

Abstract
Alphaviruses are a family of positive-strand RNA viruses that circulate on all continents between mosquito vectors and vertebrate hosts. Despite a significant public health threat, their biology is not sufficiently investigated, and the mechanisms of alphavirus replication and virus-host interaction are insufficiently understood. In this study, we have applied a variety of experimental systems to further understand the mechanism by which infected cells detect replicating alphaviruses. Our new data strongly suggest that activation of the antiviral response by alphavirus-infected cells is determined by the integrity of viral genes encoding proteins with nuclear functions, and by the presence of two cellular pattern recognition receptors (PRRs), RIG-I and MDA5. No type I IFN response is induced in their absence. The presence of either of these PRRs is sufficient for detecting virus replication. However, type I IFN activation in response to pathogenic alphaviruses depends on the basal levels of RIG-I or MDA5.
Keywords: Alphaviruses, virus-host interactions, innate immunity, pattern recognition receptors, RIG-I, MDA5, type I interferon
INTRODUCTION
The Alphavirus genus in the Togaviridae family contains a wide variety of important human and animal pathogens (Strauss and Strauss, 1994b), which represent a public health threat on all continents. These viruses are transmitted by mosquito vectors between vertebrate hosts (Brown and Condreay, 1986), in which they cause diseases of varying symptoms and severity (Strauss and Strauss, 1994a). Some of the Old World (OW) alphaviruses, such as Sindbis virus (SINV), induce rash, fever and polyarthritis. For chikungunya virus (CHIKV) (Halstead et al., 1969a; Halstead et al., 1969b; Rao, 1966), polyarthritis is associated with excruciating pain and can continue for months (Griffin, 2001). The New World (NW) alphaviruses, represented by Venezuelan (VEEV), eastern (EEEV) and western (WEEV) equine encephalitis viruses, circulate in Central, South and North Americas. These viruses, and VEEV in particular, cause periodic, extensive equine epizootics and epidemics of highly debilitating diseases in humans, which often lead to encephalitis with frequent lethal outcome and neurological sequelae (Dal Canto and Rabinowitz, 1981). Importantly, VEEV is also recognized as a potential biological warfare agent due to the ease with which it can be cultured and aerosolized.
One of the most important characteristics of alphaviruses is their replication to very high titers in cell culture and rapid development of viremia in vivo. The robust replication and infection spread are not only the results of highly efficient synthesis of virus-specific RNA and proteins, but are also determined by the ability of alphaviruses to efficiently interfere with the development of the innate immune response. The hallmark of the innate immune response is the induction of type I interferons (IFN), which functions both in para- and autocrine modes (Katze et al., 2002; Sen, 2001; Stark et al., 1998). The IFN response can affect already established virus replication and also activates the antiviral state in as yet uninfected cells.
Replication of the alphavirus genomes proceeds through synthesis of double-stranded RNA intermediates (Frolova et al., 2010; Gorchakov et al., 2008a), which represent common virus-specific pathogen-associated molecular patterns (PAMP). The dsRNAs can be efficiently detected by cellular pattern recognition receptors (PRRs), such as RIG-I, MDA5 or TLR3 (Habjan and Pichlmair, 2015; Loo and Gale, 2011; Pichlmair and Reis e Sousa, 2007). Interaction of the indicated receptors with dsRNA leads to activation of the IRF3-dependent signaling pathways and ultimately, type I IFN induction. However, in alphavirus-infected cells, the dsRNA are isolated into membrane spherules at the plasma and endosomal membranes, which are connected with cytoplasm by very narrow necks (Frolova et al., 2010; Froshauer et al., 1988; Gorchakov et al., 2008b). Thus, RNAs in spherule cavities are likely to be poorly accessible to cellular PRRs. Moreover, alphaviruses have developed additional efficient means of interfering with the antiviral response. The OW alphaviruses employ their nonstructural protein nsP2 to induce rapid degradation of Rpb1, the catalytic subunit of cellular DNA-dependent RNA polymerase II. In cultured cells of vertebrate origin, this very efficient Rpb1 degradation leads to shutoff of cellular transcription within a few hours post infection (Akhrymuk et al., 2012). The NW alphaviruses utilize their capsid protein, but not nsP2, to block nucleocytoplasmic trafficking, and this also results in rapid transcription inhibition (Atasheva et al., 2010a; Atasheva et al., 2008). Thus, replication of both groups of alphaviruses induces the same phenomenon, transcriptional shutoff, which prevents activation of type I IFN (Garmashova et al., 2007b). Moreover, this transcription inhibition interferes with activation of interferon-stimulated genes (ISGs) even if cells are treated with IFN after the beginning of virus replication (Frolov et al., 2012).
Previous studies with VEEV and SINV viruses did not provide compelling evidence that these two representative members of the NW and the OW alphaviruses are capable of inducing type I IFN response in cultured cells: SINV usually induced no detectable levels of IFN in commonly used cell lines (Frolova et al., 2002), and VEEV was able to induce only low levels of type I IFN and only at late times post infection (Atasheva et al., 2012; Atasheva et al., 2010b). Thus, the data suggested that inhibition of cellular transcription and translation by alphaviruses in cultured cells (Gorchakov et al., 2005) controls the development of the antiviral response even if cellular receptors are capable of detecting viral dsRNA. However, despite demonstrating profound IFN inhibitory functions during replication in vitro, many alphaviruses efficiently induce type I IFN in small animal models, although the level of IFN induction depends on the virus and mouse strain involved (Aguilar et al., 2005; Cruz et al., 2010; Gardner et al., 2008; Gardner et al., 2011; Gardner et al., 2009). Mice with defects in type I IFN signaling succumb to alphavirus infections dramatically faster (White et al., 2001). These data suggest that there is a discrepancy between the data generated in the in vitro and in vivo experimental systems. Thus, in spite of expressing proteins with strong transcription inhibitory functions, alphaviruses do not always downregulate the innate immune response to undetectable levels. However, so far, it remains unclear which particular PRR or combination of PRRs is involved in sensing of alphaviruses.
The currently available data regarding activation of the innate immune response by different alphaviruses are very fragmented and highly contradictory. They have mostly been generated using fibroblasts derived from RIG-I−/− or MDA5−/− mice of different genetic backgrounds or MEFs with incomplete knockdown of these proteins by siRNAs. Based on these data, the role of different RLRs (RIG-I like receptors) seems to depend on the specific virus, genetic background of the mice and the applied experimental approach. SINV and SFV have been observed to induce reduced levels of IFN production in MDA5 KO, but not RIG-I KO or KD cells (Burke et al., 2009; Pichlmair et al., 2009; Schulz et al., 2010). However, the effect was very small. Induction of type I IFN by CHIKV in primary RIG-I or MDA5 KO fibroblasts was noted to be dependent on the genetic background of the mice, and RIG-I appears to be a stronger inducer (Schilte et al., 2010). Information about the roles of RLRs in the induction of response to the NW alphaviruses (VEEV, EEEV and WEEV) is also insufficient. Thus, to date, it remains unclear whether RIG-I or MDA5 or both proteins are involved in primary sensing of alphavirus replication.
In the new study, we have applied a variety of experimental systems to demonstrate that isolation of alphavirus dsRNAs into membrane spherules is likely incomplete, and replication remains detectable by the cytoplasmic receptors. Two cellular PRRs, RIG-I and MDA5 were found to play equally important roles in the induction of the primary anti-alphavirus response within the first hours post infection. Both RIG-I and MDA5 can sense replicating alphaviruses and determine activation of the antiviral defense mechanisms. Their sensing functions are critically dependent on the concentration of both PRRs at the time of infection, and this determines the rates, time and scale of type I IFN induction.
