

Abstract
DNA repair pathways play a critical role in maintaining cellular homeostasis by repairing DNA damage induced by endogenous processes and xenobiotics, including environmental chemicals. Induction of DNA damage may lead to genomic instability, disruption of cellular homeostasis and potentially tumours. Isogenic chicken DT40 B-lymphocyte cell lines deficient in DNA repair pathways can be used to identify genotoxic compounds and aid in characterising the nature of the induced DNA damage. As part of the US Tox21 program, we previously optimised several different DT40 isogenic clones on a high-throughput screening platform and confirmed the utility of this approach for detecting genotoxicants by measuring differential cytotoxicity in wild-type and DNA repair-deficient clones following chemical exposure. In the study reported here, we screened the Tox21 10K compound library against two isogenic DNA repair-deficient DT40 cell lines (KU70 −/−/RAD54 −/− and REV3 −/−) and the wild-type cell line using a cell viability assay that measures intracellular adenosine triphosphate levels. KU70 and RAD54 are genes associated with DNA double-strand break repair processes, and REV3 is associated with translesion DNA synthesis pathways. Active compounds identified in the primary screening included many well-known genotoxicants (e.g. adriamycin, melphalan) and several compounds previously untested for genotoxicity. A subset of compounds was further evaluated by assessing their ability to induce micronuclei and phosphorylated H2AX. Using this comprehensive approach, three compounds with previously undefined genotoxicity—2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether—were identified as genotoxic. These results demonstrate the utility of this approach for identifying and prioritising compounds that may damage DNA.
Introduction
Genotoxic chemicals can generate a variety of DNA lesions, such as single-strand DNA breaks, double-strand DNA breaks (DSBs), alkylation of DNA bases and covalent links between bases [intrastrand and interstrand crosslinks (ICLs)]. Damage left unrepaired or repaired incorrectly might lead to genetic mutations and/or instability and increase the risk of carcinogenesis (1).
To reduce the risk of exposure to toxic chemicals, newly developed chemicals and established chemicals that have not been studied previously require comprehensive toxicological characterisation, including an assessment of genotoxic potential. Traditionally, in vitro, chemical-induced DNA damage has been assessed by a battery of independent assays, including the Ames test, mouse lymphoma assay, micronucleus test, chromosomal aberration test and/or the comet assay (2). However, none of these assays is well-suited for high-throughput screening (HTS) using large chemical libraries on a robotic platform because of their complexity and specific protocol features, including duration of exposure, number of cells per sample needed and the use of liver S9 mix for metabolic activation.
To overcome the limitations of traditional genotoxicity tests for adaption to HTS platforms and to increase testing throughput, we previously developed and optimised a new screening system using a group of DNA repair-deficient chicken B-lymphocyte DT40 cell lines on a quantitative HTS (qHTS) platform (3), as part of the US Tox21 program (4,5). With this screening system, we measured differential cytotoxicity using a cell viability assay (CellTiter-Glo) that measures intracellular adenosine triphosphate (ATP) levels in wild-type versus six different DNA repair-deficient DT40 cell lines (ATM −/−, FANCC −/−, POLβ −/−, KU70 −/−/RAD54 −/−, REV3 −/− and UBC13 −/−), following exposure to a library of 1408 compounds. We observed that the combination of the KU70 −/−/RAD54 −/− and REV3 −/− cell lines provided the highest sensitivity to known genotoxic chemicals, such as actinomycin D, adriamycin, alachlor, benzotrichloride and melphalan, compared with any other combination of DNA repair-deficient clones (3).
In the present study, we screened the Tox21 10K compound library against the KU70 −/−/RAD54 −/− and REV3 −/− DT40 cell lines and the parental wild-type cell line using the same cell viability assay described previously (3). In this assay system, active (i.e. genotoxic) compounds are those that reduce cell proliferation to a greater extent in the DNA repair-deficient cell lines compared with the parental, isogenic wild-type cell line (6).
KU70 and RAD54 participate in DSB repair by non-homologous end joining (NHEJ) and homologous recombination (HR), respectively (7,8). REV3 is the catalytic subunit of translesion DNA synthesis (TLS) polymerase ζ (9,10), can bypass a wide variety of DNA lesions to maintain progression of DNA replication (11), and may play a dominant role in TLS-mediated mutagenesis in mammalian cells (12). In addition to TLS, REV3 may operate within the Fanconi anemia DNA-repair pathway to eliminate ICLs (13,14).
In the primary screening of the Tox21 10K compound library, we identified several well-known genotoxic compounds (e.g. adriamycin, melphalan) that induced significantly greater cytotoxicity in the DNA repair-deficient cell lines compared with wild-type cell line. Moreover, several compounds previously untested for genotoxicity were identified as potential direct-acting genotoxicants in our assay. In follow-up studies, selected compounds were evaluated further for genotoxicity using a high content micronucleus (MN) assay and phosphorylated H2AX (γH2AX) immunostaining. Using this approach (Figure 1), we confirmed several known and novel genotoxic chemicals. The results presented in this study demonstrate the utility of this approach for evaluating the genotoxic activity of chemicals in a qHTS format and for acquiring information on the type(s) of DNA damage induced by these chemicals.
Figure 1.

Flow chart for the identification of genotoxic compounds. One hundred and nineteen compounds with ≥3-fold increase in cytotoxicity (P < 0.05) in the KU70 −/−/RAD54 −/− and/or REV3 −/− cells compared with wild-type cells were identified in the primary screening. Sixty-three of the 119 compounds were confirmed in a replicate qHTS cells viability assay using the same 3-fold differential cytotoxicity measure. Eight compounds were selected from the 63 based on their novelty, commercial availability and potency of the differential cytotoxicity response for further testing in in vitro γH2AX immunostaining and MN assays.
Materials and methods
Tox21 10K compound library and chemicals
The Tox21 10K compound library containing >8300 unique compounds has been previously described (4).
For the follow-up studies, adriamycin [Chemical Abstract Services Registry Number (CASRN) = 25316-40-9], cyclophosphamide (CASRN = 6055-19-2), melphalan (CASRN = 148-82-3), mitomycin C (CASRN = 50-07-7), sobuzoxane (CASRN = 98631-95-9), tetraoctylammonium bromide (CASRN = 14866-33-2), tetraphenylolethane glycidyl ether (CASRN = 7328-97-4), trifluridine (CASRN = 70-00-8) and 2-oxiranemethanamine (CASRN = 28768-32-3) were purchased from Sigma–Aldrich (St Louis, MO, USA). AD-67 (CASRN = 71526-07-3) was obtained from Ark Pharm (Libertyville, IL, USA). 4-Hydroperoxy cyclophosphamide (CASRN = 39800-16-3) was obtained from Toronto Research Chemicals (North York, ON, Canada). All chemicals were dissolved in dimethyl sulfoxide (DMSO, Fischer Scientific, Pittsburgh, PA, USA) and prepared as 20mM stock solutions prior to use.
Cell culture
DNA repair-deficient DT40 cell lines, developed at Kyoto University, Japan (8,11,15), and the isogenic wild-type cell line were cultured in RPMI 1640 medium (Life Technologies, Grand Island, NY, USA) supplemented with 10% FBS (Gemini Bio-Products, West Sacramento, CA, USA), 1% chicken serum (Life Technologies), 50 µM β-mercaptoethanol (Sigma–Aldrich), 100U/ml penicillin and 100 μg/ml streptomycin (Life Technologies).
