

Abstract
The α7 nicotinic acetylcholine receptor (nAChR) is a target for control of inflammation-related phenomena via compounds that are able to selectively induce desensitized states of the receptor. Compounds that selectively desensitize, without facilitating significant channel activation, are termed “silent agonists” because they can be discriminated from antagonists by the currents evoked with co-application with type II positive allosteric modulators (PAMs). One example is N, N-diethyl-N′-phenyl-piperazine (diEPP) (Papke, R.L.; Chojnacka, K.; Horenstein, N.A. J. of Pharm. and Exp. Ther. 2014, 350, 665). We used Ullmann-type aryl amination to synthesize a panel of 27 compounds related to diEPP by substitutions at the aryl ring and in the linkage between the piperazine and phenyl rings. Two-electrode voltage clamping of the human α7 nAChR expressed in Xenopus oocytes revealed that it was possible to tune the behavior of compounds to show enhanced desensitization without corresponding partial agonist activity such that trifluoromethyl and carboxamide aryl substituents showed 33 to 46-fold larger PAM-dependent net-charge responses, indicating selective partitioning of the ligand receptor complexes into the desensitized state.
Graphical abstract
1. Introduction
The homopentameric α7 nicotinic acetylcholine receptor (nAChR) is a ligand-gated ion channel1 characterized by a unique form of concentration-dependent rapid desensitization.2, 3 The receptor belongs to the large superfamily of ligand-gated ion channels1 that are all characterized by a disulfide-constrained “Cys-loop”, which is thought to be involved in the conformational changes linking ligand binding and ion channel activation. Nicotinic acetylcholine receptors are validated therapeutic targets for several pathologies of the central and peripheral nervous system including Alzheimer's and Parkinson's diseases, addiction disorders, schizophrenia, pain management, and inflammation-mediated processes. Recent reports have provided evidence for expression of this receptor subtype in non-neuronal cells including lymphocytes, macrophages, and intestinal and lung endothelial and epithelial cells,4-7 indicating a non-synaptic role for the receptor. Moreover, α7 is a key part of the cholinergic anti-inflammatory response8 in which levels of pro-inflammatory cytokines are decreased,9 making this receptor of great interest considering the wide range of diseases in which systemic inflammation is present. Further, nicotine and other α7 agonists10 have been effective in models of inflammation, inhibiting local leukocyte recruitment and reducing endothelial cell activation, implicating α7 nAChR involvement in regulation of inflammatory processes.
All these data makes the α7 receptor a promising drug target for the treatment of inflammatory diseases and several neurological disorders including chronic pain. Notably, anti-inflammatory effects have been associated with desensitized, nonconducting states of the receptor.11, 12 Thus, compounds that are able to selectively place the receptor into a desensitized state rather than act as partial agonists are of considerable interest, and have shown analgesic activity in the mouse that was directly associated with α7, via α7 knockout mice controls.12
For the α7 receptor, two distinct desensitized states have been identified.13 They differ in being sensitive (Ds) or insensitive (Di) to conversion to open states by type II positive allosteric modulators (PAMs), which reduce desensitization.14 One of the most well-studied PAMs is PNU-120596.15 The binding site for this very effective allosteric enhancer of channel activation appears to be within the receptor's transmembrane domain.16, 17 Silent agonists are desensitizing compounds that have been identified and designed to selectively induce the Ds state in the α7 nAChR with extremely low or absent partial agonist activity. While technically in the absence of a PAM, one may consider a silent agonist to simply be a competitive antagonist, this is not the full story. A classical competitive antagonist would not respond to a type II PAM, because they are simply competing for a binding site, but not inducing desensitization. Again, this is a key point because it is via the desensitized (and non-conductive state) that we believe anti-inflammatory signal transduction occurs. The archetype compound, NS6740, lacking in the ability to generate an α7 ion current, is an example of a silent agonist associated with anti-inflammatory activity; however, even the simple tetraethyl ammonium cation is a silent agonist for the α7 nAChR.12 We found that as the steric bulk around the quaternary ammonium group of simple α7 agonists was increased, agonism activity was lost but desensitization, as evidenced by robust responses when co-applied with type II PAM, was observed.18 We reported that the compound N, N-diethyl-N′-phenyl piperazine (diEPP) is also active as a silent agonist and it too followed the same trend regarding steric factors at the quaternary nitrogen.18 It should be noted that diMPP (N,N-dimethyl-N′-phenyl piperazine) is not a silent agonist, but is a known ganglionic-active agonist. Interestingly, the conversion to the N,N-diethyl homolog diEPP abolishes ganglionic activity and confers α7 nAChR selectivity. Thus the diEPP core structure lends itself well to explore the potential for further enhancement and control of silent agonist activity in this and closely related molecular structures.
In this study, we varied the structure of the basic diEPP framework in three ways. We explored how phenyl ring substituents affect activity, based on the identity of the functional groups and their position. We varied the nature of the linkage between the two rings, to test the essentiality of the piperazine N-aryl linkage, and we also utilized monoethyl tertiary amine analogs of diEPP to test if a positive charge at a quaternary nitrogen was required for silent agonism, or if tertiary amines, which are capable of being charged in their ammonium form, would suffice.
2. Materials and Methods
The Supplemental section contains extensive details regarding compound syntheses and characterization. Briefly, N-ethyl-N'-aryl piperazines were assembled by Ullman-type coupling of N-ethyl piperazine with iodo- or bromo-substituted benzenes, followed by alkylation as outlined in Schemes 1 and 2. Final compounds were purified by a combination of chromatography and recrystallization, and obtained in greater than 95% purity and fully characterized by NMR and mass spectrometric methods.
