

Abstract
Treating bacterial infections can be difficult due to innate or acquired resistance mechanisms, and the formation of biofilms. Cyclic lipopeptides derived from fusaricidin/LI-F natural products represent particularly attractive candidates for the development of new antibacterial and antibiofilm agents, with the potential to meet the challenge of bacterial resistance to antibiotics. A positional-scanning combinatorial approach was used to identify the amino acid residues responsible for driving antibacterial activity, and increase the potency of these cyclic lipopeptides. Screening against the antibiotic resistant ESKAPE pathogens revealed the importance of hydrophobic as well as positively charged amino acid residues for activity of this class of peptides. The improvement in potency was especially evident against bacterial biofilms, since the lead cyclic lipopeptide showed promising in vitro and in vivo anti-biofilm activity at the concentration far below its respective MICs. Importantly, structural changes resulting in a more hydrophobic and positively charged analog did not lead to an increase in toxicity toward human cells.
Keywords: combinatorial library, cyclic lipopeptides, biofilm, porcine model, resistance, toxicity
Graphical Abstract
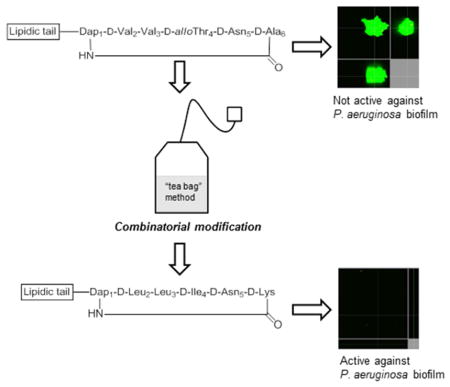
1. Introduction
Pathogens such as Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter sp.[1, 2] (collectively called the ESKAPE pathogens) are the leading causes of hospital-acquired infections, and are resistant to the most commonly used antibiotics due to their acquisition of resistance genes, and their ability to form biofilms; leaving very few therapeutic options.[3, 4] The discovery of novel antibiotics has historically focused on bacteria growing in a planktonic state, however it has recently been indicated that novel therapies addressing bacterial infections must account for the presence of biofilms.[5, 6] It has been estimated that bacterial biofilms are responsible for approximately 60–80% of all chronic infections.[7] The important characteristic of chronic biofilm-associated infections is an insensitivity to both the host immune response and antimicrobial intervention.[8–11] For example, bacteria within a biofilm are up to 1,000 times more resistant to the effect of antibacterial agents than the same organism circulating in a planktonic (free swimming) state.[12–14] Poor penetration of the drug through the biofilm, slow growth of the biofilm due to nutrient limitation, the activation of general stress response pathways, the emergence of a biofilm-specific phenotype and the presence of persister cells have been suggested as mechanisms of biofilm resistance.[15–19] Chronic wounds, such as diabetic ulcers, pressure ulcers and venous leg ulcers, are particularly susceptible to the development of biofilm-associated infections due to impaired healing properties of patients.[20] These infections are typically polymicrobial, with S. aureus and P. aeruginosa most frequently isolated from such wounds.[21–23] In addition, A. baumannii has emerged as a significant nosocomial pathogen, and is responsible for an increasing number of infections among immunocompromised and trauma patients.[24–26] A number of antibiotics have been evaluated for the prevention and disruption of biofilms, however few have proven to be effective.[27, 28]
Although limited therapeutic options are presently available, in order to provide effective treatments, new and innovative anti-infectives are needed, preferably with novel modes of action and/or belonging to novel classes of drugs. To this end, the structurally unique LI-F family of natural products, including fusaricidins, represent attractive candidates for the discovery and development of new antibacterial agents, capable of treating complicated infections caused by multidrug-resistant bacteria, including those found within biofilms.[29–33] Fusaricidins/LI-Fs are cyclic lipodepsipeptide antifungal antibiotics isolated from Paenibacillus sp. Amongst the isolated fusaricidin/LI-F antibiotics, fusaricidin A or LI-F04a has been shown to possess the most potent in vitro antimicrobial activity, targeting a variety of fungi and Gram-positive bacteria (MICs ranging from 0.78–3.12 μg/mL).[31, 32]
Structural modifications of fusaricidin A/LI-F04a that include incorporation of a simpler lipidic tail, and substitution of an ester bond with an amide bond, resulted in comparably potent analogs, with improved proteolytic stability under physiologically relevant conditions, and greatly decreased nonspecific cytotoxicity.[34] Structures of the most potent fusaricidins/LI-Fs synthetic analogs, 1 and 2, are shown in Figure 1. In addition to being active against planktonic Gram-positive bacteria, cyclic lipopeptides 1 and 2 showed promising activity against bacterial biofilms as well. We have previously demonstrated that 1 and 2 very efficiently inhibit the growth of S. aureus biofilm in vitro, at concentrations corresponding to their MICs, and that depsipeptide 1 reduced the proliferation of community-associated MRSA (USA300) in an in vivo porcine wound model.[35] However, depsipeptide 1 exhibited significant in vitro toxicity toward human liver and red blood cells.[34] Although the mode of action of this class of antibacterial peptides is not yet fully understood, the bacterial membrane has been suggested as a potential target based on analysis of the Bacillus subtilis transcriptome after treatment with the fusaricidin/LI-F natural product mixtures.[36] Our initial mechanistic studies showed that 1 and 2 are able to depolarize the cytoplasmic membranes of Gram-positive bacteria in a concentration-dependent manner. However, a lack of correlation between membrane depolarization and cell lethality suggested that membrane-targeting activity is not the primary mode of action for this class of antibacterial peptides.[37] Due to its improved stability and lower cytotoxicity, cyclic lipopeptide 2 may have significant advantages over naturally occurring fusaricidin A/LI-F04a and its depsipeptide analogs as a lead structure for the development of new antibacterial agents. In addition, amide analogs are synthetically more accessible than the parent depsipeptides, allowing for further structural optimization using a combinatorial chemistry approach. Indeed, given the 20 amino acid building blocks, even small cyclic peptides such as 2 offer enormous diversity, and the potential for discovery of more potent analogs. Among combinatorial chemistry strategies, positional scanning synthetic combinatorial libraries (PS-SCL) offer a unique and rapid approach for peptide sequence optimization.[38–40]
Figure 1.

Sequences of cyclic lipopeptides 1–12 and control lipopeptide 13.
In the pilot study reported herein, we have generated a positional scanning combinatorial library of cyclic lipopeptide 2, containing 130,321 cyclic lipopeptides to screen for enhanced antibacterial activity. A lead cyclic lipopeptide was identified and assessed for its antibacterial/antibiofilm activities and nonspecific toxicity.
2. Results and discussion
To gain further insight into the amino acid requirements for antibacterial activity and toxicity of the fusaricidin/LI-F class of cyclic lipopeptides, and to identify analog(s) with improved activities, we prepared a combinatorial library of cyclic lipopeptide 2. Cyclic lipopeptide 2 was used as a model compound due to its low nonspecific toxicity, promising antibacterial/antibiofilm activities, and ease of synthesis.
