

Abstract
Radioisotopes of arsenic are of considerable interest to the field of nuclear medicine with unique nuclear and chemical properties making them well-suited for use in novel theranostic radiopharmaceuticals. However, progress must still be made in the production of isotopically pure radioarsenic and in its stable conjugation to biological targeting vectors. This work presents the production and irradiation of isotopically enriched 72Ge(m) discs in an irrigation-cooled target system allowing for the production of isotopically pure 72As with capability on the order of 10 GBq. A radiochemical separation procedure isolated the reactive trivalent radioarsenic in a small volume buffered aqueous solution, while reclaiming 72Ge target material. The direct thiol-labeling of a monoclonal antibody resulted in a conjugate exhibiting exceptionally poor in vivo stability in a mouse model. This prompted further investigations to alternative radioarsenic labeling strategies, including the labeling of the dithiol-containing chelator dihydrolipoic acid, and thiol-modified mesoporous silica nanoparticles (MSN-SH). Radioarsenic-labeled MSN-SH showed exceptional in vivo stability toward dearsenylation.
INTRODUCTION
In recent years there has been considerable interest in the development of “theranostic pharmaceuticals” allowing for the initial diagnostic probing of a specific pharmacological target, followed by the subsequent administration of a therapeutic dose of the agent.1 Such theranostics promise advances to the field of personalized medicine, improving therapy selection, determination of optimal therapeutic dose, minimizing adverse effects, and gauging treatment response. In nuclear medicine, theranostics may consist of a biological targeting vector made with a radionuclide pair having similar chemical properties but different radioactive decay properties, one with a positron- or gamma-emitting nuclide for positron-emission tomography (PET) or single photon emission computed tomography diagnostic imaging and one with an electron-, alpha- or Auger-emitting nuclide for therapy. Examples include radio-pharmaceuticals that pair diagnostic 111In/68Ga with therapeutic 90Y/177Lu or incorporate different isotopes of iodine, with diagnostic 123I/124I and therapeutic 131I.2,3 However, these approaches suffer from limitations due to the potential for differences in diagnostic/therapeutic agent biodistribution resulting from the use of different radiolabel elements or, in the case of the 124I/131I pair, problems with deiodination and subsequent thyroid localization and damage following therapeutic dose administration.
Radioisotopes of arsenic offer an alternative radionuclide-based theranostic system. As summarized in Table 1, the decay properties, production methods, and potential nuclear medicine use of these isotopes are well suited for a variety of diagnostic and therapeutic applications. Multiple arsenic isotopes exist with a wide range of positron-emission half-lives, ranging from an hour to weeks and electron-emission half-lives with radiotherapeutic potential. These isotopes can each be produced in existing medical cyclotrons with solid target capability, and the most promising diagnostic/therapeutic pair, 72As and 77As, can also be produced through alternative means. Arsenic-72 can be produced through electron capture decay of 72Se (t1/2 = 8.5 d), allowing for a 72Se/72As generator system4,5 and 77As through β− decay of 77Ge (t1/2 = 11.3 h), allowing for its no-carrier-added production by neutron capture on natural germanium.6
Table 1.
Summary of Decay Properties, Radionuclide Production Methods, and Potential Medical Use of Selected Radioarsenic Isotopes
| isotope | half-life (h) | decay propertiesa | production method | medical use |
|---|---|---|---|---|
| 70As | 0.877 | 90% β+, 10% ec | Medical cyclotron | PET imaging |
| 71As | 65.30 | 28% β+, 72% ec | Medical cyclotron | PET imaging |
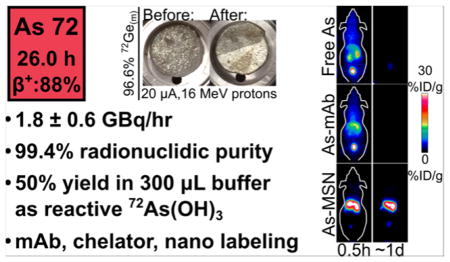
| 72As | 26.0 | 88% β+, 12% ec | Medical cyclotron, 72As/72Se generator | PET imaging |
| 74As | 426.5 | 29% β+, 36% ec, 34% β− | Medical cyclotron | PET imaging |
| 76As | 25.867 | 100% β− | Medical cyclotron | Radiotherapy |
| 77As | 38.79 | 100% β− | Medical cyclotron, neutron capture | Radiotherapy |
β+: positron emission, ec: electron capture, β−: electron emission.
In addition to these nuclear decay properties, arsenic has unique chemical and biochemical properties that make it notable for novel diagnostic or therapeutic radiopharmaceuticals. The most significant of these properties is the element’s high toxicity, which was first reported in third and fourth century B.C. Chinese and Greek writings and has been used for millennia as both a poison and a medicine.7 In recent years, arsenic trioxide (ATO), which dissolves as As(OH)3, has been clinically used to treat refractory acute promyelocytic leukemia (APL) through the irreversible inhibition of thioredoxin reductase.8 The success of ATO for treatment of APL has led to the continued development of arsenic-containing small molecules and drug delivery vehicles to mitigate systemic toxicity and allow for arsenic-based treatments for other cancer indications.9,10 The biologic activity of trivalent arsenic is largely due to the high sulfur affinity, allowing for its covalent binding to thiol/sulfhydryl groups, such as those cysteine side chains. These chemical and biochemical properties make radioarsenic uniquely suited for theranostic applications, through both the production of diagnostic PET analogues of arsenic-based chemotherapy agents and the use of covalent arsenic-thiol linkages to radiolabel biological targeting vectors.
While the production of diagnostic and therapeutic isotopes of arsenic with existing medical cyclotrons is feasible, it also is considerably challenging. First, the cyclotron irradiation of germanium with high intensity proton and deuteron beams poses technical difficulties due to the poor electrical and thermal properties associated with its physical forms. Pressed germanium oxide irradiation targets11–14 are characterized by limited electrical and thermal conductivity and are incapable of withstanding high current (>10 μA) irradiation. Targets of elemental crystalline germanium, which is brittle, has a low thermal conductivity and a melting point of 940 °C, have been utilized12,15–19 to greater success. Additionally, the cyclotron production of any isotopically pure arsenic radionuclide necessitates the use of isotopically enriched germanium target material, further complicating the target fabrication process. To date, there are no reports of isotopically pure radioarsenic production using a cyclotron. A second challenge lies in the radiochemical isolation of arsenic from irradiated germanium target material. While a wide variety of isolation procedures are reported and well reviewed,19 there remains a need for an isotopically enriched-target-compatible procedure that isolates the final radioarsenic product as reactive trivalent arsenic in a small volume of buffered solution, while reclaiming the expensive germanium target material for reuse. Additionally, the radioisolation procedure should minimize physical manipulations keeping radiation dose to the chemist as low as reasonably achievable.
The present study had two main goals. The first was to develop a cyclotron production and radiochemical isolation procedure capable of sustained, high proton intensity (>15 μA) irradiations of isotopically enriched 72Ge targets and isolating GBq-scale quantities of isotopically pure 72As. The second was to investigate established and novel labeling strategies to functionalize the radioarsenic product into biologically relevant targeting vectors. The accomplishment of these goals will further foster the development of radioarsenic-based theranostic radiopharmaceuticals.
RESULTS AND DISCUSSION
Target Preparation of Metallic 72Ge Targets
Isotopically enriched metallic germanium powder was produced through the reduction of 72GeO2 with hydrogen gas. The reduction proceeded as detailed in the literature.20 At 600 °C and 200 mL/min H2(g) flow rate, the reduction took place quantitatively in 5 h with no measurable mass lost to GeO(g) sublimation. The dark gray/black 72Ge(m) powder produced in this reduction was effectively molded into a metallic disc by heating to 1050 °C in a lidded boron nitride (BN) crucible with typical mass loss of (5 ± 3)% (n = 13) as shown in Figure 1. The discs were ~145 mg, 7–9 mm in diameter, and ~0.5 mm thick with a measured density of ~4.5 g/mL, notably lower than the established polycrystalline germanium density of ~5.5 g/mL.
