

Abstract
Background: Aberrant expression of RON, a MET family receptor tyrosine kinase, has been correlated to tumor growth and metastasis. Intense research efforts are on to target RON using small molecule tyrosine kinase inhibitors or specific antibodies. However, progress towards specific targeting of RON is hampered by a lack of understanding of the nature and number of isoforms of RON expressed by tumors. We hypothesize that formation of different isoforms via alternative splicing may be fundamental to the tumor promoting functions associated with aberrantly expressed RON in cancers. Methods: In this study, we analyzed the transcript sequence variations caused by alternative splicing in the C-terminal region of RON cDNA by PCR amplification and sequencing of five small cell lung carcinoma (SCLC) and seven non-small cell lung carcinoma (NSCLC) cell lines. Results: Results revealed the presence of two alternatively spliced variants, each caused by unique exon(s) deletion: a previously known transcript variant lacking exon 19 and a novel one lacking exons 18+19. The two alternatively spliced variants together with the wild-type transcript were detected in each of the 12 lung cancer cell lines analyzed. Combined loss of exons 18+19 results in an in-frame deletion of 303 nucleotides corresponding to 101 amino acids of the tyrosine kinase domain. Translation products of transcript variants lacking exons 18 and 19 are expected to dominant negatively inhibit ligand stimulated RON signaling. Conclusions: The ubiquitous presence of alternatively spliced transcripts and their translation products may affect quantitative expression analysis, either by immunological or PCR methods, by interfering with estimation of normal RON, leading to exaggerated values. Besides, RON isoforms with dominant negative activities may interfere with siRNA based functional analysis of wild-type RON.
Keywords: RON, MST1R, alternative splicing, RON isoforms, receptor tyrosine kinase, macrophage stimulating protein
Introduction
Several RTKs and their ligands have been targeted using small molecule inhibitors and/or antibodies in lung and other cancers with varying degrees of success [1-3]. RON is a member of Met family of receptor tyrosine kinases (RTKs), and overexpression of RON has been reported to correlate to tumor stage and malignancy in several cancers [4-7]. Currently, therapies targeted towards RON RTK are in various stages of development [8,9]. However, targeting RON for therapy is complicated due to the presence of multiple isoforms, which, despite having similar sequence structures, exhibit vastly different, and in a few cases even opposing, functions [10,11].
Various structural features of RON, such as the presence of large number of exons and variety of functional motifs, combined with differential splicing may affect the functionality of the resultant isoforms in a number of ways (Figure 1). Hitherto identified transcript variants and/or protein isoforms of RON are listed in Table 1. Isoform products of differentially spliced transcripts have been found to localize differently resulting in intracellular, plasma membrane bound and extracellular detection of RON [12,13]. At a functional level, both constitutively active [14] and dominant negative [12] isoforms of RON have been identified. RON isoforms caused epithelial cell transformation in in-vitro, produced invasive phenotypes in vivo [15] and promoted tumor progression towards malignancy [6,16]. In our previous study, western blotting analysis of several lung cancer cell lines indicated the presence of isoforms, whereas the full length RON protein was not detected, implying important tumor promoting functions for isoforms [17]. A recent study demonstrated the futility of targeting wild type RON using monoclonal antibodies (mAbs) as they failed to stop tumor progression [18].
Figure 1.