2.1. Library design and synthesis
The general strategy for cyclic lipopeptide combinatorial library preparation and library deconvolution is outlined in Scheme 1. The cyclic lipopeptide synthetic combinatorial library was generated by the process of divide, couple and recombine (tea bag“ method) using a previously developed Fmoc SPPS chemistry.[34, 41, 42] In brief, our synthetic strategy included attachment of the C-terminal amino acid Fmoc-D-Asp5-OAllyl to a PEG-based amide resin via the side chain, use of a combination of orthogonal protecting groups, stepwise solid-phase assembly of a linear precursor, attachment of a lipidic tail followed by coupling of the N-terminal Fmoc-Dap1(Mtt)-OH and on-resin cyclization. To maintain the same order of D- and L-amino acids as they appear in the sequences of the fusaricidin/LI-F natural products in the synthesized cyclic lipopeptides, D-Val2, D-Thr4, and D-Ala6 were replaced with D-amino acid mixtures, whereas L-Val3 was similarly replaced with an L-amino acid mixture. Fmoc deprotection, amino acid coupling reactions and final cyclization steps were monitored by a ninhydrin colorimetric test.[43, 44] As depicted in Scheme 1, four sets of 19 cyclic lipopeptide sub-libraries (Cys is omitted because of its propensity to form disulfide bonds) were prepared. The first set of 19 sub-libraries had specifically defined amino acids in position 4 of the sequence (one of the 19 commercially available D-amino acids), whereas the remaining three positions consisted of mixtures of the 19 D- or 19 L-amino acids. The next set of cyclic peptide sub-libraries were defined by the amino acid in position 3 (one of the 19 commercially available L-amino acids), and the remaining positions contained all possible amino acid combinations. This process was thus repeated for the remaining amino acid residues. Variable positions were incorporated by coupling of a mixture of 19 Fmoc-protected amino acids in predetermined molar ratios to compensate for different coupling rates.[42] Protocols for the removal of Allyl and Mtt protecting groups, on-resin cyclization, and cleavage of cyclic lipopeptide mixtures from the resin were identical to those previously described for the synthesis of cyclic lipopeptide 2.[34, 37] Using a PS-SCL approach, we generated a combinatorial library containing 130,321 cyclic lipopeptides.
Scheme 1.

Positional scanning synthetic combinatorial library of cyclic lipopeptides. Each set contains 19 separate mixtures. (X) represents position composed of a mixture of 19 amino acids (redox-sensitive Cys is omitted). (Y) represents position defined with an individual amino acid.
2.2. Library screening and assessment antibacterial and antibiofilm activities of individual peptides
To demonstrate the feasibility of the PS-SCL approach to identify novel cyclic lipopeptides with improved antibacterial and antibiofilm activities, we screened the cyclic lipopeptide library for activity against the ESKAPE pathogens. Screening assays were performed according to standard microbroth dilution methods, using a tier of three different concentrations (100, 50 and 25 μg/mL) in 96-well plates.[45, 46] As indicated from screening assays, Figure 2, the sequences of cyclic lipopeptides for activity against both Gram-positive and Gram-negative bacteria were identified to contain neutral and positively charged Lys and/or Arg amino acids. Therefore, we synthesized the nine peptides with sequences shown in Figure 1. Cyclic lipopeptides 1, 2 and linear lipopeptide 13 were used for comparison purposes.
Figure 2.

Screening of cyclic lipopeptide positional scanning-synthetic combinatorial library for activity against the ESKAPE pathogens. Nineteen amino acids were tested for activity at each of the four varying positions. A = R2; B = R3; C = R4; and D = R6. The height for each individual bar is determined by dividing 100 μg/mL (the maximum concentration tested) by the individual MIC for each agent against the respective pathogen. Libraries are then given a scaled score for each pathogen, and these are then stacked to determine the amino acid that displays the broadest activity, at the lowest concentration.
All individual peptides 1–13 were synthesized as described previously.[34, 37] The antibacterial in vitro activity of peptides 1–13 against ESKAPE pathogens was assessed in an identical fashion as above, and those data are shown in Table 1. Cyclic lipopeptide 3 was active against all ESKAPE pathogens at 110 μM (100 μg/mL); whereas at a much lower concentration of 28 μM (25 μg/mL), this peptide exhibits antibacterial activity against VRE, MRSA and A. baumannii. In addition, cyclic lipopeptide 3 was active against MRSA and P. aeruginosa at 22 μM (20 μg/mL) and 55 μM (50 μg/mL) respectively. In comparison to depsipeptide 1 and parent amide 2, these data demonstrate significant extension of the antibacterial spectrum of 3.
Table 1.
Activities of lipopeptides 1–13 and control antibiotics against ESKAPE pathogens.
| Concentration (μg/mL)[a] | ||||||
|---|---|---|---|---|---|---|
| Peptide | 100 | 50 | 35 | 25 | 10 | 5 |
| 1 | SA[b] | ES | nt[d] | ES | S | -[e] |
| 2 | ESA | E | nt | S[c] | - | - |
| 3 | ESKAPE | ESK[c] AE[c] | nt | ESA | - | - |
| 4 | ESKA | ESKA | ESA | A | - | - |
| 5 | S | S | S | - | - | - |
| 6 | S | S | S | - | - | - |
| 7 | EA | EA | A | A | - | - |
| 8 | S | S | S | - | - | - |
| 9 | S | S | S | - | - | - |
| 10 | S | S | S | - | - | - |
| 11 | S | S | S | S | - | - |
| 12 | S | S | - | - | - | - |
| 13 | - | - | nt | - | - | - |
| Vancomycin | SA | S | nt | S | S | S |
| Colistin | ESKAPE | EAPE | nt | EAPE | EAP | AP |
| Nisin | - | - | nt | - | - | - |
Concentrations of antimicrobials were expressed in μM and μg/mL throughout the text.
Abbreviations: E: Enterococcus faecium (VRE), S: Staphylococcus aureus (MRSA), K: Klebsiella pneumoniae (CRE), A: Acinetobacter baumannii, P: Pseudomonas aeruginosa and E: Enterobacter cloacae (CRE).
Observed partial inhibition of bacterial cells growth.
not tested.
No antibacterial activity was observred.
As shown in Table 1, depsipeptide 1 was active against VRE and MRSA at concentrations of 30 μM (25 μg/mL) and 12 μM (10 μg/mL), respectively, whereas amide 2 at 62 μM (50 μg/mL) showed activity only against MRSA. Among other synthesized cyclic lipopeptides, 4 exhibits activity against VRE, MRSA and A. baumannii at 36 μM (35 μg/mL) and against K. pneumoniae at 52 μM (50 μg/mL). No activity against P. aeruginosa and E. cloacae was observed for this peptide at concentrations ≤104 μM (100 μg/mL), as shown in Table 1. All other cyclic lipopeptides 5–12 did not show appreciable antibacterial activity against the tested ESKAPE pathogens. Possible synergy of multiple peptides present in mixtures (76 mixtures each composed of 6,859 cyclic lipopeptides) containing these peptides may explain the observed antibacterial activity for mixtures, as well as loss of activity in the case of the individual peptides,[47, 48] More accurate identification of amino acids that drive antibacterial activity of the described cyclic lipopeptides can be achieved by synthesis and screening of a less complex, second generation PS-SCL composed only of the identified hydrophobic and positively charged residues, thus reducing the possibility for synergistic effect. Quite interestingly, an increase in a peptide’s cationicity, as exemplified by peptides 7 and 10, did not improve antibacterial activity. This observation is consistent with the literature, and suggests that the balance of charge and hydrophobicity is delicate, and has to be determined for each class of antimicrobial peptide.[49] Taking into consideration that peptide 3 exhibits the most broad and potent antibacterial activity in this series of cyclic lipopeptides, we selected it for further studies.
