

Abstract
During DNA replication, thousands of replication origins are activated across the genome. Chromatin architecture contributes to origin specification and usage, yet it remains unclear which chromatin features impact on DNA replication. Here, we perform a RNAi screen for chromatin regulators implicated in replication control by measuring RPA accumulation upon replication stress. We identify six factors required for normal rates of DNA replication and characterize a function of the bromodomain and PHD finger‐containing protein 3 (BRPF3) in replication initiation. BRPF3 forms a complex with HBO1 that specifically acetylates histone H3K14, and genomewide analysis shows high enrichment of BRPF3, HBO1 and H3K14ac at ORC1‐binding sites and replication origins found in the vicinity of TSSs. Consistent with this, BRPF3 is necessary for H3K14ac at selected origins and efficient origin activation. CDC45 recruitment, but not MCM2‐7 loading, is impaired in BRPF3‐depleted cells, identifying a BRPF3‐dependent function of HBO1 in origin activation that is complementary to its role in licencing. We thus propose that BRPF3‐HBO1 acetylation of histone H3K14 around TSS facilitates efficient activation of nearby replication origins.
Keywords: BRPF, DNA replication, HBO1, H3K14ac, origin activation
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; DNA Replication, Repair & Recombination; Post-translational Modifications, Proteolysis & Proteomics
Introduction
In S phase of the cell cycle, the genome must be faithfully duplicated in order to maintain genome integrity across cell generations. To ensure complete replication of the entire human genome, replication initiates from thousands of origins in a well‐defined manner. Tight regulation of replication initiation is central to maintain genome integrity and prevent tumorigenesis (Alver et al, 2014). Origin hyper‐activation in response to oncogenic signalling can drive genome instability (Halazonetis et al, 2008; Alver et al, 2014), and cell type‐specific deficits in origins at so‐called fragile sites can contribute to DNA breakage and chromosomal rearrangements (Letessier et al, 2011; Debatisse et al, 2012).
Initiation of DNA replication can be divided into three tightly regulated steps. First, ORC (origin recognition complex) binding specifies replication origins as cells exit mitosis (Bell & Dutta, 2002; MacAlpine & Bell, 2005; Mechali, 2010; Alabert & Groth, 2012). Second, in a process termed licencing, pre‐RCs (pre‐replication complexes) are assembled via CDT1‐ and CDC6‐dependent loading of the hexameric MCM2‐7 complex (Bell & Dutta, 2002; Remus & Diffley, 2009; Mechali, 2010). Third, in S phase, origins are activated by the dual action of CDK and DDK kinases that by phosphorylation of the pre‐RC and accessory fork components recruit CDC45 and the GINS complex to activate the replicative helicase (Labib & Gambus, 2007; Remus & Diffley, 2009). To ensure that initiation happens only once per cell cycle, these events are restricted to specific phases of the cell cycle. However, to provide backup sites for replication initiation in case nearby replication forks are compromised, a large number of excess origins are licensed during the G1 phase (Woodward et al, 2006; Ge et al, 2007). Only a fraction of these origins are activated during S phase, and the rest remain dormant and are passively replicated by forks arriving from nearby active origins (reviewed in Alver et al, 2014). If fork progression is impaired due to DNA damage or reduced dNTP supply, nearby dormant origins can be activated. It is not clear what determines whether an origin is dormant or not. In part, which origins are activated or remain dormant appear to be stochastic, reflecting the intrinsic inefficiency of origin activation. Single‐molecule analysis has established that checkpoint kinases control origin firing and dormant origin activation (Ge et al, 2007; Maya‐Mendoza et al, 2007; Petermann et al, 2010b). Whether chromatin context directly influences origin efficiency remains unclear (Mechali, 2010), but chromosomal architecture is important for confining origin firing to a distinct spatiotemporal pattern during S phase (Gilbert et al, 2010; Pope et al, 2010). This replication‐timing programme is cell type specific, and chromatin context provides a potential key to understand cell type‐specific origin usage and thus replication timing (Gilbert et al, 2010; Pope et al, 2010). In general, active regions of the genome replicate early and silenced domains replicate late (Goren & Cedar, 2003). Recently, it was shown that replication timing correlates directly with the three‐dimensional organization of the genome in topological domains (Pope et al, 2014), which translates into open and closed chromatin compartments.
Mounting evidence indicates that chromatin environment contributes to origin specification and licensing. Genomewide analysis has revealed that regions of low nucleosome occupancy are preferred binding sites for ORC (Lubelsky et al, 2011). Features typical of active chromatin such as histone acetylation, the histone variant H3.3 and recruitment of chromatin remodellers generally demarcate ORC‐binding sites (MacAlpine et al, 2010; Mechali, 2010). Methylation of histone H4 at lysine 20 can also facilitate ORC1 recruitment by serving as a recognition site for ORC1 and co‐factors (Beck et al, 2012; Kuo et al, 2012). Subsequently, acetylation of histone H4 at lysine 5, 8 and 12 by the lysine acetyltransferase (KAT) HBO1 contributes to MCM2‐7 loading (Miotto & Struhl, 2010). HBO1 is required for DNA replication and interacts with ORC1, Cdt1 and MCM2‐7 (Iizuka & Stillman, 1999; Burke et al, 2001; Doyon et al, 2006; Miotto & Struhl, 2008, 2010). Notably, artificial tethering of the H4K20me1 enzyme SET8 and HBO1 can promote recruitment of ORC and MCM2‐7 (Aggarwal & Calvi, 2004; Tardat et al, 2010; Chen et al, 2013).
The HBO1 protein was initially characterized as the catalytic subunit of the main histone H4 acetyltransferase complex in human 293T cells, and it is enriched near the transcription start site of active genes (Doyon et al, 2006; Avvakumov et al, 2012). In this complex, PHD finger‐containing subunits JADE1/2/3(PHF15/16/17) and ING4/5 enable HBO1 activity towards chromatin substrates and H3K4me3‐bearing nucleosomes, respectively (Saksouk et al, 2009). It was recently discovered that HBO1 also exists in native complexes containing BRPF1/2 scaffold subunits instead of JADEs, shifting the specificity of chromatin acetylation to histone H3 instead of H4 (Mishima et al, 2011; Lalonde et al, 2013, 2014). HBO1 knockout in mice led to a dramatic reduction of histone H3 acetylation at K14 and severe defects in embryonic development (Kueh et al, 2011). BRPF1 has been shown to be important for brain development (You et al, 2015a,c), and deletion of the mouse BRPF1 gene causes embryonic lethality (You et al, 2015b). Similar to HBO1‐knockdown erythroblasts, BRPF2‐deficient mice exhibited impaired global H3K14 acetylation and decreased H3K14 acetylation at the promoters of dysregulated erythroid developmental regulator genes (Mishima et al, 2011). However, the function of BRPF3 remains uncharacterized. Given that H4 acetylation by HBO1‐JADE is important for DNA replication (Doyon et al, 2006; Miotto & Struhl, 2008, 2010; Swarnalatha et al, 2012), whether BRPFs function with HBO1 in DNA replication is unclear.
To uncover novel chromatin‐based mechanisms controlling DNA replication, we established a siRNA screen based on the phenotype of cells lacking the ASF1 histone H3‐H4 chaperone. ASF1‐depleted cells fail to expose single‐stranded DNA (ssDNA) at replication forks upon treatment with hydroxyurea (HU) (Groth et al, 2007). ssDNA accumulates as a result of uncoupling between the replicative helicase and DNA polymerases (Walter & Newport, 2000; Pacek & Walter, 2004; Cimprich & Cortez, 2008; Toledo et al, 2013), representing a hallmark of stalled replication forks as well as a very robust read‐out for screening. We reasoned that lack of ssDNA accumulation in S‐phase cells treated with HU could result from either a DNA unwinding defect or a reduced number of active forks (Groth et al, 2007; Mejlvang et al, 2014). Using a customized library targeting chromatin factors, we identified 20 factors required for HU‐induced ssDNA accumulation. A subset of these was also required for efficient DNA replication, including the multi‐domain scaffold protein BRPF3. We show that BRPF3 forms a complex with HBO1, which specifically acetylates H3K14 and is required for efficient activation of licenced origins in S phase.
Results
siRNA screen identifies candidate genes involved in DNA replication
To identify chromatin factors involved in regulation of DNA replication, we performed a siRNA screen for proteins that are required for HU‐induced ssDNA exposure similar to the ASF1 histone chaperone (Groth et al, 2007). We used a customized library containing three independent siRNAs targeting 219 putative chromatin factors (Table EV1). To monitor ssDNA exposure in S‐phase cells, we used a reporter cell line stably expressing GFP‐RPA1 together with RFP‐PCNA (Mejlvang et al, 2014). To focus on DNA unwinding at stalled replication forks, rather than collapsed forks, we treated cells short term with HU (2 h) (Petermann et al, 2010a). Furthermore, we removed soluble proteins prior to fixation to measure chromatin‐bound GFP‐RPA1 and RFP‐PCNA. siRNAs targeting ASF1 (a and b) were used as a positive control, efficiently inhibiting GFP‐RPA1 accumulation as shown previously (Fig 1A, Groth et al, 2007; Mejlvang et al, 2014). We performed the screen in two biological replicas and ranked all genes in our library by the collective activities of the multiple siRNAs using redundant siRNA activity (RSA) analysis (Konig et al, 2007; Beck et al, 2010). The top hits were derived by the Fisher method, ranking genes with combined P‐values (Kost & McDermott, 2002; Menzel et al, 2011) (Fig 1B and Table EV2). On this basis along with available literature, we selected 20 genes for further validation and assembled a sub‐library containing the two highest scoring siRNAs targeting each candidate. We then carried out systematic functional analyses by high‐content imaging. We focused on S‐phase cells, and therefore, only PCNA‐positive cells were included in the following assays. Firstly, we verified that siRNA depletion of our candidate genes attenuated GFP‐RPA1 accumulation in response to HU (Fig 1C). siRNAs against ASF1 (a and b) and CDC45 were included as positive controls (Walter & Newport, 2000; Pacek & Walter, 2004; Groth et al, 2005, 2007; Toledo et al, 2013). Given that lack of HU‐induced RPA1 accumulation may result from a cell cycle defect such as G1 arrest, we also quantified PCNA‐positive cells. However, the proportion of PCNA‐positive cells was largely unchanged upon siRNA depletion of the candidate genes (Appendix Fig S1A), arguing that they are either not required for S‐phase entry or that the siRNA knockdown is partial. Importantly, this ruled out that impaired ssDNA accumulation was secondary to a cell cycle arrest, supporting that the 20 identified genes could play a role in DNA replication.