Scheme 1. Synthesis of diEPP Derivatives.
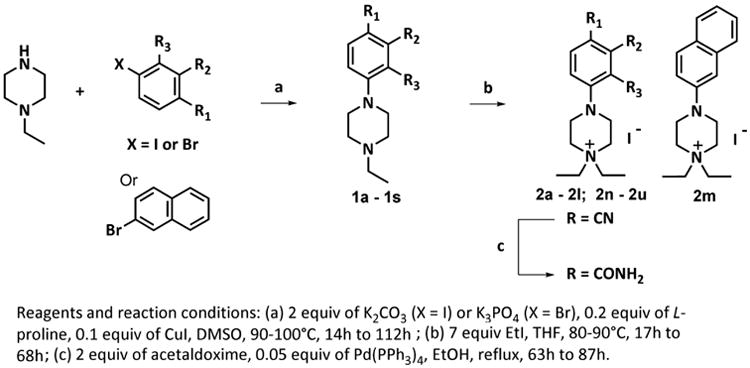
Synthetic approach to compounds 1a-s and 2a-u.
Scheme 2. Synthesis of diEPP Analogues.
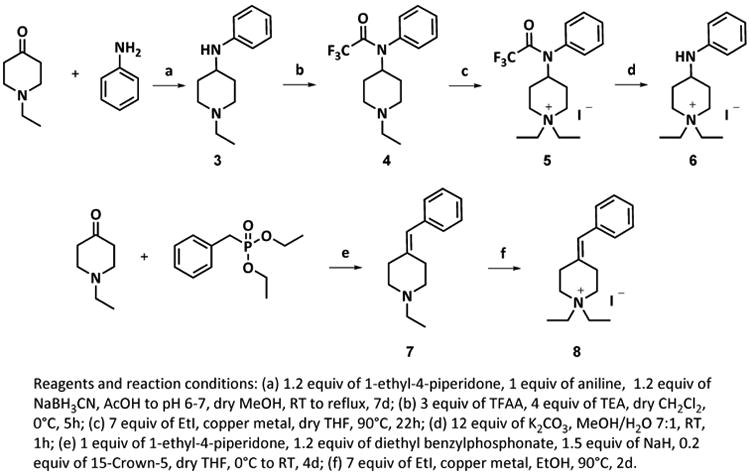
Synthetic approach to compounds 6 and 8.
2.1 Electrophysiology
New compounds were assayed for activity with the human α7 nAChR expressed in Xenopus oocytes using two-electrode voltage clamping as previously described18 and compared to responses evoked by 60 μM ACh. The compound set was assayed at a concentration of 30 μM to provide a standard comparison benchmark. Based on earlier experience this concentration is high enough to provide an observable response and low enough to avoid possible complications such as channel block. The net-charge response for each compound application is reported relative to those for ACh control applications. Data were expressed as the mean ± S.E.M. from at least four oocytes for each experiment and were plotted by Kaleidagraph (Abelbeck Software, Reading, PA).
Multi-cell averages were also calculated for comparisons of responses. The averages of normalized data were calculated using an Excel (Microsoft) template for each of the 10,500 points in each of the 210 s traces (acquired at 50 Hz). Following subtraction of the basal holding current, data from each cell, including the ACh controls, were normalized by dividing each point by the peak of the ACh control from the same cell. The normalized data were then averaged and standard errors of the mean (SEM) for the multi-cell averages calculated on a point-by-point basis. The dark lines in the plots represent the average normalized currents and the shaded areas the range of the SEM.
3. Results and Discussion
3.1 Compound syntheses
The general approach used for the synthesis of diEPP derivatives 2 is depicted in Scheme 1 and proceeds in two steps. The key step in the synthesis is a copper-catalyzed Ullmann-type aryl amination19 promoted and accelerated by the α-amino acid L-proline. Even though N-methylglycine was reported more effective than L-proline, it was also more reactive toward coupling with aryl halides, so L-proline was selected as the ligand.19 The catalytic coupling allowed us to avoid typical drawbacks such as stoichiometric amounts of copper reagents and the high costs for palladium catalysts and their phosphine ligands.20 We found that both electron-rich and electron-deficient aryl halides were successfully coupled with 1-ethylpiperazine at 90 °C in DMSO using 10 mol % CuI and 20 mol % L-proline as the catalytic system.20 Generally, the coupling reactions proceeded smoothly, but the range of yields of this key step was quite wide (from 5 to 83%), and some of our observations follow. The coupling reaction was found to be inhibited by ortho-substituents (compounds 1h and 1j), presumably due to steric interference within the intermediate addition and eliminations in the copper complex; moreover, aryl iodides gave better yields (52-83%) than aryl bromides (20-64 %) discounting the ortho-substituted halides. Though aryl bromides were usually less costly, aryl iodides were therefore first choice whenever possible. Once obtained, the N-phenyl-N′-ethyl piperazines 1 were then converted into the quaternary ammonium salts 2 by reacting them with ethyl iodide in tetrahydrofuran and then purified by chromatography and/or recrystallization to afford the final compounds as pure crystalline products (yields ranged from 15-76%). In a few cases, the crystallization resulted in formation of co-crystals between the desired compound and the crystallization solvent, so that solvent was found to be present in the final compound even after prolonged evaporation under vacuum. In two cases, we were unable to recrystallize compounds 2n (p-trifluoromethyl) and 2s (meta-fluoro) despite a broad survey of different solvent systems.
