

Abstract
Background
Although the HKT transporter genes ascertain some of the key determinants of crop salt tolerance mechanisms, the diversity and functional role of group II HKT genes are not clearly understood in bread wheat. The advanced knowledge on rice HKT and whole genome sequence was, therefore, used in comparative gene analysis to identify orthologous wheat group II HKT genes and their role in trait variation under different saline environments.
Results
The four group II HKTs in rice identified two orthologous gene families from bread wheat, including the known TaHKT2;1 gene family and a new distinctly different gene family designated as TaHKT2;2. A single copy of TaHKT2;2 was found on each homeologous chromosome arm 7AL, 7BL and 7DL and each gene was expressed in leaf blade, sheath and root tissues under non-stressed and at 200 mM salt stressed conditions. The proteins encoded by genes of the TaHKT2;2 family revealed more than 93 % amino acid sequence identity but ≤52 % amino acid identity compared to the proteins encoded by TaHKT2;1 family. Specifically, variations in known critical domains predicted functional differences between the two protein families. Similar to orthologous rice genes on chromosome 6L, TaHKT2;1 and TaHKT2;2 genes were located approximately 3 kb apart on wheat chromosomes 7AL, 7BL and 7DL, forming a static syntenic block in the two species. The chromosomal region on 7AL containing TaHKT2;1 7AL-1 co-located with QTL for shoot Na+ concentration and yield in some saline environments.
Conclusion
The differences in copy number, genes sequences and encoded proteins between TaHKT2;2 homeologous genes and other group II HKT gene families within and across species likely reflect functional diversity for ion selectivity and transport in plants. Evidence indicated that neither TaHKT2;2 nor TaHKT2;1 were associated with primary root Na+ uptake but TaHKT2;1 may be associated with trait variation for Na+ exclusion and yield in some but not all saline environments.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-016-0714-7) contains supplementary material, which is available to authorized users.
Keywords: Group II HKT, IWGS, Rice genome, Na+ exclusion
Background
Response to high saline conditions results from interaction of several biological processes controlled by multiple genes. Increasing evidence indicated that Na+ exclusion from the transpiration stream is an important mechanism associated with salt tolerance [1]. Na+ exclusion when measured as Na+ and/or K+ content in tissues or organs, is a robust and highly heritable trait in bread wheat [2]. The high affinity potassium transporter (HKT) genes are one of the most studied groups of membrane transporters in plants and the group I HKT genes that encode Na+ selective transporter proteins act in cohesion with the salt overly sensitive (SOS) pathway [3] identifying a major role in Na+ exclusion [2, 4] in wheat and other species [5–7]. Several group I HKT transporters are associated with retrieval of Na+ from xylem in root or sheath restricting transport and accumulation of salt in sensitive leaf tissues [1, 8, 9]. Grass species evolved a second class of HKT proteins encoded by group II HKT genes that function as Na+ and K+ permeable transporters [10]. A single member of this group, TaHKT2;1, has been identified from bread wheat that encoded a protein presumed to function in Na+ uptake from external medium through the roots [11, 12]. A recent study, however, showed that TaHKT2;1 is a multigene family consisting of four functional genes and pseudogenes located on the long arm of homeologous group 7 chromosomes with evidence that the individual genes were not involved in controlling Na+ influx from the external medium into roots but may have a role in excluding Na+ from leaves or possibly involved in maintaining K+ status in the plant [13]. Therefore, a probable new capacity for group II HKT genes has been recognized where further research is warranted to gain additional insights into the role and potential of these genes in trait variation under different saline environments.
The most comprehensive analysis of group II HKT genes has been in rice (Oryza sativa L.) with up to four genes OsHKT2;1; OsHKT2;2, OsHKT2;3, and OsHKT2;4 characterized for gene structure, expression and function. Some of the functional genes such as OsHKT2;2 in Indica rice Pokkali [14] was identified as a chimeric gene, No-OsHKT2;2/1, in japonica rice Nona Bokra [15] and a truncated pseudogene in Nipponbare [14] that provided evidence of recent evolutionary changes in group II HKT genes in modern rice accessions. Phylogenetic relationships between rice group II HKT genes showed evidence of gene duplication and divergence, identifying two distinct clusters whereby the genes OsHKT2;1 and functional OsHKT2;2 diverged and clustered separately from OsHKT2;3 and OsHKT2;4 with >91 % DNA sequence identity within but only 40–50 % identity between clusters [5, 16–18]. Transcripts of the rice genes OsHKT2;1 and OsHKT2;2 were detected in roots with variable tissue expression in tolerant and susceptible rice varieties [19] whereas OsHKT2;3 and OsHKT2;4 transcripts accumulated in the shoot [17, 20]. Although OsHKT2;1 is down regulated under saline conditions [21], there was no evidence to indicate a significant effect on the expression of the remaining rice group II HKT genes [17]. Most of the functional group II HKT genes in rice serve as Na+/K+ co-transporters with a role in maintaining K+/Na+ homeostasis in plants [20, 22, 23]. However, OsHKT2;1 is an exception and presumed to function as a Na+ selective transporter with a putative role in “nutritional Na+ uptake” under K+ starvation [21]. The extensive knowledge on structure, expression and function of rice group II HKT genes, therefore, can be effectively used to identify and characterize gene orthologs in wheat based on comparative gene studies.
Phylogenetic relatedness of genes and whole genome sequence provides opportunities to identify gene orthologs across species. The advanced genomic resources available for rice including genome sequence data for 95 % of the 389 Mb genome with 37,544 annotated protein-coding genes [24] and integrated search tools that allow user-friendly access to genomic data enable a robust application of the rice genome in comparative gene studies with other cereal species. Although not as advanced as rice, the draft sequence of the 17Gb bread wheat genome identified >124,000 annotated and ordered gene loci [25] which has expedited comparative gene studies between rice and wheat to identify wheat genes and their association with biological processes controlling trait variation [26–30]. More specifically, the high degree of sequence conservation between HKT genes [10] allowed a comparative gene analysis within and across multigene families in the same [13], or different grass species [31]. Therefore, whole genome sequence from wheat and rice can be exploited in data mining and detailed comparative gene analysis for identification of wheat orthologs of the rice HKT genes.
While comparative gene analysis between rice and wheat enabled gene identification and characterization, determining function of wheat HKT orthologs defines their contribution towards improving salt tolerance. Quantitative trait loci (QTL) studies for salt tolerance in wheat [32–36] can be strategically utilized to investigate genes that may be functionally associated with trait variation. In particular, the doubled haploid (DH) mapping population derived from wheat cultivars Berkut and Krichauff as parents detected a number of QTL associated with physiological and yield related traits in controlled and field saline environments including 17 QTL for Na+ exclusion measured as leaf or shoot Na+ concentration in different environments [33, 37]. Interestingly, a member of the TaHKT2;1 gene family was located in a similar region on chromosome 7AL [13] as QTL for shoot Na+ concentration and seedling biomass under controlled (hydroponics) saline conditions and in similar chromosomal regions for variation for yield components under moderate saline field environments [33, 37]. Therefore, QTL information can be used to make inferences on the role of wheat group II HKT gene orthologs in controlling phenotypes expressed under different saline environments, providing insights into their possible role in contributing towards improving salinity tolerance.
Although one multigene family TaHKT2;1 was well characterized from wheat [13], given the fact that four individual genes exists in rice [17] it is reasonable to assume that wheat may have evolved multiple copies of more than one group II HKT gene family. The aim of this study, therefore, was to apply whole genome sequence in a comparative gene analysis to identify and characterize wheat orthologs of rice group II HKT genes. The association of wheat gene orthologs with trait variation was investigated by QTL analysis under different saline environments using the Berkut/Krichauff DH mapping population. The outcome of this study will increase our knowledge on the group II HKT genes in wheat and their significance on trait variation under different saline environments.
Results
Wheat genes orthologous to rice group II HKTs
Full length cDNA (FL-cDNA) of the rice genes OsHKT2;1, OsHKT2;2, OsHKT2;3 and OsHKT2;4 were used as query sequence in blastn and tblastx search of the IWGS survey sequence database in order to identify related wheat gene sequences. The four rice FL-cDNAs identified related wheat sequences on seven scaffolds (Table 1). The closely related FL-cDNA of OsHKT2;1 and OsHKT2;2, both identified hits on six wheat scaffolds, three on wheat chromosome 7AL, two on 7BL and one on 7DL with up to 76 % DNA identity (Table 1) and in the same region as the known wheat group II HKT, TaHKT2;1. TaHKT2;1 had 75–76 % DNA identity to both OsHKT2;1 and OsHKT2;2. Similarly, FL-cDNA of OsHKT2;3 and OsHKT2;4 both had 80 % DNA sequence identity in the same region on three wheat scaffolds (Table 1). Therefore, a wheat group II HKT gene distinctly different to TaHKT2;1 had identity with both OsHKT2;3 and OsHKT2;4. The wheat scaffolds having sequence similarity with OsHKT2;3 and OsHKT2;4, #4510252; #6569883 and #3312548, on chromosomes 7AL, 7BL and 7DL, respectively, were selected for further investigation.
Table 1.
Wheat sequence scaffolds retrieved from IWGS having significant DNA identity (E = 0) with the FL-cDNA of rice group II HKT genes
| Scaffolds number (chromosome arm) | Scaffold size (bp) | OsHKT2;1 (Genbank #AB061311) | OsHKT2;2 (Genbank #AB061313) | OsHKT2;3 (Genbank #AJ491819) | OsHKT2;4 (Genbank #AJ491854) |
|---|---|---|---|---|---|
| #4510252 (7AL) | 10,610 | 3767–4922 (76 %) | 3767–4922 (76 %) | 8794–9915 (80 %) | 8804–9915 (80 %) |
| #4434238 (7AL) | 7049 | 3531–4667 (75 %) | 3531–4667 (75 %) | * | * |
| #4523843 (7AL) | 7561 | 4043–5179 (75 %) | 4043–5179 (75 %) | * | * |
| #6657249 (7BL) | 6544 | 2431–3586 (76 %) | 2431–3586 (76 %) | * | * |
| #6748027 (7BL) | 7204 | 3091–4246 (76 %) | 3019–4246 (76 %) | * | * |
| #6569883 (7BL) | 5298 | * | * | 1575–448 (80 %) | 1565–448 (80 %) |
| #3312548 (7DL) | 22,420 | 16,269–15,114 (76 %) | 16,269–15,114 (76 %) | 11,081–9960 (80 %) | 11,071–9960 (80 %) |
The region of similarity in the wheat scaffold is shown in bp and DNA sequence identity is in parenthesis. Scaffolds with no significant DNA sequence identity with rice FL–cDNA are shown by an asterix
The wheat sequences with identity to OsHKT2;3 and OsHKT2;4 on 7AL, 7BL and 7DL were analysed in multiple sequence alignments using the gene on 7AL as a reference to ascertain sequence variants that enabled design of gene specific reverse transcription polymerase chain reaction (RT-PCR) primer pairs (Table 2) to amplify FL-cDNA. The RT-PCR primers were strategically positioned against putative wheat exons assuming similar gene structure to OsHKT2;3 and OsHKT2;4 (Fig. 1a). Since the scaffold on 7BL did not appear to contain sequence corresponding to the full length sequence of OsHKT2;3 or OsHKT2;4, the 3’-region of the gene on 7BL was amplified using primers designed from similar regions on 7AL and 7DL (Fig. 1a). Primer pairs showing sub-genome specificity were used to amplify partial but overlapping gene transcripts specifically from 7A, 7B and 7D and confirmed by nullisomic-tetrasomic (NT) analysis (data not shown). Subsequently, overlapping gene-specific cDNA from chromosome 7AL, 7BL and 7DL were amplified from root tissue, sequenced and assembled (Genbank accession numbers KR422354, KR422355 and KR422356 respectively). The FL-cDNA assembled for each gene on 7AL, 7BL and 7DL was aligned against the cognate genomic sequence from scaffolds #4510252; #6569883 and #3312548, respectively, to determine the intron-exon structure (Fig. 1b). The genes on chromosomes 7AL, 7BL and 7DL had similar structure to the OsHKT2;3 and OsHKT2;4 including 3 exons interrupted by 2 introns with intron splice sites having conserved motif GT and AG at the 5’ and 3’ boundaries, respectively. The cDNA of each gene had <64 % DNA sequence identity with TaHKT2;1 gene family confirming that the two gene families were distinctly different. Therefore, the genes on 7AL, 7BL and 7DL were designated as TaHKT2;2 7AL-1, TaHKT2;2 7BL-1 and TaHKT2;2 7DL-1, respectively. When compared with TaHKT2;2 7AL-1, all variants within exons were SNPs, totalling 69 for TaHKT2;2 7BL-1 and 39 for TaHKT2;2 7DL-1 whereas the comparison between TaHKT2;2 7BL and TaHKT2;2 7DL identified 76 SNPs.
Table 2.