Figure 1. siRNA screen identifies BRPF3 as a novel regulator of DNA replication.
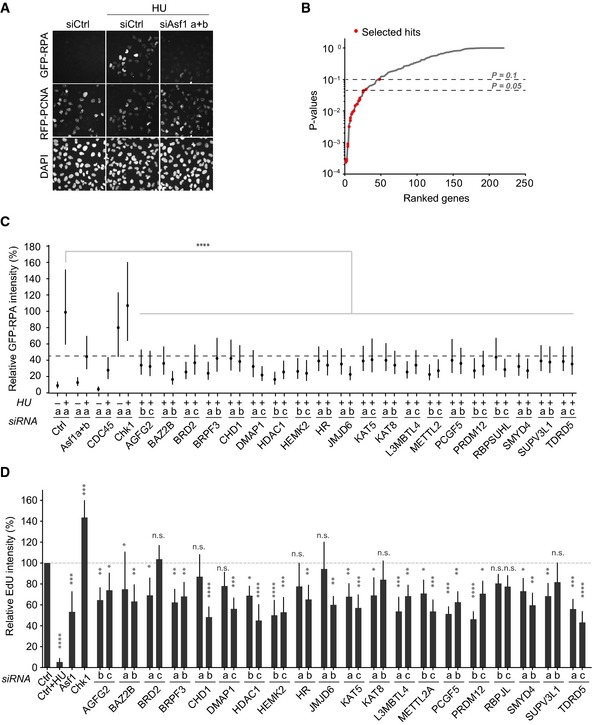
- Representative images of controls from the siRNA screen for HU‐induced RPA accumulation. GFP‐RPA1 and RFP‐PCNA reporter cells were transfected with siRNAs and 48 h later treated with HU (3 mM) for 2 h prior to pre‐extraction, fixation and imaging.
- Gene ranking based on siRNA scores from two independent screens. A total of 219 genes were targeted by three individual siRNAs and ranked based on P‐values derived from RSA and Fisher's analysis. Selected hits are highlighted.
- Validation of selected genes by single‐cell analysis of GFP‐RPA1 intensity in pre‐extracted cells. Only S‐phase cells positive for RFP‐PCNA were analysed. Median with interquartile range of relative intensity per cell is shown, n > 4,000. Mann–Whitney: ****P < 10−4, n.s. non‐significant. One representative experiment out of two biological replicas is shown.
- Analysis of DNA synthesis rate. Cells were pulsed 15 min with EdU, and EdU intensity was analysed in S‐phase cells positive for RFP‐PCNA. EdU incorporation is shown relative to siCtrl. The average and standard deviation from three biological replicas is shown. n > 2,000. ANOVA t‐tests: ****P < 10−4, ***P < 10−3, **P < 10−2, *P ≤ 0.05; n.s., non‐significant.
Next, we measured the rate of DNA synthesis by quantifying ethynyl deoxyuridine (EdU) incorporation in siRNA‐depleted cells. Here, siRNA against ASF1 (a and b) served as a positive control, repressing DNA replication as previously reported (Groth et al, 2007). Although the degree of inhibition varied, we found that several of the siRNAs significantly impaired EdU incorporation (Fig 1D). By correlating this phenotype with knockdown efficiency measured by RT‐qPCR (Appendix Fig S2A–O), we identified six genes acting as positive regulators of DNA replication (AGFG2, BRPF3, HEMK2, KAT5, KAT8 and PRDM12). However, reduced DNA replication could be a secondary effect of high loads of DNA damage. To address this point, we screened for γH2AX, a hallmark of DNA damage signalling (Harrison & Haber, 2006; Harper & Elledge, 2007). However, none of the siRNAs significantly increased the γH2AX level, arguing that the identified factors are required for normal rates of DNA replication independent of DNA damage signalling (Appendix Fig S1B).
BRPF3 is required for DNA replication and H3K14 acetylation
We decided to focus on the high‐ranking candidate, BRPF3 (bromodomain‐ and PHD finger‐containing protein 3), whose function is largely unaddressed. The BRPF family of proteins contains typical domains found in chromatin proteins, including PHD fingers, a bromodomain and a chromo/Tudor‐related PWWP domain (Doyon et al, 2006). BRPF3, along with its paralogs BRPF1 and BRPF2, was initially identified as potential scaffold proteins in large MOZ/MORF KAT complexes containing ING5 (Doyon et al, 2006). From our siRNA screen data, we noted that BRPF1 and BRPF2 were not required for ssDNA formation in response to HU (Appendix Fig S3A), suggesting that we could separate the function of these paralogs. Consistent with this, depletion of BRPF1 and BRPF2 with verified siRNAs did not affect DNA replication, whereas depletion of BRPF3 with multiple independent siRNAs reduced DNA synthesis by 30% without activating a DNA damage response (γH2AX, P‐RPA and P‐Chk1) (Fig 2A, Appendix Figs S3B and S4A). This reduction in DNA replication was highly consistent between different BRPF3 siRNAs and significant regardless of whether EdU intensities were compared across the total cell population or in S‐phase cells (Appendix Fig S3C and D). Furthermore, the replication defect could be partially rescued by exogenous expression of siRNA‐resistant BRPF3 (Fig 2B and Appendix Fig S3E). Thus, BRPF3, but not BRPF1 or BRPF2, regulates DNA replication.
Figure 2. BRPF3‐HBO1 regulates DNA replication and acetylates H3K14ac.
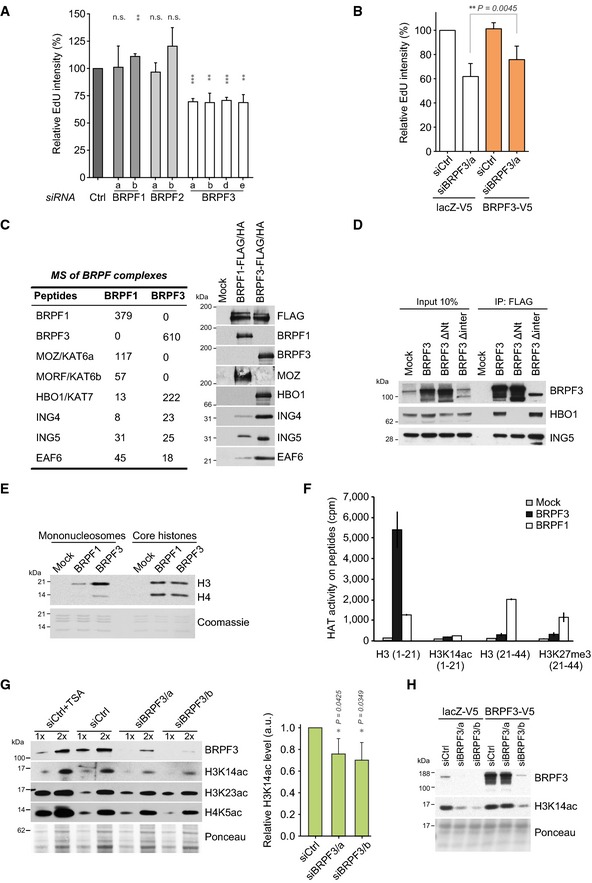
- DNA replication measured by EdU incorporation. RPF‐PCNA reporter cells were transfected with the indicated siRNAs and pulsed with EdU for 20 min. S‐phase cells positive for RFP‐PCNA were analysed. n > 150. Error bars, SD; n = 3 biological replicas. One‐sample t‐test, ***P < 0.001, **P < 0.01, n.s., non‐significant.
- Complementation analysis of DNA synthesis. Stable cell lines expressing BRPF3 resistant to the BRPF3/a siRNA and control (lacZ‐V5) were siRNA transfected and analysed for EdU incorporation as in (A). Error bars, SD; n = 6 biological replicas. Two‐tailed t‐test, **P < 10−2.
- Comparison of native BRPF1 and BRPF3 complexes purified from K562 cells. (left) Mass spectrometric analysis (spectral counts/total peptides identified) and (right) verification of associated protein by Western blot.
- Immunoprecipitation analysis of BRPF3 deletion mutants. One representative experiment out of two is shown.
- Histone acetyltransferase assay (HAT) with purified BRPF1/3 complexes on free histones and mononucleosomes.
- HAT assay with purified BRPF1/3 complexes on H3 peptides unmodified or acetylated (ac)/methylated (me) at K14 and K27, respectively. Mean ± SD is shown, n = 4.
- Analysis of histone acetylation in BRPF3‐depleted cells. (left) Western blot of siRNA‐treated U‐2‐OS cells. TSA treatment (1 h) was included as a positive control. 2x, double amount of extract loaded as in 1x. (right) H3K14 acetylation levels quantified relative to total H3. Error bars, SD; n = 3 biological replicates. One‐sample t‐test.
- Complementation analysis of H3K14ac. Total cell extracts of siRNA‐treated lacZ‐V5 and BRPF3‐V5 (siBRPF3/a resistant) expressing cells were analysed by Western blotting. One representative experiment out of four biological replicas is shown.
