

TEXT
Molecular biology has made a tremendous impact on the diagnosis and treatment of liver diseases[1]. This review will provide several recent examples. Emphasis will be placed on how molecular biology has influenced the diagnosis of viral hepatitis, autoimmune and metabolic liver diseases. The use of recombinant DNA technology for drug development and the possibility of gene therapy as a treatment modality will then be discussed.
DIAGNOSIS OF VIRAL HEPATITIS
Hepatitis B
Approximately 350000000 individuals are chronically infected with the hepatitis B virus (HBV) worldwide and the disease is endemic in eastern Asia including China[2]. As viral protein antigens can be readily detected in serum, chronic HBV infection can usually be diagnosed by serological assays for the presence of hepatitis B surface antigen (HBsAg). HBsAg is detectable in virtually all infected individuals and the hepatitis B e antigen (HBeAg) is detected in most individuals with high levels of viral replication. Serological assays, usually enzyme linked immunosorbant assays (ELISAs), can utilize either proteins purified from serum or recombinant proteins expressed in yeast or tissue culture cells. ELISAs for detection of antigens can utilize monoclonal antibodies.
Because HBV infection can usually be diagnosed by serological assays, molecular biological methods to measure or detect nucleic acids are usually not necessary in routine clinical diagnosis. In some instances, however, measurement of viral nucleic acids from serum may be helpful in the assessment of patients with chronic hepatitis B. Viral nucleic acid concentrations in serum can be assessed by hybridization to complementary DNA sequences and by a branched chain DNA (bDNA) assay[3]. Such assays can be quantitated to provide estimates of the concentration of viral DNA in serum. As serum viral DNA concentrations may be predictive of prognosis or of response to treatment and prognosis, estimation of viral loads may be useful clinically.
Polymerase chain reaction (PCR) amplification[4,5] of HBV DNA is also relatively easy as HBV is a double stranded DNA virus whose genome is fairly stable in blood and tissue. Sequencing of amplified DNA can be performed to identify mutant viruses of clinical significance. Truncating mutations in the HBV precore gene have been identified that prevent secretion of the e antigen but allow the continued assembly of infectious virus[6,7]. Such mutant strains may be actively replicating even though HBeAg is not detectable using serological assays.
Hepatitis C
In Western countries, molecular biology has arguably had its greatest clinical impact with regards to liver diseases in the diagnosis of viral hepatitis C. Although the virus that causes hepatitis C cannot be propagated in cell culture, molecular biological methods have enabled the identification of this virus and determination of the sequence of its entire genome. This has revolutionized the practice of hepatology.
The hepatitis C virus (HCV) was identified in 1989 by investigators at Chiron Corporation[8]. HCV was identified by antibody screening of cDNA expression libraries made from DNA and RNA from the plasma of chimpanzees infected with serum from humans with what was then called non-A, non-B hepatitis. The expression library was screened with serum antibodies from patients with non-A, non-B hepatitis. This work led to the isolation of cDNA clones that were derived from portions of the HCV genome and encoded fragments of viral polypeptides. The authors also showed that HCV was a positive stranded RNA virus[8] and that the vast majority of individuals with chronic non-A, non-B hepatitis had antibodies against this virus[9].
Following the identification of fragments of the HCV genome, the entire genome was cloned and sequenced in several laboratories[10-13]. This work showed the HCV genome to be a positive stranded RNA of approximately 10000 nucleotides with a single open reading frame encoding a polyprotein of 3010 to 3033 depending upon the strain (Figure 1). The polyprotein is processed by host cell and virally encoded proteases into several structural and non-structural polypeptides (Figure 1). The structural proteins are the core and two envelope polypeptides. The several non structural proteins have various different enzymatic functions.
Figure 1.
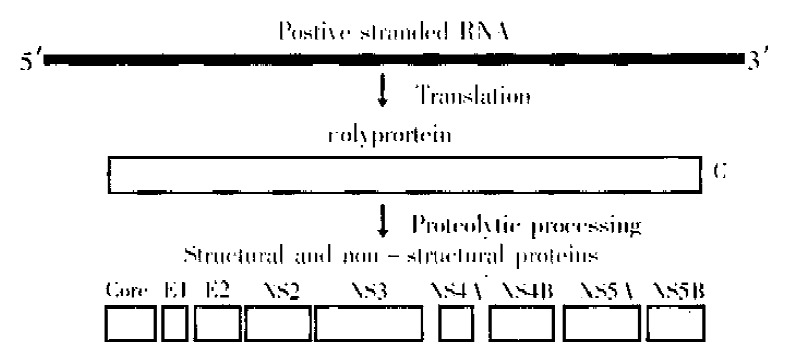
Schematic diagram of the HCV genome and proteins. The positive stranded RNA of about 10000 nucleotides is translated into a polyprotein of approximately 3000 amino acids. This polyprotein is proteolytically cleaved into several smaller proteins. Core, E1 and E2 are structural polypeptides. Core protein is the virus nucleocapsid and E1 and E2 are viral envelope proteins. A small polypeptide known as P7 (not shown) is also produced by additional cleavage between E2 and NS2. The major non-structural proteins are NS2, NS3, NS4 and NS5. NS4 is further processed into NS4A and NS4B and NS5 into NS5A and NS5B. NS2 and part of NS3 are proteases that process the viral polyprotein. NS3 also has RNA helicase activity. NS4A is a cofactor for the NS3 protease and NS5B is an RNA-dependent, RNA polymerase. The functions of NS4B and NS5A are less well understood but NS5A is thought to play a role in determining sensitivity to interferon.
ELISAs utilizing recombinant HCV polypeptides have been available since the discovery of the virus to detect antibodies against HCV. In individuals with risk factors, the presence of anti-HCV antibodies and an elevated serum alanine aminotransferase activity is highly sensitive for the diagnosis of infection. However, false positive antibody tests are not uncommon and some individuals with HCV in fection develop antibodies at low titers that may not be detected.