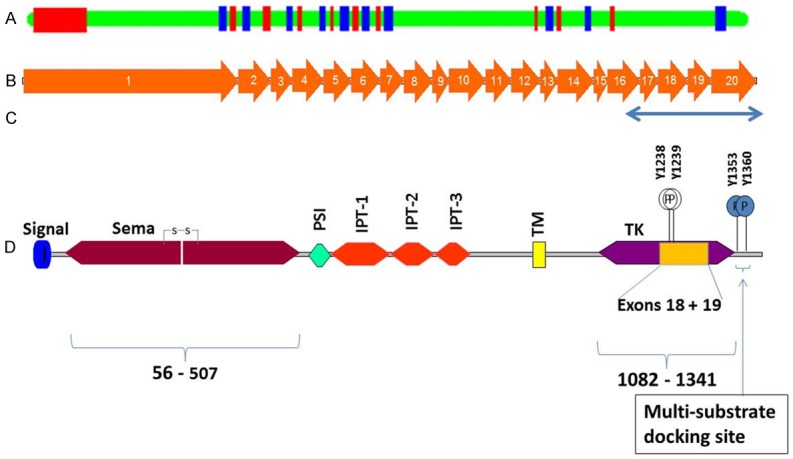
Schematic diagram showing exons, structure-function domains and important amino acid residues of RON coding sequence. A. RON gene with exons shown in red and blue and introns and untranslated regions shown in green. B. 20 coding exons of RON are shown in proportion to sequence length; C. Region PCR amplified and sequenced in the present study (RON coding sequence lying between exons 14 and 20); starting nucleotide numbers are given for each exon. D. Various domains of RON protein and two juxtaposed tyrosine residues at position 1238 and 1239 respectively (Y-Y) in the kinase domain; carboxy-terminal docking site for multiple substrates with src homology 2 (SH2) domain contains two phosphorylation sites for tyrosine at amino acid positions 1353 and 1360; the other important motifs/domains are secretory peptide signal sequence, sema, plexin, semaphorin and integrin (PSI), Ig-like, plexins, transcription factors (IPT), transmembrane (TM) and tyrosine kinase (TK).
Table 1.
Previously characterized isoforms/transcripts of RON
| Isoform | Sequence change | Frame-shift | Affected Amino Acids | Affected domain(s) | Function | Cellular localization | Reference |
|---|---|---|---|---|---|---|---|
| RON 170 | Exon 19 deletion | Yes | 1271-1400 | TK, C-terminal docking site | Dominant negative | Transmembrane | [19] |
| RON 165 | Exon 11 deletion | No | 884-932 | IPT-4 | Constitutively active | Cytoplasmic | [19] [6,14] |
| RON 160 | Exons 5 & 6 deletion | No | 574-682 | IPT-1 | Constitutively active | Transmembrane | [19] [6] [23] |
| RON 155 | Exons 5, 6 & 11 deletion | No | 574-682; 884-932 | IPT-1, 4 | Constitutively active | Cytoplasmic | [19] [6] |
| RON 90 | Exon 6 deletion | Yes | 627-1400 | TM, TK, C-terminal adapter | Antagonist to MSP/RON signaling | Secreted | [12] |
| RON 85 | Insertion of 49 bases between exons 5 and 6 | Yes | 627-1400 | TM, TK, C-terminal adapter | Antagonist to MSP/RON signaling | Secreted | [20] |
|
| |||||||
| Additional forms of RON | |||||||
| sf-RON (RON 52) | Alternative promoter initiated | 1-883 | Sema, PSI, IPT-1 | Constitutively active | Cytoplasmic | [21] [27] | |
| RON 110 | Product of protease action | 1-631 | Sema, PSI, IPT-1 | Constitutively active | Transmembrane | [19] [15] | |
|
| |||||||
| RON transcripts | |||||||
| RON-495 | Retention of 69 bases of intron 2 | Yes | 475-1400 | TM, TK, C-terminal adapter | Antagonist to MSP/RON signaling? | Secreted | [28] |
| RON-541 | Del: exons 2, 3; retention of 64 bps of intron 5 | Yes | 411-516; 627-1400 | Sema, PSI, IPT-1-4, TM, TK, C-terminal adapter | Antagonist to MSP/RON signaling? | Secreted | [28] |
| RON-908 | Del: exon 11; retention of 75 bps of intron 11 | Yes | 884-1400 | IPT-1-4, TM, TK, C-terminal adapter | Antagonist to MSP/RON signaling? | Secreted | [28] |
| RON-647 | Retention of 64 bps of intron 5 | Yes | 627-1400 | IPT-1-4, TM, TK, C-terminal adapter | Antagonist to MSP/RON signaling? | Secreted | [28] |
Quantification and functional analysis of aberrantly expressed RON using methods lacking isoform specificity has led to the general belief that RON overexpression may be the driver of various cancers. We believe that isoform specific quantification and functional determination are important prerequisites for understanding the deregulated RON signaling in cancers. Understanding and targeting aberrantly expressed RON for tumor treatments requires identification of all the isoforms as well as knowledge of their distribution in cancers. In this study, we applied a sensitive method to screen lung cancer cell lines for novel RON transcripts and detected ubiquitous presence of two alternatively spliced transcript variants of RON. The ubiquitous presence of these two transcript variants, potentially coding for dominant negative isoforms, suggest a tumor suppressor role for wild-type RON in lung cancer.
