

Abstract
Kaposi’s sarcoma (KS) is the most common cancer in HIV-infected untreated individuals. Kaposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) is the infectious cause of this neoplasm. In this Review we describe the epidemiology of KS and KSHV, and the insights into the remarkable mechanisms through which KSHV can induce KS that have been gained in the past 16 years. KSHV latent transcripts, such as latency-associated nuclear antigen (LANA), viral cyclin, viral FLIP and viral-encoded microRNAs, drive cell proliferation and prevent apoptosis, whereas KSHV lytic proteins, such as viral G protein-coupled receptor, K1 and virally encoded cytokines (viral interleukin-6 and viral chemokines) further contribute to the unique angioproliferative and inflammatory KS lesions through a mechanism called paracrine neoplasia.
Introduction
The pathobiology of Kaposi’s sarcoma (KS) retraces the history of modern viral oncology. From an oncological curiosity described more than 100 years ago to an AIDS-defining cancer, the discovery of Kaposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) and its oncogenic enigmas has enlightened many fields of tumour biology and viral oncogenesis. KS was first described as a skin cancer affecting elderly, mainly Jewish men of Ashkenazi origin in Vienna, Austria1. In 1981, physicians in New York, USA, and Los Angeles, USA, observed an epidemic of this disease affecting young, homosexual men2, 3. This KS epidemic was the first indicator of a devastating pandemic to follow, caused by the soon to be discovered HIV1 (Refs 4, 5). In 1994, a group at Columbia University, New York, USA, used representational difference analysis6 (a PCR-based technique) to make a seminal discovery: DNA sequences of a new γ-herpesvirus were invariably present in KS lesions, but not in unaffected skin or most other diseased tissues, thereby coining the term KSHV7. KSHV fulfils most of the modern-day Koch’s postulates8 causally linking this oncogenic virus with a human cancer. The full genome of KSHV has been sequenced9 and studies on individual open reading frames (ORFs) have led to the discovery of new mechanisms for oncogenesis (such as the putative role of transiently expressed viral genes) and novel functions for cellular orthologues that are encoded by this virus. KS remains one of the most important and interesting viral-induced cancers that affects humans, and the discovery of KSHV7 heralded a bourgeoning area for epidemiological, molecular and clinical research in viral oncology (Timeline). In the 15 years since its discovery, we have gone from not knowing what causes KS, to having a substantial understanding of the causative agent, its major virological and pathophysiological features, and potential rational targets for intervention.
Timeline.

A history of KS KSHV
But many key questions regarding this pathogen and its associated tumours remain unanswered. As a result of the AIDS pandemic, KS has become one of the commonest cancers affecting men and children in many subequatorial African countries, where it is associated with significant morbidity and mortality10, 11. Although the incidence of AIDS-associated KS (AIDS-KS) in the Western world has declined since the widespread implementation of highly active antiretroviral treatment (HAART), up to 50% of patients with AIDS-KS never achieve total remission12. Furthermore, although treatments for KS exist, none is curative. In this Review we discuss the current knowledge of KS oncogenesis and approaches towards rationally designed therapies and prevention that could affect the large AIDS-KS health burden.
Epidemiology of KS
KS is grouped into four epidemiological forms13: classic KS affecting elderly men of Mediterranean or eastern European Jewish ancestry; endemic KS, existing in parts of Central and Eastern Africa, described long before the HIV pandemic and often affecting children with disseminated lymphadenopathy14, 15, 16; iatrogenic KS, developing in immunosuppressed individuals after an organ transplant, for example17; and epidemic or AIDS-KS, a major AIDS-defining malignancy. In the Western world, AIDS-KS predominantly affects HIV-infected homosexual men. However, in Africa, since the spread of HIV, epidemic KS has become more common in both sexes, with a dramatic lowering of the male to female ratio, especially in East Africa18.
KSHV-specific antibody titres correlate with viral load, and individuals with a low viral load consequently have lower antibody titres that might be missed by current serological assays. Therefore, there is the possibility of underestimating overall prevalence. However, despite this caveat and some regional exceptions, there is a strong concordance between KSHV seroprevalence rates and the incidence of KS (Fig. 1).
FIGURE 1. Geographical prevalence of KS and seroprevalence of KSHV.

The standardized incidence of Kaposi’s sarcoma (KS) is depicted for males, and was obtained from the International Agency for Research on Cancer Cancer Incidence in Five Continents publication (see Further information). The rate provided for the United States is an average, but rates in some States (including, California, New York, Georgia and the District of Columbia) can be as high as 6 in some subpopulations. According to Surveillance Epidemiology and End Results (SEER; see Further information) overall rates in the United States among non-Hispanic caucasians is 0.8, among caucasian Hispanics 1.4, and among African Americans is 2.4. In Italy, rates also vary by region, being as low as 0.2 in Umbria but 2.2 in Brescia. Incidences in Africa are taken from the Globocan database (see Further information). b | Seroprevalence rates were compiled from multiple studies. When different rates from the same country are reported, an average was taken. Values represent those in the general population, usually blood donors, and cohorts comprising of HIV-infected individuals were excluded. The seroprevalence of KSHV infection in northern Europe, Asia and the United States is less than 10%, but in most of sub-Saharan Africa, overall seroprevalence is more than 50%. The Mediterranean region has intermediate seroprevalence rates of 10–30%175.
In Mediterranean populations, where classic KS exists, and in sub-Saharan Africa, where childhood KS occurs, mother-to-child transmission of KSHV through saliva is the most likely route of transmission19, 20. In HIV-infected homosexual men, where the risk for transmission is associated with the number of sexual partners, it is likely that the most probable route of transmission is also through saliva21, 22. These epidemiological associations concur with the observation that KSHV can replicate in vitro in primary oral-derived epithelial cells23. Although KSHV transmission by blood transfusion or transplanted organs is documented, based on cost–benefit analyses most countries do not yet routinely screen blood or organ donors for KSHV infection.
Although KSHV infection is necessary for KS to develop, it is not sufficient and cofactors exist. The most important cofactor is HIV infection. KS incidence is 1 in 100,000 in the general population, but in HIV-infected individuals it is around 1 in 20 (Ref. 13), climbing to almost 1 in 3 in HIV-infected homosexual men before the introduction of HAART24. There has been extensive debate regarding whether immunodeficiency itself is the main determinant of KS, or whether HIV has a more direct role. Individuals with iatrogenic immunosuppression, particularly patients with renal transplants, also have an increased risk for KS, but this increase is not as great as that seen with HIV infection. This may reflect differences in KSHV infection rates, rather than HIV-specific causes, or differences in immune dysfunction, although a role for HIV as a cofactor has not been excluded. Individuals acquiring KSHV infection with pre-existing HIV infection have a significantly higher risk of developing KS; almost 50% develop KS, indicating that in this setting KSHV is one of the most oncogenic human viruses currently known25. This suggests that an already damaged immune system predisposes to a higher KSHV load, with subsequent KS development. Countries in which KS was endemic before the AIDS epidemic have seen a dramatic increase in the incidence of KS. Currently, KS is one of the most common cancers in certain sub-Saharan African countries18, 26 where 89% of all KS cases occur, and only ~12% of patients are alive at 5 years after diagnosis10.
The observation that in higher incidence groups and endemic areas, most HIV-negative KSHV-infected individuals never develop KS suggests that host factors have an important role. Studies have started to explore the potential contribution of host genetic factors, including genetic polymorphisms of inflammatory and immune-response genes. Classic KS risk is associated with diplotypes of interleukin-8 receptor-β (IL8RB), IL-13 (Ref. 27) and certain human leukocyte antigen (HLA) haplotypes28, 29. Transplant KS risk is associated with an IL6 promoter polymorphism30, and genotypes of FcγRIIIA influence the development of KS in HIV-infected men31. So far, these association studies have been small, and only show a slight overall increased risk.
These data suggest that common host genetic variants, in addition to environmental factors, timing and possibly routes of infection, all contribute to the oncogenic outcome of KSHV infection.
KS histogenesis
The histological features of the four epidemiological forms of KS are indistinguishable. They consist of spindle cells (the tumour cell), a proliferation of abnormal and leaky vessels and extravasated red blood cells with haemosiderin deposits3, 32, 33. A prominent inflammatory infiltrate is also present early in the development of these lesions (Fig. 2). Clinically, lesions have been described as patch, plaque, nodule and tumour stages, but as the same patient can have different types of lesions, and flat lesions (patch or plaque) can occupy extensive areas, and raised lesions (nodule or tumour) can be localized, the AIDS Clinical Trials Group (ACTG) tumour staging classification is more often used as a measure of the extent of disease in clinical studies34, 35. AIDS-associated KS can present as an aggressive disseminated disease affecting skin, lymph nodes and visceral organs.
Figure 2. Cellular Heterogeneity in Kaposi’s sarcoma.

A biopsy sample from a nodular KS lesion showing numerous spindle cells in the dermis. Immunohistochemical staining shows Kaposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) latency-associated nuclear antigen (LANA) in spindle cells lining vascular spaces. The lymphatic marker D2-40 is also localized to vascular spaces. Kaposi’s sarcoma (KS) lesions are composed of various cell types, including vascular (CD34) and lymphatic endothelial cells (D2-40), macrophages (lysozyme), lymphocytes, plasma cells and red blood cells. The inflammatory infiltrate is both inside and outside well-formed or poorly defined vascular spaces. These images highlight the complexity of KS lesions and the presence of virus in only a variable proportion of cells in the lesions, which is consistent with an important role of paracrine angiogenic and inflammatory signals. Magnifications: for hematoxylin and eosin (H&E) x10 and x40; for LANA x40; for CD34, D2-40 and lysozyme x20.
The cellular origin and neoplastic nature of KS remains contentious. The most common cell type in nodular lesions is the spindle-shaped cells (also known as KS cells). The vast majority of these spindle cells express endothelial markers, including CD31, CD34 and Factor VIII, but also markers of lymphatic endothelium, such as vascular endothelial growth factor receptor 3 (VEGFR3), lymphatic vessel endothelial hyaluronan 1 (LYVE1), D2-40 and podoplanin36, 37 (Fig. 2). However, a few spindle cells also express markers of dendritic cells (Factor XIII), macrophages (CD68) or smooth muscle cells (SMA), leading to the idea that these cells do not represent a uniform cell type38. Ultrastructurally, spindle cells have features of both the lymphatic and vascular endothelium39. The observation that KSHV infection of blood vessel endothelial cells in vitro induces lymphatic endothelial markers, and infection of lymphatic cells leads to reprogramming towards blood vessel cells40, 41, adds to the complexity. We suspect that KSHV infects circulating endothelial precursor cells, driving them towards a lymphatic lineage. Other oncogenic viruses such as Epstein–Barr virus (EBV) and human papilloma virus (HPV) are known to infect either B cell precursors or keratinocyte precursors, respectively, and exploit their differentiation for viral maturation and replication. Circulating vascular progenitors have certain KS spindle cell markers36, 42, and infection of these cells would be consistent with the multifocal presentation of advanced KS43, 44, 45 and the reported donor origin of post-transplant KS46.