In the past few years, assays have become routinely available for the detection of HCV RNA in serum. In the bDNA assay, viral RNA is captured by virus-specific nucleotide probes followed by hybridization to branched DNA molecules which are detected by a chemiluminescent substrate system[3,14]. The bDNA assay can be quantitated and is relatively easy to perform in the routine clinical laboratory. However, bDNA is of lower sensitivity than PCR that is now available to detect HCV RNA is serum and tissue. Since the HCV genome is RNA, reverse transcription must be performed before HCV complementary DNA sequences can be amplified by PCR. The amplified cDNA products are separated by agarose gel electrophoresis and detected by a variety of methods such as ethidium bromide staining or Southern hybridization. Care must be taken in the clinical laboratory to avoid contamination when using reverse transcription-PCR to detect HCV RNA, however, the standardization of this method in excellent clinical laboratories has made it the gold-standard for the detection of HCV infection. PCR has even been made semi-quantitative by using competitive inhibitors or highly standardized assay procedures.
The ability to readily reverse transcribe, amplify and s equence HCV RNA has led to the identification of several HCV genotypes[15]. By using different PCR primers and probes to amplify and detect different sequences from various isolates, routine HCV genotype analysis is possible in the clinical laboratory[16,17]. Different genotypes may differ in the severity of disease they cause and the response to treatment[18].
Hepatitis G
Sensitive molecular biological techniques led to the identification of a new virus in 1995 and 1996 in individuals with hepatitis[19,20]. This virus, termed both hepatitis G virus (HGV) and GB-C virus, is a novel Flavivirus with approximately 25% sequence identity to HCV. The initial identification of this virus was originally met with considerable enthusiasm as it was proposed to be a relatively common cause of non-A, non-B, non-C acute and chronic hepatitis. However, subsequent studies[21,22] have questioned the etiological role of HGV/GB-C as a cause of serious liver disease. Most investigators now feel that HGV/GB-C infection is quite common but that it does not cause clinically significant liver disease.
AUTOIMMUNE LIVER DISEASES
Autoimmune liver diseases such as primary biliary cirrhosis, autoimmune hepatitis and sclerosing cholangitis are associated with the presence of variousauto antibodies. Although the pathophysiological significance of these autoantibodies are generally unclear, their detection is central to diagnosis. Basic molecular biological methods have made possible the identification and cDNA cloning of some of the intracellular protein antigens and the predominant epitopes recognized by disease-specific autoantibodies. This work has led to the development of assays for their detection that can be used in the clinical laboratory.
Illustrative examples on the identification of intracellular antigens recognized by autoantibodies in a liver disease come from work on primary biliary cirrhosis (PBC). Almost all individuals with this disease have autoantibodies directed against the E2 subunits of mitochondrial oxo-acid dehydrogenases[23-25] and about 25% of patients have autoantibodies against gp210, an integral membrane protein of the nuclear pore complex[26,27]. Autoantibodies of these specificities are virtually 100% specific for primary biliary cirrhosis.
Screening of bacteriophage lambda cDNA expression libraries with autoantibodies from patients with PBC was used to identify the E2 subunits of the pyruvate dehydrogenase complex, the branched chain 2-oxo-acid dehydrogenase complex and the 2-oxo-glutarate dehydrogenase complex as the mitochondrial autoantigens in this disease[24,25,28-30]. ELISAs utilizing expressed recombinant proteins have been devised that are sensitive and specific for the detection of antibodies against these proteins[23,31,32]. Determination of the immunodominant epitopes the E2 subunits of all three oxo acid dehydrogenases has allowed for the construction a designer hybrid clone that contains the major epitope of each[32]. An immunoassay utilizing a polypeptide expressed from this hybrid clone is highly sensitive and specific for the diagnosis of PBC[32].
In 1990, nuclear pore membrane glycoprotein gp210 was identified as an autoantigen recognized by approximately 25% of individuals with PBC[33]. Using overlapping cDNA for gp210, its immunodominant epitope which was mapped to a stretch of 15 amino acids in this protein of over 1880 amino acids[34]. Based on this information, two ELISAs have been developed for the detection of autoantibodies, one which utilizes a recombinant HJfusion protein expressed in bacteria[35] and the other which uses a synthetic polypeptide[36].
As PBC is a relative uncommon diseases, assays to detect autoantibodies again oxo-acid dehydorgenase subunits and gp210 have not received much commercial attention. These examples nonetheless demonstrate how molecular biology can impact on the diagnosis of a relatively rare disease.
GENE DISCOVERY AND METABOLIC LIVER DISEASES
Advances in positional cloning and genomics have led to the identification of the genes responsible for some of the major metabolic diseases that affect the liver. In the past decade, genes mutated in Wilson disease, hereditary hemochromatosis, congenital hyperbilirubinemias and inherited cholestatic disorders have been discovered (Table 1). These discoveries will permit the use of molecular diagnostic methods to diagnose these disorders. An understanding of the defective genes will also lead to new treatment options. This section reviews some of the major discoveries of genes responsible for metabolic liver diseases in the 1990s. The entry number for On line Mendelian Inheritance in Man (OMIM; http://www3.ncbi.nlm.nih.gov/Omim/) is given for each diseased that is discussed.
Table 1.
Some inherited diseases that affect the liver and the genes that were indentified in the 1990s by molecular biological and genetic methods.
| Disease | Gene |
| Wilson disease | Cu-transporting ATPase |
| Hereditary hemochromatosis | HFE |
| Crigler-Najjar syndrome | UDP-glucuronosyltransferase |
| Dubin-Johnson syndrome | cMOAT |
| Benign recurrent intrahepatic cholestasis | P-type ATPase |
| Progressive familial Intrahepatic Cholestasis Type 1 | P-type ATPase |
Wilson disease (OMIM # 277900)
Wilson disease is an autosomally inherited disorder that causes changes in the basal ganglia and liver that respectively lead to neuropsychiatric disease, hepatitis and cirrhosis. Abnormalities in serum ceruloplasmin, urinary copper excretion and copper accumulation in the liver have for many years suggested a primary defect in copper metabolism as the cause. In 1985, the Wilson disease gene was shown to be linked to the esterase D locus on chromosome 13[37]. Over the next eight years, the Wilson disease locus was more precisely localized by genetic analysis of various families and individuals and was placed at the junction of band q14.3 and q21.1 on chromosome 13 in 1993 by using fluorescence in situ hybridization studies of chromosomal aberrations[38]. Finally, in 1993, several yeast artificial chromosomes spanning this region were molecularly cloned and a gene encoding a P-type ATPase that was mutated in individuals with Wilson disease was identified[39-41]. This ATPase was highly similar to the ATPase previously shown to be responsible for Menke disease, another disorder of copper metabolism, and is thought to be a copper-translocating ATPase. At least 70 different mutations have since been described in this copper-ATPase gene in individuals with Wilson disease[42].
