- Nucleosome position frequency values for the coding regions of 4,013 Schizosaccharomyces pombe genes were k‐means clustered (k = 9) using the abo1∆ data from biorep1 (low MNase) and displayed with positive values coloured yellow and other values coloured blue (left‐hand panel). The cluster order was then used to display the equivalent wild‐type frequency values in the right‐hand panel.
- Level of histone gene mRNAs was determined by qRT–PCR. Data are the mean of four independent biological repeats, and error bars are ± SEM. Two‐tailed unpaired t‐tests showed no significant differences (P > 0.05) between wild‐type and abo1Δ cells.
- Whole‐cell extracts were subjected to Western blotting with histone H3 (Abcam), histone H2A (Abcam) and tubulin antibodies. Examples of the primary data are shown (left) along with a quantification of histone H2A and H3 levels normalised to tubulin (right). Data are the mean of at least three independent repeats, and error bars represent ± SEM. P‐values were calculated using a two‐tailed unpaired t‐test.
Source data are available online for this figure.
To examine whether or not a specific set of genes was affected by loss of Abo1, we used k‐means clustering to define nine arbitrary nucleosome position peak profiles for S. pombe protein‐coding regions in the abo1Δ (low MNase digestion) data set (Fig 2B). When this clustering was used to order the nucleosome position peak profiles from the wild‐type data set, no one cluster was found to exhibit a distinct dependence or independence on Abo1. A similar result was observed when this analysis was applied to the high MNase digestion data set (Appendix Fig S1). In both cases, all of the clustered peak profiles identified in the abo1Δ mutant resolved more clearly with the wild‐type data set, suggesting that most S. pombe protein‐coding gene reading frames exhibit changes to nucleosome organisation to some extent in the absence of Abo1.
To determine whether loss of Abo1 affects nucleosome occupancy as well as organisation, we compared histone levels in wild‐type and abo1Δ. Although steady‐state histone mRNA levels were not affected by loss of Abo1 when measured using both the microarray data described above and qRT–PCR (Fig 2C), Western analyses revealed significant drops in both histone H3 and H2A protein levels in abo1Δ cells (Fig 2D). This indicates that total histone dosage and thus nucleosome occupancy at a global level are lowered in the absence of Abo1. Consistent with this, we found that abo1Δ cells are sensitive to DNA‐damaging agents, and deletion of the H3–H4 gene pair hht2 +–hhf2 + exacerbates this phenotype (Fig EV3A).
Figure EV3. Abo1 physically and genetically interacts with histones and FACT .
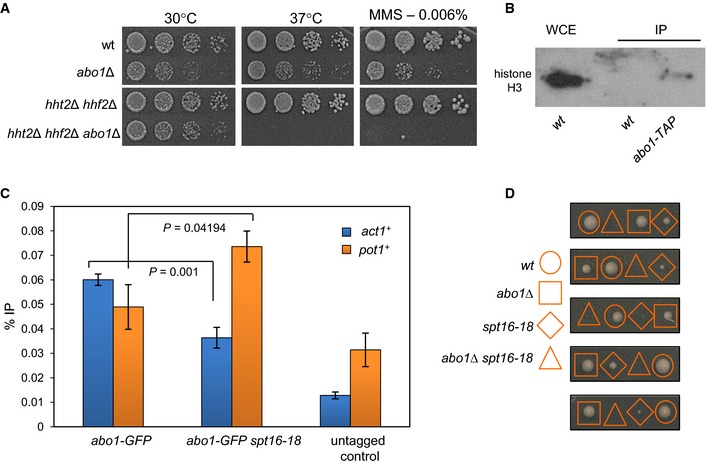
- Deletion of the histone H3–H4 gene pair hht2 + –hhf2 + exacerbates phenotypes associated with deletion of abo1 +. The indicated strains were grown to mid log phase, subjected to five‐fold serial dilution and spotted onto YES5 plates supplemented as indicated. Plates were incubated for 2 days (37°C) or 3–5 days (30°C). Images are representative of three biological repeats. All strains were present on the same agar plates.
- Whole‐cell extracts prepared from the indicated strains were partially purified using IgG sepharose and analysed by Western blotting. Data are representative of two biological repeats.
- ChIP–qPCR analysis was used to detect Abo1‐GFP enrichment at the indicated loci in mid log‐phase cells grown at 30°C. Data are the mean of three independent repeats, and error bars denote ± SEM. P‐values were calculated using a two‐tailed unpaired t‐test. GFP‐tagged strains exhibit significant enrichment (P < 0.05) at both loci relative to the untagged control.
- Tetrad dissection of a genetic cross between abo1Δ and spt16‐18. The genotypes of colonies arising from the spores of five asci are shown. Analysis of a total of 28 tetrads from two independent genetic crosses failed to identify a viable double mutant strain.
Abo1 associates with the FACT histone chaperone and suppresses cryptic transcription
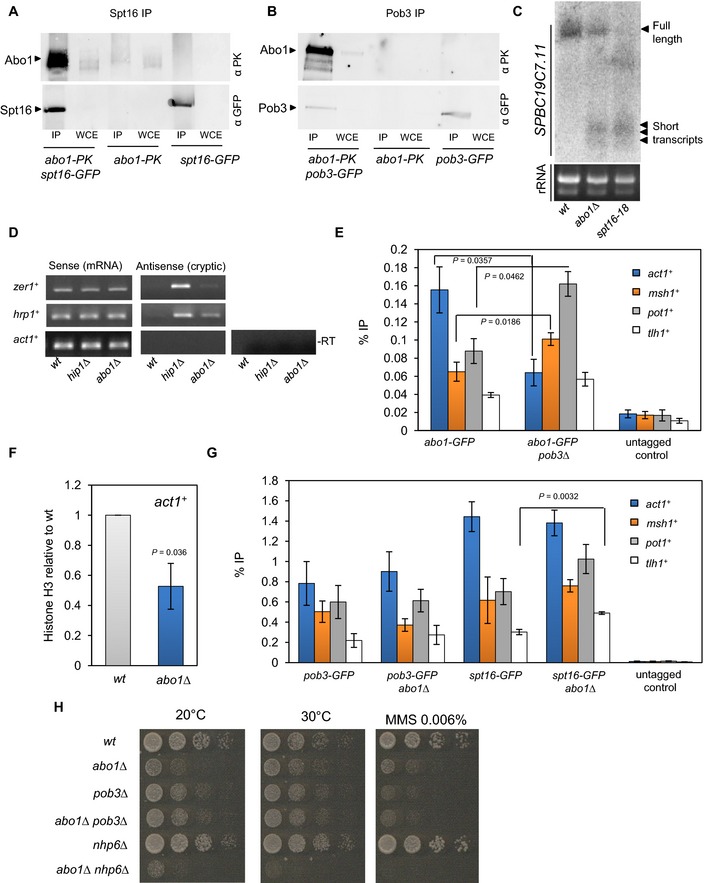
As Abo1 is required to maintain nucleosome architecture in gene sequences, we hypothesised that it may interact with a histone chaperone that mediates RNAP II transcription‐coupled nucleosome assembly. Indeed, preliminary analysis of Abo1 affinity purifications revealed the presence of subunits of the FACT complex. Therefore, this interaction was probed using co‐immunoprecipitation experiments. Abo1 was found to co‐purify with both Pob3 and Spt16 subunits, confirming an association with FACT (Fig 3A and B). As expected, Abo1 also co‐purified with histone H3 (Fig EV3B).
Figure 3. Abo1 associates with FACT and suppresses cryptic transcription.
-
A, BWhole‐cell extracts (WCE) were prepared from the indicated strains, immunoprecipitated (IP) with anti‐GFP antibody and subjected to Western blotting with anti‐V5‐Pk (Serotec) and anti‐GFP (Life Technologies) antibodies. Data are representative of three independent biological repeats.
-
CRNA purified from wild‐type, abo1∆ and spt16‐18 cells was analysed by Northern blotting using a probe to the 3′ end of SPBC19C7.11 (top panel). RNA (5 μg) used for Northern blotting was analysed on an ethidium bromide‐stained 1% TAE agarose gel (bottom panel). Data are representative of two independent biological repeats.
-
DRNA purified from wild‐type and abo1∆ cells was analysed by strand‐specific RT–PCR. RNA from hip1∆ cells was analysed as a control. One primer, complementary to either the forward or reverse transcripts, was included during the reverse transcription step, and the second primer was then added during PCR amplification. Control reactions omitting the reverse transcription step (−RT) were included to demonstrate the absence of contaminating genomic DNA. Data are representative of two independent biological repeats.
-
EThe indicated strains were subjected to ChIP analysis with anti‐GFP antibodies, and the resulting DNA was analysed by qPCR for the indicated locus. Data are the mean of three independent biological repeats, and error bars represent ± SEM. P‐values were calculated using a two‐tailed unpaired t‐test. Significant (P < 0.05) differences are indicated.
