

ABSTRACT
Human leukocyte antigen (HLA) class I-associated polymorphisms in HIV-1 that persist upon transmission to HLA-mismatched hosts may spread in the population as the epidemic progresses. Transmission of HIV-1 sequences containing such adaptations may undermine cellular immune responses to the incoming virus in future hosts. Building upon previous work, we investigated the extent of HLA-associated polymorphism accumulation in HIV-1 polymerase (Pol) through comparative analysis of linked HIV-1/HLA class I genotypes sampled during historic (1979 to 1989; n = 338) and modern (2001 to 2011; n = 278) eras from across North America (Vancouver, BC, Canada; Boston, MA; New York, NY; and San Francisco, CA). Phylogenies inferred from historic and modern HIV-1 Pol sequences were star-like in shape, with an inferred most recent common ancestor (epidemic founder virus) sequence nearly identical to the modern North American subtype B consensus sequence. Nevertheless, modern HIV-1 Pol sequences exhibited roughly 2-fold-higher patristic (tip-to-tip) genetic distances than historic sequences, with HLA pressures likely driving ongoing diversification. Moreover, the frequencies of published HLA-associated polymorphisms in individuals lacking the selecting HLA class I allele was on average ∼2.5-fold higher in the modern than in the historic era, supporting their spread in circulation, though some remained stable in frequency during this time. Notably, polymorphisms restricted by protective HLA alleles appear to be spreading to a greater relative extent than others, though these increases are generally of modest absolute magnitude. However, despite evidence of polymorphism spread, North American hosts generally remain at relatively low risk of acquiring an HIV-1 polymerase sequence substantially preadapted to their HLA profiles, even in the present era.
IMPORTANCE HLA class I-restricted cytotoxic T-lymphocyte (CTL) escape mutations in HIV-1 that persist upon transmission may accumulate in circulation over time, potentially undermining host antiviral immunity to the transmitted viral strain. We studied >600 experimentally collected HIV-1 polymerase sequences linked to host HLA information dating back to 1979, along with phylogenetically reconstructed HIV-1 sequences dating back to the virus' introduction into North America. Overall, our results support the gradual spread of many—though not all—HIV-1 polymerase immune escape mutations in circulation over time. This is consistent with recent observations from other global regions, though the extent of polymorphism accumulation in North America appears to be lower than in populations with high seroprevalence, older epidemics, and/or limited HLA diversity. Importantly, the risk of acquiring an HIV-1 polymerase sequence at transmission that is substantially preadapted to one's HLA profile remains relatively low in North America, even in the present era.
INTRODUCTION
Human leukocyte antigen (HLA) class I-restricted CD8+ cytotoxic T-lymphocyte (CTL) responses against HIV-1 exert selection pressure on the pool of viruses present in an individual, driving the selection of escape variants capable of evading CTL recognition (1–4). CTL escape in HIV-1 is broadly reproducible based on the HLA alleles expressed by the host, and the locations and mutational pathways of CTL escape and other HLA-associated viral polymorphisms have been mapped across the HIV-1 genome (5–8). Following transmission to hosts lacking the restricting HLA allele, some of these HIV-1 polymorphisms revert, usually to the subtype consensus residue (9–12), likely as a result of fitness costs (10, 13–15). However, HLA-associated HIV-1 polymorphisms that carry no fitness costs, or where such costs are compensated for by the presence of secondary mutations, can in some cases stably persist following transmission (11, 16–18). If so, certain viral polymorphisms could gradually spread in the population as the epidemic progresses (17, 19–22). Analogous to the negative effects of transmitted drug resistance on treatment efficacy (23), acquisition of HIV-1 “preadapted” to (i.e., harboring escape mutations specific for) an individual's HLA alleles could compromise cellular immune responses to the incoming virus. This is supported by the observation that individuals infected with HIV-1 harboring polymorphisms associated with HLA alleles they share with their donors (their mothers in the case of vertical transmission or partners in the case of horizontal transmission) display suboptimal CTL responses and/or adverse clinical outcomes (19, 24–27).
The extent to which HLA-associated polymorphisms in HIV-1 are accumulating at the population level remains incompletely understood, though evidence suggests it is occurring. Analysis of a European cohort supported the accumulation of HIV-1 adaptations to CTL responses over the period 1985 to 2005 (28), as did a South American study of HIV-1 subtypes B and F over the same time period (29). More strikingly, a recent study of 9 global cohorts revealed a strong positive correlation between the prevalence of HLA-B*51 and that of its associated HIV-1 reverse transcriptase (RT) I135X polymorphism in circulating sequences (22). In particular, in Japan, where the frequency of B*51 exceeds 20%, the frequency of RT I135X exceeds 70%, supporting the spread of the polymorphism at the population level in the country. Studies in Europe (30), Asia (31), and Africa (32) have also reported (or inferred) decreases in HIV-1 replication capacity over the course of their respective epidemics, presumably as a result of HIV-1 adaptation to host pressures. A recent study of HIV-1 Gag and Nef evolution in North America by our group also indicated that HLA-associated polymorphisms have spread in circulation over time (33). However, the extent to which this is occurring in North America appears more modest than in high-seroprevalence settings, such as Botswana and South Africa (32), or in populations with relatively limited HLA diversity, such as Japan (22, 34).
The present study extends our previous work (33) by investigating changes in the frequencies of HLA-associated polymorphisms at the population level in North America from 1979 to the present in HIV-1 polymerase (Pol), a conserved viral protein targeted by CD8+ T-cell responses (35–37) that was included as an immunogen in both the STEP (38) and RV144 (39) HIV-1 vaccine trials. Pol was investigated as there is strong evidence that both natural (8, 40) and vaccine-induced (41) CD8+ T-cell responses exert potent selective pressures on the protein. To investigate the extent to which HLA-associated polymorphisms in this viral protein may be spreading in North America, we characterized plasma/serum specimens obtained from 1979 to 1989 (historic era) and 2001 to 2011 (modern era) for HIV-1 RNA polymerase sequence and host HLA class I type and performed a comparative analysis. Ancestral phylogenetic reconstruction was used to infer the epidemic founder virus sequence (estimated to be the most recent common ancestor [MRCA] at the root of the tree), as well as HIV-1 sequences circulating prior to 1979. Overall, we observed that HIV-1 polymerase sequences have diversified roughly 2-fold between the historic and modern eras, in part due to HLA-mediated selection pressures. HLA-associated polymorphisms have also increased approximately 2.5-fold in frequency at the population level during this time. Despite this, the extent of adaptation of the “average” circulating HIV-1 polymerase sequence to the majority of North American hosts remains relatively low. Taken together, our results suggest that, similar to HIV-1 Gag and Nef (33), population-level accumulation of HLA-associated polymorphisms in North America is occurring, but to a lesser extent than in some other global regions.
MATERIALS AND METHODS
Historic and modern cohorts.
The historic cohort comprised 338 participants of observational cohort studies of men who have sex with men (MSM) established in 4 key cities in the North American epidemic for whom a plasma or serum sample collected between 1979 and 1989 was available and HIV-1 Pol and HLA class I genotyping were successful (see below and reference 33 for more details). They included 239 individuals from the Fenway Community Health Clinic in Boston, MA (1985 to 1989) (42, 43); 65 individuals from the New York Blood Center in New York, NY (1979 to 1988); 26 individuals from the San Francisco Department of Public Health in San Francisco, CA (1984) (44–46); and 8 individuals from the Vancouver Lymphadenopathy-AIDS Study (VLAS) in Vancouver, BC, Canada (1984 to 1988) (47–49). Of these, 67 (19.8%) individuals were sampled <18 months following their estimated infection dates (“early infection”); the remainder were known or presumed to have chronic infections. As CTL escape mutations in HIV-1 polymerase tend to arise later in infection than those in Gag and Nef (50), differences in the clinical stage could influence escape mutation prevalence in individuals expressing the relevant HLA. Thus, individuals with known or suspected early infection were excluded from analyses where appropriate (as indicated in the figure legends).
The modern cohort comprised 278 participants of observational cohort studies from the same four cities for whom a plasma specimen collected between 2001 and 2011 was available and HIV-1 Pol and HLA class I genotyping were successful. They included 209 individuals from the British Columbia HAART Observational Medical Evaluation and Research (HOMER) study and Vanguard cohorts in Vancouver, 17 individuals from the Massachusetts General Hospital (MGH) in Boston, 21 individuals from the Aaron Diamond AIDS Research Center (ADARC) in New York, and 31 individuals in the Research in Access to Care Among the Homeless (REACH) Study in San Francisco (51). Of these, 16 (5.8%) were sampled during early infection; the remainder were known or presumed to have chronic infections. The modern cohort comprised various risk groups, though MSM predominated. Approximately 95% of the individuals were antiretroviral naive; the remainder were untreated at the time of sampling.
All the participants were enrolled under Institutional Review Board (IRB)-approved protocols and provided written informed consent to participate in the original studies for which the specimens were collected. Ethical approval to conduct this study was obtained from the Institutional Review Boards at Providence Health Care/University of British Columbia and Simon Fraser University.
Host and viral genotyping.