Reactive inflammation or true neoplasm?
The propensity of KS lesions to localize to scar tissue or sites of inflammation (known as the Koebner phenomenon)47 provided one of the first clues that KS tumour cells are attracted by certain chemokines and flourish in a cytokine-rich microenvironment. In many early KS lesions the spindle cells are outnumbered by inflammatory cells. Therefore, before the discovery of KSHV, AIDS-KS research focused on the role of AIDS-associated cytokines and HIV-encoded proteins in driving inflammation and spindle cell proliferation (reviewed in Refs 48, 49). Various inflammatory cytokines increase in HIV pathogenesis50, including IL-1, tumour necrosis factor-α and interferon-γ, promote KS spindle cell proliferation, induce spindle-like cell morphology in endothelial cells and spindle-like differentiation in circulating endothelial progenitor cells43. Explanted KS spindle cells differ from most other tumour cells as they are dependent on external cytokines and growth factors to grow in vitro and do not induce tumours in nude mice, unlike truly transformed cells. However, in the presence of inflammatory cytokines, cells isolated from KS lesions are able to induce KS-like lesions in immunodeficient mice51. Cytokines and growth factors with autocrine and paracrine growth effects in spindle cells include T helper 1 (TH1) inflammatory cytokines, and cytokines and growth factors with pro-angiogenic activity such as IL-6, Oncostatin M52, hepatocyte growth factor (also known as scatter factor)53, fibroblast growth factor 2 and VEGF54. As discussed below, these cytokines are key mechanistic components in KS pathogenesis because their secretion is induced by KSHV infection, and they are necessary as autocrine and paracrine factors for driving KSHV oncogenesis.
Clinical observations are consistent with a deregulated inflammatory-driven angiogenic process, as KS seems to be more multifocal than metastatic, and regression of AIDS- and transplant-KS can occur when immune responses are partially restored during HAART or when immunosuppression is reduced. Adding controversy to the inflammatory versus neoplastic debate, X chromosome inactivation studies in single lesions as well as comparisons of multiple lesions from a single patient support a clonal origin in a subset of advanced cases only55. Most lesions are polyclonal, and multiple lesions from the same individual are also mainly polyclonal56, 57. The currently accepted interpretation of these data is that KS starts as a hyperplastic polyclonal lesion that is associated with inflammation and KSHV infection that could give rise, under specific circumstances like immunosuppression or other selective pressures, to clonal metastatic lesions. Supporting the idea that KS is only truly neoplastic in advanced stages is the observation that cellular oncogenic alterations, such as p53 and KRAS mutations or BCL-2 overexpression, as well as gene copy number changes, occur only in late-stage advanced disease58, 59, 60. Therefore, KS has features reminiscent of post-transplant lymphoproliferative disorders, which are EBV-driven B cell proliferations progressing from polyclonal hyperplasia to monoclonal tumours, and eventually to malignant lymphoma with oncogene and tumour suppressor alterations61. The proliferative nature of the spindle cells in the lesion, driven by latent viral proteins affecting cellular proliferation and survival (see below), concurs with many of the existing paradigms of cancer. However, the heterogeneous, multiclonal cellular composition of KS, the involution of KS following an immune response, the probably important role of infiltrating inflammatory cells, and the key role that the release of inflammatory and angiogenic mediators by KSHV-infected cells has in driving KS tumorigenesis (see below), exemplify an inflammatory-driven oncogenic process or paracrine neoplasia62.
Mechanisms of KSHV-induced oncogenesis
KSHV infection of endothelial cells or circulating endothelial and/or haematopoietic progenitors45, 63 leads to changes in their morphology64, glucose metabolism65 growth rate, lifespan and gene expression40, 41, 66, 67, resulting in the precipitation of KS. KSHV oncogenicity is reflected by the numerous pro-angiogenic molecules that are induced after infection of endothelial cells, including members of the VEGF–VEGFR family, angiopoietin family, cyclooxygenase 2 (COX2) and angiogenin41, 68, 69, 70, 71. However, in most experimental systems, in vitro infection of endothelial cells with KSHV leads to morphological changes and an extended lifespan and provides a survival advantage in response to apoptotic stimuli, but not full neoplastic transformation. Moreover, although KSHV encodes oncogenic genes that could potentially induce all KS-related malignant phenotypes (see below), KSHV infection in the general population rarely leads to KS. This underscores the existence of cofactors, such as HIV or drug-induced immunosuppression, that are required for the virus to induce a tumour.
Although the vast majority of KS spindle cells are latently infected with the virus37, 72 (Box 1), in a small proportion of infected cells the virus undergoes lytic replication leading to the production of mature virus and cell lysis. Apart from the viral cyclin (vcyclin) and viral FLICE inhibitory protein (vFLIP), the other cellular orthologues that are encoded by KSHV are early lytic genes that are generally expressed only in cells in which the virus is undergoing lytic replication. However, certain KSHV immunoregulatory and growth-promoting genes, including vIL-6, vMIR3 and vMIR5, could be activated by Notch signalling independently of the lytic transactivator RTA73. Moreover, limiting-dilution reverse transcription-PCR analysis shows that transcripts such as vIL-6 can be expressed in KSHV latency in a context-dependent manner; for example, in B cell lines74. This implies that the expression of KSHV genes might not be restricted by the classic herpesviral paradigm of latent or lytic infection, as lytic genes can be expressed without the full execution of the lytic cycle75. This is an important observation when we consider the complementary role of latent and lytic gene-expressing cells in KS pathogenesis.
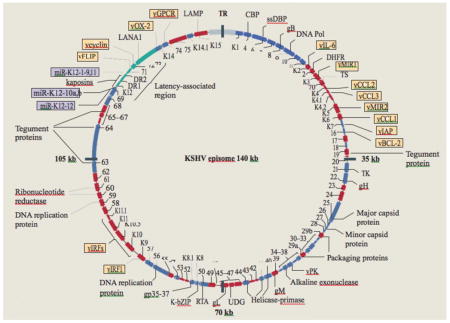
Box 1. The KSHV episome.
Kaposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) has at least four major subtypes that track human migration through Africa (predominantly subtype B and A5); the mediterranean (subtype C); northern Europe and North and South America (A); and the Far East (D)162. KSHV encodes 87 open reading frames (ORFs) and at least 17 microRNAs (purple boxes), 14 of which are co-expressed as a cluster. A striking feature of KSHV is the number (at least 14) of ORFs that encode cellular orthologues. Identified ORFs and encoded proteins are indicated in the figure. Putative latent transcripts are indicated in green, and cellular orthologues in yellow. Infection occurs when mature virions anchor to specific cellular receptors. After viral glycoprotein binding to the necessary receptors, clathrin-mediated endocytosis facilitates entry into cells163. Following infection, rapid circularization of the viral genome occurs164 and, like other herpesviruses, KSHV exists as an episome (double-stranded circular DNA) within the host nucleus. Reactivation can occur when the promoter of ORF50 is activated (by demethylation, for example)165, resulting in the expression of replication and transcription activator (RTA), the main regulator for the viral lytic replication programme77, 166. Early lytic genes include those encoding viral proteins required for DNA replication or viral gene expression, whereas late lytic genes are those encoding viral structural proteins, such as envelope and capsid proteins, that are required for assembly of viral particles (virions).
Primary effusion lymphoma (PEL) cells are latently infected with KSHV76 and were instrumental in classifying KSHV genes as latent or lytic, and identifying the major effectors of latent and lytic replication77, 78. Many observations made from PEL-derived tumours, such as the switch to lytic replication that occurs during in vivo growth79, are also observed in KS models, indicating that PEL is a valid model to study aspects of KSHV biology. Simple in vitro models, such as NIH3T3 and 293 cells, together with more sophisticated single gene transgenic mice have been instrumental in gaining molecular and functional information on individual viral genes. The potential contributions of individual latent and lytic viral infection to KS oncogenesis, together with emerging theories on how they collaborate (Fig. 3) are summarized below. The mechanisms by which KSHV perturbs normal immune responses and evades host immunity, are reviewed elsewhere80, 81.
FIGURE 3. Proposed mechanism of KSHV-induced sarcoma.


a | In lytic or abortive lytic-infected cells, expression of Kaposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) early lytic genes (such as viral G protein-coupled receptor (vGPCR), K1, viral interleukin-6 (vIL-6) and K15; shown in red) subvert host signalling pathways, leading to the expression and secretion of angiogenic, inflammatory and proliferative factors (including, vascular endothelial growth factor (VEGF), platelet-derived growth factor-β (PDGFB), angiopoietin 2 (ANGPT2), IL-6 and IL-8). This can occur together with intracrine activity and the secretion of vIL-6. b | Secreted factors stimulate their receptors in latently infected cells through a paracrine mechanism, complementing the autocrine (such as the secretion of cytokines by viral FLICE inhibitory protein (vFLIP)) and direct pro-oncogenic activities of KSHV latent genes, such as vFLIP, vcyclin and latency-associated nuclear antigen (LANA), as well as the KSHV-encoded microRNAs. β-cat, β-catenin; CDK, cyclin-dependent kinase; GSK3β, glycogen synthase kinase 3β; HIF, hypoxia-inducible factor; IAPs, inhibitor of apoptosis proteins; NF-κB, nuclear factor-κB; PKC, protein kinase C; PLC, phospholipase C; ROS, reactive oxygen species.