Results
Development of experimental systems for studying the roles of PRRs
Infection of vertebrate cells such as NIH 3T3 cells, which are competent in type I IFN expression and signaling, by VEEV TC-83 or SINV does not lead to efficient induction of IFN-β (Garmashova et al., 2007b). This fact correlates with the ability of these viruses to efficiently inhibit cellular transcription (Garmashova et al., 2007b). Thus, we tested whether noncytopathic variants of VEEV and SINV (VEEV/GFP/Cm and SINV/G/GFP) (Fig. 1A), which are not capable of inducing transcriptional shutoff, would induce IFN-β in infected cells. VEEV/GFP/Cm encodes a capsid protein variant with mutations in its nuclear localization signal, which abolish its ability to inhibit nucleocytoplasmic traffic (Atasheva et al., 2010a; Atasheva et al., 2010b). SINV/G/GFP contains a previously described, attenuating P726G mutation in nsP2, which affects the ability of nsP2 to induce degradation of the Rpb1 subunit of cellular DNA dependent RNA polymerase II (Akhrymuk et al., 2012; Frolov et al., 1999b). These mutations had either negligible (SINV/G/GFP) or no effect (VEEV/GFP/Cm) on virus replication (Fig. 1B), and so far, there is no experimental evidence that either formation of replication complexes or their compartmentalization was affected. However, these small modifications in the nsP2 or capsid genes of SINV and VEEV, respectively, transformed the viruses into very potent type I IFN inducers in NIH 3T3 and other cell lines with intact IFN signaling (Fig. 1B). This is a strong indication that replication of VEEV and SINV generates PAMPs, which can be recognized by cellular PRRs. Thus, VEEV/GFP/Cm and SINV/G/GFP mutants, which demonstrate no nuclear functions, but have no defects in other aspects of virus replication, can be used as an experimental system for identification and studying the functions of specific PRRs in sensing alphavirus replication. The results also suggested that NIH 3T3 cells contain fully functional PRRs, which are capable of sensing replicating VEEV and SINV, and activation of type I IFN expression.
Fig. 1.

Alphaviruses deficient in nuclear functions, but not their wt variants, induce high levels of IFN-β. (A) The schematic representation of alphavirus genomes encoding wt and mutated nsP2- and capsid-coding genes (SINV/G/GFP and VEEV/GFP/Cm, respectively). (B) NIH 3T3 cells were infected with the indicated viruses at an MOI of 20 PFU/cell. Media were harvested at 16 h post infection and used for assessment of both virus titers and IFN-β levels. Means of three biological repeats with SD are presented.
In the last number of years, the cytoplasmic DExD/H box helicase family members, RIG-I and MDA5, have been shown to be critical determinants of the development of the innate immune response against numerous RNA viruses (Brubaker et al., 2015; Sparrer and Gack, 2015; Weber and Weber, 2014; Wilkins and Gale, 2010). These proteins, termed RIG-I-like receptors, are the primary sensors of virus-specific dsRNA ligands, and their activation initiates signaling cascades through the MAVS adaptor, which ultimately induce type I IFN expression. It has been also demonstrated that cell lacking MAVS failed to release IFN-β in response to chikungunya virus, CHIKV (Schilte et al., 2010). Therefore, we next focused on defining a particular sensor, which is involved in alphavirus detection. NIH 3T3 cells, which were used in this study, express both cytosolic PRRs, RIG-I and MDA5. Thus, we have developed stable cell lines, in which RIG-I (KD RIG-I) or MDA5 (KD MDA5) expression was strongly downregulated by specific shRNAs. In the parental NIH 3T3 cells, both RIG-I and MDA5 proteins were present at low levels and were poorly detectable on Western blot by commercially available antibodies. Therefore, downregulation of RIG-I and MDA5 expression in KD cells was evaluated after treatment of KD and the parental NIH 3T3 cells with IFN-β (500 IU/ml) for 24 h (Fig. 2A). The efficiency of RIG-I or MDA5 knockdown was determined by Western blot and clones that demonstrated reduction of RIG-I or MDA5 protein expression by more than 90% were selected for this study (Fig. 2A). The residual expression of the knocked down genes in IFN-β-treated cells was examined by RT-qPCR (Fig. 2B). Both tests demonstrated that expression of RIG-I and MDA5 genes was reduced by more than 10-fold, and their products were almost undetectable even after IFN treatment. We have also identified a clone expressing shRNA against RIG-I, in which expression of MDA5 was not detectable due to an unknown defect. This clone was designated as a double knockdown cell line, dKD (Figs. 2A and B).
Fig. 2.

RIG-I and MDA5 expression is strongly inhibited in the selected KD cell lines. (A) Equal numbers of NIH 3T3 cells and indicated clone-derived cell lines were treated with mouse IFN-β (500 IU/ml) for 24 h or remained untreated. Cell lysates, corresponding to equal numbers of cells were analyzed by SDS-PAGE, followed by Western blot using MDA5- and RIG-I-specific, and tubulin-specific antibodies in conjunction with infrared dye-labeled secondary antibodies. Membranes were scanned on a LI-COR imager. (B) The indicated clone-derived cell lines were treated with mouse IFN-β (500 IU/ml) for 24 h. RNAs were isolated, and relative concentrations of RIG-I- and MDA5-specific mRNAs were determined by RT-qPCR as described in the Materials and Methods.
Knockdown of a single PRR does not have a deleterious effect on IFN-β induction
The developed cell lines were further used for identification of a murine RLR capable of detecting alphavirus infection and inducing a type I IFN response. The KD RIG-I, KD MDA5, dKD and the parental NIH 3T3 cells were infected with VEEV/GFP/Cm and SINV/G/GFP viruses. The results presented in Fig. 3A demonstrate that upon infection, both single KD cell lines (KD RIG-I and KD MDA5) were still capable of IFN-β expression to levels comparable to those detected in infected NIH 3T3 cells. In contrast, the dKD cells completely lost the ability to respond to replication of both mutant viruses. At all times post infection, IFN-β was undetectable in the media, despite sensitivity of the assay was ~1 pg/ml. Similarly, in the control experiments, in contrast to the parental NIH 3T3, the dKD cells demonstrated no ability to respond to poly(I:C) delivered directly into the cytoplasm by different transfection reagents (Fig. 3B). Both NIH 3T3 and the KD cells failed to produce IFN after poly(I:C) treatment in the absence of transfection reagent, indicating that TLR3-mediated signaling is not activated in the NIH 3T3 cells.
Fig. 3.

Single PRR KD, but not dKD, cells respond to alphavirus replication by IFN-β release. (A) Parental NIH 3T3, KD RIG-I, KD MDA5 and dKD cells were infected with SINV/G/GFP and VEEV/GFP/Cm viruses at an MOI of 20 PFU/cell. Media was replaced at 24 h post infection and harvested again at 48 h post infection. Concentration of IFN-β was assessed as described in the Materials and methods. Sensitivity of the assay was ~1 pg/ml. Means of three biological repeats with SD are presented. (B) NIH 3T3 and dKD cells were transfected with poly(I:C) using the indicated transfection reagents or treated with poly(I:C)-containing media as described in the Materials and Methods. Media were harvested at 18 h post transfection, and concentration of IFN-β was determined as indicated above. This experiment was repeated twice with reproducible results. (C) NIH 3T3 and dKD cells were infected with SINV/G/GFP and VEEV/GFP/Cm viruses at an MOI of 20 PFU/cell for 8 h or treated with IFN-β at a concentration of 500 IU/ml for 30 min. Cell lysates were analyzed by Western blot using p-STAT1-, STAT1-, GFP- and β-tubulin-specific antibodies.