In the case of cyclic lipopeptide 3, the greatest improvement in activity was observed against bacterial biofilms. The in vitro ability of peptides 1–3 to decrease the number of cells within biofilms, and disrupt mature 2-day old biofilms formed by MRSA and P. aeruginosa, was assessed using flow cell chamber methodology, as described previously.[50] In each experiment, all cells within biofilms were stained green with the fluorescent dye Syto-9, and dead cells were stained red with propidium iodide (merge shown as yellow to red) for subsequent visualization using confocal microscopy. Representative confocal microscopy reconstructed images of S. aureus and P. aeruginosa biofilms untreated or treated with cyclic lipopeptodes 1–3 are shown in Figure 3. The most potent activity against biofilms formed by both organisms was observed for cyclic lipopeptide 3. Treatment with 3 at a concentration of 4 μg/mL resulted in complete inhibition of biofilm formation, as well as complete eradication of mature biofilms formed by both bacterial species. As shown in Figure 3, depsipeptide 1 at 4 μg/mL was comparably effective in prevention and eradication of Gram-positive (S. aureus) biofilms, whereas this peptide was slightly less effective against Gram-negative (P. aeruginosa) biofilms. Conversely, the parent amide 2 at the same concentration was able to prevent biofilm formation by S. aureus and P. aeruginosa, but was unable to completely eradicate existing biofilms of these two organisms. Even though amide 2 did not eradicate mature S. aureus biofilms, it did substantially trigger biofilm cell death under these conditions, as shown by the bacterial uptake of propidium iodide that stains cells red (merged with Syto-9 shows as yellow) in Figure 3. The observed improved antibiofilm activity of 3 is consistent with its activity against bacteria in a planktonic state and can be attributed to an increase in the peptide’s overall hydrophobicity and cationicity relative to the parent peptide 2. The more hydrophobic and cationic cyclic lipopeptide 3 more efficiently penetrates into bacterial biofilm matrices and consequently more effectively reduces bacterial viability.
Figure 3.

Cyclic lipopeptides prevented biofilm formation and eradicated existing biofilms of Gram-negative and Gram-positive bacterial pathogens. Inhibition of biofilm formation was tested by immediately adding 4.4 μM (4 μg/mL) of each cyclic lipopeptide 1–3 into the flow-through medium of the flow cell apparatus, and subsequently monitoring biofilm formation for a total of 3 days. Eradication experiments involved growing biofilms untreated for two days before the addition of peptides into flow-through medium. After 3 days, all bacteria were stained green with stain Syto-9, while dead-bacteria were stained red with propidium iodide (merge shown as yellow to red) prior to confocal imaging. Each panel shows reconstructions from the top in the large panel and sides in the right and bottom panels (xy, yz and xz dimensions). (A to C) shows inhibition of biofilm formation. (D–F) shows eradication of preformed biofilms. (G) shows untreated bacteria.
We and others have shown that substitution of D-Ala6 residue with either Gly6 or L-Ala6 causes a significant loss in antimicrobial activity of the fusaricidin/LI-F class of cyclic lipopeptides.[34, 51] However, the fact that cyclic lipopeptide 3 with D-Lys in position 6 exhibits improved antibacterial activity, suggests that incorporation of a positively charged amino acid into the sequence of fusaricidin/LI-F cyclic lipopeptides is well tolerated. This finding is of particular practical significance for further optimization of cyclic lipopeptide 3 and related peptides, considering that the net charge, and the number of positively charged residues, has been strongly correlated with biological activity for antimicrobial peptides.[52, 53] Predictably, control linear peptide 4 did not show activity against any of the ESKAPE bacteria, Table 1, supporting our previous study that indicated the importance of the cyclic structure for antibacterial activity.[34]
2.3. In vitro assessment of the potential for resistance development to 3
Serial exposure of bacteria to antibacterial agents can be used to assess the propensity of an organism to develop resistance.[54] Therefore, we investigated the potential of 3 to select for resistance by initially passaging bacteria at sub-inhibitory concentrations, and then increasing this by 2-fold each day. MRSA and P. aeruginosa were selected for these studies as representative Gram positive and Gram-negative species because of the promising inhibitory activity demonstrated thus far. In addition, these strains are clinical isolates, and drug resistance amongst these organisms is wide-spread, substantially limiting available treatment options. Vancomycin and ciprofloxacin were used as reference antibiotics. After eight passages, the MIC of 3 increased approximately 8-fold against MRSA and P. aeruginosa, Figure 4. In contrast, under the same experimental conditions, MICs obtained for vancomycin and ciprofloxacin increased 64-fold in both tested bacteria. These findings demonstrate limited ability of our clinical, broadly drug-resistance isolates, to develop resistance towards the cyclic lipopeptides, compared to clinically relevant antibiotics. As such, this indicates the potential for very low rates of resistance development to 3, and related cyclic lipopeptides.
Figure 4.

Resistance acquisition during serial passaging in the presence of sub-MIC levels of antimicrobials. (A) MRSA CBD-635 and (B) P. aeruginosa USF-1423. MICs are: a) cyclic lipopeptide 3, 22 μM (20 μg/mL) for S. aureus and 55 μM (50 μg/mL) for P. aeruginosa; b) vancomycin, 7 μM (10 μg/mL); c) ciprofloxacin, 30 μM (10 μg/mL). The figure is representative of three independent experiments.
2.4. Cytotoxicity
It has been well established in the literature that the net positive charge (which facilitates interaction with anionic phospholipids in the bacterial membrane), conformational flexibility (allowing transition from a given peptides solution conformation to its active conformation), hydrophobicity and amphipathicity (required for membrane penetration) are major factors that affect the activity of antimicrobial peptides.[55–57] However, the enhancement of hydrophobicity and cationicity can decrease selectivity between bacterial and human cells.[57] Incorporation of more hydrophobic amino acids such as Leu and Ile, and positively charged Lys into the cyclic lipopeptide sequence, increased its overall hydrophobicity and net positive charge. In the case of cyclic lipopeptides 1–3, the relative overall hydrophobicity order was 2<1<3, as indicated by their HPLC retention times (15.1, 15.8 and 16.9 min, respectivly). Since 3 is the most hydrophobic cyclic lipopeptide in the series, as a measure of toxicity, the hemolysis of human erythrocytes and toxicity towards BJ human skin fibroblasts induced by 3 was assessed.
Hemolytic activity was determined against human erythrocytes (0.5 % in PBS), Figure 5. PBS and 1 % Triton X-100 were used as controls for 0 and 100 % hemolysis, respectively. Despite the increase in hydrophobicity and positive charge in comparison to 1 or 2, cyclic lipopeptide 3 did not show hemolytic activity at the highest tested concentration of 564 μM (512 μg/mL), Figure 5. In comparison, low hemolytic activity was reported for cyclic lipopeptide 2, as no appreciable hemolysis was observed at a concentration of 320 μM (260 μg/mL), whereas cyclic lipodepsipeptide 1 was shown to be highly hemolytic at a concentration of 155 μM (128 μg/mL).[34] DMSO (1% v/v final concentration) used to solubilize cyclic lipopeptide 3 did not cause hemolysis under the conditions described in the Experimental Section.
Figure 5.