Materials and methods
SCLC cell lines (H249, H69, H526, H524 and H82) and NSCLC cell lines (SKMES, H1703, H1437, H1993, SKLU1, H358 and A549) were obtained from ATCC (Manassas, VA) and were cultured in RPMI 1640 medium (Gibco/BRL) supplemented with 10% (v/v) fetal bovine serum, L-glutamine and 1% (v/v) penicillin/streptomycin at 37°C with 5% CO2. Total RNA from the cell lines was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following manufacturer’s instructions. cDNA was generated from 1 µg of total RNA and oligo dT primer using Single Strand cDNA Synthesis Kit (Clontech, Palo Alto, CA, USA). cDNAs were PCR amplified using RON specific PCR primers covering 755 bps of RON reference mRNA sequence (NM_000247) using forward primer (located in exon 16), 5’-CCCTATATGTGCCACGGTGA-3’, and reverse primer (located in exon 20), 5’-CAAGGCAGCTAAGCAGGTCCAG-3’. PCR reactions were carried out using Phusion high fidelity DNA polymerase (New England Biolabs, MA) in a final volume of 20 mL reagent. PCR conditions were: initial denaturation at 98°C for 30 sec followed by 30 cycles of i) denaturation at 98°C for 10 sec, ii) annealing at 60°C for 20 sec and iii) extension at 72°C for 15 sec. PCR products were treated with EXO-SAP-IT (USB, Cleveland, OH) to remove excess primers, and sequenced bi-directionally using the same PCR amplification primers. Sequencing was performed by employing Big Dye Terminator Chemistry (Applied Biosystems, Weiterstadt, Germany). Sequence deletions in the PCR products were identified manually by aligning sequencing chromatograms with reference RON sequence using Mutation Surveyor version 3.1 software (SoftGenetics, State College, PA). The nucleotide positions numbering was done relative to the first base of the translational initiation codon according to the full-length RON coding sequence (CCDS 2807.1).
Results
Owing to the presence of a large number of exons and reports of the existence several isoforms of RON, we applied a novel and sensitive method to identify novel splicing variations in the C-terminal kinase coding region of RON. cDNAs prepared from lung cancer cell lines were used to PCR amplify a 755 bp region of RON, located between exons 16 to 20. Amplification of this short region, as compared to >4200 bp full length RON, was expected to yield better quality products for sequencing and detection of all possible splicing variants in this region of mRNA.
Sequencing of PCR products from cell cline H249 with the forward primer yielded RON reference sequence as the predominant transcript type. The sequencing chromatogram also showed an additional overlapping sequence starting from nucleotide 3645 (Figure 2A). The overlapping sequence ended at nucleotide 3934 of the reference sequence indicating the deletion of exons 18+19, corresponding to absence of 303 base pairs in the shortened product (not shown in the figure). Alignment and analysis of the sequencing chromatogram obtained using the reverse sequencing primer indicated the presence of two alternatively spliced transcripts, in addition to wild type RON transcript, one lacking exons 18+19 and the other lacking only exon 19 (Figure 2B). The overlapping sequences start at nucleotide 3947 of the reference sequence. One of the overlapping sequences starts (from 3’ end) with bases corresponding to exons 18 (indicating deletion of exon 19) and the other overlapping sequence starts with bases corresponding to exon 17 (indicating deletion of exons 18+19). This confirmed the presence of two alternatively spliced transcripts in the PCR mixture, one involving deletion of exons 18+19 and the other involving deletion of exon 19. From these results it was inferred that the same cell line, A249, contained 3 transcript variants, which includes the full length RON. The two alternatively spliced transcripts (Del: exons 18+19 and Del: exon 19) together with the normal transcript of RON were also found in SCLC cell lines H69, H526, H524 and H82 (Figure 3) and all the seven NSCLC cell lines (Supplementary Figure 1) used in this study.
Figure 2.