Hereditary hemochromatosis (OMIM #235200)
Hereditary hemochromatosis is the most common inherited disease in individuals of European descent[43]. Homozygous individuals suffer from the complications of excess iron deposition in the liver, heart, joints and some endocrine organs. Excessive hepatocyte iron causes hepatitis and cirrhosis. The disease often goes undiagnosed which is especially unfortunate because phlebotomy is effective treatment.
In the 1970’s, hereditary hemochromatosis was linked to the HLA-A locus on chromosome[44-46]. In 1996, investigators at Mercator Genetics[47] used linkage disequilibrium and full haplotype analysis to identify a candidate gene for hemochromatosis on chromosome 6. They termed this gene HLA-H because of its homology to other MHC class I family members; the current accepted designation of this gene is HFE. A guanine to adenine transition at coding nucleotide 845 of this gene resulted in a cysteine to tyrosine substitution at amino acid residue 282 in the protein in 85% of 178 patients they examined. Several other studies subsequently confirmed the mutations identified by the group at Mercator Genetics[48-50] and the cysteine to tyrosine substitution in HFE is now felt to be present in between 70% and 100% of individuals with hereditary hemochromatosis[51]. Another mutation in HFE that converts histidine 63 to aspartate also increases the relative risk of for the development of hemochromatosis in individuals who are heterozygous for the cysteine to tyrosine substitution at amino acid 282[47,52].
When the HFE gene was discovered, it was unclear how a protein with homology to MHC class I proteins could be involved in the regulation of iron metabolism. Recently, however, it has been shown that the protein encoded by HFE associates with the transferrin receptor and decreases the affinity of transferrin receptor for iron bound transferrin[53-55]. The cysteine to tyrosine mutant protein does not interact with transferrin receptor and hence does not decrease its affinity for iron-bound transferrin, while the histidine to aspartate mutant protein associates with transferrin receptor but does not decrease its affinity for iron bound transferrin as much as the wild type protein[54]. These findings can explain how mutations in the HFE gene product cause iron overload.
Crigler-Najjar syndrome type 1 (OMIM #218800)
Individuals with Crigler-Najjar syndrome type 1 have a complete absence of activity of the UDP-glucuronosyltransferase isoform that catalyzes the conjugation of bilirubin to mono and diglucuronides[56,57]. Affected individuals present with severe childhood disease manifested by jaundice, kernicterus, resultant abnormal neurologic development and early death. Patients with Crigler-Najjar syndrome type 2 have a partial deficiency of this enzyme activity and generally survive through adulthood without significant problems[57]. A single gene of chromosome 2 called UGT1 encodes bilirubin, phenol, and other UDP glucuronosyltransferase isozymes with identical carboxyl termini[58]. Several different mutations in UGT1 have since been described in individuals with Crigler Najjar syndrome type 1[58-61].
Dubin-Johnson syndrome (OMIM #237500)
Dubin-Johnson syndrome results form the inability of conjugated bilirubin to be secreted from hepatocytes. A cDNA for a rat multispecific organic anion transporter (cMOAT), a protein homologous to the multidrug resistance proteins that is located in the canalicular membrane of hepatocytes, was characterized in 1996[62]. In 1997, the human orthologue of rat cMOAT was mapped to chromosome 10q24[63]. The human cMOAT cDNA was cloned and sequenced in 1997[64] and mutations in this gene have been identified in individuals with Dubin Johnson syndrome[64,65].
Benign recurrent intrahepatic cholestasis (OMIM #243300) and progressive familial intrahepatic cholestasis type 1 (OMIM #211600)
Benign recurrent cholestasis type 1 or Summerskill syndrome is characterized by intermittent episodes of cholestasis without extrahepatic bile duct obstruction. The symptoms usually spontaneously resolve after periods of weeks to months and patients generally have a good prognosis. In 1994, a candidate gene was localized by linkage disequilibrium to chromosome 18[66], and in 1997, the localization was further refined to chromosome 18q21[67]. Progressive intrahepatic cholestasis or Byler disease was first described in the OLd Order Amish. This condition is severe and usually leads to death within the first decade of life. Because both benign recurrent intrahepatic cholestasis type 1 and progressive familial intrahepatic cholestasis have similar symptoms, albeit different prognoses, it was suspected that they may be caused by different mutations in the same gene. In 1995, the gene for progressive familial intrahepatic cholestasis was mapped to chromosome 18q21-22, the benign recurrent intrahepatic cholestasis region, in an extended Amish kindred[68]. In 1998, a gene at this locus that encodes a P-type ATPase was identified[69]. Different mutations in the gene encoding this P-type ATPase were identified in individuals with benign recurrent intrahepatic cholestasis and progressive familial intrahepatic cholestasis type 1. The P-type ATPases responsible for these two diseases likely actively transports bile salts across cell membranes. Depending upon the specific mutation in this P-type ATPase, a cholestatic disorder with a relatively benign or very severe phenotype can result.
MOLECULAR BIOLOGY AND DRUG DEVELOPMENT
Recombinant vaccines to prevent viral hepatitis
Molecular biology has had an impact on the development of safe vaccines to prevent viral hepatitis. The greatest impact has been in the development of vaccines against HBV. Highly effective HBV vaccines have been produced by recombinant DNA technology[70,71]. For these preparations, HBV surface antigen is generally produced as a recombinant protein in yeast. In a landmark study published in 1997, it was demonstrated that universal hepatitis B vaccination reduced the incidence of hepatocellular carcinoma in children in Taiwan[72]. Universal vaccination with relatively inexpensive preparations produced by recombinant DNA technology could lead to the eradication of this disease. The use of recombinant proteins for vaccination against hepatitis viruses other than HBV has lagged. Preliminary results in monkeys suggest that vaccination with a recombinant protein containing part of the viral capsid antigen may be protective against hepatitis E[73].