Figure 1.

Boron nitride crucible containing germanium metal powder before melting at 1050 °C (left) and germanium metal disc after heating (right).
Irradiation of Metallic 72Ge Targets
The 72Ge(m) discs were well suited for irradiation in a custom flowing-liquid-cooled cyclotron target. The liquid coolant for the target was water with 5 mM ethanol to ensure 13N produced through the 16O(p,α) nuclear reaction on water was effectively rinsed from the target as 13NH4+.21 In the majority of irradiations, the 72Ge(m) disc showed only slight discoloration as a result of the 20 μA of 16 MeV protons as shown in Figure 2. During one irradiation, the disc shattered into six or seven pieces, highlighting the brittle nature of the germanium target material and the turbulent flow pattern in the irrigated irradiation cavity. As this did not impede coolant flow or the integrity of the niobium containment foil, the irradiation continued, but resulted in a ~50% decrease in 72As production yield. Overall radionuclide production yields for long-lived nuclear reaction products on the 72Ge(m) targets were quantified through high purity germanium gamma spectroscopy and shown in Table 2. The relatively short-lived 70As (t1/2 = 53 min) is also produced through the 70Ge(p,n) reaction. Based on sensitivity-corrected dose calibrator measurements, the 70As radioactivity was roughly 7% that of 72As at the end of a 1 h bombardment. The produced 72As had a radionuclidic purity of 99.4% 24 h after the end of bombardment. A 5 h irradiation at 20 μA of 16 MeV proton current would result in the production of up to 10 GBq of 72As activity, enough to envision local or regional distribution of this long-lived PET radionuclide.
Figure 2.

Germanium metal disc in irradiation pocket before (left) and after (right) flowing-liquid-cooled irradiation with 20 μA of 16 MeV protons for 1 h.
Table 2.
Long-Lived Radionuclide Production Yields for Proton Irradiation of (144 ± 7) mg 72Ge(m) Targets
| radionuclide | yield (MBq/(μA*h)) |
|---|---|
| 72As | 90 ± 30 |
| 71As | 0.3 ± 0.1 |
| 73As | 0.05 ± 0.02 |
| 74As | 0.03 ± 0.02 |
| 67Ga | 0.005 ± 0.002 |
Radiochemical Purification of 72As
The investigated metallic germanium target dissolution, distillation, anion exchange (AX) chromatography, and reduction procedures were adapted from the literature.19 The initial dissolution of the irradiated 72Ge(m) target proceeded rapidly in 2.6 mL of hot aqua regia and was characterized by vigorous bubbling and evolution of brown fumes. The dissolution of the disc slowed as a white precipitate (likely GeO2) was formed but was completed with increased heat and the distillation of the remaining aqua regia. The two subsequent additions of hydrochloric acid and hydrogen peroxide served a dual purpose. First, they successfully distilled the germanium target material as 72GeCl4, while keeping the radioarsenic in its nonvolatile As(V) oxidation state. All visible GeO2 precipitate had disappeared by the evaporation of the first HCl/H2O2 addition. Second, these evaporation steps effectively removed the oxidizing HNO3, which caused problems in the post-AX reduction of As(V) to As(III) and subsequent solid phase extraction (SPE) procedure. Over the course of the distillation, white precipitate was deposited in the gas exhaust lines carrying the argon sweep gas and distillate. Care was taken to ensure these lines did not clog and result in overpressurization leading to an uncontrolled vapor release from the distillation vessel. The dissolution and distillation procedure was observed to be very conservative of 72As activity, with (3 ± 1)% of the activity lost to the distillate, and highly effective at removing all detectable germanium target material with a microwave plasma atomic emission spectrometry- (MP-AES-) measured Ge/As decontamination factor of >1.2 × 104.
Anion exchange chromatography effectively isolated 72As from trace germanium target material and 67Ga. In 10 M HCl, germanium and gallium, forming anionic complexes, were strongly retained to the resin with distribution coefficients (Kd) of 200 and 104.5, respectively, while As(V) was not retained (Kd ~4).22,23 The conditions utilized in this work resulted in high yields of 72As, with (93 ± 2)% of activity in the 9 mL As-rich elution fraction. No 67Ga activity was detected in the 72As elution peak or extended elution tail. The elution of 67Ga was only observed with the addition of 1 M HCl where gallium Kd < 1.
The reduction and SPE procedures detailed in this manuscript were adapted from Jahn et al.19 and Jennewein et al.,13 respectively. The result is significantly more automatable and resulted in less radiation worker dose exposure compared with the liquid-phase solvent extraction procedure adapted from Jahn et al.19 and Shehata et al.14 The conditions utilized in this work resulted in reasonable yields of 72As in a small volume of buffered eluant, with (57 ± 5)% yield in the first 300 μL of 1 M HEPES. Yields were only modestly improved with additional elution volume, with (6 ± 5)% of total loaded 72As eluting in the second 300 μL of 1 M HEPES. Additional losses were (11 ± 3)% in loading, (9 ± 1)% in the 10 M HCl rinse, and (16 ± 5)% remaining bound to the SPE resin. This radiochemical yield is modestly lower than the (73 ± 8)% observed for the liquid–liquid extraction procedure. Despite eluting in 1 M HEPES, the eluant had pH < 0 as a result of residual HCl on the SPE column. Under these low pH conditions, the reoxidation of the trivalent 72As(OH)3 eluant to the pentavalent 72AsO(OH)3 occurred over the course of minutes. The post elution addition of hydroxylamine (HA) and ethylenediamine-tetraacetic acid (EDTA), and neutralization with NaOH significantly slowed this oxidation to occur over the course of hours to days. This lability for arsenite ions to reoxidize to arsenate is of particular import as radioarsenic labeling strategies often involve thiol reactivity exclusive to the As(III) oxidation state.
Table 3 summarizes the purpose, 72As yield, and Ge/As decontamination factor for each of the steps of the radiochemical purification procedure. The procedure’s overall decay-corrected chemical yield for 72As was measured to be (50 ± 1)% with the majority of the loss to the load, rinse, and irreversible trapping on the SPE column. The final product contained quantities of arsenic and germanium below MP-AES detectable limits. The quantity of copper in the final ~360 μL, neutral, buffered 72As(OH)3 solution was (2.4 ± 0.7) μg. Therefore, the SPE procedure exhibited a decontamination factor of the Cu(I) reducing agent of (6 ± 2) × 104.
Table 3.
Summary of Results from 72As Radiochemical Purification Procedure
| procedure | purpose | 72As yield | Ge/As decon. factora |
|---|---|---|---|
| Distillation | Bulk Ge target removal | (97 ± 1)% | >1.2 × 104 |
| AX column | Trace Ge, 67Ga removal | (93 ± 2)% | n/a |
| SPE column | Reduction to As(III), concentration in buffered formulation | (57 ± 5)% | n/a |
| Overall | Isolate 72As(OH)3 | (50 ± 1)% | >1.2 × 104 |
Decontamination factor determined through MP-AES.
Reclamation and reprocessing of 72Ge Irradiation Target Material
The 72Ge target material was isolated through the neutralization of the acid fume condensate effectively, albeit laboriously. Care was taken to use the basic gas scrubber neutralizing solution to dissolve all the white germanium precipitate in the gas line connecting the distillation vial and the condensate trap. On several occasions, attempts to melt reclaimed 72Ge(m) powder into a 72Ge(m) disc were unsuccessful due to residual salt from the acid neutralization. In these cases, the misformed, salt-encrusted 72Ge(m) pieces were removed from the crucible, sonicated in water to remove salt, and remelted in a subsequent target preparation step. The overall, nonoptimized reclamation yield of the entire process, 72Ge(m) target disc to reclaimed 72Ge(m) target disc, was found to be ~60%. Unreclaimed 72Ge remained dissolved in the neutralized condensate and salt-removal wash solutions. Further optimization of the reclamation yield will proceed through the collection and evaporation of all 72Ge-contacted solutions, followed by the washing of the NaCl/72GeO2 residue with a minimum volume of water to isolate the less soluble 72GeO2 from the bulk NaCl.