In our previous study, we have demonstrated that even very low doses of IFN-β, below 1 IU/ml, could play critical roles in development of the antiviral state (Frolov et al., 2012). Such low concentrations of IFN-β cannot be conclusively detected by ELISA or by standard biological tests, but the sensitivity of IFN-β detection could be increased by assessing the levels of phosphorylated STAT1. Phosphorylation of STAT1 is readily detectable in the presence of IFN-β in the media at concentrations of 0.1 IU/ml and even lower (Frolov et al., 2012). Nevertheless, even in this test, the dKD cells demonstrated no detectable levels of p-STAT1 at 8 h (Fig. 3C) or 24 h (data not shown) post infection with the indicated viruses. Importantly, IFN-β treatment induced phosphorylation of STAT1 in dKD cells suggesting that, at least the early steps in type I IFN signaling remained functional (Fig. 3C).
Taken together, the results of these experiments suggest that only knock down of both RIG-I and MDA5 expression simultaneously, but not inhibition of expression of either gene in isolation, makes NIH 3T3 cells completely incapable of IFN-β induction in response to alphavirus replication. Moreover, RIG-I and MDA5 appear to be the only PRRs capable of alphavirus sensing in this experimental system, and other receptors, such as PKR and TLR3 do not play a significant role in activation of IFN-β expression during alphavirus replication.
RIG-I and MDA5 expression determines spread of alphaviruses and their clearance
To further demonstrate that RIG-I and MDA5 proteins have redundant functions, and that each of these two RLRs is efficient in detecting alphaviruses, we i) compared the abilities of the mutant viruses to develop spreading infections in NIH 3T3, KD RIG-I, KD MDA5 and dKD cells and ii) assessed the ability of these cell lines to clear their replication.
The indicated cell lines were infected at the same low MOI by VEEV/GFP/Cm or SINV/G/GFP, and we evaluated the infection spread by comparing the sizes of foci of GFP-positive cells formed under agarose cover (Fig. 4A). The parental NIH 3T3 cells normally respond to replication of VEEV/GFP/Cm and SINV/G/GFP mutants by efficient release of IFN-β (Fig. 1) (Atasheva et al., 2010b), which rapidly activates an antiviral state in the surrounding and as yet uninfected cells, and makes them resistant to the next rounds of infection. Thus, in the NIH 3T3 cells, both mutants formed only small foci of GFP-positive cells (Fig. 4A). In the dKD cell line, both viruses demonstrated more efficient spread and formed very large GFP-positive foci, indicating that cells remained fully susceptible to subsequent rounds of infection (Fig. 4A). The single KD cells produced foci of a size similar to that in the original NIH 3T3 cells. However, the foci developed by both viruses in KD RIG-I cells were noticeably larger than those in the KD MDA5 cell line. Their increased size did not correlate with higher levels of IFN-β release by these cells in response to virus replication at 24 h post infection (Fig. 3A). These data suggested that the effect of type I IFN induction on infection spread/foci size was determined by difference in kinetics of IFN release rather than by the magnitude of the response. The experiments presented in the following sections support the hypothesis that both RIG-I and MDA5 induce IFN-β with different kinetics in response to alphavirus replication.
Fig. 4.

Single PRR KD cells are capable of both downregulating the spread of mutant alphaviruses and inhibiting already established virus replication. (A) NIH 3T3, KD RIG-I, KD MDA5 and dKD cells were seeded into 6-well Costar plates at a concentration of 5×105 cells per well. SINV/G/GFP and VEEV/GFP/Cm virus stocks were serially diluted and used for infection of the indicated cells with different numbers of infectious units. After 1-h-long incubation at 37°C, the virus-containing media were replaced with 2 ml of media supplemented with 0.6% agarose. After incubation at 37°C for 48 h, cells were fixed, and plates were scanned on a Typhoon imager to enumerate and assess foci of GFP-expressing cells. Images represent wells infected with the same numbers of infectious units. (B) Subconfluent monolayers of NIH 3T3, KD RIG-I, KD MDA5 and dKD cells were infected with SINV/G/GFP and VEEV/GFP/Cm at an MOI of 20 PFU/cell. Media were replaced every day and cells were split upon reaching confluency, usually every 24 hours. Virus titers in harvested media were determined by plaque assay on BHK-21 cells. Dashed lines represent the limits of detection. This experiment was repeated twice with similar results.
In parallel experiments, the NIH 3T3 cells and indicated KD cell lines were infected at high MOI in liquid media with VEEV/GFP/Cm and SINV/G/GFP, and virus release was examined for 10 days (Fig. 4B). At 24 h post infection, all of the cells expressed GFP, indicative of intracellular virus replication. NIH 3T3 and single KD cells infected with mutant viruses, released high concentrations of IFN-β (data not shown and see Fig. 3), and within 6–8 days post infection, reduced virus replication to undetectable levels without CPE development. Both VEEV/GFP/Cm and SINV/G/GFP variants persistently replicated in dKD cells for the duration of the experiments, in a similar fashion to what we previously described for STAT1−/− and IFN-α/βR−/− cells (Atasheva et al., 2010b) (Fig. 4B). This persistent replication correlated with the inability of the dKD cells to respond to infection by IFN-β expression and mount the antiviral state resulting from the autocrine effect of IFN (Fig. 3).
Thus, the data demonstrated that dKD cells did not respond to virus replication with type I IFN release and apparently could no longer sense infection caused by VEEV and SINV noncytopathic mutants. However, these data did not necessarily mean that this was the only defect. Other steps in activation of the antiviral response and ISG activation may have been compromised in the clonal cell lines. Therefore, we evaluated their ability to develop the antiviral state upon IFN-β treatment. First, the parental NIH 3T3, KD RIG-I and dKD cells were treated with 500 IU/ml of mouse IFN-β for 20 h or mock treated, and then infected with SINV/GFP, which encodes wt nsP2 (Fig. 5A). This virus replicated equally efficiently in all of the mock-treated cells, and it was unable to replicate in either IFN-treated cell line. In a complementing experiment, dKD cells were infected with VEEV/GFP/Cm and at 5 days post infection, after establishment of persistent replication, they were further incubated in medium supplemented with IFN-β at a concentration of 1,000 IU/ml. After 5 days of continuous IFN treatment, virus release was no longer detected and cells expressed no virus-encoded GFP, indicative of virus clearance (Fig. 5B). Thus, taken together, the experimental data demonstrate that despite the fact that dKD cells were deficient in sensing alphavirus infection and developing the type I IFN response, they remained fully competent in activation of the antiviral state in response to IFN-β treatment. The IFN pre-treatment made them resistant to alphavirus replication, and presence of IFN-β in the media made dKD cells capable of clearing replication of the attenuated virus.
Fig. 5.

Single KD and dKD cells remain fully capable of responding to IFN-β treatment and activating the antiviral state. (A) NIH 3T3, KD RIG-I and dKD cells were treated with IFN-β at a concentration of 500 IU/ml for 20 h or mock-treated and then infected with SINV/GFP at an MOI of 5 PFU/cell. Media were replaced at the indicated time points and virus titers were determined by plaque assay on BHK-21 cells. (B) dKD cells were infected with the VEEV/GFP/Cm mutant at an MOI of 5 PFU/cell and incubated for 5 days with periodic splitting. After 5 days, the persistently infected cells were either treated with IFN-β at concentration of 1000 IU/ml or mock treated. Titers of released virus were assessed by plaque assay on BHK-21 cells. The arrow indicates the beginning of IFN treatment. The dashed line indicates the limit of detection.
Higher levels of either RIG-I or MDA5 make cells more efficient in IFN-β expression, in response to alphavirus replication
The experiments described above demonstrate that both RIG-I and MDA5 are capable of detecting replicating mutant alphaviruses and activating IFN-β expression. Moreover, they also appear to be the only alphavirus-specific PRRs, at least in NIH 3T3 cells, which activate the IFN-β response to replication of both SINV and VEEV mutants. However, in contrast to the VEEV and SINV variants used in those experiments, wt VEEV TC-83 and SINV induce very low or no IFN production, respectively, in NIH 3T3 (Fig. 1B) and other continuous cell lines (data not shown). On the other hand, infected mice respond with high levels of IFN to infection with the same viruses (Frolova et al., 2002; White et al., 2001). One of the possible explanations for such a discrepancy between the in vivo and in vitro data is that permissive cell lines, which are used in general research, express very low basal levels of RLRs. Indeed, in the NIH 3T3 cells and immortalized MEFs, we were able to detect RIG-I and MDA5 by Western blotting only after their treatment with IFN-β (Fig. 6A). Thus, we hypothesized that higher basal concentration of RLRs could lead to earlier and more efficient type I IFN induction upon infection even with wt viruses.