-
FHistone H3 levels at act1 + were determined by ChIP–qPCR. Data are the mean of three independent biological repeats, and error bars represent ± SEM. P‐value was calculated using a two‐tailed unpaired t‐test.
-
GThe indicated strains were subjected to ChIP analysis as described for (E).
-
HLog‐phase cells were subjected to five‐fold serial dilution and spotted onto rich (YE5S) agar or agar supplemented with MMS (0.006%). Plates were incubated for 3–5 days at 30°C or 7 days at 20°C. Images are representative of three independent biological repeats.
Source data are available online for this figure.
An increased level of cryptic intragenic transcripts is a hallmark of impaired transcription‐coupled chromatin assembly 1. To determine whether abo1Δ mutants experience increased levels of spurious transcription, we examined the SPBC19C7.11 locus using Northern blotting. As previously reported 33, mutations in spt16 + resulted in high levels of short abnormal transcripts from this gene and furthermore, similar small transcripts were observed in the abo1Δ background (Fig 3C). To further investigate this result, we examined some other genes (hrp1 + and zer1 +) where disruption to RNAP II‐coupled chromatin reassembly is known to result in increased levels of cryptic antisense transcription 24, 34. Strand‐specific RT–PCR confirmed that increased levels of cryptic transcripts were detectable in the absence of abo1 + (Fig 3D). As such, we conclude that like FACT, Abo1 is required to limit cryptic intragenic transcription.
ChIP analysis revealed that Abo1 was readily detected at ORFs (Fig 3E) ranging from the highly expressed act1 + gene to the silenced tlh1 + gene 22. This is consistent with Abo1 playing a direct role in maintaining nucleosome architecture in transcribed regions, and indeed, histone H3 levels at act1 + were significantly reduced in the abo1∆ background (Fig 3F). As Abo1 co‐immunoprecipitates with both Spt16 and Pob3, we next determined whether FACT influences the recruitment of Abo1 to chromatin or vice versa. Levels of Pob3 and Spt16 at ORFs were not affected by abo1 + deletion with the exception of tlh1 + which exhibited significantly increased levels of Spt16 in the abo1Δ background (Fig 3G). As discussed further below, we find that Abo1 is required to silence tlh1 + expression and therefore, the increase in Spt16 levels at this locus may simply reflect increased RNAP II (and thus FACT) recruitment. To determine the influence of FACT upon Abo1 recruitment, we first utilised a pob3Δ mutant. Deletion of pob3 + did not affect Abo1 levels at tlh1 +, but a significant reduction was observed at act1 +. In contrast, at both msh1 + and pot1 +, Abo1 levels were significantly increased (Fig 3E). Since spt16 + is an essential gene, it was not possible to determine the impact of its deletion upon Abo1 levels. However, ChIP experiments with the spt16‐18 allele showed a similar pattern to the pob3Δ mutant (Fig EV3C). Thus, it appears that the recruitment of Abo1 to transcription units does not depend upon FACT, but the chromatin distribution of Abo1 is perturbed by mutations in this histone chaperone.
We next examined genetic interactions between abo1 + and genes encoding subunits of the FACT complex. Analysis of the progeny resulting from genetic crosses between spt16‐18 and abo1∆ strains indicated that the double mutant is not viable indicating that Abo1 is essential when Spt16 function is impaired (Fig EV3D). In contrast, deletion of abo1 + did not exacerbate the growth or DNA damage sensitivity phenotypes associated with pob3∆ (Fig 3H), suggesting that Abo1 functions on the same pathway as Pob3. We also examined the interaction of abo1∆ with nhp6∆ (SPAC57A10.09c) because in S. cerevisiae, Nhp6 proteins are loosely associated with Spt16‐Pob3 and promote FACT function in vivo 35. Interestingly, we found a strong negative genetic interaction between abo1∆ and nhp6∆ alleles (Fig 3H).
Nucleosomes in intergenic regions are disrupted in abo1Δ
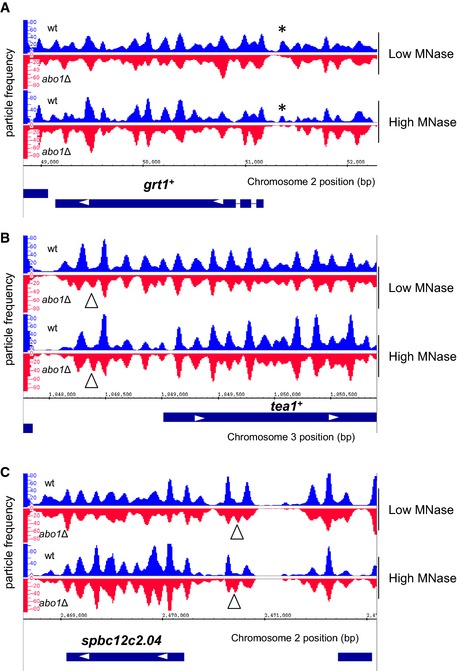
Although average nucleosome positions surrounding TSSs from wild‐type and abo1Δ mutant data sets revealed a relatively normal NDR and upstream gene regulatory chromatin structure (Fig 2A), we were also able to detect Abo1‐dependent changes in chromatin structures within intergenic regions. For instance, inspection of the nucleosome peak profiles of grt1 +, which was identified by microarray analysis as an Abo1‐repressed gene, revealed that the −1 nucleosome peak is lost in abo1Δ cells (Fig EV4A). We also found examples of promoters (e.g. tea1 + and SPBC12C2.04) where loss of Abo1 resulted in additional mis‐positioned nucleosome peaks (Fig EV4B and C). Thus, Abo1 can also affect the chromatin associated with specific intergenic regions.
Figure EV4. Abo1 is required for the organisation of chromatin in promoters.
-
A–CNucleosome (150 ± 30 bp size class) read profile over the indicated chromosomal loci. Data from low MNase (biorep1) are shown in the top panel and data from high MNase (biorep2) in the bottom panel. Missing or additional peaks in the abo1∆ background are marked with asterisks and triangles, respectively.
Abo1 is required for heterochromatin
As FACT is required for heterochromatin in S. pombe 36, we determined whether the function of Abo1 also extended to heterochromatic regions such as the pericentromeric otr (dh‐dg) and outer imr repeats. ura4 + reporter genes located in these repeats are transcriptionally silenced, thereby rendering cells resistant to 5‐FOA. While deletion of abo2 + did not alter the resistance of either the otr::ura4 + or imr::ura4 + reporter strains to 5‐FOA, deletion of abo1 + resulted in 5‐FOA sensitivity. Therefore, pericentromeric silencing is impaired by loss of Abo1 (Fig 4A and B).
Figure 4. Abo1 is required for heterochromatic silencing.
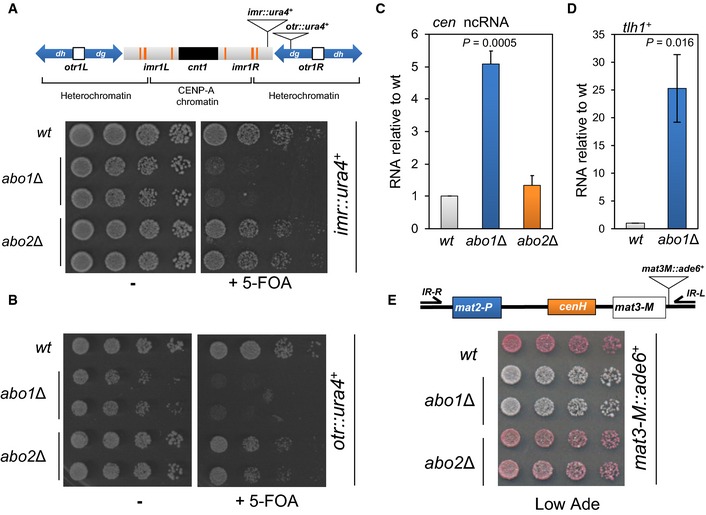
- The position of ura4 + reporter alleles in centromere 1 is shown in the top panel. Strains containing the imr::ura4 + allele were grown to log phase in YE5S medium, subjected to five‐fold serial dilutions and spotted onto YE5S agar or YE5S agar supplemented with 5‐FOA (1 mg/ml). Data are representative of three independent biological repeats.
- Strains carrying otr::ura4 + were analysed as described for (A).
- RNA was purified from the indicated strains and the level of centromeric transcripts determined by qRT–PCR. Data are the mean of three independent repeats, and error bars indicate ± SEM. P‐value was calculated using a two‐tailed unpaired t‐test.
- RNA was purified from the indicated strains and subjected to qRT–PCR for the subtelomeric gene, tlh1 +. Data are the mean of three biological repeats, and error bars indicate ± SEM. P‐value was calculated using a two‐tailed unpaired t‐test.