The para- and meta- carboxamides 2t and 2u (Scheme 1) were readily obtained by hydration of the corresponding nitrile diEPP derivatives (2b and 2i) by reacting them with acetaldoxime in the presence of tetrakis Pd(0) as catalyst. In this reaction, acetaldoxime acts as an efficient water donor for delivery to the nitrile. That, together with its commercial availability at low price, and the easy separation of product from the side products (acetonitrile and acetamide), made acetaldoxime our reagent of choice for this transformation. The synthetic routes to diEPP analogues 6 and 8 are shown in Scheme 2. Compound 6, 1,1-diethyl-4-(phenylamino)piperidinium iodide, was synthesized by N-alkylation of the protected reductive amination product of 1-ethyl-4-piperidone with aniline (Scheme 2). The reductive amination proceeded in fair yield (69 %) as a one-pot reaction in dry methanol/AcOH using sodium cyanoborohydride as reducing agent to afford 3. Complete consumption of the starting materials was not obtained, even after having heated the reaction to reflux. Attempts to optimize the reaction via isolation of the imine intermediate, or use of different anhydrous solvents (CH2Cl2, toluene, THF), and water-scavenging agents like MgSO4, with or without acetic acid addition to the reaction mixture, were unsuccessful. Compound 3 was then protected at the aromatic amino group by reaction with trifluoroacetic anhydride in triethylamine in dry dichloromethane at 0 °C. Compound 4 was reacted with iodoethane in THF dry to afford the quaternary ammonium corresponding derivative 5, which was then refluxed in a mixture of methanol-water in the presence of potassium carbonate to cleave the trifluoroacetyl protecting group to produce 6.
The 4-benzylidine substituted piperidinium salt 8 was obtained after a two-step synthesis. The key step utilized Wittig-Horner reaction of 1-ethyl-4-piperidone and diethyl benzylphosphonate, both commercially available, in the presence of sodium hydride and [15-crown-5] to provide the alkene intermediate 7 in 45 % yield. We found that use of the crown ether in the reaction is important to accelerate the reaction; because omission of the crown ether yielded an incomplete reaction after 3 days. After purification, the olefinated product was ethylated to the corresponding quaternary ammonium salt (8) by reacting it with iodoethane in dry THF, affording, after careful silica chromatography and recrystallization, the desired final product in 3 % yield. We attribute this poor yield to the difficulty in removing an unidentified impurity that, even if present in small amount, co-eluted with the desired product.
3.2 Electrophysiology-General observations
The panel of compounds synthesized was assayed using human α7 nAChR expressed in Xenopus oocytes and two-electrode voltage clamping. The profile of compound activity is presented in Table 1. We evaluated the partial agonism activity, quantified as net-charge2 for 30 μM applications of compounds, measured relative to the control response to applications of 60 μM ACh. The induction of PAM-sensitive desensitization was detected as the net-charge responses when compounds were co-applied with the PAM PNU-120596 (30 μM compound + 10 μM PNU-120596), measured relative to responses to 60 μM ACh applied alone. It is evident that the diEPP series of compounds 2 (structures Figure 1) exhibit an exceptional sensitivity to the nature and position of the aryl substituent, as evidenced by the broad range of activities against the α7 nAChR, which included partial agonism, varying degrees of silent agonism, and compounds that were effectively inactive, with no agonism and very weak silent agonism.
Table 1. Tabulated Responses.
| Compound | Relative response* | Potentiated response* |
|---|---|---|
| diEPP | 0.002 ± 0.003 | 1.3 ± 0.3 |
| 1f, m-Chloro PEP | 0.009 ± 0.008 | 0.1 ± 0.1 |
| 1n, p-CF3 PEP | 0.000 ± 0.004 | 1.4 ± 0.7 |
| 1p, m-Bromo PEP | 0.000 ± 0.003 | 6.9 ± 2.4 |
| 1r, p-fluoro PEP | 0.000 ± 0.003 | 0.0 ± 0.0 |
| 2a, p-methyl | 0.011 ± 0.003 | 3.6 ± 2.0 |
| 2b, p-cyano | 0.234 ± 0.024 | 68.0 ± 25.4 |
| 2c, p-methoxy | 0.063 ± 0.014 | 11.6 ± 0.3 |
| 2d, m-methyl | 0.001 ± 0.000 | 0.2 ± 0.1 |
| 2e, p-chloro | 0.234 ± 0.048 | 18.3 ± 6.2 |
| 2f, m-chloro | 0.020 ± 0.003 | 4.2 ± 1.0 |
| 2g, m-methoxy | 0.011 ± 0.011 | 0.6 ± 0.2 |
| 2h, o-methyl | 0.319 ± 0.066 | 16.4 ± 2.4 |
| 2i, m-cyano | 0.022 ± 0.024 | 1.5 ± 0.7 |
| 2j, o-chloro | 0.367 ± 0.054 | 36.6 ± 6.1 |
| 2k, m-OH | 0.283 ± 0.013 | 31.4 ± 5.1 |
| 2l, p,o-dimethoxy | 0.014 ± 0.009 | 1.1 ± 0.1 |
| 2m, 2-naphthalene | 0.143 ± 0.018 | 15.9 ± 2.3 |
| 2n, p-CF3 | 0.032 ± 0.003 | 61.8 ± 7.7 |
| 2o, m-CF3 | 0.000 ± 0.002 | 3.1 ± 2.1 |
| 2p, m-bromo | 0.040 ± 0.006 | 10.0 ± 1.7 |
| 2q, p-bromo | 0.270 ± 0.017 | 12.3 ± 3.5 |
| 2r, p-fluoro | 0.010 ± 0.002 | 22.9 ± 5.8 |
| 2s, m-fluoro | 0.061 ± 0.089 | 0.2 ± 0.1 |
| 2t, p-CONH2 | 0.065 ± 0.009 | 43.9 ± 8.5 |
| 2u, m-CONH2 | 0.056 ± 0.022 | 50.7 ± 8.5 |
| 6, diEPP analogue NH | 0.002 ± 0.004 | 5.6 ± 2.1 |
| 8, diEPP analogue | 0.071 ± 0.027 | 9.9 ± 3.1 |
Values are the mean ± SEM for N ≥ 4 experiments. All data are relative to the response of the receptor to 60 μM control applications of ACh. “Relative response” refers to receptor response to a 30 μM application of test compound. Potentiated response refers to receptor response with 30 μM test compound and 10 μM PNU-120596 co-application. Refer to experimental section for details.