Chinese hamster ovary (CHO-K1, Catalog number CCL61) cells, purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), were cultured in F-12K Nutrient Mixture (Life Technologies) supplemented with 10% FBS (HyClone Laboratories, Logan, UT, USA) and 100U/ml penicillin and 100 μg/ml streptomycin (Life Technologies). All cell cultures were maintained at 37°C under a humidified atmosphere and 5% CO2.
Southern blot
Restriction enzyme-digested genomic DNA was run on a 0.7% agarose gel. The gel was then transferred to nylon membrane. Purified probes are labelled using AlkPhos Direct Labelling system (GE Healthcare Life Sciences, Pittsburgh, PA, USA), and hybridised to the membrane at 60°C overnight. After hybridisation, the membrane was exposed to X-ray film, following incubation with CDP-Star Detection Reagent (GE Healthcare Life Sciences). The details of the construction of the gene knockouts have been described previously (8,11,15). The 0.4kb fragment from the RAD54 cDNA digested by PstI was used as a probe for RAD54 knockout Southern blot analysis. The size of the hybridising fragments of the wild-type locus is 12kb and the RAD54 knockout locus is 7kb. The 0.5kb fragment from the genomic DNA amplified using the primers 5′- GAATTCAGCATTGGGTCATTTAC -3′ and 5′-CCAAGGATGCGAAGTTTGAAGCA -3′ was used as a probe for KU70 knockout Southern blot analysis. The size of the hybridising fragments of the wild-type locus is 5.5kb and the KU70 knockout locus is 2.8kb. The 0.8kb fragment from the genomic DNA amplified using the primers 5′-ATTACGTTAGCCGGGTCCATGGG -3′ and 5′- AGAACAGCGTTGCTGTAGAAGCGGG-3′ was used as a probe for REV3 knockout Southern blot analysis. The size of the hybridising fragments for the wild-type locus is 4.4kb and for the REV3 knockout locus is 3.8 and 2.0kb.
Cell viability assay
DT40 cells (1.5×104 cells/ml) were irradiated with a 137Cs γ-ray source (0.02 Gy/s, Gammacell 40, Atomic Energy of Canada Limited, Ontario, Canada) for 50, 100 or 150 s to provide doses of 1, 2 or 3 Gy, respectively. Following irradiation, cells were cultured for 48h in the complete medium and cell viability evaluated using the CellTiter-Glo reagent (Promega, Madison, WI, USA) to measure intracellular ATP levels. The percentage of surviving cells was calculated as a ratio of the ATP level in irradiated cells relative to untreated cells.
In the primary qHTS as previously described (16), DT40 cells were dispensed at 300 cells/5 µl/well in 1536-well white wall/solid bottom plates (Greiner Bio-One, North America, NC, USA) at the speed of 40 s per assay plate using 8-tip multidrop reagent dispenser (Thermo Fisher Scientific, Waltham, MA, USA) at room temperature, followed by addition of 23 nl of each compound in the Tox21 10K compound library at 15 concentrations ranging from 0.6nM to 92 µM into the assay plates using a Pintool station (Kalypsys, San Diego, CA, USA). DMSO was used as a vehicle control. Tetraoctylammonium bromide was used as a positive control. A number of known genotoxic compounds (e.g. adriamycin, cisplatin, etoposide, mitomycin C) were present in the 10K library and therefore, no single positive genotoxic compound was selected to serve as a designated positive control in this primary screen. After the assay plates were incubated for 40h at 37°C, 5 µl per well of CellTiter-Glo reagent was added into each well of the assay plate. The plates were incubated for 30min at room temperature and the luminescence intensity of the plates was measured using a ViewLux plate reader (Perkin Elmer, Shelton, CT, USA).
For the confirmation study conducted with 119 compounds identified in the primary screen and replated from the stock solutions stored at the laboratory, the assay protocol was the same as the one used for the primary screening, with a slight adjustment to the dose range. In this confirmatory study, each compound was tested over a concentration range of 0.04nM to 92 µM at 11 concentrations. Two known genotoxic compounds that were clearly active in the primary screening—adriamycin (enhanced cytotoxicity observed in KU70 −/−/RAD54 −/− cells compared with wild-type and REV3 −/− cells) and melphalan (enhanced cytotoxicity observed in REV3 −/− cells compared with wild-type and KU70 −/− /RAD54 −/− cells)—were used as positive controls for differential cytotoxicity in this confirmation study. Adriamycin interferes with the dissociation of topoisomerase II from DNA during replication and repair, leading to the generation of DSBs (17). Adriamycin also induces oxygen radicals and formation of DNA adducts (18). Melphalan damages DNA by forming mono-adducts and cross-links (19). Tetraoctylammonium bromide was chosen as the cytotoxicity positive control in the study because it induced 100% cytotoxicity in the wild-type DT40 cells (half maximal inhibitory concentration, IC50 = 0.45 μM) and gave no indication of differential cytotoxicity in either mutant clone at any concentration (IC50 = 0.42 μM for REV3 −/− cells and IC50 = 0.43 μM for KU70 −/− /RAD54 −/− cells).
Sixty-three of 119 compounds were identified as active in the confirmation screening, and these were tested again in a second confirmation assay as described above. From this screen, eight compounds were selected for in-depth follow-up testing in orthogonal assays (i.e. γH2AX immunostaining and MN assay) to confirm genotoxicity. These eight compounds were ordered as powders from commercial sources as described above and were again examined for their ability to induce differential cytotoxicity in one or both of the DT40 mutant cell lines, to ensure that the reconstituted compound solutions gave the same responses as the solutions generated from the Tox21 10K compound library stock solutions.
Colony formation assay
To establish plating efficiency and demonstrate the functional independence of the three DT40 cell lines in response to a potent genotoxic agent, a colony formation assay was conducted. Serially diluted DT40 cells were plated in triplicate onto six-well plates in 4ml/well of D-MEM/F-12 (Life Technologies), 15% FBS, 1.5% chicken serum, 2mM l-Glutamine and 50 μM β-mercaptoethanol. Because DT40 cells are non-adherent, the culture medium also contained 1.5% (w/v) methylcellulose (Wako, Osaka, Japan) to allow colony formation. Subsequently, cells were irradiated using a 137Cs γ-ray source (0.02 Gy/s, Gammacell 40) for 50, 100 or 150 s as described above. Colonies were counted 7 days after irradiation. The percentage of surviving colonies after irradiation was determined relative to the percentage of surviving untreated colonies. The mean plating efficiencies for the control (0 Gy) plates were 73% in wild-type, 59% in KU70 −/− /RAD54 −/− and 38% in REV3 −/− cells.
qHTS data analysis
Analysis of compound concentration–response data was performed as described previously (20). Briefly, raw plate reads for each titration point were first normalised relative to the reference cytotoxic compound (tetraoctylammonium bromide, 92 µM, −100%) and DMSO-only wells (0%), and then corrected by applying a NCGC in-house pattern correction algorithm using compound-free control plates (i.e. DMSO-only plates) at the beginning and end of the compound plate stack. Concentration–response titration points for each compound were fitted to a four-parameter Hill equation (21) yielding concentrations of half-maximal inhibition (IC50) and maximal response (efficacy) values. Compounds were designated as Class 1–5 according to the type of concentration–response curve observed (20,22). Curve classes are heuristic measures of data confidence, classifying concentration–responses on the basis of efficacy, the number of data points observed above background activity, and the quality of fit. Active compounds were identified as those with significantly different IC50 values [at least a 3-fold difference in the average IC50, P < 0.05 (t-test)] between the concentration–response curves for cytotoxicity in the wild-type cell line and either of the two isogenic DNA repair-deficient cell lines.