Latent KSHV infection
The major latency viral transcripts expressed in KS spindle cells are from the same genomic region and include the expression of the latency-associated nuclear antigen (LANA), vcyclin, vFLIP, viral-encoded microRNAs (miRNAs), as well as kaposins (Box 1). These transcripts endow growth and proliferative signals, evasion of apoptosis, pro-angiogenic and inflammatory signals, as well as limitless replicative potential. However, together these viral transcripts have not been shown to transform endothelial or any other cells in vitro. Although the main function of LANA is to maintain the viral episome78, LANA can also interfere with important anti-tumorigenic pathways (Fig. 3): LANA inhibits the activities of the p53 (Refs 82, 83) and the RB–E2F tumour suppressor pathways84. In addition, LANA has been shown to deregulate Wnt signalling by nuclear trapping of glycogen synthase kinase 3β (GSK3β), thereby stabilizing β-catenin85, and to inhibit anti-proliferative transforming growth factor-β (TGFβ) signalling by epigenetic suppression of TGFβ receptors86. LANA might contribute to angiogenesis, by stabilizing hypoxia-inducible factor 1α (HIF1α) and by targeting von Hippel Lindau (VHL) for degradation87. LANA is also an activator of telomerase reverse transcriptase (TERT) expression88 and can increase the lifespan of human umbilical vascular endothelial cells (HUVECs)89.
vcyclin90 is a constitutive activator of cyclin-dependent kinase 6 (CDK6)91 and, strikingly, has activities that are not restricted to cellular D-type cyclins92, 93. vcyclin expression leads to cytokinesis defects and polyploidy, which activates p53. However, in the absence of functional p53, such cells survive, exposing the oncogenic potential of vcyclin94. The exact function of this viral protein, in the context of all other viral proteins, is still not clear, but it is likely that vcyclin drives cellular proliferation and so promotes viral replication. It has been suggested that genomic instability is an inevitable consequence of latent KSHV infection, owing to vcyclin–CDK6-mediated phosphorylation of nucleophosmin (NPM1)95, 96.
vFLIP binds to inhibitor of κB kinase-γ (IKKγ), leading to the direct activation of nuclear factor-κB (NF-κB)97, 98 (Fig. 3). A large number of cytokines, including chemokines implicated in KS pathogenesis, are induced in endothelial cells by vFLIP activation of NF-κB99, 100. In addition to cytokine secretion, constitutive NF-κB activation by vFLIP could have important anti-apoptotic roles in oncogenesis by leading to the induction of proteins that inhibit apoptosis, such as BCL-2 and BCL-XL101. Interestingly, vFLIP also suppresses autophagy, an important pro-oncogenic activity, by preventing ATG3 from binding and processing LC3 (Ref. 102). vFLIP is also responsible for the spindle cell morphological transformation of endothelial cells in vitro103.
Kaposins are proteins that are encoded by the alternatively spliced ORF K12 (Ref. 104). Kaposin A is a latent protein with transforming potential in rodent fibroblasts105. Kaposin B affects signalling by binding to MK2, a MAPK-associated protein kinase. Kaposin B-mediated activation of MK2 blocks the decay of mRNAs with AU-rich elements (AREs) in their 3′ untranslated regions. As several cytokine mRNAs have ARE elements, kaposin B expression results in an increase in the production of pro-inflammatory cytokines106. Therefore, vFLIP, kaposin A and kaposin B are likely to contribute to the inflammatory microenvironment of KS.
Mice that express a LANA transgene using the natural viral promoter, a vFLIP transgene (using an H2κB promoter and IgH enhancer), or vcyclin transgene using the Eμ promoter and enhancer, develop lymphoid malignancies with low frequency and after a long latency108, 109, 110. Although these findings support the idea that LANA, vcyclin and vFLIP could drive B cell proliferation and survival, the induced tumours fail to exhibit characteristics of KSHV lymphoproliferations, such as a plasmablastic phenotype.
Certain transgenic models develop lesions with KS characteristics. For example, mice expressing vcyclin targeted to lymphatic endothelium using a Vegfr3 promoter develop lymphatic abnormalities and oedema111. Concurring with in vitro data, there is no obvious proliferation or transformation of lymphatic endothelial cells in vivo, only their aberrant development and leakiness. Although transgenic animals that express individual viral ORFs provide useful information about the in vivo functions of a specific viral protein, these viral ORFs are expressed out of the context of global viral replication and persistence, and so data must be interpreted with caution.
The KSHV-encoded miRNAs are expressed in latently infected cells112, 113, 114 and are thought to be involved in suppressing the lytic reactivation of the virus, and are thought to influence endothelial cell differentiation and angiogenesis. One viral miRNA, miR-K1, targets IκBα, an inhibitor of NF-κB. NF-κB inhibits the activation of lytic viral promoters115; therefore, by activating NF-κB, this miRNA suppresses viral lytic replication, maintaining latent infection116. The viral miRNAs also inhibit the anti-angiogenic molecule thrombospondin 1 (Ref. 117), possibly contributing to KS-related angiogenesis. At least four of the viral miRNAs, including the orthologue of cellular miR-155, target the cellular oncogene MAF to induce reprogramming of lymphatic endothelial cells118. By studying the viral miRNAs, MAF was identified as a potential transcriptional repressor that functions in endothelial cells. These viral miRNAs could thus influence the differentiation of infected endothelial cells, contributing to KS development. KSHV also induces cellular miRNAs. One of the most upregulated miRNAs after the infection of endothelial cells and in KS lesions, is miR-132 (Ref. 119). This miRNA not only inhibits anti-viral innate immune responses119, but intriguingly also induces abnormal endothelial cell proliferation120. Therefore, this miR-132 could link two important features of KS: viral immune escape and angiogenesis.
Lytic KSHV infection
Many lytic viral proteins such as K1, the viral interferon response factors (vIRFs), vIL-6, the viral-encoded chemokines (vCCLs), viral G protein-coupled receptor (vGPCR) and K15, which are expressed by a proportion of cells in KS lesions, have impressive putative tumorigenic activities and so could contribute to the angiogenic and inflammatory phenotype of KS lesions. By activating RAC1 and RHOA, vGPCR induces the activation of MAPKs, AKT and NF-κB, resulting in cell proliferation121, 122, secretion of VEGF122, 123 and other pro-angiogenic and inflammatory cytokines such as angiopoietin 2 (ANGPT2)124, IL-6 and IL-8 (Refs 125, 126).
Mice expressing transgenic vGCPR develop angiogenic lesions that resemble KS127, 128, as vGPCR is only expressed in a proportion of cells, and these cells drive VEGF-mediated angiogenesis using paracrine mechanisms62. As vGPCR is not expressed during latency and therefore not by most tumour cells, paracrine models more accurately reflect its role in KS biology129.
A transgenic model of endothelial cell-specific transduction using an avian retrovirus and testing several KSHV lytic and latent genes, showed that vGPCR is the only ORF that has the ability to initiate angiogenic lesions130. This system identified RAC1 as a mediator of vGCPR oncogenesis131 and added to the evidence that vGCPR activates TSC2–mTOR signalling, making mTOR a potential therapeutic target in KS132. RAC1 is involved in the production of reactive oxygen species (ROS) through NADPH oxidases and is overexpressed in all KSHV-infected cells in KS lesions. Moreover, constitutive activation of RAC1 induces angiogenic lesions in mice133. Overall, vGPCR could promote infected endothelial cell proliferation, angiogenesis and the recruitment of an inflammatory infiltrate through autocrine and paracrine mechanisms (Box 2; Fig. 3).
Box 2. Animal models for virally induced KS.
Animal models of γherpesvirus infection and pathogenesis include the mouse herpesvirus 68 (MHV68)167 and the Rhesus rhadinovirus (RRV)168 models. Although these viruses are related to Kaposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)), they generate B cell lymphoproliferation, and are of limited use to study Kaposi’s sarcoma (KS) pathogenesis. The squirrel monkey γherpesvirus, herpesvirus saimirii, induces lymphomas when infecting non-natural hosts such as owl monkeys, but never endothelial tumours. The retroperitoneal fibromatosis herpesvirus (RFHV) induces retroperitoneal fibrosis (RF) in animals that become immunodeficient after infection with a simian virus. The spindle-shaped cells in these RF lesions express the latency-associated nuclear antigen (LANA) orthologue of RFHV169, but these spindle cells belong to the mesenchymal rather than the endothelial lineage. These lesions are not relevant to KS biology.
Two cell lines bearing the KSHV genome generate KSHV-infected tumours. One is based on HUVECs that express telomerase (TIVE-LTC)170 and the other is based on an infectious bacterial artificial chromosome (KSHVBac36) transfected into normal mouse bone marrow endothelial lineage cells (mECK36)171 (Fig. 4). Both systems suggest that KSHV tumour formation requires both latent and lytic viral gene expression. This observation contrasts with mechanisms underlying classic viral-induced tumours, such as Epstein–Barr virus-driven lymphoma or human papilloma virus-associated cervical cancer, in which latent viral proteins (including viral oncogenes) are expressed in clonally derived cells, and lytic or abortive infection plays little, if any, role. The mECK36 system has viral and host transcriptome characteristics that are related to those found in KS. This model demonstrates the de novo tumorigenicity of KSHV infection in normal mouse cells, showing that KSHV provides a survival advantage to cells in vivo, and a role for viral G protein-coupled receptor in vascular endothelial growth factor-mediated angiogenesis in the context of KSHV-induced tumours. Some studies have used different types of humanized immunodeficient mice to establish a more physiologically relevant in vivo model of KSHV infection of human cells, but these models fail to generate KS-like tumours172, 173. Common marmosets are susceptible to KSHV infection, and one of the infected animals developed a KS-like tumour, expressing both latent and lytic viral proteins174.
Other notable lytic viral proteins that could have a role in KS pathogenesis include the cellular orthologues vIL-6, vBCL-2, vIRFs and vCCLs. The gp130-binding IL-6R modulated vIL-6 virokine134 is angiogenic135 and might induce spindle cell proliferation and survival. The vCCLs are angiogenic in various experimental systems, promoting endothelial cell proliferation and migration, and could also curtail the local immune response against virally infected cells136, 137. vBCL-2 (Ref. 138) inhibits apoptosis139 through the inhibition of pro-apoptotic BH3 domain-containing proteins140. Several vIRFs inhibit p53-induced apoptosis141. In particular, vIRF1 inhibits DNA damage-induced apoptosis by inhibiting ATM activation of p53, a mechanism that could lead to resistance to genotoxic drugs and the accumulation of mutations and genetic instability142. vIRF3 activates VEGF secretion by stabilizing HIF1α143.
The first ORF of KSHV, K1, has substantial diversity between viral isolates, and is used to sub-classify KSHV into A, B, C and D strains (Box 1). All K1 subtypes seem to function similarly. K1 activates the PI3K–AKT anti-apoptotic pathway, inducing survival factors such as VEGF that can function in an autocrine and paracrine manner144. K1 can also suppress CD95-mediated apoptosis145. The main role of lytic viral proteins inhibiting apoptosis (like vBCL-2, vIRF1 and K1) could be to delay apoptosis during lytic replication, thus providing time for virion production and assembly, before cell lysis. Like K1, the SH2 and SH3 domain-containing K15 also flanks the terminal repeat region of the KSHV episome (Box 1), activates the NF-κB and MAPK pathways and induces the expression of several inflammatory and angiogenic genes146, 147.