To gain molecular insight into the different function of the BRPF paralogs, we isolated BRPF1 and BRPF3 complexes from cells carrying a single‐copy ZFN integrated transgene and identified interaction partners by mass spectrometry. This revealed that the two paralogs were part of distinct KAT complexes; BRPF3 associated exclusively with HBO1, while BRPF1 formed complex with MOZ/MORF as described previously (Doyon et al, 2006; Lalonde et al, 2013) (Fig 2C). These results were confirmed by co‐immunoprecipitation analysis in U‐2‐OS cells (Fig 2D, Appendix Fig S3F and G). Analysis of BRPF3 deletion mutants showed that a small N‐terminal region was required for HBO1 binding, while deletion of an internal region specific to BRPF3 (BRPF3Δinter) was dispensable (Fig 2D and Appendix Fig S3H). Of note, both BRPF3 mutants interact with ING5 (Fig 2C and D), consistent with previous data showing that the BRPF scaffold binds the KAT and ING proteins independently (Doyon et al, 2006; Ullah et al, 2008). HBO1 is part of the MYST family of KATs and has previously been linked to acetylation of histone H4 K5/8/K12, and histone H3 K14 and K23 (Doyon et al, 2006; Kueh et al, 2011; Mishima et al, 2011; Lalonde et al, 2013). In particular, it was recently proposed that the scaffold partner protein could determine the target specificity of HBO1 (Lalonde et al, 2013). We thus addressed the specificity of the BRPF3‐HBO1 complex. The purified BRPF3 complex could acetylate both histones H3 and H4 when presented as core histones in solution, but on a mononucleosome substrate, the BRPF3 complex preferentially acetylated histone H3 (Fig 2E) as reported previously for the BRPF1 complex (Lalonde et al, 2013). BRPF3 complexes specifically acetylated H3 peptides spanning K4, K9 and K14 (1–21), but had no activity on an identical peptide already acetylated at K14 (Fig 2F). BRPF1 complexes showed activity against K14 in the same assay, but, in contrast to BRPF3, also acetylated peptides spanning K23 and K27 (21–44) regardless of whether K27 was blocked by methylation (Fig 2F). This shows that BRPF3 complexes preferentially target H3K14ac in vitro, and consistent with this, H3K14ac (but not H3K23ac or H4K5ac) was significantly reduced upon BRPF3 depletion in vivo (Fig 2G). Furthermore, the loss of H3K14ac could be rescued by expression of siRNA‐resistant BRPF3 (Fig 2H). BRPF1 complexes could target both H3 K14 and K23 in vitro, but depletion of BRPF1 mainly affected H3K23ac levels in vivo (Appendix Fig S4A). Collectively these data suggest that acetylation of H3K14 by BRPF3‐HBO1 could be important for DNA replication. In complex with another scaffold, JADE1, HBO1 can acetylate histone H4 (Doyon et al, 2006; Foy et al, 2008) and this has been shown to facilitate origin licensing (Miotto & Struhl, 2010). However, given that H4K5ac is not altered upon BRPF3 knockdown, we anticipated that the function of the BRPF3‐HBO1 complex would be distinct from the role of HBO1‐JADE1 in licensing.
BRPF3 is regulating replication origin activation
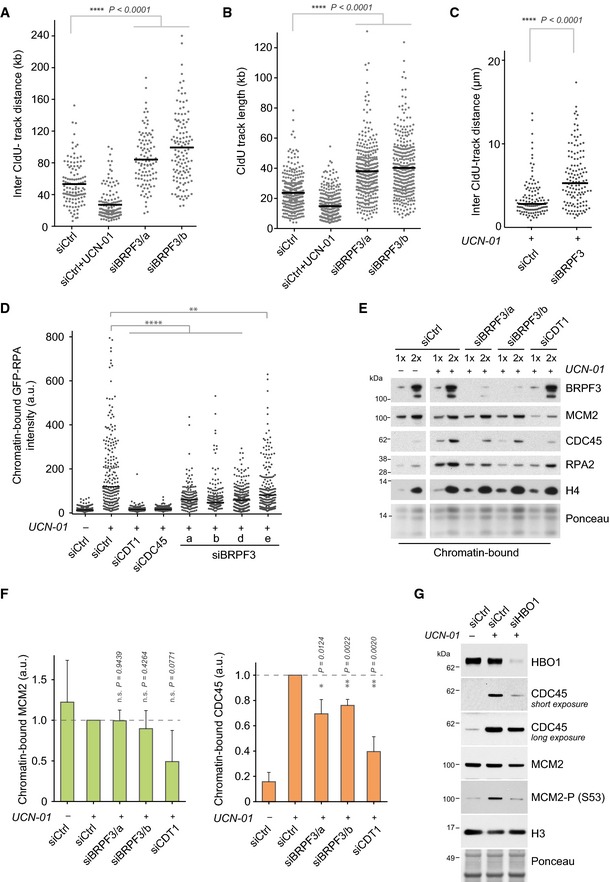
To understand whether BRPF3 regulates DNA replication at the level of elongation or new origin firing, we investigated replication at the single‐molecule level by molecular DNA combing (Mejlvang et al, 2014). Newly synthesized DNA was labelled by consecutive pulses of IdU and CldU to allow identification of independent forks by selection of CldU tracks flanked by IdU. First, we measured inter‐track distances to evaluate the number of active origins and used the Chk1 inhibitor 7‐hydroxystaurosporine (UCN‐01) as a control for increased origin firing. Chk1 inhibition activates dormant origins and consistently inter‐track distances were reduced (Ge et al, 2007; Maya‐Mendoza et al, 2007; Petermann et al, 2010b) (Fig 3A). In contrast, BRPF3‐depleted cells showed significantly longer inter‐track distances as compared to control cells (Fig 3A). Concomitantly, the length of CIdU‐labelled tracks was increased in cells lacking BRPF3 (Fig 3B and Appendix Fig S4B) and similar results were observed by DNA fibre assay (Appendix Fig S4C). Collectively, these data suggest that BRPF3 depletion reduces origin firing and, likely as part of a compensatory mechanism (Blow et al, 2011), increases fork elongation rate. This phenocopies cdc7 mutants in yeast, which show reduced origin firing, fewer but faster forks and reduced Rad53 signalling (Zhong et al, 2013).
Figure 3. BRPF3 is required for replication origin firing.
-
A, BSingle‐molecule analysis of DNA replication by DNA combing of siRNA‐transfected cells pulse‐labelled with IdU (10 min) and CIdU (20 min). (A) Distribution of inter‐track distances and (B) size distribution of CldU track length. Bars represent the median. n > 100 (A), n > 300 (B). Mann–Whitney: ****P < 0.0001. One representative experiment out of two biological replicas is shown.
-
CDistribution of intertrack distances measured by IdU/CIdU pulse labelling and DNA fibre analysis. Bars represent the median. n > 110. Mann–Whitney: ****P < 10−4. One representative experiment out of two biological replicas is shown.
-
DScatter plot of chromatin‐bound GFP‐RPA1 intensity per cell. GFP‐RPA1 and RFP‐PCNA reporter cells were transfected with the indicated siRNAs and treated for 2 h with UCN‐01 (300 nM) prior to pre‐extraction and fixation. GFP‐RPA1 levels were measured in PCNA‐positive cells. Lines represent medians. n > 150. t‐tests: ****P < 10−4, **P < 0.01; a.u., arbitrary unit.
-
EAnalysis of replication factor loading. Soluble proteins from siRNA‐transfected cells treated as in (D) where removed by CSK‐T extraction and chromatin pellets analysed by Western blotting. One representative experiment out of three independent biological replicas is shown.
-
FQuantification of chromatin‐bound MCM2 (left) and CDC45 (right) from the three independent experiments described in (E). MCM2 and CDC45 levels were normalized to histone H4 levels and shown relative to untreated control. t‐tests: **P < 0.01, *P < 0.05; n.s., non‐significant.
-
GAnalysis of chromatin‐bound proteins in HBO1‐depleted cells as in (E). One representative experiment out of two biological replicas is shown.
To further dissect the function of BRPF3 in replication initiation, we examined fork density in response to UCN‐01 treatment, which triggers origin hyper‐activation including dormant origin firing (Ge et al, 2007; Maya‐Mendoza et al, 2007; Petermann et al, 2010b). Inter‐track distances were substantially increased in BRPF3‐depleted cells treated with UCN‐01 as compared to control cells (Fig 3C and Appendix Fig S4D), indicating that dormant origins were more refractory to activation. We also established a complementary assay to measure origin hyper‐activation in response to UCN‐01 treatment based on quantification of GFP‐RPA1 accumulation on ssDNA (Syljuasen et al, 2005; Maya‐Mendoza et al, 2007; Petermann et al, 2010b) (Fig 3D and Appendix Fig S4E). In this assay, depletion of the initiation factors CDT1 and CDC45 as well as inhibition of CDK activity by roscovitine blocked GFP‐RPA1 accumulation (Fig 3D and Appendix Fig S4E). In contrast, repression of DNA replication without changing origin density, as can be achieved by depletion of FLASH or SLBP to block histone supply (Mejlvang et al, 2014), did not affect GFP‐RPA1 accumulation in response to UCN‐01 (Appendix Fig S4E). Importantly, BRPF3 depletion by four independent siRNAs significantly reduced accumulation of chromatin‐bound GFP‐RPA1 in this setting, corroborating that BRPF3 regulates dormant origins firing (Fig 3D). We next proceeded to dissect molecularly at which step in replication initiation BRPF3 functions. We analysed the loading of MCM2 and CDC45 onto chromatin, as markers of origin licensing and activation, respectively (Remus & Diffley, 2009; Mechali, 2010). RPA2 is recruited after CDC45‐dependent activation of the MCM2‐7 helicase and was included to follow DNA unwinding and for comparison with our imaging‐based GFP‐RPA1 assay (Fig 3D). Origin hyper‐activation in response to UCN‐01 triggered a substantial increase in chromatin‐bound CDC45 and RPA2, whereas MCM2 loading was largely unaffected (Fig 3E and F). Depletion of CDT1, required for MCM2‐7 loading during Pre‐RC assembly, reduced the levels of chromatin‐bound MCM2 and CDC45 as expected. In contrast, BRPF3 depletion impaired CDC45 and RPA loading without affecting MCM2 levels on chromatin (Fig 3E and F). Loss of HBO1 also reduced CDC45 loading on chromatin upon UCN‐01 treatment (Fig 3G). In addition, HBO1 depletion also reduced the level of MCM2 and DDK phosphorylated MCM2 on chromatin, consistent with previous reports (Miotto & Struhl, 2008, 2010). Taken together, these data indicate that the BRPF3‐HBO1 complex regulates the ability of licenced origins to be activated. As anticipated, this function of BRPF3‐HBO1 is distinct from and complementary to the previously described role of HBO1‐JADE1 in origin licencing, in which HBO1‐dependent acetylation of histone H4 facilitates loading of the MCM2‐7 complex in G1 (Iizuka et al, 2006; Miotto & Struhl, 2008, 2010).