Micronucleus assay
Although our qHTS assay protocols do not include a method to supply metabolic activation, MN induction was assessed using a high-content assay format, with and without exogenous metabolic activation (Aroclor 1254-induced rat liver S9) (MUTAZYME™ S9 Mix, Moltox, Boone, NC, USA), as per the recommended procedure in OECD Guideline 487 (23). In the absence of S9, CHO-K1 cells were plated onto collagen I coated 384-well black wall/clear bottom plates (Corning Incorporated, Tewksbury, MA, USA) at 750 cells/well, and incubated at 37°C for 4h followed by the addition of compound. Incubation with compound continued for an additional 24h. After 24h, medium plus compound was removed and replaced with fresh medium containing cytochalasin B (cytoB, 3 μg/ml, Sigma–Aldrich); incubation continued for 24h.
In the experiments with S9, CHO-K1 cells were plated onto collagen I coated 384-well black wall/clear bottom plates at 4500 cells/well, and incubated at 37°C for 4h followed by addition of compound and S9 mix (2% final concentration). Incubation continued for an additional 4h. After 4h, medium containing chemical plus S9 was removed and replaced with fresh medium, and incubation continued for 20h, and then the medium was removed and replaced with fresh medium containing cytoB (3 μg/ml). Incubation continued for an additional 24h.
After cytoB treatment in both assay conditions (+/− S9), medium was removed and cells were fixed with 4% paraformaldehyde (24) containing 0.05% Hoechst 33342 (Life Technologies), 0.01% Red Cell Mask (HCS CellMask™ Red stain, Life Technologies) and 0.1% Cell Event™ Caspase-3/7 Green Detection Reagent (Life Technologies) for 30min, followed by three washes with Hanks’ balanced salt solution (HBSS, Life Technologies). Cell Event™ Caspase-3/7 Green Detection Reagent detects activated caspase3/7, and is used to label apoptotic cells. At least 1050 binucleated cells per compound treatment were imaged with an ImageXpress Micro Widefield High Content Screening System (Molecular Devices, Sunnyvale, CA) using DAPI (to acquire nuclear images), FITC (to acquire apoptosis images) and Texas Red (to acquire the whole cell images) filter sets. The collected images were analysed using the Micronuclei application module, a proprietary analysis protocol with MetaXpress software. The analysis module identifies individual Hoechst-stained nuclei based on the size, intensity and distance from adjacent cells. The nuclei from the total number of cells in the well are classified as mononucleated, binucleated multinucleated or mitotic. Micronuclei are identified based on the size, intensity and distance from the main nucleus. A small nuclear mass that is contiguous or attached to a main nucleus will not be identified as a MN. The image analysis software provides information on the number of micronuclei in mononucleated, binucleated and multinucleated cells, respectively. The percent of micronuclei in the current study represents healthy binucleated cells that contain micronuclei. The number of micronucleated binucleated cells/1050 binucleated cells in treated cultures was compared with the number of micronucleated binucleated cells/1050 binucleated cells in the corresponding vehicle control culture. Data are expressed as the mean % micronucleated binucleated cells from three replicate cultures ± standard deviation. Statistical significance of the frequency of micronucleated cells in the treated cultures at each dose level compared with the control value was determined using a one-tailed t-test. For cytotoxicity assessment, the nuclear division index (NDI) from MetaXpress software was used. NDI was defined as: (M 1 + 2M 2 + 3M 3 + 4M 4)/N, where M 1–M 4 represent the number of cells with 1–4 nuclei and N is the total number of viable cells (excluding apoptotic cells). The percentage of cytotoxicity (% cytotoxicity) was defined as: 100 − 100{NDIT − 1)/(NDID − 1)}; NDIT = NDI of treated cells; NDID = NDI of DMSO control.
Detection of γH2AX foci in nuclear DNA
Wild-type (6250 cells/well), KU70 −/− /RAD54 −/− (6250 cells/well) and REV3 −/− (8750 cells/well) DT40 cells were plated onto collagen I coated 384-well black wall/ clear bottom plates and incubated overnight followed by the addition of compounds. After 24h of compound treatment, cells were fixed with 4% paraformaldehyde and 0.1% Hoechst 33342 for 10min at room temperature followed by washing with HBSS. Cells were permeabilised with 0.1% IGEPAL (Sigma–Aldrich) for 15min followed by two washes with HBSS. After blocking with HBSS containing 3% bovine serum albumin (Sigma–Aldrich), cells were treated with primary mouse monoclonal anti-phospho-Histone H2AX antibody (1:1000; #05-636; EMD Millipore, Billerica, MA, USA) (25,26) for 1h under humidified conditions at 37°C. Cells were then washed three times in HBSS and incubated with Alexa Fluor 594 goat-anti mouse IgG secondary antibody (1:1000, A-11032, Life Technologies) at 37°C for 45min. Cells were again washed three times with HBSS, and then at least 300 cells per compound treatment were imaged with an ImageXpress Micro Widefield High Content Screening System (Molecular Devices) using DAPI (to acquire nuclear images) and Texas Red (to acquire γH2AX foci) filter sets. The collected images were analysed using the Transfluor module, a proprietary analysis protocol with MetaXpress software. The analysis module identifies Hoechst-stained nuclei based on the size and intensity of the DAPI channel. Total number of nuclei in a well represents the number of cells. The foci are identified based on the size and intensity of the Texas Red channel, since the Alexa Fluor 594 secondary antibody was used to detect the primary antibody bound to γH2AX. Thus, the analysis module quantifies the number of foci per nucleus. In the current study, the average number of foci per nucleus was used. The number of γH2AX foci/ nucleus in treated cultures was compared with the number of γH2AX foci/ nucleus in the corresponding untreated control culture and statistical significance of the differences at each dose point was determined using a one-tailed t-test.
Results
Characterisation of DNA repair-deficient DT40 cell lines
To verify the molecular identity of the two isogenic DNA repair-deficient DT40 cell lines (KU70 −/−/RAD54 −/− and REV3 −/−) used in the Tox21 10K library screening, we ran Southern blots in these two mutant cell lines. Using this method, the KU70 gene in KU70 −/−/RAD54 −/− cells was detected as a 2.8-kb band instead of 5.5-kb band after using EcoRI-digested genomic DNA hybridised to a KU70 gene-specific probe (Figure 2A); the RAD54 gene was detected as a 7-kb band instead of 12-kb band after using BamHI-digested genomic DNA hybridised to a RAD54 gene-specific probe (Figure 2A). The disruption of the REV3 gene was verified by the presence of 3.8- and 2.0-kb bands instead of a 4.4-kb band after using HindIII-digested genomic DNA hybridised to a REV3 gene-specific probe (Figure 2A). After exposure to γ-radiation at various doses, the DT40 cell lines (KU70 −/−/RAD54 −/− and REV3 −/−) showed much lower cell survival rates than the parental wild-type cells in both the cell viability and the colony formation assays (Figure 2B and C, respectively). At 1 Gy of γ-radiation, the cell-survival rates of KU70 −/−/RAD54 −/− and REV3 −/− cell lines were 8–15% (cell viability assay) and 8–9% (colony formation assay), whereas the survival rates of the wild-type cell line was 60–70% (Figure 2B and C). These results are consistent with data published previously (6,8,11). Taken together, these results demonstrate that the DT40 cell lines used in this study were functionally knocked out for DNA repair and DNA damage-tolerance pathways.