Interplay between latent and lytic KSHV-infected cells in KS paracrine oncogenesis
Both latent and lytic KSHV genes contribute to the malignant phenotype of KS. However, lytic infection is unlikely to have any direct role in endothelial cell autonomous growth, transformation or immortalization, as lytic viral gene expression is generally associated with viral replication and cellular lysis. During lytic infection, KSHV ORF 37, a homologue of a DNA exonuclease, is also responsible for wide-scale cellular mRNA degradation, inhibiting host gene expression and therefore curtailing the role of cellular proteins during oncogenesis148. One theory currently gaining experimental support is that in order for latent genes to drive oncogenic cell proliferation, they need to be enhanced, in a paracrine manner, by host and viral growth factors and cytokines supplied by a minority of lytically infected cells and/or lytic gene-expressing cells that are present in KS lesions62, 122 (Fig. 3). Paracrine-acting factors such as VEGF, ANGPT2, platelet-derived growth factor (PDGF), GROa and IL-6 induced by lytic genes such as vGPCR, K1 and K15, in addition to vIL-6 and the vCCLs, could be necessary to drive latently infected cell proliferation, induce angiogenesis and inflammation, and further support the recruitment of uninfected cells, as well as the survival and immune escape of latently infected cells that form the majority of the KS tumour. The role of lytic infection in KS pathogenesis is supported by several observations. First, lytic viral proteins are expressed and virions are present in a minority of cells within KS lesions72, 149. Second, immunosuppression increases KSHV re-activation and lytic replication. Third, interrupting lytic replication by immune reconstitution or by anti-lytic herpes anti-virals, such as gancyclovir, can also inhibit or prevent KS development150, 151, 152. Fourth, lytic infection seems to be necessary to support viral episomal maintenance by the recruitment of new cells to latency to replace those that have segregated their viral episome153. Last, co-injection of vGPCR-expressing cells is necessary to induce tumour formation by cells expressing only latent genes154. The proposed molecular interplay between latent and lytic genes resulting in KS is shown in Fig. 3.
Rational treatment
Underscoring its dependence on HIV infection and immunosuppression, HAART has reduced the incidence of AIDS-KS, and can induce AIDS-KS regression. Mechanisms are likely to include immune reconstitution against KSHV152 and possibly decreasing circulating HIV-associated pro-angiogenic and inflammatory cytokines13. In addition, HIV protease inhibitors have direct antitumour activity155, although non-protease-containing HAART combinations also induce KS regression156. Despite the widespread availability of HAART in the Western world, KS remains a clinical problem with only around 50% of patients achieving complete resolution12. In addition to HAART, radiotherapy for isolated lesions and systemic chemotherapy, including liposomal daunorubicin and taxanes, are useful for disseminated disease. Our understanding of the molecular basis and biology of KS is leading to rational therapeutic trials and drug design157, 158(Table 1). Promising approaches aim to intervene in the paracrine and autocrine mechanisms depicted in Fig. 3 and include targeting the angiogenic or lymphangiogenic axis of VEGFA–VEGFR2, VEGFC–VEGFR3, as well as ANGPT2 and the proliferative and vascular growth factor PDGFβ (Fig. 4). Another angiogenesis-related target in KSHV oncogenesis is the Notch pathway159, which could potentially be targeted by γ-secretase inhibitors and inhibitors of Notch ligand interactions, including delta-like ligand 4 (DLL4)160, 161. Targeting the IKKγ–vFLIP interaction with specific small molecules should provide specific inhibition of KSHV-induced NF-κB without interfering with normal cellular NF-κB pathways. Targeting mTORC1 using rapamycin was serendipitously found to be effective in post-transplant KS and is currently being used to prevent and treat this disease. Although the exact mechanism for KS inhibition by rapamycin is still unclear, it is thought that rapamycin interferes with the dependence of KS cells on the PI3K–AKT–mTORC1 pathway, which is activated by several KSHV genes and is essential for promoting cytokine and angiogenic growth secretion that is central to KSHV-induced oncogenesis.
TABLE 1.
Potential new drugs for the treatment of KS
| Target | Biological effect | Agent | Proposed mechanism of action | Current status | Refs |
|---|---|---|---|---|---|
| VEGFC, VEGFD and VEGFR3 axis | Inhibits lymphangiogenesis | Cediranib (AZD2171) | Tyrosine kinase inhibitor blocking VEGFR3 | Phase II/III studies for various cancers, no clinical trials yet for KS | 176 |
| VEGFA | Inhibits angiogenesis | Bevacizumab, sorafenib, sunitinib and PTC299 | Monoclonal antibody bevacizumab that targets VEGF and small molecules that inhibit VEGFR (as well as KIT and PDGFR). PTC299 inhibits the production of VEGF by targeting post-transcriptional VEGF synthesis | NCI Phase II bevacizumab with liposomal doxorubicin; NCI Phase and PTC299 is in preclinical development | 177 |
| ANGPT2 | Inhibits angiogenesis and lymphangiogenesis | Blocks proliferation of KSHV-infected spindle cells that express ANGPT2 | Inhibitors in preclinical development | 41, 124, 178 | |
| DLL4 | Vascular disrupting | In preclinical studies, blocking of DLL4-NOTCH signalling results in a paradoxical increase in tumour vessel density, but causes marked growth inhibition owing to functionally defective vasculature | Inhibitors in preclinical development | 159, 160 | |
| NF-KB: vFLIP-IKKG | Inhibits inflammation, angiogenesis and cell proliferation | Small molecules currently being screened that could interfere with vFLIP-IKKG interaction | Preclinical development | 97, 98 | |
| KIT and PDGFR | Inhibits spindle cell proliferation | Imatinib | KIT and PDGFR implicated in KS cell proliferation | Phase I | 179, 180 |
| mTOR | Modulates immunity and inhibits cell proliferation | Sirolimus, tacrilimus and evirulumus | Small molecules that inhibit mTOR. KSHV infection activates mTOR signalling, which is important for cell survival and angiogenesis | Phase II/III studies | 132, 181 |
ANGPT2, angiopoietin 2; DLL4, delta-like ligand 4; IKKγ, inhibitor of κB kinase-γ; KSHV, Kaposi’s sarcoma herpesvirus; KS, Kaposi’s sarcoma; NCI, National Cancer Institutes; NF-κB, nuclear factor-κB; PDGFR, platelet-derived growth factor receptor; ROS, reactive oxygen species; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; vFLIP, viral FLICE inhibitory protein.
FIGURE 4. Mouse model of KSHV-induced KS.


a | Nude mice bearing enhanced green fluorescent protein-expressing tumours that are induced by subcutaneous injection of mouse endothelial cells transfected with Karposi’s sarcoma-associated herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) KSHV Bac 36 (mECK36). b | Immunoflurorescence analysis of latency-associated nuclear antigen (LANA; white) and the Kaposi’s sarcoma (KS) marker podoplanin (red) in mECK36 tumours showing punctuated LANA staining that is characteristic of episomal KSHV in cell nucleus (DAPI Blue). c | Activation of paracrine and autocrine endothelial stimulation as shown by gene expression data (heat maps) from AIDS-KS41 and from the mECK36 mouse KS model171. Data are grouped by ligands (left) and receptors (right), which are present in both molecular signatures. Open connectors indicate paracrine stimulation by upregulation in KS of at least one of the receptor–ligand pairs; ligand and receptor closed connectors indicate upregulation of both receptor and ligand with potential for both paracine and autocrine stimulation. ANGPT2, angiopoietin 2; CCL5, chemokine, CC motif, ligand 5; CCR5, chemokine, CC motif receptor 5; CXCL12, chemokine CXC motif, ligand 12; EdnA, Endothelin A; NRP, neuropilin; PDGF, platelet-derived growth factor; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor.
Future perspectives
KS is an unusual tumour with features pertinent to viral oncogenesis, inflammation and cancer. The large number of cellular orthologues encoded by this virus, and its ability to subvert multiple signalling pathways have attracted considerable research interest, which has been enhanced by new disease paradigms. Sixteen years after the discovery of KSHV as the causal agent of KS, and after more than 2,000 publications on this virus, KS remains a common and devastating disease in some geographic regions. Although palliative treatments exist, and the control of HIV infection is helpful in preventing KS or inducing its regression, there are no vaccines or curative drugs. Promising results in the laboratory and their successful translation to the clinic show that our improved understanding of KSHV pathobiology is leading to the development of better preventive and therapeutic approaches for KS.
“At a glance” summary.
Kaposi’s sarcoma herpesvirus (KSHV; also known as human herpesvirus 8 (HHV8)) is the causative agent of Kaposi’s sarcoma (KS) and certain lymphoproliferations, and its seroepidemiology correlates with the global incidence of KS.
KS is the most common neoplasm in untreated HIV-infected individuals, and also occurs in other states of immunosuppression, including after an organ transplant.
KSHV is transmitted through saliva and replicates in oropharyngeal cells.
Unlike most cancer cells, KS tumour cells are not fully transformed, do not show autonomous growth, and remain dependent on exogenous cytokines for in vitro growth.
KS starts as a proliferation of endothelial-type cells, with an early onset inflammatory and abnormal leaky blood vessel expansion.
KSHV is present in the vast majority of KS tumour cells (that is, spindle cells), expressing the latent viral proteins, including viral cyclin, viral FLICE inhibitory protein, latency-associated nuclear antigen (LANA) and a group of viral microRNAs.
A proportion of cells in KS lesions seem to undergo lytic replication, expressing lytic viral proteins including K1, viral interleukin-6, viral BCL-2, viral G protein-coupled receptor, K15 and viral chemokines.
KSHV latent genes drive cell proliferation and prevent apoptosis; whereas KSHV lytic genes could further contribute to KS tumorigenesis by triggering host signalling cascades that lead to cytokine and growth factor secretion.
Biological insights into KSHV oncogenesis are leading to promising rational therapeutic approaches.
Acknowledgments
E.A.M. is supported by US National Institutes of Health grants CA75918 and CA136387, and C.B. by Cancer Research UK, the Medical Research Council and the University College London and University College London Hospital Comprehensive Biomedical Research Centre.
Contributor Information
Enrique A. Mesri, Email: emesri@med.miami.edu, Viral Oncology Program, Department of Microbiology & Immunology, Sylvester Comprehensive Cancer Center, University of Miami Miller School of Medicine, 1550 NW 10th Ave, Papanicolau Bldg # 109, Miami, FL 33136, TEL 305-243-8838, FAX 305-243-8309
Ethel Cesarman, Department of Pathology and Laboratory Medicine, Weill Cornell Medical College, 1300 York Avenue, New York, NY 10065, Tel 212-746-8838, FAX 212-746-8816.
Chris Boshoff, Cancer Research UK Viral Oncology Group, UCL Cancer Institute, Huntley Street, University College London, London, UK, WC1E 6BT.