Recombinant interferons for viral hepatitis
Interferon alpha is used in the treatment of chronic hepatitis B and C[74]. Cloning of the cDNA for interferon alpha has made it possible to produce it using recombinant DNA technology. Several slightly different recombinant preparations with various amino acid substitions are available. Although much leaves to be desired regarding long-term cure rates of treated individuals, recombinant interferons currently provide a partially effective first line treatment option for individuals with hepatitis B and C.
Rational drug design
Determination of the three-dimensional structures of target molecules facilitates the design of compounds that inhibit or possibly enhance the functions of the target. For example, inhibitors of an enzyme can be rationally designed if the three-dimensional structure of the active site is known. Structure based design can be combined with high throughput combinatorial methods to rapidly identify compounds that are effective against a target.
The ability to express portions of proteins from recombinant cDNA clones has tremendously simplified the ability to purify starting materials for structural studies. Improvements in methods for X-ray crystallography and nuclear magnetic resonance spectroscopy have also reduced the amount of time and starting material required for accurate structure determination. These advances have recently had an impact in the rational design of drugs to treat liver diseases as exemplified by the structure determinations of several non structural proteins of HCV (Table 2). The NS3 polypeptides has two functional domains, one with protease activity that cleaves the viral polyprotein at several sites and the other with RNA helicase activity that unwinds viral RNA. Both of these activities are necessary for viral replication. Expression of recombinant polypeptides of the protease and helicase domains of NS3, as well as the protease domain complexed with its co-factor NS4A, has permitted their three dimensional structures to be determined[75-78]. Based on this knowledge, pharmaceutical and biotechnology companies are in the process of rationally designing inhibitors of these essential viral enzymatic functions.
Table 2.
HCV non-structural proteins with three-dimensional structures determined and published in the literature
| Polypeptide | Function | Reference |
| NS3 protease domain | Protease that cleaves HCV polyprotein at several locations | 75, 76 |
| NS4A (complexed to NS3) | Cofactor for NS3 protease | 76 |
| NS3 RNA helicase domain | Unwinds viral RNA | 77, 78 |
Novel processes have also recently been used to determine the structures of hepatitis viral proteins. Despite many efforts, the core protein of HBV has eluded crystallization. However, high resolution cryoelectron microscopy has recently been used to establish the three-dimensional structure of complexes of this protein[79,80]. This knowledge can also provide a rational foundation to develop inhibitors of HBV particle assembly.
With regards to liver diseases, rational drug design is not limited to hepatitis virus. For example, the domain of the hemochromatosis protein HFE that binds to transferrin receptor has been expressed as a recombinant protein and its three-dimensional structure determined by X-ray crystallography[55]. This knowledge can lead to the potential design of drugs that bind to transferrin receptor in a similar fashion to inhibit cellular iron uptake that may be increased in hereditary hemochromatosis because of defective HFE binding.
Gene therapy
Gene therapy for gastrointestinal and liver diseases have been reviewed in detail elsewhere[81,82]. A detailed discussion of hepatic gene therapy is beyond the scope of this paper and only some of the major areas are touched upon here. Hepatic gene therapy will probably have the greatest initial impact in the treatment of inherited metabolic diseases but variations on classical techniques may also find utility in the treatment of viral diseases, cancer and in the production of vaccines.
In ex vivo hepatic gene therapy, hepatocytes are removed from the patient, cultured in vitro and the desired expression vector is introduced into the cultured hepatocytes. The transduced or transfected hepatocytes are then introduced into the portal vein and lodge in the patient’s liver. Hepatocyte directed ex vivo gene therapy has already been performed in human subjects with familial hypercholesterolemia using the low density lipoprotein (LDL) receptor gene[83]. Several treated patients had persistent and significant reductions in serum cholesterol and LDL concentrations.
In vivo gene therapy is also an option for the treatment of liver diseases. In in vivo gene therapy, gene transfer vectors are introduced directly into the patient and taken up by the liver. Vectors for hepatic in vivo gene therapy include DNA complexed with proteins, DNA in liposomes, naked DNA and hepatotropic viral vectors such as those based on adenovirus.
Besides the transfer of human genes, other nucleic acid based therapies may be useful in the treatment of liver diseases. Antisense oligonucleotides[84] can be used to inhibit the expression of genes such as those essential for the replication of hepatitis viruses. Ribozymes are catalytic RNA molecules that can also be used for similar purposes[85]. Direct injection of DNA vectors into human cells may also be used for the production of vaccines against viral hepatitis[86]. All of these techniques are in their infancy, however, clinical trials will likely be common in the next decade.
CONCLUSIONS
Molecular biology will continue to have a tremendous impact on the diagnosis and treatment of liver diseases. The basic research conducted in the past three decades will revolutionize the way hepatology, and all of medicine, is practiced. As a result, common conditions such as viral hepatitis B and C and inherited metabolic diseases affecting the liver such as hemochromatosis will hopefully vanish from the world.