RON transcripts lacking exons 18 and 19 in cell line A249. A. PCR product sequenced from 5’ end showing the presence of wild type transcript and an alternatively spliced transcript lacking exons 18+19. B. PCR product sequenced from 3’ end showing the presence of two alternatively spliced transcripts having deletions of exons 18+19 and exon 19 together with the native form. In each case, corresponding region of reference RON sequence with numbering is shown at the top of each panel.
Figure 3.

Alternatively spliced RON transcripts from four SCLC cell lines showing deletion of exons 18+19 and exon 19. cDNAs from cell lines were PCR amplified and sequenced from 3’ end using reverse primer as described in methods. Reference sequence and the numbering of base pairs are given at the top.
A search of human EST database for partial RON transcripts resulting from deletion of exons 18+19 failed to bring up any positive result indicating that this is a novel transcript. Transcript variant lacking exon 19, however, is represented by EST sequence AW009348.
Loss of 303 bases, from nucleotide 3645 to 3947, caused by deletion of exons 18+19, corresponds to loss of 101 amino acids from 1216 to 1316 of the reference RON sequence (Supplementary Figure 2). Amino acids coded by exons 18 and 19 form part of tyrosine kinase domain (amino acids 1082 to 1341 of RON) and deletion of exons 18+19 does not cause reading-frame shift. This region of the tyrosine kinase domain has tyrosine residues Y1238 and Y1239 that are phosphorylated (Figure 1) following binding of RON by its ligand, macrophage-stimulating protein (MSP). Exon 19 is 137 bases long and consists of nucleotides 3811 to 3947 of the reference RON transcript sequence, and deletion of exon 19 leads to a shift in reading frame causing changes to amino acid sequence after nucleotide 1270 and the appearance of a termination codon.
Discussion
Since isoforms of RON have diverse, and sometimes even opposing, functions, it is imperative to identify and characterize the individual isoforms, both structurally and functionally, for target validation and therapy. In this study, by sequencing the C-terminal region of RON transcripts of SCLC and NSCLC cell lines, we discovered widespread occurrence of two alternatively spliced transcripts, one lacking exons 18+19 and the other lacking only exon 19. The wild type and the two alternatively spliced transcript sequences were present in all the 12 cell lines studied. Of the two, RON transcript variant lacking exons 18+19 is a novel one. Combined deletion of exons 18+19 results in an in-frame deletion of 303 bases (or 101 amino acids) of the catalytic kinase domain of RON and the isoform derived from this transcript is expected to act in a dominant negative fashion by blocking MSP stimulated RON kinase activity. Ubiquitous presence of these two transcripts in tumor cells may interfere with quantitative expression and functional analysis of normal RON.
RON transcript variant lacking exon 19 and its isoform product RONΔ170 have been reported previously [19]. Exon 19 consists of 137 nucleotides and skipping of exon 19 resulted in loss of 46 amino acids (correspond to exon 19) together with the appearance of a premature termination codon due to frame-shift. This splicing change results in the loss of part of the kinase domain and disappearance of the multi-functional C terminal docking site. RONΔ170, capable of binding MSP as well as dimerizing with normal RON, was shown to act as a dominant negative receptor by negatively regulating biochemical and biological activities initiated by MSP binding to RON [19].
The novel transcript lacking exons 18+19, identified in this study, can’t be a substrate for nonsense mediated decay (NMD), since no premature non-sense codons are generated from alternative splicing, and hence, is expected to be translated into a functional protein. Skipping of exons 18 and 19 results in an in-frame deletion of 303 nucleotides corresponding to a loss of 101 amino acids in the kinase domain. The translation product of this novel transcript is expected to behave in a dominant negative fashion similar to RONΔ170.
Several dominant negative isoforms of RON, either secreted or intracellular, have previously been reported in cancers. The variant RONΔ85 was a soluble/secreted protein derived from a differentially spliced mRNA transcript arising out of retention of 49 nucleotide intronic sequence between exons five and six [20]. RONΔ90 was another secreted RON isoform, coded by a transcript lacking exon six and N-terminally truncated as a result [12]. Both these isoforms were detected in conditioned media in in vitro cell culture studies and were shown to be capable of binding both MSP and RON and block MSP/RON signaling.