References
- 1.Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syph. 1872;4 Original description of KS. [Google Scholar]
- 2.Centers for Disease Control. Kaposi’s sarcoma and Pneumocystis pneumonia among homosexual men- New York City and California. MMWR Morb Mortal Wkly Rep. 1981;30:305–308. [PubMed] [Google Scholar]
- 3.Gottlieb GJ, et al. A preliminary communication on extensively disseminated Kaposi’s sarcoma in young homosexual men. Am J Dermatopathol. 1981;3:111–114. doi: 10.1097/00000372-198100320-00002. [DOI] [PubMed] [Google Scholar]
- 4.Barre-Sinoussi F, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS) Science. 1983;220:868–871. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 5.Gelmann EP, et al. Proviral DNA of a retrovirus, human T-cell leukemia virus, in two patients with AIDS. Science. 1983;220:862–865. doi: 10.1126/science.6601822. [DOI] [PubMed] [Google Scholar]
- 6.Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- 7.Chang Y, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. The molecular discovery of KS-associated herpesvirus. [DOI] [PubMed] [Google Scholar]
- 8.zur Hausen H. Oncogenic DNA viruses. Oncogene. 2001;20:7820–7823. doi: 10.1038/sj.onc.1204958. [DOI] [PubMed] [Google Scholar]
- 9.Russo JJ, et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8) Proc Natl Acad Sci USA. 1996;93:14862–14867. doi: 10.1073/pnas.93.25.14862. The original description of the KSHV genome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parkin DM. The global health burden of infection- associated cancers in the year 2002. Int J Cancer. 2006;118:3030–3044. doi: 10.1002/ijc.21731. [DOI] [PubMed] [Google Scholar]
- 11.Sinfield RL, et al. Spectrum and presentation of pediatric malignancies in the HIV era: experience from Blantyre, Malawi, 1998–2003. Pediatr Blood Cancer. 2007;48:515–520. doi: 10.1002/pbc.20917. [DOI] [PubMed] [Google Scholar]
- 12.Nguyen HQ, et al. Persistent Kaposi sarcoma in the era of highly active antiretroviral therapy: characterizing the predictors of clinical response. AIDS. 2008;22:937–945. doi: 10.1097/QAD.0b013e3282ff6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gallo RC. The enigmas of Kaposi’s sarcoma. Science. 1998;282:1837–1839. doi: 10.1126/science.282.5395.1837. [DOI] [PubMed] [Google Scholar]
- 14.Oettle AG. Geographical and racial differences in the frequency of Kaposi’s sarcoma as evidence of environmental or genetic causes. Acta Unio Int Contra Cancrum. 1962;18:330–363. [PubMed] [Google Scholar]
- 15.Slavin G, Cameron HM, Forbes C, Mitchell RM. Kaposi’s sarcoma in East African children: a report of 51 cases. J Pathol. 1970;100:187–199. doi: 10.1002/path.1711000307. [DOI] [PubMed] [Google Scholar]
- 16.Bhagwat GP, Naik KG, Sachdeva R, Bhushan V. Disseminated lymphadenopathic Kaposi’s sarcoma in Zambian children. Med J Zambia. 1980;14:61–63. [PubMed] [Google Scholar]
- 17.Siegel JH, et al. Disseminated visceral Kaposi’s sarcoma. Appearance after human renal homograft operation. JAMA. 1969;207:1493–1496. First description of KS from New York, USA, after an organ transplant. [PubMed] [Google Scholar]
- 18.Wabinga HR, Parkin DM, Wabwire-Mangen F, Mugerwa JW. Cancer in Kampala, Uganda, in 1989–1991: changes in incidence in the era of AIDS. Int J Cancer. 1993;54:26–36. doi: 10.1002/ijc.2910540106. [DOI] [PubMed] [Google Scholar]
- 19.He J, et al. Seroprevalence of human herpesvirus 8 among Zambian women of childbearing age without Kaposi’s sarcoma (KS) and mother-child pairs with KS. J Infect Dis. 1998;178:1787–1790. doi: 10.1086/314512. [DOI] [PubMed] [Google Scholar]
- 20.Davidovici B, et al. Seroepidemiology and molecular epidemiology of Kaposi’s sarcoma-associated herpesvirus among Jewish population groups in Israel. J Natl Cancer Inst. 2001;93:194–202. doi: 10.1093/jnci/93.3.194. [DOI] [PubMed] [Google Scholar]
- 21.Pauk J, et al. Mucosal shedding of human herpesvirus 8 in men. N Engl J Med. 2000;343:1369–1377. doi: 10.1056/NEJM200011093431904. [DOI] [PubMed] [Google Scholar]
- 22.Martro E, et al. Risk factors for human Herpesvirus 8 infection and AIDS-associated Kaposi’s sarcoma among men who have sex with men in a European multicentre study. Int J Cancer. 2007;120:1129–1135. doi: 10.1002/ijc.22281. [DOI] [PubMed] [Google Scholar]
- 23.Duus KM, Lentchitsky V, Wagenaar T, Grose C, Webster-Cyriaque J. Wild-type Kaposi’s sarcoma- associated herpesvirus isolated from the oropharynx of immune-competent individuals has tropism for cultured oral epithelial cells. J Virol. 2004;78:4074–4084. doi: 10.1128/JVI.78.8.4074-4084.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 1990;335:123–128. doi: 10.1016/0140-6736(90)90001-l. One of the first studies to highlight that KS could be caused by an infectious agent other than HIV. [DOI] [PubMed] [Google Scholar]
- 25.Martin JN, et al. Sexual transmission and the natural history of human herpesvirus 8 infection. N Engl J Med. 1998;338:948–954. doi: 10.1056/NEJM199804023381403. [DOI] [PubMed] [Google Scholar]
- 26.Parkin DM, et al. Part I: cancer in indigenous Africans-burden, distribution, and trends. Lancet Oncol. 2008;9:683–692. doi: 10.1016/S1470-2045(08)70175-X. [DOI] [PubMed] [Google Scholar]
- 27.Brown EE, et al. Associations of classic Kaposi sarcoma with common variants in genes that modulate host immunity. Cancer Epidemiol Biomarkers Prev. 2006;15:926–934. doi: 10.1158/1055-9965.EPI-05-0791. [DOI] [PubMed] [Google Scholar]
- 28.Cottoni F, et al. Susceptibility to human herpesvirus-8 infection in a healthy population from Sardinia is not directly correlated with the expression of HLA-DR alleles. Br J Dermatol. 2004;151:247–249. doi: 10.1111/j.1365-2133.2004.06060.x. [DOI] [PubMed] [Google Scholar]
- 29.Dorak MT, et al. HLA-B, -DRB1/3/4/5, and -DQB1 gene polymorphisms in human immunodeficiency virus-related Kaposi’s sarcoma. J Med Virol. 2005;76:302–310. doi: 10.1002/jmv.20361. [DOI] [PubMed] [Google Scholar]
- 30.Gazouli M, et al. The interleukin-6-174 promoter polymorphism is associated with a risk of development of Kaposi’s sarcoma in renal transplant recipients. Anticancer Res. 2004;24:1311–1314. [PubMed] [Google Scholar]
- 31.Lehrnbecher TL, et al. Variant genotypes of FcγRIIIA influence the development of Kaposi’s sarcoma in HIV- infected men. Blood. 2000;95:2386–2390. [PubMed] [Google Scholar]
- 32.Friedman-Kien AE. Disseminated Kaposi’s sarcoma syndrome in young homosexual men. J Am Acad Dermatol. 1981;5:468–471. doi: 10.1016/s0190-9622(81)80010-2. One of the original descriptions of KS from New York, USA, heralding the AIDS pandemic. [DOI] [PubMed] [Google Scholar]
- 33.Safai B, et al. The natural history of Kaposi’s sarcoma in the acquired immunodeficiency syndrome. Ann Intern Med. 1985;103:744–750. doi: 10.7326/0003-4819-103-5-744. [DOI] [PubMed] [Google Scholar]
- 34.Krown SE, Testa MA, Huang J. AIDS-related Kaposi’s sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol. 1997;15:3085–3092. doi: 10.1200/JCO.1997.15.9.3085. [DOI] [PubMed] [Google Scholar]
- 35.El Amari EB, et al. Predicting the evolution of Kaposi sarcoma, in the highly active antiretroviral therapy era. AIDS. 2008;22:1019–1028. doi: 10.1097/QAD.0b013e3282fc9c03. [DOI] [PubMed] [Google Scholar]
- 36.Jussila L, et al. Lymphatic endothelium and Kaposi’s sarcoma spindle cells detected by antibodies against the vascular endothelial growth factor receptor-3. Cancer Res. 1998;58:1599–1604. [PubMed] [Google Scholar]
- 37.Dupin N, et al. Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma. Proc Natl Acad Sci USA. 1999;96:4546–4551. doi: 10.1073/pnas.96.8.4546. First antibody-based study to describe the distribution of latent KSHV in KS, PEL and multicentric Castleman’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roth WK, Brandstetter H, Sturzl M. Cellular and molecular features of HIV-associated Kaposi’s sarcoma. AIDS. 1992;6:895–913. doi: 10.1097/00002030-199209000-00001. [DOI] [PubMed] [Google Scholar]
- 39.Orenstein JM. Ultrastructure of Kaposi sarcoma. Ultrastruct Pathol. 2008;32:211–220. doi: 10.1080/01913120802343871. [DOI] [PubMed] [Google Scholar]
- 40.Hong YK, et al. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nature Genet. 2004;36:683–685. doi: 10.1038/ng1383. [DOI] [PubMed] [Google Scholar]
- 41.Wang HW, et al. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nature Genet. 2004;36:687–693. doi: 10.1038/ng1384. [DOI] [PubMed] [Google Scholar]
- 42.Beckstead JH, Wood GS, Fletcher V. Evidence for the origin of Kaposi’s sarcoma from lymphatic endothelium. Am J Pathol. 1985;119:294–300. [PMC free article] [PubMed] [Google Scholar]
- 43.Browning PJ, et al. Identification and culture of Kaposi’s sarcoma-like spindle cells from the peripheral blood of human immunodeficiency virus-1-infected individuals and normal controls. Blood. 1994;84:2711–2720. [PubMed] [Google Scholar]
- 44.Parsons CH, Szomju B, Kedes DH. Susceptibility of human fetal mesenchymal stem cells to Kaposi sarcoma-associated herpesvirus. Blood. 2004;104:2736–2738. doi: 10.1182/blood-2004-02-0693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Della Bella S, et al. Peripheral blood endothelial progenitors as potential reservoirs of Kaposi’s sarcoma-associated herpesvirus. PLoS ONE. 2008;3:e1520. doi: 10.1371/journal.pone.0001520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barozzi P, et al. Post-transplant Kaposi sarcoma originates from the seeding of donor-derived progenitors. Nature Med. 2003;9:554–561. doi: 10.1038/nm862. Study proposes that KS tumour cells could be transmitted during an organ transplant. [DOI] [PubMed] [Google Scholar]
- 47.Niedt GW, Prioleau PG. Kaposi’s sarcoma occurring in a dermatome previously involved by herpes zoster. J Am Acad Dermatol. 1988;18:448–451. doi: 10.1016/s0190-9622(88)70068-7. [DOI] [PubMed] [Google Scholar]
- 48.Miles SA. Pathogenesis of HIV-related Kaposi’s sarcoma. Curr Opin Oncol. 1994;6:497–502. doi: 10.1097/00001622-199409000-00009. [DOI] [PubMed] [Google Scholar]
- 49.Ensoli B, et al. Biology of Kaposi’s sarcoma. Eur J Cancer. 2001;37:1251–1269. doi: 10.1016/s0959-8049(01)00121-6. [DOI] [PubMed] [Google Scholar]
- 50.Lane HC, Fauci AS. Immunologic abnormalities in the acquired immunodeficiency syndrome. Annu Rev Immunol. 1985;3:477–500. doi: 10.1146/annurev.iy.03.040185.002401. [DOI] [PubMed] [Google Scholar]
- 51.Salahuddin SZ, et al. Angiogenic properties of Kaposi’s sarcoma-derived cells after long-term culture in vitro. Science. 1988;242:430–433. doi: 10.1126/science.2459779. One of the first studies to suggest that KS tumour cells secrete and are dependent on angiogenic factors for survival. [DOI] [PubMed] [Google Scholar]