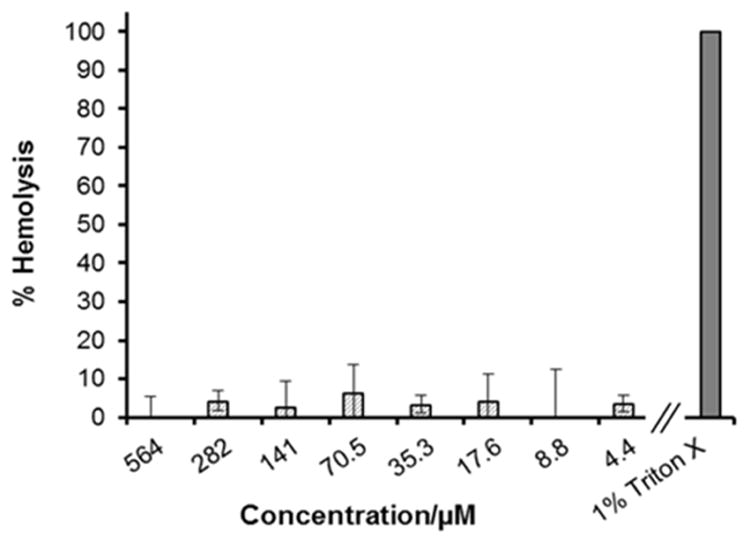
Hemolytic activity of cyclic lipopeptide 3. 100% hemolysis was determined by incubating hRBCs with 1% Triton X.
Toxicity of cyclic lipopeptides 1–3 toward human BJ cells is shown in Figure 6. The known cytotoxic drug doxorubicin (Adriamycin) and 20% DMSO were used as positive and negative controls in these assays. Low toxicity of 2 and 3 was observed against BJ cells; with inordinately high concentrations required for both peptides 2 (250 μM, 203 μg/mL) and 3 (250 μM, 227 μg/mL) to induce a significant loss in cell viability. In contrast, no viable cells were detected in cultures after incubation with depispeptide 1 at a concentration of 150 μM (124 μg/mL). This is consistent with our previous finding, showing lower cytotoxicity of cyclic lipopeptides (amide analogs) in comparison to their depsipeptide counterparts.[34]
Figure 6.
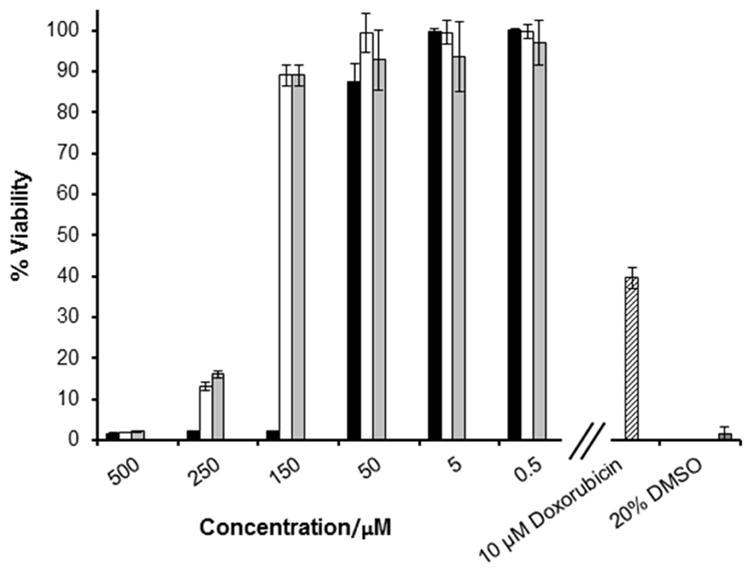
Toxicity of cyclic lipopeptides 1–3 toward BJ human skin fibroblasts. Doxorubicin and DMSO were used as positive controls. 1 (■), 2 (□), 3 (
 ).
).
2.5. In vivo study
The ability of cyclic lipopeptide 3 to reduce the proliferation of A. baumannii was assessed in a porcine model of deep partial thickness wound, Figure 7. Wound infections caused by this pathogen are difficult to treat due to a high level of resistance to multiple antibiotics and its capacity to form a biofilm.[58] In addition, the recent reports indicated an increasing identification of A. baumannii from wound, skin and soft-tissue infections,[24, 25] with a higher incidence rate among hospitalized patients and injured military service members.[26] Swine were chosen for this experiment due to the morphological and biochemical similarity with human skin.[59]
Figure 7.

Effect of 3 on A. baumannii 19606 in vivo using a deep partial thickness porcine wound model. A. baumannii CFUs were determined from porcine wounds 24h post infection and topical treatment with 3 (n=3). Silver sulfadiazine was used as 1% cream.
Wounds were inoculated with 25 μL of 106 CFU/mL of A. baumannii immediately after treatment application and covered with a polyurethane film dressing to allow for biofilm formation as previously described.[60] Infected wounds were treated with cyclic lipopeptide 3 at concentrations of 5.5, 11, 22 mM (5, 10, and 20 mg/mL), DMSO solutions. As shown in Figure 6, peptide 3 reduced the number of A. baumannii in wounds in a concentration-dependent manner. At the highest tested concentration of 22 mM (20 mg/mL), a decrease of approximately 3 log-units in the number of viable A. baumannii in comparison to untreated wound was observed, indicating an antimicrobial effect of cyclic lipopeptide 3 on A. baumannii in vivo. The same effect on A. baumannii was observed upon wound treatment with silver sulfadiazine (1% cream), widely used to prevent and treat wound infections in patients. Importantly, cyclic lipopeptide 3 at 22 mM (20 mg/mL) does not exhibit any observable adverse effects, which is consistent with the low toxicity observed for this peptide in the in vitro experiments.
3. Conclusion
In summary, we have demonstrated that modifying the amino acid sequences of cyclic lipopeptides belonging to the fusaricidin/LI-F class of antimicrobial peptides seemingly leads to analogs with improved anibacterial/antibiofilm activities and markedly decreased nonspecific toxicity. The sequence of cyclic lipopeptide 2 was modified in a positional scanning format, allowing for the rapid identification of amino acid residues responsible for driving antibacterial activity at each position. Of note, screening against the ESKAPE pathogens revealed the importance of hydrophobic as well as positively charged amino acid residues for activity of this class of antibacterial peptides. An increase in overall hydrophobicity of cyclic lipopeptides, and their net positive charge, resulted in improved antibacterial activity. This improvement is particularly important for activity against bacterial biofilms, as the lead peptide 3 showed potent broad-spectrum antibiofilm activity. In contrast to the parent cyclic lipopeptide 2, peptide 3 at a concentration of 4.4 μM (4 μg/mL) effectively inhibited biofilm formation and was able to fully eradicate mature biofilms formed by both MRSA and P. aeruginosa. In addition, a serial passage in vitro resistance development study shows that peptide 3 has a low propensity to select for resistance compared with vancomycin and ciprofloxacin. Promising in vivo antibacterial activity of 3 was demonstrated by prevention of A. baumannii growth in a porcine model of deep partial thickness wounds. Importantly, structural changes resulting in an increase of overall hydrophobicity and cationicity did not lead to increased nonspecific toxicity. By identifying peptide 3 possessing amino acid sequences that are not found in either the parent peptide 2, or in any of the fusaricidin/LI-F natural products, we have demonstrated that our approach may go beyond fusaricidin/LI-F natural product sequences in the discovery of novel and more potent antibacterial peptides of this class.
Clearly, our findings warrant further investigation of the fusaricidin/LI-F class of cyclic lipopeptide structure-activity relationship and their modification to find the optimal balance between antibacterial activity and toxicity. Synthesis of additional cyclic lipopeptides based on our screening assay against ESKAPE bacteria, and assesment of their antibacterial/antibiofilm activities and toxicity, is currently underway by our group.
