

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease that is characterized by selective loss of motor neurons in brain and spinal cord. TAR DNA‐binding protein 43 (TDP‐43) was identified as a major component of disease pathogenesis in ALS, frontotemporal lobar degeneration (FTLD), and other neurodegenerative disease. Despite the fact that TDP‐43 is a multi‐functional protein involved in RNA processing and a large number of TDP‐43 RNA targets have been discovered, the initial toxic effect and the pathogenic mechanism underlying TDP‐43‐linked neurodegeneration remain elusive. In this study, we found that loss of TDP‐43 strongly induced a nuclear translocation of TFEB, the master regulator of lysosomal biogenesis and autophagy, through targeting the mTORC1 key component raptor. This regulation in turn enhanced global gene expressions in the autophagy–lysosome pathway (ALP) and increased autophagosomal and lysosomal biogenesis. However, loss of TDP‐43 also impaired the fusion of autophagosomes with lysosomes through dynactin 1 downregulation, leading to accumulation of immature autophagic vesicles and overwhelmed ALP function. Importantly, inhibition of mTORC1 signaling by rapamycin treatment aggravated the neurodegenerative phenotype in a TDP‐43‐depleted Drosophila model, whereas activation of mTORC1 signaling by PA treatment ameliorated the neurodegenerative phenotype. Taken together, our data indicate that impaired mTORC1 signaling and influenced ALP may contribute to TDP‐43‐mediated neurodegeneration.
Keywords: ALS, Autophagy, mTORC1, TDP‐43, TFEB
Subject Categories: Autophagy & Cell Death, Neuroscience, RNA Biology
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disorder that is characterized by degeneration of motor neurons, leading to progressive muscle weakness, atrophy, and eventually fatal paralysis and respiratory failure. Approximately 90% of ALS cases are sporadic and the remaining 10% are familial. SOD1 gene that encodes superoxide dismutase 1 (SOD1) was the first discovered causative gene of ALS, and a breakthrough of ALS research began with the discovery of TAR DNA‐binding protein‐43 (TDP‐43) that is encoded by TARDBP gene (Ling et al, 2013). TDP‐43 was identified as a key component of the ubiquitin‐positive inclusions in both ALS and frontotemporal lobar degeneration (FTLD) patients (Neumann et al, 2006), and the clinical and pathological overlap between ALS and FTLD indicated a central role for TDP‐43 in diverse disease pathogenesis. TDP‐43 is a RNA‐binding protein, which contains two RNA recognition motifs (RRM1 and RRM2) that allow TDP‐43 to bind to nucleic acids and a C‐terminal glycine‐rich domain (GRD) that mediates protein–protein interactions. TDP‐43 functions in multiple steps of RNA processing and homeostasis including transcription, splicing, and transport of target mRNAs, such as CFTR, HDAC6, SMN, and Nefl (Bose et al, 2008; Volkening et al, 2009; Buratti & Baralle, 2010; Fiesel et al, 2010; Polymenidou et al, 2011; Alami et al, 2014). It is therefore conceivable that defects in RNA processing, due to loss of nuclear TDP‐43, could be linked to neurodegeneration (Xu, 2012; Diaper et al, 2013; Vanden Broeck et al, 2013, 2014).
Autophagy–lysosome pathway (ALP) is a critical cellular quality control system that is tightly associated with various neurodegenerative diseases especially in ALS, since mutations in ALP‐associated genes, such as p62/SQSTM1, OPTN, and FIG 4, are the genetic cause of ALS, indicating a critical linkage between ALP and ALS disease pathogenesis (Ling et al, 2013). Recently, the transcription factor EB (TFEB) was identified as a master regulator of ALP that controls ALP by driving the expressions of autophagic gene products such as ATG5, Beclin‐1, and ATG9B, as well as lysosomal gene products such as LAMP1, cathepsins, and subunits of vacuolar ATPases (Sardiello et al, 2009; Settembre et al, 2011). The cellular localization and activity of TFEB are mainly controlled by Ragulator‐Rag‐mTORC1 (mTOR complex 1) (Pena‐Llopis et al, 2011; Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Settembre et al, 2012; Martina & Puertollano, 2013), an important amino acid‐sensing complex on the lysosomal surface that controls cell growth, proliferation, and autophagy (Zoncu et al, 2011b; Bar‐Peled & Sabatini, 2014). It has been shown that ALP and TFEB functions are affected by proteins in association with neurodegenerative diseases. For examples, presenilins in Alzheimer's disease, α‐synuclein in Parkinson's disease, and polyglutamine expanded androgen receptor in spinal and bulbar muscular atrophy could affect cargo recognition, autophagosome–lysosome fusion, and TFEB nuclear localization in ALP (Wong & Cuervo, 2010; Decressac et al, 2013; Chua et al, 2014; Cortes et al, 2014). In addition, autophagic vesicle accumulation is a common feature of neurodegenerative diseases including ALS (Iguchi et al, 2013; Ling et al, 2013), indicating that ALP is involved in neurodegeneration. However, it remains unclear whether the accumulation of autophagic vesicles reflects a compensatory neuroprotective response or results from a failure of autophagic degradation.
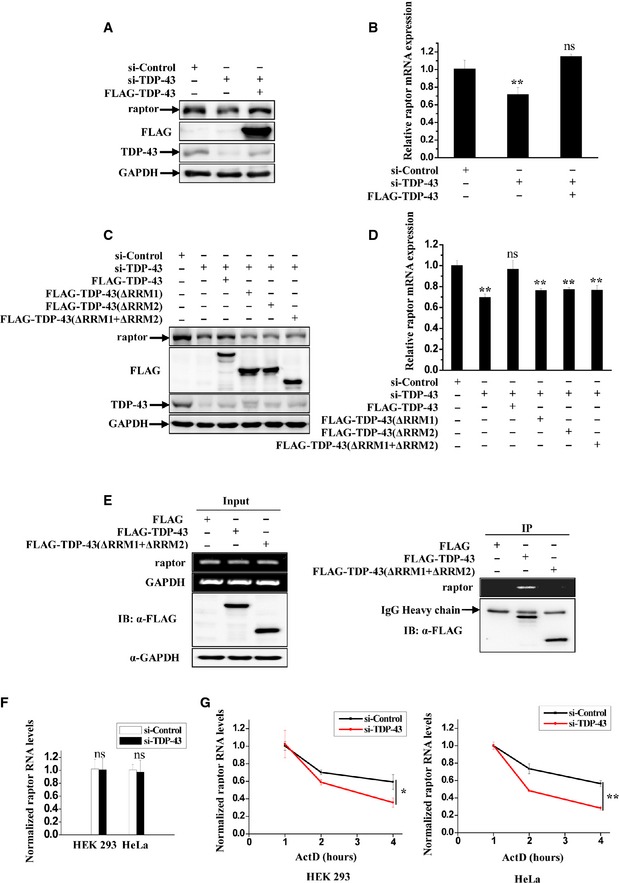
Figure 4. TDP‐43 specifically binds to raptor mRNA and regulates the expression of raptor.
-
A–DHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with FLAG‐tagged wild‐type or mutant TDP‐43 for 24 h. (A, C) Cell lysates were subjected to immunoblot analysis using anti‐raptor, ‐FLAG, ‐TDP‐43, and ‐GAPDH antibodies. (B, D) Cells were processed for qRT–PCR analysis. The level of raptor mRNA was quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
ESimilar transfection as in (C) was performed, and transfected cells were processed for RIP analysis. The inputs and RIP products were subjected to RT–PCR amplifying raptor and GAPDH, and immunoblot analysis using anti‐FLAG and ‐GAPDH antibodies.
-
FHEK 293 and HeLa cells were transfected with the indicated siRNAs. After 72 h, the newly transcribed RNAs in cells were metabolically labeled and isolated by Click‐iT and then subjected to qRT‐PCR analysis. The level of newly transcribed raptor RNA was quantified. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; one‐way ANOVA.
-
GSimilar transfection as in (F) was performed. The cells were treated with ActD for indicated times and then were processed for qRT–PCR analysis. The level of raptor mRNA was quantified and normalized relative to GAPDH mRNA. Data from three independent experiments represented as means ± S.E.M.; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
To investigate the molecular mechanism of ALP in association with loss of TDP‐43 function, we comprehensively studied the effects of TDP‐43 on ALP and TFEB in cellular and fly models with TDP‐43 loss of function. We found that loss of TDP‐43 strongly induced a nuclear translocation of TFEB by directly targeting raptor, a key component of mTORC1. TDP‐43‐mediated mTORC1 dysregulation could affect the expression of ALP genes and the neurotoxicity in cells and in vivo. The current study reveals a role of mTORC1 dysfunction, abnormal altered autophagy, and lysosomal biogenesis in TDP‐43‐mediated neurodegeneration.
Results
TFEB nuclear translocation in TDP‐43‐depleted cells
Given that ALP, the critical cellular quality control system, is involved in ALS (Ling et al, 2013) and that TFEB is a master regulator of ALP and is functionally associated with neurodegenerative diseases as recently reported (Sardiello et al, 2009; Settembre et al, 2011; Tsunemi et al, 2012; Decressac et al, 2013; Spampanato et al, 2013), we wonder whether TDP‐43 is able to regulate TFEB. We knocked TDP‐43 down in HeLa (non‐neuronal) and SH‐SY5Y (neuronal) cells and examined TFEB nuclear translocation, a process that is associated with autophagy activation and lysosomal biogenesis. We observed that knockdown of TDP‐43 resulted in a dramatic increase of nuclear and a decrease of cytoplasmic GFP‐tagged TFEB in those cells (Fig 1A and B). Similar results were obtained using non‐GFP‐tagged TFEB in other types of cells (Appendix Fig S1). Consistently, similar results were obtained using subcellular fractionation assays (Fig 1C).
Figure 1. Effect of TDP‐43 on TFEB nuclear translocation.
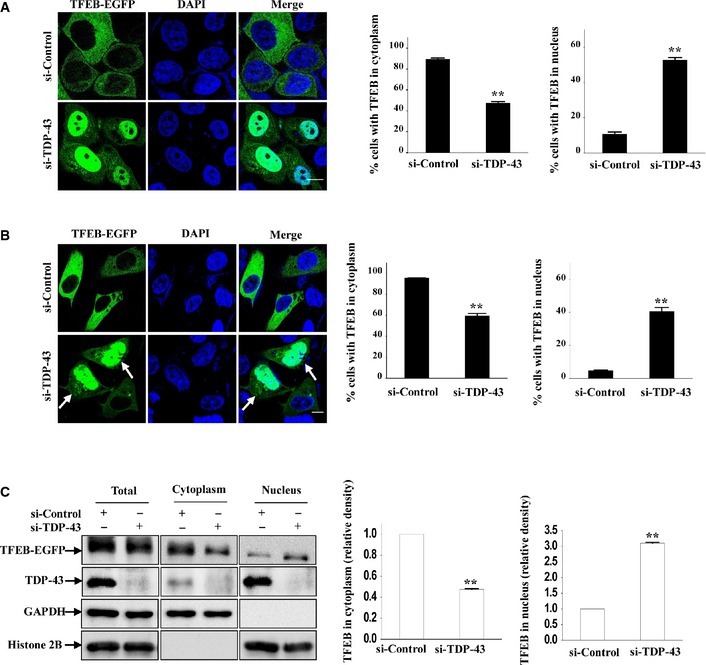
- HeLa cells were transfected with the indicated siRNAs for 48 h, and then were re‐transfected with TFEB‐EGFP. Twenty‐four hours after transfection, the cells were fixed. DAPI (blue) was used for nuclear staining. Cells were visualized using confocal microscopy. Scale bar, 5 μm. The quantification of TFEB localization in cytoplasm and nucleus is shown on the right side. Data are from three independent experiments, means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
- Similar experiments as in (A) were performed in SH‐SY5Y cells. Scale bar, 5 μm. The quantification data are shown on the right side. Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA. White arrowheads indicate nuclear localization of TFEB.
- HEK 293 cells were similarly transfected as in (A). Lysates from the cells were separated into cytoplasmic and nuclear fractions and then subjected to immunoblot analysis using anti‐GFP, ‐GAPDH, and ‐histone 2B antibodies. The relative densities are shown on the right side. The data from three independent experiments are presented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
TDP‐43 is required for mTOR lysosomal localization and mTORC1 activity
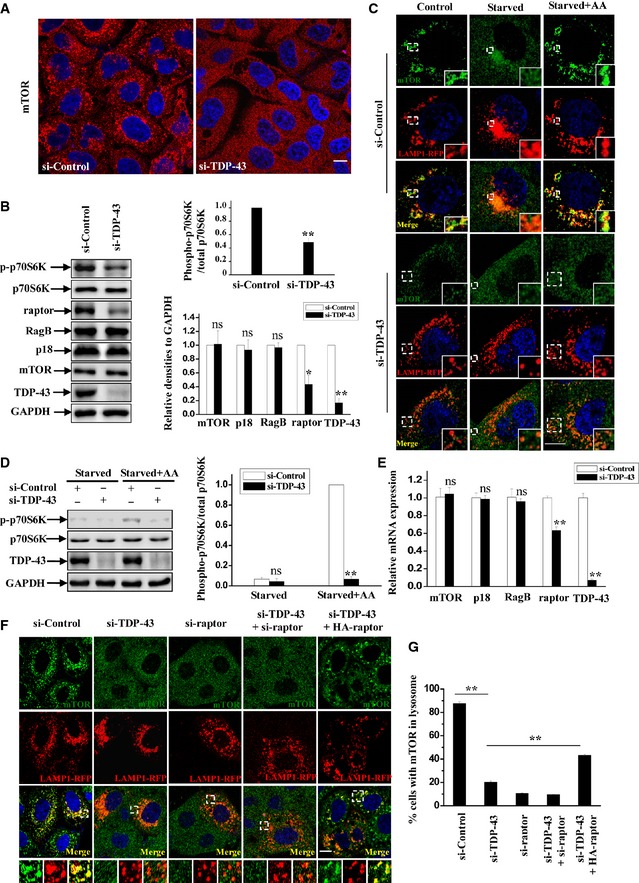
Based on the observations that mTORC1 plays an important role in the regulation of TFEB (Pena‐Llopis et al, 2011; Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Settembre et al, 2012; Martina & Puertollano, 2013), we hypothesized that TDP‐43 may regulate TFEB nuclear translocation in an mTORC1‐dependent manner. To test this possibility, we knocked TDP‐43 down and checked the cellular localization of mTOR, which reflects the activity of mTORC1. TDP‐43‐depleted cells showed a dramatic reduction of the punctate mTOR distribution that could indicate its lysosomal localization according to the previous reports (Fig 2A). Furthermore, the activity of mTORC1 was suppressed after knockdown of TDP‐43, indicated by the phosphorylation of mTORC1 substrates p70S6K (Fig 2B). Given that amino acid is a well‐established regulator of mTORC1 and promotes mTOR translocation to the lysosomal surface, we next examined the intracellular localization of mTOR under starvation or re‐addition of amino acids in cells after starvation with or without TDP‐43 knockdown. Under normal conditions, mTOR was co‐localized with a lysosomal‐specific marker LAMP1, indicating that mTOR is indeed localized to the lysosomal surface (Fig 2C). The enrichment of mTOR on lysosomes was impaired once cells were starved, but was restored after amino acids re‐feeding (Fig 2C, upper panel). Meanwhile, mTOR failed to localize to lysosomes in TDP‐43‐depleted cells regardless of the presence or absence of amino acids (Fig 2C, lower panel). Consistent with these data, biochemical assays showed that the level of phosphorylated p70S6K was relatively low after starvation. Re‐treatment of amino acids failed to restore p70S6K phosphorylation in TDP‐43‐depleted cells, whereas it significantly induced p70S6K phosphorylation in control cells (Fig 2D).