BRPF3‐HBO1 acetylates H3K14 in chromatin surrounding replication origins
Based on the data above, we envisioned that the BRPF3‐HBO1 complex facilitates origin activation through a mechanism that involves H3K14 acetylation. To investigate this model, we performed chromatin immunoprecipitations followed by high‐throughput sequencing (ChIP‐seq) to precisely map BRPF3‐binding sites on the human genome. We included chromatin samples from asynchronous cells as well as cells synchronized in G1/S by mimosine and cells released into S in the presence of HU to track changes during replication. Over 12,000 BRPF3‐bound regions were identified in all conditions with no substantial differences between different cell cycle stages (Fig 4A, Appendix Fig S5A and Table EV3). We found BRPF3 at 8 well‐characterized replication origins, including the MCM4, Lamin B2, LUC7L, MEN1, PIP5K1A, TOP1, SNHG12 and AXIN1 origins (Fig 4A and Appendix Fig S5A). Using published ChIP‐seq analysis of ORC1 (Dellino et al, 2013), HBO1 (Avvakumov et al, 2012) and H3K14ac (accession number: GSM521881) in human cells, we see a strong correlation between occupancy of BRPF3, ORC1, HBO1 and H3K14ac levels (Fig 4A–D and Appendix Fig S5A). We also compared the occupancy of BRPF3, HBO1 and H3K14ac with active origins identified by short nascent strand (SNS) purification (Besnard et al, 2012; Picard et al, 2014) (Appendix Fig S5B and C). BRPF3, HBO1 and H3K14ac were highly enriched at active origins, showing a more prominent overlap than for putative origins identified by ORC1 (68% of BRPF3 peaks overlap with SNS sites, while 29% overlap with ORC1) (Fig 4B and Appendix Fig S5B). Moreover, 86% of BRPF3‐binding sites in asynchronous cells overlap with previously identified HBO1 sites in the same cell line (Fig 4B) (Avvakumov et al, 2012), supporting a function of the BRPF3‐HBO1 acetyltransferase complex in regulation of origin activation. Consistent with this notion, we found that BRPF3 and HBO1 depletion reduced the level of H3K14ac at several well‐characterized origins (Fig 4E).
Figure 4. BRPF3‐HBO1 acetylates H3K14 at replication origins.
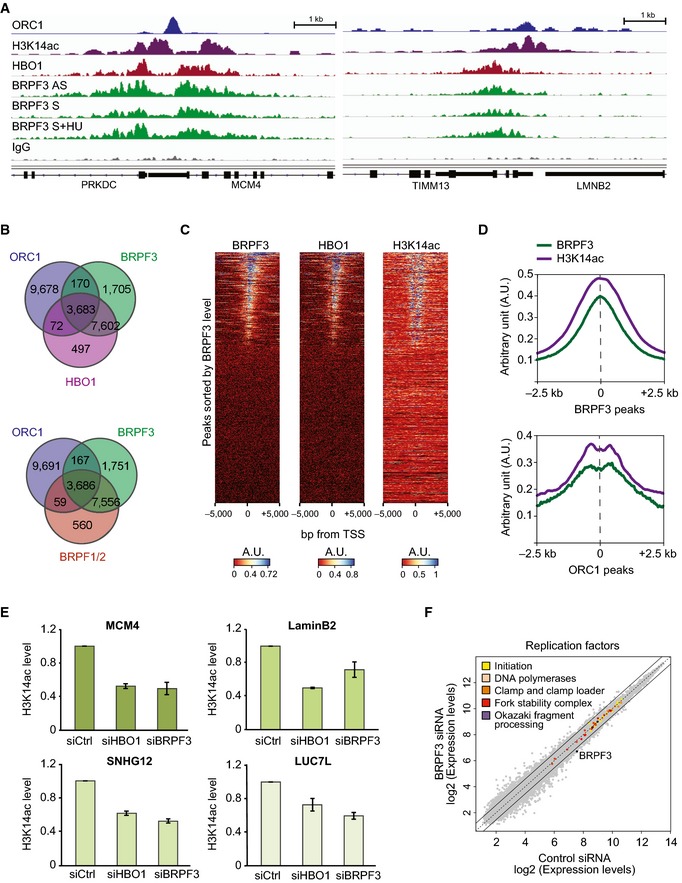
- ChIP‐seq analysis of BRPF3 binding on the human genome. BRPF3 enrichment signal obtained by ChIP‐seq with asynchronous (AS), synchronized (S) and released into HU (S+HU) RKO cells, on the MCM4 (left) and Lamin B2 (right) replication origins. Signals were compared to the published enrichment signals for ORC1 in HeLa cells (Dellino et al, 2013; GSE37583), HBO1 in RKO cells (Avvakumov et al, 2012; GSE33221) and H3K14ac in IMR90 cells (GSM521881). The negative control IgG signal shown is from the S+HU chromatin.
- Venn diagrams illustrating the overlap of genome binding sites between (top) BRPF3, HBO1 and ORC1, and (bottom) BRPF3, BRPF1/2 and ORC1 in human cells.
- Heatmaps of BRPF3, HBO1 and H3K14ac ChIP‐seq signal on ± 5 kb surrounding the TSS of genes. Genes were sorted by BRPF3 abundance from high to low. A.U., arbitrary unit.
- Average profiles of BRPF3 and H3K14ac ChIP‐seq signal on ± 2.5 kb around the centre of (top) BRPF3 and (bottom) ORC1 genome binding sites.
- ChIP–qPCR analysis of H3K14ac on well‐characterized replication origins. H3K14ac enrichments measured relative to IgG control are shown relative to control siRNA‐treated cells. Error bars, range; n = 2 biological replicas.
- Expression levels of DNA replication factors were not altered by the lack of BRPF3. Each point represents the expression levels of a gene in control and BRPF3‐depleted cells (GSE65065). Replication factors were classified as described previously (Alabert et al, 2014). Total genes (grey), DNA replication factors (colours) and BRPF3 (black) expression levels are shown. Continuous black lines mark 1.5‐fold change in expression levels.
BRPF3‐ and HBO1‐binding sites along with H3K14ac levels are highly enriched at transcription start sites (TSS) (Fig 4C). This suggests that BRPF3‐HBO1 would mainly regulate the many replication origins found at TSS (Miotto & Struhl, 2008, 2010; Tardat et al, 2010; Besnard et al, 2012; Dellino et al, 2013; Picard et al, 2014). This is in part specific for BRPF3, as BRPF1/2‐binding sites are also highly enriched at TSS (Lalonde et al, 2013, Fig 4B) without influencing DNA replication (Fig 2A). However, given that BRPF3 overlaps both TSS as well as replication origins, the genomewide analysis cannot separate functions in transcription and DNA replication. To address the possibility that BRPF3 is controlling the expression of replication factors, we performed transcriptome analysis on BRPF3‐depleted cells. Overall BRPF3 depletion only affected 4.3% of the transcripts of the genome with 2.3% downregulated and 2% upregulated (Table EV4). Importantly, Gene Ontology (GO) analysis of genes with significantly expression changes did not identify categories related to DNA replication (Appendix Fig S5D and Table EV5), and focused analysis of characterized replication factor genes (Alabert et al, 2014) showed no significant expression differences upon BRPF3 loss (Fig 4F). Collectively, our genomewide analyses and interrogation of H3K14ac at origins in BRPF3‐depleted cells support the view that H3K14ac by BRPF3‐HBO1 demarcates origins in the vicinity of TSS to influence origin activation.
BRPF3 depletion protects cells against replication stress‐induced DNA damage
We initially identified BRPF3 due to its role in ssDNA formation upon HU treatment (Fig 1C and Appendix Fig S3A). ssDNA coated with RPA, along with primer‐template junctions, is required for activation of the ATR‐Chk1 checkpoint signalling pathway (Zou & Elledge, 2003; Cimprich & Cortez, 2008; Van et al, 2010). We thus predicted that cells would show reduced checkpoint signalling. Consistent with this, BRPF3 depletion reduced Chk1 activation and accumulation of DNA damage markers (γH2AX and P‐RPA) in response to short‐ and long‐term HU treatment (Fig 5A). This suggested that either BRPF3 is required for checkpoint signalling or BRPF3‐depleted cells simply sustain less DNA damage because fewer forks are active and/or dormant origin firing is restricted. The latter would be consistent with recent reports that dormant origin firing and competition for RPA contribute to replication fork collapse (Toledo et al, 2013; Dungrawala et al, 2015). Furthermore, this scenario predicts that BRPF3 depletion could be advantageous in the face of replication stress. To test this, we released cells from long‐term HU treatment and followed cell cycle progression by measuring EdU incorporation and DNA content. After release, BRPF3‐depleted cells synthesized DNA more efficiently and progressed faster through the cell cycle as compared to control cells (Fig 5B and C, Appendix Fig S6A and B). Taken together, these results argue that restriction of origin firing can be an advantage under high doses of genotoxic stress.
Figure 5. Lack of BRPF3 improves recovery after replication stress.
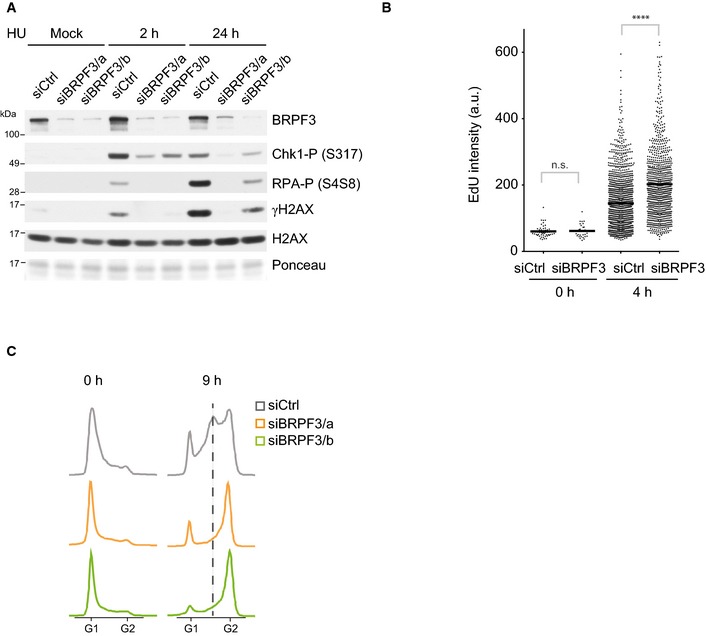
-
AWestern blot of DNA damage signalling in BRPF3‐depleted cells. siRNA‐transfected U‐2‐OS cells were treated with HU for 2 or 24 h. One representative experiment out of two independent biological replicas is shown.