Figure 2.
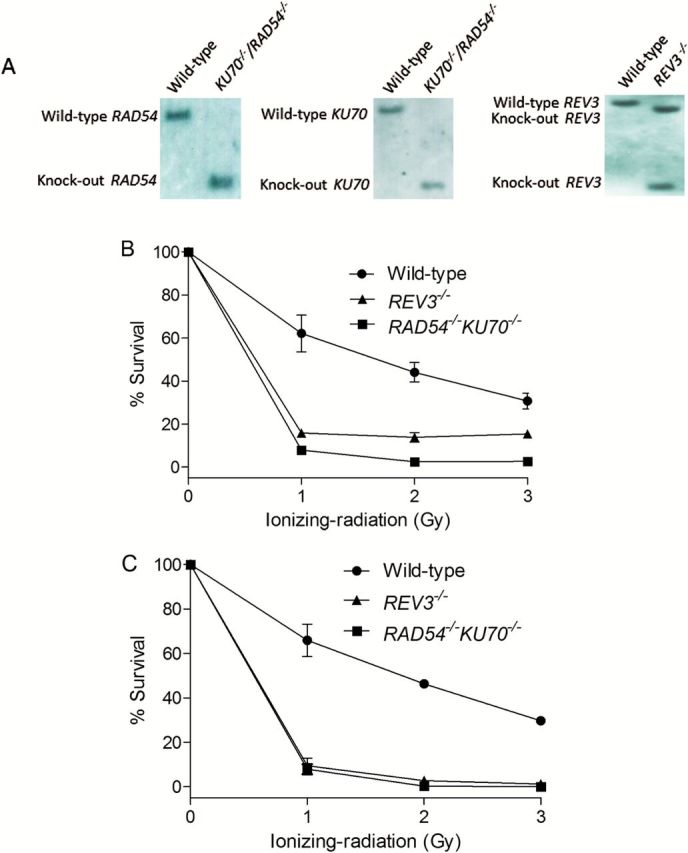
Confirmation of genotype of DT40 clones that were used in this study. (A) Southern blot analysis of indicated gene disrupted DT40 clones using the gene specific probes. (B) Liquid survival assay after exposure to ionising radiation. Cells were cultured for 48h after IR. Cellular ATP level was used to measure cell survival. The survival of untreated cells was set as 100%. (C) Colony survival assay after exposure to IR. Percent survival was determined relative to numbers of colonies from untreated cells. For both B and C, error bars represent SD from at least three independent experiments.
Identification of chemicals with differential cytotoxicity
In the primary screening, we tested each compound from the Tox21 10K compound library against the DNA repair-deficient DT40 cell lines (KU70 −/−/RAD54 −/− and REV3 −/−) and the wild-type cell line using a cell viability assay that measures intracellular ATP levels. Compounds that were not cytotoxic up to the 92 µM top concentration were considered inactive. Approximately 60% of the library compounds were found to be inactive in each cell line and 50% of the library compounds were inactive in all cell lines (wild-type and the two mutant cells lines). Compound potencies were quantitated using IC50 values from the DNA repair-deficient cell lines compared with those of the wild-type cells. A total of 119 compounds with more than a 3-fold difference in IC50 values (mutant < wild-type) (P < 0.05) in the KU70 −/−/RAD54 −/− and/or REV3 −/− cell lines compared with wild-type cells were identified. Among these compounds, eight were more cytotoxic to both mutant cell lines compared with wild-type cells, 66 were more cytotoxic only in the REV3 −/− cells and 45 were more cytotoxic only in the KU70 −/−/RAD54 −/− cells when compared with wild-type cells.
In the confirmation study, these 119 chemicals were re-tested under the same assay conditions as the primary screening. Of the 119 compounds, differential cytotoxicity >3-fold was confirmed for 63 compounds in KU70 −/−/RAD54 −/− and/or REV3 −/− cells compared with wild-type cells (Supplementary Table 1, available at Mutagenesis Online), resulting in a 53% confirmation rate. Among the confirmed compounds were many known genotoxicants. In addition to pre-selected positive controls (melphalan and adriamycin), known genotoxic compounds such as etoposide (27) and N-methyl-N′-nitro-N-nitrosoguanidine (28) were also confirmed. Moreover, several compounds with no published genotoxicity data, such as 2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether, were identified as potential direct-acting genotoxic chemicals in our qHTS assay.
Confirmation of differential cytotoxicity of compounds
A total of nine compounds (8 compounds from 63 that were confirmed in the re-testing, plus tetraoctylammonium bromide, used as a reference cytotoxic compound) were selected for additional evaluation based on their novelty, commercial availability and potency of their differential cytotoxicity response. These nine compounds were purchased from commercial vendors and re-tested for cytotoxicity against the three DT40 cell lines. Consistent with the results in the primary screening and the confirmation assay, adriamycin significantly reduced the viability of KU70 −/−/RAD54 −/− cell line, with an IC50 of 5.8nM (Figure 3A), a value that was 6-fold more potent than in REV3 −/− (IC50 = 35.7nM) and in the wild-type cell lines (IC50 = 43.3nM). On the other hand, melphalan was much more cytotoxic in the REV3 −/− (IC50 = 0.1 µM) mutant compared with the KU70 −/−/RAD54 −/− (IC50 = 3.0 µM) or wild-type cell lines (IC50 = 3.4 µM) (Figure 3B). As expected, tetraoctylammonium bromide (Figure 3C) had similar potencies in all three cell lines, consistent with the knowledge that the cytotoxicity of this compound is not mediated via DNA damage (29).
Figure 3.

Sensitivity of DT40 DNA repair-deficient and the parental cell lines to chemical compounds selected from the primary screening. Cellular survival was determined using CellTiter-Glo after 40h exposure to adriamycin (positive control) (A), melphalan (positive control) (B), tetraoctylammonium bromide (C), trifluridine (D), 4-hydroperoxy cyclophosphamide (E), sobuzoxane (F), 2-oxiranemethanamine (G), AD-67 (H) and tetraphenylolethane glycidyl ether (I). Error bars represent SD from at least three independent experiments.
Three additional known genotoxic compounds—trifluridine, 4-hydroperoxy cyclophosphamide and sobuzoxane—were also confirmed in this study. Trifluridine showed similar increased cytotoxic potency in both the KU70 −/−/RAD54 −/− and the REV3 −/− cell lines (Figure 3D). 4-Hydroperoxy cyclophosphamide was more cytotoxic to the REV3 −/− cell line than to either the KU70 −/−/RAD54 −/− or the wild-type cell lines (Figure 3E), while sobuzoxane was more potent in KU70 −/−/RAD54 −/− cell line compared with both REV3 −/− and wild-type cell lines (Figure 3F). Three compounds with no pre-existing genotoxicity data—2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether—were also identified in this study as potential genotoxicants. Interestingly, all three of these compounds significantly reduced the viability of the REV3 −/− cell line compared with the KU70 −/−/RAD54 −/− and wild-type cells (Figure 3G, H and I), indicating that the compounds likely created DNA lesions that require REV3-mediated TLS for survival.