Figure 2. TDP‐43 regulates mTORC1 activity and localization.
-
AHeLa cells were transfected with the indicated siRNAs. After 72 h, the cells were subjected to immunofluorescence assay using antibody against mTOR (red). DAPI (blue) was used for nuclear staining. The stained cells were visualized using confocal microscopy. Scale bar, 5 μm.
-
BHeLa cells were transfected as in (A). After 72 h, cells lysates were subjected to immunoblot analysis using anti‐TDP‐43, ‐p70S6K, ‐phosphorylated p70S6K (T389), ‐mTOR, ‐RagB, ‐P18, ‐raptor, and ‐GAPDH antibodies. The relative densities of phosphorylated/total p70S6K are shown in the upper‐right panel, and the relative densities of mTOR, RagB, P18, raptor, and TDP‐43 to GAPDH are shown in the lower right panel. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
-
CHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with LAMP1‐RFP (red) for 24 h. The cells were then incubated with Earle's balanced salt solution (Starvation) for 2 h or re‐stimulated with amino acids (starvation + amino acids) for 30 min. The cells were stained with mTOR (green) and DAPI (blue) and then visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
-
DSimilar transfection and treatment as in (C) were performed, and the lysates from starved and re‐feeding cells were subjected to immunoblot analysis using anti‐TDP‐43, ‐p70S6K, ‐phosphorylated p70S6K, and ‐GAPDH antibodies. The relative densities are shown on the right side. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
ESimilar transfection as in (B) was performed, and transfected cells were processed for qRT–PCR analysis. The level of TDP‐43, mTOR, raptor, RagB, and P18 mRNA was quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
F, GHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with LAMP1‐RFP (red), with or without HA‐raptor for 24 h. The cells were stained with anti‐mTOR (green) antibody and DAPI (blue) and then visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm. The quantification of mTOR on lysosomes is shown in (G). Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
Raptor, the adaptor of mTORC1, is involved in TDP‐43‐mediated mTOR lysosomal localization
Previous studies have shown that the subcellular localization and activity of mTOR are highly controlled by mTORC1 adaptor protein raptor, Rag GTPases, and Ragulator complex which is comprised of p18, p14, and MP1 proteins that reside directly on the lysosomal membrane surface (Sancak et al, 2008, 2010; Zoncu et al, 2011a; Bar‐Peled et al, 2012). In Ragulator–Rag–mTORC1 complex, Ragulator anchors the Rag GTPases to lysosomes, which is essential for Rag GTPases activation through an amino acids‐dependent manner. Meanwhile, raptor interacts with active Rag GTPase heterodimers in which GTP‐bound RagA or RagB (very similar to RagA) associates with GDP‐bound RagC or RagD (similar to RagC), and raptor targets mTOR to lysosome surface where mTOR could be activated by its activator Rheb on the lysosomes. We examined the cellular localization and expression level of components of Ragulator–Rag–mTORC1 complex in TDP‐43‐deficient cells. TDP‐43‐depleted cells showed a decrease of raptor, but not mTOR, p18, and RagB, compared with control cells (Fig 2B). Moreover, knockdown of TDP‐43 did not affect the cellular localization of Rag GTPases (Fig 3D). To further investigate the effects of TDP‐43 on the expression of raptor, we performed quantitative RT–PCR assays. Downregulation of raptor mRNA was observed in TDP‐43‐depleted cells, whereas mRNA levels of other known members of mTOR‐associated machinery on the lysosomes, such as mTOR itself, RagB, p18, MP1, p14, HBXIP, C7orf59, RagA, RagC, RagD, and FLCN, were not changed (Figs 2E and EV1C). The cells treated with siRNA against raptor had diminished mTOR localization to lysosomes, showing a similarity to the cells treated with siRNA against TDP‐43 (Figs 2F and G, and EV1A and B). Similar effects were also observed in cells treated with siRNAs against both TDP‐43 and raptor (Figs 2F and G, and EV1A and B). Given that raptor is the key adaptor of mTORC1 and directly regulates mTOR lysosomal localization, we wondered whether TDP‐43 regulates mTOR lysosomal localization in a raptor‐dependent manner. In TDP‐43‐depleted cells, mTOR was recruited to lysosomes, indicated by a co‐localization of lysosomal marker LAMP1 and CD63 after raptor re‐transfection (Figs 2F and G, and EV1D and E), suggesting a specific dependence of raptor in TDP‐43‐mediated mTOR lysosomal localization.
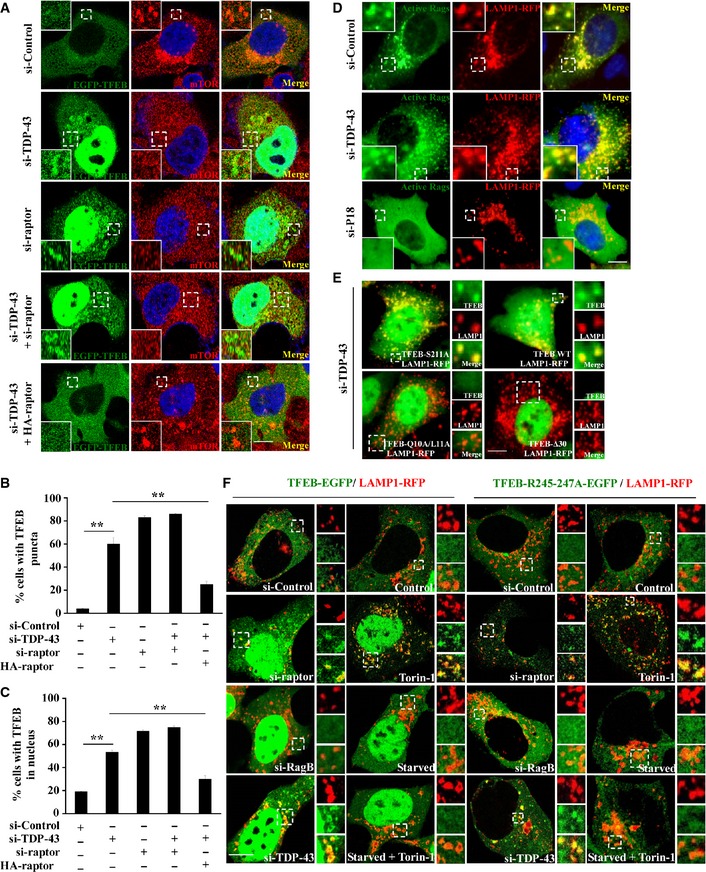
Figure 3. TDP‐43 regulates TFEB lysosomal localization.
-
A–CHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with TFEB‐EGFP (green) for 24 h. The cells were stained with anti‐mTOR (red) antibody and DAPI (blue) and then visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm. The quantification data of TFEB puncta and nuclear localization are shown in (B and C), respectively. Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
DHEK 293 cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with LAMP1‐RFP (red) and constitutively active HA‐GST‐tagged Rag GTPase mutants (RagA Q66L + RagC S75L = RagAGTP + RagCGDP) for 24 h. The cells were stained with anti‐HA (green) antibody and DAPI (blue). Cells were visualized using microscope IX71. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
-
EHEK 293 cells were transfected with siRNA targeting TDP‐43. After 48 h, the cells were re‐transfected with EGFP‐tagged TFEB‐WT, TFEB‐S211A, TFEB‐Q10A/L11A, or TFEB‐Δ30, along with LAMP1‐RFP for 24 h. Cells were visualized using microscope IX71. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
-
FHEK 293 cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with EGFP‐tagged TFEB‐WT or TFEB‐R245‐247A, along with LAMP1‐RFP for 24 h. Then, the cells were incubated with Torin‐1 (250 nM) for 1 h, Earle's balanced salt solution (starvation) for 2 h or incubated with both. Cells were fixed and visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
Figure EV1. TDP‐43 regulates mTOR lysosomal localization and mTORC1 activity in a raptor‐dependent manner.
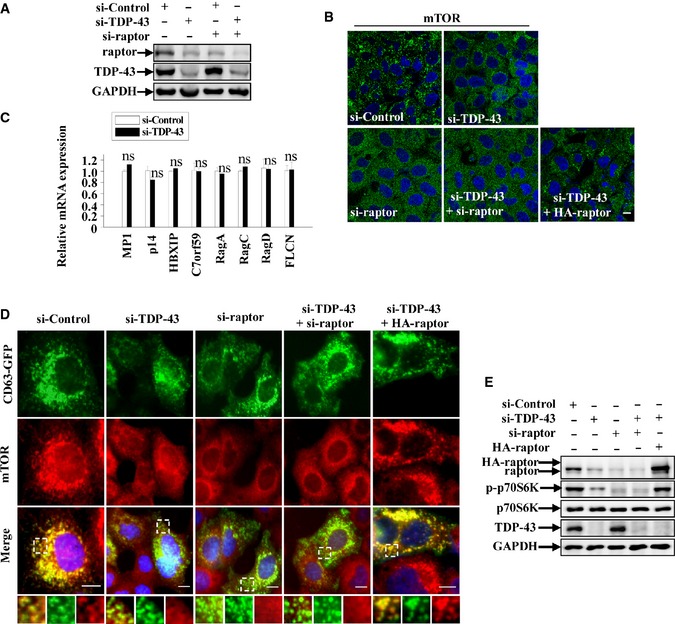
- HeLa cells were transfected with the indicated siRNAs. After 72 h, cell lysates were subjected to immunoblot analysis using anti‐TDP‐43, ‐raptor, and ‐GAPDH antibodies.
- HeLa cells were transfected with the indicated siRNAs. After 72 h, the cells were stained with anti‐mTOR (green) antibody and DAPI (blue). Cells were fixed and visualized using confocal microscopy. Scale bar, 5 μm.
- Similar transfection as in (A) was performed, and transfected cells were processed for qRT–PCR analysis. The level of MP1, p14, HBXIP, C7orf59, RagA, RagC, RagD, and FLCN mRNA was quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; one‐way ANOVA.
- HeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with CD63‐GFP (green), with or without HA‐raptor for 24 h. The cells were stained with anti‐mTOR (red) antibody and DAPI (blue). Cells were fixed and visualized using microscope IX71. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
- Similar transfection as in (D) was performed, and transfected cells were subjected to immunoblot analysis using anti‐TDP‐43, ‐p70S6K, ‐p‐p70S6K, ‐raptor, and ‐GAPDH antibodies.
TDP‐43 regulates the localization of TFEB by targeting raptor, but not Rag GTPases
As we have shown that TDP‐43 controls TFEB nuclear translocation and regulates the activity of mTORC1, we wonder how TDP‐43 mediates TFEB localization. Recent studies showed that in fully fed cells, most TFEB appear to diffusely distributed in the cytoplasm and a little fraction of TFEB is recruited to activated Rag GTPases on the lysosomal surface, where TFEB is phosphorylated by mTORC1 on serine 211 (S211). Phosphorylated TFEB then interacts with cytosolic 14‐3‐3, resulting in the sequestration of TFEB in the cytosol. However, TFEB would transport into the nucleus once the interaction is abolished, resulting in an increased expression of TFEB targeting genes (Pena‐Llopis et al, 2011; Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Settembre et al, 2012; Martina & Puertollano, 2013). In consistence with those findings, we observed that TFEB showed a diffuse cytoplasmic distribution in si‐control cells, whereas mTOR was localized to lysosomes (Fig 3A–C). In contrast, in TDP‐43 knockdown cells, TFEB and mTOR localization was dramatically changed, showing that TFEB was translocated from the cytosol to the nucleus and formed lysosomal puncta in cytoplasm, whereas mTOR had a diffusively cytoplasmic distribution (Figs 3A and EV2), suggesting an involvement of mTORC1 in TDP‐43‐mediated TFEB nuclear translocation. Moreover, similar results were obtained in raptor knockdown cells as well as in TDP‐43 and raptor double‐knockdown cells (Figs 3A and EV2). To further test whether TDP‐43‐induced redistribution of TFEB and mTOR depends on raptor, we performed raptor re‐transfection assay in TDP‐43‐depleted cells and examined the subcellular localization of TFEB and mTOR. After a restoration of raptor, but not active Rag GTPases (RagAGTP/RagCGDP), TFEB shuttled back to the cytoplasm (Figs 3A, EV2, and EV3A and B), suggesting a critical role of raptor in TDP‐43‐mediated regulation of TFEB. Based on the studies that Rag GTPases functions in the recruitment of TFEB and mTOR to lysosomes (Martina & Puertollano, 2013), we next tested whether TDP‐43 could influence the lysosomal localization of Rag GTPases. The enrichment of the active Rag GTPases (RagAGTP/RagCGDP) (Martina & Puertollano, 2013) on lysosomes was not disturbed after knockdown of TDP‐43, but was strongly abolished after knockdown of p18, which anchors Rag GTPases to lysosomes (Fig 3D), indicating that loss of TDP‐43 does not affect the localization of the Rag GTPases.
Figure EV2. TDP‐43 regulates TFEB cellular localization by targeting raptor.