- 52.Miles SA, et al. Oncostatin M as a potent mitogen for AIDS-Kaposi’s sarcoma-derived cells. Science. 1992;255:1432–1434. doi: 10.1126/science.1542793. [DOI] [PubMed] [Google Scholar]
- 53.Naidu YM, et al. Role of scatter factor in the pathogenesis of AIDS-related Kaposi sarcoma. Proc Natl Acad Sci USA. 1994;91:5281–5285. doi: 10.1073/pnas.91.12.5281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Masood R, et al. Vascular endothelial growth factor/vascular permeability factor is an autocrine growth factor for AIDS-Kaposi sarcoma. Proc Natl Acad Sci USA. 1997;94:979–984. doi: 10.1073/pnas.94.3.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rabkin CS, et al. Monoclonal origin of multicentric Kaposi’s sarcoma lesions. N Engl J Med. 1997;336:988–993. doi: 10.1056/NEJM199704033361403. [DOI] [PubMed] [Google Scholar]
- 56.Gill PS, et al. Evidence for multiclonality in multicentric Kaposi’s sarcoma. Proc Natl Acad Sci USA. 1998;95:8257–8261. doi: 10.1073/pnas.95.14.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duprez R, et al. Evidence for a multiclonal origin of multicentric advanced lesions of Kaposi sarcoma. J Natl Cancer Inst. 2007;99:1086–1094. doi: 10.1093/jnci/djm045. [DOI] [PubMed] [Google Scholar]
- 58.Nicolaides A, Huang YQ, Li JJ, Zhang WG, Friedman-Kien AE. Gene amplification and multiple mutations of the K-ras oncogene in Kaposi’s sarcoma. Anticancer Res. 1994;14:921–926. [PubMed] [Google Scholar]
- 59.Pillay P, Chetty R, Reddy R. Bcl-2 and p53 immunoprofile in Kaposi’s sarcoma. Pathol Oncol Res. 1999;5:17–20. doi: 10.1053/paor.1999.0017. [DOI] [PubMed] [Google Scholar]
- 60.Pyakurel P, et al. CGH of microdissected Kaposi’s sarcoma lesions reveals recurrent loss of chromosome Y in early and additional chromosomal changes in late tumour stages. AIDS. 2006;20:1805–1812. doi: 10.1097/01.aids.0000244199.72887.3d. [DOI] [PubMed] [Google Scholar]
- 61.Knowles DM, et al. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood. 1995;85:552–565. [PubMed] [Google Scholar]
- 62.Cesarman E, Mesri EA, Gershengorn MC. Viral G protein-coupled receptor and Kaposi’s sarcoma: a model of paracrine neoplasia? J Exp Med. 2000;191:417–422. doi: 10.1084/jem.191.3.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu W, et al. KSHV/HHV-8 infection of human hematopoietic progenitor (CD34+) cells: persistence of infection during hematopoiesis in vitro and in vivo. Blood. 2006;108:141–151. doi: 10.1182/blood-2005-04-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moses AV, et al. Long-term infection and transformation of dermal microvascular endothelial cells by human herpesvirus 8. J Virol. 1999;73:6892–6902. doi: 10.1128/jvi.73.8.6892-6902.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Delgado T, et al. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc Natl Acad Sci USA. 2010;107:10696–10701. doi: 10.1073/pnas.1004882107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flore O, et al. Transformation of primary human endothelial cells by Kaposi’s sarcoma-associated herpesvirus. Nature. 1998;394:588–592. doi: 10.1038/29093. [DOI] [PubMed] [Google Scholar]
- 67.Ciufo DM, et al. Spindle cell conversion by Kaposi’s sarcoma-associated herpesvirus: formation of colonies and plaques with mixed lytic and latent gene expression in infected primary dermal microvascular endothelial cell cultures. J Virol. 2001;75:5614–5626. doi: 10.1128/JVI.75.12.5614-5626.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ye FC, et al. Kaposi’s sarcoma-associated herpesvirus promotes angiogenesis by inducing angiopoietin-2 expression via AP-1 and Ets1. J Virol. 2007;81:3980–3991. doi: 10.1128/JVI.02089-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sivakumar R, et al. Kaposi’s sarcoma-associated herpesvirus induces sustained levels of vascular endothelial growth factors A and C early during in vitro infection of human microvascular dermal endothelial cells: biological implications. J Virol. 2008;82:1759–1776. doi: 10.1128/JVI.00873-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sadagopan S, et al. Kaposi’s sarcoma-associated herpesvirus upregulates angiogenin during infection of human dermal microvascular endothelial cells, which induces 45S rRNA synthesis, antiapoptosis, cell proliferation, migration, and angiogenesis. J Virol. 2009;83:3342–3364. doi: 10.1128/JVI.02052-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma-Walia N, et al. Kaposi’s sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog. 2010;6:e1000777. doi: 10.1371/journal.ppat.1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Staskus KA, et al. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. Analysis of KSHV latent and lytic gene expression in KS spindle cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang H, Dittmer DP, Shin YC, Hong Y, Jung JU. Role of Notch signal transduction in Kaposi’s sarcoma-associated herpesvirus gene expression. J Virol. 2005;79:14371–14382. doi: 10.1128/JVI.79.22.14371-14382.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chandriani S, Ganem D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus. J Virol. 2010;84:5565–5573. doi: 10.1128/JVI.02723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okuno T, et al. Activation of human herpesvirus 8 open reading frame K5 independent of ORF50 expression. Virus Res. 2002;90:77–89. doi: 10.1016/s0168-1702(02)00142-9. [DOI] [PubMed] [Google Scholar]
- 76.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. Original study describing the association of KSHV with PEL. [DOI] [PubMed] [Google Scholar]
- 77.Sun R, et al. A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc Natl Acad Sci USA. 1998;95:10866–10871. doi: 10.1073/pnas.95.18.10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ballestas ME, Chatis PA, Kaye KM. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science. 1999;284:641–644. doi: 10.1126/science.284.5414.641. Analysis of the role of LANA in maintaining the KSHV episome. [DOI] [PubMed] [Google Scholar]
- 79.Staudt MR, et al. The tumor microenvironment controls primary effusion lymphoma growth in vivo. Cancer Res. 2004;64:4790–4799. doi: 10.1158/0008-5472.CAN-03-3835. [DOI] [PubMed] [Google Scholar]
- 80.Coscoy L. Immune evasion by Kaposi’s sarcoma-associated herpesvirus. Nature Rev Immunol. 2007;7:391–401. doi: 10.1038/nri2076. [DOI] [PubMed] [Google Scholar]
- 81.Areste C, Blackbourn DJ. Modulation of the immune system by Kaposi’s sarcoma-associated herpesvirus. Trends Microbiol. 2009;17:119–129. doi: 10.1016/j.tim.2008.12.001. [DOI] [PubMed] [Google Scholar]
- 82.Friborg J, Kong W, Hottiger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 83.Si H, Robertson ES. Kaposi’s sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen induces chromosomal instability through inhibition of p53 function. J Virol. 2006;80:697–709. doi: 10.1128/JVI.80.2.697-709.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nature Med. 2000;6:1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 85.Fujimuro M, et al. A novel viral mechanism for dysregulation of β-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nature Med. 2003;9:300–306. doi: 10.1038/nm829. [DOI] [PubMed] [Google Scholar]
- 86.Di Bartolo DL, et al. KSHV LANA inhibits TGF-β signaling through epigenetic silencing of the TGF-β type II receptor. Blood. 2008;111:4731–4740. doi: 10.1182/blood-2007-09-110544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cai QL, Knight JS, Verma SC, Zald P, Robertson ES. EC5S ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and p53 tumor suppressors. PLoS Pathog. 2006;2:e116. doi: 10.1371/journal.ppat.0020116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verma SC, Borah S, Robertson ES. Latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor Sp1. J Virol. 2004;78:10348–10359. doi: 10.1128/JVI.78.19.10348-10359.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Watanabe T, et al. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen prolongs the life span of primary human umbilical vein endothelial cells. J Virol. 2003;77:6188–6196. doi: 10.1128/JVI.77.11.6188-6196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang Y, et al. Cyclin encoded by KS herpesvirus. Nature. 1996;382:410. doi: 10.1038/382410a0. First characterization of the viral-encoded cyclin, revealing that vcyclin could function as a D type cyclin. [DOI] [PubMed] [Google Scholar]
- 91.Godden-Kent D, et al. The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1. J Virol. 1997;71:4193–4198. doi: 10.1128/jvi.71.6.4193-4198.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mittnacht S, Boshoff C. Viral cyclins. Rev Med Virol. 2000;10:175–184. doi: 10.1002/(sici)1099-1654(200005/06)10:3<175::aid-rmv283>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 93.Ojala PM, et al. The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2. Nature Cell Biol. 2000;2:819–825. doi: 10.1038/35041064. [DOI] [PubMed] [Google Scholar]
- 94.Verschuren EW, Klefstrom J, Evan GI, Jones N. The oncogenic potential of Kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo. Cancer Cell. 2002;2:229–241. doi: 10.1016/s1535-6108(02)00123-x. [DOI] [PubMed] [Google Scholar]
- 95.Cuomo ME, et al. p53-Driven apoptosis limits centrosome amplification and genomic instability downstream of NPM1 phosphorylation. Nature Cell Biol. 2008;10:723–730. doi: 10.1038/ncb1735. [DOI] [PubMed] [Google Scholar]
- 96.Sarek G, et al. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLoS Pathog. 2010;6:e1000818. doi: 10.1371/journal.ppat.1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu L, et al. The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Iκ B kinase complex. J Biol Chem. 2002;277:13745–13751. doi: 10.1074/jbc.M110480200. [DOI] [PubMed] [Google Scholar]
- 98.Bagneris C, et al. Crystal structure of a vFlip-IKKγ complex: insights into viral activation of the IKK signalosome. Mol Cell. 2008;30:620–631. doi: 10.1016/j.molcel.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 99.Sun Q, Matta H, Lu G, Chaudhary PM. Induction of IL-8 expression by human herpesvirus 8 encoded vFLIP K13 via NF-κB activation. Oncogene. 2006;25:2717–2726. doi: 10.1038/sj.onc.1209298. [DOI] [PubMed] [Google Scholar]