Fig. 6.

Ectopic expression of RIG-I or MDA5 in dKD cells restores their ability to rapidly respond to replication of alphavirus mutants. (A) Comparative levels of RIG-I and MDA5 expression in the parental NIH 3T3 cells and MEFs, treated with IFN-β at a concentration of 500 IU/ml for 24 h, and in selected clones of KI cells. (B) NIH 3T3, dKD, KI RIG-I and KI MDA5 cells were infected with VEEV/GFP/Cm and SINV/G/GFP viruses at an MOI of 20 PFU/cell. Concentrations of released IFN-β were assessed at the indicated times post infection. Means of three biological repeats with SD are presented. (C) The NIH 3T3, KI RIG-I, KI MDA5 and dKD cells were seeded into 6-well Costar plates at a concentration of 5×105 cells per well. SINV/G/GFP and VEEV/GFP/Cm virus stocks were serially diluted and used for infection of indicated cells with different numbers of infectious units. After 1 h incubation at 37°C the virus-containing media were replaced with 2 ml of media supplemented with 0.6% agarose. After incubation at 37°C for 48 h, cells were fixed and plates were scanned on a Typhoon imager to enumerate and assess foci of GFP-expressing cells. Images represent wells infected with the same numbers of infectious units. (D) NIH 3T3, dKD, KI RIG-I and KI MDA5 cells were infected with VEEV/GFP/Cm at an MOI of 20 PFU/cell. Media were replaced every 24 h and cells were split upon reaching confluency in a 1:2 ratio. Virus titers were determined by plaque assay on BHK-21 cells. Dashed line represents the limit of detection. This experiment was repeated twice with similar results.
To experimentally test this possibility, we used the dKD cells to generate stable knock-in cell lines, which expressed RIG-I and MDA5 at levels higher than those detected in the parental NIH 3T3 cells (see Materials and Methods for details). Several clones were analyzed, and we selected clones producing RLRs at levels, which were similar to those detected in IFN-β-treated MEFs (Fig. 6A). The dKD-derived cell lines producing RIG-I or MDA5 were termed KI RIG-I and KI MDA5, respectively.
The single KI cell lines became capable of IFN-β induction in response to replication of SINV/G/GFP and VEEV/GFP/Cm. The MDA5-producing cell lines reproducibly expressed IFN-β earlier than those expressing RIG-I, but to detectably lower final concentrations measured at 24 h post infection (Fig. 6B). The applied viruses became incapable of forming large foci of GFP-positive cells under agarose cover, suggesting that infected cells rapidly released IFN-β, which made the as yet uninfected cells resistant to the next rounds of infection (Fig. 6C). The KI cells also gained back the ability to strongly suppress virus replication (Fig. 6D). At 24 h post infection, all of the cells were infected and demonstrated GFP expression, but within the next 5 days, titers of the released viruses fell below the detection limit. After the dramatic decrease in replication, VEEV/GFP/Cm and SINV/G/GFP were able to develop chronic infection, characterized by low level of IFN-β release and a small percentage of infected cells (Fig. 6D and data not shown). Interestingly, the time required to re-establish persistent virus infection reproducibly differed in parental NIH 3T3, KI RIG-I and KI MDA5 cells. This phenomenon requires further investigation.
We next investigated whether the single KI cells with higher basal levels of PRR expression would become capable of more efficient response to replication of the wild type alphavirus. Indeed, KI MDA5 and KI RIG-I cells were more efficient in IFN-β induction than parental NIH 3T3 cells (Fig. 7A). At all times post infection, KI MDA5 cells reproducibly produced almost 100-fold more INF-β than NIH 3T3 cells (P<0.0001). The difference between NIH 3T3 and KI RIG-I was not so pronounced but statistically significant (5 fold difference at 10 h post infection, P<0.002). We next evaluated the rates of VEEV/GFP replication in liquid media. Cells were infected at an MOI of 20, and 100% were GFP-positive by 6 h post infection. Media was collected at 10 and 24 h post infection, and no significant differences in virus accumulation were found (Fig. 7B). This suggested that the more efficiently released IFN-β could not affect virus replication in the already infected cells due to global transcriptional shutoff induced by replicating viruses. However, the released type I IFN readily established an antiviral state in as yet uninfected cells under agarose cover. In plaque assay, IFN-β released from infected cells, protected the surrounding single KI cells from new rounds of infection by VEEV/GFP, and caused it to form dramatically smaller plaques (Fig. 7C). Similar results were generated on these cells using SINV/GFP, expressing wt nsP2 protein (data not shown). Taken together, these data demonstrate that higher concentrations of either RIG-I or MDA5 at the time of alphavirus infection have a very strong positive impact on type I IFN release and, consequently, on the infection spread.
Fig. 7.
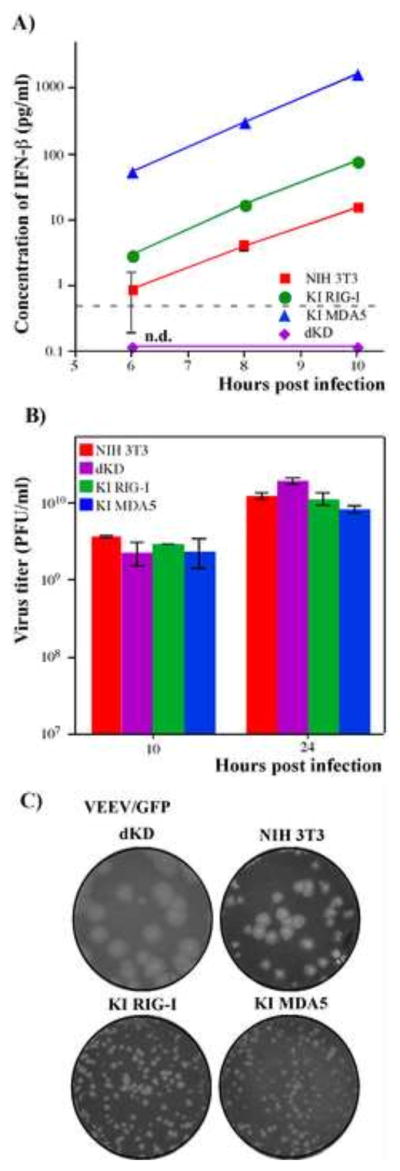
Ectopic expression of RIG-I or MDA5 in dKD cells leads to efficient IFN induction in response to replication of VEEV/GFP, which encodes wt capsid protein. (A) NIH 3T3, dKD, KI RIG-I and KI MDA5 cells were infected with VEEV/GFP at an MOI of 20 PFU/cell, and accumulation of IFN-β in the media was assessed at the indicated time points. Means of three biological repeats with SD are presented. (B) NIH 3T3, KI RIG-I, KI MDA5 and dKD cells were infected with VEEV/GFP at an MOI of 20 PFU/cell. Media were replaced at the indicated time points and titers were determined by plaque assay on BHK-21 cells. Means of three biological repeats with SD are presented. (C) NIH 3T3, KI RIG-I, KI MDA5 and dKD cells were seeded into 6-well Costar plates at a concentration of 5×105 cells per well. VEEV/GFP virus stock was serially diluted and used for infection of indicated cells with different numbers of PFUs. After 1 h incubation at 37°C the virus-containing media were replaced with 2 ml of media supplemented with 0.6% agarose. After incubation at 37°C for 48 h, cells were fixed and stained with Crystal Violet. Images represent wells infected with the same numbers of PFUs.