RON and a number of other RTKs have also been reported to be localized in the nucleus in normal as well as cancer cells [21]. The nuclear localized isoform of EGFR, which lacked kinase activity, was reported to be involved in DNA binding and transactivation functions [22]. RON was shown to be localized in the nucleus under hypoxic conditions where it served to upregulate the expression of c-Jun by binding its promoter, leading to increased cell proliferation, survival adaptation, migration and tumorigenicity [22]. Hence, the dominant negative isoforms of RON, lacking functional kinase domain, may be localized differently and mediate transcriptional or other functions.
While dominant negative isoforms were shown to be produced by tumors, cancer cells also formed constitutively active isoforms which could act without stimulation by the ligand, MSP. Three splicing variants of RON, namely RONΔ165, RONΔ160, and RONΔ155, detected in two primary colon cancer samples were generated by deletions in different regions in extracellular domains of the RON beta chain; RONΔ165 and RONΔ155 were intracellular isoforms lacking exon 11 and exons 5, 6 and 11, respectively, which were also constitutively phosphorylated; RONΔ160 resulted from an in-frame deletion of 109 amino acids in the RON β-chain extracellular domain leading to auto-phosphorylation and increased kinase activity [23]. Overexpression of RON Δ160 isoform caused abnormal accumulation of β-catenin leading to tumorigenic phenotypes [24]. We speculate that accelerated cellular proliferation and migration by constitutively active forms and suppression of MSP dependent RON activation through dominant negative forms may be just some of the pro-tumor functions of aberrantly expressed isoforms of RON.
Based on results of studies performed using reagents that lacked isoform specificity, reduced survival and increased apoptosis were attributed to overexpression of wild type RON [25,26], even though constitutively activated isoforms of RON have also been shown to accelerate cell proliferation in various cancers [14,19]. In a recent study, three high-affinity monoclonal antibodies against human RON were found to efficiently block ligand (MSP) binding and RON dimerization, but none of the antibodies inhibited tumor growth when tested in epithelial tumor xenografts in nude mice prompting the authors to suggest that properties other than blocking MSP ligand binding may be essential for anti-RON mAbs to exert antitumor effects in vivo [18]. Due to the dominant negative role of some of the RON isoforms produced by cancer cells, we hypothesize that inhibiting normal RON or MSP for therapeutic purposes may not neutralize the tumor promoting role of aberrantly expressed RON.
Conclusions
Alternative splicing involving skipping of exons is common in lung cancer. Due to the large sequence similarity among transcript variants and isoforms, conventional methods used for quantifying wild type RON in tumors by immunohistochemical or RT-PCR methods, which lack isoform/transcript variant specificity, may not give true estimates. Further, siRNA studies performed without due consideration for each of the differently spliced transcripts may lead to incorrect functional correlations. Hence, we urge due consideration for the presence of transcript variants lacking one or more exons and the dominant negative functions of their translation products in future target validation studies.
Acknowledgements
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, Kingdom of Saudi Arabia (Award Number 11-MED-2086-02).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Dhomen NS, Mariadason J, Tebbutt N, Scott AM. Therapeutic targeting of the epidermal growth factor receptor in human cancer. Crit Rev Oncog. 2012;17:31–50. doi: 10.1615/critrevoncog.v17.i1.40. [DOI] [PubMed] [Google Scholar]