- The position of insertion of ade6 + reporter allele in the silent mat locus is shown in the top panel. Log‐phase cells containing the mat3‐M::ade6 + allele were subjected to five‐fold serial dilution and spotted onto YE5S agar plates lacking adenine (Low Ade). The plates were incubated for 4 days at 30°C. Data are representative of at least three independent biological repeats. Note, microarray analysis indicates that deletion of abo1 + does not influence the expression of ade6 + (or ura4 +) when these genes are present at their normal genomic loci.
Silencing at pericentromeric regions is dependent upon the RNAi machinery. The dg‐dh repeats are bi‐directionally transcribed at low levels by RNAP II, giving rise to long ncRNAs which are processed by Dicer to form siRNAs which promote heterochromatin assembly 37, 38. As the establishment of heterochromatin limits the transcription of repeat sequences, mutations that disrupt its integrity result in the accumulation of long ncRNAs. Consistent with the results of the reporter gene silencing assays, deletion of abo1 + resulted in a significant (~five‐fold) increase in these ncRNAs relative to wild‐type cells (Fig 4C).
To determine whether Abo1 is also required for heterochromatin at other loci, we examined the tlh1 + gene which is one of a family of telomere‐linked helicase genes that contain dg‐dh repeats similar to those in pericentromeric regions 39. Deletion of abo1 + resulted in a large (> 20 fold) increase in tlh1 + expression, indicating that Abo1 is required for subtelomeric heterochromatin silencing (Fig 4D). We also examined the cryptic mating (mat) locus as heterochromatin assembly at this site is only partially dependent upon the RNAi pathway due to the existence of a parallel pathway that utilises the Atf1/Pcr1 bZIP transcription factor 40, 41. Wild‐type cells that contain a silenced mat3‐M::ade6 + reporter form red colonies on adenine‐limiting media (Fig 4E). Silencing was maintained in the abo2Δ background, but abo1Δ cells formed light pink/white colonies, indicating that silencing had been impaired. Thus, Abo1 is also required for mat locus silencing, suggesting that it contributes to heterochromatin structures independently of the RNAi pathway.
To address the mechanism by which Abo1 contributes to heterochromatin, we analysed the transcript levels of 65 genes whose products are involved in its assembly and maintenance (Appendix Fig S2). No marked changes in the expression of these genes were observed in abo1Δ cells, suggesting that Abo1 regulates heterochromatin directly. Furthermore, ChIP analysis revealed that Abo1 is enriched at dh, dg and imr repeats (Fig 5A), consistent with a direct contribution to pericentromeric heterochromatin. However, deletion of abo1 + did not result in a reduction in pericentromeric H3K9me2 (Fig EV5A) or perturbations to the level or localisation of the HP1 homolog Swi6 (Figs 5B and EV5B and C). Furthermore, analysis of H3K9me2 levels associated with central core sequences of centromere 1 (cnt1) revealed no difference between wild‐type cells and the abo1Δ mutant, suggesting that Abo1 is also not required to prevent the spread of heterochromatin into the CENP‐A containing chromatin in the central core (cnt) (Fig EV5D).
Figure 5. Deletion of abo1 + perturbs centromeric heterochromatin.
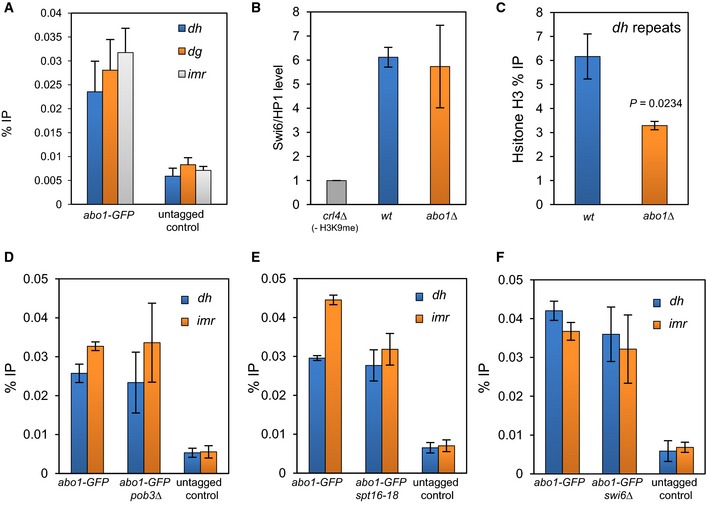
- ChIP analysis was performed on wild‐type (untagged) and abo1‐GFP cells and the resulting DNA analysed by qPCR for centromeric (dh, dg and imr) repeat sequences. Data are the mean of four independent biological repeats, and error bars represent ± SEM. P‐values calculated using a two‐tailed unpaired t‐test indicate that all loci are significantly enriched (P < 0.05) relative to the untagged control.
- The indicated strains expressing GFP‐Swi6 were subjected to ChIP analysis. The level of centromeric (dg) repeat sequences relative to the euchromatic control locus, adh1 +, was determined by qPCR and scaled to a clr4Δ (‐H3 K9me control) mutant. Data are the mean of two independent ChIP experiments, and error bars represent the range of the data.
- Histone H3 levels at centromeric (dh) repeat sequences were determined by ChIP–qPCR. Data are the mean of three independent ChIP experiments, and error bars are ± SEM. P‐values were calculated using a two‐tailed unpaired t‐test.
- ChIP analysis of Abo1‐GFP at centromeric (dh and imr) repeat sequences regions in wild‐type and pob3Δ cells. Data are the mean of three independent experiments, and error bars represent ± SEM. P‐values, calculated using a two‐tailed unpaired t‐test, indicated no significant difference (P > 0.05) for wild‐type and pob3Δ cells.
- ChIP analysis of Abo1‐GFP at centromeric (dh and imr) repeat sequences in wild‐type and pob3Δ cells. Data are the mean of duplicate experiments, and error bars represent the range of the data.
- ChIP analysis of Abo1‐GFP over the dh and imr regions in wild‐type and swi6Δ cells. Data are the mean of three independent experiments, and error bars represent ± SEM. P‐values, calculated using a two‐tailed unpaired t‐test, indicated no significant differences (P > 0.05) for wild‐type and swi6Δ cells.
Figure EV5. Loss of Abo1 does not reduce H3K9me2 or Swi6/HP1.
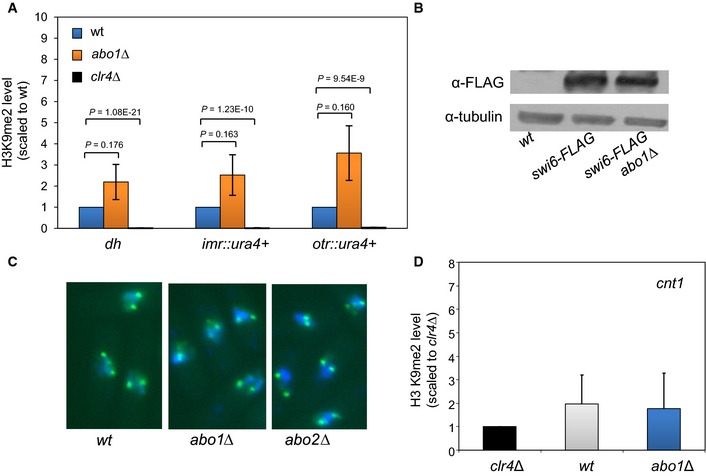
- H3K9me2 levels at dh repeats and a centromeric ura4 + marker gene were determined by ChIP–qPCR. H3K9me2 enrichment relative to adh1 + was determined and levels scaled to wild‐type. Data are the mean of three independent biological replicates, and error bars represent ± SEM. P‐values were calculated using a two‐tailed unpaired t‐test.
- Whole‐cell extracts prepared from the indicated strains were analysed by Western blotting with the indicated antibodies. Data are representative of two independent biological repeats.
- Cell expressing GFP‐Swi6 were stained with DAPI and visualised using fluorescence microscopy. Data are representative of two independent biological repeats.
- Loss of Abo1 does not result in the inappropriate spread of H3K9me2 into the centromeric central core. ChIP analysis of H3K9me2 levels at central core (cnt) sequences of chromosome 1. H3K9me2 enrichment relative to adh1 + was determined and levels scaled to the clr4Δ (‐H3 K9me2) mutant. Data are the mean of three independent ChIP experiments, and error bars are ± SEM.
Nucleosome position frequency values observed in the MNase‐seq data within the pericentromeric repeat regions were relatively low compared to those found associated with arrays of positioned nucleosomes associated with ORF 5′ regions suggesting a more disorganised nucleosome positioning environment. No consistent changes in nucleosome position were evident between the two MNase‐seq experiments. However, ChIP analysis did reveal a significant reduction in histone H3 levels at centromeric (dh) repeats (Fig 5C), indicating that Abo1 is required for normal levels of nucleosome occupancy in pericentromeric heterochromatin.