Radioarsenic Labeling and Evaluation of TRC105
Motivated by reports utilizing trivalent radioarsenic to label monoclonal antibodies,19,24 initial studies using the *As(OH)3 (* = 72, 74, 76, 71) product attempted to directly label the monoclonal antibody TRC105. This angiogenesis-targeted, antiendoglin (CD105) human/murine chimeric IgG1 monoclonal antibody is currently undergoing multiple phase I and II studies for various cancer indications.25–27 TRC105 was chosen for these radioarsenic labeling studies because of previous experience in its 64Cu- and 89Zr-radiolabeling and well characterized in vivo biodistribution.28,29 Some studies investigating the labeling chemistry of radioarsenic utilized *As(OH)3 produced through the irradiation of natGe target material as opposed to isotopically pure 72As(OH)3 produced with enriched 72Ge as detailed above. Initial results from the radiolabeling of TRC105 were favorable as measured by high purity germanium (HPGe) gamma analysis of the size exclusion chromatography (SEC) purification fractions. Radiolabeling resulted in 69% of the radioarsenic eluting with the high-molecular-weight (MW) fraction following reaction with tris(2-carboxyethyl)phosphine- (TCEP-) reduced TRC105. Similarly, 94% of the radioarsenic eluted with the high-MW fractions following the labeling of Traut-modified, TCEP-reduced TRC105. While the labeling yields observed here are lower than those reported in the literature,19,24 the results are in overall agreement.
The biodistribution of *As–S-TRC105 was compared with that of free *As as shown through serial maximum intensity projection PET images in Figure 3. The biodistribution of the positron emitting radiotracer in the *As–S-TRC105 preparation was observed to be nearly identical to that of the free *As, which was characterized by rapid renal excretion. Additionally, the biodistribution for *As–S-TRC105 labeled under TCEP-reduced conditions exhibited indistinguishable in vivo behavior to those prepared under Traut-modified, TCEP-reduced conditions. The *As–S-TRC105 results stand in stark contrast to those of 64Cu-TRC105 and 89Zr-TRC105, which show long blood circulation half-lives and significant blood pool, liver, and 4T1 tumor uptake (5–15%ID/g) 24 h post injection (p.i.).28,29 The disparity between the *As–S-TRC105 biodistribution and those reported for 64Cu-DOTA-/89Zr-Df-TRC105 could be as a result of several confounding factors. One such factor could be that the TCEP-reducing labeling conditions necessary to attach *As to TRC105 reduced the disulfide bonds connecting the heavy and light chains of TRC105, thereby breaking up the antibody and destroying its specificity for the CD105 protein. However, it has been shown that incubation of antibodies in concentrations of TCEP ten times higher than those used in this study only reduced immunoreactivity to 85%.30 Also, it is unlikely that *As-bound mAb fragments would have rapid renal clearance as observed in this study. Another possible explanation is that the 2.6 GBq/μmol specific activity of *As–S-TRC105 used in this study is significantly lower than the 440 GBq/μmol 64Cu-DOTA-TRC10528 and the 15–74 GBq/μmol 89Zr-Df-TRC105,29 thus creating a self-blocking effect limiting tumor uptake. This is unlikely, since the biodistribution of 64Cu-DOTA-/89Zr-Df-TRC105 following administration of a 2 mg blocking dose of TRC105 clearly shows high hepatic uptake and retention of the radiotracer, not rapid renal clearance.28,29
Figure 3.
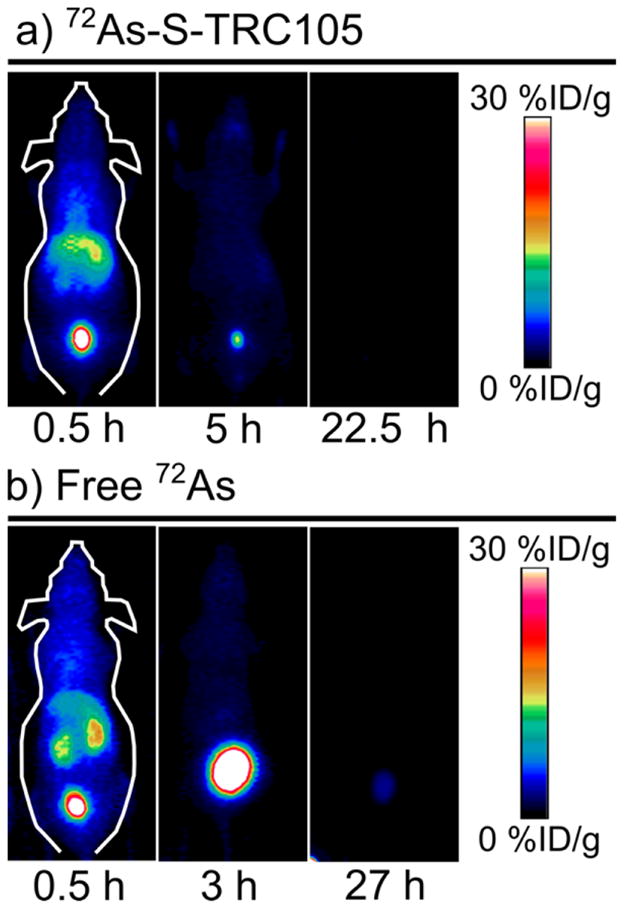
Representative serial PET maximum intensity projection images taken at three times following the injection of (a) 72As–S-TRC105 and (b) free 72As.
A third explanation for these results is that the covalent bond between arsenic and the TRC105 thiol is degrading in vivo resulting in the liberation of the *As radiolabel. This could occur as a result of transchelation of the *As with plasma proteins, such as albumin, or small molecules such as reduced glutathione and cysteine, which have been shown to bind to As(III).31–33 Rapid in vivo dearsenylation of *As-TRC105 would fully explain the observed experimental results. This hypothesis is further supported by silica plate TLC results analyzing the *As-TRC105 labeling solutions as a function of time after mixing *As(OH)3 and TRC105-SH. These results showed a decrease of As(III) (Rf = 0.8) over time, with a corresponding increase of activity with intermediate retention factor (Rf = 0.2–0.8). As one would expect the high-molecular-weight antibody to stay at the origin of the TLC plate (Rf = 0), these results suggest that the tartrate ions in the mobile phase were a strong enough arsenic-binding chelator to dislodge the “labeled” *As from TRC105, resulting in *As with an intermediate Rf.