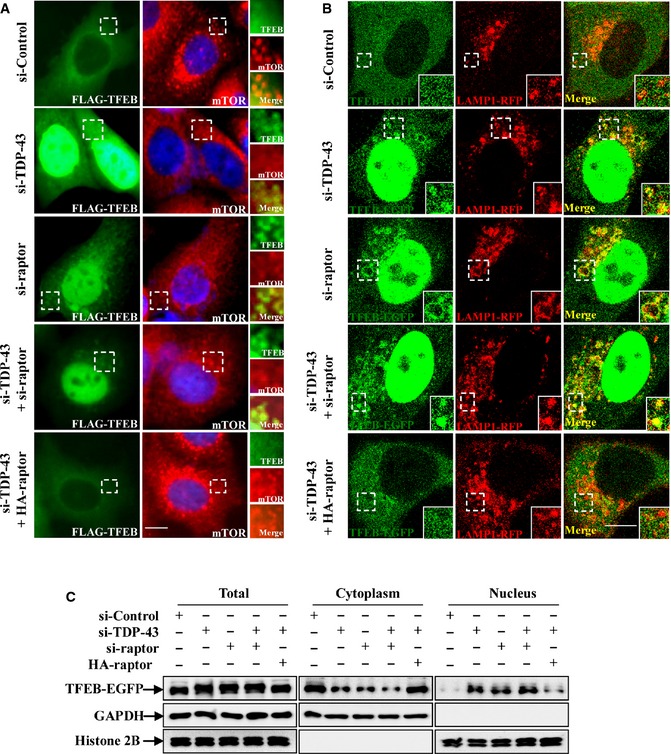
- HeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with FLAG‐TFEB, with or without HA‐raptor for 24 h. The cells were stained with anti‐FLAG (green) and ‐mTOR (red) antibodies and DAPI (blue), and then the stained cells were visualized using microscope IX71. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 µm.
- HeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with TFEB‐EGFP (green) and LAMP1‐RFP (red), with or without HA‐raptor for 24 h. Cells were fixed and visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
- HeLa cells were similarly transfected as in (B). Lysates from the cells were separated into cytoplasmic and nuclear fractions and then subjected to immunoblot analysis using anti‐GFP, ‐GAPDH, and ‐histone 2B antibodies.
Figure EV3. TDP‐43 does not regulate TFEB cellular localization through Rag GTPases, and effects of TDP‐43 on TFEB mutants.
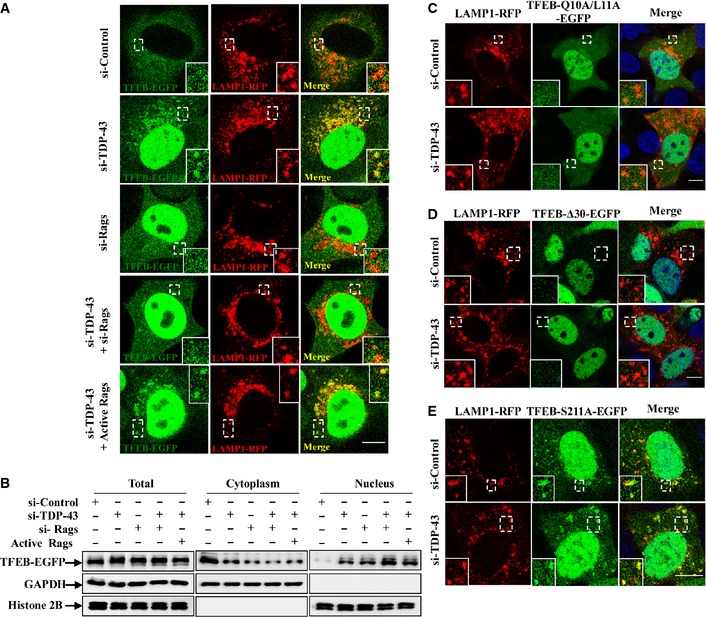
-
AHeLa cells were transfected with the indicated siRNAs. Note that si‐Rags indicates a combination of siRNAs targeting RagA and RagB. After 48 h, the cells were transfected with TFEB‐EGFP (green) and LAMP1‐RFP (red), with or without constitutively active HA‐GST‐tagged Rag GTPase mutants (RagA Q66L + RagC S75L = RagAGTP + RagCGDP) for 24 h. Cells were fixed and visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
-
BHeLa cells were similarly transfected as in (A). Lysates from the cells were separated into cytoplasmic and nuclear fractions and then subjected to immunoblot analysis using anti‐GFP, ‐GAPDH, and ‐histone 2B antibodies.
-
C–EHEK 293 cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with EGFP‐tagged TFEB‐Q10A/L11A, TFEB‐Δ30, or TFEB‐S211A, along with LAMP1‐RFP for 24 h. The cells were stained with DAPI (blue), and then the stained cells were visualized using confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm.
The conversion of serine 211 to alanine in TFEB results in a loss of phosphorylation of TFEB by mTORC1, which in turn blocks the formation of the TFEB‐14‐3‐3 complex in the cytosol, leading to a constitutive nuclear localization, as well as an increased lysosomal localization of TFEB (Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Martina & Puertollano, 2013). In contrast, TFEB‐Δ30 mutant (deletion of the first 30 amino acids) and TFEB‐Q10A/L11A mutant could not associate with Rag GTPases and lysosomes, leading to a failure of phosphorylation of Ser211 by mTORC1 and subsequently being transported easily into the nucleus (Martina et al, 2012; Roczniak‐Ferguson et al, 2012). In our observations, knockdown of TDP‐43 significantly increased wild‐type (WT) TFEB, but not TFEB‐Δ30 or TFEB‐Q10A/L11A lysosomal localization (Figs 3E and EV3C and D), suggesting that Rag GTPases, but not TDP‐43, are critical for the association of TFEB with lysosomes. Meanwhile, knockdown of TDP‐43 had no effects on TFEB‐S211A localization (Figs 3E and EV3E), suggesting that TDP‐43 may affect TFEB phosphorylation by mTORC1 to modulate TFEB lysosomal distribution, similarly as raptor does.
We further comprehensively analyzed the localization of TFEB under various conditions. In agreement with other studies (Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Settembre et al, 2012; Martina & Puertollano, 2013), TFEB was dramatically translocated to the nuclei and lysosomes after the treatment of Torin‐1, a specific mTORC1 inhibitor. In contrast, starvation or depletion of RagB led to an increase of nuclear, but not lysosomal translocation of TFEB, even if the cells were subsequently incubated with Torin‐1 (Fig 3F). Meanwhile, the localization of TFEB in TDP‐43‐ or raptor‐depleted cells was similar to that in Torin‐1‐treated cells, but not in starved or RagB‐depleted cells (Fig 3F). Similar results were obtained using cells that were transfected with the TFEB‐R245‐247A mutant, which lacks a nuclear localization signal but still has the lysosomal localization signal by targeting Rag GTPases (Fig 3F). In those cells, raptor‐ or TDP‐43 depletion, or Torin‐1 treatment still induced an increased translocation of TFEB‐R245‐247A to lysosomes (Fig 3F). All together, our data reveal that TDP‐43 regulates the localization of TFEB by affecting raptor and mTORC1, but not Rag GTPases.
TDP‐43 directly binds to raptor mRNA through its RRM domains and regulates raptor mRNA stability
To confirm that the decreased raptor level following TDP‐43 depletion was a specific effect, we re‐expressed wild‐type TDP‐43 after the cells were treated with siRNA targeting the 3′UTR of TARDBP, which does not affect the re‐transfected TARDBP with coding sequences. The mRNA and protein levels of raptor were restored upon re‐expression with wild‐type TDP‐43 in TDP‐43‐deficient cells, indicating a role of TDP‐43 in the regulation of raptor gene expression level (Fig 4A and B). TDP‐43 is a DNA/RNA‐binding protein containing two RNA recognition motifs (RRM1 and RRM2) and a C‐terminal glycine‐rich domain (GRD) that allow its binding to nucleic acids and mediate protein–protein interactions. To further verify which domains could affect the regulation of raptor by TDP‐43, we constructed TDP‐43 deletion mutants that lack the domains of RRM1 (ΔRRM1), RRM2 (ΔRRM2), or both RRM1 and RRM2 (ΔRRM1+ΔRRM2), respectively. In TDP‐43 depletion cells, both mRNA and protein levels of raptor failed to be restored by these TDP‐43 deletion mutants (Fig 4C and D). Similarly, in TDP‐43‐depleted cells, raptor levels and phosphorylated p70S6K levels were not restored by a 25‐kDa C‐terminal fragment of TDP‐43 (named TDP‐25) lacking the RRMs, which could form cytoplasmic aggregates in cells (Appendix Fig S2A–C), suggesting that the cytoplasmic TDP‐43 aggregates themselves cannot affect raptor level or mTORC1 activity. Therefore, our results indicate that the raptor level and mTORC1 activity are regulated by the functions of endogenous TDP‐43 in cells. In addition, we found that GRD deletion also failed to restore the raptor levels in TDP‐43 depletion cells (Appendix Fig S2D and E). Since the GRD domain of TDP‐43 could interact with other key regulators (such as hnRNPs) that are critical for the TDP‐43 function, these factors could cooperate together with TDP‐43 to regulate TDP‐43 RNA targets such as raptor.
Given that the mutation sites of above TDP‐43 deletion mutants were RNA‐binding domains, we suspected that TDP‐43 could bind to raptor mRNA directly. In agreement with this, RNA immunoprecipitation (RIP) analysis showed that wild‐type TDP‐43, but not the RRM1 and RRM2 domain deletion mutant, could bind to raptor mRNA (Fig 4E). Therefore, our data suggest that RRM domains of TDP‐43 specifically bind to raptor mRNA and mediate the regulation of raptor mRNA level.
To further explore how TDP‐43 mediates the regulation of raptor mRNA, we examined the transcription and the stability of raptor in TDP‐43‐deficient cells. We used Click‐iT technology to label the newly transcribed RNA in TDP‐43‐depleted cells and control cells, and then, the labeled RNA was selected and subjected to qRT–PCR. Results showed that the level of newly synthesized raptor was not affected in TDP‐43‐deficient cells (Fig 4F). To test whether TDP‐43 can affect the stability of raptor mRNA, we analyzed half‐lives of raptor mRNA by inhibiting transcription with the treatment of actinomycin D (ActD). Importantly, we observed that the mRNA stability of raptor, but not RagB, was decreased in TDP‐43‐deficient cells (Fig 4G and Appendix Fig S3). Taken together, our results suggest that TDP‐43 can bind to raptor mRNA and regulate raptor mRNA stability at the post transcriptional level.
TDP‐43 regulates lysosomal and autophagosomal biogenesis in a TFEB‐dependent manner
As nuclear translocation of TFEB is associated with TFEB transcriptional activity, which is tightly involved in the regulation of lysosomal biogenesis and autophagic level, we wonder whether TDP‐43 could regulate ALP. To address this, we first employed Lysotracker (a specific lysosomal fluorescent probe) as a marker to monitor lysosomal biogenesis. In TDP‐43‐depleted cells, the fluorescent intensity of Lysotracker was markedly increased (Fig 5A and Appendix Fig S4A and B). We next broadly assessed the expression levels of lysosomal and autophagic genes in TDP‐43‐depleted cells. TDP‐43‐depleted cells showed a global increase of both mRNA and protein expression levels of a set of TFEB target genes, including lysosomal and autophagic genes, such as genes involved in lysosomal biogenesis and function (LAMP1, cathepsins, and genes encoding subunits of vacuolar ATPases), and genes involved in autophagy (ATG5, Beclin‐1, and ATG9B; Fig 5B and C). Similar results were obtained in various cell lines, such as SH‐SY5Y (Fig 5D and E). However, the relative genes of ALP were no longer upregulated in TDP‐43‐depleted cells if TFEB was silenced (Fig 5F and G).
Figure 5. TDP‐43 regulates global gene expression levels in ALP through TFEB .
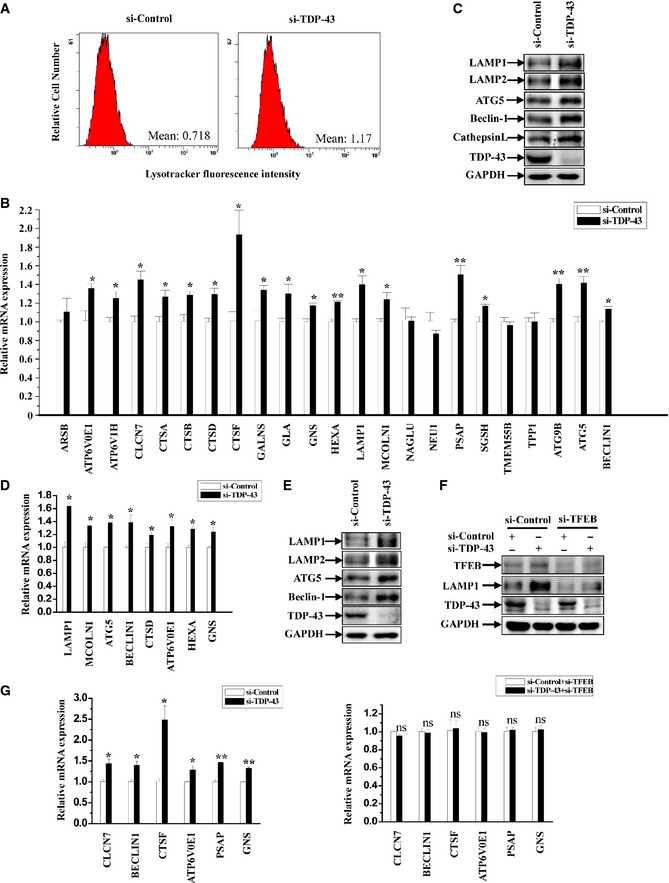
- HeLa cells were transfected with the indicated siRNAs. After 72 h, the cells were stained with Lysotracker (red). Then, the cells were harvested and processed for flow cytometry analysis.
- Similar transfection as in (A) was performed, and transfected cells were processed for qRT–PCR analysis. The mRNA levels of lysosomal and autophagic genes were quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
- HeLa cells were similarly transfected as in (A), and the cell lysates were subjected to immunoblot analysis using anti‐TDP‐43, ‐LAMP1, ‐LAMP2, ‐ATG5, ‐Beclin‐1, ‐cathepsin L, and ‐GAPDH antibodies.
- SH‐SY5Y cells were similarly transfected as in (A) and processed as in (B). Data from three independent experiments represented as means ± S.E.M.; *, P < 0.05; one‐way ANOVA.
- Similar experiments as in (C) were performed in SH‐SY5Y cells.
- HeLa cells were transfected with the indicated siRNAs. Cell lysates were subjected to immunoblot analysis using anti‐TDP‐43, ‐TFEB, ‐LAMP1, and ‐GAPDH antibodies.