We next determined whether the recruitment of Abo1 to heterochromatin repeats was dependent upon FACT. Mutations in spt16 + and pob3 + did not influence the level of Abo1 at centromeric repeats, suggesting that its recruitment to heterochromatin is FACT independent (Fig 5D and E). Moreover although Abo1 has previously been show to co‐purify with Swi6/HP1 42, deletion of swi6 + did not alter the level of Abo1 enrichment at centromeric repeats (Fig 5F). Taken together, our data indicate that Abo1 is recruited to heterochromatin in a FACT‐ and Swi6‐independent manner where it is required to maintain proper nucleosome occupancy.
Abo1 represses LTR retrotransposons
The microarray analyses suggested that Abo1 also represses the expression of Tf2 LTR retrotransposons. Although these retroelements are silenced, they are not enriched with H3K9me 43 and are instead subjected to a distinct form of silencing 34, 44, 45, 46, 47, 48. Quantitative RT–PCR and reporter gene analyses confirmed that the suppression of Tf2 mRNA levels is also dependent upon Abo1 (Fig 6A and B). The 13 Tf2 elements are the only full‐length retrotransposons in the genome of the S. pombe reference strain, although a related element, Tf1, has been present during its evolution and intact Tf1 elements can be isolated from other wild‐type S. pombe strains 49. To address the regulation of Tf1 elements, we constructed a strain with an integrated Tf1‐lacZ reporter and determined that this “extinct” retrotransposon is also silenced in an Abo1‐dependent manner (Fig 6C).
Figure 6. Abo1 represses LTR retrotransposons.
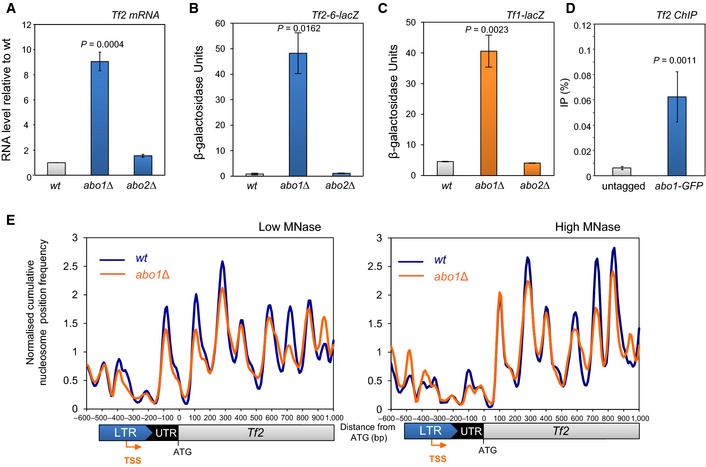
-
ARNA was extracted from mid log‐phase cells, and Tf2 mRNA levels were determined by qRT–PCR, normalised to act1 + mRNA and scaled relative to the wild‐type level. Data are the mean of three independent biological repeats, and error bars represent ± SEM.
-
B, CMid log‐phase cells with the indicated integrated lacZ reporter were subjected to quantitative β‐galactosidase assays. Data are the mean of three independent biological repeats, and error bars represent ± SEM.
-
DChIP DNA samples from wild‐type (untagged) and abo1‐GFP cells were analysed by qPCR for Tf2 LTR. Data are the mean of four independent biological repeats, and error bars represent ± SEM.
-
ENormalised cumulative nucleosome (150 ± 30 bp) position frequency profiles for Tf2 LTR retrotransposons aligned at the ATG plotted from low MNase (biorep1) and high MNase (biorep2) data sets.
ChIP analysis confirmed that Abo1 is associated with Tf2 LTR retrotransposons (Fig 6D) and so we determined the impact of Abo1 on the MNase‐seq profiles associated with these elements (Fig 6E). The peak downstream of the TSS was found to be highly sensitive to MNase digestion, suggesting that this nucleosome is labile. In the abo1∆ background, changes in the amplitude of specific peaks were observed in both data sets consistent with a perturbation to local nucleosome architecture at Tf2 LTR retrotransposons.
Abo1 is required for accurate chromosome segregation
Our findings predicted that deletion of abo1 + will lead to dysfunctional centromeres. Therefore, we analysed the sensitivity of abo1Δ cells to thiabendazole (TBZ), a drug which depolymerises microtubules and impairs mitotic spindle function. In common with other mutants that have defective centromeres, abo1Δ cells were sensitive to TBZ (Fig 7A). To further investigate the requirement of Abo1 in chromosome segregation, we analysed the mitotic stability of a 0.5‐Mbp linear mini‐chromosome, Ch16 50. Deletion of abo1 + resulted in a ~25‐fold increase in the loss rate of Ch16, confirming that it is required for accurate mitotic chromosome segregation (Fig 7B).
Figure 7. Abo1 is necessary for accurate chromosome segregation.
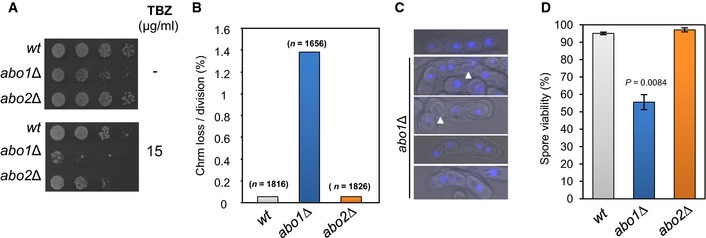
- Log‐phase cells were subjected to five‐fold serial dilution and spotted onto rich (YE5S) agar or agar supplemented with TBZ (15 μg/ml) and incubated for 3–5 days at 30°C. Data are representative of three independent biological repeats.
- The frequency of Ch16 minichromosome loss was assayed by plating cells onto adenine‐limiting agar and determining the frequency of half‐sectored (red‐white) colonies. At least two independently derived strain isolates were used for each genotype, and the total number of colonies counted for each genotype is indicated in parentheses.
- Heterothallic (h 90) strains were cultured on nitrogen‐limiting (EMMG) medium for 3 days at 25°C to induce mating and meiosis. The resulting asci were stained with DAPI and visualised by fluorescence microscopy. Spores lacking a DAPI signal are indicated by arrowheads. Data are representative of three biological repeats.
- Spore viability was measured by dissecting spores onto YE5S agar followed by incubation for 4–5 days at 30°C. Data are the mean of three independent biological repeats, and error bars represent ± SEM. P‐value was calculated using a two‐tailed unpaired t‐test.
To analyse the role of Abo1 in meiosis, the abo1Δ allele was introduced into a homothallic (h 90) background. Then mating and meiosis were induced by starving cells for nitrogen. The h 90 abo1Δ cells also showed no obvious defect in their ability to conjugate, but they frequently produced asci with an aberrant number of spores, a proportion of which failed to display a DAPI signal (Fig 7C). To quantify this defect, spore viability was determined and was found to be significantly reduced (~50%) in abo1Δ (Fig 7D). Therefore, the changes in chromatin structure that result from loss of Abo1 function lead to major defects in both the mitotic and meiotic cell cycles.
Discussion
Here, we report the global chromatin functions of a bromodomain AAA‐ATPase. Cells lacking abo1 + have perturbed nucleosome organisation in transcribed regions and a corresponding increase in cryptic transcription which are features of mutants that are defective in transcription‐coupled chromatin assembly. Consistent with this, Abo1 interacts with histone H3 and the essential histone chaperone FACT, and furthermore, the level of Abo1 at some genes is altered by mutations in FACT subunits. However, the impact of Abo1 upon nucleosome structure was not restricted to transcribed gene sequences, and so like FACT, it may also promote the proper re‐establishment of chromatin in other contexts such as replication.
Our finding that Abo1 promotes nucleosome occupancy was unexpected given previous analysis of S. cerevisiae, which indicates that Yta7 facilitates nucleosome removal 19, 20. Nucleosome density within genes downstream of the +1 nucleosome increases in the absence of Yta7, and furthermore, the phenotypes associated with its loss can be suppressed by reducing histone H3–H4 dosage. In contrast, there is an overall reduction in histone content in cells lacking Abo1 and we also find that reducing histone H3–H4 gene dosage exacerbates the temperature and DNA damage‐sensitive phenotypes of abo1Δ cells. At first glance, it is difficult to reconcile the disparate phenotypes that result from loss of bromodomain AAA‐ATPases in budding and fission yeast. However, it is becoming increasingly evident that ATP‐independent histone chaperones that mediate nucleosome assembly are also capable of promoting the reverse reaction of disassembly 51. Likewise, bromodomain AAA‐ATPases may participate in both assembly and disassembly reactions in a context‐dependent manner. It is also possible that Abo2 mediates nucleosome removal; however, Abo2 is expressed only at very low levels. Indeed, in a recent proteomic analysis, Abo2 was not detectable, while Abo1 is relatively abundant (~1,600 molecules per cell) 22. Furthermore, our microarray analysis indicates that loss of Abo1 does not lead to an increase in abo2 + expression. Therefore, it is highly unlikely that the reduced nucleosome levels in abo1Δ result from the up‐regulation of Abo2.