The result in which the *As–S-mAb radioimmunoconjugates formed with these methods exhibit exceptionally poor in vivo radiostability is in direct contradiction to a conclusion reached in the literature with similarly labeled 74/77As–S-bavituximab.24 It is possible that this apparent disagreement is a result of a number of experimental differences, including the different radioarsenic labeling synthon (AsI3 versus As(OH)3), different method of thiol incorporation (N-succinimidyl S-acetylthioacetate modification, SATA, yielding 3.5 thiol/mAb versus Traut reagent, yielding 8 thiol/mAb), the significantly higher specific activity (>100 GBq/μmol versus 2.6 GBq/μmol), the different mAb targeting vectors offering distinct conformational stability of *As-thiol bonds (bavituximab versus TRC105), or different model organisms (rat versus mouse). However, it is also possible that the literature’s conclusion of high in vivo radiostability was not actually supported by their results. This conclusion was based on the fact that very little nonspecific uptake of radioactivity was detected in the liver at 24–72 h p.i.24 The free *As(III) biodistribution results in mice shown here in Figure 3b demonstrate that free or plasma-chelated radioarsenic is in fact rapidly renally excreted, not hepatically sequestered and retained. In addition, comparison of the 74/77As–S-bavituximab biodistribution results in AT1-tumor bearing rats with recent biodistribution results for 64Cu-labeled bavituximab in LNCaP-tumor bearing mice34 shows significant disagreement. At 48 h p.i. 74/77As–S-bavituximab was observed with uptake of 0.25%ID/g in the tumor and 0.125%ID/g in the liver, while 64Cu-bavituximab exhibited 3.2%ID/g in the tumor and 20.6%ID/g in the liver.24,34 While comparison of these results makes a strong case that 74/77As–S-bavituximab is, indeed, seeing significant dearsenylation in vivo, it fails to negate the fact that the radiotracer does offer impressive tumor to liver and tumor to muscle ratios at 72 h p.i. (22 and 470, respectively).24 This result can be explained by the possibility that the radiotracer can be more rapidly dearsenylated when sequestered by the liver than when specifically bound to the tumor, allowing radiotracer to flush from the nonspecifically bound, liver-sequestered antibody.
Radioarsenic Labeling and Evaluation of Dihydroli-poic Acid
The results support the conclusion that thiol-modified antibodies labeled directly with trivalent *As(OH)3 do not exhibit adequate in vivo stability. Thus, investigations into alternative methods of arsenic radiolabeling of biological targeting vectors is a worthy and necessary pursuit to bring radioarsenic to its full potential as a theranostic agent. One avenue for stabilizing such a radiopharmaceutical toward in vivo dearsenylation would be through the use of a chelating ligand with high affinity for arsenic. The use of chelators to radiolabel biological targeting vectors is well developed with radiometal-based imaging agents using, for example, 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetraacetic acid (DOTA) to chelate 64Cu and 68Ga and desferrioxamine (DFO) to chelate 89Zr as recently reviewed.35 Owing to the fact that trivalent arsenic preferentially forms tridentate, trigonal pyramidal complexes with sulfur-containing ligands, the most stable chelators would likely contain three free thiols capable of binding in such a geometry. Recent work has begun to explore this promising approach.36,37 The present work adopts a simpler strategy to arsenic chelation through the use of 1,2-dithiolane-3-pentaoic acid (lipoic acid, LA), a disulfide-bond-containing, reduction-/oxidation-active eight carbon fatty acid that is endogenously produced in cells and is a cofactor to several mitochondrial enzyme complexes.38 Through the reduction of the disulfide bond, dihydrolipoic acid (DHLA) is produced with two thiol groups capable of forming a bidentate six-membered ring structure with trivalent arsenic, shown in Figure 4. The particular stability of the trivalent arsenic–DHLA complex has been measured and shown compared with other dithiol ligands.33 In addition, following activation with a carbodiimide compound such as 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC), the carboxylic acid functional group of DHLA could be bioconjugated to primary amines present on biological targeting vectors.
Figure 4.

Molecular schematic of reduction of (1) lipoic acid to (2) dihydrolipoic acid and subsequent chelation of arsenic trihydroxide to form (3) As-DHLA.
The preparation of DHLA from LA through sodium borohydride reduction and solvent extraction was performed with 75% chemical yield. The final DHLA product was a viscous, clear, slightly yellow liquid with ~85% chemical purity as determined by finding 1.7 thiols per DHLA through its quantification using 5,5′-dithiobis(2-nitrobenzoic acid) according to literature procedures.39 Care was taken to limit the exposure of DHLA solutions to atmospheric oxygen by purging vials and solvents with argon to minimize reoxidation of DHLA to LA. The chelation of carrier-free and carrier-added *As(OH)3 after 10 min and 18 h of contact with DHLA was measured using reversed phase thin layer chromatography (RP-TLC) as shown in Figure 5a. Under the RP-TLC conditions used, free arsenic predominantly traveled to high retention factors (Rf = 0.8–0.9), with a small fraction remaining at Rf = 0, while As-DHLA was highly retained by the plate with Rf = 0. The RP-TLC profile of carrier-added free arsenic is shown in the top two profiles. The Rf = 0.9 to Rf = 0 ratios determined in these profiles were used to subtract the contribution of free arsenic to the Rf = 0 peak and measure the fraction of radioarsenic bound to DHLA. Both no-carrier-added and carrier-added *As(OH)3 were observed to very rapidly form As-DHLA complexes with ~90% yield within 10 min of contact. The carrier-added radioarsenic was observed to decrease its labeling fraction between 10 min and 18 h of contact. This may be the result of the formation of 2:3 As:DHLA complexes such as those reported in the literature,33 resulting in lower labeling yields for solutions with a molar equivalent of arsenic and DHLA. The results of the DHLA reactivity titration of 72As(OH)3 after 18 h of labeling are shown in Figure 5b,c. In these results, the Rf = 0.9 to Rf = 0 ratio of free arsenic was determined from the profile of the solution with the smallest amount (10 pmol) of DHLA. When plotted as As-DHLA labeling percent versus amount of DHLA, a sigmoidal curve is elucidated with its point of inflection at 20 nanomol of DHLA. The DHLA effective specific activity for 72As(OH)3 was measured to be 6.4 MBq/40 nanomol = 0.2 GBq/μmol at 45 h after the end of bombardment. Future work will include efforts to improve this value to the range of 10–100s of GBq/μmol, which are typical 64Cu/89Zr radiometal chelator effective specific activities.
Figure 5.

(a) Radioarsenic activity profiles of RP-TLC analysis 10 min (left) and 18 h (right) after mixture of solutions containing carrier-added *As(OH)3 (top), carrier-added *As(OH)3 and DHLA (middle), and no-carrier-added *As(OH)3 and DHLA (bottom). (b) RP-TLC profile analysis of no-carrier-added 72As(OH)3 combined with various amounts of DHLA. (c) Plot of 72As-DHLA labeling percent as a function of moles of DHLA from results shown in Figure 4b.
Radioarsenic Labeling and Evaluation of Thiol-Modified Mesoporous Silica Nanoparticles
An alternative method for functionalizing radioarsenic to biological targeting vectors is through the versatility of nanomedicine. In the past 20 years, the use of nanotechnology in the development of novel cancer imaging agents and therapeutics has reached considerable maturity and offers great flexibility for radio-labeling strategies. Such strategies have recently been used to functionalize radioarsenic to thiol-containing polymer nanoparticles40 and to superparamagnetic iron oxide nanoparticles (SPION).41 Mesoporous silica nanoparticles (MSN) are characterized by their good biocompatibility, high surface area, and versatility toward surface modifications and have previously been used for in vivo imaging and drug delivery studies.42,43 A common surface modification for MSN is the addition of thiol groups which are used to surface functionalize the nanoparticles with polyethylene glycol (PEG) and biological targeting vectors through the use of Michael-reaction thiol-maleimide coupling chemistry. The high surface area and thiol density of such thiol-modified MSN (MSN-SH) offers the promise of a particularly efficient and stable radiolabeling strategy for trivalent radioarsenic. In addition, recent work performing serial in vivo PET imaging of 89Zr-labeled MSN has shown that such nonfunctionalized, negatively charged MSN are rapidly and irreversibly sequestered by the liver and spleen for the entire length of the study (21 days post injection).44 The stark difference between this behavior of MSN with that of rapid renal clearance for free radioarsenic shown in Figure 3b allows for effective characterization of the stability of radio-arsenic-labeled thiol-modified MSN(*As–S-MSN) toward in vivo dearsenylation.