- Similar transfection as in (F) was performed, and transfected cells were processed for qRT–PCR analysis as in (B). The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
As the protein level of LC3‐II and the numbers of LC3‐labeled vesicles reflect the autophagosome numbers, we used biochemical and immunofluorescent assays to determine whether autophagy is induced by TDP‐43 depletion. Indeed, we observed a significant enhancement of LC3‐II levels in 8 various types of TDP‐43‐depleted cell lines, including HeLa (human cervical), A549 (human epithelial), HEK 293 (human kidney), HT22 (mouse neuronal), PC12 (rat neuronal), and Atg5 wild‐type (autophagy‐competent) mouse embryonic fibroblasts (ATG5‐WT MEFs), but not in Atg5 knockout (autophagy‐deficient) mouse embryonic fibroblasts (ATG5‐KO MEFs) (Fig EV4A), suggesting that TDP‐43 could strikingly affect autophagosome formation across cell types of diverse species and tissues with functional autophagy. Consistent with these results, immunofluorescent assay showed a dramatic increased punctuate LC3 in the absence of TDP‐43 (Fig EV4B and C). Knockdown of TDP‐43 also increased punctuate p62 (an autophagic substrate and marker protein) and LC3 that were co‐localized in cells (Fig EV4D and E). Moreover, immunoblot and electron microscopy (EM) analyses demonstrated that loss of TFEB could abolish the increases of levels of LC3‐II, autophagosomal, and lysosomal numbers in TDP‐43‐depleted cells (Fig 6A and B). Taken together, our data suggest that TDP‐43 regulates lysosomal and autophagosomal biogenesis in a TFEB‐dependent manner.
Figure EV4. TDP‐43 influences autophagosome formation.
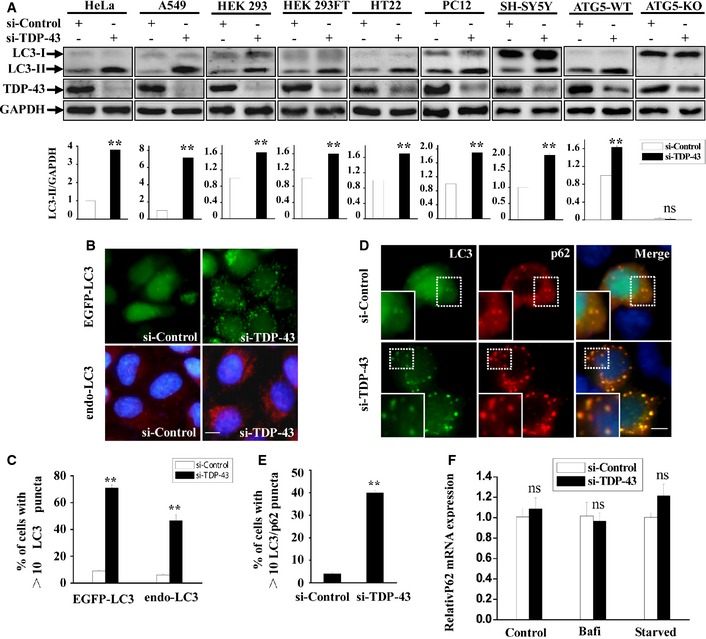
-
AHeLa, A549, HEK 293, HEK 293FT, HT22, PC12, SH‐SY5Y, ATG5‐WT, and ATG5‐KO cells were transfected with the indicated siRNAs. After 72 h, lysates from the cells were subjected to immunoblot analysis using anti‐TDP‐43, LC3, and GAPDH antibodies. The relative densities of LC3‐II to GAPDH are shown in the lower panel. Data from three independent experiments represented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
B, CHeLa cell line stably expressing EGFP‐LC3 or HeLa cells was transfected with the indicated siRNAs. After 72 h, the cells were stained with anti‐rabbit Rhodamine (Santa Cruz Biotechnology) following treatment with anti‐LC3 (red) antibody and DAPI (blue), and then, the stained cells were visualized using microscope IX71. Scale bar, 5 μm. The quantification data of punctuate LC3 are shown in Figure EV4C. Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
D, EHEK 293 cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with EGFP‐LC3 and FLAG‐p62 for 24 h. The cells were stained with anti‐FLAG (red) antibody and DAPI (blue), and then, the stained cells were visualized using microscope IX71. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 µm. The quantification data of punctuate LC3/p62 are shown in (E). Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
FHeLa cells were transfected with the indicated siRNAs for 48 h. For bafilomycin A1 treatment, the cells were then incubated with bafilomycin A1 (100 nM) for 24 h. For starvation treatment, the cells were cultured for another 22 h and then treated with Earle's balanced salt solution for 2 h. The cells were subjected to qRT–PCR analysis. The mRNA level of p62 was quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different.
Figure 6. Effect of TDP‐43 on autophagic flux and cell death.
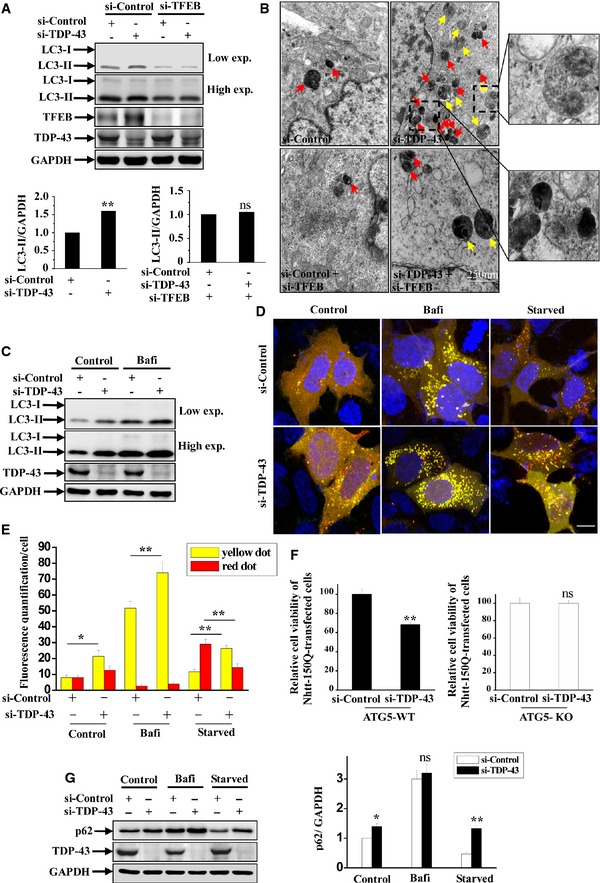
-
AHeLa cells were transfected with the indicated siRNAs. Cell lysates were subjected to immunoblot analysis using anti‐TDP‐43, ‐TFEB, ‐LC3, and ‐GAPDH antibodies. The relative densities of LC3‐II to GAPDH are shown on the lower side. Data from three independent experiments represented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
BFor EM analysis, HeLa cells were transfected with the indicated siRNAs. Double‐membrane autophagosomes are indicated with yellow arrows, and autolysosomes are indicated with red arrow. Regions within the dotted boxes are magnified in the insets. Scale bar, 250 nm.
-
CHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were incubated with bafilomycin A1 (100 nM) for 24 h. Then, lysates from the cells were subjected to immunoblot analysis using anti‐TDP‐43, ‐LC3, and ‐GAPDH antibodies.
-
D, EHEK 293 cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with mCherry‐EGFP‐LC3. For bafilomycin A1 treatment, the cells were then incubated with bafilomycin A1 (100 nM) for 24 h. For starvation treatment, the cells were cultured for 22 h and then treated with Earle's balanced salt solution for 2 h. Cells were fixed and visualized using confocal microscopy. Scale bar, 5 μm. The quantification data of yellow (autophagosomes) or red (autolysosomes) are shown in (E). Data from three independent experiments represented as means ± S.E.M.; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
-
FATG5‐WT and ATG5‐KO MEFs were transfected with Nhtt‐150Q‐EGFP. After 6 h, the cells were transfected with the indicated siRNAs for 5 days, and then, the cells were subjected to MTT assays. The relative cell viability from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
GSimilar transfection as in (C) was performed, and the transfected cells were incubated with bafilomycin A1 or Earle's balanced salt solution (starvation) as in (D). Lysates from the cells were subjected to immunoblot analysis using anti‐TDP‐43, ‐p62, and ‐GAPDH antibodies. The relative densities are shown on the right side. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
Accumulation of immature autophagic vesicles and overwhelmed autophagic flux in TDP‐43‐depleted cells
Given that the protein level of LC3‐II reflects both autophagosome synthesis and degradation, we employed bafilomycin A1, a V‐ATPase inhibitor which blocks the fusion of autophagosome with lysosome and ultimately leads to the inhibition of autophagosome degradation, to monitor autophagosome synthesis in TDP‐43‐depleted cells. The level of LC3‐II was also increased in TDP‐43‐depleted cells treated with bafilomycin A1 compared with control cells treated with bafilomycin A1 (Fig 6C), indicating that loss of TDP‐43 enhanced autophagosome synthesis. To better study the effect of TDP‐43 on autophagic flux and function, we tested mCherry‐EGFP‐LC3 as a reporter of autophagic flux. An significant increase of autophagosomes (yellow dots with both mCherry and EGFP signals) was observed in TDP‐43‐depleted cells compared with control cells, but the formation of autolysosomes (red dots with only mCherry signal since quenching of the EGFP signal in the acidic lysosomes) was not enhanced to a same extent as autophagosomes (Fig 6D and E). In addition, formation of autophagosomes (yellow) was also enhanced in TDP‐43‐depleted cells treated with bafilomycin A1 (Fig 6D and E). Notably, under starvation condition, we observed an increase of autophagosomes and a decrease of autolysosomes in TDP‐43‐depleted cells compared with control cells, suggesting that knockdown of TDP‐43 could impair autophagosome–lysosome fusion in a mTORC1‐independent manner, thereby inhibiting autophagic flux under starvation condition (Fig 6D and E). In supporting with this notion, the protein level of autophagic substrate p62 was upregulated in TDP‐43‐deficient cells under basal and starvation conditions (Fig 6G), whereas the mRNA level of p62 was not changed in those cells (Fig EV4F).
To further explore the role of altered ALP in TDP‐43‐mediated cytotoxicity, we examined the cell viability in TDP‐43‐depleted cells that were transfected with EGFP‐tagged Nhtt‐150Q, a N‐terminal truncated huntingtin known as a autophagy substrate that could damage ALP function and increase the sensitivity of cell death induced by autophagic dysfunction (Wong & Cuervo, 2010). Knockdown of TDP‐43 induced a significant cell death in ATG5‐WT MEFs that were transfected with Nhtt‐150Q, but not in ATG5‐KO MEFs (Fig 6F). Taken together, our data indicate that abnormal accumulation of autophagic cargoes and overwhelmed ALP function could trigger cytotoxicity in TDP‐43‐depleted cells.
To study how TDP‐43 affects autophagosome–lysosome fusion, we examined a set of genes involved in autophagosome–lysosome fusion, including syntaxin‐17 (Itakura et al, 2012), SNAP‐29 (Diao et al, 2015), VAMP8 (Jean et al, 2015), VPS33A (Jiang et al, 2014), VPS16 (Jiang et al, 2014), hVPS41 (Jiang et al, 2014), Rab7, DCTN1, MYO1C (Brandstaetter et al, 2014), DYNC1H1 (Jahreiss et al, 2008), DNAI1/DIC, DYNLL1, DYNC1LI2, ARL8B, myrlysin, SERCA (Mauvezin et al, 2015), PLEKHM1 (McEwan et al, 2015). Our results showed that the gene expression level of DCTN1, which encodes dynactin 1, was significantly decreased in TDP‐43‐depleted cells (Fig EV5A and B). Furthermore, protein expression level of dynactin 1, but not dynein heavy and intermediate chains (DHC and DIC), was decreased in those cells (Fig 7A). Dynactin 1 is a motor protein component of the dynein–dynactin complex which mediates the retrograde transport of lysosomes, and dysfunction of dynein–dynactin complex results in abnormality of lysosomal positioning and autophagosome–lysosome fusion as reported (Jahreiss et al, 2008; Korolchuk et al, 2011). Under starvation condition, we could observe a dramatic increase of perinuclear lysosomes in si‐controls cells (Fig EV5C) as reported (Korolchuk et al, 2011), and we found more dispersed distributed lysosomes in si‐TDP‐43 cells, as well as si‐dynactin 1 or si‐DHC cells, but not si‐KIF5B (KIF5B mediates the anterograde transport) cells (Figs 7B and C, and EV5D–H). Importantly, we observed significant decreased LC3/LAMP1 co‐localization (autophagosome–lysosome fusion) in dynactin 1‐deficient cells, and loss of TDP‐43 was not able to decrease LC3/LAMP1 co‐localization in dynactin 1‐deficient cells (Fig 7D and E). Meanwhile, re‐transfection of dynactin 1 could increase LC3/LAMP1 co‐localization in TDP‐43‐depleted cells (Fig 7F and G), indicating that dynactin 1 deficiency contributes to the impairment of autophagosome–lysosome fusion in TDP‐43‐depleted cells. Taken together, these data suggest that TDP‐43 could regulate lysosomal positioning and autophagosome–lysosome fusion by targeting dynactin 1.
Figure EV5. TDP‐43 regulates gene expression of DCTN1, which encodes dynactin 1, and influences lysosome positioning.
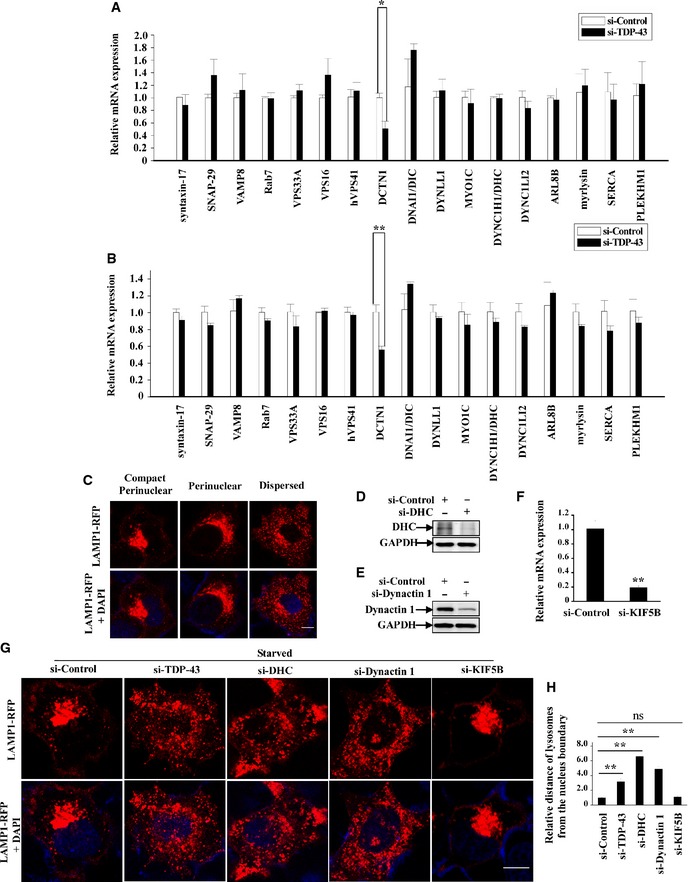
-
AHeLa cells were transfected with the indicated siRNAs. After 72 h, the transfected cells were processed for qRT–PCR analysis. The mRNA levels of autophagosome/endosome/lysosome fusion‐related proteins were quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; *, P < 0.05; one‐way ANOVA.