Chromatin‐seq analysis revealed that Abo1 impacts upon nucleosome positioning in coding sequences. Indeed, the peak profiles suggest that in abo1Δ cells, many nucleosomes become “fuzzy” and shift from occupation of a single favoured position. Regular nucleosome spacing over genes sequences is controlled by ISWI and CHD chromatin remodelling complexes 30, 52, 53. In S. pombe (which lacks ISWI complexes), loss of the CHD remodelers Hrp1 and Hrp3 abolishes regularly spaced nucleosomal arrays over transcribed sequences 30. Given that Abo1 is an AAA‐ATPase, it is unlikely that it has any direct nucleosome spacing/sliding activity. Furthermore, nucleosomal arrays are not completely lost in abo1Δ, as is the case in hrp3Δ hrp1Δ mutants, suggesting that Abo1 is not necessary for the function of these CHD remodelers. However, we cannot exclude the possibility that Abo1 may promote proper localisation of these or other chromatin modifying complexes. In this context, it is interesting that human ATAD2 controls the chromatin association of the MLL histone methyltransferase complex 6.
The phenotypes of the abo1Δ strain are reminiscent of S. cerevisiae mutants which lack the HMGB proteins, Nhp6a and Nhp6b. These mutants experience a substantial drop in nucleosome numbers and also a proportion of the remaining nucleosome positions become fuzzy 54. On this basis, it has been argued that Nhp6 proteins provide a chaperone‐like activity for nucleosome assembly 54. An alternative interpretation is that Nhp6 proteins function to increase nucleosome stability 55. Equally, the abo1Δ phenotypes are open to both interpretations. While these modes of action are not mutually exclusive, a prediction of the stabilisation model is that nucleosomes would become increasingly labile in the absence of Abo1 and that rates of H2A–H2B dimer exchange may increase. It is therefore noteworthy that photobleaching experiments suggest that H2A turnover is increased by knockdown of human ATAD2 12. The impact of S. pombe Nhp6 on global nucleosome occupancy and organisation has not yet been determined. However, that the loss of Nhp6 enhances the growth and DNA damage phenotypes associated with Abo1 inactivation suggests that they contribute to the maintenance of chromatin via distinct pathways.
Saccharomyces cerevisiae Yta7 was originally identified as a component of a tRNA gene‐containing barrier element that prevents the inappropriate spread of silencing from HMR 17. While the S. pombe barriers that prevent the invasion of heterochromatin into the central core of centromere 1 are also dependent upon tRNA genes 56, they apparently do not require Abo1. Instead, our finding that Abo1 is required for silencing provides the first demonstration that bromodomain AAA‐ATPases can contribute to the function of heterochromatin. Heterochromatin is characterised by the association of the HP1 proteins which form a platform for the assembly of an array of additional silencing factors (such as histone chaperones, histone deacetylases and ATP‐dependent chromatin remodelling enzymes). Although Abo1 also appears to function as one of these silencing factors and is known to interact with Swi6/HP1 42, its recruitment to heterochromatin is not dependent upon Swi6/HP1. Furthermore, the dysfunction of centromeric heterochromatin in the abo1Δ background does apparently result from reduced levels of Swi6/HP1. However, ChIP analysis revealed that loss of Abo1 leads to changes in nucleosome occupancy over centromeric repeats. Furthermore, recent studies indicate that relatively subtle changes to either nucleosome positioning or occupancy in centromeric heterochromatin can be sufficient to alleviate silencing and drive chromosome mis‐segregation events 30, 57.
Histone post‐translational modification and ATP‐dependent chromatin remodelling represent key mechanisms for modulating chromatin structure and function. However, controlling whether or not a particular DNA sequence is assembled into a nucleosome represents an even more profound mechanism for influencing chromatin structure. Thus, the regulation of nucleosome occupancy may be considered a primary layer of chromatin control. In this respect, it is now apparent that nucleosome number within cells is not fixed 54. For instance, both ageing yeast and mammalian cells exhibit reduced histone and nucleosome content 58, 59. Here, we demonstrate that a bromodomain AAA‐ATPase is required for global nucleosome occupancy, indicating that these factors play a key role in regulation of this primary layer of chromatin control.
Materials and Methods
Schizosaccharomyces pombe strains
Routine culture and genetic manipulation were performed as previously described 60. The strains used in this study are listed in Appendix Table S1. Strains carrying kanMX replacements of ORFs SPAC31G5.19 and SPBP22H7.05c were purchased from Bioneer, genotyped by PCR analysis and backcrossed with the appropriate strains. A strain expressing Pk‐tagged Abo1 was constructed by cloning a PCR fragment (amplified with primers CAGTTCGGATCCTTGTAGTGCATTAATCATAAACTC and GTTCACCTGCAGAATTAAGGCACGGAAAGCTTC) into the PstI and BamHI sites of pRip42‐Pk 61. The resulting plasmid was digested with BclI and transformed into S. pombe. A strain expressing Abo1 tagged with GFP was constructed using the approach described in 62. Phenotypic analysis demonstrated that epitope‐tagged versions of Abo1 are functional (Appendix Fig S3). A strain with a Tf1‐lacZ reporter integrated into chromosome II (base pairs 1,877,855–1,878,381) was constructed by PCR amplifying a DNA fragment using template DNA derived from wild strain NCYC132 and with primers (GCTAAGCTGCAGTGTCAGCAATACTACACTACGCTA) and (GGAT GCCTGCAGGCTGTTCAGTTGAATATCTATCGG). The fragment was cleaved with PstI and cloned into the PstI site of pSPI356ChrmIICDAB 34. The resulting plasmid was linearised with BamHI and transformed into the appropriate strain S. pombe cells. Quantitative β‐galactosidase assays were performed as previously described 63.
Microarrays
RNA was extracted using hot phenol and purified over RNase easy columns (Qiagen) as previously described 64. A 15‐μg aliquot of total RNA was prepared for microarray analysis using the Superscript Plus Direct labelling kit (Invitrogen, Life Technologies), in two biological repeats with dye swaps. Paired samples were hybridised over two 44K Custom microarrays (Agilent) overnight at 65°C, scanned using GenePix 4000B scanner (Axon Instruments) and analysed using Genepix Pro 6.0 software(Axon Instruments). In house normalisation, scripts were applied (to gpr files) and the data uploaded into Genespring 7.3.1 software for analysis. Microarray data can be accessed at ArrayExpress accession E‐MTAB‐3455.
Histone levels
Approximately 4 × 107 cells were harvested following the addition of trichloroacetic acid (TCA) to a final concentration of 10%. Cells were resuspended in 200 μl 10% TCA and then disrupted using a beadbeater with 0.75 ml of glass beads using two pulses of 15 s with 1 min on ice in between. A 500‐μl aliquot of 10% TCA was added, and the lysate was recovered from the beads which was then clarified by spinning at 15,000 × g in a microcentrifuge. The resulting pellet was washed three times in acetone, dried and resuspended in 30 μl 100 mM Tris–HCl (pH 8.0), 1% w/v SDS and 1 mM EDTA. Samples were analysed on SDS–PAGE gels and subjected to Western blotting using anti‐histone H3 (Abcam ab1791), anti‐histone H2A (Abcam ab13923) and anti‐tubulin (TAT‐1) antibodies. Western blots were visualised using a Typhoon FLA9500 (GE Healthcare) and band intensities quantified using ImageQuant.
RT–PCR and Northern analyses
RNA was purified as described for microarray analysis and subjected to RT–PCR using a One Step RT‐PCR kit (Qiagen). For strand‐specific RT–PCR, one primer complementary to the sense or antisense transcript was added during first‐strand cDNA synthesis, while the second primer was added prior to the PCR amplification steps. cDNA for quantitative (real‐time) RT–PCR was made using a Superscript II kit (Invitrogen). Real‐time PCRs were performed using a LightCycler 2.0 PCR system (Roche) and SYBR Green mix (Molecular Probes) using the appropriate primers. Reactions were normalised using primers specific to act1 +. Northern analysis was performed as previously described 65.