Figure 1.

The structures of PEP compounds 1a-1s, diEPP compounds 2a-2u and analogs discussed in this study.
3.2.1 Partial agonists
Figure 2 presents a summary of compounds, o-chloro(2j), o-methyl(2h), m-hydroxy(2k), naphthalene(2m), p-chloro(2e) and p-cyano(2b) (Figure 2A), that were classified as partial agonists based on their ability to stimulate a net-charge response that was greater than a threshold value of 1/10th the ACh control, when the compounds were applied alone (Figure 2B). It is a unique property of α7 orthosteric agonist-induced currents that they vary systematically in the relationship between peak current and net charge as a function of the effective concentration applied.2, 3 This is due to the concentration-dependent desensitization of the receptors, which is likely to be associated with high levels of agonist binding site occupancy21 and is observed even when agonists are rapidly applied to small cells22 or acutely dissociated neurons.23 The control ACh concentration used for these experiments is roughly the EC80 for the net-charge responses (not shown). By normalizing both the peak currents and net-charge measurements of the drug responses at the probe concentration of 30 μM (Figure 2B) to those of the ACh control, we can estimate the relationship between the probe concentration and the EC80 for each of the drugs.24 At a concentration where the ratio of the normalized measures equaled one, the drug would be at the same effective concentration as 60 μM ACh. As shown in Table 2, the peak current to net charge ratio for each of the experimental drugs at 30 μM was less than one. Responses to 2m and 2e had the highest peak currents to net charge ratios, indicating that they are the most potent of the drugs tested. The estimated rank potency for the other compounds is given in Table 2.
Figure 2.
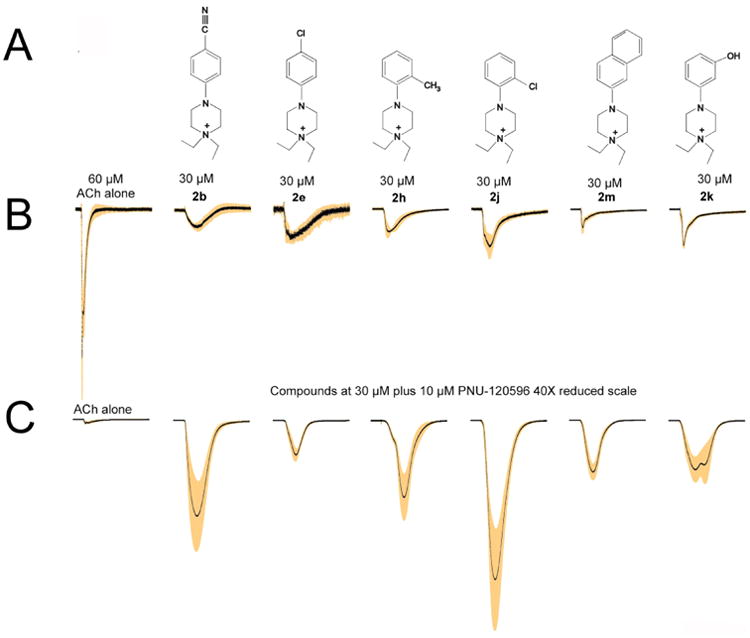
A) The structures of six test compounds that were classified as partial agonists. B) The responses of oocytes expressing α7 to the application of the compounds at the probe concentration of 30 μM. In each trace, the black line is the average of the normalized response of at least four cells, and the shaded areas are the range of the standard error of the mean for each point in the averaged traces. The data were obtained at 50 Hz and the traces are each 55 seconds in duration (2750 points in each trace). Two control responses to 60 μM ACh were first obtained from each cell for purposes of normalization. The drug-evoked responses were scaled in amplitude to the average of the two ACh-evoked control responses. A representative set of averaged ACh responses are shown on the left to provide scale. Those shown are the ACh responses obtained prior to the application of 30 μM p-cyano. Note that the drug-evoked responses vary in amplitude and also in regard to the ratio of peak currents to net charge (area) calculated relative to the peak and net charge of the ACh controls (Table 2). This ratio is indicative of the drug's potency, i.e. the relationship between the test concentration and EC50. 24 C) Responses of oocytes expressing α7 to the application of the compounds at the probe concentration of 30 μM co-applied with 10 μM PNU-120596. Data were normalized and averaged as described for panel B. For comparison and scale, shown on the left is the averaged ACh response obtained prior to the application of 30 μM p-cyano plus 10 μM PNU-120596. All of the traces in panel C are reduced 40-fold relative to those in panel B.