References
- 1.Worman HJ. Molecular biological methods in diagnosis and treatment of liver diseases. Clin Chem. 1997;43:1476–1486. [PubMed] [Google Scholar]
- 2.Mamiya N, Worman , HJ Epidemiology, prevention, clinical features and therapy of hepatitis B, C, and G. Curr Opin Infect Dis. 1997;10:390–397. [Google Scholar]
- 3.Urdea MS. Synthesis and characterization of branch DNA for the direct and quantitative detection of CMV, HBV, HCV and HIV. Clin Chem. 1993;39:725–726. [Google Scholar]
- 4.Saiki RK, Scharf S, Faloona F, Mullis KB, Horn GT, Erlich HA, Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 1985;230:1350–1354. doi: 10.1126/science.2999980. [DOI] [PubMed] [Google Scholar]
- 5.Mullis KB, Faloona FA. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 1987;155:335–350. doi: 10.1016/0076-6879(87)55023-6. [DOI] [PubMed] [Google Scholar]
- 6.Carman WF, Jacyna MR, Hadziyannis S, Karayiannis P, McGarvey MJ, Makris A, Thomas HC. Mutation preventing formation of hepatitis B e antigen in patients with chronic hepatitis B infection. Lancet. 1989;2:588–591. doi: 10.1016/s0140-6736(89)90713-7. [DOI] [PubMed] [Google Scholar]
- 7.Ulrich PP, Bhat RA, Kelly I, Brunetto MR, Bonino F, Vyas GN. A precore-defective mutant of hepatitis B virus associated with e antigen-negative chronic liver disease. J Med Virol. 1990;32:109–118. doi: 10.1002/jmv.1890320208. [DOI] [PubMed] [Google Scholar]
- 8.Choo QL, Kuo G, Weiner AJ, Overby LR, Bradley DW, Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 9.Kuo G, Choo QL, Alter HJ, Gitnick GL, Redeker AG, Purcell RH, Miyamura T, Dienstag JL, Alter MJ, Stevens CE. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244:362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 10.Kato N, Hijikata M, Ootsuyama Y, Nakagawa M, Ohkoshi S, Sugimura T, Shimotohno K. Molecular cloning of the human hepatitis C virus genome from Japanese patients with non-A, non-B hepatitis. Proc Natl Acad Sci USA. 1990;87:9524–9528. doi: 10.1073/pnas.87.24.9524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choo QL, Richman KH, Han JH, Berger K, Lee C, Dong C, Gallegos C, Coit D, Medina-Selby R, Barr PJ. Genetic organization and diversity of the hepatitis C virus. Proc Natl Acad Sci USA. 1991;88:2451–2455. doi: 10.1073/pnas.88.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Okamoto H, Okada S, Sugiyama Y, Kurai K, Iizuka H, Machida A, Miyakawa Y, Mayumi M. Nucleotide sequence of the genomic RNA of hepatitis C virus isolated from a human carrier: comparison with reported isolates for conserved and divergent regions. J Gen Virol. 1991;72(Pt 11):2697–2704. doi: 10.1099/0022-1317-72-11-2697. [DOI] [PubMed] [Google Scholar]
- 13.Takamizawa A, Mori C, Fuke I, Manabe S, Murakami S, Fujita J, Onishi E, Andoh T, Yoshida I, Okayama H. Structure and organization of the hepatitis C virus genome isolated from human carriers. J Virol. 1991;65:1105–1113. doi: 10.1128/jvi.65.3.1105-1113.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Urdea MS. Branched DNA signal amplification. Biotechnology (N Y) 1994;12:926–928. doi: 10.1038/nbt0994-926. [DOI] [PubMed] [Google Scholar]
- 15.Simmonds P, Smith DB, McOmish F, Yap PL, Kolberg J, Urdea MS, Holmes EC. Identification of genotypes of hepatitis C virus by sequence comparisons in the core, E1 and NS-5 regions. J Gen Virol. 1994;75(Pt 5):1053–1061. doi: 10.1099/0022-1317-75-5-1053. [DOI] [PubMed] [Google Scholar]
- 16.Lau JY, Mizokami M, Kolberg JA, Davis GL, Prescott LE, Ohno T, Perrillo RP, Lindsay KL, Gish RG, Qian KP. Application of six hepatitis C virus genotyping systems to sera from chronic hepatitis C patients in the United States. J Infect Dis. 1995;171:281–289. doi: 10.1093/infdis/171.2.281. [DOI] [PubMed] [Google Scholar]
- 17.Lau JY, Davis GL, Prescott LE, Maertens G, Lindsay KL, Qian K et al. Distri-bution of hepatitis C virus genotypes determined by line probe assay in pa-tients with chronic hepatitis C seen at tertiary referral centers in the United States. Ann Intern Med. 1996;124:868–876. doi: 10.7326/0003-4819-124-10-199605150-00002. [DOI] [PubMed] [Google Scholar]
- 18.Zein NN, Rakela J, Krawitt EL, Reddy KR, Tominaga T, Persing DH. Hepatitis C virus genotypes in the United States: epidemiology, pathogenicity, and response to interferon therapy. Collaborative Study Group. Ann Intern Med. 1996;125:634–639. doi: 10.7326/0003-4819-125-8-199610150-00002. [DOI] [PubMed] [Google Scholar]
- 19.Simons JN, Leary TP, Dawson GJ, Pilot-Matias TJ, Muerhoff AS, Schlauder GG, Desai SM, Mushahwar IK. Isolation of novel virus-like sequences associated with human hepatitis. Nat Med. 1995;1:564–569. doi: 10.1038/nm0695-564. [DOI] [PubMed] [Google Scholar]
- 20.Linnen J, Wages J, Zhang-Keck ZY, Fry KE, Krawczynski KZ, Alter H, Koonin E, Gallagher M, Alter M, Hadziyannis S, et al. Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science. 1996;271:505–508. doi: 10.1126/science.271.5248.505. [DOI] [PubMed] [Google Scholar]
- 21.Alter MJ, Gallagher M, Morris TT, Moyer LA, Meeks EL, Krawczynski K, Kim JP, Margolis HS. Acute non-A-E hepatitis in the United States and the role of hepatitis G virus infection. Sentinel Counties Viral Hepatitis Study Team. N Engl J Med. 1997;336:741–746. doi: 10.1056/NEJM199703133361101. [DOI] [PubMed] [Google Scholar]