Gene specific primers and PCR annealing temperatures to amplify genomic and cDNA sequence from members of the TaHKT2;2 gene family and qRT-PCR for TaHKT2;1 and TaHKT2;2 genes
| Forward primer | Reverse Primer | ||||
|---|---|---|---|---|---|
| Gene | Primer | Primer sequence (5’-3’) | Primer | Primer sequence (5’-3’) | Annealing Temperature °C |
| TaHKT2;2 7AL-1 | 2;2AF1 | CCATCTATCTAACTCCAATGACTG | 2;2AR1 | CTCTCAGTGCCCATACCAT | 54 |
| TaHKT2;2 7AL-1 | 2;2AF2 | TCAATACCATGCTCTTCACG | 2;2AR2 | AGACGCTCTGCTTCTCTGC | 55–50 |
| TaHKT2;2 7AL-1 | 2;2AF3 | GGGGCAAACAAGAGAAGAA | 2;2ADR1 | CTAGCTCCTCTGCCTGTG | 55–50 |
| TaHKT2;2 7BL-1 | 2;2CF1 | CTATCTAACTCCAATGCCTATC | 2;2BR1 | GCAAGTGGTTGAAACTCACAC | 60–50 |
| TaHKT2;2 7BL-1 | 2;2BF2 | CTGTTGGCATACATGGTATC | 2;2BR2 | ATGGACAGTCCTACGTTCATA | 65–55 |
| TaHKT2;2 7BL-1 | 2;2BF3 | CCGTTGCCATCACACTCTTG | 2;2ADR1 | CTAGCTCCTCTGCCTGTG | 60–50 |
| TaHKT2;2 7DL-1 | 2;2ADCF1 | CACACTGCCATCTATCTAACTC | 2;2DR1 | ACAGTCTTCTCTTGTTCGCT | 55–50 |
| TaHKT2;2 7DL-1 | 2;2DF2 | TGCTTTTCTCCATACCCA | 2;2ADR1 | CTAGCTCCTCTGCCTGTG | 55–50 |
| TaHKT2;1 7AL-1 | 2;1AF1 | TCGGCTCTTATCAGAACACA | 2;1AR1 | CCACACGTTGATAGATAATGTC | 55 |
| TaHKT2;1 7AL-1 | qPCR2;1AF1 | ATGTCCCCTGCCATTGTAGAAT | qPCR2;1AR1 | CGTGTTCTCATTGGTGGTTTTACTG | 60 |
| TaHKT2;1 7AL-1 | qPCR2;1BF1 | TGCGTTTTGCTAATTTGCCTG | qPCR2;1BR1 | GATAAGAGCTGAGCCCATCCAAG | 60 |
| TaHKT2;1 7DL-1 | qPCR2;1DF1 | ACTGTTTTTCTCTCCTCAACGCTT | qPCR2;1DR1 | TGCCTTTTGTGCTCGCTTC | 60 |
| TaHKT2;2 7AL-1 | qPCR2;2AF1 | GATATGGGCACTGAGAGGACTATGA | qPCR2;2AR1 | AAACAGCATTTTATTCAGCGAGAT | 60 |
| TaHKT2;2 7BL-1 | qPCR2;2BF1 | GCTGCTTGAACTGGAATGCG | qPCR2;2BR1 | GTGTTCTGTGATGCCCCTCTTGT | 60 |
| TaHKT2;2 7DL-1 | qPCR2;2DF1 | GATTCACTTGTCCTATTTTGTTGTCG | qPCR2;2DR1 | GCAGGGAAACAAACATCTCTCTG | 58 |
| TaActin | TaActin_qR | TGGCACCCGAGGAGCACCCTG | TaActin_qR | GCGACGTACATGGCAGGAACA | 60 |
| GAPDH | GAPDHF | CGCCAGGGTTTTCCCAGTCACGAC | GAPDHR | TCAC ACAGGAAACAGCTATGAC | 60 |
| TaEFA | TaEFA_qF | GATTGGCAACGGCTACG | TaEFA_qR | CGGACAGCAAAACGACC | 60 |
Fig. 1.
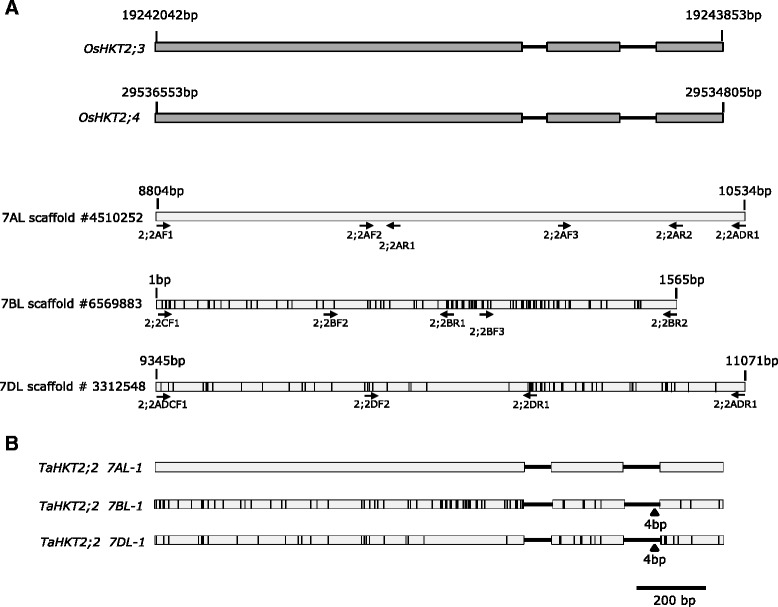
Wheat genes orthologous to rice OsHKT2;3 and OsHKT2;4. a Gene structure of rice OsHKT2;3 and OsHKT2;4 showing 3 exons (grey boxes) interrupted by 2 introns (black lines). Wheat genomic sequence on scaffolds from 7AL, 7BL and 7DL with 80 % DNA identity to rice FL-cDNA of OsHKT2;3 and OsHKT2;4 are shown below the rice gene structures. The numbers at the top of each sequence represents the position of the gene on each scaffold. SNPs between wheat 7BL and 7DL relative to 7AL are indicated by black vertical lines. Position and direction of PCR primers to amplify wheat FL-cDNA are indicated by arrows. b Structure of the wheat genes, TaHKT2;2 7AL-1, TaHKT2;2 7BL-1 and TaHKT2;2 DL-1 deduced from the alignment of FL-cDNA sequences with cognate genomic scaffold sequences. Exons are shown in grey boxes and the introns in horizontal bars. SNPs are indicated by black vertical lines whereas black triangles represent INDELS relative to the gene on chromosome 7AL
Analysis of proteins encoded by TaHKT2;2 genes
Sequencing of FL-cDNA for TaHKT2;2 enabled the predicted translation and subsequent characterization of encoded proteins. Each TaHKT2;2 gene on chromosomes 7AL, 7BL and 7DL encoded a predicted protein of 508 amino acids. Combined analysis of hydrophobicity plots (Fig. 2a) and predicted 3D- structures (Fig. 2b) of the proteins encoded by TaHKT2 genes allowed identification of protein folding patterns and modelling of protein topology. Superimposing models identified a number of differences in folding structure between each respective proteins encoded by TaHKT2;1 and TaHKT2;2 genes (Fig. 2b). The proteins encoded by each gene modelled a topology consisting of four sequentially arranged membrane-pour-membrane domains resembling core protein structure typical of HKT proteins (domains I–IV in Fig. 2c). The protein encoded by each TaHKT2;2 gene was highly similar with 93–97 % amino acid identity and a high degree of sequence conservation in P-loops (Fig. 3). In each protein the conserved glycine molecules at Gly80, Gly221, Gly349 and Gly453 amino acid positions comprised the putative cation selectivity filter (Fig. 3). However, amino acid substitutions predicted variation in physical properties of the proteins including hydrophobic regions in the NH2-terminus encoded by TaHKT2;2 7AL-1 and TaHKT2;2 7DL-1 compared to TaHKT2;2 7BL-1 (Figs. 2a and 3).
Fig. 2.
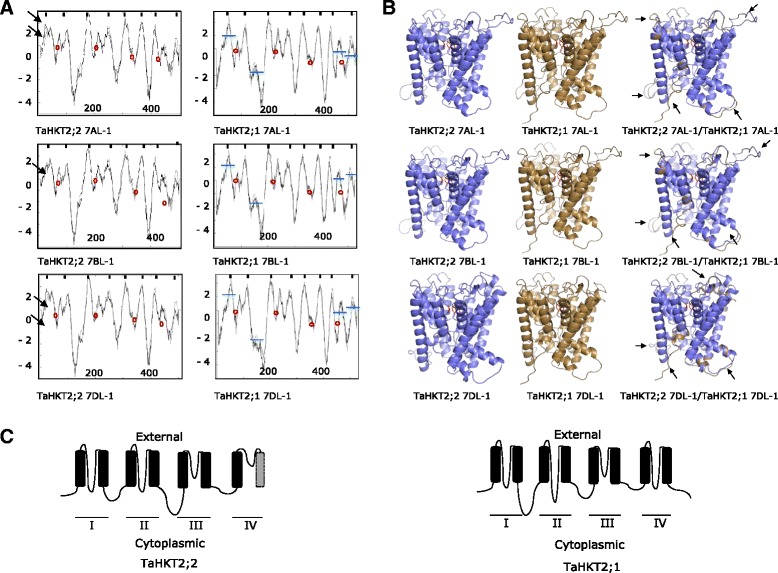
Predictions of transmembrane domains (TM) in proteins encoded by TaHKT2;2 and TaHKT2;1 genes. a Hydrophobicity plots of proteins encoded by each member of the TaHKT2;2 and TaHKT2;1 multigene families. The horizontal axis represents amino acid position and the vertical axis represents hydrophobicity value. TMs are indicated as black boxes on top of each graph. The region of the glycine filter domains are circled in red. Physical differences in the N-terminus shown by peak variation are indicated by black arrows. Differences in peak structures TaHKT2;1 relative to TaHKT2;2 proteins are indicated by horizontal blue lines. b The 3-D protein models of individual proteins encoded by TaHKT2;1 and TaHKT2;2 families and superimposed 3-D models of predicted proteins encoded by members of TaHKT2;2 and TaHKT2;1 from the same chromosome. Black arrows on the superimposed 3-D models represent different folding domains between proteins encoded by TaHKT2;1 and TaHKT2;2 counterparts on each chromosome. Conserved filter glycines are indicated in red. c Schematic diagram of the general model predicting protein topology deduced from proteins encoded by TaHKT2;2 and TaHKT2;1 genes. Black filled rectangles represent TMs and the lines cytoplasmic, external and P-loop domains. Putative TM (hydrophobicity value <0) is indicated by grey rectangle
Fig. 3.
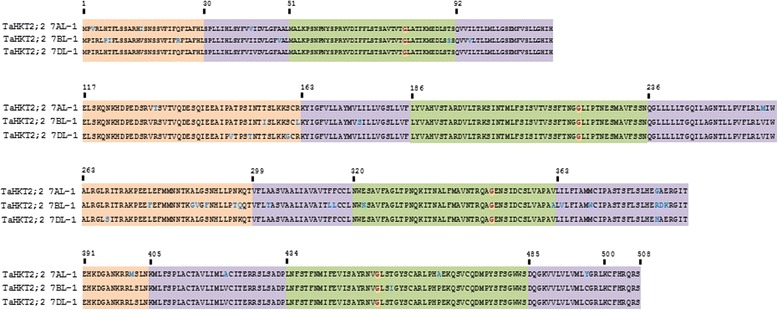
Multiple sequence alignment of predicted proteins encoded by TaHKT2;2 genes from homeologous chromosome 7AL, 7BL and 7DL containing four membrane-pore-membrane structures (I to IV). Cytoplasmic, trans-membrane, and p-loop domains are in pink, purple and green backgrounds respectively. Conserved glycine residues representative of the cation selectivity filter are indicated in red. Differences in amino acid residues are shown in blue
The proteins encoded by TaHKT2;2 and TaHKT2;1 were compared to ascertain similarities and differences. The proteins encoded by TaHKT2;2 revealed less than 52 % amino acid identity with the proteins encoded by TaHKT2;1 (Additional file 1: Figure S1), indicating that they are distinctly different. Although both protein families had a conserved structural core, protein topology and important functional domains, the protein encoded by TaHKT2;2 relative to TaHKT2;1 had truncated NH2-terminus, second cytoplasmic domain and carboxy-terminus, in addition to a four amino acid insertion in the third cytoplasmic domain (Additional file 1: Figure S1). Significant differences were identified in the carboxy-terminus whereby both hydrophobicity based analysis and in homology based 3D structures did not predict a transmembrane inner helix of 4th membrane-pore-membrane structure but a membrane embedded carboxy end in the proteins encoded by TaHKT2;2 genes (Additional file 1: Figure S1). This is in contrast to the cytoplasm bound carboxy terminal in the proteins encoded by TaHKT2;1. In addition regions of amino acid variability in external and cytoplasmic caused distinct structural differences and these are depicted in Fig. 2c. Despite the different group II HKT proteins having conserved structural core and G-G-G-G type cation selectivity filter domains, variation in amino acid composition in the filter region (Additional file 1: Figure S1) and differences in cytoplasmic and external domains implied variability in functional characteristics.