-
B, CRecovery from replication stress. siRNA‐treated cells were treated for 24 h with HU (0 h) and released into normal medium for the indicated times (see also Appendix Fig S6A and B). (B) DNA replication rate measured by EdU incorporation. Cells were pulsed with EdU for 15 min and gated as shown in Appendix Fig S6A to analyse EdU‐positive cells. n > 1,000. Mann–Whitney: ****P < 10−4. (C) Cell cycle progression measured by FACS analysis of DNA content. One representative experiment out of two independent biological replicas is shown.
Discussion
siRNA screen for chromatin regulators of DNA replication
Here, we establish a screen to identify chromatin‐based mechanisms that control DNA replication. We use RPA1 accumulation in response to short‐term replication stress as read‐out for perturbed DNA replication. This read‐out is in our hands more robust than EdU incorporation measurements and, importantly, it is sensitive to alterations in both DNA unwinding (Syljuasen et al, 2005; Groth et al, 2007) fork progression (Mejlvang et al, 2014) and fork density (this work). We identify 20 genes that are required for HU‐induced RPA1 accumulation and show that 6 of these are required to sustain normal rates of DNA synthesis (AGFG2, BRPF3, HEMK2, KAT5, KAT8 and PRDM12). In the course of our investigation, two other hits were also linked to DNA replication [SUPV3L1 and L3MBTL1 (Gurvich et al, 2010; Veno et al, 2011)], further validating our screen. Interestingly, we identify 3 regulators of histone acetylation, BRPF3, KAT5 and KAT8. We provide evidence that BRPF3 forms a complex with HBO1, which acetylates histone H3K14 and is required for efficient origin activation (as discussed below). KAT5 (TIP60) and KAT8 (MOF) can both acetylat e histone H4 at lysine 16. KAT8 is moderately enriched at active replication forks (Alabert et al, 2014), and both KATs play important roles in the DNA damage response (Ikura et al, 2000; Kusch et al, 2004; Sun et al, 2005; Taipale et al, 2005; Murr et al, 2006; Sharma et al, 2010; Krishnan et al, 2011; Gupta et al, 2014). Our screening data argue that it will be important to address how KAT5 and KAT8 influence the replication programme in human cells, also given that H4K16ac is required for early replication of the male X chromosome in Drosophila (Bell et al, 2010; Lubelsky et al, 2014).
HBO1 uses distinct scaffolds to control consecutive steps in replication initiation
We propose that BRPF3 in complex with HBO1 facilitates origin activation through a mechanism that involves H3K14 acetylation (Fig 6). We present several independent lines of evidence in support of this model. First, BRPF3 (not the two paralogs BRPF1/2) is required for HU‐induced RPA1 accumulation and to sustain normal rates of DNA replication. Second, BRPF3 is found in a complex with HBO1, ING4/5 and EAF6, unlike BRPF1 that partners with MOZ. Importantly, BRPF3 directs HBO1 to preferentially acetylate histone H3K14 in vitro and in vivo. Third, genomewide analyses establish that BRPF3, HBO1 and H3K14ac are found at a large fraction of replication origins and, further, H3K14ac is reduced at selected origins upon HBO1 and BRPF3 depletion. Fourth, origin density is significantly reduced in BRPF3‐depleted cells, which also fail to activate dormant origins and load CDC45 onto chromatin upon acute treatment with UCN‐01.
Figure 6. Model of HBO1 function in replication initiation.
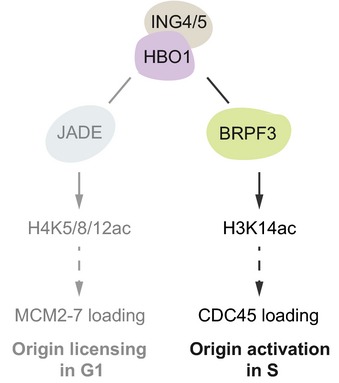
HBO1, as part of two distinct KAT complexes, occupies the chromatin environment surrounding a large fraction of replication origins found close to TSSs. The BRPF3 scaffold directs HBO1 specificity towards H3K14ac, which in turn facilitates the activation of replication origins. Previous work has shown that the HBO1‐JADE complex promotes H4K5/8/12 acetylation and facilitates MCM2‐7 loading (Miotto & Struhl, 2008, 2010). Thus, by partnering with different scaffolds, HBO1‐mediated acetylation of chromatin facilitates two consecutive steps, licensing and activation, in replication initiation.
HBO1 is part of KAT complexes in which interactions with ING4/5 are bridged by a scaffold protein, JADE1/2/3 (Avvakumov et al, 2012) or BRPF1/2/3 (Lalonde et al, 2013; Mishima et al, 2011, this work). Whereas ING proteins together with the scaffold determine the chromatin‐binding properties (Saksouk et al, 2009), the choice of scaffold protein has unexpectedly been found to direct substrate specificity (Lalonde et al, 2013). HBO1‐JADE1 preferentially acetylates the H4 tail (Doyon et al, 2006; Foy et al, 2008), whereas BRPF factors stimulate histone H3 acetylation (Doyon et al, 2006; Lalonde et al, 2013; this work). HBO1 is required for MCM2‐7 loading during replication licencing and has been shown to acetylate H4 at K5, K8 and K12 at well‐characterized mammalian replication origins (Iizuka et al, 2006; Miotto & Struhl, 2008, 2010). We show that BRPF3 is dispensable for MCM2‐7 loading, but important for the subsequent activation of pre‐RCs in S phase. Histone H3K14ac, not H4K5ac, was reduced in cells lacking BRPF3, and accordingly purified BRPF3‐HBO1 complex preferentially acetylated H3K14. In HBO1‐depleted cells H3K14ac, H4K5ac and H4K12ac were reduced, consistent with previous work (Doyon et al, 2006; Foy et al, 2008; Miotto & Struhl, 2010; Kueh et al, 2011; Mishima et al, 2011). Collectively, this argues that the function of HBO1‐BRPF3 in origin firing is distinct from the role of HBO1‐JADE1 in origin licensing (Fig 6), illustrating how subunit composition can directly impact on target specificity and cellular function of KAT enzymes. Intriguingly, these two complexes act in a highly complementary manner, facilitating two consecutive steps in replication initiation. The function of HBO1 in origin activation could thus only be unmasked by targeting BRPF3 as origin licensing would be dominant over and thus conceal functions later in replication.
Our work also shows that the BRPF paralogs have separate functions despite their high similarity and large overlap in chromatin binding (85% of BRPF3 peaks overlap with BRPF1/2). In a recent study, BRPF2 was found to form a complex with HBO1 and be required for H3K14ac in mice (Mishima et al, 2011). Our knockdown data show that in U‐2‐OS cells, BRPF3 is important for global levels of H3K14ac, whereas BRPF2 depletion leads to a modest reduction in acetylation at this site. In contrast, BRPF1 is mainly required for H3K23ac. Consistent with distinct substrate specificity, BRPF1 is almost exclusively part of MOZ KAT complex, whereas BRPF3 associates with HBO1 and not MOZ. Genomewide profiling of these scaffold proteins shows a high degree of overlap. However, differences in BRPF1 and BRPF2 localization cannot currently be resolved due to the absence of specific antibodies, thus we can only conclude that BRPF3 shows a large overlap with BRPF1/2. Nevertheless, depletion of BRPF1 and BRPF2 do not impair DNA replication, demonstrating a clear separation of function for these paralogs. We favour the view that BRPF3‐HBO1 regulates origin activation through a mechanism that involves H3K14ac on nucleosomes surrounding the origin. We cannot exclude that acetylation of a non‐histone substrate is involved or that changes in H3K14ac at other places in the genome could indirectly influence origin firing as reported for yeast rpd3 (Yoshida et al, 2014). However, we show that BRPF3‐HBO1 and H3K14ac occupy the surroundings of a large fraction of origins and demonstrate that BRPF3 and HBO1 are required for H3K14ac at several well‐established origins. In yeast, H3K14ac is associated with efficient origin activation (Unnikrishnan et al, 2010) and targeting of GCN5, which can acetylate H3K14, to late firing origins accelerates activation (Vogelauer et al, 2002). H3K14ac can facilitate recruitment of the RSC chromatin remodeller complex (Kasten et al, 2004), which has been shown to influence recovery of DNA replication after UV (Niimi et al, 2012). Recent models suggest that many MCM2‐7 double hexamers might be loaded at each ORC‐binding site and that these hexamers move away and distribute to form a broad initiation zone (Powell et al, 2015). In this light, it is tempting to speculate that H3K14ac would contribute to a more open chromatin environment that could allow MCM2‐7 spreading and perhaps facilitate clearance of nearby nucleosomes upon pre‐RC activation.
Fork rate and density, a balancing act
Cells lacking BRPF3 had fewer, but faster replication forks (Appendix Fig S6C). This is in line with findings in yeast where reduced origin firing is accompanied by elevated fork elongation rates in mutants lacking the CDC7 kinase (Zhong et al, 2013). Balancing fork rate and density probably serves as a general compensatory mechanism that gives robustness to the replication process (Alver et al, 2014), explaining why DNA synthesis rates are only moderately affected by BRPF3 depletion. In response to replication inhibitors, BRPF3‐depleted cells suffered a reduced load of DNA damage as indicated by a dampened checkpoint response and RPA loading (Appendix Fig S6C). In turn, BRPF3‐depleted cells were more apt to recover and progress through the cell cycle, suggesting that a moderate replication initiation defect can protect cells from replication stress‐induced DNA damage. This corroborates recent reports showing that new origin firing and exhaustion of RPA pools can contribute to fork collapse (Toledo et al, 2013; Dungrawala et al, 2015). However, reduced licensing upon partial depletion of MCM2‐7 leads to increased sensitivity to replication stress, elevated DNA damage signalling and genome instability (Ge et al, 2007; Ibarra et al, 2008). Partial MCM2‐7 depletion mainly impairs dormant origin activation without affecting origin density and fork speed. Our data thus suggest that deficient origin firing and lack of dormant origins are not necessarily comparable. Although mechanistically clearly different, this has some resemblance to the observation that defects in origin licensing in Meier–Gorlin syndrome mutations (ORC1, CDT1, CDC6) do not show genome instability or sensitivity to replication stress (Alver et al, 2014). An important future challenge is thus to resolve how the balance between origin usage, fork speed and dormant origin activation affects susceptibility to replication stresses, because this has significant implications for cancer development and therapy.