- 22.Alter HJ, Nakatsuji Y, Melpolder J, Wages J, Wesley R, Shih JW, Kim JP. The incidence of transfusion-associated hepatitis G virus infection and its relation to liver disease. N Engl J Med. 1997;336:747–754. doi: 10.1056/NEJM199703133361102. [DOI] [PubMed] [Google Scholar]
- 23.Van de Water J, Cooper A, Surh CD, Coppel R, Danner D, Ansari A, Dickson R, Gershwin ME. Detection of autoantibodies to recombinant mitochondrial proteins in patients with primary biliary cirrhosis. N Engl J Med. 1989;320:1377–1380. doi: 10.1056/NEJM198905253202104. [DOI] [PubMed] [Google Scholar]
- 24.Gershwin ME, Coppel RL, Mackay IR. Primary biliary cirrhosis and mitochondrial autoantigens--insights from molecular biology. Hepatology. 1988;8:147–151. doi: 10.1002/hep.1840080128. [DOI] [PubMed] [Google Scholar]
- 25.Gershwin ME, Mackay IR. Primary biliary cirrhosis: paradigm or paradox for autoimmunity. Gastroenterology. 1991;100:822–833. doi: 10.1016/0016-5085(91)80033-6. [DOI] [PubMed] [Google Scholar]
- 26.Worman HJ, Courvalin JC. Autoantibodies against nuclear envelope proteins in liver disease. Hepatology. 1991;14:1269–1279. [PubMed] [Google Scholar]
- 27.Courvalin JC, Worman HJ. Nuclear envelope protein autoantibodies in primary biliary cirrhosis. Semin Liver Dis. 1997;17:79–90. doi: 10.1055/s-2007-1007185. [DOI] [PubMed] [Google Scholar]
- 28.Coppel RL, McNeilage LJ, Surh CD, Van de Water J, Spithill TW, Whittingham S et al. Primary structure of the human M2 mitochondria autoantigen of primary biliary cirrhosis: dihydrolipoamide acetyltransferase. Proc Natl Acad Sci USA. 1988;85:7317–7321. doi: 10.1073/pnas.85.19.7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fussey SP, Guest JR, James OF, Bassendine MF, Yeaman SJ. Identification and analysis of the major M2 autoantigens in primary biliary cirrhosis. Proc Natl Acad Sci USA. 1988;85:8654–8658. doi: 10.1073/pnas.85.22.8654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surh CD, Danner DJ, Ahmed A, Coppel RL, Mackay IR, Dickson ER et al. Reactivity of primary biliary cirrhosis sera with a human fetal liver cDNA line of branched chain alpha keto acid dehydrogenase dihydrolipoamide acetyltransferase, the 52 kD mitochondrial antigen. Hepatology. 1989;9:63–68. doi: 10.1002/hep.1840090110. [DOI] [PubMed] [Google Scholar]
- 31.Leung PS, Iwayama T, Prindiville T, Chuang DT, Ansari AA, Wynn RM, Dickson R, Coppel R, Gershwin ME. Use of designer recombinant mitochondrial antigens in the diagnosis of primary biliary cirrhosis. Hepatology. 1992;15:367–372. doi: 10.1002/hep.1840150302. [DOI] [PubMed] [Google Scholar]
- 32.Moteki S, Leung PS, Coppel RL, Dickson ER, Kaplan MM, Munoz S, Gershwin ME. Use of a designer triple expression hybrid clone for three different lipoyl domain for the detection of antimitochondrial autoantibodies. Hepatology. 1996;24:97–103. doi: 10.1002/hep.510240117. [DOI] [PubMed] [Google Scholar]
- 33.Courvalin JC, Lassoued K, Bartnik E, Blobel G, Wozniak RW. The 210-kD nuclear envelope polypeptide recognized by human autoantibodies in primary biliary cirrhosis is the major glycoprotein of the nuclear pore. J Clin Invest. 1990;86:279–285. doi: 10.1172/JCI114696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nickowitz RE, Worman HJ. Autoantibodies from patients with primary biliary cirrhosis recognize a restricted region within the cytoplasmic tail of nuclear pore membrane glycoprotein Gp210. J Exp Med. 1993;178:2237–2242. doi: 10.1084/jem.178.6.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tartakovsky F, Worman HJ. Detection of Gp210 autoantibodies in primary biliary cirrhosis using a recombinant protein containing the predominant autoepitope. Hepatology. 1995;21:495–500. [PubMed] [Google Scholar]
- 36.Bandin O, Courvalin JC, Poupon R, Dubel L, Homberg JC, Johanet C. Specificity and sensitivity of gp210 autoantibodies detected using an enzyme-linked immunosorbent assay and a synthetic polypeptide in the diagnosis of primary biliary cirrhosis. Hepatology. 1996;23:1020–1024. doi: 10.1002/hep.510230512. [DOI] [PubMed] [Google Scholar]
- 37.Frydman M, Bonné-Tamir B, Farrer LA, Conneally PM, Magazanik A, Ashbel S, Goldwitch Z. Assignment of the gene for Wilson disease to chromosome 13: linkage to the esterase D locus. Proc Natl Acad Sci USA. 1985;82:1819–1821. doi: 10.1073/pnas.82.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kooy RF, Van der Veen AY, Verlind E, Houwen RH, Scheffer H, Buys CH. Physical localisation of the chromosomal marker D13S31 places the Wilson disease locus at the junction of bands q14.3 and q21.1 of chromosome 13. Hum Genet. 1993;91:504–506. doi: 10.1007/BF00217780. [DOI] [PubMed] [Google Scholar]
- 39.Bull PC, Thomas GR, Rommens JM, Forbes JR, Cox DW. The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet. 1993;5:327–337. doi: 10.1038/ng1293-327. [DOI] [PubMed] [Google Scholar]
- 40.Petrukhin K, Fischer SG, Pirastu M, Tanzi RE, Chernov I, Devoto M, Brzustowicz LM, Cayanis E, Vitale E, Russo JJ. Mapping, cloning and genetic characterization of the region containing the Wilson disease gene. Nat Genet. 1993;5:338–343. doi: 10.1038/ng1293-338. [DOI] [PubMed] [Google Scholar]
- 41.Tanzi RE, Petrukhin K, Chernov I, Pellequer JL, Wasco W, Ross B, Romano DM, Parano E, Pavone L, Brzustowicz LM. The Wilson disease gene is a copper transporting ATPase with homology to the Menkes disease gene. Nat Genet. 1993;5:344–350. doi: 10.1038/ng1293-344. [DOI] [PubMed] [Google Scholar]