Quantitative expression of members of TaHKT2;1 and TaHKT2;2 multigene families
TaHKT2;1 and TaHKT2;2 genes on chromosome 7AL, 7BL and 7DL were analysed for gene specific transcripts in roots, sheaths and leaf blade tissues of wheat seedlings (Chinese Spring). Significantly higher Na+ levels in leaf (cv = 3.26 %, P < 0.0001), root (cv = 3.35 %, P < 0.0003) and sheath (cv = 7.73 %, P < 0.0015) were observed for samples treated with 200 mM NaCl for 72 h compared with untreated control samples for each tissue (Fig. 4a) and, therefore, suitable for expression using quantitative RT-PCR (qRT-PCR). Sub-genome specific primer pairs (Table 2) were designed based on unique polymorphic sites and specificity was confirmed by NT analysis (Fig. 4b). The qRT-PCR analysis showed that members of the TaHKT2;2 gene family and TaHKT2;1 7AL-1 and TaHKT2;1 7BL-1 were expressed in untreated root, sheath and leaf blade tissues and in the same tissue under NaCl stressed conditions (Fig. 4c). The exception, however, was TaHKT2;1 7DL-1 where transcripts were detected in both sheath and root samples but not in the detectable limits of the qRT-PCR assay for the leaf blade in either the control or NaCl treated samples. Expression in control and 200 mM NaCl treated leaf blade (cv = 11.39 %, P > 0.88), root (cv = 13.75 %, P > 0.16) and sheath (cv = 15.97 %, P > 0.11) tissue samples showed that TaActin was suitable for gene expression normalization and was a reliable internal reference gene for quantification of HKT transcripts under salt stress. Fold change between control and NaCl treatments (∆∆CT = -1 to +1) using TaActin as an internal reference showed no difference in expression of individual genes and it is likely that these genes were not salt responsive. However, a two-fold down regulation in TaHKT2;1 7DL-1 expression and up to three-fold down regulation in TaHKT2;2 7AL-1 expression levels indicated that these genes were differentially regulated in control and NaCl treated conditions (Fig. 4c).
Fig. 4.
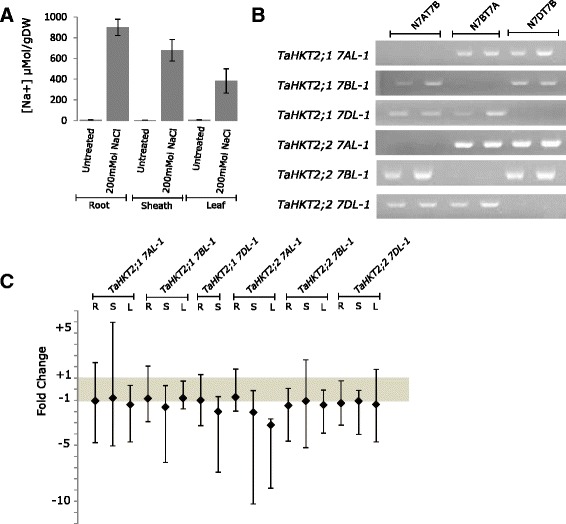
Expression of TaHKT2;2 genes under non-treated and saline conditions. a Na+ concentration of tissue samples for qRT-PCR analysis. Na+ ion concentrations were measured in triplicate from four biological replicates for untreated (control) and 200 mM NaCl treated tissue samples. b Agarose gel electrophoresis showing sub-genome specificity of primer pairs used in qRT-PCR of group II HKT gene members using NT lines. Each line is in duplicate (c). Bar graph representing fold change estimated using the ∆∆CT method. The error bars represent possible range of relative quantity values, RQmax and RQmin, defined by the standard error of the ∆CT s qRT-PCR for transcript analysis of group II HKT gene family members in wheat (Chinese Spring) using cDNA from root (R), sheath (S) and leaf (L) tissue samples untreated and treated for 72 h in 200 mM NaCl. The internal control genes TaActin was used to normalize for variability in initial RNA template for each reaction. The grey shaded region within the graph highlights the fold range that has no significant difference
Physical mapping of wheat group II HKT genes and the promoter region of TaHKT2;2 gene family
Analysis of the pseudomolecule for rice chromosome 6L identified that OsHKT2;1 (Ordered Locus Name, LOC_Os06g48810) and OsHKT2;4 (LOC_Os06g48800) were separated by a physical distance of 2441 base pairs (Fig. 5). Similarly, TaHKT2;1 and TaHKT2;2 were identified on the same sequence scaffolds from chromosome 7AL (scaffold #4510252) and 7DL (scaffold #3312548) with intergenic distances of 3154 base pairs and 3330 base pairs, respectively (Fig. 5). TaHKT2;2 7BL-1 and TaHKT2;1 7BL-1 genes were separated by an intergenic distance of 3302 base pairs, however, only partial sequence of TaHKT2;1 7BL-1, including the 3rd exon, 2nd intron and partial sequence of 2nd exon were identified on scaffold #6569883 (Fig. 5). Although a second copy of TaHKT2;1 7BL-2 was retrieved from contig 3599841+ assembled in two different scaffolds (#6657249 and #16748027), neither of the scaffolds identified TaHKT2;2 7BL-1, hence, the wheat genome survey sequence did not contain a single scaffold with the entire length of both TaHKT2;1 and TaHKT2;2 genes on 7BL. Nevertheless, it was evident that TaHKT2;1 and TaHKT2;2 genes in wheat are physically linked on 7AL, 7BL and 7DL with intergenic distances, comparable to OsHKT2;1 and OsHKT2;4 genes on rice chromosome 6L.
Fig. 5.
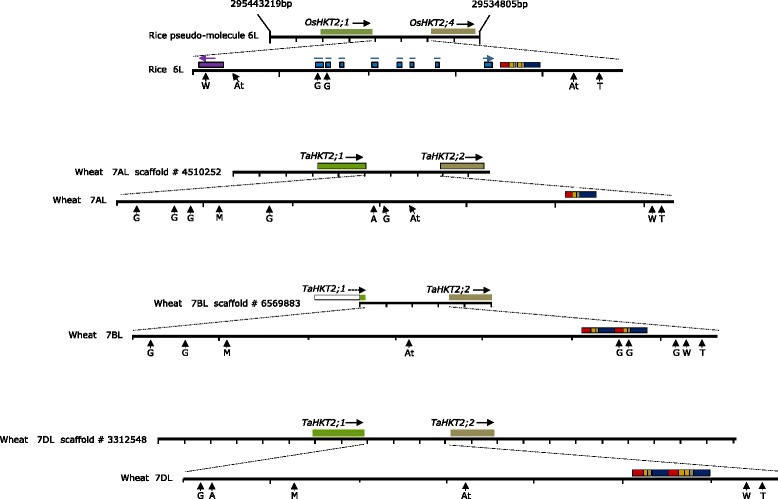
Comparative physical map of OsHKT2;1 - OsHKT2;4 intergenic region on rice chromosome 6L and TaHKT2;1 - TaHKT2;2 intergenic region on wheat chromosomes 7AL, 7BL and 7DL. Sequence is represented by chromosome region from pseudomolecule of rice chromosome 6L and scaffold sequences from wheat chromosomes 7AL, 7BL and 7DL where the scale intervals represents 1 kb. The direction of gene transcription is shown by black arrows. The intergenic region is expanded below each sequence or scaffold where scale intervals represent 500 base pairs. Tandem repeat units to the right of each expanded intergenic region are indicated by coloured boxes where each colour represents an individual repeat motif. PIF/ Harbinger type MITE elements are shown in purple whereas remnants of a CACTA type transposon “DTC_Isidor” are indicated in blue boxes. Salt induced cis-acting elements and other regulatory motifs identified on the OsHKT2;4 and TaHKT2;2 promoter regions are indicated by arrows, including ABRE (A), AtMYC2 (At), MYCATERD1 (M), GT-1 box (G), W-box (W) and TATA box (T)
The similar size of the intergenic regions between the rice genes OsHKT2;1 and OsHKT2;4 and wheat genes TaHKT2;1 and TaHKT2;2 prompted a further comparison of the nucleotide sequences. The intergenic region on 7AL, 7BL and 7DL had <40.8 % DNA identity to the intergenic region between OsHKT2;1 and OsHKT2;4 in rice, indicating significant diversity in this region. A blastn and tblastx analysis in NCBI did not identify sequence similarity with any expressed genes in the intergenic regions for rice or wheat. However, a self-by-self blastn search identified direct, tandem imperfect repeats in the intergenic region (approximately 400 base pairs upstream of the translation start site of TaHKT2;2) of rice OsHKT2;4 and wheat TaHKT2;2 -7BL and TaHKT2;2 -7DL where each repeat motif was less than 70 base pairs in size (Fig. 5, Additional file 2: Figure S2). However, only one copy of two imperfect motifs was represented in the intergenic region on 7AL (Fig. 5, Additional file 2: Figure S2). Furthermore, a complete PIF/Harbinger type miniature inverted transposon element (MITE), DTH_Ors48, was identified in the 5’ region of the rice intergenic sequence and remnants of a DTC_Isidor type DNA transposon (TREP accession number TREP3425) in the central region (Fig. 5, Additional file 2: Figure S2) but no transposon related elements were identified in the intergenic region of wheat 7AL, 7BL or 7DL.
The TaHKT2;1 and TaHKT2;2 intergenic region also represented the promoter region for TaHKT2;2 genes on 7AL, 7BL and 7DL and enabled comparative analysis of putative stress regulatory elements for genes on homoeologous chromosomes. The wheat TaHKT2;2 promoter region revealed 67–88 % sequence identity between promoters of genes on homoeologous chromosomes (Additional file 2: Figure S2). Conserved cis-acting regulatory elements (CRE) were identified including three major salt induced CREs, W-box, GT-box and AtMYC2 in the promoter regions on wheat chromosomes 7AL, 7BL and 7DL with similar elements also represented in the promoter region of rice OsHKT2;4 (Fig. 5, Additional file 2: Figure S2). Furthermore, the TaHKT2;2 promoter identified additional CRE including MYCATRED1 and ABRE on 7AL, 7BL and 7DL but were not evident in the promoter region of rice (Fig. 5, Additional file 2: Figure S2). Therefore, the CRE elements indicated a role for salt- activated transcription factors (TF) in regulation of TaHKT2;2 genes and orthologs in rice but with variable frequency of the individual elements.
Functional Association of wheat group II HKT genes
An association of TaHKT2;2 genes with trait variation under salt stress was investigated using deletion and genetic mapping. Deletion line analysis showed that TaHKT2;2 genes were assigned to the distal end of chromosomes 7AL and 7DL and the proximal region of 7BL (data not shown) and the same chromosome bins as TaHKT2;1 [13]. Therefore, functional analysis based on deletion line analysis previously reported [13] indicated that TaHKT2;2 is associated with similar phenotypes as TaHKT2;1 under salt stress. In order to further discriminate whether TaHKT2;1 and TaHKT2;2 were associated with specific trait variation under salt stress in different environments, QTL detected in response to low and high salt stress in Berkut/Krichauff DH mapping population [33, 37] were integrated onto the deletion map based on the chromosomal bin location of flanking markers. A total of six QTL were aligned to the distal bin on chromosome 7AL and in the same region as TaHKT2;1 7AL-1 and TaHKT2;2 7AL-1, however, neither TaHKT2;1 nor TaHKT2;2 genes were co-located with QTL in the same chromosomal bin on 7DL (Fig. 6) and QTL were not identified on 7BL. Therefore, the co-location of genes and QTL in the same chromosomal region indicated that TaHKT2;1 7AL-1 and TaHKT2;2 7AL-1 may contribute to variation for leaf/shoot Na+ concentration, 1000 grain weight, grain number per m2 and seedling biomass on 7AL in response to specific saline environments.
Fig. 6.