- 2.Wykosky J, Fenton T, Furnari F, Cavenee WK. Therapeutic targeting of epidermal growth factor receptor in human cancer: successes and limitations. Chin J Cancer. 2011;30:5–12. doi: 10.5732/cjc.010.10542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med. 2013;19:1389–1400. doi: 10.1038/nm.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Toole JM, Rabenau KE, Burns K, Lu D, Mangalampalli V, Balderes P, Covino N, Bassi R, Prewett M, Gottfredsen KJ, Thobe MN, Cheng Y, Li Y, Hicklin DJ, Zhu Z, Waltz SE, Hayman MJ, Ludwig DL, Pereira DS. Therapeutic implications of a human neutralizing antibody to the macrophage-stimulating protein receptor tyrosine kinase (RON), a c-MET family member. Cancer Res. 2006;66:9162–9170. doi: 10.1158/0008-5472.CAN-06-0283. [DOI] [PubMed] [Google Scholar]
- 5.Camp ER, Yang A, Gray MJ, Fan F, Hamilton SR, Evans DB, Hooper AT, Pereira DS, Hicklin DJ, Ellis LM. Tyrosine kinase receptor RON in human pancreatic cancer: expression, function, and validation as a target. Cancer. 2007;109:1030–1039. doi: 10.1002/cncr.22490. [DOI] [PubMed] [Google Scholar]
- 6.Zhou YQ, He C, Chen YQ, Wang D, Wang MH. Altered expression of the RON receptor tyrosine kinase in primary human colorectal adenocarcinomas: generation of different splicing RON variants and their oncogenic potential. Oncogene. 2003;22:186–197. doi: 10.1038/sj.onc.1206075. [DOI] [PubMed] [Google Scholar]
- 7.Maggiora P, Marchio S, Stella MC, Giai M, Belfiore A, De Bortoli M, Di Renzo MF, Costantino A, Sismondi P, Comoglio PM. Overexpression of the RON gene in human breast carcinoma. Oncogene. 1998;16:2927–2933. doi: 10.1038/sj.onc.1201812. [DOI] [PubMed] [Google Scholar]
- 8.Feng L, Yao HP, Wang W, Zhou YQ, Zhou J, Zhang R, Wang MH. Efficacy of anti-RON antibody Zt/g4-drug maytansinoid conjugation (Anti-RON ADC) as a novel therapeutics for targeted colorectal cancer therapy. Clin Cancer Res. 2014;20:6045–6058. doi: 10.1158/1078-0432.CCR-14-0898. [DOI] [PubMed] [Google Scholar]
- 9.Guin S, Yao HP, Wang MH. RON receptor tyrosine kinase as a target for delivery of chemodrugs by antibody directed pathway for cancer cell cytotoxicity. Mol Pharm. 2010;7:386–397. doi: 10.1021/mp900168v. [DOI] [PubMed] [Google Scholar]
- 10.Haendeler J, Mlynek A, Buchner N, Lukosz M, Graf M, Guettler C, Jakob S, Farrokh S, Kunze K, Goy C, Guardiola-Serrano F, Schaal H, Cortese-Krott M, Deenen R, Kohrer K, Winkler C, Altschmied J. Two isoforms of Sister-Of-Mammalian Grainyhead have opposing functions in endothelial cells and in vivo. Arterioscler Thromb Vasc Biol. 2013;33:1639–1646. doi: 10.1161/ATVBAHA.113.301428. [DOI] [PubMed] [Google Scholar]
- 11.Hakre S, Tussie-Luna MI, Ashworth T, Novina CD, Settleman J, Sharp PA, Roy AL. Opposing functions of TFII-I spliced isoforms in growth factor-induced gene expression. Mol Cell. 2006;24:301–308. doi: 10.1016/j.molcel.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 12.Eckerich C, Schulte A, Martens T, Zapf S, Westphal M, Lamszus K. RON receptor tyrosine kinase in human gliomas: expression, function, and identification of a novel soluble splice variant. J Neurochem. 2009;109:969–980. doi: 10.1111/j.1471-4159.2009.06027.x. [DOI] [PubMed] [Google Scholar]
- 13.Smith LD, Lucas CM, Eperon IC. Intron retention in the alternatively spliced region of RON results from weak 3’ splice site recognition. PLoS One. 2013;8:e77208. doi: 10.1371/journal.pone.0077208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collesi C, Santoro MM, Gaudino G, Comoglio PM. A splicing variant of the RON transcript induces constitutive tyrosine kinase activity and an invasive phenotype. Mol Cell Biol. 1996;16:5518–5526. doi: 10.1128/mcb.16.10.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang MH, Wang D, Chen YQ. Oncogenic and invasive potentials of human macrophagestimulating protein receptor, the RON receptor tyrosine kinase. Carcinogenesis. 2003;24:1291–1300. doi: 10.1093/carcin/bgg089. [DOI] [PubMed] [Google Scholar]