In the next experiments, we determined whether RIG-I and MDA5 could synergistically function in regulation/inhibition of alphavirus infection. We have developed double KI cells (dKI), which stably express both RIG-I and MDA5. Both proteins were expressed at similar levels to those detected in IFN-β-treated MEFs (Fig. 8A). This cloned cell line and the original dKD cells were compared for their ability to support replication and produce plaques upon infection with different alphaviruses (Fig. 8B). Both VEEV/GFP and SINV/GFP became unable to produce either plaques or GFP-positive foci in dKI cells (Fig. 8B). Surprisingly, CHIKV and SFV, in particular, were detectably more resistant to high levels of RIG-I and MDA5 and remained capable of plaque formation in the dKI cell line (Fig. 8B). This higher resistance of CHIKV and SFV is an interesting phenomenon and will be further investigated. We also investigated the effects of RIG-I and MDA5 on a number of unrelated viruses. VSV was also found to be noticeably sensitive to ectopic expression of both RIG-I and MDA5. In contrast, EMCV produced similar plaques in both dKD and dKI cells.
Fig. 8.

Ectopic, simultaneous expression of both RIG-I and MDA5 in dKD cells (dKI cell line) differentially affects replication of alpha- and other viruses. (A) Comparative levels of RIG-I and MDA5 expression in MEFs, mock-treated or treated with IFN-β at a concentration of 500 IU/ml for 24 h, and in stable dKI cells. (B) The original dKD and stable dKI cells were seeded into 6-well Costar plates at a concentration of 5×105 cells per well. Stocks of the indicated viruses were serially diluted and used for infection of cells with different numbers of PFUs. After 1 h incubation at 37°C the virus-containing media were replaced with 2 ml of media supplemented with 0.6% agarose. After incubation at 37°C for 48 h, cells were fixed and stained with Crystal Violet. Images represent wells infected with the same numbers of PFUs.
Discussion
Alphavirus infection results in synthesis of the negative strand of viral genome, which remains associated with the positive-stranded RNA and is present in the cells as a dsRNA intermediate (Frolova et al., 2010). This dsRNA represents a virus-specific PAMP, which can be sensed by a variety of cellular, dsRNA-specific PRRs, such as MDA5, RIG-I or PKR, and thus, induces signaling pathways that stimulate the antiviral response (Habjan and Pichlmair, 2015; Loo and Gale, 2011). Alphaviruses have developed at least two very efficient means of avoiding activation of the antiviral response during their intracellular replication. First, similarly to other RNA+ viruses, they isolate the synthesized dsRNA intermediates into membrane compartments, termed spherules, which are likely to make them very poorly accessible for the cellular PRRs (Frolova et al., 2010). These membranous, dsRNA-containing structures are initially formed at the plasma membrane. Some of spherules are later transported into the cytoplasm as a component of the endosome and lysosome membranes (Frolova et al., 2010). Spherule necks are also plugged by nsP1–4-containing protein complexes (Frolova et al., 2010), which additionally complicate detection of dsRNA by PRRs. Importantly, both at the plasma and endosome membranes, the dsRNAs in the spherules are not exposed to TLR3, TLR7 or TLR8, which could also mediate activation of the antiviral response by sensing the ds and ssRNAs inside the endosomes.
The ability of alphaviruses to induce global transcriptional shutoff within 6–8 h post infection is another potent mechanism of inhibiting the development of the cellular antiviral response (Akhrymuk et al., 2012; Atasheva et al., 2010a; Garmashova et al., 2007a; Garmashova et al., 2006; Garmashova et al., 2007b). The NW and OW alphaviruses achieve transcription inhibition by using different genome-encoded proteins, capsid and nsP2, respectively (Akhrymuk et al., 2012; Atasheva et al., 2010a; Garmashova et al., 2007a; Garmashova et al., 2006; Garmashova et al., 2007b). However, the time between the beginning of virus replication and profound inhibition of cellular transcription represents a short window of opportunity, which can be utilized by infected cells for induction of cell signaling and type I IFN release.
Continuous cell lines traditionally used in laboratory practice, do not efficiently respond by type I IFN production to replication of wt alphaviruses with type I IFN production (Fig. 1). Thus, it remained unclear whether all of the initially synthesized dsRNA molecules are completely isolated inside the spherules or can become exposed to RIG-I/MDA5. Moreover, it is important to acknowledge that in addition to viral RNAs, the cellular mRNA pool can serve as another source of dsRNAs (Nikonov et al., 2013), and thus, cellular RNA templates can be additional active players in the induction of the RIG-I/MDA5-mediated cellular response. Most cellular mRNAs are shorter than alphavirus-specific genomic and even alphavirus defective interfering (DI) RNAs (Levis et al., 1986). Thus, they are likely less efficient in spherule-formation, whose size was found to be dependent on the length of RNA templates (Kallio et al., 2013). Thus, the role of cellular RNAs in dsRNA PAMP formation during alphavirus replication should not be ignored.
It has been previously demonstrated that many alphaviruses induce type I IFN during in vivo replication. However, the transcriptional and translational shutoff systems inherent to these viruses complicated investigation of the mechanism in vitro. The nature of the PRRs responsible for alphavirus sensing has been addressed in very few previous studies. Our experiments with KD cells suggest that MDA5 is the major PRR for alphaviruses, and its deletion reduces IFN production from infected cells (Fig. 3). This was also suggested by previous studies utilizing attenuated SINV (Burke et al., 2009). Other studies with chikungunya virus found that level of IFN-β RNA was significantly reduced in RIG-I−/− MEFs, but the effect of MDA5 knockdown on IFN-β activation was strongly dependent on the genetic background (Schilte et al., 2010). These studies led to different conclusions, and the mechanism of IFN induction remained unclear.
Here we have analyzed the roles of cellular PRRs in the cells of the same genetic background. First, we have assessed the induction of antiviral response in NIH 3T3 cells, in which RIG-I and/or MDA5 expression was very strongly inhibited by corresponding shRNAs. Then, cells lacking both receptors (dKO cells) were used to develop stable cell lines expressing RIG-I or MDA5 at higher, but biologically relevant levels, and induction of type I IFN was re-examined. To demonstrate the ability of PRRs to sense alphavirus replication, we applied viruses with wt structural and nonstructural genes, and mutants, which had no defects in RNA replication, but lacked transcription inhibitory functions. The experiments were performed on two distantly related, geographically separated OW (SINV) and NW (VEEV) alphaviruses, and thus, the results are likely applicable to other members of the genus.
There are two main types of cellular PRRs that sense RNA replication: cytosolic receptors, RIG-I and MDA5, and membrane-bound receptors, TLR3, TLR7 and TLR8 (Brubaker et al., 2015; Chow et al., 2015). TLR7 and TLR8 are not expressed in used fibroblasts (EIF, unpublished), and TLR3 is not functional in NIH 3T3 cells (Fig. 3B); therefore, at this point, we cannot rule out the possibility of sensing alphaviruses by TLRs in other cell types. However, our data unambiguously demonstrate that at least in NIH 3T3 cells, both RIG-I and MDA5 efficiently detect SINV and VEEV RNA replication and induce INF-β production. Cells lacking both RIG-I and MDA5 did not produce any detectable levels of INF-β in response to either replication of alphavirus mutants or transfection of poly(I:C). Thus, in the cells utilized in this study, RIG-I and MDA5 are the only sensors of alphavirus-specific PAMPs.