- 100.Sakakibara S, Pise-Masison CA, Brady JN, Tosato G. Gene regulation and functional alterations induced by Kaposi’s sarcoma-associated herpesvirus-encoded ORFK13/vFLIP in endothelial cells. J Virol. 2009;83:2140–2153. doi: 10.1128/JVI.01871-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Guasparri I, Keller SA, Cesarman E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J Exp Med. 2004;199:993–1003. doi: 10.1084/jem.20031467. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 102.Lee JS, et al. FLIP-mediated autophagy regulation in cell death control. Nature Cell Biol. 2009;11:1355–1362. doi: 10.1038/ncb1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grossmann C, Podgrabinska S, Skobe M, Ganem D. Activation of NF-κB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype. J Virol. 2006;80:7179–7185. doi: 10.1128/JVI.01603-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sadler R, et al. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J Virol. 1999;73:5722–5730. doi: 10.1128/jvi.73.7.5722-5730.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Muralidhar S, et al. Identification of kaposin (open reading frame K12) as a human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) transforming gene. J Virol. 1998;72:4980–4988. doi: 10.1128/jvi.72.6.4980-4988.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCormick C, Ganem D. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science. 2005;307:739–741. doi: 10.1126/science.1105779. [DOI] [PubMed] [Google Scholar]
- 107.Prakash O, et al. Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene. J Natl Cancer Inst. 2002;94:926–935. doi: 10.1093/jnci/94.12.926. [DOI] [PubMed] [Google Scholar]
- 108.Verschuren EW, et al. The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis. Cancer Res. 2004;64:581–589. doi: 10.1158/0008-5472.can-03-1863. [DOI] [PubMed] [Google Scholar]
- 109.Chugh P, et al. Constitutive NF-κB activation, normal Fas-induced apoptosis, and increased incidence of lymphoma in human herpes virus 8 K13 transgenic mice. Proc Natl Acad Sci USA. 2005;102:12885–12890. doi: 10.1073/pnas.0408577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fakhari FD, Jeong JH, Kanan Y, Dittmer DP. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J Clin Invest. 2006;116:735–742. doi: 10.1172/JCI26190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sugaya M, et al. Lymphatic dysfunction in transgenic mice expressing KSHV k-cyclin under the control of the VEGFR-3 promoter. Blood. 2005;105:2356–2363. doi: 10.1182/blood-2004-08-3364. [DOI] [PubMed] [Google Scholar]
- 112.Cai X, et al. Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc Natl Acad Sci USA. 2005;102:5570–5575. doi: 10.1073/pnas.0408192102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pfeffer S, et al. Identification of microRNAs of the herpesvirus family. Nature Methods. 2005;2:269–276. doi: 10.1038/nmeth746. [DOI] [PubMed] [Google Scholar]
- 114.Samols MA, Hu J, Skalsky RL, Renne R. Cloning and identification of a microRNA cluster within the latency-associated region of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2005;79:9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005. References 112, 113 and 114 described the identification of KSHV-encoded miRNAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brown HJ, et al. NF-κB inhibits γherpesvirus lytic replication. J Virol. 2003;77:8532–8540. doi: 10.1128/JVI.77.15.8532-8540.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lei X, et al. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nature Cell Biol. 2010;12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Samols MA, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007;3:e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hansen A, et al. KSHV-encoded miRNAs target MAF to induce endothelial cell reprogramming. Genes Dev. 2010;24:195–205. doi: 10.1101/gad.553410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lagos D, et al. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Natures Cell Biol. 2010;12:513–519. doi: 10.1038/ncb2054. [DOI] [PubMed] [Google Scholar]
- 120.Anand S, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nature Med. 2010;16:909–914. doi: 10.1038/nm.2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation. Nature. 1997;385:347–350. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- 122.Bais C, et al. G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator. Nature. 1998;391:86–89. doi: 10.1038/34193. References 121 and 122 report the initial charaterization of the vGPCR. [DOI] [PubMed] [Google Scholar]
- 123.Sodhi A, et al. The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 2000;60:4873–4880. [PubMed] [Google Scholar]
- 124.Vart RJ, et al. Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6 and G-protein-coupled receptor regulate angiopoietin-2 expression in lymphatic endothelial cells. Cancer Res. 2007;67:4042–4051. doi: 10.1158/0008-5472.CAN-06-3321. [DOI] [PubMed] [Google Scholar]
- 125.Schwarz M, Murphy PM. Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor constitutively activates NF-κ B and induces proinflammatory cytokine and chemokine production via a C-terminal signaling determinant. J Immunol. 2001;167:505–513. doi: 10.4049/jimmunol.167.1.505. [DOI] [PubMed] [Google Scholar]
- 126.Shepard LW, et al. Constitutive activation of NF-κ B and secretion of interleukin-8 induced by the G protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus involve Gα13 and RhoA. J Biol Chem. 2001;276:45979–45987. doi: 10.1074/jbc.M104783200. [DOI] [PubMed] [Google Scholar]
- 127.Yang TY, et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J Exp Med. 2000;191:445–454. doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guo HG, et al. Kaposi’s sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J Virol. 2003;77:2631–2639. doi: 10.1128/JVI.77.4.2631-2639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Grisotto MG, et al. The human herpesvirus 8 chemokine receptor vGPCR triggers autonomous proliferation of endothelial cells. J Clin Invest. 2006;116:1264–1273. doi: 10.1172/JCI26666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Montaner S, et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi’s sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23–36. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- 131.Montaner S, et al. The small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia. Blood. 2004;104:2903–2911. doi: 10.1182/blood-2003-12-4436. [DOI] [PubMed] [Google Scholar]
- 132.Sodhi A, et al. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell. 2006;10:133–143. doi: 10.1016/j.ccr.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 133.Ma Q, et al. Antitumorigenesis of antioxidants in a transgenic Rac1 model of Kaposi’s sarcoma. Proc Natl Acad Sci USA. 2009;106:8683–8688. doi: 10.1073/pnas.0812688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nicholas J, et al. Kaposi’s sarcoma-associated human herpesvirus-8 encodes homologues of macrophage inflammatory protein-1 and interleukin-6. Nature Med. 1997;3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- 135.Aoki Y, et al. Angiogenesis and hematopoiesis induced by Kaposi’s sarcoma-associated herpesvirus-encoded interleukin-6. Blood. 1999;93:4034–4043. [PubMed] [Google Scholar]
- 136.Boshoff C, et al. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science. 1997;278:290–294. doi: 10.1126/science.278.5336.290. [DOI] [PubMed] [Google Scholar]
- 137.Stine JT, et al. KSHV-encoded CC chemokine vMIP-III is a CCR4 agonist, stimulates angiogenesis, and selectively chemoattracts TH2 cells. Blood. 2000;95:1151–1157. [PubMed] [Google Scholar]
- 138.Sarid R, Sato T, Bohenzky RA, Russo JJ, Chang Y. Kaposi’s sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nature Med. 1997;3:293–298. doi: 10.1038/nm0397-293. [DOI] [PubMed] [Google Scholar]
- 139.Cheng EH, et al. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci USA. 1997;94:690–694. doi: 10.1073/pnas.94.2.690. References 138 and 139 characterized the viral encoded BCL-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Flanagan AM, Letai A. BH3 domains define selective inhibitory interactions with BHRF-1 and KSHV BCL-2. Cell Death Differ. 2008;15:580–588. doi: 10.1038/sj.cdd.4402292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Seo T, Park J, Lee D, Hwang SG, Choe J. Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J Virol. 2001;75:6193–6198. doi: 10.1128/JVI.75.13.6193-6198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shin YC, et al. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol. 2006;80:2257–2266. doi: 10.1128/JVI.80.5.2257-2266.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Shin YC, Joo CH, Gack MU, Lee HR, Jung JU. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 3 stabilizes hypoxia-inducible factor-1 α to induce vascular endothelial growth factor expression. Cancer Res. 2008;68:1751–1759. doi: 10.1158/0008-5472.CAN-07-2766. [DOI] [PubMed] [Google Scholar]
- 144.Tomlinson CC, Damania B. The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway. J Virol. 2004;78:1918–1927. doi: 10.1128/JVI.78.4.1918-1927.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Berkova Z, et al. Mechanism of Fas signaling regulation by human herpesvirus 8 K1 oncoprotein. J Natl Cancer Inst. 2009;101:399–411. doi: 10.1093/jnci/djn516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Brinkmann MM, et al. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein. J Virol. 2003;77:9346–9358. doi: 10.1128/JVI.77.17.9346-9358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Brinkmann MM, Pietrek M, Dittrich-Breiholz O, Kracht M, Schulz TF. Modulation of host gene expression by the K15 protein of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2007;81:42–58. doi: 10.1128/JVI.00648-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Glaunsinger B, Ganem D. Lytic KSHV infection inhibits host gene expression by accelerating global mRNA turnover. Mol Cell. 2004;13:713–723. doi: 10.1016/s1097-2765(04)00091-7. [DOI] [PubMed] [Google Scholar]
- 149.Orenstein JM, et al. Visualization of human herpesvirus type 8 in Kaposi’s sarcoma by light and transmission electron microscopy. AIDS. 1997;11:F35–F45. doi: 10.1097/00002030-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 150.Martin DF, et al. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group. N Engl J Med. 1999;340:1063–1070. doi: 10.1056/NEJM199904083401402. Clinical study indicating that anti-herpesviral agents could prevent KS development. [DOI] [PubMed] [Google Scholar]