Thiol-modified MSN were synthesized and characterized to have a radius of (65 ± 5) nm, a pore diameter of 2–3 nm, a zeta potential of (−44 ± 2) mV in pH 5–6 H2O, a surface area per particle of 3.5 × 104 nm2, and 5.6 × 106 thiols per particle. Following 12 h of contact with *As(OH)3, a MSN-SH labeling yield of 44% was observed, with ~5% of the labeled activity lost with each of the two washing steps. The biodistribution of *As–S-MSN in healthy nude mice is shown through serial maximum intensity projection PET images and region of interest quantification results shown in Figure 6. As expected for nonfunctionalized, negatively charged MSN, the radiotracer is very rapidly sequestered by the liver and spleen with (32 ± 3) %ID/g and (11 ± 3) %ID/g, respectively. This liver distribution remains largely stagnant for the first 3 days before tapering slightly to (21 ± 6) %ID/g at 7 days post injection. Spleen uptake remains constant over the investigated seven day period. Bladder uptake was also observed at all time points with (4 ± 2) %ID/g. With the knowledge that MSN are retained in the liver and spleen for greater than 7 days and that free radioarsenic is rapidly and nearly quantitatively renally excreted, these results show that *As–S-MSN is largely resistant to in vivo dearsenylation. The small, consistent bladder uptake and slightly decreasing liver uptake after 3 days does, however, indicate a small amount of radioarsenic desorption from the MSN-SH. The observed in vivo radiostability of *As–S-MSN is dramatically improved over that shown in Figure 3a for a directly labeled thiol-modified monoclonal antibody. This improvement is likely due to the high thiol density on the MSN surface allowing for the binding of trivalent radioarsenic to two or three thiol groups, resulting in a significantly stronger arsenic sequestration. Additional characterization of the arsenic–MSN binding properties and stability will be reported in a forthcoming manuscript. Through the use of standard methods to surface functionalize *As–S-MSN with PEG to improve blood circulation lifetime and biological targeting vectors to give tumor specificity,43 this extremely high in vivo radiostability can be conveyed to a new class of tumor-targeted, drug-delivery-capable, radioarsenic-based theranostic agents.
Figure 6.

(a) Representative serial PET maximum intensity projection images taken at six times following the injection of *As–S-MSN. (b) PET-quantified percent injected dose per gram (%ID/g) for regions of interests around the liver (red diamonds), bladder (blue squares), and spleen (yellow triangles) as a function of time since injection of *AsS-MSN.
CONCLUSION
Radioisotopes of arsenic have the potential for use in novel theranostic radiopharmaceuticals due to their unique nuclear and chemical properties. However, significant hurdles must still be overcome related to the production of isotopically pure radioarsenic and its stable incorporation into biological targeting vectors. Through producing isotopically enriched 72Ge(m) discs, followed by irradiation in a simple custom irrigation-cooled target system, isotopically pure 72As was produced in large enough quantities to provide multiple human doses or allow for local or regional distribution. The radiochemical isolation procedure, suitable for automation, resulted in acceptable radiochemical yield of reactive 72As-(OH)3 in a small volume of buffered aqueous solution, while reclaiming and reprocessing 72Ge target material.
Investigations into established literature procedures for radioarsenic labeling of antibodies yielded results that initially seemed in disagreement previous reports.24 Upon closer inspection and comparison with recent studies,34 the results of these studies are brought into agreement with the conclusion that antibodies directly labeled with radioarsenic exhibit exceptionally poor in vivo stability toward dearsenylation. To address this issue, two alternative methods were investigated. The chelation of trivalent radioarsenic was achieved with DHLA, a dithiol-containing small molecule that is capable of biofunctionalization to antibodies and proteins. Additionally, thiol-modified MSN were effectively labeled by radioarsenic and exhibit exceptional in vivo stability toward dearsenylation. These explorations into radioarsenic labeling strategies for biological targeting vectors highlight the challenges and illuminate several promising avenues of investigation toward realizing the potential of radioarsenic for theranostic pharmaceuticals.
EXPERIMENTAL PROCEDURES
Materials
Unless otherwise stated, reagents were obtained from commercial vendors and used as received. Isotopically enriched 72GeO2 (96.59% 72Ge, 0.35% 70Ge, 2.86% 73Ge, 0.20% 74Ge, 0.01% 76Ge) was obtained from Isoflex USA (San Francisco, CA, USA). Argon (industrial grade) and hydrogen gases (research grade, 99.9999% purity) were used. The human/murine chimeric IgG1 monoclonal antibody TRC105 was provided by TRACON Pharmaceuticals Inc. (San Diego, CA, USA). Dihydrolipoic acid was prepared from (±)-α-lipoic acid by sodium borohydride reduction according to established literature procedures.33
Target Preparation of Metallic 72Ge Discs
Germanium-72 dioxide (500–800 mg) was weighed and sealed in a glass test tube fitted with 1/16″ polytetrafluoroethylene gas lines. The tube was purged thrice with alternating Ar and low vacuum (20 kPa absolute) and thrice with alternating H2 and low vacuum, followed by the constant flow of 200 mL/min H2 gas. Under the reducing hydrogen atmosphere, the GeO2 was heated to 600 °C in a vertically oriented tube furnace for 2.5–5 h with occasional gentle agitation until the GeO2 to metallic Ge(m) conversion was complete, as measured by stoichiometric mass change.
Metallic 72Ge powder (~150 mg) was weighed into a BN crucible with 9.5-mm-diameter, 1.9-mm-deep circular pocket and lightly compressed with a BN lid with 9.45-mm-diameter, 1.3-mm-tall tamper. The filled crucible and lid were then placed under a ~100 g tantalum weight and sealed in a tube furnace. The furnace was purged thrice with alternating Ar and low vacuum (20 kPa absolute), followed by the constant flow of 200 mL/min Ar gas and subsequent 100 min temperature ramp to 1050 °C where it was held for 60 min, followed by passive cooling. Once the oven temperature was below 100 °C the BN crucible was removed, carefully opened, and the 72Ge(m) disc dislodged from its molding pocket and subjected to dimensional and mass measurements.
Cyclotron Irradiation of Metallic 72Ge Targets
Metallic 72Ge discs were proton-irradiated in a custom irrigation-cooled target system using a PETtrace cyclotron (GE Healthcare, Little Chalfont, Buckinghamshire, England). The target system consisted of a water-cooled aluminum body with a 1-cm-diameter, 3.2-mm-deep pocket, an O-ring, a bolt circle, and two 1.5-mm-diameter channels leading to exterior 1/16″ Swagelok fittings. A (144 ± 7) mg 72Ge(m) disc was placed in the pocket and sealed with a 51-μm-thick niobium foil with a clamp ring. Ethanol (5 mM) was flowed at 4 mL/min through the target chamber during irradiation with 20 μA of 16 MeV protons for 1 h. After irradiation, the target was disassembled and the 72Ge(m) disc removed and quantified for 72As radioactivity using a dose calibrator (CRC-15, Capintec, Inc., Ramsey, NJ, USA) with setting #970, which was determined through efficiency-calibrated HPGe gamma analysis.
Radiochemical Purification of 72As
The 72Ge(m) disc was dissolved in 2.6 mL of 3:1::10 M HCl:15 M HNO3 (aqua regia) at 90 °C under flowing argon. The sweep gas and fumes were passed through 1/8″ tubing connecting a series of gas scrubbers including an initial condensation collection vessel, a second and third vessel each containing 15 mL of 6 M NaOH, and a fourth vessel with 15 mL of H2O. After bubbling subsided from the 72Ge(m) disc, the temperature was increased to 130 °C to initiate the distillation of the aqua regia. Upon reaching dryness, the vessel was distilled to dryness with two subsequent quantities of 2.5 mL 10 M HCl, 50 μL 30% H2O2, effectively distilling the 72Ge target material as 72GeCl4.