- 42.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: spectrum of mutations and their consequences. Nat Genet. 1995;9:210–217. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- 43.Edwards CQ, Griffen LM, Goldgar D, Drummond C, Skolnick MH, Kushner JP. Prevalence of hemochromatosis among 11,065 presumably healthy blood donors. N Engl J Med. 1988;318:1355–1362. doi: 10.1056/NEJM198805263182103. [DOI] [PubMed] [Google Scholar]
- 44.Simon M, Bourel M, Fauchet R, Genetet B. Association of HLA-A3 and HLA-B14 antigens with idiopathic haemochromatosis. Gut. 1976;17:332–334. doi: 10.1136/gut.17.5.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevens FM, Walters JM, Watt DW, McCarthy CF. Inheritance of idiopathic haemochromatosis. Lancet. 1977;1:1107. [Google Scholar]
- 46.Cartwright GE, Skolnick M, Amos DB, Edwards CQ, Kravitz K, Johnson A. Inheritance of hemochromatosis: linkage to HLA. Trans Assoc Am Physicians. 1978;91:273–281. [PubMed] [Google Scholar]
- 47.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Ellis MC, Fullan A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 48.Beutler E, Gelbart T, West C, Lee P, Adams M, Blackstone R, Pockros P, Kosty M, Venditti CP, Phatak PD, et al. Mutation analysis in hereditary hemochromatosis. Blood Cells Mol Dis. 1996;22:187–194; discussion 194a-194b. doi: 10.1006/bcmd.1996.0027. [DOI] [PubMed] [Google Scholar]
- 49.Jazwinska EC, Cullen LM, Busfield F, Pyper WR, Webb SI, Powell LW, Morris CP, Walsh TP. Haemochromatosis and HLA-H. Nat Genet. 1996;14:249–251. doi: 10.1038/ng1196-249. [DOI] [PubMed] [Google Scholar]
- 50.Jouanolle AM, Gandon G, Jézéquel P, Blayau M, Campion ML, Yaouanq J, Mosser J, Fergelot P, Chauvel B, Bouric P, et al. Haemochromatosis and HLA-H. Nat Genet. 1996;14:251–252. doi: 10.1038/ng1196-251. [DOI] [PubMed] [Google Scholar]
- 51.Cuthbert JA. Iron, HFE, and hemochromatosis update. J Investig Med. 1997;45:518–529. [PubMed] [Google Scholar]
- 52.Beutler E. The significance of the 187G (H63D) mutation in hemochromatosis. Am J Hum Genet. 1997;61:762–764. [PMC free article] [PubMed] [Google Scholar]
- 53.Parkkila S, Waheed A, Britton RS, Bacon BR, Zhou XY, Tomatsu S, Fleming RE, Sly WS. Association of the transferrin receptor in human placenta with HFE, the protein defective in hereditary hemochromatosis. Proc Natl Acad Sci USA. 1997;94:13198–13202. doi: 10.1073/pnas.94.24.13198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feder JN, Penny DM, Irrinki A, Lee VK, Lebrón JA, Watson N, Tsuchihashi Z, Sigal E, Bjorkman PJ, Schatzman RC. The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding. Proc Natl Acad Sci USA. 1998;95:1472–1477. doi: 10.1073/pnas.95.4.1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lebrón JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111–123. doi: 10.1016/s0092-8674(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 56.Crigler JF, NAJJAR VA. Congenital familial nonhemolytic jaundice with kernicterus. Pediatrics. 1952;10:169–180. [PubMed] [Google Scholar]
- 57.Arias IM, Gartner LM, Cohen M, Ben-Ezzer J, Levi AJ. Chronic nonhemolytic unconjugated hyperbilirubinemia with glucuronyl transferase deficiency: clinical, biochemical, pharmacologic and genetic evidence for heterogeneity. Am J Med. 1969;47:395–409. doi: 10.1016/0002-9343(69)90224-1. [DOI] [PubMed] [Google Scholar]
- 58.Ritter JK, Chen F, Sheen YY, Tran HM, Kimura S, Yeatman MT, Owens IS. A novel complex locus UGT1 encodes human bilirubin, phenol, and other UDP-glucuronosyltransferase isozymes with identical carboxyl termini. J Biol Chem. 1992;267:3257–3261. [PubMed] [Google Scholar]
- 59.Bosma PJ, Chowdhury JR, Huang TJ, Lahiri P, Elferink RP, Van Es HH, Lederstein M, Whitington PF, Jansen PL, Chowdhury NR. Mechanisms of inherited deficiencies of multiple UDP-glucuronosyltransferase isoforms in two patients with Crigler-Najjar syndrome, type I. FASEB J. 1992;6:2859–2863. doi: 10.1096/fasebj.6.10.1634050. [DOI] [PubMed] [Google Scholar]
- 60.Ritter JK, Yeatman MT, Kaiser C, Gridelli B, Owens IS. A phenylalanine codon deletion at the UGT1 gene complex locus of a Crigler-Najjar type I patient generates a pH-sensitive bilirubin UDP-glucuronosyltransferase. J Biol Chem. 1993;268:23573–23579. [PubMed] [Google Scholar]
- 61.Aono S, Yamada Y, Keino H, Sasaoka Y, Nakagawa T, Onishi S, Mimura S, Koiwai O, Sato H. A new type of defect in the gene for bilirubin uridine 5'-diphosphate-glucuronosyltransferase in a patient with Crigler-Najjar syndrome type I. Pediatr Res. 1994;35:629–632. doi: 10.1203/00006450-199406000-00002. [DOI] [PubMed] [Google Scholar]
- 62.Paulusma CC, Bosma PJ, Zaman GJ, Bakker CT, Otter M, Scheffer GL, Scheper RJ, Borst P, Oude Elferink RP. Congenital jaundice in rats with a mutation in a multidrug resistance-associated protein gene. Science. 1996;271:1126–1128. doi: 10.1126/science.271.5252.1126. [DOI] [PubMed] [Google Scholar]
- 63.van Kuijck MA, Kool M, Merkx GFM, Geurts van Kessel A, Bindels RJM, Deen PMT et al. Assignment of the canalicular multispecific organic anion transporter gene (CMOAT) to human chromosome 10q24 and mouse chro-mosome 19D2 by fluorescent in situ hybridization. Cytogenet Cell Genet. 1997;77:285–287. doi: 10.1159/000134599. [DOI] [PubMed] [Google Scholar]