- 151.Wilkinson J, et al. Identification of Kaposi’s sarcoma-associated herpesvirus (KSHV)-specific cytotoxic T-lymphocyte epitopes and evaluation of reconstitution of KSHV-specific responses in human immunodeficiency virus type 1-Infected patients receiving highly active antiretroviral therapy. J Virol. 2002;76:2634–2640. doi: 10.1128/JVI.76.6.2634-2640.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bourboulia D, et al. Short-and long-term effects of highly active antiretroviral therapy on Kaposi sarcoma-associated herpesvirus immune responses and viraemia. AIDS. 2004;18:485–493. doi: 10.1097/00002030-200402200-00015. [DOI] [PubMed] [Google Scholar]
- 153.Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest. 2004;113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Montaner S, et al. The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor as a therapeutic target for the treatment of Kaposi’s sarcoma. Cancer Res. 2006;66:168–174. doi: 10.1158/0008-5472.CAN-05-1026. [DOI] [PubMed] [Google Scholar]
- 155.Monini P, Sgadari C, Toschi E, Barillari G, Ensoli B. Antitumour effects of antiretroviral therapy. Nature Rev Cancer. 2004;4:861–875. doi: 10.1038/nrc1479. [DOI] [PubMed] [Google Scholar]
- 156.Bower M, Palmieri C, Dhillon T. AIDS-related malignancies: changing epidemiology and the impact of highly active antiretroviral therapy. Curr Opin Infect Dis. 2006;19:14–19. doi: 10.1097/01.qco.0000200295.30285.13. [DOI] [PubMed] [Google Scholar]
- 157.Sullivan R, Dezube BJ, Koon HB. Signal transduction targets in Kaposi’s sarcoma. Curr Opin Oncol. 2006;18:456–462. doi: 10.1097/01.cco.0000239884.05914.13. [DOI] [PubMed] [Google Scholar]
- 158.Dittmer DP, Krown SE. Targeted therapy for Kaposi’s sarcoma and Kaposi’s sarcoma-associated herpesvirus. Curr Opin Oncol. 2007;19:452–457. doi: 10.1097/CCO.0b013e3281eb8ea7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Emuss V, et al. KSHV manipulates Notch signaling by DLL4 and JAG1 to alter cell cycle genes in lymphatic endothelia. PLoS Pathog. 2009;5:e1000616. doi: 10.1371/journal.ppat.1000616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Curry CL, et al. γ secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi’s sarcoma tumor cells. Oncogene. 2005;24:6333–6344. doi: 10.1038/sj.onc.1208783. [DOI] [PubMed] [Google Scholar]
- 161.Liu R, et al. KSHV-induced notch components render endothelial and mural cell characteristics and cell survival. Blood. 2010;115:887–895. doi: 10.1182/blood-2009-08-236745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Hayward GS. KSHV strains: the origins and global spread of the virus. Semin Cancer Biol. 1999;9:187–199. doi: 10.1006/scbi.1998.0116. [DOI] [PubMed] [Google Scholar]
- 163.Greene W, Gao SJ. Actin dynamics regulate multiple endosomal steps during Kaposi’s sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog. 2009;5:e1000512. doi: 10.1371/journal.ppat.1000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Dezube BJ, Zambela M, Sage DR, Wang JF, Fingeroth JD. Characterization of Kaposi sarcoma-associated herpesvirus/human herpesvirus-8 infection of human vascular endothelial cells: early events. Blood. 2002;100:888–896. doi: 10.1182/blood.v100.3.888. [DOI] [PubMed] [Google Scholar]
- 165.Chen J, et al. Activation of latent Kaposi’s sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc Natl Acad Sci USA. 2001;98:4119–4124. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 167.Simas JP, Efstathiou S. Murine γherpesvirus 68: a model for the study of γherpesvirus pathogenesis. Trends Microbiol. 1998;6:276–282. doi: 10.1016/s0966-842x(98)01306-7. [DOI] [PubMed] [Google Scholar]
- 168.Orzechowska BU, et al. Rhesus macaque rhadinovirus-associated non-Hodgkin lymphoma: animal model for KSHV-associated malignancies. Blood. 2008;112:4227–4234. doi: 10.1182/blood-2008-04-151498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bruce AG, et al. High levels of retroperitoneal fibromatosis (RF)-associated herpesvirus in RF lesions in macaques are associated with ORF73 LANA expression in spindleoid tumour cells. J Gen Virol. 2006;87:3529–3538. doi: 10.1099/vir.0.82339-0. [DOI] [PubMed] [Google Scholar]
- 170.An FQ, et al. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi’s sarcoma-associated herpesvirus latency in vitro and in vivo. J Virol. 2006;80:4833–4846. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mutlu AD, et al. In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma. Cancer Cell. 2007;11:245–258. doi: 10.1016/j.ccr.2007.01.015. A KSHV-BAC-infected line induces lesions resembling KS in a murine model. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Dittmer D, et al. Experimental transmission of Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) to SCID-hu Thy/Liv mice. J Exp Med. 1999;190:1857–1868. doi: 10.1084/jem.190.12.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Parsons CH, et al. KSHV targets multiple leukocyte lineages during long-term productive infection in NOD/SCID mice. J Clin Invest. 2006;116:1963–1973. doi: 10.1172/JCI27249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chang H, et al. Non-human primate model of Kaposi’s sarcoma-associated herpesvirus infection. PLoS Pathog. 2009;5:e1000606. doi: 10.1371/journal.ppat.1000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Boshoff C, Weiss R. AIDS-related malignancies. Nature Rev Cancer. 2002;2:373–382. doi: 10.1038/nrc797. [DOI] [PubMed] [Google Scholar]
- 176.Heckman CA, et al. The tyrosine kinase inhibitor cediranib blocks ligand-induced vascular endothelial growth factor receptor-3 activity and lymphangiogenesis. Cancer Res. 2008;68:4754–4762. doi: 10.1158/0008-5472.CAN-07-5809. [DOI] [PubMed] [Google Scholar]
- 177.Ardavanis A, Doufexis D, Kountourakis P, Rigatos G. A Kaposi’s sarcoma complete clinical response after sorafenib administration. Ann Oncol. 2008;19:1658–1659. doi: 10.1093/annonc/mdn528. [DOI] [PubMed] [Google Scholar]
- 178.Brown LF, Dezube BJ, Tognazzi K, Dvorak HF, Yancopoulos GD. Expression of Tie1, Tie2, and angiopoietins 1, 2, and 4 in Kaposi’s sarcoma and cutaneous angiosarcoma. Am J Pathol. 2000;156:2179–2183. doi: 10.1016/S0002-9440(10)65088-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Moses AV, et al. Kaposi’s sarcoma-associated herpesvirus-induced upregulation of the c-kit proto-oncogene, as identified by gene expression profiling, is essential for the transformation of endothelial cells. J Virol. 2002;76:8383–8399. doi: 10.1128/JVI.76.16.8383-8399.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Koon HB, et al. Imatinib-induced regression of AIDS-related Kaposi’s sarcoma. J Clin Oncol. 2005;23:982–989. doi: 10.1200/JCO.2005.06.079. [DOI] [PubMed] [Google Scholar]
- 181.Stallone G, et al. Sirolimus for Kaposi’s sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–1323. doi: 10.1056/NEJMoa042831. Original description of striking responses of KS lesions after treatment with an mTOR inhibitor. [DOI] [PubMed] [Google Scholar]
- 182.Giraldo G, Beth E, Haguenau F. Herpes-type virus particles in tissue culture of Kaposi’s sarcoma from different geographic regions. J Natl Cancer Inst. 1972;49:1509–1526. doi: 10.1093/jnci/49.6.1509. [DOI] [PubMed] [Google Scholar]
- 183.Huebner K, Ferrari AC, Delli Bovi P, Croce CM, Basilico C. The FGF-related oncogene, K-FGF, maps to human chromosome region 11q13, possibly near int-2. Oncogene Res. 1988;3:263–270. [PubMed] [Google Scholar]
- 184.Cesarman E, et al. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi’s sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood. 1995;86:2708–2714. [PubMed] [Google Scholar]
- 185.Soulier J, et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood. 1995;86:1276–1280. First paper to link KSHV to the pathogenesis of multicentric Castleman’s disease. [PubMed] [Google Scholar]
- 186.Boshoff C, et al. Kaposi’s sarcoma-associated herpesvirus infects endothelial and spindle cells. Nature Med. 1995;1:1274–1278. doi: 10.1038/nm1295-1274. Original paper showing KSHV is present in each tumour (spindle cell) in KS lesions. [DOI] [PubMed] [Google Scholar]
- 187.Renne R, et al. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nature Med. 1996;2:342–346. doi: 10.1038/nm0396-342. First paper describing in vitro lytic growth of the virus. [DOI] [PubMed] [Google Scholar]
- 188.Gao SJ, et al. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nature Med. 1996;2:925–928. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- 189.Kedes DH, et al. The seroepidemiology of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nature Med. 1996;2:918–924. doi: 10.1038/nm0896-918. [DOI] [PubMed] [Google Scholar]
- 190.Moore PS, Boshoff C, Weiss RA, Chang Y. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science. 1996;274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 191.Neipel F, et al. Human herpesvirus 8 encodes a homolog of interleukin-6. J Virol. 1997;71:839–842. doi: 10.1128/jvi.71.1.839-842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Coscoy L, Ganem D. Kaposi’s sarcoma-associated herpesvirus encodes two proteins that block cell surface display of MHC class I chains by enhancing their endocytosis. Proc Natl Acad Sci USA. 2000;97:8051–8056. doi: 10.1073/pnas.140129797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ishido S, et al. Inhibition of natural killer cell-mediated cytotoxicity by Kaposi’s sarcoma-associated herpesvirus K5 protein. Immunity. 2000;13:365–374. doi: 10.1016/s1074-7613(00)00036-4. [DOI] [PubMed] [Google Scholar]
- 194.Ishido S, Wang C, Lee BS, Cohen GB, Jung JU. Downregulation of major histocompatibility complex class I molecules by Kaposi’s sarcoma-associated herpesvirus K3 and K5 proteins. J Virol. 2000;74:5300–5309. doi: 10.1128/jvi.74.11.5300-5309.2000. References 192, 193 and 194 characterized ORFs K3 and K5, which led to the discovery of cellular MIR proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Akula SM, Pramod NP, Wang FZ, Chandran B. Integrin α3β1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell. 2002;108:407–419. doi: 10.1016/s0092-8674(02)00628-1. [DOI] [PubMed] [Google Scholar]
- 196.Sun Q, Matta H, Chaudhary PM. The human herpes virus 8-encoded viral FLICE inhibitory protein protects against growth factor withdrawal-induced apoptosis via NF-κ B activation. Blood. 2003;101:1956–1961. doi: 10.1182/blood-2002-07-2072. [DOI] [PubMed] [Google Scholar]
- 197.Kaleeba JA, Berger EA. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT. Science. 2006;311:1921–1924. doi: 10.1126/science.1120878. [DOI] [PubMed] [Google Scholar]