The dried 72As was then reconstituted in 1 mL of 10 M HCl for anion exchange chromatographic purification to remove trace germanium and 67Ga impurities. Anion exchange resin (~350 mg, AG 1 × 8, 200–400 mesh, Bio-Rad Laboratories, Hercules, CA, USA) was slurry loaded into a 1 mL, 9.5-mm-inner-diameter fritted cartridge and preconditioned with 10 mL of 10 M HCl. The 72As in HCl was then flowed through the AX column, followed by a second 1 mL 10 M HCl rinse of the distillation vial and 25 mL of 10 M HCl. Following the initial elution of ~400 μL, the subsequent 9 mL of 72As-containing eluant was collected as a single fraction. Following the 10 M HCl elution, 20 mL of 1 M HCl was added to elute 67Ga and trace germanium.
The purified 72As was isolated as trivalent 72As(OH)3 in a small-volume, buffered solution through reduction, followed by either SPE or liquid–liquid extraction (LLE). Copper(I) chloride (~200 mg) was added to the 9 mL 72As-rich 10 M HCl AX fraction and incubated at 60 °C for greater than 30 min. In the SPE procedure, polystyrene–divinylbenzene SPE resin (~100 mg, Chromabond HR-P, 1200 m2/g, 50–100 μm, Macherey-Nagel Inc., Bethlehem, PA, USA) was dry loaded into a fritted 5.6-mm-inner-diameter column and preconditioned immediately before use with 10 mL of H2O and 10 mL of 10 M HCl. The 72As in 0.2 M CuCl, 10 M HCl was then flowed through the column extracting 72AsCl3, followed by its subsequent rinsing with 10 mL of 10 M HCl and 20 mL of air. The 72As was then eluted with 300 μL of 1 M 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer at pH 9. Hydroxylamine·HCl, EDTA, and sodium hydroxide were added to the 72As eluant to bring the solution to 0.5 M HA, 25 mM EDTA, and pH 7 immediately following elution. In the LLE procedure, the 72As in 0.2 M CuCl, 10 M HCl was shaken twice with an equal volume of cyclohexane, extracting 72AsCl3. The combined organic phases were then shaken with 500 μL of 0.5 M HA, 25 mM EDTA, phosphate buffered saline (PBS) at pH 7.1, back-extracting 72As(OH)3 into the small volume buffered aqueous phase.
Quality Analyses of Isolated 72As(OH)3 Product
The efficacy of this radiochemical procedure and the purity of the 72As(OH)3 product was determined through a variety of methods. Trace metal analysis was performed using MP-AES (4200, Agilent Technologies, Santa Clara, CA, USA). Limits of detection for arsenic, germanium, and copper in the final 72As(OH)3 product were found to be 10 ppm, 1 ppm, and 10 ppb, respectively. The oxidation state of 72As was monitored with the thin layer chromatography (TLC) procedure using a silica stationary phase, 3:1::0.01 M sodium tartrate:methanol as mobile phase,19 and visualized by a Cyclone Plus storage phosphor autoradiography (AR) system (PerkinElmer, Waltham, MA, USA).
Reclamation and Reprocessing of 72Ge Irradiation Target Material
The 72GeCl4/HCl condensate resulting from the 72As isolation procedure was chemically processed to reclaim 72Ge for future use. Using the 6 M NaOH from the first gas scrubber vessel, the condensate was neutralized to pH 7, resulting in the precipitation of 72GeO2. The slurry was centrifuged at 3 krpm for 10 min, the clear supernatant decanted, and the pellet washed with 10 mL of cold H2O by centrifugation/decantation. The pellet was dried with heat and argon flow and reduced to 72Ge(m) as described above. Following reduction, the 72Ge(m) powder was further rinsed to remove sodium chloride and nitrate salts by repeatedly soaking in H2O, followed by centrifugation/decantation. The rinsed 72Ge(m) powder was then molded into 72Ge(m) discs as described above.
Radioarsenic Labeling and Evaluation of TRC105
Initial experiments to functionalize the isolated trivalent radioarsenic to a biologically relevant targeting vector involved direct labeling of thiol groups on the monoclonal antibody TRC105. These studies utilized *As(OH)3, (* = 72, 74, 76, 71) produced from the proton irradiation of natGeO2 through the cyclotron production and initial chemical isolation steps detailed previously.45 The final *As(OH)3 sample preparation involved the AX and LLE procedures detailed above. Immediately following the LLE procedure, *As(OH)3 was combined with 1.25 mg TRC105 in 1 mL of 0.25 M HA, 12.5 mM EDTA, 0.35 mM TCEP, PBS at pH 7.1. After 1 h of incubation at room temperature, the mixture was purified using a PD-10 SEC column (GE Healthcare Bio-Sciences, Pittsburgh, PA, USA), collecting the 0.5 mL PBS fraction most rich in *As-labeled TRC105. Two mice bearing 4T1 murine breast cancer xenografts were each injected with 250 μL of this fraction, containing *As–S-TRC105 in PBS (4.3 MBq, 240 μg TRC105, 2.7 Gbq/μmol). Following injection, serial PET images were obtained at 0.5, 4, and 24 h p.i. using an Inveon microPET scanner (Siemens Medical Solutions USA, Inc., Malvern, PA, USA) at the University of Wisconsin Carbone Cancer Center Small Animal Imaging Facility. The images were reconstructed with an OSEM3D algorithm with neither attenuation nor scatter correction. This labeling and PET imaging study was repeated using 1.0 mg of TRC105 that had been modified to exhibit 8 thiol groups per TRC105-SH molecule using 2-iminothiolane (Traut’s reagent). This TRC105-SH was prepared through a 2 h room temperature incubation with a 1:25 molar ratio of TRC105 to Traut’s reagent followed by SEC purification, as previously described.39 Two healthy nude mice were each injected with 200 μL of SEC-purified, TCEP-reduced, thiol-modified *As–S-TRC105 in PBS (2.7 MBq *As, 160 μg TRC105, 2.5 GBq/μmol), followed by their serial PET imaging at 1 and 18 h p.i. The biodistribution of free *As was determined by injecting three healthy nude mice with 200 μL of 12 MBq *As, 6 mM HEPES, 31 mM HA, 2 mM EDTA in PBS at pH 7.4, followed by serial PET imaging at 0.5, 3, and 27 h p.i. HPGe gamma analysis of each injection solution allowed for accurate quantification of the radioisotopic purity of injected *As, which contained a mixture of positron-emitting radioactivity at injection, including 79% 72As, 9.7% 74As, 2.5% 71As with the remaining 8.9% the electron-emitting 76As. This quantified isotopic information was used in concert with dose calibrator measurements to display PET images in terms of percent injected dose per gram (%ID/g). This additional complication of quantifying and accounting for the presence of multiple positron-emitting radioarsenic isotopes is mitigated through the production of isotopically pure 72As through the use of isotopically enriched 72Ge target material as detailed above.
Radioarsenic Labeling and Evaluation of Dihydroli-poic Acid
Chelation was determined in 0.4 M HEPES, 25 mM EDTA, 0.5 M HA, pH ~7 solutions (300 μL) containing *As(OH)3 isolated from the SPE procedure described above with and without 22 mM natAs(OH)3, and 22 mM DHLA (added in 10 μL CH3CN). The labeling efficacy 10 min and 18 h after mixing was quantified through reversed-phase- (RP-) TLC using octadecyl-functionalized silica stationary phase and 50:50::acetonitrile:20 mM ammonium acetate as mobile phase. Additionally, the DHLA reactivity of 72As(OH)3 was measured by titration by preparing 1 mL labeling solutions containing ~11 MBq of 72As(OH)3, 0.11 M HEPES, 0.5 M HA, 25 mM EDTA, and 10 mM, 1 mM, 100 μM, 10 μM, 1 μM, 100 nM, or 10 nM DHLA (added in 10 μL CH3CN) at pH 7.4. TLC analysis after 18 h allowed for the generation of a plot with moles of DHLA as ordinate and As-DHLA labeling percent as abscissa with the results forming a sigmoidal curve. The inflection point of this curve corresponds to the nDHLA, the moles of DHLA necessary to reach 50% labeling of A72As MBq of 72As. This allows for the calculation of the DHLA effective specific activity (ESADHLA) of the 72As(OH)3 product, with ESADHLA = A72As/(2 · nDHLA).