Table 2. Partial agonist properties.
| Estimated Rank | ||||||
|---|---|---|---|---|---|---|
| Compound | Peak currenta | Net Chargea | Ratio | potency | efficacy | PNU |
| responsea | ||||||
| o-chloro(2j) | 0.23 ± 0.03 | 0.37 ± 0.05 | 0.63 | 4 | 2 | 37 ± 6 |
| o-methyl(2h) | 0.15 ± 0.02 | 0.32 ± 0.07 | 0.46 | 5 | 1 | 16 ± 2 |
| m-hydroxy(2k) | 0.19 ± 0.02 | 0.28 ± 0.02 | 0.67 | 3 | 4 | 31 ± 5 |
| naphthalene(2m) | 0.10 ± 0.02 | 0.14 ± 0.02 | 0.71 | 1 | 6 | 16 ± 2 |
| p-chloro(2e) | 0.17 ± 0.02 | 0.23 ± 0.05 | 0.71 | 1 | 5 | 18 ± 6 |
| p-cyano(2b) | 0.10 ± 0.01 | 0.23 ± 0.02 | 0.43 | 6 | 3 | 68 ± 25 |
Relative to 60 μM ACh controls (n ≥ 4)
Potency and efficacy can, of course, vary independently. Compounds that are likely to be relatively potent (based on peak-current to net-charge ratios) but produced relatively small net-charge responses at the test concentration are likely to be less efficacious than less potent compounds such as 2h and 2j, which produced larger responses at the test concentration. Based on these considerations, estimated rank efficacies are also provided in Table 2. Of the partial agonists, the ortho-substituted ones showed the highest responses, whether non-polar (Me) or polar (Cl). It is evident that, while these compounds were weak as agonists, their response to application with PNU-120596 was quite strong (Figure 2C).
3.2.2 Silent agonists
Several substitutions yielded compounds that were candidate silent agonists with little or no partial agonism (i.e. above our detection threshold, but too small for accurate characterization), comparable to, or perhaps slightly greater than the unsubstituted parent diEPP compound (Table 1, and Figure 3): m-trifluoromethyl (2o), p-methyl (2a), m-chloro(2f), m-bromo(2p), p-methoxy(2c), p-fluoro(2r), p- and m-carboxamide (2t, 2u), and p-trifluoromethyl(2n). However, four compounds showed no significant activity as either partial agonists or as silent agonists at the concentration tested: m-methoxy(2g), m-methyl(2d), m-fluoro(2s) and m-cyano(2i) derivatives. We also prepared two analogs of diEPP (Scheme 2) in which the N-aryl linkage was modified to test the importance of this position. One, compound 6, replaced the piperazine ring with a piperidine ring linked to an aniline group; the other completely eliminated the aniline nitrogen, replacing the aniline group with a benzylidine residue, compound 8 (Figure 1). Compounds 6 and 8 both showed enhanced PAM-dependent responses relative to the parent diEPP compound (Figure 3C), suggesting that the nitrogen atom of the N-aryl bond is not essential for silent agonist activity within the diEPP framework.
Figure 3.

Activity of diEPP compounds and analogs with the α7 nAChR. The left vertical axis refers to the net-charge response of compounds when compounds (30 μM) were co-applied with 10 μM PNU-120596, relative to ACh controls. Experimental values are the average of at least 4 independent measurements, and the error bars reflect the calculated standard deviation of the mean. In all cases these compounds showed less than 10% of the control ACh response when they were applied alone to the receptor. For reference, all figures include the reference response of diEPP. A) Data for the meta-substituted compounds 2. B) Data for the para substituted compounds 2. C) Data for selected compounds 1r,f,n,p (left side) and compounds 6 and 8 (right side).
For some quaternary ammonium diEPP derivatives shown to be silent agonists, we also investigated the activity of the corresponding tertiary amine, i.e. select ethyl phenyl piperazine derivatives, Figure 1. Specifically, we examined the meta-bromo, meta-chloro, para-trifluoromethyl and para-fluoro compounds (1f, 1p, 1n, 1r). Interestingly, the tertiary amine 1p (meta-bromo) showed an approximately 5-fold enhanced response relative to diEPP and was only reduced by 0.69-fold relative to its corresponding quaternary salt 2p. This result indicates the quaternary ammonium positive charge is not an absolute requirement for silent agonist activity.
With regard to receptor subtype specificity, none of the compounds tested were partial agonists for α3β4, which is the heteromeric nAChR with a pharmacophore closest to that of α7.25 Only one compound (2g) showed significant antagonism at 100 μM (data not shown). Select compounds (2p, 2n) were also screened against α4β2 and found neither to be partial agonists nor to have significant antagonist activity at 100 μM (data not shown). Historically, compounds in this structural family, as exemplified by diMPP have been developed in the context of their function as agonists for a variety of targets.26-30 The emphasis on agonism in prior work underscores the necessity of determining what is required to make a silent agonist, silent and desensitizing. We find that diEPP compounds show clear selectivity towards the α7 receptor, and some are capable of placing it into a desensitized state. As mentioned earlier, α7 desensitizers are strongly associated with anti-inflammatory activity making these new investigations into how to control desensitization an important area of inquiry.