- 64.Paulusma CC, Kool M, Bosma PJ, Scheffer GL, ter Borg F, Scheper RJ, Tytgat GN, Borst P, Baas F, Oude Elferink RP. A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology. 1997;25:1539–1542. doi: 10.1002/hep.510250635. [DOI] [PubMed] [Google Scholar]
- 65.Wada M, Toh S, Taniguchi K, Nakamura T, Uchiumi T, Kohno K, Yoshida I, Kimura A, Sakisaka S, Adachi Y, et al. Mutations in the canilicular multispecific organic anion transporter (cMOAT) gene, a novel ABC transporter, in patients with hyperbilirubinemia II/Dubin-Johnson syndrome. Hum Mol Genet. 1998;7:203–207. doi: 10.1093/hmg/7.2.203. [DOI] [PubMed] [Google Scholar]
- 66.Houwen RH, Baharloo S, Blankenship K, Raeymaekers P, Juyn J, Sandkuijl LA, Freimer NB. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat Genet. 1994;8:380–386. doi: 10.1038/ng1294-380. [DOI] [PubMed] [Google Scholar]
- 67.Sinke RJ, Carlton VE, Juijn JA, Delhaas T, Bull L, van Berge Henegouwen GP, van Hattum J, Keller KM, Sinaasappel M, Bijleveld CM, et al. Benign recurrent intrahepatic cholestasis (BRIC): evidence of genetic heterogeneity and delimitation of the BRIC locus to a 7-cM interval between D18S69 and D18S64. Hum Genet. 1997;100:382–387. doi: 10.1007/s004390050520. [DOI] [PubMed] [Google Scholar]
- 68.Carlton VEH, Knisely AS, Freimer NB. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the be-nign recurrent intrahepatic cholestasis region. Hum Molec Genet. 1995;4:1049–1053. doi: 10.1093/hmg/4.6.1049. [DOI] [PubMed] [Google Scholar]
- 69.Bull LN, van Eijk MJ, Pawlikowska L, DeYoung JA, Juijn JA, Liao M, Klomp LW, Lomri N, Berger R, Scharschmidt BF, et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18:219–224. doi: 10.1038/ng0398-219. [DOI] [PubMed] [Google Scholar]
- 70.McAleer WJ, Buynak EB, Maigetter RZ, Wampler DE, Miller WJ, Hilleman MR. Human hepatitis B vaccine from recombinant yeast. Nature. 1984;307:178–180. doi: 10.1038/307178a0. [DOI] [PubMed] [Google Scholar]
- 71.Lemon SM, Thomas DL. Vaccines to prevent viral hepatitis. N Engl J Med. 1997;336:196–204. doi: 10.1056/NEJM199701163360307. [DOI] [PubMed] [Google Scholar]
- 72.Chang MH, Chen CJ, Lai MS, Hsu HM, Wu TC, Kong MS, Liang DC, Shau WY, Chen DS. Universal hepatitis B vaccination in Taiwan and the incidence of hepatocellular carcinoma in children. Taiwan Childhood Hepatoma Study Group. N Engl J Med. 1997;336:1855–1859. doi: 10.1056/NEJM199706263362602. [DOI] [PubMed] [Google Scholar]
- 73.Tsarev SA, Tsareva TS, Emerson SU, Govindarajan S, Shapiro M, Gerin JL, Purcell RH. Successful passive and active immunization of cynomolgus monkeys against hepatitis E. Proc Natl Acad Sci USA. 1994;91:10198–10202. doi: 10.1073/pnas.91.21.10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoofnagle JH, di Bisceglie AM. The treatment of chronic viral hepatitis. N Engl J Med. 1997;336:347–356. doi: 10.1056/NEJM199701303360507. [DOI] [PubMed] [Google Scholar]
- 75.Love RA, Parge HE, Wickersham JA, Hostomsky Z, Habuka N, Moomaw EW, Adachi T, Hostomska Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell. 1996;87:331–342. doi: 10.1016/s0092-8674(00)81350-1. [DOI] [PubMed] [Google Scholar]
- 76.Kim JL, Morgenstern KA, Lin C, Fox T, Dwyer MD, Landro JA, Chambers SP, Markland W, Lepre CA, O'Malley ET, et al. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–355. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 77.Yao N, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- 78.Kim JL, Morgenstern KA, Griffith JP, Dwyer MD, Thomson JA, Murcko MA, Lin C, Caron PR. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure. 1998;6:89–100. doi: 10.1016/s0969-2126(98)00010-0. [DOI] [PubMed] [Google Scholar]
- 79.Böttcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;386:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- 80.Conway JF, Cheng N, Zlotnick A, Wingfield PT, Stahl SJ, Steven AC. Visualization of a 4-helix bundle in the hepatitis B virus capsid by cryo-electron microscopy. Nature. 1997;386:91–94. doi: 10.1038/386091a0. [DOI] [PubMed] [Google Scholar]
- 81.Wilson JM, Askari FK. Hepatic and gastrointestinal gene therapy. In: Yamada T, ed , editors. Textbook of Gastroenterology (Yamada T, ed. Gastroenterology Updates, Vol. 1) Philadelphia: J.B. Lippincott Co; 1996. pp. 1–20. [Google Scholar]
- 82.Xu CT, Pan BR. Current status of gene therapy in gastroenterology. World J Gastroenterol. 1998;4:85–89. doi: 10.3748/wjg.v4.i1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grossman M, Raper SE, Kozarsky K, Stein EA, Engelhardt JF, Muller D, Lupien PJ, Wilson JM. Successful ex vivo gene therapy directed to liver in a patient with familial hypercholesterolaemia. Nat Genet. 1994;6:335–341. doi: 10.1038/ng0494-335. [DOI] [PubMed] [Google Scholar]
- 84.Askari FK, McDonnell WM. Antisense-oligonucleotide therapy. N Engl J Med. 1996;334:316–318. doi: 10.1056/NEJM199602013340508. [DOI] [PubMed] [Google Scholar]
- 85.Christoffersen RE, Marr JJ. Ribozymes as human therapeutic agents. J Med Chem. 1995;38:2023–2037. doi: 10.1021/jm00012a001. [DOI] [PubMed] [Google Scholar]
- 86.McDonnell WM, Askari FK. DNA vaccines. N Engl J Med. 1996;334:42–45. doi: 10.1056/NEJM199601043340110. [DOI] [PubMed] [Google Scholar]