Total nucleic acids were extracted from plasma/sera using standard methods. Two HIV-1 polymerase fragments comprising protease (PR) and the first 400 codons of reverse transcriptase (RT), denoted PRRT, and all of integrase (INT) were amplified using nested RT-PCR with gene-specific primers. Amplicons were bulk (directly) sequenced on a 3130xl or 3730xl automated DNA sequencer (Applied Biosystems). Chromatograms were analyzed using Sequencher version 5.0.1 (Gene Codes) or RECall (52), with nucleotide mixtures called if the height of the secondary peak exceeded 25% of the height of the dominant peak (Sequencher) or 20% of the dominant peak area (RECall). All sequences were confirmed as subtype B using the Recombinant Identification Program (RIP) 3.0 (http://www.hiv.lanl.gov/content/sequence/RIP/RIP.html). Sequences were aligned using Clustal Omega (53), and maximum-likelihood (ML) phylogenetic trees were constructed using PhyML (54) and visualized using FigTree (version 1.3.1; http://tree.bio.ed.ac.uk/software/figtree/) to ensure the uniqueness of sequences. Linked HIV and HLA data are available upon request, in accordance with the Providence Health Care/University of British Columbia Institutional Review Board protocols.
HLA class I sequence-based typing was performed using an in-house protocol and interpretation algorithm (55). Where necessary, HLA types were imputed using a machine learning algorithm trained on more than 13,000 individuals of known ethnicity and HLA-A, -B, and -C types (56). Intercohort differences in HLA allele frequency were assessed using HLA Comparison (http://www.hiv.lanl.gov/content/immunology/hla/hla_compare.html), hosted by the Los Alamos HIV immunology database.
Phylogenetic ancestral reconstruction and diversification analyses.
The MRCA dates and sequences of our PRRT and INT sequence data sets were estimated using the software program Bayesian evolutionary analysis by sampling trees (BEAST) (57) with constrained input phylogenies. To do this, historic and modern PRRT and INT sequences were aligned against their respective regions in the HXB2 reference genome (GenBank accession no. K03455) using the Gotoh pairwise alignment algorithm implemented in HyPhy (58) with affine gap penalties of 20 and 1 for opening and extension, respectively, and nucleotide scores of +5 for a match and −4 for a mismatch. Codons associated with antiretroviral resistance (59) were removed from the alignment, and sequences were annotated with sampling dates. ML phylogenetic trees for PRRT and INT alignments for input into BEAST were generated using RAxML (version 8.1.3) under a GTRCAT model with otherwise default settings (60). ML trees were initially rooted and binarized using an in-house script implemented in the R package ape (61) and then rerooted and time scaled using Path-O-Gen (http://tree.bio.ed.ac.uk/software/pathogen/), with some in-house modifications. BEAST XML files were generated, with rooting and time scaling fixed by disabling tree modification operators. Both strict and relaxed (uncorrelated lognormal) clock models were run with a chain length of 107 (appropriate due to fixed input trees) and results logged every 105 steps. Based on prior experience, the following parameters were used: TN93 substitution model, generalized skyline coalescent model with 5 size classes, and partitioned rates and transition biases by codon positions 1&2 and 3. Model comparison was performed using Akaike's information criterion in a Bayesian Monte Carlo context (AICM) (62) implemented in Tracer (version 1.6; http://beast.bio.ed.ac.uk/Tracer). This revealed that a strict clock was favored for PRRT, while an uncorrelated lognormal clock was favored for INT. “Consensus trees” were generated by setting node heights to the median of the respective posterior distributions. Ancestral sequences were reconstructed by fitting a Muse-Gaut codon model crossed with the Tamura-Nei nucleotide substitution model in HyPhy (58) to the consensus trees.
For analyses of epidemic sequence diversification, tip-to-tip distances were extracted from Newick tree files using PATRISTIC (63). Intercohort differences in the Shannon entropy score at each HIV-1 polymerase residue were determined using Entropy Two (http://www.hiv.lanl.gov/content/sequence/ENTROPY/entropy.html) with 1,000 randomizations with replacement. Nonsynonymous versus synonymous substitution rates (β and α, respectively) were estimated using fast unconstrained Bayesian approximation for inferring selection (64), implemented on the Datamonkey Web server (65; http://www.datamonkey.org/). The β/α ratio is proportional to the more conventional ratio of scaled substitution counts, dN/dS. Codons at which the posterior probability of β being greater than α exceeded a threshold of 0.9 were interpreted as undergoing significant positive selection.
Definitions of HLA-associated polymorphisms.
HLA-associated polymorphisms in HIV-1 polymerase were defined according to a published reference list derived from phylogenetically informed analysis of a multicenter cohort of >1,800 chronically subtype B-infected individuals from Canada, the United States, and Australia recruited in the 1990s and 2000s (8). The published cohort did not overlap those analyzed here. We employed two definitions of HLA-associated polymorphisms, one “comprehensive” and one “conservative.” The comprehensive definition comprised all published HLA-associated polymorphisms in PRRT and INT identified at a false-discovery rate (q value) of <0.2 or <0.05 in the original study (8) (the q value cutoff used is specified for each analysis). However, since the cohort originally used to identify HLA-associated polymorphisms (8) was roughly 6-fold larger, and thus better powered, than the cohorts analyzed here, many comprehensively defined polymorphisms were not, or were only rarely, observed in the present cohorts. As such, a conservative list of HLA-associated polymorphisms, limited to only those detectable at a P value of <0.05 in both our historic and modern cohorts, was generated as follows. For each association that was significant at a q value of <0.05 in the original study (n = 129 in total [8]), the identified HLA class I allele, amino acid residue, covariates (other HLA alleles and covarying sites), and model were refitted to either the historic or modern data, using maximum-likelihood trees estimated separately for the two cohorts. A total of 20 associations that were significant at a q value of <0.05 in the published analysis and a P value of <0.05 in both the modern and historic cohorts comprised our conservative polymorphism list (Table 1).
TABLE 1.
Conservatively defined HLA-associated HIV-1 polymerase polymorphismsa
| Polymerase protein | HIV-1 polymorphism | HLA allele |
P value |
|
|---|---|---|---|---|
| Historic cohort | Modern cohort | |||
| Protease | T12A | B*52:01 | 0.023 | 0.003 |
| I15V | B*51:01 | 0.027 | 0.011 | |
| E35D | B*44:02 | 1.12 × 10−5 | 6.40 × 10−6 | |
| I93L | B*15:01 | 4.28 × 10−8 | 1.53 × 10−8 | |
| Reverse transcriptase | D123E | B*35:01 | 1.12 × 10−6 | 2.95 × 10−4 |
| I135T | B*51:01 | 1.15 × 10−11 | 6.10 × 10−6 | |
| S162C | B*07:02 | 0.02 | 0.011 | |
| D177E | B*35:01 | 0.008 | 0.007 | |
| K277R | A*03:01 | 2.07 × 10−15 | 5.45 × 10−19 | |
| T468A | B*13:02 | 0.003 | 1.33 × 10−4 | |
| Integrase | E10D | B*44:03 | 0.001 | 0.04 |
| E11D | B*44:02 | 1.37 × 10−4 | 4.98 × 10−7 | |
| S24G | B*51:01 | 2.49 × 10−5 | 0.008 | |
| V32I | B*51:01 | 3.91 × 10−6 | 6.15 × 10−7 | |
| L45V | B*51:01 | 0.031 | 0.004 | |
| M50I | C*16:01 | 0.001 | 0.002 | |
| T122I | B*57:01 | 0.027 | 2.28 × 10−4 | |
| T122I | C*05:01 | 2.58 × 10−8 | 4 × 10−4 | |
| T124N | B*57:01 | 0.002 | 6.24 × 10−4 | |
| G193E | B*27:05 | 2.18 × 10−8 | 7.47 × 10−4 | |
Published polymorphisms identified via phylogenetically informed methods (8) detectable in both cohorts at a P value of <0.05 were included in the conservative definition.
The same phylogenetically corrected methods (8) were also used to investigate the presence of novel HLA-associated polymorphisms in the historic and modern cohorts.
Nucleotide sequence accession numbers.
The historic HIV-1 Pol sequences have been deposited in GenBank (accession no. KT167847 to KT168174 for PRRT and KT167560 to KT167846 for INT).
RESULTS
Phylogenetic reconstruction of HIV-1 polymerase evolution in North America.
We began by inferring a time-dated phylogeny of HIV-1 in North America from our historic and modern sequence data sets (Fig. 1A and data not shown). We also estimated the date and sequence of the MRCA at the tree root, representing the inferred epidemic founder virus. The estimated MRCA dates differed slightly based on the gene region analyzed (1965 for PRRT versus 1972 for INT); the former is likely a more reliable estimate due to the longer length and greater sequence variability in the PRRT data set. Regardless, both dates are broadly consistent with estimates of the arrival of HIV-1 in North America sometime in the 1960s (66, 67), as well as our previous estimates from HIV-1 Gag and Nef sequences from similar cohorts (33).
FIG 1.

Phylogenetic ancestral reconstruction of HIV-1 in North America. (A) Time-scaled maximum-likelihood phylogenetic tree inferred from historic and modern PRRT sequences. The MRCA, representing the epidemic founder virus, is indicated by the black dot at the root. (B) Amino acid alignments of the inferred PRRT (PR, red; RT, blue) MRCA sequence at the root of the tree (ANC), the global HIV-1 subtype B consensus (Los Alamos database; GLOBAL_CONSB), the North American HIV-1 subtype B consensus (Los Alamos database; N.AM_CONSB), the consensus of our historic cohort (HIS_CONS), and the consensus of our modern cohort (MOD_CONS). Note that PRRT codon numbering in the alignment begins from PR and continues through RT; as such, RT 207 is PRRT codon 306.