While in both single KD and single KI cell lines, RIG-I and MDA5 induced IFN-β production upon infection with mutant viruses to comparable levels (Figs. 3A, 6B), the kinetics of IFN release strongly differed. In MDA5-expressing cells, IFN-β was detected in the media as early as 4 h post infection with either virus, and its production ceased by 16 h post infection. In contrast, in KI RIG-I cells, IFN-β release started later, but reached higher levels by 24 h post infection. It has been shown that MDA5 recognizes long dsRNA, which are formed during replication of many ssRNA(+) viruses (Feng et al., 2012; Kato et al., 2006). In alphavirus infections, the dsRNA are indeed produced in the early stages of replication, and after 4 h post infection, the negative-strand RNAs (in form of dsRNAs) are no longer synthesized (Sawicki and Sawicki, 1980). Thus, earlier, rapid production of IFN in alphavirus-infected KI MDA5 cells strongly correlates with accumulation of dsRNA intermediates. Most of the dsRNAs are normally isolated in membranous spherules. However, early activation of the MDA5-mediated IFN-β response, detected in our experiments, suggests that at some point during their synthesis, dsRNAs are accessible to MDA5, or some of the dsRNA-containing replication complexes fail to form spherules, and dsRNA intermediates remain in the cytoplasm.
Activation of IFN-β by RIG-I was found to begin later in the infection (Fig. 6). By 6–8 h post infection, alphavirus replication switches to synthesis of large amounts of single-stranded genomic and subgenomic ssRNAs. These newly synthesized ssRNAs are only partially capped (Sokoloski et al., 2015), and a large fraction of them contains triphosphate at the 5′UTRs, folded into stable secondary structures, and thus representing excellent RIG-I-specific PAMPs (Kulasegaran-Shylini et al., 2009). The differing kinetics of ds and ssRNA synthesis, therefore, strongly correlate with variations in RIG-I- and MDA5-mediated IFN-β induction. Thus, our data suggest that alphavirus dsRNA is a primary PAMP for MDA-5, while viral ssRNAs are likely recognized by RIG-I. It has been also recently proposed that alphavirus polymerase can utilize cellular mRNAs as substrates for synthesis of dsRNA (Nikonov et al., 2013). This cellular mRNA derived dsRNAs would be relatively short and contain 5′-PPP, which represent a good substrate for both RIG-I and MDA5. It remains to be shown which of these RNA species are primary PAMPs for RIG-I and MDA5 in alphavirus-infected cells.
Our data suggest that the ability to induce type I IFN is strongly dependent on the levels of RIG-I and MDA5 present at the time of infection. In NIH 3T3 cells, basal levels of these PRRs are very low, but are strongly increased during infection (Atasheva et al., 2012). MDA5 transcription is activated by both type I IFN treatment and by viral replication in the absence of IFN stimulation, while RIG-I is activated by type I IFN (Atasheva et al., 2012). These activation mechanisms are likely to be additional contributors to very high levels of IFN-β release detected in the experiments with alphavirus mutants having no transcription inhibitory functions. On the other hand, higher levels of PRRs in KI cells, made these cell lines capable of responding to IFN-β even to replication of the wt viruses.
In this study, we compared the abilities of RIG-I and MDA5 to sense two very distantly related alphaviruses, SINV and VEEV. Both viruses demonstrated similar patterns of IFN-β induction in the presence of a single PRR. They exhibited differences only in the final levels of IFN-β response, which may be the result of either distinct kinetics of RNA synthesis or the existence of additional, virus-specific means of evading of the antiviral response. Thus, while varying alphavirus species certainly differ in replication mechanisms and virus-host interactions, it appears that their presence and activity in the cell is sensed by both RIG-I and MDA5 for all members of the genus. So far, there is also no experimental basis to expect that human RIG-I and MDA5 are different from those of mouse origin in terms of detecting alphavirus replication and, thus, our finding can be applied to human cells.
In conclusion, the accumulated data strongly suggest that replication of alphaviruses and infection spread strongly depend on the ability of viruses to inhibit transcription of cellular genes and thus, to downregulate the development of the innate immune response. Activation of the antiviral response and, primarily, induction of IFN-β is determined a) by the viral capsid or nsP2 proteins, which demonstrate nuclear functions, and b) by the presence of two cellular PRRs, RIG-I and MDA5 in the infected cells. The latter two proteins appear to be the only sensors of alphavirus replication in mouse fibroblasts. No type I IFN response is induced in their absence, but each of them alone is capable of initiating IFN-β expression in response to replication of alphavirus-specific RNAs, albeit they promote the induction with different kinetics. RIG-I and MDA5 function in concentration-dependent modes: their presence at low concentration in standard continuous cell lines is sufficient for IFN induction to the replication of attenuated, but not wt alphaviruses. However, expression of either RIG-I or MDA5 to higher levels leads to early IFN-β activation even in response to wt alphavirus infections. The dependence of the induction of the antiviral response on the PRRs’ concentrations provides at least partial explanation for the discrepancy of the data generated in in vivo and in vitro studies. High expression of RIG-I and/or MDA5 in primary cells in vivo would promote induction of type I IFN by wt alphaviruses.
Experimental procedures
Cell cultures
The BHK-21 cells were kindly provided by Paul Olivo (Washington University, St. Louis, Mo). The NIH 3T3 cells were obtained from the American Type Culture Collection (Manassas, VA). These cell lines were maintained at 37°C in alpha minimum essential medium (αMEM) supplemented with 10% fetal bovine serum (FBS) and vitamins.
Plasmid constructs
Plasmids encoding VEEV TC-83 genomes, pVEEV/GFP, pVEEV/GFP/Cm, and SINV Toto1101 genomes, pSINV/GFP and pSINV/G/GFP (see Fig. 1 for details), were described elsewhere (Atasheva et al., 2010b; Frolova et al., 2002). pVEEV/GFP/Cm contains mutations in the capsid protein’s nuclear localization signal (see VEEV/C1/GFP in (Atasheva et al., 2010b)). pSINV/G/GFP contains a P726G mutation in the nsP2-coding gene (Frolov et al., 1999a). RIG-I and MDA5 genes were synthesized by RT-PCR using RNA isolated from NIH 3T3 cells. These genes were cloned into modified PiggyBAC plasmids (System Bioscience, Inc). To prevent degradation of ectopically expressed RIG-I RNA by shRNAs, the target sequence was modified by clustered mutations, which did not change the encoded amino acids.
Generation of stable knock-down and knock-in cells
Stable knock-down (KD) cell lines were generated using shRNA-expressing lentiviruses according to the manufacturer’s instructions (TRCN0000378444 for RIG-I and TRCN0000103648 for MDA5, Sigma). Clones of PurR cells were analyzed in terms of RIG-I and MDA5 expression before and after IFN-β treatment by Western blotting and RT-qPCR, and the expression levels were compared to those in IFN-β- and mock-treated NIH 3T3 cells.
Stable knock-in (KI) cell lines were generated by transfection of double KD cells, which had been developed for this study, with PiggyBAC-based plasmids (System Biosciences) encoding RIG-I or MDA5 and the integrase-encoding helper plasmid. After blasticidin or G418 selection, clones of the stable single or double KI cells were analyzed for the levels of RIG-I and MDA5 expression. Cloned cells demonstrating levels of protein expression similar to those found in IFN-β-treated MEFs were used in the following experiments.
RNA transcriptions
Plasmids were purified by centrifugation in CsCl gradients. They were linearized using the MluI or XhoI restriction sites located downstream of the poly(A) sequence of viral genomes. RNAs were synthesized by SP6 RNA polymerase in the presence of a cap analog using previously described conditions (Rice et al., 1987). The yield and integrity of transcripts were analyzed by gel electrophoresis under non-denaturing conditions, and transcription reactions were used for electroporation without additional purification (Liljeström et al., 1991). Released viruses were harvested at 24 h post electroporation, and titers were determined by plaque assay on BHK-21 cells (Lemm et al., 1990).