Verification of compound genotoxicity
To further characterise the potential genotoxicity of the eight compounds that demonstrated differential cytotoxicity in the DT40 cell assays, we evaluated induction of phosphorylated H2AX S139 (γH2AX) foci in the three DT40 cell lines as well as the frequency of micronucleated CHO-K1 cells following exposure to these compounds over a broad dose range. Both of these endpoints are commonly used in standard assays for genotoxicity (23,30). The number of γH2AX foci was determined following exposure to the chemicals for 24h. All eight compounds induced γH2AX foci in a concentration-dependent manner, indicating that these compounds induce DNA damage (Figures 4 and 5A).
Figure 4.

Induction of γH2AX foci in wild-type and DNA repair-deficient DT40 clones after 24h chemical treatment. The y-axis represents the average number of γH2AX foci per nucleus from three independent experiments. Error bars represent SEM. *P < 0.05, **P < 0.01 and ***P < 0.001.
Figure 5.

Generation of γH2AX foci in DT40 cells and micronucleus formation in CHO-K1 cells treated with 2-oxiranemethanamine. (A) Dose-dependent γH2AX foci (red) formation in DT40 cells. The indicated DT40 clones were treated with 2-oxiranemethanamine for 24h. Images were acquired in ImageXpress using a 40× objective. Hoechst staining (blue) indicates DNA. (B) Micronucleus formation in CHO-K1 cells. CHO-K1 cells were treated with 2-oxiranemethanamine for 24h without S9 treatment. Hoechst staining (blue) indicates DNA. Images were acquired in ImageXpress using a 20× objective. Arrows indicate a cell with MN.
The ability of the eight compounds to induce MN was determined in CHO-K1 cells rather than in the DT40 cells because the low cytoplasm to nuclear ratio makes detection of MN in chicken cells more difficult. To ensure that MN were observed in CHO-K1 cells that had completed one mitotic division (necessary for the formation of MN), cytoB was used as a cytokinesis blocker, and only binucleated cells were evaluated for presence of MN. Cyclophosphamide and mitomycin C were used as positive controls for S9 treated and untreated cells, respectively. All eight compounds induced MN (Table 1), and the increases in MN were concentration-dependent (Figures 6 and 7). Compounds that were not reported previously to be genotoxic, such as 2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether, also induced MN. 2-Oxiranemethanamine (Figures 5B and 6H) and AD-67 induced MN in the absence of S9 (Figure 6I) and tetraphenylolethane glycidyl ether induced MN both with and without S9 (Figures 6J and 7J).
Table 1.
Micronucleus induction in CHO-K1 cells by eight selected compounds identified as active in the qHTS assays with DT40 cells
| Chemicala | CASRN | S9 (−) | S9 (+) | ||||
|---|---|---|---|---|---|---|---|
| Concentration range (μM)b | % MN binucleated cellsc | Fold-increase over DMSO controld | Concentration range (μM) | % MN binucleated cells | Fold-increase over DMSO control | ||
| Mitomycin C (positive control) | 50-07-7 | 0.00059–1.2 | 10.94±2.68 | 7.93* | 0.00059–1.2 | 6.47±1.72 | 1.01 |
| Cyclophosphamide (positive control) | 6055-19-2 | 0.0022–17.9 | 1.71±0.65 | 1.24 | 0.0022–17.9 | 21.80±5.01 | 3.40* |
| Adriamycin | 25316-40-9 | 0.0028–0.1 | 10.31±2.27 | 7.47* | 0.0028–0.2 | 12.46±5.76 | 1.94* |
| Melphalan | 148-82-3 | 0.0028–5.8 | 13.59±2.96 | 9.85* | 0.0028–2.9 | 20.83±5.27 | 3.25* |
| Trifluridine | 70-00-8 | 0.0028–92.0 | 8.51±2.60 | 6.17* | 0.0028–92.0 | 11.76±5.40 | 1.83* |
| 4-Hydroperoxy cyclophosphamide | 39800-16-3 | 0.0028–5.8 | 13.32±1.31 | 9.65* | 0.0028–23.0 | 23.02±4.21 | 3.59* |
| Sobuzoxane | 98631-95-9 | 0.0028–92.0 | 10.92±1.95 | 7.91* | 0.0028–92.0 | 13.74±4.69 | 2.14* |
| 2-Oxiranemethanamine | 28768-32-3 | 0.0028- 5.8 | 9.65±2.95 | 6.99* | 0.0028–92.0 | 8.23±1.75 | 1.28 |
| AD-67 | 71526-07-3 | 0.0028–46.0 | 2.40±1.27 | 1.74* | 0.0028–92.0 | 7.44±4.65 | 1.16 |
| Tetraphenylolethane glycidyl ether | 7328-97-4 | 0.0028–1.4 | 31.22±7.33 | 22.62* | 0.0028–1.4 | 14.62±3.80 | 2.28* |
S9 (−), S9 untreated; S9 (+), S9 treated.
aMitomycin C and cyclophosphamide were used as positive controls for S9 (−) and S9 (+), respectively.
bRange of concentrations with acceptable levels of cytotoxicity (% cytotoxicity below 60%) for which data are reported.
c% MN binucleated cells determined from scoring at least 1050 binucleated cells at the highest concentration tested with acceptable cytotoxicity levels. Values are the mean ± SD of the results from three experiments.
dCalculated by dividing the % MN binucleated cells at the highest dose tested by the % MN binucleated cells of the DMSO control [1.38±0.81% for S9 (−), 6.06±2.48% for S9 (+)]. Significant increases are marked with an asterisk (one-tailed t-test, P < 0.05). Dose–response data are represented in Figures 6 and 7.
Figure 6.

The frequency of micronucleated cells after chemical treatment in the absence of S9. The line chart represents the percentage of binucleated cells with MN out of the total number of binucleated cells evaluated. The bar chart represents the NDI-based cytotoxicity. Data represents the mean ± SEM from three experiments.
Figure 7.

The frequency of micronucleated cells after chemical treatment in the presence of S9. The line chart represents the percentage of binucleated cells with MN out of the total number of binucleated cells evaluated. The bar chart represents the NDI-based cytotoxicity. Data represents the mean ± SEM from three experiments.
Discussion
In this study, we used isogenic chicken DT40 cell lines deficient in specific DNA repair pathways to screen the Tox21 10K compound library. Potentially genotoxic compounds were identified by measuring differential cytotoxicity between wild-type and DNA repair-deficient cell lines (KU70 −/−/RAD54 −/− and REV3 −/−) following exposure to chemicals. Differential cytotoxicity was quantified for each compound based on the IC50 values in the DNA repair-deficient cell lines and the wild-type cells (wild-type IC50/mutant IC50). In the primary qHTS screening, 119 of >8300 unique compounds tested (1.4%) showed possible genotoxic activity based on a ≥3-fold difference in IC50 values between mutant and wild-type cells. In the follow-up study, differential cytotoxicity (≥3-fold difference in IC50 values) was confirmed for 63 compounds, giving a confirmation rate of 53%. Among these 63 compounds are many well-known genotoxic compounds as well as compounds with no pre-existing genotoxicity data, such as 2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether. The genotoxicity of 8 of these 63 compounds was further evaluated using two in vitro genotoxic assays: γH2AX immunostaining and the MN assay.