Moreover, the reconstructed PRRT and INT MRCA amino acid sequences were nearly identical to the modern global and North American subtype B consensus sequences (Fig. 1B and data not shown), differing at only 2 (of 499) PRRT and 3 (of 289) INT codons, all variable residues. Of note, two of these five differences from the consensus (RT 207E and INT 72V) are adapted forms associated with HLA-B*15:01 and B*15:10/B*53:01, respectively (8), and are discussed later. The reconstruction of a “consensus-like” MRCA sequence is consistent with the star-like appearance of phylogenies inferred from historic and modern sequence data sets (Fig. 2). Together, these observations suggest that widespread selective sweeps (host immune driven or otherwise) in HIV-1 have not occurred since the virus' establishment in North America, though sweeps occurring before the collection of our earliest sequences cannot be ruled out.
FIG 2.

HIV-1 polymerase diversity in the historic (1979 to 1989) and modern (2001 to 2011) cohorts. Shown are unrooted maximum-likelihood phylogenetic trees of historic (left) and modern (right) PRRT sequences drawn to the same genetic-distance scale. The sequences are color coded by sampling date. North American PRRT sequences retrieved from the Los Alamos database are shown in gray. The HXB2 reference sequence is indicated.
Despite the close resemblance between the inferred epidemic founder virus and the modern subtype B consensus, HIV-1 sequences have clearly diversified between the historic and modern eras (Fig. 2). Specifically, the mean patristic (tip-to-tip) distance, inferred from maximum-likelihood phylogenies, was 0.025 (standard deviation [SD], 0.007) substitutions per nucleotide site in historic PRRT sequences compared to 0.055 (SD, 0.010) in modern sequences. Likewise, the mean patristic distance in historic INT sequences was 0.021 (SD, 0.007) substitutions per nucleotide site compared to 0.044 (SD, 0.009) in modern sequences. These results are consistent with HIV-1 diversity in North America approximately doubling between the 1980s and the 2000s.
HLA pressures drive HIV-1 polymerase sequence diversification.
In a diversifying epidemic, genetic variation increases over time. We hypothesized that HLA-mediated selection pressures have contributed substantially to HIV-1's ongoing diversification. We initially investigated this by determining differences in Shannon entropy (Δentropy) between historic and modern amino acid Pol codon alignments (Fig. 3A and B). As Pol is generally highly conserved, we focused our subsequent analysis on only “variable” codons (those that exhibited >1% sequence variation in historic and modern cohorts, which included 263 of 788 [33.4%]) codons in PRRT and INT. Of these 263 codons, 71 (27%) exhibited significantly higher entropy in modern than in historic sequences, while only 3 (1.1%) exhibited the opposite (P < 0.001 for all). The remaining 189 (71.9%) codons did not exhibit significant Δentropy between eras. Moreover, of these 263 codons, 129 (49%) are known to be under HLA-mediated selection pressure (comprehensive definition at a q value of <0.2 [8]) (see Materials and Methods) (Fig. 3A and B). Stratifying HIV-1 Pol codons with respect to Δentropy (significantly different between eras versus not significantly different) and their HLA association status revealed a significant positive association: 49 of 129 (38%) HLA-associated codons exhibited a significant change in entropy between eras compared to 25 of 134 (18.7%) non-HLA-associated codons (P = 0.0006) (Fig. 3C).
FIG 3.

Diversifying polymerase codons are enriched for HLA-associated sites. (A) Per-codon Δentropy values calculated from historic and modern polymerase sequence alignments. HIV protein codon numbering is indicated on the x axis. The region without sequence coverage (RT codons 401 to 560) is indicated with a gray hatched bar. The red and blue bars denote significant (P < 0.001) and nonsignificant Δentropy values, respectively. The black dots denote polymerase codons known to be under HLA-mediated selection (comprehensive definition, q < 0.2 [8]). The green dots denote codons with dN/dS ratios of >1. (B) Same as panel A, but with Δentropy values ranked in descending order rather than by codon. (C) Proportions of HLA-associated and non-HLA-associated polymerase codons exhibiting significant (red) versus nonsignificant (blue) intercohort Δentropy. (D) Proportions of polymerase codons with and without significant intercohort Δentropy exhibiting dN/dS ratios of >1 (green) versus not (gray). (E) Proportions of HLA-associated and non-HLA-associated codons with dN/dS ratios of >1 (green) versus not (gray). Only variable (<99% conserved) polymerase codons were included in the analyses for panels C, D, and E. P values were determined using Fisher's exact test.
We also calculated dN/dS ratios in our historic and modern sequence alignments, which provide a more direct way to investigate elevated substitution rates within the phylogeny than entropy-based approaches. Of the 263 variable Pol codons, 46 (17.5%) exhibited dN/dS values of >1 (posterior probability of positive selection > 0.9) (Fig. 3A and B). As expected, codons exhibiting significant intercohort Δentropy values also tended to exhibit dN/dS ratios of >1 (P = 0.018) (Fig. 3D), consistent with positive selection driving some of the diversification in HIV-1 polymerase. Importantly, codons with dN/dS ratios of >1 also tended to be HLA associated: 35 of 129 (27.1%) HLA-associated codons had dN/dS ratios of >1 compared to 11 of 134 (8.2%) non-HLA-associated codons (P < 0.0001) (Fig. 3E). Taken together, the results support HLA class I-mediated immune pressures as significant drivers of population-level HIV-1 diversification.
HLA-associated polymorphisms in HIV-1 polymerase have spread during the epidemic.
We next turned to our main objective of investigating the extent to which known HLA-associated polymorphisms in HIV-1 polymerase have increased in circulation between the historic and modern eras. Despite being convenience samples, our historic and modern cohorts are nevertheless well matched in terms of their HLA allele frequencies. These correlated robustly between cohorts (Pearson's R = 0.96; P < 0.0001), and of 49 HLA alleles observed at >1% in both cohorts, only one (A*11:01) differed significantly in frequency between them (P < 0.05; q < 0.05) (data not shown). Matching of HLA frequencies between cohorts is important, as the circulating prevalence (and thus the transmission frequency) of HLA-associated HIV-1 polymorphisms can in some cases be influenced by the frequency of the restricting HLA in the population (22, 68).
We began by investigating the frequencies of the subset of 20 published HLA-associated polymerase polymorphisms strongly detectable by statistical association in data sets of the present size (conservative definition) (see Materials and Methods and Table 1) in individuals with and without the restricting HLA by era. All 20 polymorphisms were cases where the HLA-associated HIV-1 amino acid was a residue other than the population consensus (“nonconsensus” polymorphisms). As expected, the frequencies of these HIV-1 polymorphisms in individuals expressing the relevant HLA allele were higher than in individuals lacking the HLA, regardless of era (compare Fig. 4A and B). Also as expected, the frequencies of these HIV-1 polymorphisms among individuals carrying the relevant HLA were not significantly different between eras (median, 42.3% [interquartile range {IQR}, 25.6 to 71.5%] in the historic cohort versus 51.5% [IQR, 38.9 to 79.2%] in the modern cohort; P = 0.17) (Fig. 4A). This result was also expected because, regardless of the epidemic era, HLA-associated polymorphisms should be reproducibly selected in individuals expressing the relevant HLA.
FIG 4.

HLA-associated polymorphism frequencies in HLA+ and HLA− individuals, by era. (A) Frequencies of select nonconsensus HLA-associated polymorphisms (conservative definition [see Materials and Methods and Table 1]) among individuals expressing the relevant HLA allele (HLA+) in the historic versus modern cohorts. Polymorphisms of particular interest are colored. (B) Frequencies of the same conservatively defined HLA-associated polymorphisms in individuals lacking the relevant HLA allele (HLA−) by era. (C) Frequencies of 120 comprehensively defined nonconsensus HLA-associated polymorphisms (q < 0.05 [8]) in individuals lacking the restricting HLA allele (HLA−) between historic and modern eras. Polymorphisms of particular interest are colored. P values were determined using the Wilcoxon matched-pairs test. Individuals with known/suspected early infection were excluded from the analyses.
In contrast, in individuals lacking the restricting HLA allele, the frequencies of these HIV-1 polymorphisms were significantly higher in the modern (median, 16.8% [IQR, 5.8 to 19.7%]) than in the historic (median, 6.6% [IQR, 1.4 to 14.2%]) cohort (P = 0.0004) (Fig. 4B). This indicates that, on average, HLA-associated polymorphisms have spread approximately 2.5-fold in the general population between the historic and modern eras in North America. Despite this, the frequencies of some polymorphisms nevertheless remained stable between eras. Examples are the B*51:01-restricted I135T substitution and the A*03:01-restricted K277R substitution, both in reverse transcriptase.