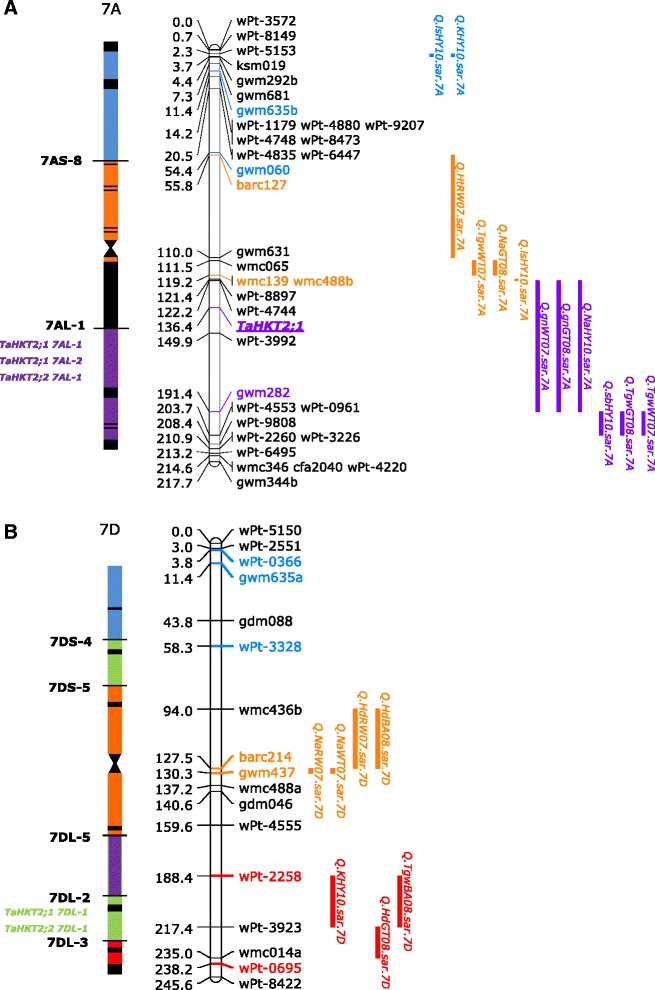
Deletion bin and genetic maps of chromosome 7A (a) and 7D (b) and allocation of TaHKT2;1 and TaHKT2;2 genes and QTL in similar regions under different saline environments. Allocation of TaHKT2;1 and TaHKT2;2 in the deletion bin map for each chromosome is presented on the left whereas the chromosomal linkage map with position of TaHKT2;1 (bold and underlined) and the QTL positions derived from Berkut/Krichauff DH mapping population [33, 37] is represented on the right. TaHKT2;1 and TaHKT2;2 genes, QTLs and flanking markers are colour coded according to their allocation within deletion bins [79, 80]. QTL detected for phenotypes in different saline environments include shoot Na+ concentration (Q.Na), thousand grain weight (Q.Tgw), seedling biomass (Q.sb), grain number per m2 (Q.gn), leaf symptoms (Q.ls), plant height (Q.Ht), Heading date (Q.Hd) and shoot K+ concentration (Q.K) as previously reported [37]
The association of TaHKT2;1 7AL-1 and TaHKT2;2 7AL-1 with specific trait variation was further investigated using genetic mapping analysis. TaHKT2;1 7AL-1 and TaHKT2;2 7AL-1 were PCR amplified and sequenced from Berkut and Krichauff to identify polymorphism and to develop gene specific markers for mapping in the DH population. Single nucleotide polymorphism (SNP) or insertion-deletion (INDEL) polymorphisms between Berkut and Krichauff were not identified in exons or introns for TaHKT2;2 7AL-1, indicating that no protein differences encoded by this gene were associated with trait variation on chromosome 7AL and, therefore, was excluded from further analysis in the DH population. However, TaHKT2;1 7AL-1 identified 11 SNPs between Berkut (Genbank accession number KR422357) and Krichauff (Genbank accession number KR422358) (Fig. 7a). A SNP at 1230 base pairs from translation start site identified a restriction site for Xmn1 enzyme in Berkut but not in Krichauff and this was used to develop a marker for TaHKT2;1 7AL-1 (Fig. 7a). A cleaved amplified polymorphic sequence (CAPS) marker was developed using a 3’ mismatch PCR primer pair, 2;1AF1 and 2;1AR1 (Table 2), designed to amplify a 1103 base pairs fragment containing the SNP at 1230 base pairs The sub-genome specificity of the PCR fragment was confirmed by NT analysis (Fig. 7b). The TaHKT2;1 7AL-1 specific CAPS marker identified a 922 base pairs DNA fragment for Berkut and 1103 base pairs DNA fragment for Krichauff parents following digestion with Xmn1 (Fig. 7b). The Berkut/Krichauff mapping population (150 DH lines) was genotyped for the TaHKT2;1 7AL-1 specific CAPS marker and the data was integrated into existing genetic map containing 34 markers on chromosome 7A. TaHKT2;1 7AL-1 mapped on chromosome 7AL at a genetic distance of 136.4 cM and flanked by markers wpt-4744 (122.2 cM) and wpt-3992 (149.9 cM) and within the QTL intervals for shoot Na+ concentration (Q.NaHY10.sar.7A) and grain number per m2 (Q.gnWT07.sar.7A, Q.gnGT08.sar.7A) detected under different saline environments (Fig. 6a). Therefore, based on genetic mapping and QTL analysis, it is reasonable to assume that TaHKT2;1 7AL-1 was associated with specific trait variation but not under all saline environments.
Fig. 7.
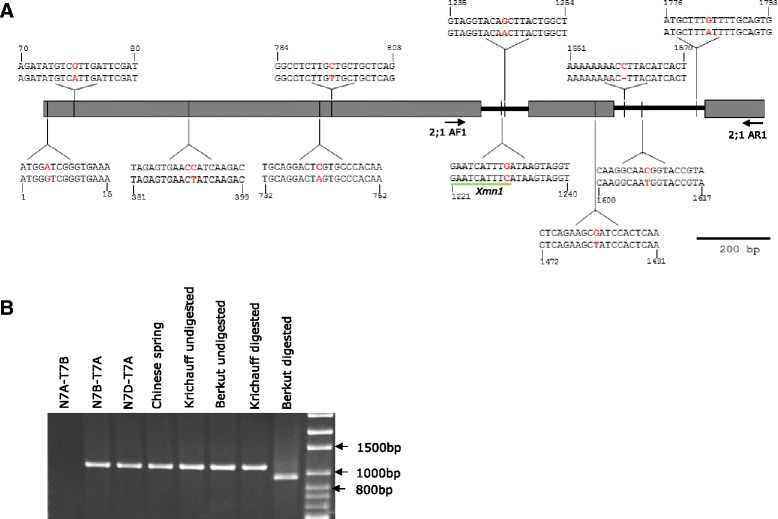
DNA sequence variability in the TaHKT2;1 7AL-1 gene between wheat cultivars Krichauff and Berkut. a Intron-exon structure where exons are represented as grey boxes and introns represented by black lines. SNPs are indicated by black vertical lines. DNA sequence and position (in base pairs) flanking each SNP is shown, with top sequence representing Krichauff and bottom sequence representing Berkut. SNP variation within the restriction enzyme recognition site, Xmn1, is underlined. Location of gene specific PCR primer pair for the TaHKT2;1 7AL-1, (2;1 AF1 and 2;1 AR1) to amplify the SNP at 1230 base pairs is shown by black arrows. b Agarose gel electrophoresis of TaHKT2;1 7AL-1 gene specific CAPS marker showing specificity to chromosome 7A using NT analysis and size difference of amplicons for Berkut and Krichauff parents following digestion with XmnI. The DNA ladder is shown to the right of the figure
Since TaHKT2;2 7AL-1 did not show any differences in amino acid residues, the predicted proteins encoded by Berkut and Krichauff focused on TaHKT2;1 7AL-1 alleles and were further analysed to identify protein folding differences that may be related to trait variation. The encoded proteins were predicted from gene sequences derived from each parental allele and using FL-cDNA from Chinese Spring as a reference. Four amino acid substitutions between Berkut and Krichauff were identified at amino acid residues 2, 27, 131 and 452 (Fig. 8a). The 3-D protein modelled by superimposing amino acid sequences from Berkut and Krichauff predicted differences in cytoplasmic (p.H131Y) and the p-loop (p.D453Y) domains (Fig. 8b) that anticipated functional differences between the proteins.
Fig. 8.
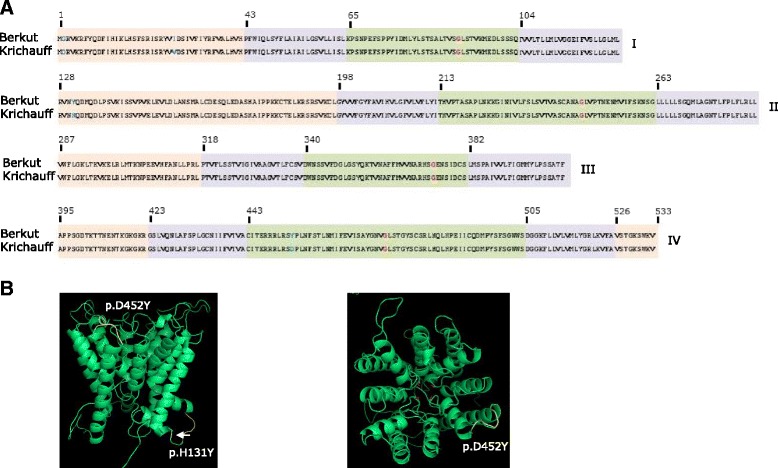
Predicted proteins encoded by TaHKT2;1 7AL-1 in cultivars Krichauff and Berkut. a The cytoplasmic, trans-membrane, and p-loop domains are identified in pink, purple and green backgrounds respectively. Conserved glycine residues representative of the cation selectivity filter are indicated in red. Amino acid differences at residues 2, 27, 131 and 452 are in blue. b Predicted 3D models of superimposed proteins encoded by TaHKT2;1 from Krichauff and Berkut, side view (right) and top view (left). The yellow regions indicate differences in the p-loop (p.D452Y) and cytoplasmic (p.H131Y) domains between Berkut and Krichauff and the position of cation selectivity filter glycine molecules are indicated in red
Discussion
Rice contains four individual group II HKT genes and it was expected that a total of at least 12 orthologs would be identified in wheat assuming one copy of each ortholog on the A, B and D genomes, with relatively similar sequences but not necessarily identical function. Instead, however, a genome-wide comparative gene analysis in this study identified only two distinct group II HKT gene families in wheat including the previously described TaHKT2;1 family [13] and a new gene family, TaHKT2;2, that is distinctly different from TaHKT2;1. Therefore, it appears that wheat and rice group II HKT genes evolved by gene duplication but diverged independently possibly reflecting their involvement in different biological processes across species.
The common structure in barley, rice and Arabidopsis [10] makes it reasonable to hypothesise that the ancestral HKT gene of plants consisted of three exons. The comparative gene analysis in wheat and rice described in this study supports this hypothesis and the ancestral gene subsequently duplicated and diverged independently in the two species. This study revealed that the two rice group II HKT gene clusters, with each comprising of two genes based on phylogenetic relationships [5, 16, 17], have orthologous gene families in wheat. The wheat TaHKT2;2 gene family had only one copy on each sub-genome and did not show evidence of gene duplication prior to or after polyploidization. However, duplication with subsequent translocation and divergence was evident for the rice ortholog OsHKT2;3 on pseudomolecule 3 and OsHKT2;4 on pseudomolecule 6. On the other hand, evidence indicated tandem gene duplication in A and B progenitor genomes predated polyploidization resulting in multiple copies of TaHKT2;1 gene family on wheat chromosome arms 7AL and 7BL including divergence and pseudogenization [13] similar to the orthologous rice genes OsHKT2;1 and OsHKT2;2 on chromosome 6. Therefore, it is apparent that gene duplication of a common ancestral HKT gene was a key mechanism contributing in the creation of group II HKT, however, gene copies were possibly subjected to different selective constraints whereby divergence did not evolve in parallel during domestication of modern wheat and rice.
Analysis of the intergenic regions between TaHKT2;2 and TaHKT2;1 and the orthologous rice genes OsHKT2;1 and OsHKT2;4 provided further insights on evolution of group II HKT genes across species. Three typical structural organization categories in grass genomes have been identified based on analysis of DNA content within large genomic fragments between genes [38]. The physical size of intergenic regions can vary significantly between conserved coding regions in two or more grass species, ranging from a few to a tens of kilobases in size largely due to differential accumulation of transposable and other repetitive elements [39]. Alternatively, regions of the genome can have high gene content whereby coding regions and the size of the physical distance between genes are conserved [40], indicating that genome regions may not necessarily be prone to proliferation of transposable and other repetitive elements. Finally, there are both conserved high gene density with similar intergenic regions across species interspersed with large intergenic regions varying in size [41]. This study identified that the group II HKT genes in wheat and rice have similar physical distance in the intergenic region and, therefore, evolutionary mechanisms are proposed to maintain genes physically close together across species possibly as a means to preserve gene regulation, co-expression or co-functionality [42, 43]. A MITE and remnants of a transposable element was identified in the intergenic region between OsHKT2;1 and OsHKT2;4 in rice indicating that retrotransposition events may have been evident but were not sustained and, consequently, eliminated during evolution leaving a transposon “footprint”. Remnants of transposable elements were not evident in the orthologous region in wheat but instead, low complexity imperfect repeats were the major features in the intergenic region between wheat TaHKT2;1 and TaHKT2;2 in wheat 7BL, 7DL and in rice but absent in wheat 7AL. The differences observed in short repeat motifs between chromosomes and species can arise from replication misalignment and polymerase slippage resulting in deletion or addition of repeat units [44, 45]. It is, therefore, evident that the intergenic region between TaHKT2;1 and TaHKT2;2 in wheat and the orthologous region between OsHKT2;1 and OsHKT2;4 in rice is not conducive to major changes in size and conservation of physical distance within and across species may be implicated in the preservation of some gene functions.