Radioarsenic Labeling and Evaluation of Thiol-Modified Mesoporous Silica Nanoparticles
MSN were synthesized, functionalized with thiol groups using (3-mercaptopropyl)trimethoxysilane, physically characterized, and analyzed for thiol concentration according to literature porcedures.43 Surface area per nanoparticle was calculated using the Brunauer–Emmett–Teller (BET) surface area and pore volume, the transmission electron microscopy (TEM) radius, and an assumed silica density of 2.2 g/mL according to literature methods.46,47 For labeling, MSN (2.8 mg Si, 180 nmol thiol) were pelletized by centrifugation at 14 krpm for 10 min followed by decantation and reconstituted in 250 μL 0.1 M HEPES and combined with 300 μL of LLE-prepared *As(OH)3, 0.5 M HA, 25 mM EDTA, 0.1 M HEPES at pH 9. The MSN and *As(OH)3 were incubated overnight at room temperature and then labeling efficacy was determined by dose calibrator measurement of the *As–S-MSN pellet and free *As supernatant following centrifugation/decantation. The *As–S-MSN pellet was washed twice with 500 μL of water through subsequent sonication-aided reconstitution and centrifugation/decantation with the final pellet reconstituted in 450 μL of PBS. Three healthy nude mice were each injected with 90–150 μL *As–S-MSN (2.5–4.4 MBq *As, 0.6–1.0 mg Si) in PBS, followed by their serial PET imaging at 0.5 h, 6 h, 1 day, 3 days, 5 days, and 7 days, p.i. as described above.
Acknowledgments
Portions of this work were supported by the University of Wisconsin - Madison, U.S. Department of Energy under Grant DE-SC0005281 and DE-SC0008384, National Institutes of Health under Grant T32CA009206 and 1R0-1CA169365, the American Cancer Society under Grant 125246-RSG-13-099-01-CCE, and the University of Wisconsin Carbone Cancer Center Support Grant P30 CA014520.
Footnotes
Notes
The authors declare the following competing financial interest(s): Charles P. Theuer is an employee of TRACON Pharmaceuticals.
References
- 1.Kelkar SS, Reineke TM. Theranostics: Combining Imaging and Therapy. Bioconjugate Chem. 2011;22:1879–1903. doi: 10.1021/bc200151q. [DOI] [PubMed] [Google Scholar]
- 2.Werner RA, Bluemel C, Allen-Auerbach MS, Higuchi T, Herrmann K. 68Gallium- and 90Yttrium-/177Lutetium: “theranostic twins” for diagnostic and treatment of NETs. Ann Nucl Med. 2015;29:1–7. doi: 10.1007/s12149-014-0898-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silberstein EB. Radioiodine: The Classic Theranostic Agent. Semin Nucl Med. 2012;42:164–170. doi: 10.1053/j.semnuclmed.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 4.Ballard B, Wycoff D, Birnbaum ER, John KD, Lenz JW, Jurisson SS, Cutler CS, Nortier FM, Taylor WA, Fassbender ME. Selenium-72 formation via natBr(p,x) induced by 100 MeV Protons: Steps toward a novel 72Se/72As generator system. Appl Radiat Isot. 2012;70:595–601. doi: 10.1016/j.apradiso.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Chajduk E, Doner K, Polkowska-Motrenko H, Bilewicz A. Novel radiochemical separation of arsenic from selenium for 72Se/72As generator. Appl Radiat Isot. 2012;70:819–822. doi: 10.1016/j.apradiso.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 6.Bokhari TH, Mushtaq A, Khan IU. Separation of no-carrier-added arsenic-77 from neutron irradiated germanium. Radiochim Acta. 2009;97:503–506. [Google Scholar]
- 7.Zhu J, Chen Z, Lallemand-Breitenbach V, de Thé H. How acute promyelocytic leukaemia revived arsenic. Nat Rev Cancer. 2002;2:705–713. doi: 10.1038/nrc887. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci U S A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dilda PJ, Hogg PJ. Arsenical-based cancer drugs. Cancer Treat Rev. 2007;33:542–564. doi: 10.1016/j.ctrv.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Swindell EP, Hankins PL, Chen H, Miodragović ĐU, O’Halloran TV. Anticancer Activity of Small-Molecule and Nanoparticulate Arsenic(III) Complexes. Inorg Chem. 2013;52:12292–12304. doi: 10.1021/ic401211u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Billinghurst MW, Abrams DN, Cantor S. Separation of Radioarsenic from a Germanium Dioxide Target. Appl Radiat Isot. 1990;41:501–507. [Google Scholar]
- 12.Tolmachev V, Lundqvist H. Separation of arsenic from germanium oxide targets by dry distillation. J Radioanal Nucl Chem. 2001;247:61–66. [Google Scholar]
- 13.Jennewein M, Qaim SM, Hermanne A, Jahn M, Tsyganov E, Slavine N, Seliounine S, Antich PA, Kulkarni PV, Thorpe PE, et al. A new method for radiochemical separation of arsenic from irradiated germanium oxide. Appl Radiat Isot. 2005;63:343–351. doi: 10.1016/j.apradiso.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 14.Shehata MM, Scholten B, Spahn I, Coenen HH, Qaim SM. Separation of radiarsenic from irradiated germanium oxide targets for the production of 71As and 72As. J Radioanal Nucl Chem. 2011;287:435–442. [Google Scholar]
- 15.Green M, Kafalas JA. Preparation and Isolation of Carrier Free As-74 from Germanium Cyclotron Targets. J Chem Phys. 1954;22:760. [Google Scholar]
- 16.Gruverman IJ, Kruger P. Cyclotron-produced Carrier-free Radioisotopes. Int J Appl Radiat Isot. 1959;5:21–31. doi: 10.1016/0020-708x(59)90347-3. [DOI] [PubMed] [Google Scholar]
- 17.Saito K, Ikeda S, Saito M. Separation of Radioactive Arsenic from Germanium Irradiated with Protons. Bull Chem Soc Jpn. 1960;33:884–887. [Google Scholar]
- 18.Guin R, Das SK, Saha SK. Separation of carrier-free arsenic from germanium. J Radioanal Nucl Chem. 1998;227:181–182. [Google Scholar]
- 19.Jahn M, Radchenko V, Filosofov D, Hauser H, Eisenhut M, Rösch F, Jennewein M. Separation and purification of no-carrier-added arsenic from bulk amounts of germanium for use in radiopharmaceutical labeling. Radiochim Acta. 2010;98:807–812. [Google Scholar]
- 20.Hasegawa R, Kurosawa T, Yagihashi T. Hydrogen Reduction of Germanium Dioxide. Trans Jpn Inst Met. 1972;13:39–44. [Google Scholar]
- 21.Wieland BW, Bida GT, Hendry GO, Padgett HC. Device and process for the production of nitrogen-13 ammonium ions using a high pressure target containing a dilute solution of ethanol in water. US Patent. 1994;5:345, 477. [Google Scholar]
- 22.Kraus KA, Nelson F. Anion Exchange Studies of the Fission Products. Proc Int Conf Peaceful Uses At Energy. 1956;7:113–125. [Google Scholar]
- 23.Nelson F, Kraus KA. Anion-exchange studies. XVIII Germanium and Arsenic in HCl Solutions. J Am Chem Soc. 1955;77:4508–4509. [Google Scholar]