Our observations of significant overall spread of HLA-associated polymorphisms between the historic and modern eras were upheld even when the complete published HLA-associated polymorphism list was used (comprehensive definition at a q value of <0.05) (see Materials and Methods). The list comprised 120 nonconsensus HLA-associated polymorphisms and 9 HLA-associated polymorphisms in which the HIV-1 amino acid represented the population consensus residue (“consensus” polymorphisms). Among individuals lacking the relevant HLA allele, the median frequency of the nonconsensus polymorphisms was 1.8% (IQR, 0.5 to 5.7%) in the historic cohort compared to 4.2% (IQR, 1.4 to 12.9%) in the modern cohort (P < 0.0001) (Fig. 4C). Though these values are lower overall than those observed in the conservative analysis (many of these published polymorphisms are uncommon in, or absent from, the historic and modern cohorts), these observations are nevertheless consistent with an approximately 2.4-fold polymorphism spread between eras. Of note, the frequencies of the 9 consensus polymorphisms in individuals lacking the relevant HLA were higher in the historic cohort (median, 90.5% [IQR, 83.4 to 94.1%]) than in the modern cohort (median, 79.7% [IQR, 66.1 to 93%]) (P = 0.0039) (data not shown), suggesting that their frequencies are decreasing over time.
The historic and modern cohorts were also investigated for the presence of HLA-associated polymorphisms using phylogenetically corrected methods in a naive manner (that is, all HLA allele–HIV-1 codon pairs were examined for possible associations). However, no novel HLA-associated polymorphisms were identified in either data set at a q value of <0.05 (not shown), though the modest size of these data sets limits their power to identify such associations.
The spread of HLA-associated polymorphisms began prior to 1979.
Though our observation of >2-fold-higher HLA-associated polymorphism frequencies in modern than in historic sequences strongly supports their spread over time, this observation could be strengthened if we could demonstrate even lower frequencies in HIV-1 sequences circulating between 1965 (the earliest estimated MRCA date) and 1979 (the date of our oldest sampled sequence). To infer HIV-1 polymerase sequences from this earlier period, we reconstructed sequences at internal nodes of the time-dated phylogenies (Fig. 1A and data not shown), yielding 339 (PRRT) and 341 (INT) “reconstructed ancestral” sequences dating to between 1965 and 1979 for analysis.
We then determined the frequencies of the 20 conservatively defined HLA-associated polymorphisms in the reconstructed MRCA sequence (all zero, as this sequence contained none of the polymorphisms), the reconstructed ancestral sequences, and our historic and modern cohort sequences (Fig. 5A). This analysis was agnostic to host HLA, as this information was not available for the MRCA or reconstructed ancestral sequences. Consistent with the previous analyses, the frequencies of these HLA-associated polymorphisms increased with each passing era: the median frequency of the 20 polymorphisms in reconstructed ancestral sequences was 2.6% (IQR, 0.3 to 12.0%) compared to 9.8% (IQR, 3.4 to 19.1%) in the historic era and 21% (IQR, 8.6 to 23.6%) in the modern era (P < 0.0001) (Fig. 5A). Similar trends were observed when the comprehensive list of 120 nonconsensus HLA-associated polymorphisms was used (P < 0.0001) (Fig. 5B).
FIG 5.

Spread of HLA-associated polymorphisms evident from the earliest days of the North American epidemic. (A) Frequencies of nonconsensus conservatively defined HLA-associated polymorphisms in the inferred MRCA sequence, reconstructed ancestral sequences (1965 to 1979), historic sequences (1979 to 1989), and modern sequences (2001 to 2011). Polymorphisms of specific interest are labeled. (B) Frequencies of 120 comprehensively defined HLA-associated polymorphisms in the inferred MRCA sequence, reconstructed ancestral sequences, historic sequences, and modern sequences. Polymorphisms of specific interest are labeled. Polymorphisms RT 207E (associated with B*15:01) and INT 72V (associated with both B*15:10 and B*53:01) are present in the reconstructed MRCA sequence and decline in prevalence at the population level thereafter. Both analyses were agnostic to the individuals' HLA status, as the information was not available for the MRCA or reconstructed ancestors. The P value (P < 0.0001) for overall comparison was determined using the Friedman test. Historic and modern patients with known/suspected early infection were excluded from the analyses.
HIV-1 polymerase polymorphisms restricted by protective HLA alleles may be spreading to a greater relative extent.
We next sought to investigate whether the characteristics of a particular HLA allele might impact the spread of its associated polymorphisms. First, we investigated whether population HLA allele frequency is a correlate of HIV-1 polymorphism spread. A total of 12 HLA class I alleles (3 HLA-A and 9 HLA-B) for which ≥3 HLA-associated polymorphisms in Pol have been identified (nonconsensus polymorphisms comprehensively defined at a q value of <0.05) (see Materials and Methods) were included in this analysis. No significant correlation was observed between the frequency of an HLA allele and the median fold increase of its associated polymorphisms between the historic and modern eras (Spearman's R = −0.13; P = 0.68) (Fig. 6A). This suggests that HIV-1 polymorphism spread is not predominantly attributable to selection pressures imposed by common HLA alleles.
FIG 6.

Correlations between HLA allele features and relative spread of associated polymorphisms. (A) No significant correlation between the population HLA allele frequency and the median fold increase in its associated HIV-1 polymorphisms between the historic and modern eras was observed. (B) A significant inverse correlation between the HR-AIDS of an HLA allele and the median fold increase of its associated HIV-1 polymorphisms between eras was observed. Individuals with known/suspected early infection were excluded from the analysis.
We next investigated whether HIV-1 polymorphism spread was associated with the protective status of a given HLA allele, defined as its published hazard ratio (HR) of progression to AIDS (HR-AIDS) in historic natural history studies (69). A total of 10 alleles (2 HLA-A and 8 HLA-B) were investigated (HRs were not defined for the remaining 2). A significant inverse correlation was observed between the HR-AIDS of an allele and the median fold increase of its associated polymorphisms between the historic and modern eras (Spearman's R = −0.71; P = 0.03) (Fig. 6B). Note, however, that when the polymorphism spread was quantified in absolute as opposed to relative, i.e., fold change, terms, this association was not significant (Spearman's R = −0.28; P = 0.43) (data not shown). Moreover, for 7 of 10 alleles considered in the present analysis, the median absolute increase in polymorphism frequency was <5%. Taken together, the results suggest that polymorphisms of protective alleles are spreading in the population to a greater relative extent than those of other alleles, though in most cases, their absolute frequencies still remain low.
Preadaptation of the average circulating HIV-1 sequence to North American hosts remains low in the modern era.
While our data support the gradual spread of many HLA-associated polymorphisms in HIV-1 polymerase over time, it is important to contextualize these findings in terms of their potential consequences for host immunity. Toward this goal, we computed the extent to which the average circulating HIV-1 sequence in each era was preadapted to (i.e., contained HLA-associated polymorphisms specific for) the HLA profiles of North American hosts. This allowed us to estimate the risk of individuals acquiring an HIV-1 strain already adapted to their CTL response should they be randomly infected with a sequence from that era. To do this, we pooled the HLA profiles of the historic and modern cohort participants to generate a hypothetical North American population. Then, for each member of this population, we analyzed HIV-1 sequences from each era in the context of their HLA profile and computed the median percentage of HLA-associated sites harboring the variant adapted to the individual's alleles (“percent escaped”). Here, the comprehensive definition of HLA-associated polymorphisms was used, excluding the 9 consensus HLA-associated polymorphisms.
As expected, the inferred epidemic founder virus was 0% preadapted to >88% of North American hosts (the 12% nonzero values are attributable to hosts expressing HLA-B*15:01, B*15:10, or B*53:01, as the reconstructed MRCA harbors the B*15:01-associated RT 207E and B*15:10/B*53:01-associated INT 72V polymorphisms) (Fig. 7). In fact, the extent of preadaptation of circulating HIV-1 sequences remained generally low across all eras (Fig. 7). Specifically, reconstructed ancestral HIV-1 sequences circulating between 1965 and 1979 exhibited a median of 0% preadaptation to 97% of North American hosts (this value is higher than that of the MRCA because not all reconstructed ancestral sequences harbored RT 207E and/or INT 72V due to inferred reversion of these mutations in some lineages). In the historic (1979 to 1989) and modern (2001 to 2011) eras, HIV-1 sequences exhibited a median of 0% preadaptation to 64% and 38% of hosts, respectively.
FIG 7.
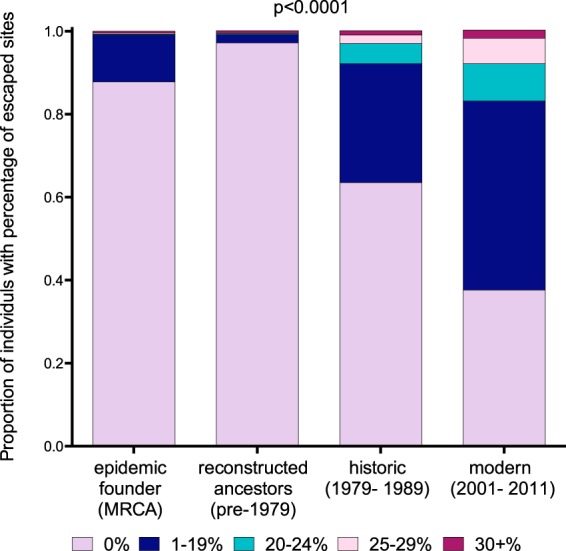
Preadaptation of the average circulating HIV-1 polymerase sequence to North Americans by era. The colored portions of the bars denote the extent to which the average circulating HIV-1 sequence was preadapted (i.e., harbored HLA-specific polymorphisms) to that proportion of North American hosts in the population. Sequences from four eras were considered: the inferred MRCA sequence (foundation of the epidemic), reconstructed ancestral sequences (1965 to 1979), historic sequences (1979 to 1989), and modern sequences (2001 to 2011). The P value (P < 0.0001) for overall comparison was determined using the Friedman test. Sequences from individuals with known/suspected early infection were excluded from the analysis.