Viral replication analysis
Cells were infected at MOIs indicated in the figures legends, washed with PBS, and overlaid with complete medium. At the times indicated in the figures, media were replaced by fresh media, and virus titers were determined by a plaque assay on BHK-21 cells as previously described (Lemm et al., 1990). In some experiments, the developed stable cell lines, expressing genes of interest, were infected with VEEV/GFP/Cm or SINV/G/GFP, which were poorly cytopathic. To analyze the efficiency of virus spread, cells were infected with different dilutions of the viruses, covered with agarose-containing media, and formation of GFP-positive foci was analyzed on a Typhoon phosphorimager (GE Healthcare Life Science).
Quantitative PCR analysis
Total RNA was isolated from cells which were either mock-treated, or treated with IFN-β as indicated in the figure legends. The relative levels of indicated RNAs were measured as previously described (Atasheva et al., 2012) using SsoFast EvaGreen Supermix (Bio-Rad) on a CFX96 real-time PCR instrument (Bio-Rad) and normalized to the level of β-actin mRNA. The primer sequences were as follows: Ifih1: forward - GGTGGACAAACTTCTGATTAACG, reverse - TCCTTCTGCACAATCCTTCTC; Ddx58: forward -TGACAGACGCTCTAAATTACCTC, reverse - GGATTCTCATTGCTGGGATCC; Actb: forward - ACCTTCTACAATGAGCTGCG, reverse - CTGGATGGCTACGTACATGG.
Western blotting
Equal amounts of proteins were separated on a 4–12% gradient NuPAGE gel (Invitrogen). After protein transfer, the membranes were incubated with primary antibodies, followed by incubation with infrared dye-labeled secondary antibodies. For quantitative analysis, membranes were scanned on the Odyssey imager (LI-COR). The following primary antibodies were used for Western blot: MDA5 (rabbit mAb D74E4, Cell Signaling), RIG-I (rabbit mAb D14G6, Cell Signaling), STAT1 (rabbit mA EPYR2154, Epitomics), pSTAT1(pY701) (MAB 4a, BD Transduction Laboratories), tubulin (rat mAb 7–20, UAB Hybridoma Core Facility).
IFN-β measurement
NIH 3T3 cells and developed stable cell lines were infected with viruses as described in the figure legends. Media were harvested at indicated times post infection, and the pH in the media was stabilized by adding HEPES buffer pH 7.5 to 0.01 M. Concentrations of IFN-β in the samples were measured with the VeriKine Mouse Interferon Beta ELISA Kit (PBL Interferon Source) according to the manufacturer’s recommendations.
Statistical analysis
Unless otherwise stated in the figure legends, experiments were repeated three times and statistical analysis of the data was performed using Graph Pad Prism. The statistical significance of differences between experimental points was determined by a two-tailed unpaired Student’s t test or a two-way ANOVA. The value p < 0.05 was considered statistically significant and degree of significance indicated as following: *p < 0.05, **p < 0.01, ***p <0.001, and ****p < 0.0001. Error bars represent standard deviation, SD.
Highlights.
Both RIG-I and MDA5 detect alphavirus replication.
Alphavirus-induced transcriptional shutoff affects type I IFN induction.
Sensing of alphavirus replication by RIG-I and MDA5 depends on their concentrations.
High basal level of RIG-I and MDA5 allows IFN induction by pathogenic alphaviruses.
This dependence determines the discrepancy between the in vivo and in vitro data.
Acknowledgments
We thank Dr. Niall J. Foy for helpful discussions, critical reading and editing of the manuscript. This work was supported by Public Health Service grants AI118867 and AI073301 to EIF and AI095449 and AI070207 to IF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilar PV, Paessler S, Carrara AS, Baron S, Poast J, Wang E, Moncayo AC, Anishchenko M, Watts D, Tesh RB, Weaver SC. Variation in interferon sensitivity and induction among strains of eastern equine encephalitis virus. J Virol. 2005;79:11300–11310. doi: 10.1128/JVI.79.17.11300-11310.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhrymuk I, Kulemzin SV, Frolova EI. Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J Virol. 2012;86:7180–7191. doi: 10.1128/JVI.00541-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasheva S, Akhrymuk M, Frolova EI, Frolov I. New PARP gene with an anti-alphavirus function. J Virol. 2012;86:8147–8160. doi: 10.1128/JVI.00733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasheva S, Fish A, Fornerod M, Frolova EI. Venezuelan equine Encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J Virol. 2010a;84:4158–4171. doi: 10.1128/JVI.02554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasheva S, Garmashova N, Frolov I, Frolova E. Venezuelan equine encephalitis virus capsid protein inhibits nuclear import in Mammalian but not in mosquito cells. J Virol. 2008;82:4028–4041. doi: 10.1128/JVI.02330-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasheva S, Krendelchtchikova V, Liopo A, Frolova E, Frolov I. Interplay of acute and persistent infections caused by Venezuelan equine encephalitis virus encoding mutated capsid protein. J Virol. 2010b;84:10004–10015. doi: 10.1128/JVI.01151-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DT, Condreay LD. Replication of Alphaviruses in Mosquito Cells. In: Schlesinger S, Schlesinger MJ, editors. The Togaviridae and Flaviviridae. Plenum Press; New York: 1986. pp. 171–207. [Google Scholar]
- Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257–290. doi: 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke CW, Gardner CL, Steffan JJ, Ryman KD, Klimstra WB. Characteristics of alpha/beta interferon induction after infection of murine fibroblasts with wild-type and mutant alphaviruses. Virology. 2009;395:121–132. doi: 10.1016/j.virol.2009.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow J, Franz KM, Kagan JC. PRRs are watching you: Localization of innate sensing and signaling regulators. Virology. 2015:479–480. 104–109. doi: 10.1016/j.virol.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CC, Suthar MS, Montgomery SA, Shabman R, Simmons J, Johnston RE, Morrison TE, Heise MT. Modulation of type I IFN induction by a virulence determinant within the alphavirus nsP1 protein. Virology. 2010;399:1–10. doi: 10.1016/j.virol.2009.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Canto MC, Rabinowitz SG. Central nervous system demyelination in Venezuelan equine encephalomyelitis infection. J Neurol Sci. 1981;49:397–418. doi: 10.1016/0022-510x(81)90030-7. [DOI] [PubMed] [Google Scholar]
- Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, Hur S, Semler BL, van Rij RP, van Kuppeveld FJ. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep. 2012;2:1187–1196. doi: 10.1016/j.celrep.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov I, Agapov E, Hoffman TA, Jr, Prágai BM, Lippa M, Schlesinger S, Rice CM. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J Virol. 1999a;73:3854–3865. doi: 10.1128/jvi.73.5.3854-3865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov I, Agapov E, Hoffman TA, Jr, Pragai BM, Lippa M, Schlesinger S, Rice CM. Selection of RNA replicons capable of persistent noncytopathic replication in mammalian cells. J Virol. 1999b;73:3854–3865. doi: 10.1128/jvi.73.5.3854-3865.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolov I, Akhrymuk M, Akhrymuk I, Atasheva S, Frolova EI. Early events in alphavirus replication determine the outcome of infection. J Virol. 2012;86:5055–5066. doi: 10.1128/JVI.07223-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova EI, Fayzulin RZ, Cook SH, Griffin DE, Rice CM, Frolov I. Roles of nonstructural protein nsP2 and Alpha/Beta interferons in determining the outcome of Sindbis virus infection. J Virol. 2002;76:11254–11264. doi: 10.1128/JVI.76.22.11254-11264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, Frolov I. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J Virol. 2010;84:11679–11695. doi: 10.1128/JVI.01441-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froshauer S, Kartenbeck J, Helenius A. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J Cell Biol. 1988;107:2075–2086. doi: 10.1083/jcb.107.6.