MNase digestion of chromatin
Cells (100 ml) were grown to OD595 = 0.75–8.0 in YE5S at 30°C, cross‐linked for 20 min at 30°C using 1% formaldehyde and quenched by the addition of glycine to 125 mM. Cells were washed once in CES buffer (50 mM citric acid/50 mM Na2HPO4 [pH 5.6], 40 mM EDTA [pH 8.0], 1.2 M sorbitol and 10 mM β‐mercaptoethanol) and resuspended in 500 μl of CES buffer with 0.5 mg Zymolase 100‐T. Cells were spheroplasted at 30°C for up to 1 h and then washed twice with ice‐cold 1.2 M sorbitol. Spheroplasts were then resuspended in 800 μl NP‐S buffer (1.2 M sorbitol, 10 mM CaCl2, 100 mM NaCl, 1 mM EDTA pH 8.0, 14 mM β‐mercaptoethanol, 50 mM Tris pH 8.0, 0.075% NP‐40, 5 mM spermidine, 0.1 mM PMSF, 1% Sigma protease inhibitors cocktail [Sigma P8215]). Spheroplasts were then divided into four 200 μl aliquots, and each aliquot was mixed with 300 μl of NP‐S buffer. Three aliquots were digested with between 75 and 187.5 units of MNase (USB) for 10 min at 37°C. The fourth was retained as an undigested control. MNase digestion was terminated by adding EDTA [pH 8.0] to a final concentration of 50 mM and SDS to 0.2%. Reactions were incubated at 65°C overnight with 0.2 mg/ml proteinase K and 10 μg RNAse. DNA was purified by extracting twice with phenol:chloroform followed by ethanol precipitation (0.1 volumes of 3 M sodium acetate followed by two volumes of ethanol). Pellets were washed in 70% ethanol and resuspended in water containing 10 μg/ml RNase and incubated at 37°C for 30 min. Triplicate digests were pooled and treated with 100 U unmodified T4 polynucleotide kinase (NEB) for 30 min at 37°C to remove 3′‐phosphate groups left by MNase. DNA was extracted once more with phenol:chloroform, re‐precipitated with sodium acetate and propan‐2‐ol, washed with 80% ethanol, dried and re‐suspended in TE (pH 7.5).
Chromatin‐seq
DNA fragments were end repaired, 3′‐adenylated and ligated to indexed adapters without size selection using Nextflex reagents (Newmarket Scientific, UK). Libraries were amplified with eight cycles PCR using Kapa HiFi PCR master mix (Anachem), primers removed with GeneRead size selection protocol (QIAgen) before quantification by Bioanalyser DNA 7500 assay. Libraries were pooled, denatured and diluted to 6 nM before clustering in a single lane of a high‐output Illumina flow cell. Sequencing (100 nt) was undertaken on a HiSeq 2500 using TruSeq SBS v3 reagents (Illumina).
Bioinformatics
Paired reads were aligned to the ASM294v1.17 reference genome using Bowtie 0.12.7 66 with command line flags: ‐n 0 –trim3 64 –maxins 5000 –fr ‐k 1 –sam. Aligned read pairs were sorted according to chromosome and then into a range of size classes based on the SAM format ISIZE value (difference between 5′ end of the mate read and the 5′ end of the first mapped read) plus or minus 20%. Mono‐nucleosome‐sized reads are, therefore, represented as 150 ± 30 bp. To define the genomic position of MNase‐resistant chromatin entities, we mapped the mid‐point position of the read pairs in a particular size class. Frequency distributions of the mid‐point positions were then calculated using 10‐bp bins. To mark positions of peak maxima, and for k‐means clustering, the frequency distributions were lightly smoothed by taking a 3‐bin moving average. For all other analyses and displays, the frequency distributions were smoothed using an Epanechnikov kernel density estimate (using the Perl module Statistics::Kernel Estimation‐0.05 with bandwidth = 30) to match the profile of the previously published S. pombe nucleosome position data set (Gene Expression Omnibus GSE40451 30). All frequency distributions were output in the zero‐referenced, chromosome base, three‐column.sgr format (chromosome number, feature/bin position, mid‐point frequency value) for rendering with the Integrated Genome Browser 67 and for further processing. Average cumulative chromatin particle position frequency distributions at, and surrounding, genomic features were calculated using the script SiteWriterCFD as described previously 28, 68, with values for each bin normalised to the average cumulative frequency value obtained for all bins within the feature window. Protein‐coding gene transcription start site (TSS) positions were taken from the previously published data set 32. Nucleosome positions in the WT biorep1 data set were defined using a simple heuristic peak summit marking process (script PeakMarker_lite; read frequency threshold set to 25) (Script EV1). Clustering was performed with Cluster 3 using k‐means and the Euclidean distance similarity metric 69. Clusters were displayed using JavaTreeView 1.1.6 with values plotted as log2; centre = 3.0; and contrast value = 10. Wild‐type DNA sequence data (low MNase digestion) can be found in the NCBI Sequence Read Archive (SRA): accession number SRS712792. Other MNase sequence data can be accessed via Gene Expression Omnibus GSE67410.
ChIP analysis
For Swi6 ChIP, strains were grown in PMG complete media to exponential growth phase at 25°C. Cells were then fixed with 3% formaldehyde for 18 min at room temperature. Chromatin immunoprecipitation was performed as previously described 70. In short, fixed cells were suspended in 400 μl ChIP lysis buffer (50 mM HEPES [pH 7.5], 140 mM NaCl, 1 mM EDTA, 0.1% DOC, 1% Triton X‐100) and disrupted by beadbeating (2 × 1 min). Chromatin was then sheared by probe sonication (3 × 20 s) and immunoprecipitated with anti‐GFP (Life Technologies). Cross‐linking was reversed by overnight incubation with TES followed by proteinase K digestion. After DNA purification, samples were subject to real‐time qPCR. Abo1‐GFP, Spt16‐GFP, Pob3‐GFP, histone H3 and H3K9me2 ChIP were performed as above but with the following modifications; cells were grown at 30°C in YE5S and were fixed by the addition of formaldehyde to a final concentration of 1% for 15 min at room temperature; chromatin was sheared using a Diagenode Bioruptor using approximately 30 cycles (30 s ON/OFF). Chromatin was immunoprecipitated with anti‐GFP (Life Technologies A11122), anti‐H3 (Abcam ab1791) or anti‐H3K9me2 (Abcam ab1220). Immunoprecipitated DNA was recovered as described previously 71.
Co‐immunoprecipitations
Whole‐cell extracts, prepared as previously described 65, were pre‐cleared with 40 μl Sepharose A beads for 1–2 h at 4°C on rotating wheel. The resulting supernatant was transferred to a fresh microcentrifuge tube and incubated with 40 μl Sepharose A beads and 1.5 μl anti‐GFP (Life Technologies) at 4°C overnight on a rotating wheel. Sepharose A beads were harvested by centrifugation and washed 3–6 times in buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 10 mM imidazole, 0.5% [v/v] NP‐40 [IGEPAL], 1 mM PMSF, 54 mM NaF, 5 μM NaVO4, 1 μl/ml aprotinin, 5 μg/ml pepstatin A and 5 μg/ml leupeptin) before being resuspended in 40 μl Laemmli buffer (0.1% β‐mercaptoethanol, 0.0005% bromophenol blue, 10% glycerol, 2% [w/v] SDS and 63 mM Tris–Hcl [pH 6.8]). Samples were then analysed on 9% SDS–polyacrylamide gels followed by Western blotting.
Microscopy
Cells were pelleted in a microcentrifuge at 3,000 × g for 2 min before pellets were resuspended in ~20 μl of medium. Microscope slides were coated in poly lysine and allowed to dry before the addition of 5 μl of the cell suspension. Cells were allowed to dry, and 5 μl DAPI containing mounting medium (Vectashield® with DAPI [Vector laboratories]) was added. Images were obtained using Zeiss Axiovert 200M with 100× oil immersion lens and Axiovision software.
Statistics
P‐values were calculated using two‐tailed unpaired t‐tests, and the statistical significance is shown in the relevant Figure. P‐values for the overlaps in microarray gene lists were calculated in Excel and are based on hypergeometric distribution using all coding genes as the background population size. For comparison of nucleosome position frequency values at −4 and +4 nucleosomes surrounding protein‐coding gene TSSs, the data were subsampled into 29 random 100 TSSs subsets to avoid the tendency of P‐values to become inaccurate with large total n 72. Normality and variance were checked using the plot command in R 3.2.2, and P‐values were calculated using Perl module Statistics::DependantTTest‐0.03.