3.3 Aryl substitution patterns control state selectivity
Hypothetically, an optimized silent agonist, by definition, would be a compound that produced no detectable channel activation, and would exclusively stabilize the receptor into a desensitized state or states that may mediate important forms of signal transduction.12, 31 In our experimental paradigm, we can detect desensitized states (Ds) that are sensitive to, and become conductive when the receptor is treated with a PAM. With this in mind, when we discuss a compound as being a good silent agonist, this implies that it produces a robust response when co-applied with PNU-120596, and shows little (if any) ability to activate the receptor on its own. With regard to discerning a pharmacophore for silent agonists, it must be noted that some substituents could have a greater effect on the diminution of agonism, or others might have a greater effect on induction and stabilization of desensitization. In some cases these two effects may work together as is evident by the data shown in Figure 2, where, relative to the parent compound diEPP, some substituents were able to facilitate both a conductive state and a non-conductive Ds state.
Starting from diEPP as the parent compound, through different modifications of the aryl ring, we were able to obtain several derivatives with enhanced silent agonism profiles (defined as large PAM-dependent currents without increased orthosteric agonism). Among the set of diEPP derivatives generated, the best were meta or para substituted, with small-to-medium size substituent groups, in particular the para-trifluoromethyl, para-fluoro, para- and meta-carboxamide derivatives 2n, 2r, 2t, and 2u. We initially asked the question as to whether a single common property of these functional groups might serve to explain their ability to enhance silent agonism. Fluorine atoms, the trifluoromethyl group, and carboxamides can be quite different in the nature of their interactions with protein binding sites, but in our case they all had enhanced silent agonism behavior compared to the parent compound diEPP, more so than other substituents. In order to interpret this observation, we considered different atomic properties and atomic interactions. Taking into account the polarity of the group as a primary feature, it is ascribable to fluorine atoms in the first two cases and to the oxygen/amino groups in the latter two. However, not all compounds containing polar groups showed great silent agonism; see for example the methoxy and hydroxy derivatives (Table 1), so that polarity on its own is insufficient to explain in a simple way the activity of the best silent agonists.
In some cases, fluorine and the trifluoromethyl groups have been considered to enhance lipophilicity,32 and if that is the case in our study, introduction of lipophilic substituents such as the methyl group might be expected to improve silent agonism compared to the parent compound diEPP. However, para-methyl and meta-methyl derivatives both failed to effectively induce the Ds state, which supports the idea that polar interactions are operative. Although hydrogen bonds are well-known to be involved in a myriad of protein-ligand interactions, fluorine rarely participates as a hydrogen-bond acceptor and it would be a weak interaction,33 however, carboxamides are known to be good H-bond acceptors. So if hydrogen bonding was the key interaction behind the silent agonism improvement of those selective compounds compared to diEPP, carboxamide derivatives would show a much greater improvement than the CF3- and F-derivatives, but this was not observed. We conclude that the basis for enhanced silent agonism of fluoro, trifluoromethyl, and carboxamide residues may be multifactorial but share in common their ability to stabilize an overall conformational state of the receptor that is desensitized yet sensitive to PNU-120596 through a variety of point-to-point interactions between the various compounds and elements in the silent agonist binding site, which we hypothesize to be an extension of the site where typical orthosteric agonists such as ACh bind.
Intriguing results were observed for the meta-substituted halogen-containing diEPP derivatives, which for the fluoro, chloro, and bromo derivatives showed increasing potentiated responses (Table 1). To describe these results, halogen bonding interactions were considered. The strength of the interaction increases in the order fluorine < chlorine < bromine < iodine.34, 35 In our meta-substituted derivatives, we observed an increase in the PAM-dependent currents (Figure 3a), moving from fluorine (2s; 0.2-fold relative to ACh) hydrogen (diEPP; 1.33-fold) to chlorine (2f; 4.23-fold) to bromine (2p; 9.98-fold), consistent with halogen-bonding interaction with a suitable electron-donor partner in the binding site of the receptor. Indeed, we would predict that electron-rich or electron-donating meta substituents might perform poorly as silent agonists, and this was the case. The meta-methoxy and meta-cyano diEPP (2g, i) derivatives, respectively showed 50% and equivalent PAM-dependent responses compared to the parent unsubstituted diEPP compound. Interestingly, the meta-hydroxy derivative, capable of acting as a hydrogen-bond donor, was found to be a partial agonist, suggesting that a unique hydrogen bond at the phenolic OH-group induces a conductive state of the receptor. Chlorine or bromine substitutions in the para or ortho positions do not yield silent agonists, since, in fact, para-chloro, para-bromo and ortho-chloro derivatives showed partial agonist activities with the α7 nAChR subtype (Table 1, Figure 2). However, in contrast with the trend highlighted among the para-halogen diEPPs, para-fluoro and para-trifluoromethyl diEPP are two of the most active compounds as silent agonists in the diEPP series. The explanation for the divergence in the activity of these para substituents must reside in the unique behavior of fluorine substituents, but this is a complex interplay of numerous effects 33, and in lieu of a high-resolution structure, becomes a speculative endeavor to determine its origin.
Both ortho-chloro and ortho-methyl derivatives are partial agonists of the α7 receptor, and these two substituents have similar Connolly-excluded molecular volumes (14.3 Å3 versus 16.9 Å3), suggesting that a steric directing effect of an ortho-substituent may facilitate entry of the receptor into a conductive state. Yet, the o,p-dimethoxy analogue 2l is not a partial agonist, suggesting that the para-substituent may supersede the putative agonism-promoting effect of an ortho substituent. The naphthalene derivative (2m) was intended to investigate the tolerance of the binding pocket of the α7 receptor towards more bulky groups, and it became a partial agonist compared to the parent diEPP. These data, compared with the activity of ortho substituted compounds, suggest that the extended point-to-point interactions between the larger naphthalene ligand and receptor are yet another way to induce conductive states of the receptor in addition to the internal conformational biasing or ortho substituents. We have previously reported on the relationship between the steric bulk around the core ammonium group as a way to convert partial agonists into silent agonists.18 In the series we describe here, substitutions of the aromatic ring are remote from the core ammonium group and do not appear to follow a simple correlation of substituent bulk with silent agonism. Thus, aromatic substituents on diEPP compounds appear to be modulating silent agonism in a different way than simple ammonium compounds do.