-
BSimilar experiments as in (A) were performed in SH‐SY5Y cells. The mRNA levels of autophagosome/endosome/lysosome fusion‐related proteins were quantified and normalized relative to GAPDH. Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
CThree representative distribution patterns of lysosome in cells: compact perinuclear, perinuclear, or dispersed. HeLa cells were transfected with LAMP1‐RFP for 24 h. DAPI (blue) was used for nuclear staining. Cells were visualized using confocal microscopy. Scale bar, 5 µm.
-
D, EHeLa cells were transfected with the indicated siRNAs. After 72 h, the cells were subjected to immunoblot analysis using anti‐DHC, dynactin 1, and GAPDH antibodies.
-
FSimilar transfection as in (E) was performed. After 72 h, the transfected cells were processed for qRT–PCR analysis. The mRNA level of KIF5B was quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
G, HHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were re‐transfected with LAMP1‐RFP (red) for 24 h. And then, the cells were incubated with Earle's balanced salt solution (starvation) for 2 h. DAPI (blue) was used for nuclear staining. Cells were fixed and visualized using confocal microscopy. Scale bar, 5 µm. The quantification of lysosomes from the nucleus boundary is shown in Figure EV5H. Data are from three independent experiments, means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
Figure 7. Altered dynactin 1‐associated lysosome positioning and autophagosome–lysosome fusion in TDP‐43‐depleted cells.
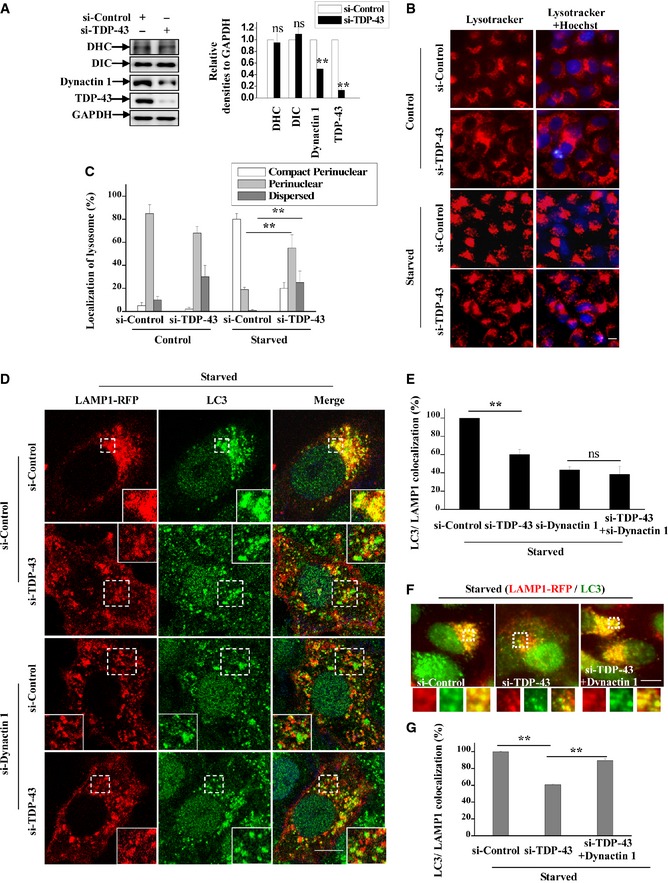
-
AHeLa cells were transfected with the indicated siRNAs. Seventy hours after transfection, the lysates from the cells were subjected to immunoblot analysis using anti‐TDP‐43, ‐DHC, ‐DIC, ‐dynactin 1, and ‐GAPDH antibodies. The relative densities are shown on the right side. Data from three independent experiments were represented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
B, CHeLa cells were transfected with the indicated siRNAs. Seventy hours after transfection, the cells were incubated with Earle's balanced salt solution (starvation) for 2 h. Then, the cells were stained with Lysotracker (red). Hoechst (blue) was used for nuclear staining. The stained cells were visualized using microscope IX71. Scale bar, 5 μm. The quantitative data of the localization of lysosomes are shown in (C). Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
D, EHeLa cells were transfected with the indicated siRNAs. Forty‐eight hours after transfection, the cells were re‐transfected with LAMP1‐RFP (red) for 24 h. And then, the cells were incubated with Earle's balanced salt solution (starvation) for 2 h. The cells were stained with anti‐LC3 antibody (green) and DAPI (blue). Cells were visualized using a confocal microscopy. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm. The quantification data of the co‐localization of LC3 and LAMP1 is shown in (E). Data from three independent experiments represented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
-
F, GHeLa cells were transfected with the indicated siRNAs. After 48 h, the cells were transfected with LAMP1‐RFP (red), with or without myc‐dynactin 1 for 30 h. And then, the cells were incubated with Earle's balanced salt solution (starvation) for 2 h. The cells were stained with anti‐LC3 (green) antibody. Cells were visualized using microscope IX71. Regions within the dotted boxes are magnified in the insets. Scale bar, 5 μm. The quantification data of the co‐localization of LC3 and LAMP1 are shown in (G). Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
Drosophila TDP‐43, TBPH, affects mTORC1 activity and expression level of lysosomal and autophagic genes
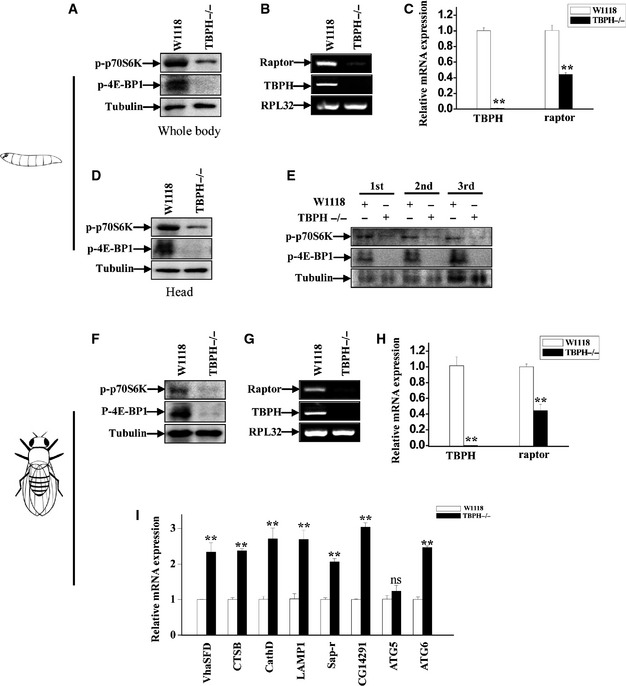
To further verify whether TDP‐43 depletion could affect mTORC1 activity in vivo, we measured mTORC1 activity by detecting the phosphorylation of S6K and 4E‐BP1 (another mTORC1 substrate) in TBPH−/− larvae and adults. In TBPH−/− larvae, compared with W1118 control larvae, the phosphorylation levels of S6K and 4E‐BP1, but not expression of S6K mRNA, were decreased (Fig 8A and D, and Appendix Fig S6A). Furthermore, the level of phosphorylated S6K and 4E‐BP1 was consistently decreased in the first‐, second‐, and third‐instar TBPH−/− larvae (Fig 8E). As we have shown that raptor is regulated by TDP‐43 in cells, we therefore examined the mRNA levels of raptor in W1118 and TBPH−/− larvae. Similarly as the changes observed in TDP‐43‐depleted cells, the expression levels of raptor were decreased (Fig 8B and C). Similar results as above were observed in TBPH−/− adult flies (Fig 8F–H). In addition, the expression level of lysosomal and autophagic genes such as VhaSFD, CTSB, CathD, LAMP1, Sap‐r, CG14291, and ATG6 was increased in TBPH‐depleted adult flies (Fig 8I). In addition, EM analysis showed a significant blockage of autophagosome–lysosome fusion in starved TBPH−/− larvae (Appendix Fig S5).
Figure 8. mTORC1 activity was strikingly impaired in TBPH‐knockout flies.
-
AW1118 and TBPH−/− larvae were blotted using antibodies against phosphorylated p70S6K (T398), phosphorylated 4E‐BP1 (T37/46), and tubulin.
-
BTotal RNA was prepared from W1118 and TBPH−/− larvae and subjected to RT–PCR analysis. Ribosomal protein L32 (RPL32) was used as a loading control.
-
CTotal RNA was prepared from W1118 and TBPH−/− larvae and subjected to qRT–PCR analysis. The level of TBPH and raptor was quantified and normalized relative to RPL32. Data from three independent experiments represented as means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
DThe heads of W1118 and TBPH−/− larvae were cut and blotted using antibodies against phosphorylated p70S6K (T398), phosphorylated 4E‐BP1 (T37/46), and tubulin.
-
EThe first‐, second‐ and third‐instar larvae of W1118 and TBPH−/− were blotted using antibodies against phosphorylated p70S6K (T398), phosphorylated 4E‐BP1 (T37/46), and tubulin.
-
F–HSimilar experiments as in (A–C) were performed in W1118 and TBPH−/− adult flies. Data from three independent experiments, means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
-
ISimilar experiments as in (C) were performed in W1118 and TBPH−/− adult flies. The mRNA levels of lysosomal and autophagic genes were quantified and normalized relative to GAPDH. The data from three independent experiments are presented as means ± S.E.M.; ns, not significantly different; **, P < 0.01; one‐way ANOVA.
Source data are available online for this figure.
Rapamycin and PA conversely affect larval locomotion and developmental viability of TBPH−/− flies
To understand the role of decreased mTORC1 activity in TDP‐43‐mediated neurodegeneration, we employed two drugs, rapamycin (an mTOR inhibitor) and PA (phosphatidic acid, an mTOR agonist), to examine their effects on TBPH−/− flies. TBPH−/− flies showed impaired developmental viability, such as defects in larval locomotion, eclosion, and motility, compared to the W1118 control flies, consistent with the findings by other investigators (Feiguin et al, 2009). Interestingly, we further observed that an administration of rapamycin strongly aggravated, whereas PA ameliorated, TBPH−/− larval locomotion defects (Fig 9A and B) Effects of rapamycin and PA on phosphorylated S6K in those flies are shown in Appendix Fig S6B and C. In addition, a higher percentage of TBPH−/− flies treated with rapamycin did not arrive to pupal, enclosed, and mature stages compared with untreated TBPH−/− flies (Fig 9C), suggesting that the decreased mTORC1 activity could worsen the neurodegenerative phenotypes, whereas PA treatment could improve the developmental viability of TBPH−/− flies (Fig 9C). Taken together, these data indicate that an enhancement of the impaired mTORC1 activity could ameliorate TDP‐43‐linked neurotoxicity.
Figure 9. Effects of rapamycin and PA treatments on TBPH −/− flies.
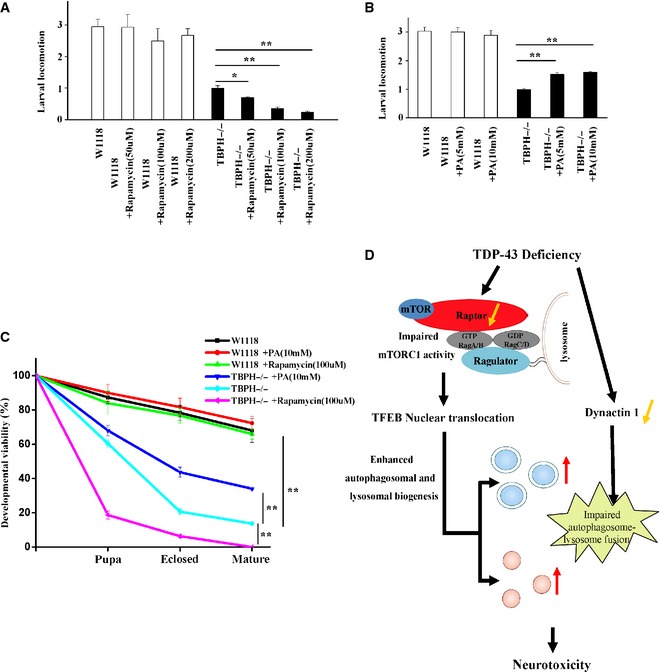
- The first‐instar TBPH−/− larvae were treated with rapamycin for 72 h. Then, the third‐instar larvae of W1118 and TBPH−/− were subjected to larval locomotion assays. Data from three independent experiments represented as means ± S.E.M.; *, P < 0.05; **, P < 0.01; one‐way ANOVA.
- Similar experiments as in (A) were performed, but using PA instead of rapamycin. Data are from three independent experiments, means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
- The first‐instar TBPH−/− larvae were selected to standard food containing rapamycin and PA. Quantitative developmental viability analysis of W1118, TBPH−/−, and TBPH−/− treated with rapamycin and PA. Data are from three independent experiments, means ± S.E.M.; **, P < 0.01; one‐way ANOVA.
- A schematic model illustrates the molecular mechanism by which loss of TDP‐43 enhances TFEB‐mediated expression of ALP genes, blocks autophagosome–lysosome fusion, and induces neurodegeneration. TDP‐43 directly targets raptor, a key component of mTORC1, to regulate mTORC1–TFEB signaling. Meanwhile, TDP‐43 can affect the autophagosome–lysosome fusion in a dynactin 1‐dependent manner. The current study highlights the role of mTORC1 and dynactin 1 dysfunction, altered autophagy and lysosomal biogenesis in TDP‐43‐mediated neurotoxicity.