The low confirmation rate (53%) for differential cytotoxicity between DNA repair-deficient (KU70 −/−/RAD54 −/− and REV3 −/−) and wild-type cell lines in the qHTS confirmation tests is likely a result of the relatively lenient cutoff of ≥3-fold, which was used as the basis for identification of active (genotoxic) compounds in the primary screening. The 3-fold differential leaves little room for accommodating a slight shift in one or both of the concentration response curves (the mutant and the wild-type), and thus, the chance that such a shift in a repeat assay would result in missing the differential cytotoxicity cutoff is substantial. In addition, the use of more lenient criteria allows for the identification of weak (borderline) actives that may not be consistently identified from run to run. Had a determination of differential cytotoxicity been based on a more stringent cutoff, for example of ≥6-fold difference in IC50 values, only 35 compounds would have been identified as active in the primary screening. Of those 35 compounds, 30 were also found to be active in the confirming experiments, yielding a confirmation rate of 86%. Most of these 35 compounds are known genotoxic compounds [e.g. bleomycin (31), chlorambucil (32), N-methyl-N′-nitro-N-nitrosoguanidine (28) and thiotepa (33)], and for those with no pre-existing genotoxicity data, their molecular structures (e.g. nitrogen mustards), strongly suggest they are genotoxic. In contrast, the 119 chemicals selected using the ≥3-fold IC50 difference cutoff included a larger number of compounds that were not previously identified as genotoxic. Therefore, using the less stringent 3-fold cutoff for initial compound selection provided a better opportunity for identifying previously unknown genotoxic compounds, which could then be confirmed in a repeat test, and further evaluated using standard assays for genotoxicity such as the MN assay.
Using qHTS assays in DT40 cells can provide a method for the rapid screening of a large chemical library for potential DNA damaging activities and facilitate the prioritisation of potential genotoxic compounds for follow-up testing. However, in our experience, these DT40 assays are fairly insensitive, although compounds that are active are likely to be true genotoxicants. For example busulfan and cisplatin (crosslinking agents), 1-ethyl-1-nitrosourea and methyl methanesulfonate (alkylating agents) and styrene oxide and hydroquinone (compounds that induce oxidative damage and/or DNA adducts) were not active these DT40 assays. There are several reasons for the low sensitivity. For example our current qHTS assays cannot detect compounds that require metabolic activation, which traditionally in in vitro tests is provided by rat liver S9 enzymes and co-factors. However, because S9 has cytotoxic effect after several hours of exposure in a homogeneous assay format, S9 cannot be used in the current qHTS system. Also, our current qHTS approach cannot detect compounds with activity at concentrations greater than 92 µM, which is the highest concentration used in our qHTS protocols. Another reason for the low sensitivity of these assays involves the rather complex data analysis methods that require clearly differential cytoxocity between the wild-type cell line and a mutant clone. A small shift in the dose response curve for either cell line that reduces the fold separation between the curves can result in a negative call with the automated analysis methods that must be used for these large data sets. Lastly, although compromising key lesion tolerance pathways such as TLS and homologous recombination should allow detection of many genotoxic agents, it is expected that not all genotoxicants will be captured by the two mutant clones used in this study (3).
Adriamycin, melphalan, trifluridine, 4-hydroperoxy cyclophosphamide and sobuzoxane, well-known genotoxic compounds, were identified from the qHTS screening and were found to be positive in both γH2AX and MN assays. Adriamycin, an anticancer drug, interferes with the dissociation of topoisomerase II from DNA during replication and repair, leading to the generation of DSBs (17). Adriamycin also induces oxygen radicals and formation of DNA adducts (18). Oxidative stress may result in induction of several types of DNA damage, including DSBs (34), and several DNA repair pathways may participate in the repair of DNA adducts, including HR (13). Adriamycin significantly reduced the viability of the KU70 −/−/RAD54 −/− cell line, compared with wild-type, consistent with the knowledge that it induces DSBs that are repaired by HR and/or NHEJ. Melphalan, another anticancer drug, damages DNA by forming mono-adducts and cross-links (19). Melphalan showed differential cytotoxicity only in REV3 −/− cell line compared with wild-type cell line, consistent with the knowledge that the damage induced by melphalan is repaired by the TLS and Fanconi Anemia DNA-repair pathways (14). The observation that KU70 −/−/RAD54 −/− cells were far more sensitive to killing by adriamycin, whereas REV3 −/− cells were far more sensitive to killing by melphalan, illustrates the utility of these mutants for elucidating the mechanism by which a chemical induces DNA damage. Trifluridine (a.k.a. trifluorothymidine) is currently used as an antiviral drug for the treatment of herpes simplex virus infection (35). Trifluridine interferes with DNA replication by inhibiting thymidylate synthase. In addition, trifluridine can be incorporated into DNA instead of thymidine by replicative DNA polymerases (36). Its genotoxicity has also been detected using the Ames test (37). In the present study, trifluridine was found to reduce the viability in both KU70 −/−/RAD54 −/− and REV3 −/− cell lines compared with wild-type cell line, suggesting that the DNA damage induced by trifluridine may be repaired through the HR and/or NHEJ and TLS pathways. Another known genotoxic compound, 4-hydroperoxy cyclophosphamide, showed differential cytotoxicity only in the REV3 −/− cell line compared with wild-type cell line, suggesting that 4-hydroperoxy cyclophosphamide induces DNA damage that must be bypassed by the TLS pathway. This notion is supported by the previous observations that 4-hydroperoxy cyclophosphamide causes alkylation of DNA and formation of ICLs (38), which can be bypassed by TLS polymerases (39). 4-Hydroperoxy cyclophosphamide causes unscheduled DNA synthesis (UDS) in human lymphocytes in vitro, indicating elimination of damaged nucleotides by excision repair pathways (40). Sobuzoxane (a.k.a. MST-16) is a topoisomerase II inhibitor used as an anticancer agent (41). Topoisomerase II inhibitors efficiently kill cycling cells by inducing DSBs (42). Sobuzoxane significantly reduced viability only in KU70 −/−/RAD54 −/− cell line compared with wild-type cells, suggesting that sobuzoxane-induced DNA damage, such as DSBs, was repaired by the NHEJ and/or HR pathways. These results indicate that qHTS assays based on the use of isogenic DT40 DNA repair-deficient cell lines can both detect genotoxic compounds and provide insight into their mechanism of action.
2-Oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether have not been tested previously for genotoxicity. However, examination of their chemical structures suggests that they would be predicted to be genotoxic (e.g. two of the compounds, 2-oxiranemethanamine and tetraphenylolethane glycidyl ether, each contain four epoxide substructures). 2-Oxiranemethanamine is a component of polymers widely used in industry (43). AD-67 is a chemical used to protect crop plants from the effects of herbicides (44). Tetraphenylolethane glycidyl ether is an epoxy resin used in the manufacture of paints, coatings and adhesives, and is classified as a US high-production volume (HPV) chemical by the US Environmental Protection Agency. In the primary screening and confirmation study, 2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether induced significantly higher cytotoxicity in the REV3 −/− cell line compared with wild-type or KU70 −/−/RAD54 −/− cell lines. The observation of reduced proliferation rates of the REV3 −/− cell line in comparison with the wild-type cell line following exposure to these three chemicals suggests that they induce DNA damage that results in stalled replication forks requiring error-prone TLS by REV3 for repair. Cytotoxicity that differentially affects the REV3 −/− cell line may be indicative of a variety of different types of chemical-induced DNA adducts because REV3, but not replicative DNA polymerases, can bypass a wide variety of DNA adducts.