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Burke CW, Tesfay MZ, Glass PJ, Klimstra WB, Ryman KD. Eastern and Venezuelan equine encephalitis viruses differ in their ability to infect dendritic cells and macrophages: impact of altered cell tropism on pathogenesis. J Virol. 2008;82:10634–10646. doi: 10.1128/JVI.01323-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Ebel GD, Ryman KD, Klimstra WB. Heparan sulfate binding by natural eastern equine encephalitis viruses promotes neurovirulence. Proc Natl Acad Sci U S A. 2011;108:16026–16031. doi: 10.1073/pnas.1110617108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner CL, Yin J, Burke CW, Klimstra WB, Ryman KD. Type I interferon induction is correlated with attenuation of a South American eastern equine encephalitis virus strain in mice. Virology. 2009;390:338–347. doi: 10.1016/j.virol.2009.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmashova N, Atasheva S, Kang W, Weaver SC, Frolova E, Frolov I. Analysis of Venezuelan equine encephalitis virus capsid protein function in the inhibition of cellular transcription. J Virol. 2007a;81:13552–13565. doi: 10.1128/JVI.01576-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmashova N, Gorchakov R, Frolova E, Frolov I. Sindbis virus nonstructural protein nsP2 is cytotoxic and inhibits cellular transcription. J Virol. 2006;80:5686–5696. doi: 10.1128/JVI.02739-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J Virol. 2007b;81:2472–2484. doi: 10.1128/JVI.02073-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Frolov I. Inhibition of transcription and translation in Sindbis virus-infected cells. J Virol. 2005;79:9397–9409. doi: 10.1128/JVI.79.15.9397-9409.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Frolova E, Sawicki S, Atasheva S, Sawicki D, Frolov I. A new role for ns polyprotein cleavage in Sindbis virus replication. J Virol. 2008a;82:6218–6231. doi: 10.1128/JVI.02624-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorchakov R, Garmashova N, Frolova E, Frolov I. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J Virol. 2008b;82:10088–10101. doi: 10.1128/JVI.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin DE. Alphaviruses. In: Knipe DM, Howley PM, editors. Fields’ Virology. 4. Lippincott, Williams and Wilkins; New York: 2001. pp. 917–962. [Google Scholar]
- Habjan M, Pichlmair A. Cytoplasmic sensing of viral nucleic acids. Curr Opin Virol. 2015;11C:31–37. doi: 10.1016/j.coviro.2015.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halstead SB, Scanlon JE, Umpaivit P, Udomsakdi S. Dengue and chikungunya virus infection in man in Thailand, 1962–1964. IV. Epidemiologic studies in the Bangkok metropolitan area. American Journal of Tropical Medicine & Hygiene. 1969a;18:997–1021. doi: 10.4269/ajtmh.1969.18.997. [DOI] [PubMed] [Google Scholar]
- Halstead SB, Udomsakdi S, Scanlon JE, Rohitayodhin S. Dengue and chikungunya virus infection in man in Thailand, 1962–1964. V. Epidemiologic observations outside Bangkok. American Journal of Tropical Medicine & Hygiene. 1969b;18:1022–1033. doi: 10.4269/ajtmh.1969.18.1022. [DOI] [PubMed] [Google Scholar]
- Kallio K, Hellstrom K, Balistreri G, Spuul P, Jokitalo E, Ahola T. Template RNA length determines the size of replication complex spherules for Semliki Forest virus. J Virol. 2013;87:9125–9134. doi: 10.1128/JVI.00660-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Katze MG, He Y, Gale M., Jr Viruses and interferon: a fight for supremacy. Nat Rev Immunol. 2002;2:675–687. doi: 10.1038/nri888. [DOI] [PubMed] [Google Scholar]
- Kulasegaran-Shylini R, Thiviyanathan V, Gorenstein DG, Frolov I. The 5′UTR-specific mutation in VEEV TC-83 genome has a strong effect on RNA replication and subgenomic RNA synthesis, but not on translation of the encoded proteins. Virology. 2009;387:211–221. doi: 10.1016/j.virol.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemm JA, Durbin RK, Stollar V, Rice CM. Mutations which alter the level or structure of nsP4 can affect the efficiency of Sindbis virus replication in a host-dependent manner. J Virol. 1990;64:3001–3011. doi: 10.1128/jvi.64.6.3001-3011.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levis R, Weiss BG, Tsiang M, Huang H, Schlesinger S. Deletion mapping of Sindbis virus DI RNAs derived from cDNAs defines the sequences essential for replication and packaging. Cell. 1986;44:137–145. doi: 10.1016/0092-8674(86)90492-7. [DOI] [PubMed] [Google Scholar]
- Liljeström P, Lusa S, Huylebroeck D, Garoff H. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J Virol. 1991;65:4107–4113. doi: 10.1128/jvi.65.8.4107-4113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–692. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov A, Molder T, Sikut R, Kiiver K, Mannik A, Toots U, Lulla A, Lulla V, Utt A, Merits A, Ustav M. RIG-I and MDA-5 detection of viral RNA-dependent RNA polymerase activity restricts positive-strand RNA virus replication. PLoS Pathog. 2013;9:e1003610. doi: 10.1371/journal.ppat.1003610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Rehwinkel J, Kato H, Takeuchi O, Akira S, Way M, Schiavo G, Reis e Sousa C. Activation of MDA5 requires higher-order RNA structures generated during virus infection. J Virol. 2009;83:10761–10769. doi: 10.1128/JVI.00770-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao TR. Recent epidemics caused by chikungunya virus in India, 1963–1965. Scientific Culture. 1966;32:215. [Google Scholar]
- Rice CM, Levis R, Strauss JH, Huang HV. Production of infectious RNA transcripts from Sindbis virus cDNA clones: Mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J Virol. 1987;61:3809–3819. doi: 10.1128/jvi.61.12.3809-3819.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawicki DL, Sawicki SG. Short-lived minus-strand polymerase for Semliki Forest virus. J Virol. 1980;34:108–118. doi: 10.1128/jvi.34.1.108-118.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilte C, Couderc T, Chretien F, Sourisseau M, Gangneux N, Guivel-Benhassine F, Kraxner A, Tschopp J, Higgs S, Michault A, Arenzana-Seisdedos F, Colonna M, Peduto L, Schwartz O, Lecuit M, Albert ML. Type I IFN controls chikungunya virus via its action on nonhematopoietic cells. J Exp Med. 2010;207:429–442. doi: 10.1084/jem.20090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz O, Pichlmair A, Rehwinkel J, Rogers NC, Scheuner D, Kato H, Takeuchi O, Akira S, Kaufman RJ, Reis e Sousa C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe. 2010;7:354–361. doi: 10.1016/j.chom.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GC. Viruses and interferons. Annu Rev Microbiol. 2001;55:255–281. doi: 10.1146/annurev.micro.55.1.255. [DOI] [PubMed] [Google Scholar]
- Sokoloski KJ, Haist KC, Morrison TE, Mukhopadhyay S, Hardy RW. Noncapped Alphavirus Genomic RNAs and Their Role during Infection. J Virol. 2015;89:6080–6092. doi: 10.1128/JVI.00553-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrer KM, Gack MU. Intracellular detection of viral nucleic acids. Curr Opin Microbiol. 2015;26:1–9. doi: 10.1016/j.mib.2015.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, and evolution. Microbiol Rev. 1994a;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss JH, Strauss EG. The alphaviruses: gene expression, replication, evolution. Microbiol Rev. 1994b;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Weber F. RIG-I-like receptors and negative-strand RNA viruses: RLRly bird catches some worms. Cytokine Growth Factor Rev. 2014;25:621–628. doi: 10.1016/j.cytogfr.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White LJ, Wang JG, Davis NL, Johnston RE. Role of alpha/beta interferon in Venezuelan equine encephalitis virus pathogenesis: effect of an attenuating mutation in the 5′ untranslated region. J Virol. 2001;75:3706–3718. doi: 10.1128/JVI.75.8.3706-3718.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins C, Gale M., Jr Recognition of viruses by cytoplasmic sensors. Curr Opin Immunol. 2010;22:41–47. doi: 10.1016/j.coi.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]