This study provided evidence to support the hypothesis that the similar physical closeness of genes in rice and wheat is an evolutionary means to maintain similar gene regulation across species. TaHKT2;2 gene family was expressed in roots, sheath and leaf blade tissues whereas TaHKT2;1 genes were expressed in roots and sheath but with a low transcript abundance in leaf blade. Orthologous rice genes showed similar expression patterns where OsHKT2;4 was expressed in root, sheath and leaf blade tissues [20] and OsHKT2;1 showed expression in roots and sheath but down regulated in leaf blades [21], despite variability in the number and types of CRE in the intergenic regions between species. Therefore, it is plausible that similar small physical distance between orthologous HKT genes could be an evolutionary mechanism to conserve gene expression in rice and wheat. Interestingly, the consistent expression and maintenance of transcript levels indicated that the two wheat group II HKT gene families may interact in coordinated manner and possibly in cohesion with other genes such as group I HKT genes regardless of saline environments. Similar gene interactions resulting in an organized response to salt stress has been observed for rice HKT genes OsHKT1;4 and OsHKT1;5 [46] providing a transcript-coordinated model for wheat HKT genes. The manner by which group II HKT genes are coordinated in a biological system in response to salinity is yet to be realized and warrants further investigation.
The intergenic region between the two group II HKT genes on each homeologous chromosome also occupy the promoter region for TaHKT2;2 genes. In addition to the variable number of CRE present in different homeologs, specific direct imperfect repeat motifs within the promoter region can act as suppressors or enhancers of gene transcription often by functioning as putative transcription factor-binding domains [47–50]. Interestingly, tandem imperfect direct repeats important for gene expression and regulation were identified in the promoter of the HKT gene from Arabidopsis thaliana, AtHKT1;1, whereby variation or absence of the repeats resulted in significant transcriptional response to elevated NaCl [51]. Therefore, further studies on the short repetitive elements is warranted to determine their role in regulation of TaHKT2;2 transcription during different stages of plant development beyond that analysed in this study and under different saline conditions.
The fold difference in transcript abundance for TaHKT2;1 7DL-1 and TaHKT2;2 7AL-1 may indicate their involvement in controlling Na+ accumulation in a tissue specific manner. Despite variable and multiple presence of salt induced CREs in promoter regions, the majority of group II HKT genes did not show a significant change in transcription in roots, sheath and leaf blades in response to NaCl treatment under the experimental conditions in this study. The lack of response to salt, therefore, raises some doubt as to the transcriptional significance of some TaHKT2 genes in Na+ transport in roots, sheath or leaf under hydroponic conditions. A similar outcome was observed for the group I HKT member where TmHKT1;4 did not show variability in transcript abundance under saline conditions but was co-located with a QTL for Na+ exclusion accounting for 25 % of the phenotypic variation indicating that transcript abundance may not solely explain gene action in controlling Na+ levels [52]. We cannot exclude the possibility that some group II HKTs are regulated under salt stress in other tissues and different environments or may, indeed, play a non-transcriptional role in controlling Na+ accumulation. Therefore, systematic and integrated studies involving whole plant analysis would provide a more comprehensive understanding of the co-ordinated transcriptional regulation and interactions of all members of the group II TaHKT gene family that control Na+ accumulation.
Distinct differences between the proteins encoded by two group II HKT gene families in wheat predicted variation in their function. The critical functional domains of the protein encoded by TaHKT2;1 7BL-1 was shown to be important for cation selectivity and ion transport properties [53–56] and used as a basis to infer functional variation in proteins encoded by TaHKT2;2. Structural modelling and superimposing the known functional domains responsible for Na+ and K+ transport in the protein encoded by TaHKT2;1 7BL-1 identified variants in proteins encoded by TaHKT2;2. The NH2 and carboxy-termini are essential functional domains in HKT proteins affecting K+ selectivity [54]. The length and amino acid composition of the NH2 and carboxy termini were distinctly different in the two protein families and it is possible that the truncated domains could result in either non-functional proteins or have significantly modified function. Highly conserved, positively charged amino acids in the inner helix of the 4th TM-P-TM domain are critical for formation of the salt bridge and K+ selective transporter activity [57, 58]. However, HKT-related proteins between species have similar structural core and these positively charged amino acids are not present in plant K+ channels [57]. Therefore, further studies such as heterologous expression would be worthwhile to decipher whether the truncated carboxy termini in proteins encoded by TaHKT2;2 have transporter activity with channel-like properties. Interestingly, protein encoded by the rice ortholog OsHKT2;4 exhibited variable kinetics in heterologous expression, including a calcium-permeable weak selective cation channel that provided evidence for alternative functional modes for group II HKT proteins [20, 22, 59]. Moreover, differences in the cytoplasmic and external domains arising from single amino acid substitutions between allelic variants were identified in the protein encoded by rice gene OsHKT1;4 [60] and Arabidopsis AtHKT1;1 [61] and similar variations predicted functional differences within the protein family encoded by TaHKT2;2 compared with proteins encoded by TaHKT2;1. In addition to a number of amino acid substitutions in the cytoplasmic and external domains, the protein encoded by TaHKT2;2 identified an alanine substitution for serine at amino acid residue 216 compared to the protein encoded by TaHKT2;1 7BL-1 that can cause significant effects on ion transport activity. The substitution of Ala216 near entrance of the pore region reduced Na+ transport capacity the protein encoded by TaHKT2;1 7BL-1 [62] and, therefore, a similar change at this residue implies a modified ion transport activity of proteins encoded by TaHKT2;2. Therefore, whilst the conserved protein structural core and the shared G-G-G-G type cation selectivity filter predicted some functional similarity between TaHKT2;2 and TaHKT2;1 (for example, ability to transport both K+ and Na+), the encoded proteins may have diversity for ion selectivity based on modified protein domains and amino acid changes in critical functional domains.
In-planta function of group II HKT transporters is poorly understood in wheat but phenotypic analysis of aenuploid lines under salt conditions provided new knowledge on their potential role in controlling ion accumulation in the plant. Wheat deletion lines null for TaHKT2;1 on chromosomes 7AL, 7BL and 7DL did not show significant difference in root Na+ concentration and, therefore, members of this gene family were proposed to be involved in Na+ transport from root to sheath and regulation of K+ in different tissues rather than import of ions from the external medium through the roots [13]. Since members of the TaHKT2;2 gene family were allocated to the same deletion bin as TaHKT2;1 on chromosomes 7AL, 7BL and 7DL, it was apparent that neither TaHKT2;2 nor TaHKT2;1 genes were responsible for primary root Na+ uptake from an external medium despite both genes being expressed in roots. However, either or both genes could be involved in regulating Na+ from root to sheath or maintaining K+ levels under untreated and salt treated conditions. Interestingly, the TaHKT2;2 ortholog in rice, OsHKT2;4 mediated a robust low affinity K+ transport in a heterologous expression system [59] yet OsHKT2;4 knockout mutants did not confer a similar phenotype under different ionic conditions [22], indicating that this gene is capable but not critical for transporting K+ in the plant and more likely reliant on the contribution of other genes and proteins in a more complex system. The functional association of TaHKT2;1 and TaHKT2;1 with trait variation, therefore, was further investigated.
Deletion bin and genetic mapping coupled with QTL analysis was used as a guide to associate group II HKT genes with trait variation under saline environments. Although these resources provided low resolution analysis, we were able demonstrate that TaHKT2;1 and TaHKT2;2 genes were not associated with some traits under saline environments on chromosomes 7BL and 7DL. However, the location of TaHKT2;2 and TaHKT2;1 on 7AL were identified in the same chromosomal deletion bin as QTL for leaf/shoot Na+ concentration and yield measurements under different saline environments [33, 37], indicating their potential involvement in phenotype variation. Exons and introns in TaHKT2;2 7AL-1 were identical between Berkut and Krichauff and, consequently, were unable to be assigned to the genetic map in the DH population. It would, however, be premature to conclude that TaHKT2;2 7AL-1 does not affect specific phenotypes as the gene may have transcriptional, post-transcriptional or post-translational regulation during different stages of plant development that could affect phenotypic variation under different saline environments. DNA sequence variability within introns and exons of TaHKT2;1 7AL-1 allowed a low resolution but genetic association with QTL, predicting a role for this gene in controlling Na+ exclusion under hydroponic and field conditions, grain number and grain weight in specific saline environments. However, the QTL has a large genetic and physical distance and we cannot exclude the possibility that genes other than TaHKT2;1 7AL-1 in the interval contribute to phenotypic variation. Fine resolution genetic mapping would confirm the role of TaHKT2;1 7AL-1and other genes in controlling variation for some traits under different saline environments. Although a further three QTL were detected within the same deletion bin, TaHKT2;1 7AL-1 did not co-locate within the same mapping interval and, therefore, unlikely to be associated with phenotypic variation for thousand grain weight and seedling biomass under different saline conditions. The genetic control of these traits, therefore, was likely caused by genes other than the TaHKT2 family. A structural model and variability for the protein encoded by Berkut allele for TaHKT2;1 7AL-1 identified a tyrosine substitution at amino acid residue 452, located near a highly conserved domain in the 4th P-loop region which assumed a larger, neutral amino acid more hydrophobic than the negatively charged aspartic acid in the protein encoded by the Krichauff. Gene ontology for biological processes [63], Interpro Protein Domains search [64] and mutation studies [53] predicted the 4th P-loop region as a critical region in Na+ binding and transport and the role of wheat HKT genes in controlling ion transport from root to leaf blade using deletion line analysis [13], providing further evidence that the protein encoded by the Berkut allele may control Na+ transport within the plant affecting some yield related traits under specific but not all saline environments.
Conclusion
Bread wheat contains two distinctly different group II HKT gene families, TaHKT2;1 and TaHKT2;2, compared with the four individual genes in rice. Therefore, duplication of a common ancestral gene containing three exons followed by independent divergence was evident during evolution and domestication of the two species. A new member of wheat group II HKT, TaHKT2;2 was characterized from homeologous chromosome group 7L and within a short (~3 k base pairs) physical distance from the related gene family TaHKT2;1. The TaHKT2;2 multigene family consisted of three putative functional genes that were expressed in root, sheath and leaf blade in control and 200 mM NaCl stress conditions with distinct differences to TaHKT2;1. The proteins encoded by TaHKT2;2 genes were similar but not identical to those encoded by TaHKT2;1. TaHKT2;2, like the TaHKT2;1 gene family, is unlikely to be responsible for root Na+ uptake from external medium but potentially involved in transporting Na+ from root to leaf blade or in regulating K+ transport and may also have broader ion selectivity and transport in the plant. Moreover, the protein encoded by TaHKT2;1 7AL-1 may be associated with yield related traits under certain saline environments but other genes in QTL intervals may also contribute to trait variation. High resolution genetic mapping and QTL analysis would provide further evidence for the role of TaHKT2;1 7AL-1 and other genes in controlling trait variation under different saline environments.
Methods
Plant material
Bread wheat variety Triticum aestivum L. cv. Chinese Spring, six nullisomic-tetrasomic lines (N7AT7B, N7AT7D, N7BT7A, N7BT7D, N7DT7A, N7DT7B), eight deletion lines (7AL-1, 7AS-8, 7BL-2, 7BL-7, 7DL-2, 7DL-3, 7DL-5, 7DS-5) and two ditelosomic lines (Dt7BL and Dt7BS) were kindly provided by Dr John Raupp, Wheat Genetic and Genomic Resources Centre, Kansas State University, USA. The DH mapping population derived from an F1 cross between Berkut and Krichuaff consisting of 150 individuals was previously reported [33].
Database search and identification of wheat orthologs of rice group II HKT genes
The group II HKT genes OsHKT2;1, OsHKT2;2, OsHKT2;3 and OsHKT2;4 and their corresponding FL-cDNA were accessed at NCBI (Genbank accession numbers AB061311, AB061313, AJ491819 and AJ491854, respectively). The rice FL-cDNA were used as query sequences in blastn and tblastx to search the wheat survey sequence database (https://urgi.versailles.inra.fr/blast/blast.php). Wheat scaffolds containing related gene sequences were identified when blast searches revealed significant hits (E = 0.0) with rice FL-cDNA sequences. Regions within each wheat scaffold having high sequence identity with FL-cDNA of rice genes were obtained and target sequences were compared with FL-cDNA of wheat group II HKTs, TaHKT2;1 (Genbank accession numbers, KJ540616, U16709, KJ540618) by local alignment using the ClustalW function in GENEIOUS 6.0.3 [65].
Primer design and PCR amplification
Multiple sequences between wheat sequences were aligned using GENEIOUS 6.0.3 [65] and sub-genome specific sequence variants were scanned manually. PCR primers were designed and positioned for 3’ mismatch [66] to obtain gene specific amplicons. NetPrimer (http://www.premierbiosoft.com/netprimer/netprlaunch/netprlaunch.html) software was used to confirm primer compatibility and annealing temperatures. Primer details are provided in Table 2.