- 16.Camp ER, Liu W, Fan F, Yang A, Somcio R, Ellis LM. RON, a tyrosine kinase receptor involved in tumor progression and metastasis. Ann Surg Oncol. 2005;12:273–281. doi: 10.1245/ASO.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 17.Kanteti R, Krishnaswamy S, Catenacci D, Tan YH, EL-Hashani E, Cervantes G, Husain AN, Tretiakova M, Vokes EE, Huet H, Salgia R. Differential expression of RON in small and nonsmall cell lung cancers. Genes Chromosomes Cancer. 2012;51:841–51. doi: 10.1002/gcc.21968. [DOI] [PubMed] [Google Scholar]
- 18.Gunes Z, Zucconi A, Cioce M, Meola A, Pezzanera M, Acali S, Zampaglione I, De Pratti V, Bova L, Talamo F, Demartis A, Monaci P, La Monica N, Ciliberto G, Vitelli A. Isolation of Fully Human Antagonistic RON Antibodies Showing Efficient Block of Downstream Signaling and Cell Migration. Transl Oncol. 2011;4:38–46. doi: 10.1593/tlo.10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lu Y, Yao HP, Wang MH. Multiple variants of the RON receptor tyrosine kinase: biochemical properties, tumorigenic activities, and potential drug targets. Cancer Lett. 2007;257:157–164. doi: 10.1016/j.canlet.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 20.Ma Q, Zhang K, Yao HP, Zhou YQ, Padhye S, Wang MH. Inhibition of MSP-RON signaling pathway in cancer cells by a novel soluble form of RON comprising the entire sema sequence. Int J Oncol. 2010;36:1551–1561. doi: 10.3892/ijo_00000642. [DOI] [PubMed] [Google Scholar]
- 21.Angeloni D, Danilkovitch-Miagkova A, Ivanova T, Braga E, Zabarovsky E, Lerman MI. Hypermethylation of Ron proximal promoter associates with lack of full-length Ron and transcription of oncogenic short-Ron from an internal promoter. Oncogene. 2007;26:4499–4512. doi: 10.1038/sj.onc.1210238. [DOI] [PubMed] [Google Scholar]
- 22.Carpenter G, Liao HJ. Receptor tyrosine kinases in the nucleus. Cold Spring Harb Perspect Biol. 2013;5:a008979. doi: 10.1101/cshperspect.a008979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang MH, Kurtz AL, Chen Y. Identification of a novel splicing product of the RON receptor tyrosine kinase in human colorectal carcinoma cells. Carcinogenesis. 2000;21:1507–1512. [PubMed] [Google Scholar]
- 24.Xu XM, Zhou YQ, Wang MH. Mechanisms of cytoplasmic {beta}-catenin accumulation and its involvement in tumorigenic activities mediated by oncogenic splicing variant of the receptor originated from Nantes tyrosine kinase. J Biol Chem. 2005;280:25087–25094. doi: 10.1074/jbc.M414699200. [DOI] [PubMed] [Google Scholar]
- 25.Park JS, Park JH, Lee S, Joo YE, Jung YD. Small interfering RNA targeting of Recepteur d’Origine Nantais induces apoptosis via modulation of nuclear factor-kappaB and Bcl-2 family in gastric cancer cells. Oncol Rep. 2010;24:709–714. [PubMed] [Google Scholar]
- 26.Logan-Collins J, Thomas RM, Yu P, Jaquish D, Mose E, French R, Stuart W, McClaine R, Aronow B, Hoffman RM, Waltz SE, Lowy AM. Silencing of RON receptor signaling promotes apoptosis and gemcitabine sensitivity in pancreatic cancers. Cancer Res. 2010;70:1130–1140. doi: 10.1158/0008-5472.CAN-09-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bardella C, Costa B, Maggiora P, Patane S, Olivero M, Ranzani GN, De Bortoli M, Comoglio PM, Di Renzo MF. Truncated RON tyrosine kinase drives tumor cell progression and abrogates cell-cell adhesion through E-cadherin transcriptional repression. Cancer Res. 2004;64:5154–5161. doi: 10.1158/0008-5472.CAN-04-0600. [DOI] [PubMed] [Google Scholar]
- 28.Jin P, Zhang J, Sumariwalla PF, Ni I, Jorgensen B, Crawford D, Phillips S, Feldmann M, Shepard HM, Paleolog EM. Novel splice variants derived from the receptor tyrosine kinase superfamily are potential therapeutics for rheumatoid arthritis. Arthritis Res Ther. 2008;10:R73. doi: 10.1186/ar2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.