To provide further evidence in support of the genotoxicity of the compounds identified in the qHTS screens, the eight compounds selected for follow-up testing were evaluated for their ability to generate DNA damage in the form of DSBs as detected by immunostaining for γH2AX foci (30,45). Unlike the direct induction of DSBs by ionising radiation, DSBs induced by chemicals are largely dependent on DNA replication. Because of the comparatively long amount of time that DT40 cells spend in S phase (~70% of cell cycle), and the lack of a G1/S checkpoint, DT40 cells should be particularly sensitive to chemicals that produce DSBs by disrupting replication forks (46,47). All eight compounds (adriamycin, melphalan, trifluridine, 4-hydroperoxy cyclophosphamide, sobuzoxane, 2-oxiranemethanamine, AD-67 and tetraphenylolethane glycidyl ether) selected for follow-up studies after qHTS testing induced γH2AX foci in a concentration-dependent manner, providing additional evidence supporting the genotoxicity of these compounds.
To further confirm the genotoxicity of these compounds, an in vitro MN test, a widely used method to evaluate the ability of a compound to induce structural or numerical chromosomal damage, was conducted in CHO-K1 cells. Micronuclei, visible in interphase cells following cell division, are small nuclear bodies, morphologically identical to the main nucleus except in size, that are formed from chromosome fragments, the result of chromosome breakage or whole chromosomes, the result of chromosome loss due to mitotic disruption (48). As shown in Table 1, adriamycin, melphalan, trifluridine, 4-hydroperoxy cyclophosphamide, sobuzoxane and tetraphenylolethane glycidyl ether induced micronuclei with and without S9. 2-Oxiranemethanamine and AD-67 induced micronuclei only in the absence of S9, confirming that all eight compounds were direct-acting genotoxicants. These results are consistent with the fact that no exogenous metabolic activation is included in the qHTS assay protocol.
In summary, we were able to identify known and potential genotoxic compounds and predict the type of DNA damage induced by these chemicals using qHTS assays in DT40 cells combined with follow-up orthogonal assays. The two DNA repair-deficient DT40 cell lines used in this study have been shown to detect the largest number of active compounds (potential genotoxicants) in our compound library compared with combinations of the other DT40 mutant clones that were investigated previously in our laboratory. This is likely due to the fact that the genes that are knocked out in these two mutant clones are vital for lesion tolerance; thus, these cells are especially sensitive to a wide variety of DNA lesions that cause DSBs by disrupting replication. The qHTS approach using these cell lines provides a useful means of identifying DNA-damaging agents from large collections of chemicals, including environmental chemicals.
Supplementary data
Supplementary Table 1 is available at Mutagenesis Online.
Funding
This work was supported by the Kyoto University Foundation, the Grant-in-Aid for JSPS Fellows (24·5986), JSPS Core-to-Core Program (Advanced Research Networks) and the interagency agreement IAG #NTR 12003 from the National Institute of Environmental Health Sciences/Division of the National Toxicology Program to the National Center for Advancing Translational Sciences, National Institutes of Health.
Supplementary Material
Acknowledgements
We thank Sam Michael and Paul Shinn for assistance with automated screening and compound management.
Conflict of interest statement: None declared.
References
- 1. Ciccia A., Elledge S. J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell, 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hendriks G., van de Water B., Schoonen W., Vrieling H. (2013) Cellular-signaling pathways unveil the carcinogenic potential of chemicals. J. Appl. Toxicol., 33, 399–409. [DOI] [PubMed] [Google Scholar]
- 3. Yamamoto K. N., Hirota K., Kono K., et al. (2011) Characterization of environmental chemicals with potential for DNA damage using isogenic DNA repair-deficient chicken DT40 cell lines. Environ. Mol. Mutagen., 52, 547–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Attene-Ramos M. S., Miller N., Huang R., et al. (2013) The Tox21 robotic platform for the assessment of environmental chemicals—from vision to reality. Drug Discov. Today, 18, 716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tice R. R., Austin C. P., Kavlock R. J., Bucher J. R. (2013) Improving the human hazard characterization of chemicals: a Tox21 update. Environ. Health Perspect., 121, 756–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ji K., Kogame T., Choi K., Wang X., Lee J., Taniguchi Y., Takeda S. (2009) A novel approach using DNA-repair-deficient chicken DT40 cell lines for screening and characterizing the genotoxicity of environmental contaminants. Environ. Health Perspect., 117, 1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goodarzi A. A., Jeggo P. A. (2013) The repair and signaling responses to DNA double-strand breaks. Adv. Genet., 82, 1–45. [DOI] [PubMed] [Google Scholar]
- 8. Takata M., Sasaki M. S., Sonoda E., Morrison C., Hashimoto M., Utsumi H., Yamaguchi-Iwai Y., Shinohara A., Takeda S. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J., 17, 5497–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gan G. N., Wittschieben J. P., Wittschieben B. Ø., Wood R. D. (2008) DNA polymerase zeta (pol zeta) in higher eukaryotes. Cell Res., 18, 174–183. [DOI] [PubMed] [Google Scholar]
- 10. Gibbs P. E., McGregor W. G., Maher V. M., Nisson P., Lawrence C. W. (1998) A human homolog of the Saccharomyces cerevisiae REV3 gene, which encodes the catalytic subunit of DNA polymerase zeta. Proc. Natl. Acad. Sci. U. S. A., 95, 6876–6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sonoda E., Okada T., Zhao G. Y., et al. (2003) Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J., 22, 3188–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibbs P. E., McDonald J., Woodgate R., Lawrence C. W. (2005) The relative roles in vivo of Saccharomyces cerevisiae Pol eta, Pol zeta, Rev1 protein and Pol32 in the bypass and mutation induction of an abasic site, T-T (6-4) photoadduct and T-T cis-syn cyclobutane dimer. Genetics, 169, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim H., D’Andrea A. D. (2012) Regulation of DNA cross-link repair by the Fanconi anemia/BRCA pathway. Genes Dev., 26, 1393–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nojima K., Hochegger H., Saberi A., et al. (2005) Multiple repair pathways mediate tolerance to chemotherapeutic cross-linking agents in vertebrate cells. Cancer Res., 65, 11704–11711. [DOI] [PubMed] [Google Scholar]
- 15. Bezzubova O., Silbergleit A., Yamaguchi-Iwai Y., Takeda S., Buerstedde J. M. (1997) Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell, 89, 185–193. [DOI] [PubMed] [Google Scholar]