Genomic DNA from wheat aneuploid lines was extracted using a phenol-chloroform based method [67] and 50 ng of genomic DNA was used as template for PCR amplification. PCR reactions contained 0.5 μM of each primer (Table 2), 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM of each deoxyribonucleotides, 1 U taq DNA polymerase (BIOTAQ™ DNA Polymerase, Bioline, Australia) in a 20 μl reaction volume. The cycle parameters for standard PCR were 35 cycles of 94 °C 30 s, primer annealing temperature (Table 2) for 30 s, 72 °C 45 s and the final extension 7 min at 72 °C; and for touch down PCR were five cycles of 94 °C 30 s, 55–50 °C 30 s, 72 °C 45 s; and then 35 cycles of 94 °C 30 s, 50 °C 30 s, 72 °C 45 s and the final extension 7 min at 72 °C. PCR products were separated on 1 % agarose gel in 0.5× Tris-acetate EDTA at constant voltage (90 V) for 30 min and the gels were stained with ethidium bromide and visualized under UV light using Gel Doc System (BioRad, Italy). Sub-genome specificity of the PCR primers were confirmed by the absence of amplicons from gene specific PCR using nullisomic-tetrasomic lines N7AT7B, N7AT7D, N7BT7A, N7BT7D, N7DT7A, N7DT7B as template DNA.
FL-cDNA cloning and sequencing
The development of plant material, total RNA extraction from root tissue and synthesis of cDNA were done as described below. Gene specific primers were designed to amplify multiple fragments of overlapping cDNA for the predicted coding regions of full length wheat genes (Table 2). Gene specific RT-PCR primers including 2;2AF1/2;2AR1, 2;2AF2/2;2AR2, 2;2CF1/2;2BR1, 2;2BF2/2;2BR2, 2;2BF3/2;2ADR1, 2;2ADCF1/2;2DR1, 2;2DF2/2;2ADR1 (Table 2) were used to amplify cDNA from root tissue. RT-PCR were done at annealing temperatures for each primer combination (Table 2) in 20 μl PCR reaction mixture containing 0.5 μM of each primer (Table 2), 1× PCR buffer, 1.5 mM MgCl2, 0.2 mM of each deoxyribonucleotides, 1 U taq DNA polymerase (BIOTAQ™ DNA Polymerase, Bioline, Australia). The thermocycling conditions for standard PCR were 35 cycles of 94 °C 30 s, primer annealing temperature (Table 2) for 30 s, 72 °C 45 s and the final extension 7 min at 72 °C; and for touch down PCR were five cycles of 94 °C 30 s, 55–50 °C 30 s, 72 °C 45 s; and then 35 cycles of 94 °C 30 s, 50 °C 30 s, 72 °C 45 s and the final extension 7 min at 72 °C. RT-PCR products were separated on 1 % agarose gel in 0.5× Tris-acetate EDTA at constant voltage (90 V) for 30 min and visualized under UV light using Gel Doc System (BioRad, Italy) after staining with ethidium bromide. The RT-PCR amplicons were excised from agarose gels, purified using Wizard® SV Gel and PCR Clean-Up System (Promega, CA, USA), and cloned into pGEM®-T Easy Vector Systems as per manufacturer’s recommendation (Promega, CA, USA). DNA templates from 3 recombinant bacterial colonies and representing each overlapping cDNA fragment were isolated and purified by Wizard® Plus SV Minipreps as per manufacturer’s recommendation (Promega, CA, USA) and sequenced using BigDye™ sequencing chemistry (Applied Biosystems, Perkin Elmer, Weiterstadt, Germany) using the M13 universal primers Forward primer (5’-CGCCAGGGTTTTCCCAGTCACGAC-3’) and Reverse primer 5‘-TCACACAGGAAACAGCTATGAC-3’). Sequences of overlapping cDNA for each wheat gene were assembled and aligned with the cognate genomic DNA sequence using GENEIOUS 6.0.3 [65].
In-silico protein analysis and 3-D modelling
Proteins were deduced from FL-cDNA of the wheat genes and amino acid sequence alignments of predicted proteins were obtained using GENEIOUS 6.0.3 [65]. The predicted proteins were analysed for hydrophobicity and membrane topology using TopRED2 [68] and MPEx 3.2 [69] software using default parameters. Three dimensional structures of the proteins were predicted by PHYRE 2 [70]. The 3-D models were analysed by the PyMOL viewer and molecular Graphics system, Version 1.5.0.4 (Schrödinger, LLC).
In-silico analysis of intergenic and promoter regions
The intergenic region between group II HKT genes were retrieved from rice genome browser release 7 (http://rice.plantbiology.msu.edu/) and the wheat sequence scaffolds from the International Wheat Genome Sequence (http://www.wheatgenome.org/). The NCBI nucleotide and protein databases were searched using blastn and tblastx and intergenic regions as query to identify DNA sequence identity with expressed sequence tags, partial or FL-cDNA with E-value = 0. Repetitive elements were identified using RepeatMasker (http://www.repeatmasker.org/) and EMBOSS einverted [71] software. Tandem repeat sequences were identified through self-by-self search in “Align sequences nucleotide BLAST” in the NCBI blastn suite. Transverse repeats were further analysed by blastn search function in TREP [72] and P-Mite (Plant MITE) [73] databases. Cis-acting regulatory elements associated with salt stress response in the promoter regions were identified by blastn search in PlantCARE [74] and PLACE [75] databases.
Hydroponic screening and development of tissue samples for expression analysis
Seeds of bread T. aestivum var. Chinese Spring were washed in 0.04 % sodium hypochlorite (42 g/L) for 30 s, thoroughly rinsed in deionized water before germinating on a floating wire mesh in aerated 0.1 strength nutrient solution under dark conditions. Nutrient composition at full strength was: (mM) K+, 3.95; Ca2+, 4; Mg2+, 0.4; NH4+, 0.625; NO3−, 4.375; SO42−, 1.9; HPO42−, 0.2; Fe-EDTA, 0.05; MES, 1.0; and micronutrients of one quarter-concentration in Hoagland solution (pH was adjusted to 6.5 using KOH). After 3 days seedlings were transferred to 0.25 strength nutrient solution in light for 1 day. Four days old seedlings were then transferred to full strength nutrient solution in aerated, foil-covered, 4.5 L pots. Seedlings were held upright by a foam holder at the stem base inserted into individual holes in the pot lids. There were four replicates per each treatment in complete randomized design. The experiment was carried out in a temperature controlled phytotron (20 ± 3 °C/15 ± 2 °C day/night) and the photosynthetically active radiation recorded at midday was at ca. 1400–1500 μmol m − 2 s − 1. Solution levels in pots were maintained by topping up with deionized water.
Ten days after initiation of the experiments and at the 2.2 Haun leaf development stage 200 mM NaCl treatments applied to designated treatment pots in 50 mM increments at 12 h intervals and the plants were maintained at the final 200 mM concentration for a full 3 days (72 h). No NaCl was added in the control pots. After 72 h samples of bulk leaf blades, sheaths and roots from each plant were sampled separately for tissue Na+ analysis. Prior to excision, roots were washed three times for 10 s each in a solution containing 4 mM CaSO4 and 368 mM mannitol (200 mM treated plants) or in 4 mM CaSO4 (plants subjected to no added NaCl). Leaves and sheath samples were rinsed in deionized water. All tissue samples for ion analyses were oven-dried at 70 °C, weighed and then ground to a powder. Samples of dried powdered tissue (100 mg) were extracted in 5 ml of 0.5 M HNO3 for 3 days on a mechanical shaker in dark conditions. Tissue Na+ concentrations were measured in technical triplicate using a flame photometer (Sherwood 410, Cambridge) and mean values represented each treatment replicate tissue sample. The reliability of the methods was confirmed by analyses of a reference tissue (broccoli, ASPAC Plant number 85) taken through the same procedures.
RNA extraction and RT-PCR
Development of unstressed and salt treated seedlings of wheat T. aestivum var. Chinese Spring was done as described above. Bulk leaf blade, sheath and roots were harvested from control and treated plants at the end of 72 h treatment period, snap frozen in liquid N2 for total RNA extraction. Total RNA was extracted from frozen tissues using a modified protocol by combining the TRIzol protocol, (Invitrogen, Carlsbad, CA) and ISOLATE II RNA Plant Kit (Bioline USA Inc, Taunton). Approximately 100 mg of tissue sample was grinded in liquid N2 and 1 mL of TRIzol reagent was added per 100 g of grinded tissue. Samples were incubated in TRIzol for 5 min at room temperature and extracted with 0.2 ml chloroform per 1 ml Trizol shaking vigorously by hand for 15–30 s. The samples were incubated at room temperature for 2 min and centrifuged at 12,000 g for 15 min at 4 °C. The aqueous phase was transferred to a clean RNase free tube avoiding the interphase and RNA was precipitated by adding 0.5 ml of isopropanol and incubating 10 min at room temperature. The isoproponal mixture was loaded on a ISOLATE II RNA Plant Column from the ISOLATE II RNA Plant Kit (Bioline USA Inc, Taunton) and the extraction was continued using the manufacturers’ recommendation. RNA purity and the quantity were determined by UV spectrophotometry at 260 and 280 nm (A260/A280 ~ 1.9 and A260/A230 > 2). First strand cDNA was synthesised using SensiFAST™ cDNA Synthesis Kit (Bioline USA Inc, Taunton) using 1 μg of total RNA in a 20 μl reaction following instructions by the manufacturer. Integrity of synthesised cDNA was verified by RT-PCR using primers designed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene (Table 2).
Quantitative real time PCR (qRT-PCR)
The qRT-PCR reactions was done in a Rotor-Gene™ 3000 (Corbett Research Ltd, UK) using SensiMix SYBR® No-ROX mix (Bioline USA Inc, Taunton) master mix. Three independent biological replicates of leaf, root and sheath tissues were used for analysis of transcript abundance of each gene under control and 200 mM NaCl treated conditions. Three technical replicates were used for each biological sample. The qRT-PCR reaction consisted of a final volume of 10 μl containing 5 μl of SensiMix SYBR® No-ROX mix (Bioline USA Inc, Taunton), 0.4 μM of each primer (Table 2) and 3 μl of a 1 in 20 dilution of synthesized cDNA and analysed using the following conditions: 3 min at 94 °C, 40 cycles at 94 °C for 15 s, 60 °C for 10s, and 72 °C for 20s. SYBR signals were acquired at the completion of the primer annealing step. Integrity and specificity of qRT-PCR was confirmed by melting curve analysis (55–95 °C with a heating rate of 0.5 °C/min) and agarose gel electrophoresis of the amplicons. The cDNA samples served as a template for stability analyses using three internal reference genes TaEFA, GAPDH and TaActin using similar qRT-PCR reaction conditions. The CT values obtained for each gene using control and 200 mM NaCl treated leaf, root and sheath cDNA were subjected to analysis of variance and genes showing stable expression in samples were selected as internal references for expression normalization of TaHKT2 genes. Primer sequences for the six genes of interest and reference genes are listed in Table 2. The relative change of transcript abundance in NaCl treated samples were estimated by 2-∆∆CT method [76]. Standard error in 2-∆∆C was estimated by predicting RQMin and RQMax [77, 78].
Deletion bin mapping
Wheat genes were bin mapped using eight deletion lines 7AL-1, 7AS-8, 7BL-2, 7BL-7, 7DL-2, 7DL-3, 7DL-5 and 7DS-5 and ditelosomic lines Dt7BL and Dt7BS. Genomic DNA from deletion and ditelosomic lines were extracted following a phenol-chloroform based method previously described [67] and used as template for PCR amplification. The location of genes within each deletion bin was determined by the presence or absence of amplicons from corresponding deletion or ditelosomic lines for gene specific PCR using the primer pairs 2;2AF1/2;2AR1, 2;2CF1/2;2BR1 and 2;2ADCF1/2;2DR1 (Table 2). The PCR reaction conditions were same as described above. PCR products were analysed on 1 % agarose gel in 0.5× Tris-acetate EDTA (constant voltage (90 V) for 30 min) and visualized under UV light using Gel Doc System (BioRad, Italy) after staining with ethidium bromide.
The chromosomal bin location of the flanking markers for the Na+ exclusion QTLs and other QTLs associated with salt related traits reported in the Berkut/ Krichauff DH population [33] were obtained from deletion bin mapping in [79, 80]. The bin location of the wmc139 SSR marker was experimentally determined by PCR using deletion lines as template DNA and primers 5’-TGTAACTGAGGGCCATGAAT-3’ and 5’-CATCGACTCACAACTAGGGT-3’ obtained from the GrainGene database (http://wheat.pw.usda.gov/GG3/). Touch down PCR was performed at 65–55 °C as described above.
Cleaved amplified polymorphic sequence (CAPS) marker development
TaHKT2;2 7AL-1 genes were amplified, cloned and sequenced from Berkut and Krichauff using gene specific primers as previously described [13]. Similarly TaHKT2;2 7AL-1 gene was cloned and sequenced from both parents using the primers 2;2AF1/2;2AR1, 2;2AF2/2;2AR2, 2;2AF3/2;2ADR1 as described above. Gene sequences were searched for DNA polymorphism by multiple sequence alignments using GENEIOUS 6.0.3 [65]. A gene specific CAPS marker polymorphic for Berkut and Krichauff parents was developed for TaHKT2;1 7AL-1 by amplifying a 1103 base pairs fragment using primers 2;1AF1 and 2;1AR1 (Table 2) in PCR reactions and thermocylcing conditions as described above. PCR products (10 μl) were digested with 10 U of Xmn1 (Promega, CA, USA) by incubation at 37 °C for 2 h in a reaction mixture as per the manufacturers recommendation (Promega, CA, USA). The digested products were separated on 1.5 % agarose at 0.5× Tris-acetate EDTA (constant voltage of 90 V for 30 min) and visualized under UV light using Gel Doc System (BioRad, Italy) after staining with ethidium bromide.