4. Experimental section
4.1. Materials and methods
TentaGel XV RAM resin was obtained from Rapp Polymere, GmbH. (Tuebingen, Germany). Fmoc-protected amino acids and coupling reagents (HOBt, HBTU, PyBOP) were purchased from Chem-Impex (Wood Dale, IL, USA). N-methylmorpholine, triethylamine and Pd(PPh3)4 was purchased from Sigma-Aldrich (St. Louis, MO, USA). All solvents were purchased from Sigma–Aldrich, and were analytical reagent grade or better. Individual peptides were synthesized on a PS3 automated peptide synthesizer (Protein Technologies Inc., Tucson, AZ, USA). Mass spectrometry was performed on MALDI-TOF Voyager-DE STR (Applied Biosystems, Foster City, CA, USA) in reflector mode using α-cyano-4-hydroxycinnamic acid as a matrix and positive mode. Analytical RP-HPLC analyses and peptide purifications were performed on 1260 Infinity (Agilent Technologies, Santa Clara, CA, USA) liquid chromatography systems equipped with a UV/Vis detector. For analytical RP-HPLC analysis, a C18 monomeric column (Grace Vydac, 250 × 4.6 mm, 5 mm, 120 Å), 1 mL/min flow rate, and an elution method with a linear gradient of 2–98 % B over 30 min, where A is 0.1 % TFA in H2O, and B is 0.08 % TFA in CH3CN were used. For peptide purification, a preparative C18 monomeric column (Grace Vydac, 250 × 22 mm, 10 mm, 120 Å) was used. Elution method was identical to the analytical method except for the flow rate, which was 19 mL/min. Cytotoxicity assays were analyzed on a Synergy H4 microplate reader (BioTek, Winooski, VT, USA). The ESKAPE pathogen isolates (Vancomycin-Resistant E. faecium strain USF-1449, Methicillin-Resistant S. aureus strain CBD-635, K. pneumoniae strain USF-1433, A. baumannii strain USF-1403, P. aeruginosa strain USF-1423 and E. cloacae strain USF-1454) used for MIC and resistance testing have been described by us previously.[61] A. baumannii (ATCC 19606) used in the in vivo study and human skin fibroblasts BJ (ATCC CRL-2522) cells were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). S. aureus (MRSA, #SAP0017) and P. aeruginosa (reference strain PA14) used in the flow cell experiments were obtained from the UBC collection. Dehydrated culture media and polystyrene plates (BD 353072 Falcon) used for antimicrobial and human cell toxicity assays were purchased from Becton Dickinson (Franklin Lakes, NJ, USA). Microscopy was performed using a Leica DMI 4000 B wide-field fluorescence microscope (Leica Microsystems Inc., Buffalo Grove, IL, USA). Control antibiotics were purchased from Sigma–Aldrich. Human red blood cells (hRBCs) were purchased from Innovative Research (Novi, MI, USA). A young female specific-pathogen-free Yorkshire pig was purchased from Looper Farms (NC, USA). Tegaderm polyurethane film dressing was purchased from 3M Health Care (St. Paul, MN, USA).
4.2. General procedure for peptide synthesis and purification
Cyclic lipopeptide PS-SCL and individual peptides 1–13 were synthesized on amide TentaGel XV RAM resin (substitution 0.26 mmol/g, 0.025 mmol scale) using standard Fmoc solid-phase chemistry as reported previously.[34, 37] In all cases the solid-phase synthesis started by attaching C-terminal Fmoc-D-Asp-OAllyl via side chain to the resin using HBTU/HOBt/NMM protocol. The same coupling protocol was used throughout, including coupling of the lipidic tail (Fmoc-12-aminododecanoic acid, 1.5 eq). In the case of depsipeptide 1, Alloc-D-Ala-OH (4 eq) was coupled via ester bond to the hydroxyl group of Fmoc-Thr using N,N′-diisopropylcarbodiimide (DIC, 4 eq.) and 4-dimethylaminopyridine (DMAP, 1 eq) coupling reagents in CH2Cl2. PS-SCL and peptides 2–13 were prepared by coupling Fmoc-Dap(Mtt)-OH instead of Fmoc-Thr-OH using the same coupling protocol as above. Selective removal of Allyl and Alloc protecting groups was performed by treatment of peptidyl-resin precursors with borane dimethylamine complex (4 eq), followed by addition of Pd(PPh3)4 (0.1 eq) in CH2Cl2 under argon. Mtt was selectively removed under mild acidic conditions (1% trifluoroacetic acid in CH2Cl2, 30 min). Solid-phase cyclization of linear precursors was carried out in a manual reaction vessel overnight using PyBOP/HOBt/DIEA (2:2:6 eq) in DMF. The conversion of the lipid tail amino group into the desired guanidino group was achieved by removal of the Fmoc protecting group using standard piperidine deprotection protocol and treatment of the peptidyl-resin with N,N′-di-Boc-N″-triflylguanidine (5 eq) and triethylamine (5 eq) in DCM as described previously.[62] The final purity of synthesized peptides 1–13 was confirmed by analytical RP-HPLC, LC ESI MS and MALDI TOF MS, and was ≥ 95% in all cases.
4.3. Screening of cyclic lipopeptide PS-SCL and assessment of antibacterial activities of peptides 1–13
Screening of cyclic lipopeptide PS-SCL for activity against the ESKAPE pathogens was performed using three different concentrations (100, 50, and 25 μg/mL) in sterile 96-well flat-bottomed polystyrene plates according to the Clinical and Laboratory Standards Institute (CLSI) guidelines.[45] Müller–Hinton broth (MHB) without dilution was used in this experiment. The cyclic lipopeptide PS-SCL was solubilized in DMSO and appropriate dilutions were made in media to a final 1% DMSO concentration (v/v). Controls on each plate were media without bacteria, bacterial inoculum without antimicrobials added, and bacterial inoculum containing methicillin, vancomycin, nisin, colistin and bacitracin. Assessment of activities of peptides 1–13 against ESKAPE bacteria was completed in an identical fashion to that described above. All experiments were performed in triplicate.
4.4. Hemolytic activity
Human red blood cells (hRBCs) were diluted in PBS to 1%. Cyclic lipopeptide 3 was dissolved in 10% DMSO/H2O (v/v) solvent mixture to concentrations of 8.8–1128 μM (8–1024 μg/mL). In each well of a clear, flat-bottom 96-well plate, 50 μL of hRBCs were placed, followed by addition of 50 μL of cyclic lipopeptide solution, to a final peptide concentration of 4.4–564 μM (4–512 μg/mL). Assays were performed in triplicate, and each experiment was repeated twice. To determine the potential effect of DMSO on hemolytic activity, controls containing 10 % DMSO in H2O (v/v) were added to the assay setup. As a positive control, 50 μL Triton X-100 in H2O was used at a final concentration of 1 % (v/v). As a negative control, 50 μL of PBS was used. Plates were incubated for 1 h at 37 °C. To each well 100 μL of PBS was added, and the plates were centrifuged for 10 min at 1000 g. Supernatants (150 μL) were transferred to a new plate, and absorbance at 405 nm was measured. No effect of 10% DMSO in H2O on hemolysis of hRBCs was observed. The degree of hemolysis of the cyclic lipopeptides was expressed in percent relative to the hemolysis caused by Triton-X.
4.5. Cytotoxicity
Cytotoxicity of peptides 1–3 was determined using Cell Titer-Glo Luminescent Cell Viability Assay (Promega). Assays were performed in flat-bottom polystyrene 96-well plates with 10,000 BJ cells (ATCC CRL-2522) per well grown in EMEM containing 10% FBS, and 5% penicillin/streptomycin (v/v). After overnight incubation at 37°C in a humidified atmosphere with 5% CO2, media was removed, and replaced with fresh media with 2% FBS containing peptides 1–3 in a concentration range of 0.5–500 μM. Peptides 1–3 were dissolved in DMSO, and appropriate dilutions were made in buffer to a final concentration of 1% DMSO (v/v). Plates were again incubated at 37°C in a humidified atmosphere with 5% CO2. As a control, 10 μM doxorubicin and 20% DMSO in H2O were used. After incubation for 48 and 72 h, respectively, media was removed, and 100 μL PBS buffer added, followed by 100 μL of Cell Titer-Glo Reagent. Plates were incubated for further 10 min at room temperature (protected from light), before luminescence readout.