Materials and Methods
Cell culture, synchronization and siRNA treatment
U‐2‐OS, 293FT, K562 and RKO cells were grown in DMEM (Gibco) containing 1% Pen/Strep and 10% FBS (Hyclone). The GFP‐RPA1/RFP‐PCNA reporter cell line was described (Mejlvang et al, 2014). Stable cell pools expressing either BRPF3‐V5 or BRPF3‐Flag were generated by lentiviral transduction of U‐2‐OS parental cells. In brief, lentivirus particles were generated in 293FT cells by co‐transfection of the plasmids pAX8 and pCMV‐VSVG along with either pLenti6‐UbC: BRPF3‐V5 siBRPF3‐aR (resistant to siBRPF3/a) or pLenti6‐UbC: lacZ‐V5 or pLenti6‐UbC: BRPF3‐Flag. Virus supernatant supplemented with polybrene (Millipore) was used to infect U‐2‐OS recipient cells. Infected cells were selected in DMEM containing blasticidin (5 μg/ml). Cells were transfected with siRNAs at 100 nM (Sigma) or 20 nM (Ambion) concentration using Oligofectamine or Lipofectamine RNAiMAX (Invitrogen) and harvested after 48 h unless otherwise indicated. Cells were synchronized at the G1/S transition by thymidine (17 h, 2 mM) and released into fresh media containing deoxycytidine (24 μM). Cells were treated as indicated with hydroxyurea (HU, 3 mM).
Robot‐automated siRNA screen
The automated screen was performed using a liquid‐handling station (STAR; Hamilton Robotics). Reporter U‐2‐OS cells expressing GFP‐RPA1 and RFP‐PCNA were reversely transfected with a siRNA library (three independent siRNAs per gene) targeting the human chromatin modifiers and remodellers (219 genes, Applied Biosystems). In brief, 4 μl siRNA was added to 2.5 μl OptiMEM (Invitrogen) to each well of a 384‐well plate (Corning). In addition, a 6.4 μl OptiMEM/0.1 μl Oligofectamine mix was added and let to incubate for 20 min. Subsequently, 27 μl of cells was added to give a total cell density of 3,300 cells per well. The final concentration of siRNA was 100 nM. Cells were incubated for 46 h, followed by hydroxyurea (3 mM, Sigma‐Aldrich) treatment for 2 h, pre‐extraction with CSK buffer (10 mM PIPES pH 7, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2) containing phosphatase inhibitors (1 mM DTT, 10 μg/ml leupeptin, 10 μg/ml pepstatin, 0.1 mM PMSF, 0.2 mM sodium vanadate, 5 mM sodium fluoride, 10 mM beta‐glycerophosphate) and 0.5% Triton for 5 min on ice, 2% paraformaldehyde fixation and Hoechst staining (bisBenzimide H 33342; Sigma‐Aldrich). Five images per well were acquired with an IN Cell Analyzer 1000 (GE Healthcare) using a 20× objective (~1,200 cells per well). Images were then analysed by the IN Cell Analyzer Workstation 3.5 software (GE Healthcare).
We evaluated the probability of a gene “hit” based on the collective activities of three siRNAs per gene using the statistical method redundant siRNA activity (RSA) analysis (Konig et al, 2007) as described (Mejlvang et al, 2014). Threshold used for selecting positive siRNA was determined based on the score of siASF1(a and b).
High‐throughput single‐cell analysis
GFP‐RPA1/RFP‐PCNA or RFP‐PCNA reporter U‐2‐OS cells were reverse transfected with siRNAs assembled into a sub‐library on 96‐well plates. Cells were pre‐extracted, fixed and stained with antibodies and DAPI 48 h after transfection. Images were acquired and analysed as described above for the robot‐automated siRNA screen. Relative fluorescence intensity of GFP‐RPA1, EdU and γH2AX was quantified in 4,000–7,000 RFP‐PCNA‐positive cells.
DNA combing
Single‐molecule analysis of DNA replication by molecular combing was performed as described in protocol 36 available from the EpiGeneSys Network of Excellence website. In brief, 48 h after siRNA transfection, U‐2‐OS cells were labelled for 10 min with 10 μM IdU (Sigma‐Aldrich) followed by 20‐min labelling with 100 μM CldU (MP Biomedicals). Cells were harvested immediately after the pulse and moulded into low‐melting agarose plugs, which were treated with proteinase K, melted at 67°C and digested by β‐agarase. DNA was combed on silanized coverslips (Genomic Vision). DNA fibres were denatured by HCl and probed by the following primary antibodies: mouse anti‐ssDNA (MAB3868; EMD Millipore) and rat anti‐BrdU (AbD Serotec). Measured distances were converted to kilobases by the constant stretching factor (1 μm = 2 kb).
DNA fibre assay
Twenty‐four hours after siRNA transfection, U‐2‐OS cells were pulsed for 10 min with 10 μM IdU (Sigma‐Aldrich) followed by 20‐min labelling with 100 μM CldU (MP Biomedicals). Two microlitres of cells resuspended in ice‐cold PBS was deposited on a microscope slide and incubated with 7 μl of spreading buffer (200 mM Tris–HCl, pH 7.5, 0.5% SDS and 50 mM EDTA) for 3 min. The slides were tilted 15° to stretch the DNA fibres (Bianco et al, 2012). After fixation with methanol/acetic acid (3:1), DNA was denatured with 2.5 M HCl and blocked (PBS with 1% BSA and 0.1% Triton X‐100) before staining with primary (anti‐CldU [AbCys SA], anti‐IdU [BD] and anti‐ssDNA [EMD Millipore]) and corresponding secondary antibodies conjugated with Alexa Fluor 488, 546 or 647 (all obtained from Invitrogen). Statistical analysis was performed using Prism 6 (GraphPad software).
Cell cycle analysis by PI/FACS
Cells were trypsinized, washed in ice‐cold PBS and fixed in ice‐cold ethanol (70%) for 1 h. To detect DNA, cells were stained with propidium iodide (50 μg/ml in PBS supplemented with 0.25 mg/ml RNase A) for 1 h at 37°C. Cell populations were analysed by flow cytometry on a BD FACSCalibur equipped with CellQuest software. Data were analysed and processed by FlowJo (ver 8.8.4).
RNA isolation, qPCR and microarray
Total RNA from cultured cells was isolated using TRIzol reagent (Invitrogen) or RNeasy Plus Mini Kit (Qiagen) according to manufacturer's instructions and reverse transcribed with TaqMan Reverse Transcription Reagents kit (ABI). Real‐time qPCRs were performed with LC480 polymerase mix in a LightCycler480 (Roche) using custom designed (Primer 3) primers. cDNA levels of target genes were analysed using comparative c(T) methods, where c(T) is the cycle threshold number and normalized to GAPDH. For microarray analysis of U‐2‐OS cells, mRNA samples were prepared according to standard protocols. Hybridization to Human Gene 2.0 ST Array (Affymetrix) and analysis was carried out at the Copenhagen University Hospital Microarray Center (http://www.genomic-medicine.dk/microarray-services/). Data are deposited in the GEO database under accession number GSE65065. Transcripts with a fold change ± 1.5 and P‐value < 0.05 are considered significant.
Purification of native BPRF1/3 complexes from human cells
Details about the system used to construct the K562 cell lines to purify stably expressed BRPF1‐3XFlag‐HA and BRPF3‐3XFlag‐HA will be published separately (Doyon and Côté, in preparation). In short, AAVS1 targeting vectors expressing BRPF1‐ or BRPF3‐3XFlag‐HA‐PuroR from a PGK promoter were integrated using zinc‐finger nuclease technology and clones were selected. A F2A peptide auto‐cleavage site between the Tags and the PuroR portion of the fusion protein allows in vivo cleavage after expression. Nuclear extracts were prepared following standard procedure (Abmayr et al, 2006) and followed by immunoprecipitations with anti‐Flag M2 agarose beads (Sigma) and eluting with Flag peptide buffer [100 mM KCl, 20 mM HEPES pH 7.5, 20% glycerol, 0.1% Triton X‐100, 400 μg/ml 3xFlag peptide, 1 mM DTT, 0.1 mM ZnCl2, 1 mM PMSF and protease cocktail inhibitors (Roche)].
HAT assays
Native human chromatin and free histone were purified as previously described (Utley et al, 1996). HAT assays with histone H3 peptides (300 ng, Millipore), core histones or H1‐depleted oligonucleosomes (500 ng) prepared from HeLa S3 cells were performed in a 15 μl reaction containing 50 mM Tris–HCl pH 8.0, 10% glycerol, 1 mM DTT, 0.1 mM EDTA, 1 mM PMSF, 10 mM sodium butyrate (Sigma) and 1.25 nCi 3H‐labelled acetyl‐CoA (Perkin Elmer Life Sciences). Samples were spotted on P81 membranes (Whatman) for liquid scintillation counting or loaded on 18% SDS–PAGE gels, Coomassie stained followed by EN3HANCE (Perkin Elmer) treatment and fluorography.
Cell synchronization, ChIP and ChIP‐seq experiments
RKO cells were synchronized with mimosin (0.5 mM) for 22 h and then harvested (M) or released in fresh media for 3 h before adding HU 3 mM for 90 min. For ChIP experiments with siRNAs, RKO cells were transfected with 20 nM of indicated siRNA and 4 nM of FITC‐labelled control siRNA (Qiagen). Forty‐eight hours post‐transfection, FITC‐positive cells were sorted and processed for ChIP. Cross‐linked chromatin preparation was done as previously described (Avvakumov et al, 2012). For immunoprecipitation of chromatin, 1 mg of chromatin was used with 1–3 μg of specific antibodies and incubated overnight at 4°C. Then, 40 μl of Protein A Dynabeads (Invitrogen) was added to each sample and incubated for 4 h at 4°C. The beads were washed extensively and eluted with 1% SDS and 0.1 M NaHCO3. Cross‐linked samples were reversed by heating overnight at 65°C in the presence of 0.2 M NaCl. Samples were then treated with RNase A and proteinase K for 2 h, and DNA was recovered by phenol–chloroform and ethanol precipitation. (ChIP‐seq analysis is included in the Appendix Supplementary Methods).