- 24.Jennewein M, Lewis MA, Zhao D, Tsyganov E, Slavine N, He J, Watkins L, Kodibagkar VD, O’Kelly S, Kulkarni P, et al. Vascular Imaging of Solid Tumors in Rats with a Radioactive Arsenic-Labeled Antibody that Binds Exposed Phosphatidylserine. Clin Cancer Res. 2008;14:1377–1385. doi: 10.1158/1078-0432.CCR-07-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD, Spencer S, Adams BJ, Alvarez D, Seon BK, et al. A Phase 1 First-in-Human Study of TRC105 (Anti-Endoglin Antibody) in Patients with Advanced Cancer. Clin Cancer Res. 2012;18:4820–4829. doi: 10.1158/1078-0432.CCR-12-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gordon MS, Robert F, Matei D, Mendelson DS, Goldman JW, Chiorean EG, Strother RM, Seon BK, Figg WD, Peer CJ, et al. An Open-Label Phase Ib Dose-Excalation STudy of TRC105 (Anti-Endoglin Antibody) with Bevacizumab in Patients with Advanced Cancer. Clin Cancer Res. 2014;20:5918–5926. doi: 10.1158/1078-0432.CCR-14-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karzai FH, Apolo AB, Cao L, Madan RA, Adelberg DE, Parnes H, McLeod DG, Harold N, Peer C, Yu Y, et al. A phase I study of TRC105 anti-endoglin (CD105) antibody in metastatic castration-resistant prostate cancer. BJU Int. 2015;116:546–555. doi: 10.1111/bju.12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong H, Yang Y, Zhang Y, Engle JW, Barnhart TE, Nickles RJ, Leigh BR, Cai W. Positron emission tomography imaging of CD105 expression during tumor angiogenesis. Eur J Nucl Med Mol Imaging. 2011;38:1335–1343. doi: 10.1007/s00259-011-1765-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong H, Severin GW, Yang Y, Engle JW, Zhang Y, Barnhart TE, Liu G, Leigh BR, Nickles RJ, Cai W. Positron emission tomography imaging of CD105 expression with 89Zr-Df-TRC105. Eur J Nucl Med Mol Imaging. 2012;39:138–148. doi: 10.1007/s00259-011-1930-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bläuenstein P, Locher JT, Seybold K, Koprivova H, Janoki GA, Mäcke HR, Hasler P, Ammann A, Novak-Hofer I, Smith A, et al. Experience with the iodine-123 and technetium-99m labelled anti-granulocyte antibody MAb47: a comparison of labelling methods. Eur J Nucl Med. 1995;22:690–698. doi: 10.1007/BF01254573. [DOI] [PubMed] [Google Scholar]
- 31.Shooshtary S, Behtash S, Nafisi S. Arsenic trioxide binding to serum proteins. J Photochem Photobiol, B. 2015;148:31–36. doi: 10.1016/j.jphotobiol.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 32.Rey NA, Howarth OW, Pereira-Maia EC. Equilibrium characterization of the As(III)-cysteine and the As(III)-glutathione systems in aqueous solution. J Inorg Biochem. 2004;98:1151–1159. doi: 10.1016/j.jinorgbio.2004.03.010. [DOI] [PubMed] [Google Scholar]
- 33.Spuches AM, Kruszyna HG, Rich AM, Wilcox DE. Thermodynamics of the As(III)–Thiol Interaction: Arsenite and Monomethylarsenite Complexes with Glutathione, Dihydrolipoic Acid, and Other Thiol Ligands. Inorg Chem. 2005;44:2964–2972. doi: 10.1021/ic048694q. [DOI] [PubMed] [Google Scholar]
- 34.Kumar A, Hao G, Liu L, Ramezani S, Hsieh JT, Öz OK, Sun X. Click-Chemistry Strategy for Labeling Antibodies with Copper-64 via a Cross-Bridged Tetraazamacrocyclic Chelator Scaffold. Bioconjugate Chem. 2015;26:782–789. doi: 10.1021/acs.bioconjchem.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Price EW, Orvig C. Matching chelators to radiometals for radiopharmaceuticals. Chem Soc Rev. 2014;43:260–290. doi: 10.1039/c3cs60304k. [DOI] [PubMed] [Google Scholar]
- 36.DeGraffenreid AJ, Cutler CS, Barnes C, Jurisson SS. Synthesis of tridentate ligand: Potential theranostic application of a radioarsenic trithiol complex. Nucl Med Biol. 2014;41:629. [Google Scholar]
- 37.DeGraffenreid A, Feng Y, Ketring AR, Barnes C, Cutler CS, Jurisson SS. Synthesis, Characterization, and Radiolabelling of Trithiol Ligands with Arsenic-77. J Labelled Compd Radiopharm. 2015;58:S58. [Google Scholar]
- 38.Packer L, Witt EH, Tritschler HJ. Alpha-Lipoic Acid as a Biological Antioxidant. Free Radical Biol Med. 1995;19:227–250. doi: 10.1016/0891-5849(95)00017-r. [DOI] [PubMed] [Google Scholar]
- 39.Hong H, Yang K, Zhang Y, Engle JW, Feng L, Yang Y, Nayak TR, Goel S, Bean J, Theuer CP, et al. In Vivo Targeting and Imaging of Tumor Vasculature with Radiolabeled Antibody-Conjugated Nanographene. ACS Nano. 2012;6:2361–2370. doi: 10.1021/nn204625e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Herth MM, Barz M, Jahn M, Zentel R, Rösch F. 72/74As-labeling of HPMA based polymers for long-term in vivo PET imaging. Bioorg Med Chem Lett. 2010;20:5454–5458. doi: 10.1016/j.bmcl.2010.07.092. [DOI] [PubMed] [Google Scholar]
- 41.Chen F, Ellison PA, Lewis CM, Hong H, Zhang Y, Shi S, Hernandez R, Meyerand ME, Barnhart TE, Cai W. Chelator-Free Synthesis of a Dual-Modality PET/MRI Agent. Angew Chem, Int Ed. 2013;52:13319–13323. doi: 10.1002/anie.201306306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang P, Gai S, Lin J. Functionalized mesoporous silica materials for controlled drug delivery. Chem Soc Rev. 2012;41:3679–3698. doi: 10.1039/c2cs15308d. [DOI] [PubMed] [Google Scholar]
- 43.Chen F, Hong H, Zhang Y, Valdovinos HF, Shi S, Kwon GS, Theuer CP, Barnhart TE, Cai W. In Vivo Tumor Targeted and Image-Guided Drug Delivery with Antibody-Conjugated, Radiolabeled, Mesoporous Silica Nanoparticles. ACS Nano. 2013;7:9027–9039. doi: 10.1021/nn403617j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen F, Goel S, Valdovinos HF, Luo H, Hernandez R, Barnhart TE, Cai W. In Vivo Integrity and Biological Fate of Chelator-Free Zirconium-89-Labeled Mesoporous Silica Nanoparticles. ACS Nano. 2015;9:7950–7959. doi: 10.1021/acsnano.5b00526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellison PA, Barnhart TE, Engle JW, Nickles RJ, DeJesus OT. Production and chemical isolation procedure of positron-emitting isotopes of arsenic for environmental and medical applications. AIP Conf Proc. 2012;1509:135–140. [Google Scholar]
- 46.Pan L, He Q, Liu J, Chen Y, Ma M, Zhang L, Shi J. Nuclear-Targeted Drug Delivery of Tat Peptide-Conjugated Monodisperse Mesoporous Silica Nanoparticles. J Am Chem Soc. 2012;134:5722–5725. doi: 10.1021/ja211035w. [DOI] [PubMed] [Google Scholar]
- 47.Lin YS, Haynes CL. Impacts of Mesoporous Silica Nanoparticle Size, Pore Ordering, and Pore Integrity on Hemolytic Activity. J Am Chem Soc. 2010;132:4834–4842. doi: 10.1021/ja910846q. [DOI] [PubMed] [Google Scholar]