Author contributions
HEM, CG, LS, AJW, KMM, KP, SC, NAK, SKW and KMC performed experiments and analysed data. JB, JFP, RCA, NAK and SKW designed experiments and analysed data. SKW and NAK wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Script EV1
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
We thank Neil Perkins and Elizabeth Veal for comments on the manuscript, Andreas Ladurner for strains, Thomas Gerland for strain construction, Michelle Wray for technical assistance and Mimoza Hoti and Babis Rallis for their help with ArrayExpress submission. HEM and AJW were supported by the BBSRC (Doctoral Training Grants), and CG was supported by the Medical Research Council and the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre based at Newcastle Upon Tyne Hospitals NHS Foundation Trust and Newcastle University. The views expressed are those of the authors and not necessarily those of the NHS, NIHR or the Department of Health. LS was supported by a Marie Curie International Incoming Fellowship (IIF275280) and an EMBO Long Term Fellowship (ALTF 1491‐2010). The Wellcome Trust supported the work of RCA (095021). The Wellcome Trust Centre for Cell Biology is supported by the Wellcome Trust core funding (092076). KMC was supported by NIA fellowship (NRSA F31‐AG038153). JFP was supported by NIH R01 grant (GM084045), Cancer Center support grant (CCSG 2 P30 CA21765), and the American Lebanese Syrian Associated Charities of St. Jude Children's Research Hospital. JB was supported by a Wellcome Trust Senior Investigator Award. KP and SKW acknowledge support from the Wellcome Trust Institutional Strategic Support Fund to the University of Exeter (WT097835MF) and Newcastle University (WT 097823/Z/11/Z), respectively.
EMBO Reports (2016) 17: 79–93
References
- 1. Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128: 707–719 [DOI] [PubMed] [Google Scholar]
- 2. Kireeva ML, Walter W, Tchernajenko V, Bondarenko V, Kashlev M, Studitsky VM (2002) Nucleosome remodeling induced by RNA polymerase II: loss of the H2A/H2B dimer during transcription. Mol Cell 9: 541–552 [DOI] [PubMed] [Google Scholar]
- 3. Schwabish MA, Struhl K (2004) Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Mol Cell Biol 24: 10111–10117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schwabish MA, Struhl K (2006) Asf1 mediates histone eviction and deposition during elongation by RNA polymerase II. Mol Cell 22: 415–422 [DOI] [PubMed] [Google Scholar]
- 5. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, Reinberg D (2003) FACT facilitates transcription‐dependent nucleosome alteration. Science 301: 1090–1093 [DOI] [PubMed] [Google Scholar]
- 6. Revenko AS, Kalashnikova EV, Gemo AT, Zou JX, Chen HW (2010) Chromatin loading of E2F‐MLL complex by cancer‐associated coregulator ANCCA via reading a specific histone mark. Mol Cell Biol 30: 5260–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hsia EY, Kalashnikova EV, Revenko AS, Zou JX, Borowsky AD, Chen HW (2010) Deregulated E2F and the AAA+ coregulator ANCCA drive proto‐oncogene ACTR/AIB1 overexpression in breast cancer. Mol Cancer Res 8: 183–193 [DOI] [PubMed] [Google Scholar]
- 8. Ciro M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J et al (2009) ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res 69: 8491–8498 [DOI] [PubMed] [Google Scholar]
- 9. Zou JX, Guo L, Revenko AS, Tepper CG, Gemo AT, Kung HJ, Chen HW (2009) Androgen‐induced coactivator ANCCA mediates specific androgen receptor signaling in prostate cancer. Cancer Res 69: 3339–3346 [DOI] [PubMed] [Google Scholar]
- 10. Zou JX, Revenko AS, Li LB, Gemo AT, Chen HW (2007) ANCCA, an estrogen‐regulated AAA+ ATPase coactivator for ERalpha, is required for coregulator occupancy and chromatin modification. Proc Natl Acad Sci USA 104: 18067–18072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boussouar F, Jamshidikia M, Morozumi Y, Rousseaux S, Khochbin S (2013) Malignant genome reprogramming by ATAD2. Biochim Biophys Acta 1829: 1010–1014 [DOI] [PubMed] [Google Scholar]
- 12. Caron C, Lestrat C, Marsal S, Escoffier E, Curtet S, Virolle V, Barbry P, Debernardi A, Brambilla C, Brambilla E et al (2010) Functional characterization of ATAD2 as a new cancer/testis factor and a predictor of poor prognosis in breast and lung cancers. Oncogene 29: 5171–5181 [DOI] [PubMed] [Google Scholar]
- 13. Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD, Chen HW (2010) ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple‐negative cells through control of B‐Myb and EZH2. Cancer Res 70: 9402–9412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leachman NT, Brellier F, Ferralli J, Chiquet‐Ehrismann R, Tucker RP (2010) ATAD2B is a phylogenetically conserved nuclear protein expressed during neuronal differentiation and tumorigenesis. Dev Growth Differ 52: 747–755 [DOI] [PubMed] [Google Scholar]
- 15. Hanson PI, Whiteheart SW (2005) AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol 6: 519–529 [DOI] [PubMed] [Google Scholar]
- 16. Gradolatto A, Smart SK, Byrum S, Blair LP, Rogers RS, Kolar EA, Lavender H, Larson SK, Aitchison JD, Taverna SD et al (2009) A noncanonical bromodomain in the AAA ATPase protein Yta7 directs chromosomal positioning and barrier chromatin activity. Mol Cell Biol 29: 4604–4611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tackett AJ, Dilworth DJ, Davey MJ, O'Donnell M, Aitchison JD, Rout MP, Chait BT (2005) Proteomic and genomic characterization of chromatin complexes at a boundary. J Cell Biol 169: 35–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kurat CF, Lambert JP, van Dyk D, Tsui K, van Bakel H, Kaluarachchi S, Friesen H, Kainth P, Nislow C, Figeys D et al (2011) Restriction of histone gene transcription to S phase by phosphorylation of a chromatin boundary protein. Genes Dev 25: 2489–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lombardi LM, Ellahi A, Rine J (2011) Direct regulation of nucleosome density by the conserved AAA‐ATPase Yta7. Proc Natl Acad Sci USA 108: E1302–E1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lombardi LM, Davis MD, Rine J (2015) Maintenance of nucleosomal balance in cis by conserved AAA‐ATPase Yta7. Genetics 199: 105–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kim DU, Hayles J, Kim D, Wood V, Park HO, Won M, Yoo HS, Duhig T, Nam M, Palmer G et al (2010) Analysis of a genome‐wide set of gene deletions in the fission yeast Schizosaccharomyces pombe . Nat Biotechnol 28: 617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marguerat S, Schmidt A, Codlin S, Chen W, Aebersold R, Bahler J (2012) Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151: 671–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hansen KR, Burns G, Mata J, Volpe TA, Martienssen RA, Bahler J, Thon G (2005) Global effects on gene expression in fission yeast by silencing and RNA interference machineries. Mol Cell Biol 25: 590–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, Grewal SI (2007) Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nat Struct Mol Biol 14: 372–380 [DOI] [PubMed] [Google Scholar]
- 25. Mandell JG, Bahler J, Volpe TA, Martienssen RA, Cech TR (2005) Global expression changes resulting from loss of telomeric DNA in fission yeast. Genome Biol 6: R1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van de Pasch LA, Miles AJ, Nijenhuis W, Brabers NA, van Leenen D, Lijnzaad P, Brown MK, Ouellet J, Barral Y, Kops GJ et al (2013) Centromere binding and a conserved role in chromosome stability for SUMO‐dependent ubiquitin ligases. PLoS ONE 8: e65628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen D, Toone WM, Mata J, Lyne R, Burns G, Kivinen K, Brazma A, Jones N, Bahler J (2003) Global transcriptional responses of fission yeast to environmental stress. Mol Biol Cell 14: 214–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kent NA, Adams S, Moorhouse A, Paszkiewicz K (2011) Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired‐end mode next‐generation DNA sequencing. Nucleic Acids Res 39: e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vera DL, Madzima TF, Labonne JD, Alam MP, Hoffman GG, Girimurugan SB, Zhang J, McGinnis KM, Dennis JH, Bass HW (2014) Differential nuclease sensitivity profiling of chromatin reveals biochemical footprints coupled to gene expression and functional DNA elements in maize. Plant Cell 26: 3883–3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shim YS, Choi Y, Kang K, Cho K, Oh S, Lee J, Grewal SI, Lee D (2012) Hrp3 controls nucleosome positioning to suppress non‐coding transcription in eu‐ and heterochromatin. EMBO J 31: 4375–4387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jiang C, Pugh BF (2009) Nucleosome positioning and gene regulation: advances through genomics. Nat Rev Genet 10: 161–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lantermann AB, Straub T, Stralfors A, Yuan GC, Ekwall K, Korber P (2010) Schizosaccharomyces pombe genome‐wide nucleosome mapping reveals positioning mechanisms distinct from those of Saccharomyces cerevisiae . Nat Struct Mol Biol 17: 251–257 [DOI] [PubMed] [Google Scholar]