These results indicate that, for a substantial proportion of the population, the risk of acquiring an HIV-1 polymerase sequence substantially preadapted to their HLA profile remained relatively low across all eras. However, it is important to note that the underlying distribution shifted markedly during this time (Fig. 7). By the historic era, 29% of hosts were at risk of acquiring an HIV-1 polymerase sequence between 1 and 19% adapted to their HLA profiles, and a further 8% were at risk of acquiring a sequence ≥20% adapted to their HLA profiles. By the modern era, these percentages increased to 46% and 17%, respectively. Despite these increases, it is worth noting that the proportion of hosts at risk of acquiring HIV-1 over 30% adapted to their HLA profile was only 2% in the modern era (Fig. 7). Taken together, while the estimated risk of acquiring an HIV-1 polymerase sequence more than 20% preadapted to their HLA profiles remains relatively low for the majority of North Americans, our data nevertheless indicate that risk of infection with a virus carrying at least some preadaptation in Pol is increasing as the epidemic progresses.
DISCUSSION
Our analysis of linked HIV-1 polymerase sequence and host HLA class I data from the historic (1979 to 1989) and modern (2001 to 2011) eras indicates that the frequencies of HLA-driven viral polymorphisms have increased, on average, approximately 2.5-fold in circulating HIV-1 sequences in North America during this time. The absolute magnitude of this increase differed based on the definition of what constituted an HLA-associated HIV-1 polymorphism. When restricted to a conservative list of 20 polymorphisms strongly detectable in the present cohorts, it translated to an average absolute increase of ∼10%. However, when a comprehensive list that included rarer polymorphisms was used (8), it amounted to an average increase of only ∼2%. In relative terms, this translated to an ∼2.5-fold increase, regardless of the definition. This, along with a median overall 2% absolute increase, is consistent with results we previously reported for HIV-1 Gag and Nef, where a similar comprehensive approach was used (33). This consistency was not unexpected. Though Gag harbors more sites under HLA-mediated selection than Pol, these sites are nevertheless generally comparable in terms of their strengths of HLA-mediated selection (8). Given that Gag and Pol exhibit similar levels of sequence conservation and that our analysis investigates individual HLA-associated polymorphisms in terms of their spread between eras, it is not surprising that, on average, both absolute and relative increases are similar across these HIV-1 proteins.
Our findings are thus consistent with the gradual spread of HLA-driven polymorphisms in North American HIV-1 sequences. However, the observation that our reconstructed epidemic founder virus sequence is essentially identical to the modern North American consensus sequence, combined with our lack of detection of novel HLA-associated polymorphisms in historic HIV-1 sequences, suggests that no HLA-associated HIV-1 polymorphism has spread in North America to an extent where it has shifted the population HIV-1 consensus. In fact, our data illuminate two possible examples of the opposite phenomenon, that is, the presence of HLA-associated polymorphisms within the epidemic founder virus sequence (B*15:01-associated RT 207E and B*15:10/B*53:01-associated INT 72V) that subsequently decline in circulating prevalence so that the consensus shifts away from these residues.
Despite overall increases, the frequencies of certain HLA-associated polymorphisms remained notably stable. Among these is B*51:01-restricted RT I135T, whose prevalence was ∼15% in B*51:01-negative persons in both the historic and modern eras. The lack of spread of RT I135T in North America over the past 30 years suggests that the polymorphism reverts upon transmission to hosts lacking the restricting HLA, though this inference is somewhat at odds with an observed absence of in vitro replicative cost (22) and the observation that the polymorphism does not revert rapidly upon transmission (12) (though some studies suggest that reversion does occur [70]). This observation is also somewhat at odds with the hypothesis that I135T is spreading in certain populations (most notably in Japan, where B*51 prevalence exceeds 20% and >70% of HIV-1 sequences express I135X [22]). We propose the following explanations for these apparent discrepancies. First, ancestral phylogenetic reconstructions revealed cases where I135T-containing ancestral nodes gave rise to I135-containing descendants (not shown), supporting the reversion of this polymorphism in at least some cases. Its inferred reversion is consistent with mathematical models indicating that any reversion, however slow, would prevent a polymorphism from reaching fixation in a population (71). This remains true even if the polymorphism is selected at high frequency in individuals expressing the relevant HLA (71). K277R in reverse transcriptase, which is highly reproducibly selected in A*03-expressing individuals (8, 50) and displays some evidence of reversion (12), illustrates this point: its frequency has remained consistent at ∼28% throughout the historic and modern eras in persons lacking this allele. Second, we hypothesize that polymorphisms are spreading differentially across global regions due to differential HLA-driven selection pressures in these populations. In Japan, for example, HLA-B*52, a relatively common allele in the population, also selects I135X (72), thereby contributing to its maintenance in circulation. Another possibility is that the Japanese HIV-1 epidemic was founded by an I135X-containing variant (73), which would also explain its persistent high prevalence.
More broadly, the overall absence of correlation between the population HLA prevalence and the extent of HIV-1 polymorphism spread suggests that the latter is not a simple result of the former. This is corroborated by a recent database-driven study reporting weak (or no) significant relationships between these two factors for the majority of HLA-associated HIV-1 polymorphisms investigated, though I135T was a notable exception (68). Rather, our observation that HIV-1 polymorphisms restricted by protective HLA alleles appear to be spreading to a greater relative extent than others, an observation that corroborates our previous analyses of HLA-associated polymorphism spread in Gag and Nef (33), is consistent with the idea that protective alleles mount strong selection pressures on HIV-1 (8) and also select escape mutations relatively early following infection in many cases, thus possibly enhancing their probability of transmission. These findings also lend credence to the idea that population-level HIV-1 adaptation to HLA may eventually diminish or eliminate the protective capacity of certain HLA alleles over time (22); indeed, recent data suggest that this phenomenon is already discernible in Botswana, a high-seroprevalence setting with a long epidemic history, where alleles such as HLA-B*57 and B*58:01 have lost their protective status (32). In contrast, in neighboring South Africa, where the epidemic is younger, the protective effects of these alleles remain intact (32). Nevertheless, even if some polymorphisms spread in circulation and such variants are acquired at transmission, it is possible that some CTL response against the incoming virus may be preserved, especially for protective HLA alleles. This is because such alleles often restrict responses to multiple epitopes and may retain cross-reactivity against viral variants (74, 75). In addition, de novo CTLs may emerge against selected variants (76, 77), and de novo epitopes may also emerge (78). It is important to emphasize that, though protective allele-associated polymorphisms have spread to a greater relative extent than others, the absolute values of these increases were generally modest.
It is also important to underscore that the estimated risk of acquiring an HIV-1 polymerase sequence substantially adapted to one's HLA alleles remains relatively low in North America, an observation that contrasts with the markedly higher levels of preadaptation of the average circulating Gag, Pol, and Nef sequences to the HLA profiles of individuals in Botswana (and, to a lesser extent, South Africa) (32). These differences suggest that viral and host genetic factors (e.g., population HLA diversity) (79), along with epidemic-specific factors, such as epidemic age, HIV incidence/prevalence, and possibly timing of transmission (i.e., whether individuals predominantly transmit during acute versus chronic infection, as more within-host escape mutations will have been selected by the latter stage), are likely to represent determinants of the tempo and impact of population-level HIV-1 adaptation to HLA.
The present study is not without limitations. Our cohorts represent a convenience sample composed predominantly of a single risk group (MSM) drawn from only four North American cities. We acknowledge the possibility of sampling biases, though the similarity between our cohort consensus and the North American HIV subtype B consensus sequence, the consistency of our MRCA date with published estimates of the arrival of HIV in North America (66, 67), and the observation that North American HIV-1 polymerase sequences obtained from the Los Alamos database (Los Alamos National Laboratory HIV Sequence Database [http://www.hiv.lanl.gov/components/sequence/HIV/search/search.html]) are interspersed with those from our cohorts in a maximum-likelihood phylogeny suggest that our data are not unrepresentative of the North American epidemic. Infection dates were unavailable for most specimens, but a greater proportion of the historic specimens derived from individuals with known or suspected early infection. Intercohort differences in clinical stage could conceivably influence the extent of CTL escape in individuals harboring the restricting HLA, though they should not affect their prevalence in the general population to any great extent. Nevertheless, to mitigate any possible biases, individuals with known or suspected early infection were excluded from relevant analyses.