3.4 Modifications at the core piperazine nitrogen atoms
As part of our work to dissect structural features within the diEPP silent agonist pharmacophore, we wished to test the importance of the nitrogen in the piperazine ring that was attached to the phenyl group and if small modifications at this point in the molecule might enhance silent agonism. Both compounds 6 and 8 were enhanced in terms of their responses when co-applied with PNU-120596, though compound 6 was superior to compound 8 from the point of view of the ratio of the amount of desensitization to residual partial agonism. While we were able to measure currents on application of 8 to α7, application of 6 to α7 resulted in no channel activation within experimental error. Compound 6 thus may serve as a suitable framework for development of cleaner, more state-selective silent agonists.
The phenyl ethyl piperazine (PEP) compounds 1f, 1n, 1p, and 1r were selected for testing because they correspond to diethylammonium compounds 2f, 2n, 2p, and 2r that had significant ability to enter Ds as evidenced by strong PAM co-application responses. The question we asked was if the positively charged quaternary ammonium functionality present in active compounds 2f, 2n, 2p, and 2r was required for silent agonism (Figure 3C). We found that, for the most part, these compounds were inactive, with no partial agonism and weak responses to PAM co-application. The exception was meta-bromo PEP 1p, which had a response nearly indistinguishable from the diethyl version 2p (6.9 ± 2.4 vs 10.0 ± 1.7, Table 1). This result provides another indication that the core minimal ammonium pharmacophore observed for simple ammonium compounds may not be required if suitable structural features remote from the core charged nitrogen are present. This observation is of particular importance for the potential therapeutic development of silent agonists that will be more likely to cross the blood-brain barrier. However, the generally greater activity of the quaternary amines can be exploited for specific indications that would not require brain penetration, such as the targeting of peripheral immune cells for anti-inflammatory activity.
A critical question that will extend the present work speaks to the mechanism by which a bound ligand is able to facilitate entry into a desensitized state or states. Available data indicate that silent agonists do not precisely place the receptor into a single desensitized state.13 Rather, the receptor silent agonist complex is in a highly dynamic series of states; depending on occupancy levels and application time, the complex can enter PAM-insensitive desensitized states (Di), which may evolve over time into PAM-sensitive desensitized states (Ds) and/or eventually dissociate from the receptor.12 Based on the results described here, it is entirely reasonable that a number of different amino acid side chains in the binding site may be utilized as interacting partners for selective entry into a Ds state. Molecular docking studies suggested that multiple binding poses in the orthosteric agonist binding site of the receptor are possible (data not shown), which leads to a number of hypotheses about specific ligand receptor interactions of interest. Ongoing studies are aimed at the identification of these critical loci within the receptor.
4. Conclusions
The diEPP molecular framework has been an excellent platform to identify some enhanced silent agonists as well as to identify new molecular features that regulate different features associated with silent agonism. We have found the p-fluoro and p-trifluoromethyl, and p- and m-carboxamide groups to provide strongly enhanced entry of α7 into the Ds state relative to the parent unsubstituted compound diEPP, and do so with little activity as partial agonists. The limited number of ortho substitutions tested yielded partial agonists. When the para position was substituted with Cl, Br, or cyano, the compounds were partial agonists with strong responses to PAM co-application; however, methoxy, fluoro, carboxamide, and trifluoromethyl substituents had diminished partial agonism and strong PAM co-application responses. The meta position is distinguished from para by the fact that fewer substitutions lead to partial agonism (meta-hydroxyl compound 2k being a noteworthy exception). The PAM co-application responses were low for most meta-group compounds, with the bromo and carboxamide being standouts for their strong Ds response to PAM application. Interestingly, the nitrogen of the N-aryl linkage is not required for activity, as exemplified by compound 8, indicating the N-aryl functionality is not a critical determinant for stabilizing desensitization. Finally, while three of the four non-quaternary PEP compounds were inactive as silent agonists, the meta-bromo PEP 1p showed modest silent agonism with little diminution relative to its quaternary ammonium analog 2p, demonstrating that a permanent positive charge is not required for activity. These compounds could prove interesting for applications requiring blood brain barrier permeability.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Health grant [GM57481] and a fellowship from the University of Milan to MQ. OpusXpress experiments were conducted by Shehd Abdullah Abbas Al Rubaiy, Lu Wenchi Corrie, and Clare Stokes.
Dedication: This manuscript is dedicated to Professor Koji Nakanishi on the occasion of his 90th birthday.