- 16. Xia M., Huang R., Witt K. L., et al. (2008) Compound cytotoxicity profiling using quantitative high-throughput screening. Environ. Health Perspect., 116, 284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tewey K. M., Rowe T. C., Yang L., Halligan B. D., Liu L. F. (1984) Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science, 226, 466–468. [DOI] [PubMed] [Google Scholar]
- 18. Yang F., Teves S. S., Kemp C. J., Henikoff S. (2014) Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta, 1845, 84–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawley P. D., Phillips D. H. (1996) DNA adducts from chemotherapeutic agents. Mutat. Res., 355, 13–40. [DOI] [PubMed] [Google Scholar]
- 20. Huang R., Xia M., Cho M. H., et al. (2011) Chemical genomics profiling of environmental chemical modulation of human nuclear receptors. Environ. Health Perspect., 119, 1142–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hill A.V. (1910) The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. (London), 40, 4–7. [Google Scholar]
- 22. Inglese J., Auld D. S., Jadhav A., Johnson R. L., Simeonov A., Yasgar A., Zheng W., Austin C. P. (2006) Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U. S. A., 103, 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.OECD. (2014) Test No. 487: In Vitro Mammalian Cell Micronucleus Test. OECD Guidelines for the Testing of Chemicals, Section 4. OECD Publishing, Paris, France. [Google Scholar]
- 24. Diaz D., Scott A., Carmichael P., Shi W., Costales C. (2007) Evaluation of an automated in vitro micronucleus assay in CHO-K1 cells. Mutat. Res., 630, 1–13. [DOI] [PubMed] [Google Scholar]
- 25. Bonner W. M., Redon C. E., Dickey J. S., Nakamura A. J., Sedelnikova O. A., Solier S., Pommier Y. (2008) GammaH2AX and cancer. Nat. Rev. Cancer, 8, 957–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Murai J., Huang S. Y., Das B. B., Renaud A., Zhang Y., Doroshow J. H., Ji J., Takeda S., Pommier Y. (2012) Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res., 72, 5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Heisig P. (2009) Type II topoisomerases—inhibitors, repair mechanisms and mutations. Mutagenesis, 24, 465–469. [DOI] [PubMed] [Google Scholar]
- 28. Wyatt M. D., Pittman D. L. (2006) Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem. Res. Toxicol., 19, 1580–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho M. H., Niles A., Huang R., Inglese J., Austin C. P., Riss T., Xia M. (2008) A bioluminescent cytotoxicity assay for assessment of membrane integrity using a proteolytic biomarker. Toxicol. In Vitro, 22, 1099–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nikolova T., Dvorak M., Jung F., Adam I., Krämer E., Gerhold-Ay A., Kaina B. (2014) The γH2AX assay for genotoxic and nongenotoxic agents: comparison of H2AX phosphorylation with cell death response. Toxicol. Sci., 140, 103–117. [DOI] [PubMed] [Google Scholar]
- 31. Garriott M. L., Phelps J. B., Hoffman W. P. (2002) A protocol for the in vitro micronucleus test. I. Contributions to the development of a protocol suitable for regulatory submissions from an examination of 16 chemicals with different mechanisms of action and different levels of activity. Mutat. Res., 517, 123–134. [DOI] [PubMed] [Google Scholar]
- 32. Moore F. R., Urda G. A., Krishna G., Theiss J. C. (1995) An in vivo/in vitro method for assessing micronucleus and chromosome aberration induction in rat bone marrow and spleen. 2. Studies with chlorambucil and mitomycin C. Mutat. Res., 335, 201–206. [DOI] [PubMed] [Google Scholar]
- 33. Zeiger E., Anderson B., Haworth S., Lawlor T., Mortelmans K. (1992) Salmonella mutagenicity tests: V. Results from the testing of 311 chemicals. Environ. Mol. Mutagen., 19(Suppl. 21), 2–141. [DOI] [PubMed] [Google Scholar]
- 34. Yan S., Sorrell M., Berman Z. (2014) Functional interplay between ATM/ATR-mediated DNA damage response and DNA repair pathways in oxidative stress. Cell. Mol. Life Sci., 71, 3951–3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Skevaki C. L., Galani I. E., Pararas M. V., Giannopoulou K. P., Tsakris A. (2011) Treatment of viral conjunctivitis with antiviral drugs. Drugs, 71, 331–347. [DOI] [PubMed] [Google Scholar]
- 36. Suzuki N., Emura T., Fukushima M. (2011) Mode of action of trifluorothymidine (TFT) against DNA replication and repair enzymes. Int. J. Oncol., 39, 263–270. [DOI] [PubMed] [Google Scholar]
- 37. Bilimoria M. H., Gupta S. V. (1986) Comparison of the mutagenic activity of 5-hydroxymethyldeoxyuridine with 5-substituted 2′-deoxyuridine analogs in the Ames Salmonella/microsome test. Mutat. Res., 169, 123–127. [DOI] [PubMed] [Google Scholar]
- 38. Fleer R., Brendel M. (1982) Toxicity, interstrand cross-links and DNA fragmentation induced by ‘activated’ cyclophosphamide in yeast: comparative studies on 4-hydroperoxy-cyclophosphamide, its monofunctional analogon, acrolein, phosphoramide mustard, and nor-nitrogen mustard. Chem. Biol. Interact., 39, 1–15. [DOI] [PubMed] [Google Scholar]
- 39. Sharma S., Canman C. E. (2012) REV1 and DNA polymerase zeta in DNA interstrand crosslink repair. Environ. Mol. Mutagen., 53, 725–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perocco P., Pane G., Santucci M. A., Zannotti M. (1985) Mutagenic and toxic effects of 4-hydroperoxycyclophosphamide and of 2,4-tetrahydrocyclohexylamine (ASTA-Z-7557) on human lymphocytes cultured in vitro. Exp. Hematol., 13, 1014–1017. [PubMed] [Google Scholar]
- 41. Andoh T., Ishida R. (1998) Catalytic inhibitors of DNA topoisomerase II. Biochim. Biophys. Acta, 1400, 155–171. [DOI] [PubMed] [Google Scholar]
- 42. Helleday T., Petermann E., Lundin C., Hodgson B., Sharma R. A. (2008) DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer, 8, 193–204. [DOI] [PubMed] [Google Scholar]
- 43. Bromberg L., Fasoli E., Alvarez M., Hatton T. A., Barletta G. L. (2010) Biguanide-, imine-, and guanidine-based networks as catalysts for transesterification of vegetable oil. React. Funct. Polym., 70, 433–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Walton J. D., Casida J. E. (1995) Specific binding of a dichloroacetamide herbicide safener in maize at a site that also binds thiocarbamate and chloroacetanilide herbicides. Plant Physiol., 109, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gerić M., Gajski G., Garaj-Vrhovac V. (2014) γ-H2AX as a biomarker for DNA double-strand breaks in ecotoxicology. Ecotoxicol. Environ. Saf., 105, 13–21. [DOI] [PubMed] [Google Scholar]
- 46. Evans T. J., Yamamoto K. N., Hirota K., Takeda S. (2010) Mutant cells defective in DNA repair pathways provide a sensitive high-throughput assay for genotoxicity. DNA Repair (Amst)., 9, 1292–1298. [DOI] [PubMed] [Google Scholar]
- 47. Löbrich M., Shibata A., Beucher A., Fisher A., Ensminger M., Goodarzi A. A., Barton O., Jeggo P. A. (2010) gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle, 9, 662–669. [DOI] [PubMed] [Google Scholar]
- 48. Fenech M. (2007) Cytokinesis-block micronucleus cytome assay. Nat. Protoc., 2, 1084–1104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.