Immunocytochemistry and microscopy
Cells were either pre‐extracted with CSK 0.5% Triton to remove soluble proteins or fixed directly with 4% formaldehyde and processed as described (Groth et al, 2005). EdU staining was performed using Click‐iT™ EdU Alexa Fluor® 488/647 High‐Throughput Imaging (HCS) Assay kit (Invitrogen) according to manufacturer's instructions. In brief, following pre‐extraction and fixation, cells were blocked with PBS containing 5% BSA and 0.1% Tween‐20 for 1 h and incubated with primary antibody for 1 h. After washing three times with PBS containing 5% BSA and 0.1% Tween‐20, secondary antibody was applied and let to incubate for 30 min. Cells were washed three times and DNA was counterstained with DAPI (Sigma). Images were collected using a Leitz DMRXE microscope (Leica) with PL Fluotar 40x/0.5‐1.00 NA oil objective lens equipped with a CCD camera (DFC340 FX, Leica), a DeltaVision system with UApo/340 40x/1.35 NA oil objective lens or a motorized IX83 wide‐field microscope (Olympus) with PlanSApo 20x/0.75 NA dry objective and analysed with SoftWoRx 5.0.0 software (Applied Precision), Volocity image analysis software (Perkin Elmer) or ScanR image analysis software (Olympus). Immersion oil (N = 1.522) was used as imaging medium. All images in the individual panels were acquired under room temperature with the same settings and adjusted for brightness and contrast identically using S Adobe Photoshop CS5. Data were visualized using Prism software (GraphPad) or Spotfire software (Tibco).
Primers, plasmids, siRNAs and antibodies used in this study are included in Appendix Tables S1–S4.
Author contributions
YF and AV carried out the majority of the experiments. XZ and JVJ analysed the screen data. CR and M‐EL carried out the cell line constructions, BRPF1/3 purifications/assays and ChIP‐seq experiments. J‐PL and A‐CG performed mass spectrometric analysis. CG‐A and EP analysed ChIP‐seq and micro‐array data. S‐BL carried out time course imaging analysis. AG, JC, YF and AV designed the strategy. X‐JY developed Brpf3 antibody used in ChIPseq. YF, AV and AG wrote the manuscript. JC, CG‐A and CA commented on the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File
Acknowledgements
We thank Zuzana Jasencakova, Anders H. Lund and Cord Brakebusch for fruitful discussions. We are grateful to Martina Hödl for help with high‐content imaging and Yannick Doyon for providing vectors and advice for construction of the AAVS1‐BRPF1/3 K562 cell lines. We appreciate the ChIP‐seq data of H3K14ac from the NIH Roadmap Epigenomics Mapping Consortium: http://nihroadmap.nih.gov/epigenomics/. Work in A.‐C.G. and J.C. laboratories is supported by the Canadian Institutes of Health Research (CIHR; MOP‐123322 and MOP‐64289). J.C. holds a Canada Research Chair. C. G‐A. is supported by a Lundbeck Foundation postdoc grant. A.G. is an EMBO Young Investigator, and her research is supported by the Danish National Research Foundation to the Center for Epigenetics (DNRF82), the European Commission ITN FP7 Nucleosome4D and aDDRess, a European Research Council Starting Grant (ERC2011StG, no. 281,765), the Danish Cancer Society, the Danish Medical Research Foundation, the Novo Nordisk Foundation and the Lundbeck Foundation. A.V. was supported by FP7 Marie Curie Actions ITN Nucleosome4D.
The EMBO Journal (2016) 35: 176–192
References
- Abmayr SM, Yao T, Parmely T, Workman JL. (2006) Preparation of nuclear and cytoplasmic extracts from Mammalian cells. Curr Protoc Mol Biol 75: Unit 12.1 [DOI] [PubMed] [Google Scholar]
- Aggarwal BD, Calvi BR (2004) Chromatin regulates origin activity in Drosophila follicle cells. Nature 430: 372–376 [DOI] [PubMed] [Google Scholar]
- Alabert C, Groth A (2012) Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13: 153–167 [DOI] [PubMed] [Google Scholar]
- Alabert C, Bukowski‐Wills JC, Lee SB, Kustatscher G, Nakamura K, de Lima Alves F, Menard P, Mejlvang J, Rappsilber J, Groth A (2014) Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16: 281–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alver RC, Chadha GS, Blow JJ (2014) The contribution of dormant origins to genome stability: from cell biology to human genetics. DNA Repair 19: 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov N, Lalonde ME, Saksouk N, Paquet E, Glass KC, Landry AJ, Doyon Y, Cayrou C, Robitaille GA, Richard DE, Yang XJ, Kutateladze TG, Cote J (2012) Conserved molecular interactions within the HBO1 acetyltransferase complexes regulate cell proliferation. Mol Cell Biol 32: 689–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, Jorgensen M, Helleday T, Syljuasen RG, Sorensen CS (2010) Regulators of cyclin‐dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol 188: 629–638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DB, Burton A, Oda H, Ziegler‐Birling C, Torres‐Padilla ME, Reinberg D (2012) The role of PR‐Set7 in replication licensing depends on Suv4‐20 h. Genes Dev 26: 2580–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- Bell O, Schwaiger M, Oakeley EJ, Lienert F, Beisel C, Stadler MB, Schubeler D (2010) Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat Struct Mol Biol 17: 894–900 [DOI] [PubMed] [Google Scholar]
- Besnard E, Babled A, Lapasset L, Milhavet O, Parrinello H, Dantec C, Marin JM, Lemaitre JM (2012) Unraveling cell type‐specific and reprogrammable human replication origin signatures associated with G‐quadruplex consensus motifs. Nat Struct Mol Biol 19: 837–844 [DOI] [PubMed] [Google Scholar]
- Bianco JN, Poli J, Saksouk J, Bacal J, Silva MJ, Yoshida K, Lin YL, Tourriere H, Lengronne A, Pasero P (2012) Analysis of DNA replication profiles in budding yeast and mammalian cells using DNA combing. Methods 57: 149–157 [DOI] [PubMed] [Google Scholar]
- Blow JJ, Ge XQ, Jackson DA (2011) How dormant origins promote complete genome replication. Trends Biochem Sci 36: 405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke TW, Cook JG, Asano M, Nevins JR (2001) Replication factors MCM2 and ORC1 interact with the histone acetyltransferase HBO1. J Biol Chem 276: 15397–15408 [DOI] [PubMed] [Google Scholar]
- Chen X, Liu G, Leffak M (2013) Activation of a human chromosomal replication origin by protein tethering. Nucleic Acids Res 41: 6460–6474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D (2008) ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 9: 616–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debatisse M, Le Tallec B, Letessier A, Dutrillaux B, Brison O (2012) Common fragile sites: mechanisms of instability revisited. Trends Genet 28: 22–32 [DOI] [PubMed] [Google Scholar]
- Dellino GI, Cittaro D, Piccioni R, Luzi L, Banfi S, Segalla S, Cesaroni M, Mendoza‐Maldonado R, Giacca M, Pelicci PG (2013) Genome‐wide mapping of human DNA‐replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res 23: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, Lane WS, Tan S, Yang XJ, Cote J (2006) ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 21: 51–64 [DOI] [PubMed] [Google Scholar]
- Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foy RL, Song IY, Chitalia VC, Cohen HT, Saksouk N, Cayrou C, Vaziri C, Cote J, Panchenko MV (2008) Role of Jade‐1 in the histone acetyltransferase (HAT) HBO1 complex. J Biol Chem 283: 28817–28826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge XQ, Jackson DA, Blow JJ (2007) Dormant origins licensed by excess Mcm2‐7 are required for human cells to survive replicative stress. Genes Dev 21: 3331–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert DM, Takebayashi SI, Ryba T, Lu J, Pope BD, Wilson KA, Hiratani I (2010) Space and time in the nucleus: developmental control of replication timing and chromosome architecture. Cold Spring Harb Symp Quant Biol 75: 143–153 [DOI] [PubMed] [Google Scholar]
- Goren A, Cedar H (2003) Replicating by the clock. Nat Rev Mol Cell Biol 4: 25–32 [DOI] [PubMed] [Google Scholar]
- Groth A, Ray‐Gallet D, Quivy JP, Lukas J, Bartek J, Almouzni G (2005) Human Asf1 regulates the flow of S phase histones during replicational stress. Mol Cell 17: 301–311 [DOI] [PubMed] [Google Scholar]
- Groth A, Corpet A, Cook AJ, Roche D, Bartek J, Lukas J, Almouzni G (2007) Regulation of replication fork progression through histone supply and demand. Science 318: 1928–1931 [DOI] [PubMed] [Google Scholar]
- Gupta A, Hunt CR, Hegde ML, Chakraborty S, Udayakumar D, Horikoshi N, Singh M, Ramnarain DB, Hittelman WN, Namjoshi S, Asaithamby A, Hazra TK, Ludwig T, Pandita RK, Tyler JK, Pandita TK (2014) MOF phosphorylation by ATM regulates 53BP1‐mediated double‐strand break repair pathway choice. Cell Rep 8: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurvich N, Perna F, Farina A, Voza F, Menendez S, Hurwitz J, Nimer SD (2010) L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc Natl Acad Sci USA 107: 22552–22557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319: 1352–1355 [DOI] [PubMed] [Google Scholar]
- Harper JW, Elledge SJ (2007) The DNA damage response: ten years after. Mol Cell 28: 739–745 [DOI] [PubMed] [Google Scholar]
- Harrison JC, Haber JE (2006) Surviving the breakup: the DNA damage checkpoint. Annu Rev Genet 40: 209–235 [DOI] [PubMed] [Google Scholar]
- Ibarra A, Schwob E, Mendez J (2008) Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci USA 105: 8956–8961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka M, Stillman B (1999) Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J Biol Chem 274: 23027–23034 [DOI] [PubMed] [Google Scholar]
- Iizuka M, Matsui T, Takisawa H, Smith MM (2006) Regulation of replication licensing by acetyltransferase Hbo1. Mol Cell Biol 26: 1098–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y (2000) Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102: 463–473 [DOI] [PubMed] [Google Scholar]
- Kasten M, Szerlong H, Erdjument‐Bromage H, Tempst P, Werner M, Cairns BR (2004) Tandem bromodomains in the chromatin remodeler RSC recognize acetylated histone H3 Lys14. EMBO J 23: 1348–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konig R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y, Chanda SK (2007) A probability‐based approach for the analysis of large‐scale RNAi screens. Nat Methods 4: 847–849 [DOI] [PubMed] [Google Scholar]
- Kost JT, McDermott MP (2002) Combining dependent P‐values. Stat Probabil Lett 60: 183–190 [Google Scholar]
- Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, Zhou Z (2011) Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24‐deficient mice. Proc Natl Acad Sci USA 108: 12325–12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kueh AJ, Dixon MP, Voss AK, Thomas T (2011) HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Mol Cell Biol 31: 845–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AJ, Song J, Cheung P, Ishibe‐Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O (2012) The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier‐Gorlin syndrome. Nature 484: 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusch T, Florens L, Macdonald WH, Swanson SK, Glaser RL, Yates JR III, Abmayr SM, Washburn MP, Workman JL (2004) Acetylation by Tip60 is required for selective histone variant exchange at DNA lesions. Science 306: 2084–2087 [DOI] [PubMed] [Google Scholar]
- Labib K, Gambus A (2007) A key role for the GINS complex at DNA replication forks. Trends Cell Biol 17: 271–278 [DOI] [PubMed] [Google Scholar]
- Lalonde ME, Avvakumov N, Glass KC, Joncas FH, Saksouk N, Holliday M, Paquet E, Yan K, Tong Q, Klein BJ, Tan S, Yang XJ, Kutateladze TG, Cote J (2013) Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev 27: 2009–2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalonde ME, Cheng X, Cote J (2014) Histone target selection within chromatin: an exemplary case of teamwork. Genes Dev 28: 1029–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letessier A, Millot GA, Koundrioukoff S, Lachages AM, Vogt N, Hansen RS, Malfoy B, Brison O, Debatisse M (2011) Cell‐type‐specific replication initiation programs set fragility of the FRA3B fragile site. Nature 470: 120–123 [DOI] [PubMed] [Google Scholar]
- Lubelsky Y, Sasaki T, Kuipers MA, Lucas I, Le Beau MM, Carignon S, Debatisse M, Prinz JA, Dennis JH, Gilbert DM (2011) Pre‐replication complex proteins assemble at regions of low nucleosome occupancy within the Chinese hamster dihydrofolate reductase initiation zone. Nucleic Acids Res 39: 3141–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubelsky Y, Prinz JA, DeNapoli L, Li Y, Belsky JA, MacAlpine DM (2014) DNA replication and transcription programs respond to the same chromatin cues. Genome Res 24: 1102–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine DM, Bell SP (2005) A genomic view of eukaryotic DNA replication. Chromosome Res 13: 309–326 [DOI] [PubMed] [Google Scholar]
- MacAlpine HK, Gordan R, Powell SK, Hartemink AJ, MacAlpine DM (2010) Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res 20: 201–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya‐Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA (2007) Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J 26: 2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechali M (2010) Eukaryotic DNA replication origins: many choices for appropriate answers. Nat Rev Mol Cell Biol 11: 728–738 [DOI] [PubMed] [Google Scholar]
- Mejlvang J, Feng Y, Alabert C, Neelsen KJ, Jasencakova Z, Zhao X, Lees M, Sandelin A, Pasero P, Lopes M, Groth A (2014) New histone supply regulates replication fork speed and PCNA unloading. J Cell Biol 204: 29–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel T, Nahse‐Kumpf V, Kousholt AN, Klein DK, Lund‐Andersen C, Lees M, Johansen JV, Syljuasen RG, Sorensen CS (2011) A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep 12: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Struhl K (2008) HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev 22: 2633–2638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miotto B, Struhl K (2010) HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell 37: 57–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, Toyoda T, Shinga J, Katsumoto T, Chiba T, Yamaguchi N, Kitabayashi I, Koseki H, Iwama A (2011) The Hbo1‐Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 118: 2443–2453 [DOI] [PubMed] [Google Scholar]
- Murr R, Loizou JI, Yang YG, Cuenin C, Li H, Wang ZQ, Herceg Z (2006) Histone acetylation by Trrap‐Tip60 modulates loading of repair proteins and repair of DNA double‐strand breaks. Nat Cell Biol 8: 91–99 [DOI] [PubMed] [Google Scholar]
- Niimi A, Chambers AL, Downs JA, Lehmann AR (2012) A role for chromatin remodellers in replication of damaged DNA. Nucleic Acids Res 40: 7393–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacek M, Walter JC (2004) A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J 23: 3667–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010a) Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 37: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, Helleday T (2010b) Chk1 promotes replication fork progression by controlling replication initiation. Proc Natl Acad Sci USA 107: 16090–16095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard F, Cadoret JC, Audit B, Arneodo A, Alberti A, Battail C, Duret L, Prioleau MN (2014) The spatiotemporal program of DNA replication is associated with specific combinations of chromatin marks in human cells. PLoS Genet 10: e1004282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Hiratani I, Gilbert DM (2010) Domain‐wide regulation of DNA replication timing during mammalian development. Chromosome Res 18: 127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, Thurman RE, Cheng Y, Gulsoy G, Dennis JH, Snyder MP, Stamatoyannopoulos JA, Taylor J, Hardison RC, Kahveci T, Ren B et al (2014) Topologically associating domains are stable units of replication‐timing regulation. Nature 515: 402–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, MacAlpine HK, Prinz JA, Li Y, Belsky JA, MacAlpine DM (2015) Dynamic loading and redistribution of the Mcm2‐7 helicase complex through the cell cycle. EMBO J 34: 531–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remus D, Diffley JF (2009) Eukaryotic DNA replication control: lock and load, then fire. Curr Opin Cell Biol 21: 771–777 [DOI] [PubMed] [Google Scholar]
- Saksouk N, Avvakumov N, Champagne KS, Hung T, Doyon Y, Cayrou C, Paquet E, Ullah M, Landry AJ, Cote V, Yang XJ, Gozani O, Kutateladze TG, Cote J (2009) HBO1 HAT complexes target chromatin throughout gene coding regions via multiple PHD finger interactions with histone H3 tail. Mol Cell 33: 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma GG, So S, Gupta A, Kumar R, Cayrou C, Avvakumov N, Bhadra U, Pandita RK, Porteus MH, Chen DJ, Cote J, Pandita TK (2010) MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double‐strand break repair. Mol Cell Biol 30: 3582–3595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Jiang X, Chen S, Fernandes N, Price BD (2005) A role for the Tip60 histone acetyltransferase in the acetylation and activation of ATM. Proc Natl Acad Sci USA 102: 13182–13187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarnalatha M, Singh AK, Kumar V (2012) The epigenetic control of E‐box and Myc‐dependent chromatin modifications regulate the licensing of lamin B2 origin during cell cycle. Nucleic Acids Res 40: 9021–9035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 25: 3553–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Rea S, Richter K, Vilar A, Lichter P, Imhof A, Akhtar A (2005) hMOF histone acetyltransferase is required for histone H4 lysine 16 acetylation in mammalian cells. Mol Cell Biol 25: 6798–6810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E (2010) The histone H4 Lys 20 methyltransferase PR‐Set7 regulates replication origins in mammalian cells. Nat Cell Biol 12: 1086–1093 [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, Tahmasebi S, Cayrou C, Doyon Y, Goh SL, Champagne N, Cote J, Yang XJ (2008) Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol Cell Biol 28: 6828–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnikrishnan A, Gafken PR, Tsukiyama T (2010) Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 17: 430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utley RT, Owen‐Hughes TA, Juan LJ, Cote J, Adams CC, Workman JL (1996) In vitro analysis of transcription factor binding to nucleosomes and nucleosome disruption/displacement. Methods Enzymol 274: 276‐291 [DOI] [PubMed] [Google Scholar]
- Van C, Yan S, Michael WM, Waga S, Cimprich KA (2010) Continued primer synthesis at stalled replication forks contributes to checkpoint activation. J Cell Biol 189: 233–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veno ST, Kulikowicz T, Pestana C, Stepien PP, Stevnsner T, Bohr VA (2011) The human Suv3 helicase interacts with replication protein A and flap endonuclease 1 in the nucleus. Biochem J 440: 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelauer M, Rubbi L, Lucas I, Brewer BJ, Grunstein M (2002) Histone acetylation regulates the time of replication origin firing. Mol Cell 10: 1223–1233 [DOI] [PubMed] [Google Scholar]
- Walter J, Newport J (2000) Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol Cell 5: 617–627 [DOI] [PubMed] [Google Scholar]
- Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ (2006) Excess Mcm2‐7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol 173: 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Bacal J, Desmarais D, Padioleau I, Tsaponina O, Chabes A, Pantesco V, Dubois E, Parrinello H, Skrzypczak M, Ginalski K, Lengronne A, Pasero P (2014) The histone deacetylases sir2 and rpd3 act on ribosomal DNA to control the replication program in budding yeast. Mol Cell 54: 691–697 [DOI] [PubMed] [Google Scholar]
- You L, Yan K, Zhou J, Zhao H, Bertos NR, Park M, Wang E, Yang XJ (2015a) The lysine acetyltransferase activator Brpf1 governs dentate gyrus development through neural stem cells and progenitors. PLoS Genet 11: e1005034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, Yan K, Zou J, Zhao H, Bertos NR, Park M, Wang E, Yang XJ (2015b) The chromatin regulator Brpf1 regulates embryo development and cell proliferation. J Biol Chem 290: 11349–11364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You L, Zou J, Zhao H, Bertos NR, Park M, Wang E, Yang XJ (2015c) Deficiency of the chromatin regulator BRPF1 causes abnormal brain development. J Biol Chem 290: 7114–7129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Nellimoottil T, Peace JM, Knott SR, Villwock SK, Yee JM, Jancuska JM, Rege S, Tecklenburg M, Sclafani RA, Tavare S, Aparicio OM (2013) The level of origin firing inversely affects the rate of replication fork progression. J Cell Biol 201: 373–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Table EV1
Table EV2
Table EV3
Table EV4
Table EV5
Review Process File