Abbreviations
- ACh
acetylcholine
- AcOH
acetic acid
- DiEPP
N,N-diethyl-N′-phenylpiperazinium
- DiMPP
N,N-dimethyl-N′-phenylpiperazinium
- PAM
positive allosteric modulator
- PEP
N-phenyl-N′ethylpiperazine
- nAChR
nicotinic acetylcholine receptor
- TFAA
trifluoroacetic anhydride
- THF
tetrahydrofuran
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Papke RL. Bio Pharm. 2014;89:1. doi: 10.1016/j.bcp.2014.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Papke RL, Papke JKP. Br J of Pharm. 2002;137:49. [Google Scholar]
- 3.Papke RL, Thinschmidt JS. Neurosci Let. 1998;256:163. doi: 10.1016/s0304-3940(98)00786-1. [DOI] [PubMed] [Google Scholar]
- 4.Parrish WR, Rosas-Ballina M, Gallowitsch-Puerta M, Ochani M, Ochani K, Yang LH, et al. Mol Med. 2008;14:567. doi: 10.2119/2008-00079.Parrish. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosas-Ballina M, Goldstein RS, Gallowitsch-Puerta M, Yang L, Valdes-Ferrer SI, Patel NB, et al. Mol Med. 2009;15:195. doi: 10.2119/molmed.2009.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Wadei MH, Al-Wadei HA, Schuller HM. Mol Can Res. 2012;10:239. doi: 10.1158/1541-7786.MCR-11-0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matteoli G, Gomez-Pinilla PJ, Nemethova A, Di Giovangiulio M, Cailotto C, van Bree SH, et al. Gut. 2014;63:938. doi: 10.1136/gutjnl-2013-304676. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, et al. Nature. 2003;421:384. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 9.Tracey KJ. The J Clin Inv. 2007;117:289. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, et al. J Exp Med. 2005;201:1113. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomsen MS, Mikkelsen JD. J Neuroimm. 2012;251:65. doi: 10.1016/j.jneuroim.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Papke RL, Bagdas D, Kulkarni AR, Gould T, AlSharari S, Thakur GA, et al. NeuroPharm. 2015;91:34. doi: 10.1016/j.neuropharm.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams DK, Wang J, Papke RL. Mol Pharm. 2011;80:1013. doi: 10.1124/mol.111.074302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gronlien JH, Haakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalakrishnan M, et al. Mol Pharm. 2007;72:715. doi: 10.1124/mol.107.035410. [DOI] [PubMed] [Google Scholar]
- 15.Hurst RS, Hajos M, Raggenbass M, Wall TM, Higdon NR, Lawson JA, et al. J Neurosci. 2005;25:4396. doi: 10.1523/JNEUROSCI.5269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young GT, Zwart R, Walker AS, Sher E, Millar NS. Proc Natl Acad Sci U S A. 2008;105:14686. doi: 10.1073/pnas.0804372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bertrand D, Bertrand S, Cassar S, Gubbins E, Li J, Gopalakrishnan M. Mol Pharm. 2008;74:1407. doi: 10.1124/mol.107.042820. [DOI] [PubMed] [Google Scholar]
- 18.Papke RL, Chojnacka K, Horenstein NA. J of Pharm and Exp Ther. 2014;350:665. doi: 10.1124/jpet.114.215236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ma D, Cai Q, Zhang H. Org lett. 2003;5:2453. doi: 10.1021/ol0346584. [DOI] [PubMed] [Google Scholar]
- 20.Zhang H, Cai Q, Ma D. J of Org Chem. 2005;70:5164. doi: 10.1021/jo0504464. [DOI] [PubMed] [Google Scholar]
- 21.Williams DK, Stokes C, Horenstein NA, Papke RL. J Gen Physiol. 2011;137:369. doi: 10.1085/jgp.201010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams DK, Peng C, Kimbrell MR, Papke RL. Mol Pharm. 2012;82:746. doi: 10.1124/mol.112.080317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uteshev VV, Meyer EM, Papke RL. Br Res. 2002;948:33. doi: 10.1016/s0006-8993(02)02946-3. [DOI] [PubMed] [Google Scholar]
- 24.Papke RL. Life Sci. 2006;78:2812. doi: 10.1016/j.lfs.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 25.Horenstein NA, Leonik FM, Papke RL. Mol Pharm. 2008;74:1496. doi: 10.1124/mol.108.048892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blanchet MR, Israel-Assayag E, Daleau P, Beaulieu MJ, Cormier Y. Am J Physiol- Lung Cell Mol Physiol. 2006;291:L757. doi: 10.1152/ajplung.00409.2005. [DOI] [PubMed] [Google Scholar]
- 27.Assayag EI, Beaulieu MJ, Cormier Y. PLoS One. 2014;9:e86091. doi: 10.1371/journal.pone.0086091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alkondon M, Pereira EF, Cortes WS, Maelicke A, Albuquerque EX. Eur J Neurosci. 1997;9:2734. doi: 10.1111/j.1460-9568.1997.tb01702.x. [DOI] [PubMed] [Google Scholar]
- 29.Yost CS, Winegar BD. Cell Mol Neurobiol. 1997;17:35. doi: 10.1023/a:1026325020191. [DOI] [PubMed] [Google Scholar]
- 30.Kristufek D, Stocker E, Boehm S, Huck S. J Physiol. 1999;516(Pt 3):739. doi: 10.1111/j.1469-7793.1999.0739u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Maanen MA, Papke RL, Koopman FA, Koepke J, Bevaart L, Clark R, et al. PLoS One. 2015;10:1. doi: 10.1371/journal.pone.0116227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bohm HJ, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, et al. ChemBiochem. 2004;5:637. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 33.Zhou P, Zou J, Tian F, Shang Z. J Chem Inf Mod. 2009;49:2344. doi: 10.1021/ci9002393. [DOI] [PubMed] [Google Scholar]
- 34.Politzer P, Lane P, Concha MC, Ma Y, Murray JS. J Mol Mod. 2007;13:305. doi: 10.1007/s00894-006-0154-7. [DOI] [PubMed] [Google Scholar]
- 35.Lu Y, Wang Y, Zhu W. Phys Chem Chem Phys. 2010;12:4543. doi: 10.1039/b926326h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.