Discussion
Since cytoplasmic TDP‐43 aggregation is accompanied by a loss of TDP‐43 function in cells, it is presently unclear whether the TDP‐43‐mediated neuropathological changes are caused by a toxic gain of function or by a loss of normal function (Lee et al, 2012). However, based on a series of recent studies, a loss‐of‐function mechanism, rather than a gain‐of‐function mechanism, may be the major factor involved in TDP‐43‐related proteinopathies (Xu, 2012; Diaper et al, 2013; Vanden Broeck et al, 2013, 2014). In supporting with this notion, numerous animal models with TDP‐43 loss of function, such as TDP‐43 knockdown or knockout mice, flies, and worms, have been demonstrated to have a neurodegenerative phenotype (Feiguin et al, 2009; Fiesel et al, 2010; Lin et al, 2011; Vaccaro et al, 2012; Wu et al, 2012; Zhang et al, 2012; Iguchi et al, 2013; Yang et al, 2014). Loss of TDP‐43 function results in a dysregulation of RNA pathways, such as RNA splicing, transport, and stability, leading to motor neuron neurodegeneration (Lee et al, 2012). Although a lot of TDP‐43 RNA targets have been identified according to the previous reports (Polymenidou et al, 2011; Ling et al, 2013), the molecular mechanism by which loss of TDP‐43 causes neurotoxicity remains elusive. Here, our findings reveal that raptor, a key component of mTORC1, as a new TDP‐43‐binding target (Fig 4). In TDP‐43‐depleted cells, raptor deficiency‐mediated mTORC1 dysfunction induces TFEB nuclear translocation (Fig 3) and enhances gene expressions in ALP (Fig 5), but not the autophagosome–lysosome fusion, which results in accumulated immature autophagic vesicles and overwhelmed overall ALP function (Fig 6). Thus, our findings provide evidence that TDP‐43‐linked neurotoxicity is triggered by a loss of normal TDP‐43 function and that raptor is the molecular target in TDP‐43‐mediated mTORC1–TFEB pathway (Figs 2, 3, 4).
The present study highlights the role of mTORC1–TFEB signaling in TDP‐43‐linked neurodegeneration (Fig 9D). TDP‐43 regulates mTORC1 activity by specifically targeting mTORC1 component raptor, but not other factors associated with mTORC1 activity, such as Ragulator or Rag GTPases (Figs 2, 3, 4). Given that Ragulator‐Rag complex can target mTOR to lysosomes and is necessary for its activation by amino acids (Fig 9D), we thought to examine the effect of TDP‐43 on Ragulator and Rag GTPases. Our data indicate that TDP‐43 does not affect the expression or cellular localization of p18, the key component of Ragulator complex, or Rag GTPases (Figs 2B and E, and 3D). Our results further suggest that TDP‐43 acts on raptor (Figs 2B and E, and 4), a component and an adaptor of mTORC1, to dissociate mTOR from lysosomes.
Raptor interacts with Rag GTPases and targets mTOR to lysosomes where mTOR can phosphorylate TFEB on serine 211, and then, phosphorylated TFEB would stay in the cytosol by interacting with 14‐3‐3 (Roczniak‐Ferguson et al, 2012; Martina & Puertollano, 2013). We note that knockdown of TDP‐43 not only leads to a nuclear transport of TFEB, but also leads to an increase of TFEB lysosomal localization (Fig 3A–C). In raptor‐deficient cells, an inactivation of mTOR inhibits the interactions between TFEB and 14‐3‐3, leading to an enrichment of TFEB in both nuclei and lysosomes (Martina & Puertollano, 2013). An explanation of this lysosomal enrichment is that dissociation of the cytosolic TFEB‐14‐3‐3 complex will cause a more stable interaction between TFEB and Rag GTPases on lysosome membrane (Martina et al, 2012; Roczniak‐Ferguson et al, 2012; Martina & Puertollano, 2013). Thus, Rag GTPases are the direct basis for TFEB lysosomal localization, and raptor indirectly modulates TFEB lysosomal localization with mTOR and 14‐3‐3. Based on this, we conclude that TDP‐43 regulates TFEB lysosomal localization by specifically targeting raptor, but not Rag GTPases. In support of this notion, the effects of TDP‐43 on TFEB lysosomal localization are diminished in raptor re‐transfected cells (Fig 3A).
As neurons are not capable of self‐renewal and cell division, they are quite susceptible to various cellular stress and stimuli. Thus, the protein quality control system such as autophagy is essential for neuronal cells to maintain cellular homeostasis to clear abnormally accumulated proteins (Feiguin et al, 2009; Wong & Cuervo, 2010; Chua et al, 2014). With this in mind, it is reasonable that neurons can exhibit a higher level of ALP‐associated autophagosome turnover, which is particular important in disease conditions (Boland et al, 2008). Although the current view of autophagy in neurons is that an induction of autophagy helps to remove cytoplasmic aggregate‐prone proteins in neurodegenerative diseases, which is considered to exhibit therapeutic benefit (Harris & Rubinsztein, 2012), the manner by which autophagy affects ALS is controversial. Recent reports showed that an induction of defective autophagy by blocking mTOR activity could actually exacerbate neurotoxicity and worsen disease in ALS (Zhang et al, 2011; Ching et al, 2013), whereas a reduction of autophagy had a rescue effect to the disease animals (Nassif et al, 2014). In accordance with these studies, our findings show that an inhibition of mTOR by rapamycin impairs larval movement and developmental viability of TBPH−/− flies, whereas mTOR activation with PA treatment benefits TBPH−/− flies (Fig 9), suggesting that a rescue of impaired mTOR activity which in turn suppresses the enhanced autophagy would have be benefit for TDP‐43‐mediated neurodegeneration.
In our observations, immature autophagic vesicles strikingly accumulated in TDP‐43‐depleted cells and flies (Fig 6B and D, and Appendix Fig S5), similar to the phenomena in other neurodegenerative disease and ALS models (Wong & Cuervo, 2010; Iguchi et al, 2013; Chua et al, 2014), suggesting that an impairment of ALP in those models. Relevant to this, we observed that a depletion of TDP‐43 has both mTORC1‐dependent and mTORC1‐independent effects on ALP (Fig 9D). Our data demonstrate that the autophagosomal and lysosomal biogenesis are increased through mTORC1 inhibition and TFEB activation in TDP‐43‐deficient cells, whereas the autophagosome–lysosome fusion, which is the later step of ALP, is impaired through a mTORC1‐independent manner (a dynactin 1‐associated manner) in those cells (Fig 7). Regarding the mTORC1‐independent manner, our data showed that re‐transfection of dynactin 1 could rescue the impaired autophagosome–lysosome fusion in TDP‐43‐deficient cells (Fig 7F and G), suggesting that TDP‐43 deficiency could impair autophagosome–lysosome fusion by affecting dynactin 1. Our data indicate that impaired autophagosome–lysosome fusion might be a cause of defective autophagic degradation in TDP‐43‐depleted cells (Fig 6G). Importantly, we reason that as autophagosome synthesis is faster than the autophagosome–lysosome fusion, chronic accumulated autophagosomes in disease progress would ultimately induce toxicity. Since increased autophagic vesicles that fail to go through lysosomal turnover would accumulate around the overwhelmed lysosomes, the accumulated autophagic cargoes such as damaged mitochondria may release toxic species such as reactive oxygen species which would impair lysosomal function. In this situation, the incoming newly synthesized autophagic vesicles could play as a cellular “stressor” that impairs cellular function including ALP function and cell homeostasis. This may just explain our data of TBPH−/− flies that an activation of autophagy by rapamycin aggravates but an inhibition of autophagy by PA ameliorates the phenotype of TBPH−/− flies. Hence, strategies to simply stop the incoming autophagic vesicles, or more efficiently, to enhance fusion of autophagosome and lysosome that could save the ALP function in the stressed cells, could be helpful for developing effective treatments.
In summary, our study demonstrates that TFEB nuclear translocation and sustained activation are caused by a decreased raptor level and an impaired mTORC1 signaling, leading to abnormal enhanced autophagy and lysosomal biogenesis in TDP‐43‐depleted cells. Moreover, more autophagosomes and lysosomes are accumulated in the cells since the fusion between autophagosomes and lysosomes is blocked due to dynactin 1 deficiency in those cells. This stressful situation in turn impairs cell homeostasis and induces neurological damage (Fig 9D). It is helpful for both understanding the basic mechanism in TDP‐43‐associated neurotoxicity and developing therapeutic treatments of related neurodegenerative disorders.
Materials and Methods
Plasmid constructs
The 3×FLAG‐TFEB plasmid (Sardiello et al, 2009) was kindly provided by Dr. Andrea Ballabio (Telethon Institute of Genetics and Medicine, Italy). EGFP‐N3‐tagged TFEB was generated by excising full‐length TFEB cDNA from 3×FLAG‐TFEB and inserting it into the pEGFP‐N3 (Clontech) vector at HindIII/BamHI sites. The point mutations and deletion mutants of 3×FLAG‐TFEB were generated by the site directed mutagenesis using MutanBEST kit (Takara) with following primers: 5′‐GACCCAGAAGCGAGAGCTCACAGAT‐3′ and 5′‐AGGTCCGCAGGGCAGGCGCTGCTGGTGA‐3′ for 3×FLAG‐TFEB S211A in which the serine 211 was converted to alanine; 5′‐GCCATGCGGGAGCAGGCG‐3′ and 5′‐CGCCATGCGCAACCCTAT‐3′ for 3×FLAG‐TFEB Q10A/L11A in which the glutamine 10 and leucine 11 were converted to alanines; 5′‐CGCTTCAATTAAGTTGTGATT‐3′ and 5′‐GCAGCAAGGTTCAACATCAAT‐3′ for 3×FLAG‐TFEB R245‐247A in which the arginine 245‐247 were converted to alanines; 5′‐ATGCATTACATGCAGCAGCA‐3′ and 5′‐CGCGGCCGCAAGCTTAATT‐3′ for 3×FLAG‐TFEB lacking 1–30 amino acids (Δ30). The point mutations and deletion mutants of EGFP‐N3‐TFEB were generated by excising TFEB mutants from above‐mentioned 3×FLAG‐TFEB constructs and inserting them into the pEGFP‐N3 vector at HindIII/BamHI sites. HA‐TDP‐43 was created by subcloning TDP‐43 PCR product from pGEX‐5x‐1‐TDP‐43 (Wang et al, 2010) into pKH3‐HA vector at HindIII/XbaI sites. FLAG‐tagged TDP‐43 was generated by excising TDP‐43 cDNA from HA‐TDP‐43 and inserting it into the p3×FLAG‐CMV24 vector at HindIII/XbaI sites. The deletion mutants of FLAG‐TDP‐43 were generated by using the following primers: 5′‐GTTCAGCATTAATCCAGCCAT‐3′ and 5′‐ACTTCTTTCTAACTGTCTATT‐3′ for FLAG‐TDP‐43 Δ274–314 (ΔGRD); 5′‐GGACGATGGTGTGACTGCAAA‐3′ and 5′‐GGATGTTTTCTGGACTGCTCT‐3′ for FLAG‐TDP‐43 Δ105–169 (ΔRRM1); 5′‐TCCAATGCCGAACCTAAGCAC‐3′ and 5′‐CACTTTTCTGCTTCTCAAAGG‐3′ for FLAG‐TDP‐43 Δ194–257 (ΔRRM2); 5′‐GGACGATGGTGTGACTGCAAA‐3′ and 5′‐GGATGTTTTCTGGACTGCTCT‐3′, 5′‐TCCAATGCCGAACCTAAGCAC‐3′ and 5′‐CACTTTTCTGCTTCTCAAAGG‐3′ for FLAG‐TDP‐43 Δ105–169 and Δ194–257 (ΔRRM1 + ΔRRM2). The following plasmids were obtained from Addgene: plasmid 1817, LAMP1‐RFP; plasmid 8513, HA‐raptor; plasmid 19300, pRK5‐HA GST RagAGTP (RagA Q66L); plasmid 19305, pRK5‐HA GST RagCGDP (RagC S75L); plasmid 22418, mCherry‐EGFP‐LC3. The CD63‐GFP plasmid was a kind gift from Dr. Shumin Duan (Zhejiang University, China), and the myc‐tagged dynactin 1 (myc‐dynactin 1) plasmid was a kind gift from Dr. Jia‐Jia Liu (Institute of Genetics and Developmental Biology, CAS, China). FLAG‐TDP‐25 was created by subcloning PCR products using primers previously described (Wang et al, 2010). The EGFP‐LC3, FLAG‐p62, and Nhtt‐150Q‐EGFP expression vectors were described previously (Ying et al, 2009; Li et al, 2010; Zhou et al, 2013).
All constructs were confirmed by sequencing.
Antibodies
The following primary antibodies were used: anti‐FLAG antibody (Sigma), anti‐GFP antibody (Santa Cruz), anti‐GAPDH antibody (Millipore), anti‐tubulin antibody (Santa Cruz), anti‐TDP‐43 antibody described previously (Wang et al, 2010), anti‐TDP‐43 antibody (Proteintech), anti‐HA (Santa Cruz), anti‐histone 2B antibody (Epitomics), anti‐phospho‐p70S6K antibody (T389) (Cell Signaling Technology), anti‐p70S6K antibody (Epitomics), anti‐mTOR antibody (Cell Signaling Technology), anti‐raptor antibody (Cell Signaling Technology), anti‐RagB antibody (Cell Signaling Technology), anti‐p18 antibody (Cell Signaling Technology), anti‐TFEB antibody (Cell Signaling Technology), anti‐LAMP1 antibody (Abcam), anti‐cathepsin L antibody (Abcam), anti‐LC3 antibody (Novus Biologicals), anti‐Beclin‐1 antibody (Abcam), anti‐LAMP2 antibody (Epitomics), anti‐ATG5 antibody (Epitomics), anti‐phospho‐drosophila‐p70S6K antibody (T398) (Cell Signaling Technology), anti‐phospho‐4E‐BP1 antibody (T37/46) (Cell Signaling Technology), and anti‐p62 antibody (Enzo life science).
The following secondary antibodies were used: horseradish peroxidase‐conjugated sheep anti‐mouse and anti‐rabbit antibodies (Amersham Pharmacia Biotech). The proteins were visualized with an ECL detection kit (Amersham Biosciences).
The following fluorescent secondary antibodies were used: anti‐rabbit Rhodamine (Santa Cruz Biotechnology), Alexa Fluor 488 donkey anti‐mouse IgG (Invitrogen), Alexa Fluor 488 donkey anti‐rabbit IgG (Invitrogen), Alexa Fluor 594‐conjugated Affinipure Donkey Anti‐mouse IgG (Invitrogen), and Alexa Fluor 594‐conjugated Affinipure Donkey Anti‐rabbit IgG (Invitrogen).