4.6. Flow cell experiments and biofilm imaging by confocal microscopy
For inhibition studies, Pseudomonas aeruginosa PA14 and Staphylococcus aureus MRSA (clinical isolate #SAP0017) biofilms were grown in BM2 minimal glucose medium for 72 h, with and without the addition of cyclic lipopeptides 1–3 (final concentration of 4.4 μM (4 μg/mL) at 37°C in flow chambers as previously described.[50] In eradication studies, bacteria were allowed to develop structured biofilms for 2 days prior to peptide treatment for 24 h. Silicone tubing (VWR, 0.062 in ID by 0.125 in OD by 0.032 in wall) was autoclaved, and the system was assembled and sterilized by pumping a 0.5% hypochlorite solution through the system at 6 rpm for 30 min using a Watson Marlow 205S multi-channel peristaltic pump. The system was then rinsed with sterile water and medium for 30 min each. Flow chambers were inoculated by injecting 400 μL of an overnight culture diluted to an OD600 of 0.05. After inoculation, chambers were left without flow for 2 h to allow bacteria to adhere to the surface of the flow cell chambers, after which medium was pumped through the system at a constant rate of 0.5 rpm (2.4 mL/h). Biofilm cells were stained using the LIVE/DEAD BacLight Bacterial Viability kit (Molecular Probes, Eugene, OR) prior to microscopy experiments. A ratio of SYTO-9 (green fluorescence, live cells) to propidium iodide (PI) (red fluorescence, dead cells) of 1:5 was used. Microscopy was done using a confocal laser scanning microscope (Olympus, Fluoview FV1000) and three-dimensional reconstructions were generated using the Imaris software package (Bitplane AG).
4.7. Serial passage experiment
Serial passage was initiated from overnight cultures of MRSA CBD-635 and P. aeruginosa USF-1423 grown in MHB to exponential growth phase and added to a fresh medium containing cyclic lipopeptide 3 or the controls vancomycin and ciprofloxacin at concentrations corresponding to ½ MICs. Final bacterial concentrations were approximately 106 CFU/mL. The bacteria were incubated overnight at 37°C and an aliquot was transferred to a new tube with 2 fold increased concentration of 3 or control antibiotics. The process was repeated for 8 passages. The MIC was determined at each step as described above.
4.8. Antibacterial activity in in vivo wound infection model
Wounding and infection
A young female specific-pathogen-free pig was used. The animal was fed a non-antibiotic chow ad libitum before the study, fasted overnight before the procedures, and housed individually in our USDA-compliant animal facilities. The experimental protocol was approved by the University of Miami Institutional Animal Care and Use Committee, and all the procedures followed the federal guidelines for the care and use of laboratory animals. Methods for the animal preparation and wounding are described previously.[23, 63–65] Briefly, the flank and the back of experimental animals were prepped on the day of the experiment. Animals were anesthetized and partial thickness wounds were made on the paravertebral area using a modified electrokeratome set at 0.5 mm deep × 10 mm × 7 mm.[63] The wounds were separated from one another by approximately 4–6 cm areas of unwounded skin. Wounds were inoculated with 25 μL of 106 CFU/mL of A. baumannii (ATCC 19606). Wounds were treated within 20 min (after inoculation) with 200 μL of peptide 3 (5, 10 and 20 mg/mL; 5.5, 11, 22 mM), vehicle (DMSO) and silver sulfadiazine (1% ointment, 150–200 mg/wound).. Control wounds were infected and no treatment was applied. All wounds, including control, were covered with polyurethane film dressing (Tegaderm) to maintain a moist environment, promote biofilm formation and prevent any cross-contamination between wounds.[60]
Bacterial recovery
Three wounds from each treatment group were cultured quantitatively using a modified scrub technique.[60] Each wound was encompassed by a sterile surgical grade steel cylinder (22 mm outside diameter), and bacteria were collected by scrubbing the wound area with a sterile Teflon spatula into 1 mL of sterile phosphate buffer saline. Serial dilutions were made and plated using a Spiral Plate System, and Leeds selective media was used to determine CFUs in the scrub solution collected from each wound. Plated bacteria were incubated aerobically for 48 h at 37°C. CFUs were determined by the standard colony counting method.
HIGHLIGHTS.
Positional-scanning combinatorial library of cyclic lipohexapeptides was prepared.
Analogs with improved antibacterial activities were identified.
The improvement in activity was especially evident against bacterial biofilms.
Hydrophobic and positively charged amino acids are crutial for activity.
Resulting structural changes did not lead to an increase in nonselective toxicity.
Acknowledgments
This work was supported in part through the Florida Drug Discovery Acceleration Program by the State of Florida, Department of Health, by grants AI103715 (to L.N.S.) and 1R21AI119288-01 (to P.C.) from the National Institute of Allergies and Infectious Diseases and by grant DM140052 (to P.C.) from the Department of Defense, Congressionally Directed Medical Research Program. R.E.W.H. holds a Canada Research Chair in Health and Genomics and his research is supported by the Canadian Institutes for Health Research. C.D.L.F.-N. received scholarships from the Fundación “la Caixa” and Fundación Canadá, and from Fundación Ramón Areces (Spain).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rice LB. Federal Funding for the Study of Antimicrobial Resistance in Nosocomial Pathogens: No ESKAPE. J Infect Dis. 2008;197:1079–1081. doi: 10.1086/533452. [DOI] [PubMed] [Google Scholar]
- 2.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert DN, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin Infec Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 3.Bales PM, Renke EM, May SL, Shen Y, Nelson DC. Purification and Characterization of Biofilm-Associated EPS Exopolysaccharides from ESKAPE Organisms and Other Pathogens. PLoS One. 2013;8:e67950. doi: 10.1371/journal.pone.0067950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11:297–308. doi: 10.1586/eri.13.12. [DOI] [PubMed] [Google Scholar]
- 5.Bjarnsholt T. The role of bacterial biofilms in chronic infections. APMIS Suppl. 2013:1–51. doi: 10.1111/apm.12099. [DOI] [PubMed] [Google Scholar]
- 6.Penesyan A, Gillings M, Paulsen IT. Antibiotic discovery: combatting bacterial resistance in cells and in biofilm communities. Molecules. 2015;20:5286–5298. doi: 10.3390/molecules20045286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.James GA, Swogger E, Wolcott R, Pulcini E, Secor P, Sestrich J, Costerton JW, Stewart PS. Biofilms in chronic wounds. Wound Repair Regen. 2008;16:37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 8.Lipsitch M. The reise and fall of antimicrobial resistance. Trends Microbiol. 2011;9:438–444. doi: 10.1016/s0966-842x(01)02130-8. [DOI] [PubMed] [Google Scholar]