- 33. Choi ES, Stralfors A, Catania S, Castillo AG, Svensson JP, Pidoux AL, Ekwall K, Allshire RC (2012) Factors that promote H3 chromatin integrity during transcription prevent promiscuous deposition of CENP‐A(Cnp1) in fission yeast. PLoS Genet 8: e1002985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anderson HE, Wardle J, Korkut SV, Murton HE, Lopez‐Maury L, Bahler J, Whitehall SK (2009) The fission yeast HIRA histone chaperone is required for promoter silencing and the suppression of cryptic antisense transcripts. Mol Cell Biol 29: 5158–5167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Formosa T (2012) The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta 1819: 247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lejeune E, Bortfeld M, White SA, Pidoux AL, Ekwall K, Allshire RC, Ladurner AG (2007) The chromatin‐remodeling factor FACT contributes to centromeric heterochromatin independently of RNAi. Curr Biol 17: 1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Creamer KM, Partridge JF (2011) RITS‐connecting transcription, RNA interference, and heterochromatin assembly in fission yeast. Wiley Interdiscip Rev RNA 2: 632–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lejeune E, Allshire RC (2011) Common ground: small RNA programming and chromatin modifications. Curr Opin Cell Biol 23: 258–265 [DOI] [PubMed] [Google Scholar]
- 39. Hansen KR, Ibarra PT, Thon G (2006) Evolutionary‐conserved telomere‐linked helicase genes of fission yeast are repressed by silencing factors, RNAi components and the telomere‐binding protein Taz1. Nucleic Acids Res 34: 78–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim HS, Choi ES, Shin JA, Jang YK, Park SD (2004) Regulation of Swi6/HP1‐dependent heterochromatin assembly by cooperation of components of the mitogen‐activated protein kinase pathway and a histone deacetylase Clr6. J Biol Chem 279: 42850–42859 [DOI] [PubMed] [Google Scholar]
- 41. Jia S, Noma K, Grewal SI (2004) RNAi‐independent heterochromatin nucleation by the stress‐activated ATF/CREB family proteins. Science 304: 1971–1976 [DOI] [PubMed] [Google Scholar]
- 42. Motamedi MR, Hong EJ, Li X, Gerber S, Denison C, Gygi S, Moazed D (2008) HP1 proteins form distinct complexes and mediate heterochromatic gene silencing by nonoverlapping mechanisms. Mol Cell 32: 778–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cam HP, Sugiyama T, Chen ES, Chen X, FitzGerald PC, Grewal SI (2005) Comprehensive analysis of heterochromatin‐ and RNAi‐mediated epigenetic control of the fission yeast genome. Nat Genet 37: 809–819 [DOI] [PubMed] [Google Scholar]
- 44. Cam HP, Noma K, Ebina H, Levin HL, Grewal SI (2008) Host genome surveillance for retrotransposons by transposon‐derived proteins. Nature 451: 431–436 [DOI] [PubMed] [Google Scholar]
- 45. Durand‐Dubief M, Sinha I, Fagerstrom‐Billai F, Bonilla C, Wright A, Grunstein M, Ekwall K (2007) Specific functions for the fission yeast Sirtuins Hst2 and Hst4 in gene regulation and retrotransposon silencing. EMBO J 26: 2477–2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Greenall A, Williams ES, Martin KA, Palmer JM, Gray J, Liu C, Whitehall SK (2006) Hip3 interacts with the HIRA proteins Hip1 and Slm9 and is required for transcriptional silencing and accurate chromosome segregation. J Biol Chem 281: 8732–8739 [DOI] [PubMed] [Google Scholar]
- 47. Li J, Liu Y, Rhee HS, Ghosh SK, Bai L, Pugh BF, Gilmour DS (2013) Kinetic competition between elongation rate and binding of NELF controls promoter‐proximal pausing. Mol Cell 50: 711–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yamane K, Mizuguchi T, Cui B, Zofall M, Noma K, Grewal SI (2011) Asf1/HIRA facilitate global histone deacetylation and associate with HP1 to promote nucleosome occupancy at heterochromatic loci. Mol Cell 41: 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bowen NJ, Jordan IK, Epstein JA, Wood V, Levin HL (2003) Retrotransposons and their recognition of pol II promoters: a comprehensive survey of the transposable elements from the complete genome sequence of Schizosaccharomyces pombe . Genome Res 13: 1984–1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Niwa O, Matsumoto T, Chikashige Y, Yanagida M (1989) Characterization of Schizosaccharomyces pombe minichromosome deletion derivatives and a functional allocation of their centromere. EMBO J 8: 3045–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Park YJ, Luger K (2008) Histone chaperones in nucleosome eviction and histone exchange. Curr Opin Struct Biol 18: 282–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gkikopoulos T, Schofield P, Singh V, Pinskaya M, Mellor J, Smolle M, Workman JL, Barton GJ, Owen‐Hughes T (2011) A role for Snf2‐related nucleosome‐spacing enzymes in genome‐wide nucleosome organization. Science 333: 1758–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pointner J, Persson J, Prasad P, Norman‐Axelsson U, Stralfors A, Khorosjutina O, Krietenstein N, Svensson JP, Ekwall K, Korber P (2012) CHD1 remodelers regulate nucleosome spacing in vitro and align nucleosomal arrays over gene coding regions in S. pombe . EMBO J 31: 4388–4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Celona B, Weiner A, Di Felice F, Mancuso FM, Cesarini E, Rossi RL, Gregory L, Baban D, Rossetti G, Grianti P et al (2011) Substantial histone reduction modulates genomewide nucleosomal occupancy and global transcriptional output. PLoS Biol 9: e1001086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dowell NL, Sperling AS, Mason MJ, Johnson RC (2010) Chromatin‐dependent binding of the S. cerevisiae HMGB protein Nhp6A affects nucleosome dynamics and transcription. Genes Dev 24: 2031–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Scott KC, Merrett SL, Willard HF (2006) A heterochromatin barrier partitions the fission yeast centromere into discrete chromatin domains. Curr Biol 16: 119–129 [DOI] [PubMed] [Google Scholar]
- 57. Gal C, Moore KM, Paszkiewicz K, Kent NA, Whitehall SK (2015) The impact of the HIRA histone chaperone upon global nucleosome architecture. Cell Cycle 14: 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, Tyler JK (2010) Elevated histone expression promotes life span extension. Mol Cell 39: 724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. O'Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J (2010) Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17: 1218–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe . Methods Enzymol 194: 795–823 [DOI] [PubMed] [Google Scholar]
- 61. Blackwell C, Martin KA, Greenall A, Pidoux A, Allshire RC, Whitehall SK (2004) The Schizosaccharomyces pombe HIRA‐like protein Hip1 is required for the periodic expression of histone genes and contributes to the function of complex centromeres. Mol Cell Biol 24: 4309–4320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, Wach A, Philippsen P, Pringle JR (1998) Heterologous modules for efficient and versatile PCR‐based gene targeting in Schizosaccharomyces pombe . Yeast 14: 943–951 [DOI] [PubMed] [Google Scholar]
- 63. Takeda T, Toda T, Kominami K, Kohnosu A, Yanagida M, Jones N (1995) Schizosaccharomyces pombe atf1+ encodes a transcription factor required for sexual development and entry into stationary phase. EMBO J 14: 6193–6208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lyne R, Burns G, Mata J, Penkett CJ, Rustici G, Chen D, Langford C, Vetrie D, Bahler J (2003) Whole‐genome microarrays of fission yeast: characteristics, accuracy, reproducibility, and processing of array data. BMC Genom 4: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Anderson HE, Kagansky A, Wardle J, Rappsilber J, Allshire RC, Whitehall SK (2010) Silencing mediated by the Schizosaccharomyces pombe HIRA complex is dependent upon the Hpc2‐like protein, Hip4. PLoS ONE 5: e13488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nicol JW, Helt GA, Blanchard SG Jr, Raja A, Loraine AE (2009) The integrated genome browser: free software for distribution and exploration of genome‐scale datasets. Bioinformatics 25: 2730–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Maruyama H, Harwood JC, Moore KM, Paszkiewicz K, Durley SC, Fukushima H, Atomi H, Takeyasu K, Kent NA (2013) An alternative beads‐on‐a‐string chromatin architecture in Thermococcus kodakarensis . EMBO Rep 14: 711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome‐wide expression patterns. Proc Natl Acad Sci USA 95: 14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Partridge JF, DeBeauchamp JL, Kosinski AM, Ulrich DL, Hadler MJ, Noffsinger VJ (2007) Functional separation of the requirements for establishment and maintenance of centromeric heterochromatin. Mol Cell 26: 593–602 [DOI] [PubMed] [Google Scholar]
- 71. Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc 1: 179–185 [DOI] [PubMed] [Google Scholar]
- 72. Lin MF, Lucas HC, Shmueli G (2013) Too big to fail: large samples and the P‐value problem. Inform Syst Res 24: 906–917 [Google Scholar]
- 73. Pearson WR, Lipman DJ (1988) Improved tools for biological sequence comparison. Proc Natl Acad Sci USA 85: 2444–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Script EV1
Review Process File
Source Data for Figure 2
Source Data for Figure 3