Cell culture, transfection, and drug treatment
Human cervical cancer cells (HeLa), HeLa cells stably expressing EGFP‐LC3 (Wang et al, 2010), human alveolar epithelial cells (A549), human embryonic kidney 293 cells (HEK 293), human embryonic kidney 293FT cells (HEK 293FT), mouse hippocampal neuronal cells (HT22), rat adrenal medulla cells (PC12), human neuroblastoma cells (SH‐SY5Y), ATG5 WT, and KO MEF cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Gibco) containing 10% fetal bovine serum (FBS) (Gibco) with penicillin (100 mg/ml) and streptomycin (100 mg/ml). Cells were transfected with siRNAs using the RNAiMAX transfection reagent upon splitting and transfected with plasmids using Lipofectamine 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Cells were starved with Earle's balanced salt solution (Gibco) for 2 h and re‐feeding with the RPMI‐1640 medium (Gibco) supplemented with 10% FBS. Cells were treated with DMSO (Sangon Biotech), Torin‐1 (250 nM) (Tocris Bioscience) for 1 h, bafilomycin A1 (100 nM) (Sigma) for 24 h or actinomycin D (1 μg/ml) (Sigma) for 1–4 h.
siRNAs
The following primers were used: human TDP‐43: 5′‐CACTACAATTGATATCAAA‐3′, mouse TDP‐43: 5′‐GGATCTGAAAGACTATTTC‐3′, rat TDP‐43: 5′‐CCAATGCTGAACCTAAGCA‐3′, human TFEB‐1: 5′‐GAGACGAAGGTTCAACATCAA‐3′, human TFEB‐2: 5′‐ GAACAAGTTTGCTGCCCACAT‐3′, human raptor‐1: 5′‐TGGCTAGTCTGTTTCGAAA‐3′, human raptor‐2: 5′‐GGGAGAAGCTGGATTATTT‐3′, human p18: 5′‐CCAAGGAGACCGUGGGCUU‐3′, human RagA‐1: 5′‐CCAACUUCGCUGCUUUCAU‐3′, human RagA‐2: 5′‐CCCGGAAACACUUUGAGAA‐3′, human RagB‐1: 5′‐GGACAUGCACUAUUACCAAT T‐3′, human RagB‐2: 5′‐GCUUGGUCCAGCAUAGUUUTT‐3′, human si‐KIF5B: 5′‐ GCCUUAUGCAUUUGAUCGG‐3′, human si‐DHC: 5′‐GAGAGGAGGTTATGTTTAA‐3′, human si‐dynactin 1: 5′‐CGAGCTCACTACTGACTTA‐3′.
Subcellular fractionation
Cells were harvested following by washing with ice‐cold PBS (pH 7.4) three times and re‐suspended with ice‐cold sucrose buffer containing 10 mM sucrose, 1 mM CaCl2, 10 mM MgAc, 2.5 mM EDTA, 1 mM DTT, 1 mM PMSF, and 0.5% NP‐40. Cells were incubated on ice for 20 min and then were separated into supernatants (cytoplasm) and pellet (nucleus) by centrifugation at 600 g for 15 min at 4°C. Subsequently, the pellet was washed twice with sucrose buffer without NP‐40 and finally re‐suspended in cell lysis buffer.
Immunoblot
Cells, Drosophila larvae, and adult flies were lysed in the cell lysis buffer containing 50 mM Tris–HCl (pH 7.6) with protease inhibitor cocktail (Roche), 150 mM NaCl, 0.5% sodium deoxycholate, and 1% Nonidet P‐40. Proteins were separated by 10% or 13.5% SDS–PAGE (polyacrylamide gel electrophoresis) and transferred onto a PVDF membrane (polyvinylidene difluoride membrane; Millipore).
Immunofluorescence and live cell staining
HeLa, HEK 293, and SH‐SY5Y cells were washed with PBS (pH 7.4) (Gibco) and fixed with 4% paraformaldehyde for 10 min at room temperature. Then, the cells were permeabilized with 0.25% Triton X‐100 and preblocked with 0.2% fetal bovine serum for 5 min. Cells were incubated with the primary antibodies for 6 h, subsequently with the fluorescent secondary antibodies for 2 h and following with DAPI (Sigma) for 5 min. The stained cells were visualized using an inverted system microscope IX71 (Olympus) or a Zeiss LSM710 confocal microscope.
The live cells were stained with Lysotracker red DND‐99 (Invitrogen) and Hoechst (Sigma) for 10 min, and then, the average Lysotracker fluorescence per cell was quantified using the ImageJ software.
Electron microscopy
HeLa cells were centrifuged at 2,000 rpm for 15 min at 4°C following by addition of a drop of glutaraldehyde to the cell suspension. Then, the cells were fixed with 2.5% glutaraldehyde in 0.2 M Na‐phosphate buffer (pH 7.4) at 4°C for 2 h. The fat bodies of starved third‐instar W1118 and TBPH−/− larvae were dissected and fixed with 2.5% glutaraldehyde in 0.2 M Na‐phosphate buffer (pH 7.4) at 4°C for 2 h.
MTT assay
Cells were washed with PBS and incubated with MTT (3‐(4,5)‐dimethylthiahiazo(‐z‐y1)‐3,5‐di‐phenytetrazoliumromide, Sigma) for 1 h. Then, the formazan crystals were dissolved in DMSO. Subsequently, the optical density (OD) was measured by a photometer at 570 nm, and background at 630 nm was subtracted.
Quantitative real‐time PCR (qRT–PCR) and RT–PCR
Total RNA from Drosophila larvae, adult flies, HeLa, and SH‐SY5Y cells were extracted with TRIzol reagent (Invitrogen) and then were reverse‐transcribed into cDNA using PrimeScript RT Master Mix (Takara). The analysis of real‐time PCR was used for SYBR Green Real‐Time PCR Master Mix (Takara) with relevant primers using a CFX96 Real‐Time System (Bio‐Rad).
The following primers were used: human GAPDH: 5′‐AAATCCCATCACCATCTTCCAG‐3′ and 5′‐AGGGGCCATCCACAGTCTTCT‐3′, human raptor: 5′‐ACTGATGGAGTCCGAAATGC‐3′ and 5′‐TCATCCGATCCTTCATCCTC‐3′, human ATG5: 5′‐TGGATTTCGTTATATCCCCTTTAG‐3′ and 5′‐CCTAGTGTGTGCAACTGTCCA‐3′, human Beclin‐1: 5′‐CAAGATCCTGGACCGTGTCA‐3′ and 5′‐TGGCACTTTCTGTGGACATCA‐3′, human ATG9B: 5′‐TGTTCCTTTGCCCTTATGGATGTG‐3′ and 5′‐GCCCAAGAAACTTAGAGCTGTGCC‐3′, human p18: 5′‐AGCCTGACCCATTGGAAGAAG‐3′ and 5′‐CTGTAGGCATAAGCAGCTATCCTG‐3′, human FLCN: 5′‐TGCAGGTGCTGGTGAAGGTAACCT‐3′ and 5′‐GGGATTGGGCAAGTCAGATGCTTG‐3′, human RagC: 5′‐ACCCAACGAGACCCTCTTTT‐3′ and 5′‐TATCAATGCTCCTGTTCCCC‐3′, human RagD: 5′‐GACAAAGTTCCTGGCTCTCG‐3′ and 5′‐TTTTTCTTCTGCAGCCGATT‐3′, human RagA: 5′‐AGAACTCTCCTGACGCCAA A‐3′ and 5′‐GACGTTCGAAAACAAGCACA‐3′, human RagB: 5′‐GTACAGGAGGATCAACGGGA‐3′ and 5′‐TTTCCAGCTGCTGAACATTG‐3′, human mTOR: 5′‐GAAACTTGGAGAGTGGCAGC‐3′ and 5′‐GTTTCTTCTTCTCATCGCGG‐3′, human p62: 5′‐GTGGTAGGAACCCGCTACAA‐3′ and 5′‐GAGAAGCCCTCAGACAGGTG‐3′, human HBXIP: 5′‐AGCAGCACTTGGAAGACACA‐3′ and 5′‐CTCCAGCATGCTCATCTGAC‐3′, human C7orf59: 5′‐GGACCTGGAGAATGATGAGC‐3′ and 5′‐CTGCCTCTTCACCACAAACA‐3′, human p14: 5′‐GCTGGCCTACTCTGGTTACG‐3′ and 5′‐ACGGTCTCCTTGGCATACAT‐3′, human MP1: 5′‐CTCCATGCCATTGTTGTGTC‐3′ and 5′‐CCAAGTTTGCTTCCTTGGTC‐3′, human p18: 5′‐AGCCTGACCCATTGGAAGAAG‐3′ and 5′‐CTGTAGGCATAAGCAGCTATCCTG‐3′, human TDP‐43: 5′‐TGGGAATCAGGGTGGATTTG‐3′ and 5′‐CCCAGCCAGAAGACTTAGAATC‐3′, human syntaxin‐17: 5′‐CTCCGATCCAATATCCGAGA‐3′ and 5′‐GATCTGGTCAAAGGAGGCTG‐3′, human SNAP‐29: 5′‐CAGGAAGCAAAGTACCAGGC‐3′ and 5′‐TTGCTGTCGATCTTCTGGTG‐3′, human VAMP8: 5′‐CATCTCCGCAACAAGACAGA‐3′ and 5′‐TGGCAAAGAGCACAATGAAG‐3′, human VPS33A: 5′‐ATTGTATCGCCCAAAAGCAC‐3′ and 5′‐CCATCCAGAGGCGTAATGTT‐3′, human VPS16: 5′‐GGTCAAGGCTTTGCTTCTTG‐3′ and 5′‐TGAATCTTGTCAGCTGTGGC‐3′, human hVPS41: 5′‐CATGGGTGTGTGTTCAGAGG‐3′ and 5′‐ATGTGGGTGCACAGCAATAA‐3′, human Rab7: 5′‐CTCTCGGTGTGGCCTTCTAC‐3′ and 5′‐CTGGCCTGGATGAGAAACTC‐3′, human DCTN1: 5′‐ATTTGCAAGGCAGAGCTGAT‐3′ and 5′‐GTGCAGCAGTTCAATGAGGA‐3′, human MYO1C: 5′‐GAAGTGGAGGACCTGCTGAG‐3′ and 5′‐GACGAGCCAGGTAAAAGTGC‐3′, human DYNC1H1: 5′‐CAGCCGGCTAAAAATGAGAG‐3′ and 5′‐TGCGTCTCCACTTCAATCTG‐3′, human DNAI1/DIC: 5′‐TCTCTGTGTCATCTGACGGC‐3′ and 5′‐TGTGGAAGTCAAAGGCAGTG‐3′, human DYNLL1: 5′‐AAAGGCCGTGATCAAAAATG‐3′ and 5′‐AGCCGCAATGTCCTTCTCTA‐3′, human DYNC1LI2: 5′‐AGTGTCCTGGAGAAGGAGCA‐3′ and 5′‐CGGCTTCACGGTTGTAAAAT‐3′, human ARL8B: 5′‐TTCGCTCTTCTGGAAGGAAG‐3′ and 5′‐TCACTGAATTGACCTGACGC‐3′, human myrlysin: 5′‐AGCTCTGCCTCCGATATCAA‐3′ and 5′‐CTTGGCGTATCTTTTCTGGC‐3′, human SERCA: 5′‐AGGCCAAGGGTGTGTATGAG‐3′ and 5′‐TGCAGTACACGGACATGGAT‐3′, human PLEKHM1: 5′‐GGCGAGTTCCTCCTTAGCTT‐3′ and 5′‐ATCTTATGGCCCGAGTGATG‐3′, the primers to analyze human genes including ARSB, ATP6V0E1, ATP6V1H, CLCN7, CTSA, CTSB, CTSD, CTSF, GALNS, GLA, GNS, HEXA, LAMP1, MCOLN1, NAGLU, NEU1, PSAP, SGSH, TMEM55B, and TPP1 were described elsewhere (Sardiello et al, 2009; Efeyan et al, 2013).
The following primers were used to analyze Drosophila genes: TBPH: 5′‐TGGCCCAGATCAAGAAGGAC‐3′ and 5′‐TTACCTCGGTGGTGTCCGTT‐3′, raptor: 5′‐CAGTGGCCTACAGGGAGAAG‐3′ and 5′‐AAAATCCACCTCCACCTCCT‐3′, VhaSFD: 5′‐GCTGAAGCAGCTATCCATCC‐3′ and 5′‐GGCGTACTCATCAAAGGAGC‐3′, CTSB: 5′‐TACACCGTCGATTATGCCAA‐3′ and 5′‐TCGTGCTGATAGACACCGTC‐3′, CathD: 5′‐ACCAAGAACGGCACTGAGTT‐3′ and 5′‐GTCCACCGAGATGGAGTTGT‐3′, LAMP1: 5′‐GCTTATTACCAGGAGCGACG‐3′ and 5′‐GCAATCGTGTCCAACTGAGA‐3′, Sap‐r: 5′‐AGCAACCAGACAGAGGAGGA‐3′ and 5′‐GTCTCCTCGGAGCTGTCATC‐3′, CG14291: 5′‐GGCAGATCAGACACTCGTGA‐3′ and 5′‐GATCTGCCAACGATTTTGGT‐3′, ATG5: 5′‐GTGACTGACAAGGTTCGCAA‐3′ and 5′‐GCTTAAGCACATCTGCCTCC‐3′, ATG6: 5′‐TTCCAGAAGGAGGTCGAGAA‐3′ and 5′‐ATTTCAGTGCCTTGGTCCAC‐3′, RPL32: 5′‐CGCCGCTTCAAGGGACAGTATC‐3′ and 5′‐CGACAATCTCCTTGCGCTTCTT‐3′, ds6k: 5′‐AGCGTGAGGGCATCTTCTTA‐3′ and 5′‐CCATGTACTCAATTGTGCCG‐3′.
RNA immunoprecipitation
HeLa cells were lysed in the polysome lysis buffer containing 100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.5% NP‐40, 1 mM DTT, 100 units/ml RNase inhibitor, and protease inhibitor cocktail. Then, cell lysates were centrifuged at 15,000 g for 15 min at 4°C following incubation on ice for 5 min. The supernatants were added to protein G beads supplemented with 5% BSA and anti‐FLAG antibody, which was previously incubated overnight at 4°C, and then, the beads mixture was incubated for 4 h at 4°C following the addition of 200 units of RNase inhibitor, 100 mM DTT, and 20 mM EDTA. Subsequently, the beads were washed with wash buffer (50 mM Tris–HCl (pH 7.4), 150 mM NaCl, 1 mM MgCl2, and 0.05% NP‐40) five times. The washed beads were re‐suspended with 100 μl of wash buffer supplemented with 30 μg of proteinase K and then incubated for 20 min at 55°C. Finally, RNA was purified by adding TRIzol reagent (Invitrogen) according to the manufacturer′s instructions and analyzed by RT–PCR.
Flow cytometry
HeLa cells were digested with 0.05% trypsin for 2 min. The cells were harvested and washed with 1× PBS three times, and then, the cells (10,000) were analyzed at 575 nm on a flow cytometer (Beckman coulter, FC500).