- 9.Costerton JW, Stewart PS, Greenberg EP. Bacterial bioflms: a common cause of persistent infections. Science. 1999;284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 10.Fluit AC, Schmitz FJ, Verhoef J. Multi-resistance to antimicrobial agents for the ten most frequently isolated bacterial pathogens. Int J Antimicrob Agents. 2001;18:147–160. doi: 10.1016/s0924-8579(01)00357-0. [DOI] [PubMed] [Google Scholar]
- 11.Donlan RM. Biofilm formation: a clinically relevant microbiological process. Clin Infect Dis. 2001;33:1387–1392. doi: 10.1086/322972. [DOI] [PubMed] [Google Scholar]
- 12.Ceri H, Olson ME, Stremick C, Read RR, Morck D, Buret A. The Calgary Biofilm Device: New technology for rapid determination of antibiotic susceptibilities of bacterial biofilms. J Clin Microbiol. 1999;37:1771–1776. doi: 10.1128/jcm.37.6.1771-1776.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Martinez JM, Pascual A. Antimicrobial resistance in bacterial biofilms. Rev Med Microbiol. 2006;17:65–75. [Google Scholar]
- 14.Potera C. Forging a link between biofilms and disease. Science. 1999;283:1837–1839. doi: 10.1126/science.283.5409.1837. [DOI] [PubMed] [Google Scholar]
- 15.Soto SM. Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence. 2013;4:223–229. doi: 10.4161/viru.23724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mah TF, O’Toole GA. Mechanisms of biofilm resistance to antimicrobial agents. Trends in microbiology. 2001;9:34–39. doi: 10.1016/s0966-842x(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 17.Balaban NQ, Merrin J, Chait R, Kowalik L, Leibler S. Bacterial Persistence as a Phenotypic Switch. Science. 2004;305:1622–1625. doi: 10.1126/science.1099390. [DOI] [PubMed] [Google Scholar]
- 18.Lewis K. Persister Cells, Dormancy and Infectious Disease. Nat Rev Microbiol. 2007;5:48–56. doi: 10.1038/nrmicro1557. [DOI] [PubMed] [Google Scholar]
- 19.Keren I, Kaldalu N, Spoering A, Wang Y, Lewis K. Persister cells and tolerance to antimicrobials. FEMS Microbiol Lett. 2004;230:13–18. doi: 10.1016/S0378-1097(03)00856-5. [DOI] [PubMed] [Google Scholar]
- 20.Percival SL, Hill KE, Williams DW, Hooper SJ, Thomas DW, Costerton JW. A review of the scientific evidence for biofilms in wounds. Wound Repair Regen. 2012;20:647–657. doi: 10.1111/j.1524-475X.2012.00836.x. [DOI] [PubMed] [Google Scholar]
- 21.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, Wolcott RD. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol. 2008;8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Falagas ME, Kapaskelis AM, Kouranos VD, Kakisi OK, Athanassa Z, Karageorgopoulos DE. Outcome of antimicrobial therapy in documented biofilm-associated infections: a review of the available clinical evidence. Drugs. 2009;69:1351–1361. doi: 10.2165/00003495-200969100-00005. [DOI] [PubMed] [Google Scholar]
- 23.Pastar I, Nusbaum AG, Gil J, Patel SB, Chen J, Valdes J, Stojadinovic O, Plano LR, Tomic-Canic M, Davis SC. Interactions of methicillin resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in polymicrobial wound infection. PLoS One. 2013;8:e56846. doi: 10.1371/journal.pone.0056846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Villegas MV, Hartstein AI. Acinetobacter outbreaks, 1977–2000. Infect Control Hosp Epidemiol. 2003;24:284–295. doi: 10.1086/502205. [DOI] [PubMed] [Google Scholar]
- 25.Scott P, Deye G, Srinivasan A, Murray C, Moran K, Hulten E, Fishbain J, Craft D, Riddell S, Lindler L, Mancuso J, Milstrey E, Bautista CT, Patel J, Ewell A, Hamilton T, Gaddy C, Tenney M, Christopher G, Petersen K, Endy T, Petruccelli B. An outbreak of multidrug-resistant Acinetobacter baumannii-calcoaceticus complex infection in the US military health care system associated with military operations in Iraq. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2007;44:1577–1584. doi: 10.1086/518170. [DOI] [PubMed] [Google Scholar]
- 26.Keen EF, 3rd, Robinson BJ, Hospenthal DR, Aldous WK, Wolf SE, Chung KK, Murray CK. Prevalence of multidrug-resistant organisms recovered at a military burn center. Burns. 2010;36:819–825. doi: 10.1016/j.burns.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 27.Parra-Ruiz J, Vidaillac C, Rose WE, Rybak MJ. Activities of High-dose Daptomycin, Vancomycin, and Moxifloxacin Alone or in Combination with Clarithromycin or Rifampin in a Novel in vitro Model of Staphylococcus aureus Biofilm. Antimicrob Agents Chemother. 2010;54:4329–4334. doi: 10.1128/AAC.00455-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raad I, Hanna H, Jiang Y, Dvorak T, Reitzel R, Chaiban G, Sherertz R, Hachem R. Comparative activities of daptomycin, linezolid, and tigecycline against catheter-related methicillin-resistant Staphylococcus bacteremic isolates embedded in biofilm. Antimicrob Agents Chemother. 2007;51:1656–1660. doi: 10.1128/AAC.00350-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bionda N, Pitteloud JP, Cudic P. Cyclic Lipodepsipeptides: A New Class of Antibacterial Agents in the Battle Against Resistant Bacteria. Future Med Chem. 2013;5:1311–1330. doi: 10.4155/fmc.13.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurusu K, Ohba K. New peptide antibiotics LI-F03, F04, F05, F07, and F08, produced by Bacillus polymyxa. I. Isolation and characterization. J Antibiot. 1987;40:1506–1514. doi: 10.7164/antibiotics.40.1506. [DOI] [PubMed] [Google Scholar]
- 31.Kajimura Y, Kaneda M. Fusaricidin A, a new depsipeptide antibiotic produced by Bacillus polymyxa KT-8. Taxonomy, fermentation, isolation. structure elucidation and biological activity. J Antibiot. 1996;49:129–135. doi: 10.7164/antibiotics.49.129. [DOI] [PubMed] [Google Scholar]
- 32.Kajimura Y, Kaneda M. Fusaricidins B, C and D, new depsipeptide antibiotics produced by Bacillus polymyxa KT-8: isolation, structure elucidation and biological activity. J Antibiot. 1997;50:220–228. [PubMed] [Google Scholar]
- 33.Kuroda J, Fukai T, Konishi M, Uno J, Kurusu K, Nomura T. LI-F Antibiotics, a Family of Antifungal Cyclic Depsipeptides Produced by Bacillus polymyxa L-1129. Heterocycles. 2000;53:1533–1549. [Google Scholar]
- 34.Bionda N, Stawikowski M, Stawikowska R, Cudic M, Lopez-Vallejo F, Treitl D, Medina-Franco J, Cudic P. Effects of Cyclic Lipodepsipeptide Structural Modulation on Stability, Antibacterial activity, and Human Cell Toxicity. Chem Med Chem. 2012;7:871–882. doi: 10.1002/cmdc.201200016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bionda N, Pastar I, Davis SC, Cudic P. In vitro and in vivo Activities of Novel Cyclic Lipopeptides Against Staphylococcal Biofilms Protein. Peptide Lett. 2014;21:352–356. doi: 10.2174/09298665113206660101. [DOI] [PubMed] [Google Scholar]