Another possible limitation is that HLA-associated polymorphisms were defined using statistical-association approaches in contemporary (1990s and 2000s) cohorts (8). It is thus conceivable that HLA-associated polymorphisms present historically that have subsequently spread substantially in the population would no longer be detectable in modern cohorts by statistical association, as they would no longer be significantly enriched among persons with the HLA. Indeed, statistical approaches favor the detection of polymorphisms that escape and revert rapidly (80) and thus are less likely to accumulate at the population level (71). Several factors mitigate these concerns. First, our comprehensive polymorphism list included rare HLA-associated polymorphisms (8). Second, we did not identify any novel HLA-associated polymorphisms in our historic cohort. Third, the near identity between our reconstructed epidemic founder virus sequence (MRCA) and the modern North American subtype B consensus sequence suggests that no polymorphisms present earlier in the epidemic have accumulated to the point where they now represent the consensus residue. In fact, the declining prevalence of consensus HLA-associated polymorphisms in circulation (e.g., RT 207E and INT 72V) suggests that they are not spreading but rather represent host adaptations present in the epidemic founder virus sequence that subsequently reverted in some hosts (33).
Finally, as we focused on the Pol gene, the introduction of antiretroviral therapies (ARVs) merits brief mention (RT inhibitors were introduced in the late 1980s and protease inhibitors in the mid 1990s). While ARV- and HLA-associated sites do not generally coincide (only 2 of 34 major PRRT drug resistance sites [RT 106 and 138] are also HLA-associated sites; 5 of 29 minor drug resistance sites are HLA associated), and even when they do, they tend to select different mutations (8, 59), and >95% of individuals in our study were drug naive, the modern era is nevertheless post-ARV. Thus, if unknown secondary or tertiary mutations selected under ARV pressures, now present in circulation, overlapped HLA-associated polymorphisms or somehow modulated their selection, then ARVs could theoretically confound our observations. We thus reanalyzed PRRT and INT fragments separately, reasoning that if ARVs were a confounder, we would observe differential evidence for HLA-associated polymorphism spread in these regions (while PR and RT inhibitors are now widely used, the first integrase inhibitor was not approved until 2007). However, doing so yielded results entirely consistent with the overall analysis (i.e., statistically significant ∼2- to 2.5-fold increases in polymorphism spread regardless of gene region or HLA polymorphism definition) (data not shown), suggesting that the introduction of ARVs is not a major confounder of our observations.
In summary, HLA-associated polymorphisms in HIV-1 polymerase, like those in Gag and Nef (33), are slowly spreading in North America. Though the majority of North American hosts remain at relatively low risk of acquiring an HIV-1 polymerase sequence at transmission that is substantially preadapted to their HLA class I profiles, even modest increases in the level of adaptation in circulating sequences could have immunologic implications. Taken together with recent studies suggesting more extensive HIV-1 adaptation to HLA in older, high-prevalence epidemics (32), the results underscore an HIV vaccine as a global priority, alongside current international efforts to achieve universal access to HIV-1 therapies as treatment and prevention (81).
ACKNOWLEDGMENTS
We thank Guinevere Lee, Celia Chui, and Conan Woods for technical and database assistance, as well as Rosemary McCloskey and Richard Liang for assistance with ancestral reconstructions. We also thank Kyle Cobarrubias, Gursev Anmole, and Mark Brockman for helpful discussions. We are greatly appreciative of those individuals who participated in the original studies, without whom this work would not have been possible.
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
This work was supported by operating grants from the Canadian Institutes for Health Research (CIHR) (MOP-93536 to Z.L.B. and HOP-115700 to Z.L.B. and A.F.Y.P.). This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research under contract no. HHSN261200800001E. This work was supported in part by the Intramural Research Program of the NIH, Frederick National Laboratory Center for Cancer Research. N.N.K. is supported by a CIHR undergraduate summer studentship. A.Q.L. was funded by a CIHR Frederick Banting and Charles Best Masters Award. P.R.H. was supported by a CIHR/GSK research chair in clinical virology. A.F.Y.P. was the recipient of a Scholar Award from the Michael Smith Foundation for Health Research (MSFHR)/St. Paul's Hospital Foundation-Providence Health Care Research Institute Career Investigator Program and a CIHR New Investigator Award. Z.L.B. was the recipient of a CIHR New Investigator Award and currently holds a scholar award from the MSFHR.
Funding Statement
The funders had no role in study design, data collection and analysis, the decision to publish, or the preparation of the manuscript.
REFERENCES
- 1.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard JA, Bangham CR, Rizza CR. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 2.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 3.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 4.Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med 193:375–386. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439–1443. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 7.Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, Matthews P, Payne R, Rolland M, Raugi DN, Maust BS, Learn GH, Nickle DC, Coovadia H, Ndung'u T, Frahm N, Brander C, Walker BD, Goulder PJR, Bhattacharya T, Heckerman DE, Korber BT, Mullins JI. 2008. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype c proteome: immune escape and viral load. J Virol 82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CKS, Cotton LA, Knapp DJHF, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL. 2012. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol 86:13202–13216. doi: 10.1128/JVI.01998-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li B, Gladden AD, Altfeld M, Kaldor JM, Cooper DA, Kelleher AD, Allen TM. 2007. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J Virol 81:193–201. doi: 10.1128/JVI.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, Evans DT, Desrosiers RC, Mothé BR, Sidney J, Sette A, Kunstman K, Wolinsky S, Piatak M, Lifson J, Hughes AL, Wilson N, O'Connor DH, Watkins DI. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat Med 10:275–281. doi: 10.1038/nm998. [DOI] [PubMed] [Google Scholar]
- 11.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJR. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 12.Brumme ZL, Brumme CJ, Carlson J, Streeck H, John M, Eichbaum Q, Block BL, Baker B, Kadie C, Markowitz M, Jessen H, Kelleher AD, Rosenberg E, Kaldor J, Yuki Y, Carrington M, Allen TM, Mallal S, Altfeld M, Heckerman D, Walker BD. 2008. Marked epitope- and allele-specific differences in rates of mutation in human immunodeficiency type 1 (HIV-1) Gag, Pol, and Nef cytotoxic T-lymphocyte epitopes in acute/early HIV-1 infection. J Virol 82:9216–9227. doi: 10.1128/JVI.01041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol 81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung C-S, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJR, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol 81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schneidewind A, Brumme ZL, Brumme CJ, Power KA, Reyor LL, O'Sullivan K, Gladden A, Hempel U, Kuntzen T, Wang YE, Oniangue-Ndza C, Jessen H, Markowitz M, Rosenberg ES, Sékaly R-P, Kelleher AD, Walker BD, Allen TM. 2009. Transmission and long-term stability of compensated CD8 escape mutations. J Virol 83:3993–3997. doi: 10.1128/JVI.01108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navis M, Matas DE, Rachinger A, Koning FA, van Swieten P, Kootstra NA, Schuitemaker H. 2008. Molecular evolution of human immunodeficiency virus type 1 upon transmission between human leukocyte antigen disparate donor-recipient pairs. PLoS One 3:e2422. doi: 10.1371/journal.pone.0002422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelissen M, Hoogland FM, Back NKT, Jurriaans S, Zorgdrager F, Bakker M, Brinkman K, Prins M, van der Kuyl AC. 2009. Multiple transmissions of a stable human leucocyte antigen-B27 cytotoxic T-cell-escape strain of HIV-1 in The Netherlands. AIDS 23:1495–1500. doi: 10.1097/QAD.0b013e32832d9267. [DOI] [PubMed] [Google Scholar]
- 19.Goulder PJ, Brander C, Tang Y, Tremblay C, Colbert RA, Addo MM, Rosenberg ES, Nguyen T, Allen R, Trocha A, Altfeld M, He S, Bunce M, Funkhouser R, Pelton SI, Burchett SK, McIntosh K, Korber BT, Walker BD. 2001. Evolution and transmission of stable CTL escape mutations in HIV infection. Nature 412:334–338. doi: 10.1038/35085576. [DOI] [PubMed] [Google Scholar]