Click‐iT technology
Newly transcribed RNA was prepared and isolated according to the manual of Click‐iT Nascent RNA Capture Kit (Life Technologies). Briefly, newly transcribed RNA in cells was metabolically labeled by the uridine analog 5‐ethynyl uridine (EU) for 1 h, and then, EU was attached to biotin. The labeled RNA was collected on streptavidin beads and subjected to qRT–PCR analysis.
Fly stocks and culture
W1118 flies were obtained from Institute of Biochemistry and Cell Biology, SIBS, CAS. TBPH−/− flies were kindly provided by Dr. Francisco Baralle (International Centre for Genetic Engineering and Biotechnology, Italy). All fly stocks were kept on standard food (yeast, cornmeal, and molasses) according to standard procedures at 25°C.
Drosophila drug treatment
Rapamycin (50 μM, 100 μM or 200 μM) (Selleck) in DMSO or PA (phosphatidic acid) (5 mM or 10 mM) (Guidechem) in chloroform was added to Drosophila food. DMSO or chloroform alone was used as a control.
Larval movement
Thirty third‐instar larvae per genotype were washed with distilled water and then placed on a LB agar plate. Larvae were allowed to recover for 30 s. Subsequently, the number of peristaltic waves was counted over two minutes. The average movement distance of the larvae per genotype was analyzed using ImageJ software.
Author contributions
QX, JH, ZY, and GW designed research; QX, HW, ZH, CF, QH, FG, HR, DC, and ZY performed experiments; QX, HW, ZY, and GW analyzed data; QX, HW, ZY, and GW wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank Dr. Francisco Baralle (International Centre for Genetic Engineering and Biotechnology, Italy) for TBPH−/− flies; Dr. Andrea Ballabio (Telethon Institute of Genetics and Medicine, Italy) for providing us the 3×FLAG‐TFEB plasmid; Dr. Shumin Duan (Zhejiang University, China) for providing us the CD63‐GFP plasmid; Dr. Jia‐Jia Liu (Institute of Genetics and Developmental Biology, CAS, China) for providing us the myc‐dynactin 1 plasmid; Dr. Ji‐Wu Wang (Shanghai Jiao Tong University, China), Dr. Shawn Ferguson (Yale University), and Dr. Rosa Puertollano (National Institutes of Health) for unpublished experimental materials and helpful suggestions; Dr. Mingyi Xie (Yale University), Dr. Haidong Xu (Soochow University), and Bin Li (Soochow University) for critical comments and suggestions on our experiments; Lujia Zhu (Soochow University) for the technical support in the Fly studies. This work was supported in part by the National Natural Sciences Foundation of China (No. 31330030), the National High‐tech Research and Development program of China 973‐projects (2012CB947602), the National Natural Sciences Foundation of China (Nos. 81371393, 31200803, and 31371072), Natural Science Foundation of Jiangsu Province (BK2012181), a Project Funded by Jiangsu Key Laboratory of Translational Research and Therapy for Neuro‐Psycho‐Diseases (BM2013003) and a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.
The EMBO Journal (2016) 35: 121–142
See also: N Skoko et al (January 2016)
References
- Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SS, Kiskinis E, Winborn B, Freibaum BD, Kanagaraj A, Clare AJ, Badders NM, Bilican B, Chaum E, Chandran S, Shaw CE, Eggan KC, Maniatis T, Taylor JP (2014) Axonal transport of TDP‐43 mRNA granules is impaired by ALS‐causing mutations. Neuron 81: 536–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Peled L, Sabatini DM (2014) Regulation of mTORC1 by amino acids. Trends Cell Biol 24: 400–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar‐Peled L, Schweitzer LD, Zoncu R, Sabatini DM (2012) Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150: 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 28: 6926–6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bose JK, Wang IF, Hung L, Tarn WY, Shen CK (2008) TDP‐43 overexpression enhances exon 7 inclusion during the survival of motor neuron pre‐mRNA splicing. J Biol Chem 283: 28852–28859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandstaetter H, Kishi‐Itakura C, Tumbarello DA, Manstein DJ, Buss F (2014) Loss of functional MYO1C/myosin 1c, a motor protein involved in lipid raft trafficking, disrupts autophagosome‐lysosome fusion. Autophagy 10: 2310–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Baralle FE (2010) The multiple roles of TDP‐43 in pre‐mRNA processing and gene expression regulation. RNA Biol 7: 420–429 [DOI] [PubMed] [Google Scholar]
- Ching JK, Elizabeth SV, Ju JS, Lusk C, Pittman SK, Weihl CC (2013) mTOR dysfunction contributes to vacuolar pathology and weakness in valosin‐containing protein associated inclusion body myopathy. Hum Mol Genet 22: 1167–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JP, Reddy SL, Merry DE, Adachi H, Katsuno M, Sobue G, Robins DM, Lieberman AP (2014) Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet 23: 1376–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, Ivanov N, Sopher BL, Carromeu C, Muotri AR, Garden GA, La Spada AR (2014) Polyglutamine‐expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci 17: 1180–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A (2013) TFEB‐mediated autophagy rescues midbrain dopamine neurons from alpha‐synuclein toxicity. Proc Natl Acad Sci USA 110: E1817–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao J, Liu R, Rong Y, Zhao M, Zhang J, Lai Y, Zhou Q, Wilz LM, Li J, Vivona S, Pfuetzner RA, Brunger AT, Zhong Q (2015) ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature 520: 563–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaper DC, Adachi Y, Sutcliffe B, Humphrey DM, Elliott CJ, Stepto A, Ludlow ZN, Vanden Broeck L, Callaerts P, Dermaut B, Al‐Chalabi A, Shaw CE, Robinson IM, Hirth F (2013) Loss and gain of Drosophila TDP‐43 impair synaptic efficacy and motor control leading to age‐related neurodegeneration by loss‐of‐function phenotypes. Hum Mol Genet 22: 1539–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM (2013) Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature 493: 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feiguin F, Godena VK, Romano G, D'Ambrogio A, Klima R, Baralle FE (2009) Depletion of TDP‐43 affects Drosophila motoneurons terminal synapsis and locomotive behavior. FEBS Lett 583: 1586–1592 [DOI] [PubMed] [Google Scholar]
- Fiesel FC, Voigt A, Weber SS, Van den Haute C, Waldenmaier A, Gorner K, Walter M, Anderson ML, Kern JV, Rasse TM, Schmidt T, Springer W, Kirchner R, Bonin M, Neumann M, Baekelandt V, Alunni‐Fabbroni M, Schulz JB, Kahle PJ (2010) Knockdown of transactive response DNA‐binding protein (TDP‐43) downregulates histone deacetylase 6. EMBO J 29: 209–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H, Rubinsztein DC (2012) Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 8: 108–117 [DOI] [PubMed] [Google Scholar]
- Iguchi Y, Katsuno M, Niwa J, Takagi S, Ishigaki S, Ikenaka K, Kawai K, Watanabe H, Yamanaka K, Takahashi R, Misawa H, Sasaki S, Tanaka F, Sobue G (2013) Loss of TDP‐43 causes age‐dependent progressive motor neuron degeneration. Brain 136: 1371–1382 [DOI] [PubMed] [Google Scholar]
- Itakura E, Kishi‐Itakura C, Mizushima N (2012) The hairpin‐type tail‐anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 151: 1256–1269 [DOI] [PubMed] [Google Scholar]
- Jahreiss L, Menzies FM, Rubinsztein DC (2008) The itinerary of autophagosomes: from peripheral formation to kiss‐and‐run fusion with lysosomes. Traffic 9: 574–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean S, Cox S, Nassari S, Kiger AA (2015) Starvation‐induced MTMR13 and RAB21 activity regulates VAMP8 to promote autophagosome‐lysosome fusion. EMBO Rep 16: 297–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Nishimura T, Sakamaki Y, Itakura E, Hatta T, Natsume T, Mizushima N (2014) The HOPS complex mediates autophagosome‐lysosome fusion through interaction with syntaxin 17. Mol Biol Cell 25: 1327–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M, Menzies FM, O'Kane CJ, Deretic V, Rubinsztein DC (2011) Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol 13: 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Lee VM, Trojanowski JQ (2012) Gains or losses: molecular mechanisms of TDP43‐mediated neurodegeneration. Nat Rev Neurosci 13: 38–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Hu Q, Wang H, Man N, Ren H, Wen L, Nukina N, Fei E, Wang G (2010) Omi/HtrA2 is a positive regulator of autophagy that facilitates the degradation of mutant proteins involved in neurodegenerative diseases. Cell Death Differ 17: 1773–1784 [DOI] [PubMed] [Google Scholar]
- Lin MJ, Cheng CW, Shen CK (2011) Neuronal function and dysfunction of Drosophila dTDP. PLoS ONE 6: e20371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79: 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Chen Y, Gucek M, Puertollano R (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8: 903–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martina JA, Puertollano R (2013) Rag GTPases mediate amino acid‐dependent recruitment of TFEB and MITF to lysosomes. J Cell Biol 200: 475–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Nagy P, Juhasz G, Neufeld TP (2015) Autophagosome‐lysosome fusion is independent of V‐ATPase‐mediated acidification. Nat Commun 6: 7007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan DG, Popovic D, Gubas A, Terawaki S, Suzuki H, Stadel D, Coxon FP, Miranda de Stegmann D, Bhogaraju S, Maddi K, Kirchof A, Gatti E, Helfrich MH, Wakatsuki S, Behrends C, Pierre P, Dikic I (2015) PLEKHM1 regulates autophagosome‐lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57: 39–54 [DOI] [PubMed] [Google Scholar]
- Nassif M, Valenzuela V, Rojas‐Rivera D, Vidal R, Matus S, Castillo K, Fuentealba Y, Kroemer G, Levine B, Hetz C (2014) Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy 10: 1256–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM (2006) Ubiquitinated TDP‐43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: 130–133 [DOI] [PubMed] [Google Scholar]
- Pena‐Llopis S, Vega‐Rubin‐de‐Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J (2011) Regulation of TFEB and V‐ATPases by mTORC1. EMBO J 30: 3242–3258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, Lagier‐Tourenne C, Hutt KR, Huelga SC, Moran J, Liang TY, Ling SC, Sun E, Wancewicz E, Mazur C, Kordasiewicz H, Sedaghat Y, Donohue JP, Shiue L, Bennett CF, Yeo GW, Cleveland DW (2011) Long pre‐mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP‐43. Nat Neurosci 14: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roczniak‐Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5: ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Bar‐Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM (2010) Ragulator‐Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141: 290–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar‐Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320: 1496–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A (2009) A gene network regulating lysosomal biogenesis and function. Science 325: 473–477 [DOI] [PubMed] [Google Scholar]
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A (2011) TFEB links autophagy to lysosomal biogenesis. Science 332: 1429–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome‐to‐nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31: 1095–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spampanato C, Feeney E, Li L, Cardone M, Lim JA, Annunziata F, Zare H, Polishchuk R, Puertollano R, Parenti G, Ballabio A, Raben N (2013) Transcription factor EB (TFEB) is a new therapeutic target for Pompe disease. EMBO Mol Med 5: 691–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, Lazarowski ER, Damian VA, Masliah E, La Spada AR (2012) PGC‐1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med 4: 142ra197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccaro A, Tauffenberger A, Ash PE, Carlomagno Y, Petrucelli L, Parker JA (2012) TDP‐1/TDP‐43 regulates stress signaling and age‐dependent proteotoxicity in Caenorhabditis elegans . PLoS Genet 8: e1002806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Broeck L, Callaerts P, Dermaut B (2014) TDP‐43‐mediated neurodegeneration: towards a loss‐of‐function hypothesis? Trends Mol Med 20: 66–71 [DOI] [PubMed] [Google Scholar]
- Vanden Broeck L, Naval‐Sanchez M, Adachi Y, Diaper D, Dourlen P, Chapuis J, Kleinberger G, Gistelinck M, Van Broeckhoven C, Lambert JC, Hirth F, Aerts S, Callaerts P, Dermaut B (2013) TDP‐43 loss‐of‐function causes neuronal loss due to defective steroid receptor‐mediated gene program switching in Drosophila . Cell Rep 3: 160–172 [DOI] [PubMed] [Google Scholar]
- Volkening K, Leystra‐Lantz C, Yang W, Jaffee H, Strong MJ (2009) Tar DNA binding protein of 43 kDa (TDP‐43), 14‐3‐3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res 1305: 168–182 [DOI] [PubMed] [Google Scholar]
- Wang X, Fan H, Ying Z, Li B, Wang H, Wang G (2010) Degradation of TDP‐43 and its pathogenic form by autophagy and the ubiquitin‐proteasome system. Neurosci Lett 469: 112–116 [DOI] [PubMed] [Google Scholar]
- Wong E, Cuervo AM (2010) Autophagy gone awry in neurodegenerative diseases. Nat Neurosci 13: 805–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LS, Cheng WC, Shen CK (2012) Targeted depletion of TDP‐43 expression in the spinal cord motor neurons leads to the development of amyotrophic lateral sclerosis‐like phenotypes in mice. J Biol Chem 287: 27335–27344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu ZS (2012) Does a loss of TDP‐43 function cause neurodegeneration? Mol Neurodegener 7: 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Wang H, Qiao T, Yang B, Aliaga L, Qiu L, Tan W, Salameh J, McKenna‐Yasek DM, Smith T, Peng L, Moore MJ, Brown RH Jr, Cai H, Xu Z (2014) Partial loss of TDP‐43 function causes phenotypes of amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 111: E1121–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, Wang G (2009) Gp78, an ER associated E3, promotes SOD1 and ataxin‐3 degradation. Hum Mol Genet 18: 4268–4281 [DOI] [PubMed] [Google Scholar]
- Zhang T, Hwang HY, Hao H, Talbot C Jr, Wang J (2012) Caenorhabditis elegans RNA‐processing protein TDP‐1 regulates protein homeostasis and life span. J Biol Chem 287: 8371–8382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Li L, Chen S, Yang D, Wang Y, Wang Z, Le W (2011) Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy 7: 412–425 [DOI] [PubMed] [Google Scholar]
- Zhou L, Wang HF, Ren HG, Chen D, Gao F, Hu QS, Fu C, Xu RJ, Ying Z, Wang GH (2013) Bcl‐2‐dependent upregulation of autophagy by sequestosome 1/p62 in vitro. Acta Pharmacol Sin 34: 651–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Bar‐Peled L, Efeyan A, Wang S, Sancak Y, Sabatini DM (2011a) mTORC1 senses lysosomal amino acids through an inside‐out mechanism that requires the vacuolar H(+)‐ATPase. Science 334: 678–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM (2011b) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12: 21–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8