Genetic mapping of group II HKTs
A total of 150 DH lines from the Berkut/Krichauff mapping population were grown in greenhouse and genomic DNA was extracted from leaf material using a phenol-chloroform based method described in [67], quantified by Nanodrop_ND_1000 v3.2 spectrometer (Thermo Fisher Inc., DE, USA) and DNA concentrations adjusted to 50 ng/μl. The 150 DH lines were genotyped for TaHKT2;1 7AL-1 gene specific CAPS marker and allelic data integrated into existing genetic maps of simple sequence repeat (SSR) and Diversity Array Technology (DArT) markers described in [33]. Marker allele data for TaHKT2;1 7AL-1 was assigned in the genetic map using the “Distribute” command,, linkage criteria = 0.01, map function = Kosambi followed by the “Ripple” command in MapManagerQTX version QTXb20 [81]. The chromosome linkage and QTL intervals were graphically presented using MapChart V2.2 [82].
Availability of supporting data
The data supporting the results of this article are included within the article and its additional files.
Acknowledgements
HACKA was funded by a Scholarship for International Research Fees (SIRF) and a University International Stipend (UIS).
Abbreviations
- CAPS
cleaved amplified polymorphic sequence
- CRE
cis-acting regulatory elements
- DArT
diversity array technology
- DH
doubled haploid
- FL-cDNA
full length cDNA
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HKT
high affinity potassium transporter
- INDEL
insertion-deletion
- MITE
miniature inverted transposon element
- NaCl
sodium chloride
- NT
nullisomic-tetrasomic
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- QTL
quantitative trait loci
- RT-PCR
reverse transcription polymerase chain reaction
- SNP
single nucleotide polymorphism
- SOS
salt overly sensitive
- SSR
simple sequence repeat
- TF
transcription factor
Additional files
Amino acid sequence alignments of TaHKT2;1 and TaHKT2;2 proteins. Glycine molecules composing the cation selectivity filter domains are indicated by a red line. (PPTX 758 kb)
DNA Sequence alignment of the OsHKT2;1-OsHKT2;4 intergenic region (A) and TaHKT2;1-TaHKT2;2 intergenic region on 7AL, 7BL and 7DL (B). The complete sequence of PIF/Harbinger type MITE is highlighted in purple, remnants of a CACTA type transposon “DTC_Isidor” in blue and individual motifs of tandem duplications are in red, yellow, green and dark blue. Boxed sequences represent salt activated cis-acting regulatory elements and other regulatory motifs in the OsHKT2;4 and TaHKT2;2 promoter regions. (RTF 545 kb)
Footnotes
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
HACK designed and conducted the experiments, generated and analyzed data, interpreted results and wrote the manuscript. KHO provided genetic map and phenotypic data of mapping population, contributed to interpretation of the data and edited the manuscript. MGF contributed to design of all experiments, data interpretation and edited the manuscript. All authors read, revised and approved the final manuscript.
Contributor Information
H. A. Chandima K. Ariyarathna, Email: 20881006@student.uwa.edu.au
Klaus H. Oldach, Email: klaus.oldach@kws.com
Michael G. Francki, Email: michael.francki@agric.wa.gov.au
References
- 1.Munns R, Tester M. Mechanisms of salinity tolerance. Annu Rev Plant Biol. 2008;59:651–81. doi: 10.1146/annurev.arplant.59.032607.092911. [DOI] [PubMed] [Google Scholar]
- 2.James RA, Davenport RJ, Munns R. Physiological characterization of two genes for Na+ exclusion in durum wheat, Nax1 and Nax2. Plant Physiol. 2006;142(4):1537–47. doi: 10.1104/pp.106.086538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Q, Chen ZZ, Zhou XF, Yin HB, Li X, Xin XF, et al. Overexpression of SOS (Salt Overly Sensitive) genes increases salt tolerance in transgenic Arabidopsis. Mol Plant. 2009;2(1):22–31. doi: 10.1093/mp/ssn058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Byrt CS, Platten JD, Spielmeyer W, James RA, Lagudah ES, Dennis ES, et al. HKT1;5-like cation transporters linked to Na+ exclusion loci in wheat, Nax2 and Kna1. Plant Physiol. 2007;143(4):1918–28. doi: 10.1104/pp.106.093476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hauser F, Horie T. A conserved primary salt tolerance mechanism mediated by HKT transporters: a mechanism for sodium exclusion and maintenance of high K+/Na+ ratio in leaves during salinity stress. Plant Cell Environ. 2010;33(4):552–65. doi: 10.1111/j.1365-3040.2009.02056.x. [DOI] [PubMed] [Google Scholar]
- 6.Moller IS, Gilliham M, Jha D, Mayo GM, Roy SJ, Coates JC, et al. Shoot Na + exclusion and increased salinity tolerance engineered by cell type-specific alteration of Na+ transport in Arabidopsis. Plant Cell. 2009;21(7):2163–78. doi: 10.1105/tpc.108.064568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rus A, Yokoi S, Sharkhuu A, Reddy M, Lee BH, Matsumoto TK, et al. AtHKT1 is a salt tolerance determinant that controls Na+ entry into plant roots. Proc Natl Acad Sci U S A. 2001;98(24):14150–5. doi: 10.1073/pnas.241501798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davenport RJ, Munoz-Mayor A, Jha D, Essah PA, Rus A, Tester M. The Na + transporter AtHKT1;1 controls retrieval of Na+ from the xylem in Arabidopsis. Plant Cell Environ. 2007;30(4):497–507. doi: 10.1111/j.1365-3040.2007.01637.x. [DOI] [PubMed] [Google Scholar]
- 9.Horie T, Sugawara M, Okunou K, Nakayama H, Schroeder JI, Shinmyo A, et al. Functions of HKT transporters in sodium transport in roots and in protecting leaves from salinity stress. Plant Biotechnol. 2008;25:233–9. doi: 10.5511/plantbiotechnology.25.233. [DOI] [Google Scholar]
- 10.Platten JD, Cotsaftis O, Berthomieu P, Bohnert H, Davenport RJ, Fairbairn DJ, et al. Nomenclature for HKT transporters, key determinants of plant salinity tolerance. Trends Plant Sci. 2006;11(8):372–4. doi: 10.1016/j.tplants.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Laurie S, Feeney KA, Maathuis FJ, Heard PJ, Brown SJ, Leigh RA. A role for HKT1 in sodium uptake by wheat roots. Plant J. 2002;32(2):139–49. doi: 10.1046/j.1365-313X.2002.01410.x. [DOI] [PubMed] [Google Scholar]
- 12.Schachtman DP, Schroeder JI. Structure and transport mechanism of a high-affinity potassium uptake transporter from higher plants. Nature. 1994;370:655–8. doi: 10.1038/370655a0. [DOI] [PubMed] [Google Scholar]
- 13.Ariyarathna HC, Ul-Haq T, Colmer TD, Francki MG. Characterization of the multigene family TaHKT 2; 1 in bread wheat and the role of gene members in plant Na+ and K+ status. BMC Plant Biol. 2014;14(1):159. doi: 10.1186/1471-2229-14-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horie T, Yoshida K, Nakayama H, Yamada K, Oiki S, Shinmyo A. Two types of HKT transporters with different properties of Na+ and K+ transport in Oryza sativa. Plant J. 2001;27(2):129–38. doi: 10.1046/j.1365-313x.2001.01077.x. [DOI] [PubMed] [Google Scholar]
- 15.Oomen RJ, Benito B, Sentenac H, Rodriguez-Navarro A, Talon M, Very AA, et al. HKT2;2/1, a K+ permeable transporter identified in a salt-tolerant rice cultivar through surveys of natural genetic polymorphism. Plant J. 2012;71(5):750–62. doi: 10.1111/j.1365-313X.2012.05031.x. [DOI] [PubMed] [Google Scholar]
- 16.Corratge-Faillie C, Jabnoune M, Zimmermann S, Very AA, Fizames C, Sentenac H. Potassium and sodium transport in non-animal cells: the Trk/Ktr/HKT transporter family. Cell Mol Life Sci. 2010;67(15):2511–32. doi: 10.1007/s00018-010-0317-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garciadeblas B, Senn ME, Banuelos MA, Rodríguez‐Navarro A. Sodium transport and HKT transporters: the rice model. Plant J. 2003;34(6):788–801. doi: 10.1046/j.1365-313X.2003.01764.x. [DOI] [PubMed] [Google Scholar]
- 18.Gomez-Porras JL, Riano-Pachon DM, Benito B, Haro R, Sklodowski K, Rodriguez-Navarro A, et al. Phylogenetic analysis of K+ transporters in bryophytes, lycophytes, and flowering plants indicates a specialization of vascular plants. Front Plant Sci. 2012;3:167. doi: 10.3389/fpls.2012.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kader MA, Seidel T, Golldack D, Lindberg S. Expressions of OsHKT1, OsHKT2, and OsVHA are differentially regulated under NaCl stress in salt-sensitive and salt-tolerant rice (Oryza sativa L.) cultivars. J Exp Bot. 2006;57(15):4257–68. doi: 10.1093/jxb/erl199. [DOI] [PubMed] [Google Scholar]
- 20.Lan WZ, Wang W, Wang SM, Li LG, Buchanan BB, Lin HX, et al. A rice high-affinity potassium transporter (HKT) conceals a calcium-permeable cation channel. Proc Natl Acad Sci U S A. 2010;107(15):7089–94. doi: 10.1073/pnas.1000698107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horie T, Costa A, Kim TH, Han MJ, Horie R, Leung HY, et al. Rice OsHKT2; 1 transporter mediates large Na+ influx component into K+ starved roots for growth. The EMBO J. 2007;26(12):3003–14. doi: 10.1038/sj.emboj.7601732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horie T, Brodsky DE, Costa A, Kaneko T, Lo Schiavo F, Katsuhara M, et al. K+ transport by the OsHKT2;4 transporter from rice with atypical Na+ transport properties and competition in permeation of K+ over Mg2+ and Ca2+ ions. Plant Physiol. 2011;156(3):1493–507. doi: 10.1104/pp.110.168047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yao X, Horie T, Xue S, Leung HY, Katsuhara M, Brodsky DE, et al. Differential sodium and potassium transport selectivities of the rice OsHKT2;1 and OsHKT2;2 transporters in plant cells. Plant Physiol. 2010;152(1):341–55. doi: 10.1104/pp.109.145722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International Rice Genome Sequencing P The map-based sequence of the rice genome. Nature. 2005;436(7052):793–800. doi: 10.1038/nature03895. [DOI] [PubMed] [Google Scholar]
- 25.Mayer KF, Rogers J, Doležel J, Pozniak C, Eversole K, Feuillet C, et al. A chromosome-based draft sequence of the hexaploid bread wheat (Triticum aestivum) genome. Science. 2014;345(6194):1251788. doi: 10.1126/science.1251788. [DOI] [PubMed] [Google Scholar]
- 26.Crawford AC, Francki MG. Chromosomal location of wheat genes of the carotenoid biosynthetic pathway and evidence for a catalase gene on chromosome 7A functionally associated with flour b* colour variation. Mol Genet Genomics. 2013;288(10):483–93. doi: 10.1007/s00438-013-0767-3. [DOI] [PubMed] [Google Scholar]
- 27.Francki MG, Hayton S, Gummer JPA, Rawlinson C, Trengove RD. Metabolomic profiling and genomic analysis of wheat aneuploid lines to identify genes controlling biochemical pathways in mature grain. Plant Biotechnol J. 2015;1–12. [DOI] [PMC free article] [PubMed]
- 28.Mackay IJ, Bansept-Basler P, Barber T, Bentley AR, Cockram J, Gosman N, et al. An eight-parent multiparent advanced generation inter-cross population for winter-sown wheat: creation, properties, and validation. Genes Genom Genet. 2014;4(9):1603–10. doi: 10.1534/g3.114.012963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasheed A, Xia X, Ogbonnaya F, Mahmood T, Zhang Z, Mujeeb-Kazi A, et al. Genome-wide association for grain morphology in synthetic hexaploid wheats using digital imaging analysis. BMC Plant Biol. 2014;14:128. doi: 10.1186/1471-2229-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Webster H, Keeble G, Dell B, Fosu-Nyarko J, Mukai Y, Moolhuijzen P, et al. Genome-level identification of cell wall invertase genes in wheat for the study of drought tolerance. Funct Plant Biol. 2012;39:569–79. doi: 10.1071/FP12083. [DOI] [PubMed] [Google Scholar]
- 31.Huang S, Spielmeyer W, Lagudah ES, Munns R. Comparative mapping of HKT genes in wheat, barley, and rice, key determinants of Na+ transport, and salt tolerance. J Exp Bot. 2008;59(4):927–37. doi: 10.1093/jxb/ern033. [DOI] [PubMed] [Google Scholar]
- 32.De León JLD, Escoppinichi R, Geraldo N, Castellanos T, Mujeeb-Kazi A, Röder MS. Quantitative trait loci associated with salinity tolerance in field grown bread wheat. Euphytica. 2011;181(3):371–83. doi: 10.1007/s10681-011-0463-5. [DOI] [Google Scholar]