- 36.Yu WB, Yin CY, Zhou Y, Ye BC. Prediction of the Mechanism of Action of Fusaricidin on Bacillus subtilis. PloS one. 2012;7:e50003. doi: 10.1371/journal.pone.0050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bionda N, Fleeman RM, Shaw LN, Cudic P. Effect of Ester to Amide or N-methylamide Substitution on Bacterial Membrane Depolarization and Antibacterial Activity of Novel Cyclic Lipopeptides. Chem Med Chem. 2013;8:1394–1402. doi: 10.1002/cmdc.201300173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Generation and Use of Synthetic Peptide Combinatorial Libraries for Basic Research and Drug Discovery. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 39.Blondelle SE, Houghten RA. Novel Antimicrobial Compounds Identified Using Synthetic Combinatorial Library Technology. Trends Biotechnol. 1996;14:60–65. doi: 10.1016/0167-7799(96)80922-X. [DOI] [PubMed] [Google Scholar]
- 40.Blondelle SE, Perez-Paya E, Houghten RA. Synthetic combinatorial libraries: novel discovery strategy for identification of antimicrobial agents. Antimicrob Agents Chemother. 1996;40:1067–1071. doi: 10.1128/aac.40.5.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bionda N, Pitteloud JP, Cudic P. Solid-phase Synthesis of Fusaricidin/LI-F Class of Cyclic Lipopeptides: Guanidinylation of Resin-bound Peptidyl Amines. Biopolymers. 2013;100:160–166. doi: 10.1002/bip.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dooly CT, Houghten RA. Synthesis and Screening of Positional Scanning Combinatorial Libraries. In: Cabilly S, editor. Combinatorial peptide library protocols, In Methods in Molacular Biology. Humana Press; Totowa, New Jersey: 1998. [DOI] [PubMed] [Google Scholar]
- 43.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color Test for Detection of Free Terminal Amino Groups in the Solid-phase Synthesis of Peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 44.Sarin VK, Kent SB, Tam JP, Merrifield RB. Quantitative Monitoring of Solid Phase Peptide Synthesis by the Ninhydrin Reaction. Anal Biochem. 1981;117:147–157. doi: 10.1016/0003-2697(81)90704-1. [DOI] [PubMed] [Google Scholar]
- 45.C.a.L.S.I. (CLSI) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically. Clinical and Laboratory Standards Institute; Wayne, PA (USA): 2011. [Google Scholar]
- 46.Van Horn KS, Burda WN, Fleeman R, Shaw LN, Manetsch R. Antibacterial Activity of a Series of N,N-Disubstituted Quinazoline-2,4-diamines. J Med Chem. 2014 doi: 10.1021/jm500039e. [DOI] [PubMed] [Google Scholar]
- 47.Houghten RA, Pinilla C, Appel JR, Blondelle SE, Dooley CT, Eichler J, Nefzi A, Ostresh JM. Mixture-based synthetic combinatorial libraries. J Med Chem. 1999;42:3743–3778. doi: 10.1021/jm990174v. [DOI] [PubMed] [Google Scholar]
- 48.Chang YP, Chu YH. Mixture-based combinatorial libraries from small individual peptide libraries: a case study on alpha1-antitrypsin deficiency. Molecules. 2014;19:6330–6348. doi: 10.3390/molecules19056330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dathe M, Wieprecht T. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim Biophys Acta. 1999;1462:71–87. doi: 10.1016/s0005-2736(99)00201-1. [DOI] [PubMed] [Google Scholar]
- 50.de la Fuente-Nunez C, Korolik V, Bains M, Nguyen U, Breidenstein EB, Horsman S, Lewenza S, Burrows L, Hancock RE. Inhibition of bacterial biofilm formation and swarming motility by a small synthetic cationic peptide. Antimicrob Agents Chemother. 2012;56:2696–2704. doi: 10.1128/AAC.00064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cochrane JR, Yoon DH, McErlean CS, Jolliffe KA. A Macrolactonization Approach to the Total Synthesis of the Antimicrobial Cyclic Depsipeptide LI-F04a and Diastereoisomeric Analogues. Beilstein J Org Chem. 2012;8:1344–1351. doi: 10.3762/bjoc.8.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jiang Z, Vasil AI, Hale JD, Hancock REW, Vasil ML, Hodges RS. Effects of Net Charge and the Number of Positively Charged Residues on the Biological Activity of Amphipathic a-helical Cationic Antimicrobial Peptides. Biopolymers. 2008;90:369–383. doi: 10.1002/bip.20911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suda S, Hill C, Cotter PD, Ross RP. Investigating the Importance of Charged Residues in Lantibiotics. Bioeng Bugs. 2010;1:345–351. doi: 10.4161/bbug.1.5.12353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez JL, Baquero F, Andersson DI. Beyond serial passages: new methods for predicting the emergence of resistance to novel antibiotics. Curr Opin Pharmacol. 2011;11:439–445. doi: 10.1016/j.coph.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 55.Jenssen H, Hamill P, Hancock RE. Peptide antimicrobial agents. Clin Microbiol Rev. 2006;19:491–511. doi: 10.1128/CMR.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chen Y, Guarnieri MT, Vasil AI, Vasil ML, Mant CT, Hodges RS. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob Agents Chemother. 2007;51:1398–1406. doi: 10.1128/AAC.00925-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin LM, Edwards MA, Li J, Yip CM, Deber CM. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J Biol Chem. 2012;287:7738–7745. doi: 10.1074/jbc.M111.303602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Navon-Venezia S, Ben-Ami R, Carmeli Y. Update on Pseudomonas aeruginosa and Acinetobacter baumannii infections in the healthcare setting. Curr Opin Infect Dis. 2005;18:306–313. doi: 10.1097/01.qco.0000171920.44809.f0. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan TP, Eaglestein WH, Davis SC, Mertz PM. The pig as a model for human wound healing. Wound Repair Regen. 2001;9:66–76. doi: 10.1046/j.1524-475x.2001.00066.x. [DOI] [PubMed] [Google Scholar]
- 60.Davis SC, Ricotti C, Cazzaniga A, Welsh E, Eaglstein WH, Mertz PM. Microscopic and physiologic evidence for biofilm-associated wound colonization in vivo. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2008;16:23–29. doi: 10.1111/j.1524-475X.2007.00303.x. [DOI] [PubMed] [Google Scholar]
- 61.Fleeman R, LaVoi TM, Santos RG, Morales A, Nefzi A, Welmaker GS, Medina-Franco JL, Giulianotti MA, Houghten RA, Shaw LN. Combinatorial Libraries As a Tool for the Discovery of Novel, Broad-Spectrum Antibacterial Agents Targeting the ESKAPE Pathogens. J Med Chem. 2015;58:3340–3355. doi: 10.1021/jm501628s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bionda N, Pitteloud JP, Cudic P. Solid-phase synthesis of fusaricidin/li-f class of cyclic lipopeptides: Guanidinylation of resin-bound peptidyl amines. Biopolymers. 2013;100:160–166. doi: 10.1002/bip.22186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pechter PM, Gil J, Valdes J, Tomic-Canic M, Pastar I, Stojadinovic O, Kirsner RS, Davis SC. Keratin dressings speed epithelialization of deep partial-thickness wounds. Wound repair and regeneration: official publication of the Wound Healing Society [and] the European Tissue Repair Society. 2012;20:236–242. doi: 10.1111/j.1524-475X.2012.00768.x. [DOI] [PubMed] [Google Scholar]
- 64.Martineau L, Davis SC, Peng HT, Hung A. Controlling methicillin resistant Staphyloccocus aureus and Pseudomonas aeruginosa wound infections with a novel biomaterial. J Invest Surg. 2007;20:217–227. doi: 10.1080/10717540701481275. [DOI] [PubMed] [Google Scholar]
- 65.Davis SC, Mertz PM. Determining the effect of an oak bark formulation on methicillin-resistant staphylococcus aureus and wound healing in porcine wound models. Ostomy Wound Manag. 2008;54:16–18. [PubMed] [Google Scholar]