- 20.Brander C, Walker BD. 2003. Gradual adaptation of HIV to human host populations: good or bad news? Nat Med 9:1359–1362. doi: 10.1038/nm941. [DOI] [PubMed] [Google Scholar]
- 21.Leslie A, Kavanagh D, Honeyborne I, Pfafferott K, Edwards C, Pillay T, Hilton L, Thobakgale C, Ramduth D, Draenert R, Le Gall S, Luzzi G, Edwards A, Brander C, Sewell AK, Moore S, Mullins J, Moore C, Mallal S, Bhardwaj N, Yusim K, Phillips R, Klenerman P, Korber B, Kiepiela P, Walker B, Goulder P. 2005. Transmission and accumulation of CTL escape variants drive negative associations between HIV polymorphisms and HLA. J Exp Med 201:891–902. doi: 10.1084/jem.20041455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung'u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daar ES, Richman DD. 2005. Confronting the emergence of drug-resistant HIV type 1: impact of antiretroviral therapy on individual and population resistance. AIDS Res Hum Retroviruses 21:343–357. doi: 10.1089/aid.2005.21.343. [DOI] [PubMed] [Google Scholar]
- 24.Milicic A, Edwards CTT, Hué S, Fox J, Brown H, Pillay T, Drijfhout JW, Weber JN, Holmes EC, Fidler SJ, Zhang H-T, Phillips RE. 2005. Sexual transmission of single human immunodeficiency virus type 1 virions encoding highly polymorphic multisite cytotoxic T-lymphocyte escape variants. J Virol 79:13953–13962. doi: 10.1128/JVI.79.22.13953-13962.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung'u T, Lakhi S, Gilmour J, Goepfert P, Walker BD, Kaslow R, Mulenga J, Allen S, Goulder PJR, Hunter E. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med 206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuhn L, Abrams EJ, Palumbo P, Bulterys M, Aga R, Louie L, Hodge T. 2004. Maternal versus paternal inheritance of HLA class I alleles among HIV-infected children: consequences for clinical disease progression. AIDS 18:1281–1289. doi: 10.1097/00002030-200406180-00006. [DOI] [PubMed] [Google Scholar]
- 27.Yue L, Prentice HA, Farmer P, Song W, He D, Lakhi S, Goepfert P, Gilmour J, Allen S, Tang J, Kaslow RA, Hunter E. 2013. Cumulative impact of host and viral factors on HIV-1 viral-load control during early infection. J Virol 87:708–715. doi: 10.1128/JVI.02118-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schellens IMM, Navis M, van Deutekom HWM, Boeser-Nunnink B, Berkhout B, Kootstra N, Miedema F, Keşmir C, Schuitemaker H, van Baarle D, Borghans JAM. 2011. Loss of HIV-1-derived cytotoxic T lymphocyte epitopes restricted by protective HLA-B alleles during the HIV-1 epidemic. AIDS 25:1691–1700. doi: 10.1097/QAD.0b013e32834981b3. [DOI] [PubMed] [Google Scholar]
- 29.Dilernia DA, Jones L, Rodriguez S, Turk G, Rubio AE, Pampuro S, Gomez-Carrillo M, Bautista CT, Bautista C, Deluchi G, Benetucci J, Lasala MB, Lourtau L, Losso MH, Perez H, Cahn P, Salomón H. 2008. HLA-driven convergence of HIV-1 viral subtypes B and F toward the adaptation to immune responses in human populations. PLoS One 3:e3429. doi: 10.1371/journal.pone.0003429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ariën KK, Troyer RM, Gali Y, Colebunders RL, Arts EJ, Vanham G. 2005. Replicative fitness of historical and recent HIV-1 isolates suggests HIV-1 attenuation over time. AIDS 19:1555–1564. doi: 10.1097/01.aids.0000185989.16477.91. [DOI] [PubMed] [Google Scholar]
- 31.Nomura S, Hosoya N, Brumme ZL, Brockman MA, Kikuchi T, Koga M, Nakamura H, Koibuchi T, Fujii T, Carlson JM, Heckerman D, Kawana-Tachikawa A, Iwamoto A, Miura T. 2013. Significant reductions in Gag-protease-mediated HIV-1 replication capacity during the course of the epidemic in Japan. J Virol 87:1465–1476. doi: 10.1128/JVI.02122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Payne R, Muenchhoff M, Mann J, Roberts HE, Matthews P, Adland E, Hempenstall A, Huang K-H, Brockman M, Brumme Z, Sinclair M, Miura T, Frater J, Essex M, Shapiro R, Walker BD, Ndung'u T, McLean AR, Carlson JM, Goulder PJR. 2014. Impact of HLA-driven HIV adaptation on virulence in populations of high HIV seroprevalence. Proc Natl Acad Sci U S A 111:E5393–E5400. doi: 10.1073/pnas.1413339111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cotton LA, Kuang XT, Le AQ, Carlson JM, Chan B, Chopera DR, Brumme CJ, Markle TJ, Martin E, Shahid A, Anmole G, Mwimanzi P, Nassab P, Penney KA, Rahman MA, Milloy M-J, Schechter MT, Markowitz M, Carrington M, Walker BD, Wagner T, Buchbinder S, Fuchs J, Koblin B, Mayer KH, Harrigan PR, Brockman MA, Poon AFY, Brumme ZL. 2014. Genotypic and functional impact of HIV-1 adaptation to its host population during the North American epidemic. PLoS Genet 10:e1004295. doi: 10.1371/journal.pgen.1004295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furutsuki T, Hosoya N, Kawana-Tachikawa A, Tomizawa M, Odawara T, Goto M, Kitamura Y, Nakamura T, Kelleher AD, Cooper DA, Iwamoto A. 2004. Frequent transmission of cytotoxic-T-lymphocyte escape mutants of human immunodeficiency virus type 1 in the highly HLA-A24-positive Japanese population. J Virol 78:8437–8445. doi: 10.1128/JVI.78.16.8437-8445.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Allen TM, Altfeld M. 2008. Crippling HIV one mutation at a time. J Exp Med 205:1003–1007. doi: 10.1084/jem.20080569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mothe B, Llano A, Ibarrondo J, Daniels M, Miranda C, Zamarreño J, Bach V, Zuniga R, Pérez-Álvarez S, Berger CT, Puertas MC, Martinez-Picado J, Rolland M, Farfan M, Szinger JJ, Hildebrand WH, Yang OO, Sanchez-Merino V, Brumme CJ, Brumme ZL, Heckerman D, Allen TM, Mullins JI, Gómez G, Goulder PJ, Walker BD, Gatell JM, Clotet B, Korber BT, Sanchez J, Brander C. 2011. Definition of the viral targets of protective HIV-1-specific T cell responses. J Transl Med 9:208. doi: 10.1186/1479-5876-9-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Borthwick N, Ahmed T, Ondondo B, Hayes P, Rose A, Ebrahimsa U, Hayton E-J, Black A, Bridgeman A, Rosario M, Hill AVS, Berrie E, Moyle S, Frahm N, Cox J, Colloca S, Nicosia A, Gilmour J, McMichael AJ, Dorrell L, Hanke T. 2014. Vaccine-elicited human T cells recognizing conserved protein regions inhibit HIV-1. Mol Ther 22:464–475. doi: 10.1038/mt.2013.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, McElrath MJ, Casimiro DR, Gottesdiener KM, Chodakewitz JA, Corey L, Robertson MN. 2008. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet 372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med 361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 40.Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, Kadie CM, Allen TM, Pereyra F, Heckerman D, Walker BD, Brockman MA. 2011. Reduced replication capacity of NL4-3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J Acquir Immune Defic Syndr 56:100–108. doi: 10.1097/QAI.0b013e3181fe9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edlefsen PT, Rolland M, Hertz T, Tovanabutra S, Gartland AJ, deCamp AC, Magaret CA, Ahmed H, Gottardo R, Juraska M, McCoy C, Larsen BB, Sanders-Buell E, Carrico C, Menis S, Bose M, Arroyo MA, O'Connell RJ, Nitayaphan S, Pitisuttithum P, Kaewkungwal J, Rerks-Ngarm S, Robb ML, Kirys T, Georgiev IS, Kwong PD, Scheffler K, Pond SLK, Carlson JM, Michael NL, Schief WR, Mullins JI, Kim JH, Gilbert PB. 2015. Comprehensive sieve analysis of breakthrough HIV-1 sequences in the RV144 vaccine efficacy trial. PLoS Comput Biol 11:e1003973. doi: 10.1371/journal.pcbi.1003973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mayer K, Appelbaum J, Rogers T, Lo W, Bradford J, Boswell S. 2001. The evolution of the Fenway Community Health model. Am J Public Health 91:892–894. doi: 10.2105/AJPH.91.6.892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seage GR, Mayer KH, Wold C, Lenderking WR, Goldstein R, Cai B, Gross M, Heeren T, Hingson R. 1998. The social context of drinking, drug use, and unsafe sex in the Boston Young Men Study. J Acquir Immune Defic Syndr Hum Retrovirol 17:368–375. doi: 10.1097/00042560-199804010-00012. [DOI] [PubMed] [Google Scholar]
- 44.van Griensven GJ, Hessol NA, Koblin BA, Byers RH, O'Malley PM, Albercht-van Lent N, Buchbinder SP, Taylor PE, Stevens CE, Coutinho RA. 1993. Epidemiology of human immunodeficiency virus type 1 infection among homosexual men participating in hepatitis B vaccine trials in Amsterdam, New York City, and San Francisco, 1978-1990. Am J Epidemiol 137:909–915. [DOI] [PubMed] [Google Scholar]
- 45.Byers RH, Morgan WM, Darrow WW, Doll L, Jaffe HW, Rutherford G, Hessol N, O'Malley PM. 1988. Estimating AIDS infection rates in the San Francisco cohort. AIDS 2:207–210. [PubMed] [Google Scholar]
- 46.Foley B, Pan H, Buchbinder S, Delwart EL. 2000. Apparent founder effect during the early years of the San Francisco HIV type 1 epidemic (1978-1979). AIDS Res Hum Retroviruses 16:1463–1469. doi: 10.1089/088922200750005985. [DOI] [PubMed] [Google Scholar]