- 33.Genc Y, Oldach K, Verbyla AP, Lott G, Hassan M, Tester M, et al. Sodium exclusion QTL associated with improved seedling growth in bread wheat under salinity stress. Theor Appl Genet. 2010;121:877–94. doi: 10.1007/s00122-010-1357-y. [DOI] [PubMed] [Google Scholar]
- 34.Ma L, Zhou E, Huo N, Zhou R, Wang G, Jia J. Genetic analysis of salt tolerance in a recombinant inbred population of wheat (Triticum aestivum L.) Euphytica. 2007;153(1–2):109–17. [Google Scholar]
- 35.Ogbonnaya FC, Huang S, Steadman E, Emebiri L, Dreccer F, Lagudah ES, et al. Mapping quantitative trait loci associated with salinity tolerance in synthetic derived backcrossed bread lines. 2008. [Google Scholar]
- 36.Quarrie SA, Quarrie PS, Radosevic R, Rancic D, Kaminska A, Barnes JD, et al. Dissecting a wheat QTL for yield present in a range of environments: from the QTL to candidate genes. J Exp Bot. 2006;57(11):2627–37. doi: 10.1093/jxb/erl026. [DOI] [PubMed] [Google Scholar]
- 37.Genc Y, Oldach K, Gogel B, Wallwork H, McDonald GK, Smith AB. Quantitative trait loci for agronomic and physiological traits for a bread wheat population grown in environments with a range of salinity levels. Mol Breed. 2013;32(1):39–59. doi: 10.1007/s11032-013-9851-y. [DOI] [Google Scholar]
- 38.Feuillet C, Keller B. Comparative genomics in the grass family: molecular characterization of grass genome structure and evolution. Ann Bot. 2002;89(1):3–10. doi: 10.1093/aob/mcf008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Appels R, Francki M, Chibbar R. Advances in cereal functional genomics. Funct Integr Genomics. 2003;3(1–2):1–24. doi: 10.1007/s10142-002-0073-3. [DOI] [PubMed] [Google Scholar]
- 40.Feuillet C, Keller B. High gene density is conserved at syntenic loci of small and large grass genomes. Proc Natl Acad Sci U S A. 1999;96(14):8265–70. doi: 10.1073/pnas.96.14.8265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tikhonov AP, SanMiguel PJ, Nakajima Y, Gorenstein NM, Bennetzen JL, Avramova Z. Colinearity and its exceptions in orthologous adh regions of maize and sorghum. Proc Natl Acad Sci U S A. 1999;96(13):7409–14. doi: 10.1073/pnas.96.13.7409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen WH, de Meaux J, Lercher MJ. Co-expression of neighbouring genes in Arabidopsis: separating chromatin effects from direct interactions. BMC Genomics. 2010;11:178. doi: 10.1186/1471-2164-11-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rustenholz C, Choulet F, Laugier C, Safar J, Simkova H, Dolezel J, et al. A 3,000-loci transcription map of chromosome 3B unravels the structural and functional features of gene islands in hexaploid wheat. Plant Physiol. 2011;157(4):1596–608. doi: 10.1104/pp.111.183921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bzymek M, Lovett ST. Instability of repetitive DNA sequences: the role of replication in multiple mechanisms. Proc Natl Acad Sci U S A. 2001;98(15):8319–25. doi: 10.1073/pnas.111008398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li YC, Korol AB, Fahima T, Beiles A, Nevo E. Microsatellites: genomic distribution, putative functions and mutational mechanisms: a review. Molecular Ecol. 2002;11(12):2453–65. doi: 10.1046/j.1365-294X.2002.01643.x. [DOI] [PubMed] [Google Scholar]
- 46.Cotsaftis O, Plett D, Shirley N, Tester M, Hrmova M. A Two-staged model of Na+ exclusion in rice explained by 3D modeling of HKT transporters and alternative splicing. PloS ne. 2012;7(7) doi: 10.1371/journal.pone.0039865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Espley RV, Brendolise C, Chagne D, Kutty-Amma S, Green S, Volz R, et al. Multiple repeats of a promoter segment causes transcription factor autoregulation in red apples. Plant Cell. 2009;21(1):168–83. doi: 10.1105/tpc.108.059329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gowik U, Burscheidt J, Akyildiz M, Schlue U, Koczor M, Streubel M, et al. cis-Regulatory elements for mesophyll-specific gene expression in the C4 plant Flaveria trinervia, the promoter of the C4 phosphoenolpyruvate carboxylase gene. Plant Cell. 2004;16(5):1077–90. doi: 10.1105/tpc.019729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thurau T, Kifle S, Jung C, Cai D. The promoter of the nematode resistance gene Hs1pro − 1 activates a nematode-responsive and feeding site-specific gene expression in sugar beet (Beta vulgaris L.) and Arabidopsis thaliana. Plant Mol Biol. 2003;52(3):643–60. doi: 10.1023/A:1024887516581. [DOI] [PubMed] [Google Scholar]
- 50.Villar CBR, Erilova A, Makarevich G, Trösch R, Köhler C. Control of PHERES1 imprinting in Arabidopsis by direct tandem repeats. Mol Plant. 2009;2(4):654–60. doi: 10.1093/mp/ssp014. [DOI] [PubMed] [Google Scholar]
- 51.Baek D, Jiang J, Chung JS, Wang B, Chen J, Xin Z, et al. Regulated AtHKT1 gene expression by a distal enhancer element and DNA methylation in the promoter plays an important role in salt tolerance. Plant Cell Physiol. 2011;52(1):149–61. doi: 10.1093/pcp/pcq182. [DOI] [PubMed] [Google Scholar]
- 52.Munns R, James RA, Xu B, Athman A, Conn SJ, Jordans C, et al. Wheat grain yield on saline soils is improved by an ancestral Na+ transporter gene. Nat Biotechnol. 2012;30(4):360–4. doi: 10.1038/nbt.2120. [DOI] [PubMed] [Google Scholar]
- 53.Diatloff E, Kumar R, Schachtman DP. Site directed mutagenesis reduces the Na+ affinity of HKT1, an Na+ energized high affinity K+ transporter. FEBS Lett. 1998;432(1):31–6. doi: 10.1016/S0014-5793(98)00833-3. [DOI] [PubMed] [Google Scholar]
- 54.Kato Y, Sakaguchi M, Mori Y, Saito K, Nakamura T, Bakker EP, et al. Evidence in support of a four transmembrane-pore-transmembrane topology model for the Arabidopsis thaliana Na+/K+ translocating AtHKT1 protein, a member of the superfamily of K+ transporters. Proc Natl Acad Sci U S A. 2001;98(11):6488–93. doi: 10.1073/pnas.101556598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maser P, Eckelmana BV, Vaidyanathan R, Horiea T, Fairbairnb DJ, Kuboc M, et al. Altered shoot/root Na+ distribution and bifurcating salt sensitivity in Arabidopsis by genetic disruption of the Na+ transporter AtHKT1. FEBS Lett. 2002;531:157–61. doi: 10.1016/S0014-5793(02)03488-9. [DOI] [PubMed] [Google Scholar]
- 56.Rubio F, Schwarz M, Gassmann W, Schroeder JI. Genetic selection of mutations in the high affinity K+ transporter HKT1 that define functions of a loop site for reduced Na+ permeability and increased Na+ tolerance. J Biol Chem. 1999;274(11):6839–47. doi: 10.1074/jbc.274.11.6839. [DOI] [PubMed] [Google Scholar]
- 57.Cao Y, Jin X, Huang H, Derebe MG, Levin EJ, Kabaleeswaran V, et al. Crystal structure of a potassium ion transporter, TrkH- supplimentary. Nature. 2011;471(7338):336–40. doi: 10.1038/nature09731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kato N, Akai M, Zulkifli L, Matsuda N, Kato Y, Goshima S, et al. Role of positively charged Amino Acids in the M2DTransmembrane Helix of Ktr/Trk/HKT type cation transporters. Channels. 2014;1(3):161–71. doi: 10.4161/chan.4374. [DOI] [PubMed] [Google Scholar]
- 59.Sassi A, Mieulet D, Khan I, Moreau B, Gaillard I, Sentenac H, et al. The rice monovalent cation transporter OsHKT2; 4: revisited ionic selectivity. Plant Physiol. 2012;160(1):498–510. doi: 10.1104/pp.112.194936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren ZH, Gao JP, Li LG, Cai XL, Huang W, Chao DY, et al. A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nature Genet. 2005;37(10):1141–6. doi: 10.1038/ng1643. [DOI] [PubMed] [Google Scholar]
- 61.Berthomieu P, Conéjéro G, Nublat A, Brackenbury WJ, Lambert C, Savio C, et al. Functional analysis of AtHKT1 in Arabidopsis shows that Na+ recirculation by the phloem is crucial for salt tolerance. EMBO J. 2003;22(9):2004–14. doi: 10.1093/emboj/cdg207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubio F, Gassmann W, Schroeder JI. Sodium-driven potassium uptake by the plant potassium transporter HKT1 and mutations conferring salt tolerance. Science. 1995;270(5242):1660–3. doi: 10.1126/science.270.5242.1660. [DOI] [PubMed] [Google Scholar]
- 63.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37(suppl 1):D211–5. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28(12):1647–9. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huang XQ, Brûlé-Babel A. Development of genome-specific primers for homoeologous genes in allopolyploid species: the waxy and starch synthase II genes in allohexaploid wheat (Triticum aestivum L.) as examples. BMC Res Notes. 2010;3(1):140. doi: 10.1186/1756-0500-3-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Francki M, Crasta O, Sharma H, Ohm H, Anderson J. Structural organization of an alien Thinopyrum intermedium group 7 chromosome in US soft red winter wheat (Triticum aestivum L.) Genome. 1997;40(5):716–22. doi: 10.1139/g97-794. [DOI] [PubMed] [Google Scholar]
- 68.Von Heijne G. Membrane protein structure prediction: hydrophobicity analysis and the positive-inside rule. J Mol Biol. 1992;225(2):487–94. doi: 10.1016/0022-2836(92)90934-C. [DOI] [PubMed] [Google Scholar]
- 69.Snider C, Jayasinghe S, Hristova K, White SH. MPEx: a tool for exploring membrane proteins. Protein Sci. 2009;18(12):2624–8. doi: 10.1002/pro.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–71. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 71.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16(6):276–7. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 72.Wicker T, Matthews DE, Keller B. TREP: a database for Triticeae repetitive elements. Trends Plant Sci. 2002;7(12):561–2. doi: 10.1016/S1360-1385(02)02372-5. [DOI] [Google Scholar]
- 73.Chen J, Hu Q, Zhang Y, Lu C, Kuang H. P-MITE: a database for plant miniature inverted-repeat transposable elements. Nucleic Acids Res. 2014;42(Database issue):D1176–81. doi: 10.1093/nar/gkt1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lescot M, Déhais P, Thijs G, Marchal K, Moreau Y, Van de Peer Y, et al. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002;30(1):325–7. doi: 10.1093/nar/30.1.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Higo K, Ugawa Y, Iwamoto M, Korenaga T. Plant cis-acting regulatory DNA elements (PLACE) database: 1999. Nucleic Acids Res. 1999;27(1):297–300. doi: 10.1093/nar/27.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Livak JK, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-∆∆CT Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 77.Eickelberg GJ, Fisher AJ. Environmental regulation of plant gene expression: An RT-qPCR laboratory project for an upper-level undergraduate biochemistry or molecular biology course. Biochem Mol Biol Educ. 2013;41(5):325–33. doi: 10.1002/bmb.20722. [DOI] [PubMed] [Google Scholar]
- 78.Schmittgen TS, Livak JK. Analyzing real-time PCR data by the comparative CT method. Nat Protoc. 2008;3(6):1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 79.Francki MG, Walker E, Crawford AC, Broughton S, Ohm HW, Barclay I, et al. Comparison of genetic and cytogenetic maps of hexaploid wheat (Triticum aestivum L.) using SSR and DArT markers. Mol Genet Genomics. 2009;281(2):181–91. doi: 10.1007/s00438-008-0403-9. [DOI] [PubMed] [Google Scholar]
- 80.Sourdille P, Singh S, Cadalen T, Brown-Guedira GL, Gay G, Qi L, et al. Microsatellite-based deletion bin system for the establishment of genetic-physical map relationships in wheat (Triticum aestivum L.) Funct Integr Genomics. 2004;4(1):12–25. doi: 10.1007/s10142-004-0106-1. [DOI] [PubMed] [Google Scholar]
- 81.Manly KF, Cudmore RH, Meer JM. Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome. 2014;12(12):930–2. doi: 10.1007/s00335-001-1016-3. [DOI] [PubMed] [Google Scholar]
- 82.Voorrips R. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–8. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]