- 47.Jeffries E, Willoughby B, Boyko WJ, Schechter MT, Wiggs B, Fay S, O'Shaughnessy M. 1985. The Vancouver Lymphadenopathy-AIDS Study. 2. Seroepidemiology of HTLV-III antibody. Can Med Assoc J 132:1373–1377. [PMC free article] [PubMed] [Google Scholar]
- 48.Schechter MT, Boyko WJ, Jeffries E, Willoughby B, Nitz R, Constance P. 1985. The Vancouver Lymphadenopathy-AIDS Study. 1. Persistent generalized lymphadenopathy. Can Med Assoc J 132:1273–1279. [PMC free article] [PubMed] [Google Scholar]
- 49.Schechter MT, Boyko WJ, Douglas B, Willoughby B, McLeod A, Maynard M, Craib KJ, O'Shaughnessy M. 1986. The Vancouver Lymphadenopathy-AIDS Study. 6. HIV seroconversion in a cohort of homosexual men. Can Med Assoc J 135:1355–1360. [PMC free article] [PubMed] [Google Scholar]
- 50.Martin E, Carlson JM, Le AQ, Chopera DR, McGovern R, Rahman MA, Ng C, Jessen H, Kelleher AD, Markowitz M, Allen TM, Milloy M-J, Carrington M, Wainberg MA, Brumme ZL. 2014. Early immune adaptation in HIV-1 revealed by population-level approaches. Retrovirology 11:64. doi: 10.1186/s12977-014-0064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robertson MJ, Clark RA, Charlebois ED, Tulsky J, Long HL, Bangsberg DR, Moss AR. 2004. HIV seroprevalence among homeless and marginally housed adults in San Francisco. Am J Public Health 94:1207–1217. doi: 10.2105/AJPH.94.7.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Woods CK, Brumme CJ, Liu TF, Chui CKS, Chu AL, Wynhoven B, Hall TA, Trevino C, Shafer RW, Harrigan PR. 2012. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J Clin Microbiol 50:1936–1942. doi: 10.1128/JCM.06689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sievers F, Higgins DG. 2014. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol Biol 1079:105–116. doi: 10.1007/978-1-62703-646-7_6. [DOI] [PubMed] [Google Scholar]
- 54.Guindon S, Dufayard J-F, Lefort V, Anisimova M, Hordijk W, Gascuel O. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 55.Cotton LA, Abdur Rahman M, Ng C, Le AQ, Milloy M-J, Mo T, Brumme ZL. 2012. HLA class I sequence-based typing using DNA recovered from frozen plasma. J Immunol Methods 382:40–47. doi: 10.1016/j.jim.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 56.Listgarten J, Brumme Z, Kadie C, Xiaojiang G, Walker B, Carrington M, Goulder P, Heckerman D. 2008. Statistical resolution of ambiguous HLA typing data. PLoS Comput Biol 4:e1000016. doi: 10.1371/journal.pcbi.1000016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Drummond AJ, Suchard MA, Xie D, Rambaut A. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pond SLK, Frost SDW, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 59.Bennett DE, Camacho RJ, Otelea D, Kuritzkes DR, Fleury H, Kiuchi M, Heneine W, Kantor R, Jordan MR, Schapiro JM, Vandamme A-M, Sandstrom P, Boucher CAB, van de Vijver D, Rhee S-Y, Liu TF, Pillay D, Shafer RW. 2009. Drug resistance mutations for surveillance of transmitted HIV-1 drug-resistance: 2009 update. PLoS One 4:e4724. doi: 10.1371/journal.pone.0004724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Paradis E, Claude J, Strimmer K. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20:289–290. doi: 10.1093/bioinformatics/btg412. [DOI] [PubMed] [Google Scholar]
- 62.Baele G, Li WLS, Drummond AJ, Suchard MA, Lemey P. 2013. Accurate model selection of relaxed molecular clocks in Bayesian phylogenetics. Mol Biol Evol 30:239–243. doi: 10.1093/molbev/mss243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fourment M, Gibbs MJ. 2006. PATRISTIC: a program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol Biol 6:1. doi: 10.1186/1471-2148-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murrell B, Moola S, Mabona A, Weighill T, Sheward D, Kosakovsky Pond SL, Scheffler K. 2013. FUBAR: a fast, unconstrained Bayesian approximation for inferring selection. Mol Biol Evol 30:1196–1205. doi: 10.1093/molbev/mst030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Delport W, Poon AFY, Frost SDW, Kosakovsky Pond SL. 2010. Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26:2455–2457. doi: 10.1093/bioinformatics/btq429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Korber B, Muldoon M, Theiler J, Gao F, Gupta R, Lapedes A, Hahn BH, Wolinsky S, Bhattacharya T. 2000. Timing the ancestor of the HIV-1 pandemic strains. Science 288:1789–1796. doi: 10.1126/science.288.5472.1789. [DOI] [PubMed] [Google Scholar]
- 67.Gilbert MTP, Rambaut A, Wlasiuk G, Spira TJ, Pitchenik AE, Worobey M. 2007. The emergence of HIV/AIDS in the Americas and beyond. Proc Natl Acad Sci U S A 104:18566–18570. doi: 10.1073/pnas.0705329104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Karakas A, Brumme ZL, Poon AFY. 2015. Global database-driven assessment of HIV-1 adaptation to the immune repertoires of human populations. J Virol 89:10693–10695. doi: 10.1128/JVI.01355-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.O'Brien SJ, Gao X, Carrington M. 2001. HLA and AIDS: a cautionary tale. Trends Mol Med 7:379–381. doi: 10.1016/S1471-4914(01)02131-1. [DOI] [PubMed] [Google Scholar]
- 70.Duda A, Lee-Turner L, Fox J, Robinson N, Dustan S, Kaye S, Fryer H, Carrington M, McClure M, McLean AR, Fidler S, Weber J, Phillips RE, Frater AJ. 2009. HLA-associated clinical progression correlates with epitope reversion rates in early human immunodeficiency virus infection. J Virol 83:1228–1239. doi: 10.1128/JVI.01545-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fryer HR, Frater J, Duda A, Roberts MG, Phillips RE, McLean AR. 2010. Modelling the evolution and spread of HIV immune escape mutants. PLoS Pathog 6:e1001196. doi: 10.1371/journal.ppat.1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yagita Y, Kuse N, Kuroki K, Gatanaga H, Carlson JM, Chikata T, Brumme ZL, Murakoshi H, Akahoshi T, Pfeifer N, Mallal S, John M, Ose T, Matsubara H, Kanda R, Fukunaga Y, Honda K, Kawashima Y, Ariumi Y, Oka S, Maenaka K, Takiguchi M. 2013. Distinct HIV-1 escape patterns selected by cytotoxic T cells with identical epitope specificity. J Virol 87:2253–2263. doi: 10.1128/JVI.02572-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, Carlson J, Yusim K, McMahon B, Gaschen B, Mallal S, Mullins JI, Nickle DC, Herbeck J, Rousseau C, Learn GH, Miura T, Brander C, Walker B, Korber B. 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315:1583–1586. doi: 10.1126/science.1131528. [DOI] [PubMed] [Google Scholar]
- 74.Turnbull EL, Lopes AR, Jones NA, Cornforth D, Newton P, Aldam D, Pellegrino P, Turner J, Williams I, Wilson CM, Goepfert PA, Maini MK, Borrow P. 2006. HIV-1 epitope-specific CD8+ T cell responses strongly associated with delayed disease progression cross-recognize epitope variants efficiently. J Immunol 176:6130–6146. doi: 10.4049/jimmunol.176.10.6130. [DOI] [PubMed] [Google Scholar]
- 75.Mothe B, Llano A, Ibarrondo J, Zamarreño J, Schiaulini M, Miranda C, Ruiz-Riol M, Berger CT, Herrero MJ, Palou E, Plana M, Rolland M, Khatri A, Heckerman D, Pereyra F, Walker BD, Weiner D, Paredes R, Clotet B, Felber BK, Pavlakis GN, Mullins JI, Brander C. 2012. CTL responses of high functional avidity and broad variant cross-reactivity are associated with HIV control. PLoS One 7:e29717. doi: 10.1371/journal.pone.0029717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allen TM, Yu XG, Kalife ET, Reyor LL, Lichterfeld M, John M, Cheng M, Allgaier RL, Mui S, Frahm N, Alter G, Brown NV, Johnston MN, Rosenberg ES, Mallal SA, Brander C, Walker BD, Altfeld M. 2005. De novo generation of escape variant-specific CD8+ T-cell responses following cytotoxic T-lymphocyte escape in chronic human immunodeficiency virus type 1 infection. J Virol 79:12952–12960. doi: 10.1128/JVI.79.20.12952-12960.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. 2013. A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 38:425–436. doi: 10.1016/j.immuni.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 78.Han C, Kawana-Tachikawa A, Shimizu A, Zhu D, Nakamura H, Adachi E, Kikuchi T, Koga M, Koibuchi T, Gao GF, Sato Y, Yamagata A, Martin E, Fukai S, Brumme ZL, Iwamoto A. 2014. Switching and emergence of CTL epitopes in HIV-1 infection. Retrovirology 11:38. doi: 10.1186/1742-4690-11-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ariën KK, Vanham G, Arts EJ. 2007. Is HIV-1 evolving to a less virulent form in humans? Nat Rev Microbiol 5:141–151. doi: 10.1038/nrmicro1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fryer HR, Frater J, Duda A, Palmer D, Phillips RE, McLean AR. 2012. Cytotoxic T-lymphocyte escape mutations identified by HLA association favor those which escape and revert rapidly. J Virol 86:8568–8580. doi: 10.1128/JVI.07020-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Montaner JSG, Lima VD, Barrios R, Yip B, Wood E, Kerr T, Shannon K, Harrigan PR, Hogg RS, Daly P, Kendall P. 2010. Association of highly active antiretroviral therapy coverage, population viral load, and yearly new HIV diagnoses in British Columbia, Canada: a population-based study. Lancet 376:532–539. doi: 10.1016/S0140-6736(10)60936-1. [DOI] [PMC free article] [PubMed] [Google Scholar]